
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel asymmetrical azines appending 1,3,4-thiadiazole sulfonamide: synthesis, molecular structure analyses, in silico ADME, and cytotoxic effect†
Samir Bondock *ab,
Tallah Albarqia,
Ibrahim A. Shaabanac and
Moaz M. Abdoud
*ab,
Tallah Albarqia,
Ibrahim A. Shaabanac and
Moaz M. Abdoud
aChemistry Department, Faculty of Science, King Khalid University, 9004 Abha, Saudi Arabia
bChemistry Department, Faculty of Science, Mansoura University, 35516 Mansoura, Egypt. E-mail: bondock@mans.edu.eg
cDepartment of Chemistry, Faculty of Science (Men's Campus), Al-Azhar University, Nasr City 11884, Cairo, Egypt
dEgyptian Petroleum Research Institute, Nasr City, 11727, Cairo, Egypt
First published on 3rd April 2023
Abstract
Toward finding potential and novel anticancer agents, we designed and prepared novel differently substituted unsymmetrical azine-modified thiadiazole sulfonamide derivatives using the “combi-targeting approach”. An efficient procedure for synthesizing the designed compounds starts with 5-acetyl-3-N-(4-sulfamoylphenyl)-2-imino-1,3,4-thiadi-azoline 4. The E/Z configuration for compound 5 was investigated based on spectral analysis combined with quantum mechanical calculation applying the DFT-B3LYP method and 6-31G(d) basis set. The computational results found that the E isomer was energetically more favorable than the Z isomer by 2.21 kcal mol−1. Moreover, 1H and 13C chemical shifts for the E and Z isomers in DMSO were predicted using the GIAO-B3LYP/6-31G(d) computations and IEF-PCM solvation model. The computed chemical shifts for both isomers are consistent with those observed experimentally, indicating that they exist in the solution phase. Moreover, the E/Z configuration for the synthesized azines 7a–c, 9, 11, 13, 15a and 15b was also studied theoretically using the DFT-B3LYP/6-31G(d) calculations. In silico prediction for the biological activities was reported regarding the HOMO–LUMO energy gaps and molecular reactivity descriptors besides the ADMT/drug-likeness properties. The cytotoxic effect of the synthesized compounds has been assayed via the determination of their IC50.
1. Introduction
Current obstacles to treating cancer include the emergence of drug resistance and unfavorable off-target effects of anticancer drugs, which energizes medicinal chemists to continuously produce novel anticancer medications with high efficacy and low toxicity.1,2One of the most promising candidates in the field of synthetic drugs is sulphonamides (Fig. 1a).3–6 The thiadiazole platform constitutes intriguing and rapidly expanding sulfonamide derivative systems. Several medicines on the market are related to this system, making it a flexible tool for drug design (Fig. 1b).7,8 According to a literature survey, azines and derivatives serve as crucial structural components in various versatile scaffolds with a wide range of drug applications (Fig. 1c).9 Azines have recently gained attention for configurations, and tautomers that profoundly affect biochemical processes.10
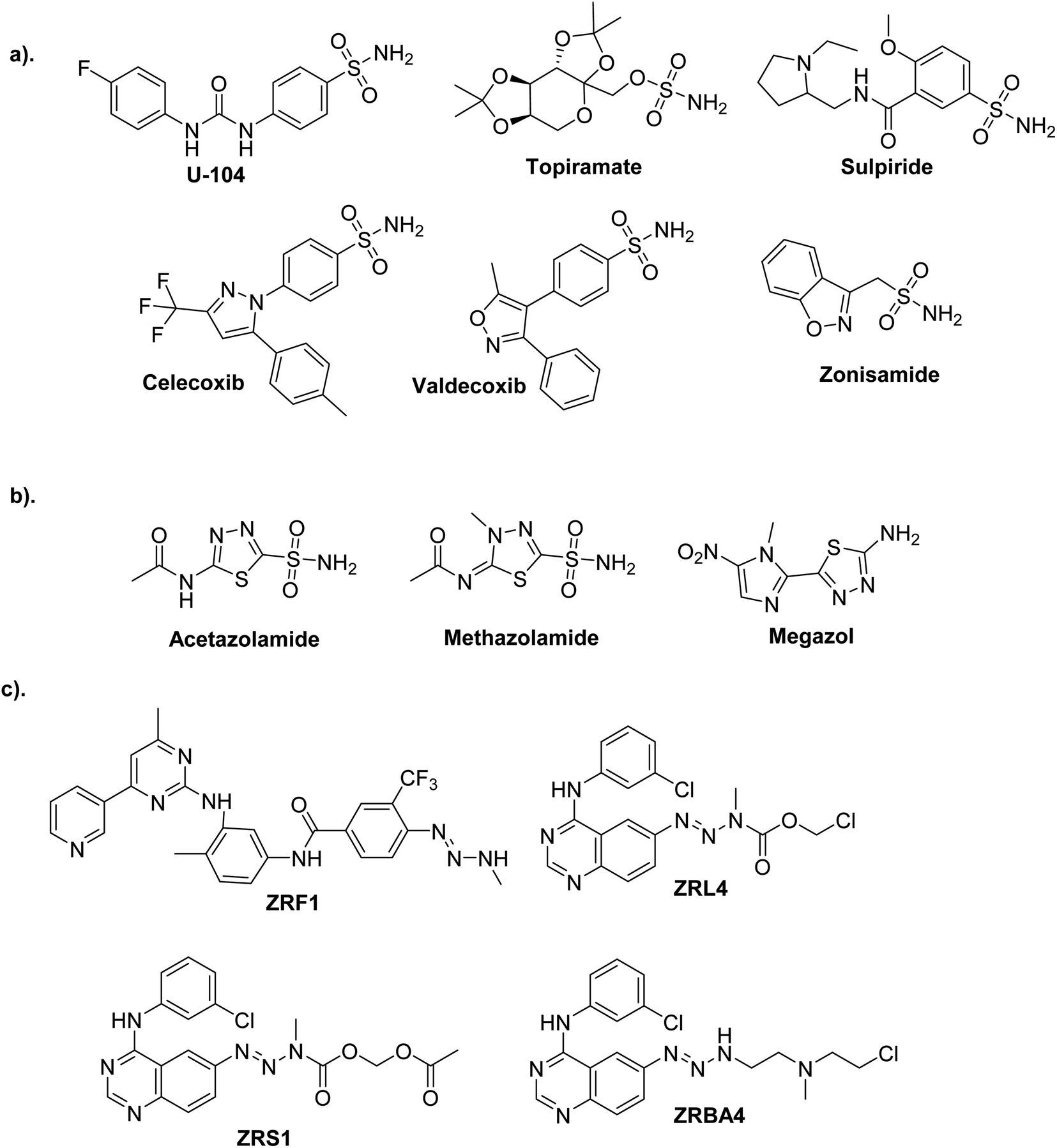 | ||
| Fig. 1 Commercial drugs on the market contain bioactive cores. (a) Sulfonamides. (b) Thiadiazole sulfonamides. (c) Asymmetrical azines. | ||
Recently, a vital approach/strategy in drug discovery is the amalgamation of two or more complete medications into a single molecular structure, known as a combi-molecule, which may be a good solution to achieve bioactive molecules, with high potency and different mechanisms of action, due to the synergistic effect.11 Hence, cohesive systems incorporating 1,3,4-thiadiazole sulfonamide with azines may help design new anticancer hybrids to improve biological properties.
To investigate the application of a combi-molecule strategy, we designed and prepared novel compounds 7a–c, 9, 11, 13 and 15a–b in this work by linking azines fragments and thiadiazole sulfonamides. These compounds were then subjected to cytotoxic assays on three cancerous cell lines. Their cytotoxic assay was comparable to the positive control staurosporine in the low micromolar region.
Moreover, we employ quantum mechanical (QM) computations to provide theoretical analyses for compound 5, including the E/Z configurations and conformational study toward free rotatable single bonds. Thus, QM calculations were reported using the density functional theory (DFT) at the level of B3LYP12 and 6-31G(d) basis set. Owing to its reliable accuracy and reduced computation time, the B3LYP/6-31G(d) calculation was widely recognized and applied for theoretical studies of organic molecules of medium-large size.13,14 The 1H and 13C NMR chemical shifts (δ, ppm) were also computed by applying the approach of gauge-invariant atomic orbitals, GIAOs,15 to investigate the E/Z configuration of 5 in the solution phase. In silico techniques have been widely applied to drug screening.16–19 Various computational tools and methods may be used to identify the candidate drug from other compounds depending on multiple features such as physicochemical/pharmacokinetic parameters and drug-likeness. Herein, QM calculations were carried out for the synthesized compounds to correlate their structures with biological activities via analysis of the FMOs and quantum chemical descriptors. To assess the synthesized compounds 7a–c, 9, 11, 13, 15a and 15b as drug candidates, the SwissADME20 and pkCSM21 servers were used to predict physicochemical characteristics, drug-likeness, and ADMET properties.
2. Results and discussion
2.1. Chemistry
To synthesize the target compounds 7a–c, 9, 11, 13, 15a and 15b the synthetic sequence starts with the preparation of 5-acetyl-3-N-(4-sulfamoylphenyl)-2-imino-1,3,4-thiadiazoline 4 as commencing material. 1,3,4-Thiadiazoline 4 was efficiently prepared via a cyclization reaction of freshly synthesized 2-oxo-N-(4-sulfamoylphenyl)propanehydrazonoyl chloride 3, obtained from the Japp–Klingemann reaction of 3-chloro-2,4-pentanedione with diazonium chloride of sulfanilamide 2 in a buffered ethanolic solution, with an aqueous ethanolic solution of ammonium thiocyanate under reflux (Scheme 1). Compound 4's structure was determined via microanalysis and spectral data. In the IR spectrum, three bands were observed at 3352, 3289, and 3277 cm−1, indicating the existence of primary and secondary N–H stretching vibration, respectively. The bands at 1692, 1330, and 1298 cm−1 indicated the existence of a carbonyl group (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and sulfonamide group (SO2NH2), respectively. The 1H-NMR spectrum displayed three singlet signals at δ 2.50, 7.45, and 9.62 ppm, characteristic for CH3, SONH2, and NH protons, respectively, as well as two doublet signals, resonating at δ 7.92 and 8.18 ppm with the identical coupling constant value (J = 9.35 Hz) and integrating for four protons indicating the existence of 4-disubstituted benzene. Its 13C-NMR spectrum showed the presence of eight signals which agrees with its molecular structure. The signals of CH3 and a carbonyl carbon resonate at 24.88 and 189.83 ppm, respectively. In the mass spectrum (MS), the molecular ion peak (M+) for 4 was found at m/z = 298, which is compatible with its molecular formula (C10H10N4O3S2).
O) and sulfonamide group (SO2NH2), respectively. The 1H-NMR spectrum displayed three singlet signals at δ 2.50, 7.45, and 9.62 ppm, characteristic for CH3, SONH2, and NH protons, respectively, as well as two doublet signals, resonating at δ 7.92 and 8.18 ppm with the identical coupling constant value (J = 9.35 Hz) and integrating for four protons indicating the existence of 4-disubstituted benzene. Its 13C-NMR spectrum showed the presence of eight signals which agrees with its molecular structure. The signals of CH3 and a carbonyl carbon resonate at 24.88 and 189.83 ppm, respectively. In the mass spectrum (MS), the molecular ion peak (M+) for 4 was found at m/z = 298, which is compatible with its molecular formula (C10H10N4O3S2).
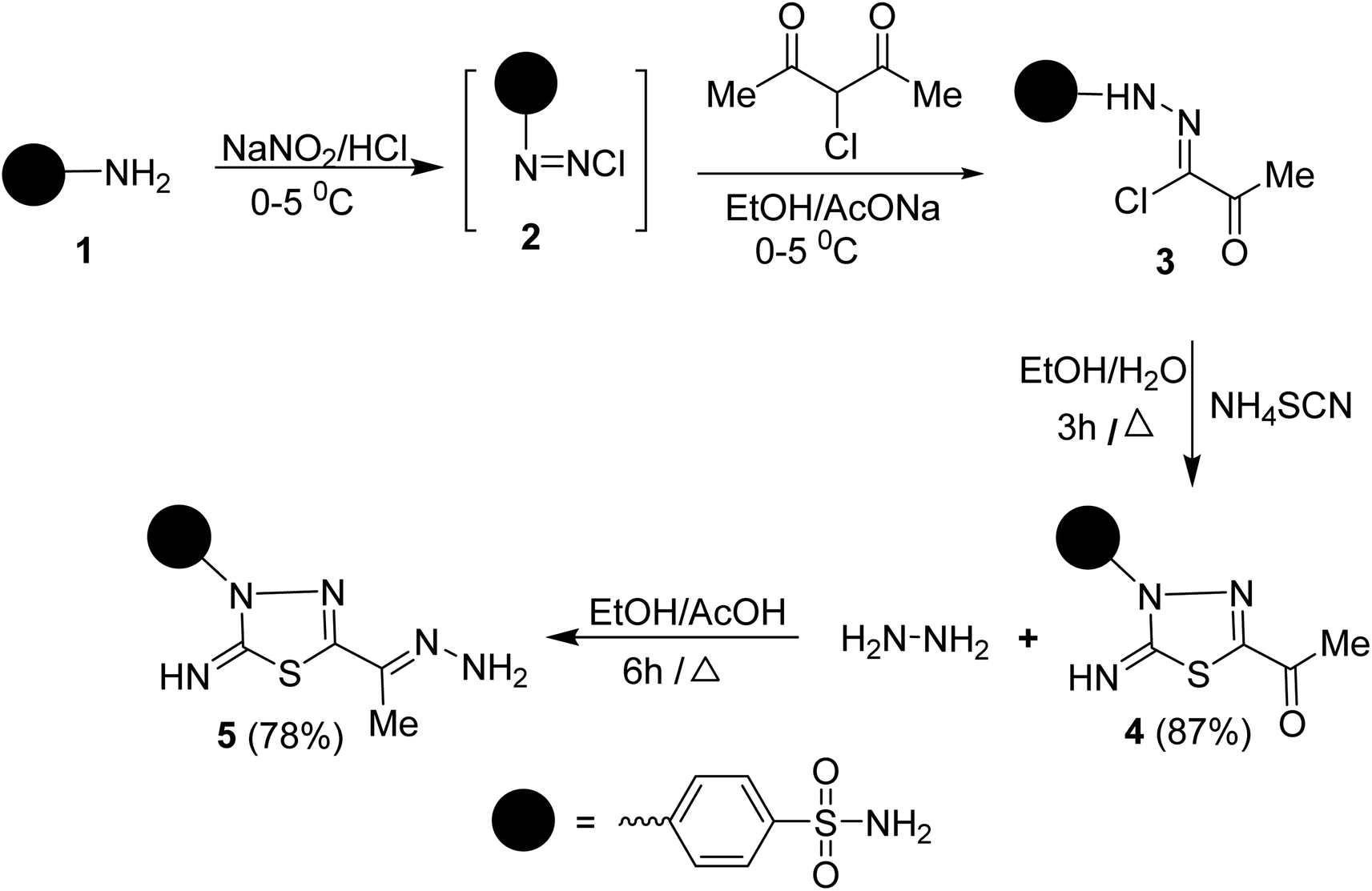 | ||
| Scheme 1 Synthesis of compound 5. | ||
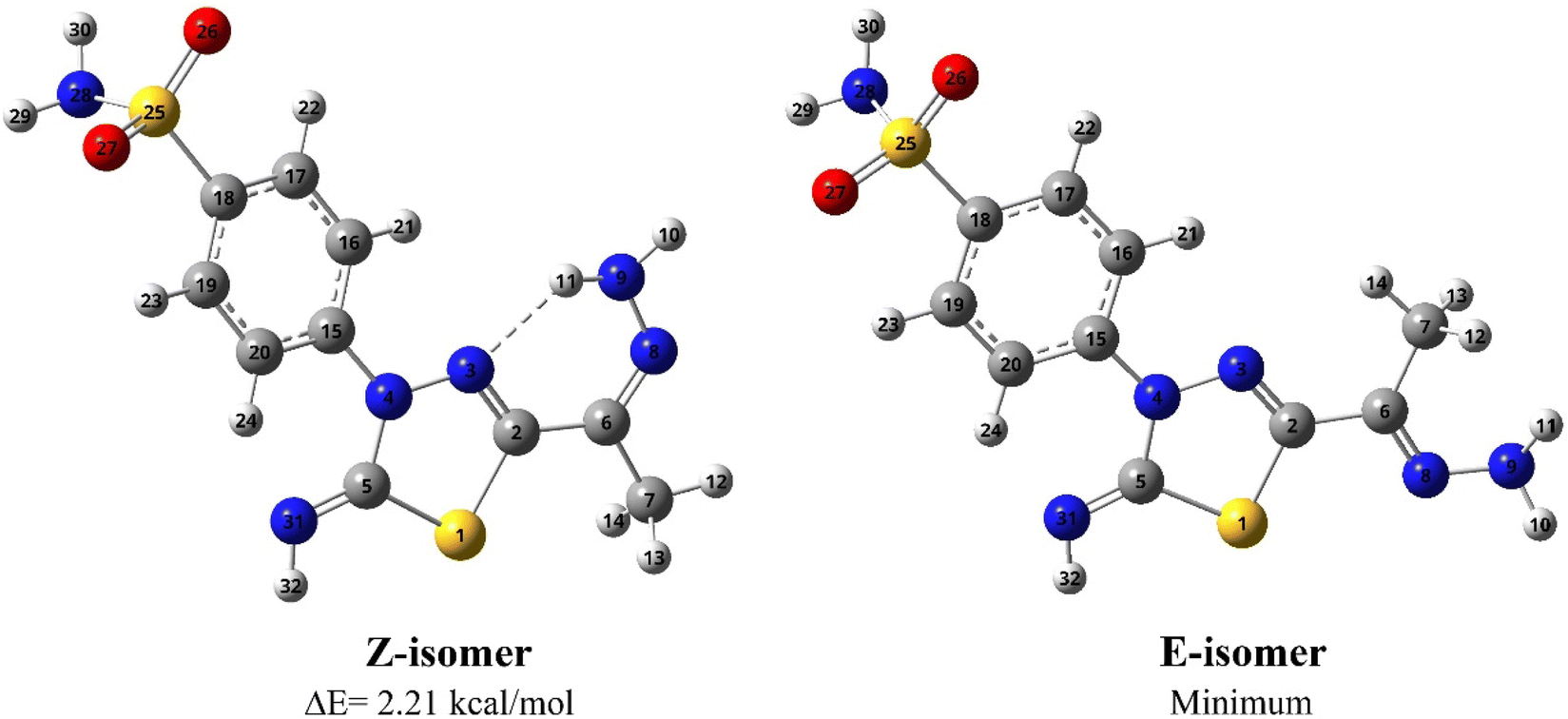
Condensation of 5-acetyl-3-N-(4-sulfamoylphenyl)-2-imino-1,3,4-thiadiazoline 4 with hydrazine afford the respective 4-(5-(1-hydrazonoethyl)-2-imino-1,3,4-thiadiazol-3(2H)-yl)benzenesulfonamide 5 (Scheme 1). The structure of later hydrazone 5 was confirmed through spectroscopic analyses. An examination of its IR spectrum revealed the lack of a carbonyl absorption band and the existence of azomethine (CN) and amino groups at wavenumbers 1645, 3416, and 3285 cm−1. Interestingly, the 1H-NMR spectrum of hydrazone 5 showed two sets of resonances that supported the presence of 5 in two isomeric forms. The separation of signals of aromatic, –NH2, and CH3 protons in the two isomers is well resolved. The population ratio of the major and minor isomers is (52![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :48). Based on previous studies, the most stable and the major isomer is assigned to the E-isomer around the CN bond.22 That is supported by the observed chemical shift value of the hydrazone-NH2 protons in E- and Z-isomers. In E-isomer, the hydrazone-NH2 protons appear as a singlet signal at δ 5.66 ppm. In comparison, Z-isomer resonates as two separate singlet signals at 7.33 and 7.32 ppm due to the possible formation of intramolecular H-bonds between N–H proton and CN group in the thiadiazole ring as shown in Fig. 2. According to MS, the M+ at m/z is the molecular weight.
:48). Based on previous studies, the most stable and the major isomer is assigned to the E-isomer around the CN bond.22 That is supported by the observed chemical shift value of the hydrazone-NH2 protons in E- and Z-isomers. In E-isomer, the hydrazone-NH2 protons appear as a singlet signal at δ 5.66 ppm. In comparison, Z-isomer resonates as two separate singlet signals at 7.33 and 7.32 ppm due to the possible formation of intramolecular H-bonds between N–H proton and CN group in the thiadiazole ring as shown in Fig. 2. According to MS, the M+ at m/z is the molecular weight.
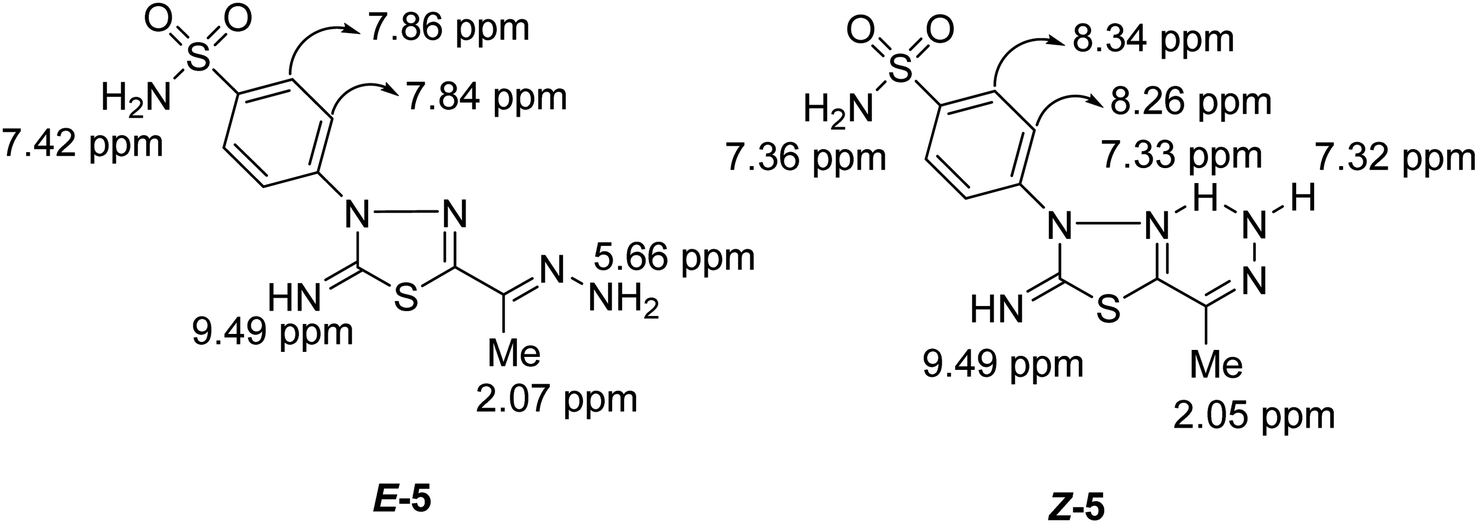 | ||
| Fig. 2 Protons chemical shift values of E- & Z-isomers of compound 5. | ||
To synthesize unsymmetrical azines, we investigated the reactivity of the terminal –NH2 group of hydrazone 5 towards aromatic aldehydes, heterocyclic aldehydes, and cyclic ketones (Scheme 2). Hence, condensation of compound 5 with aromatic aldehydes, namely, 4-methoxy benzaldehyde 6a, 4-(N,N-dimethylamino) benzaldehyde 6b, 4-nitrobenzaldehyde 6c and 2,4,6-trimethoxybenzaldehyde 8 in refluxing ethanol yielded unsymmetrical azines, 1-(4-substituted benzylidene)-2-(1-(2-imino-3-(4-sulfamoylphenyl)-1,3,4-thiadiazol-5-yl)ethylidene) hydrazine 7a–c and 1-(2,4,6-trimethoxy benzylidene)-2-(1-(2-imino-3-(4-sulfamoyl phenyl)-1,3,4-thiadiazol-5-yl)ethylidene)hydrazine 9, respectively (Scheme 2). The molecular structures of the later azines were secured from microanalyses and spectral data. As a representative example, the IR spectrum of azine 7a displayed a strong absorption band at 3360, 3255, 3110, and 1604 cm−1 due to NH2, NH, and CN functions, respectively. The 1H-NMR spectrum of 7a exhibited five characteristic singlet signals at δ 2.49, 3.84, 7.49, 8.63, 9.42 ppm corresponding to CH3, CH3O, NH2, azomethine proton (CHN), and imine (CNH) protons, respectively. Moreover, the 13C-NMR spectrum of 7a displayed fourteen carbon signals. Four characteristic peaks for four azomethine carbons showed at δ 161.91 (CHN), 162.74 (CN), 162.89 (thiadiazole-C5), and 168.75 (thiadiazole-C2) ppm. The signals at δ 13.46 and 55.85 ppm were attributed to methyl and methoxy carbons, respectively.
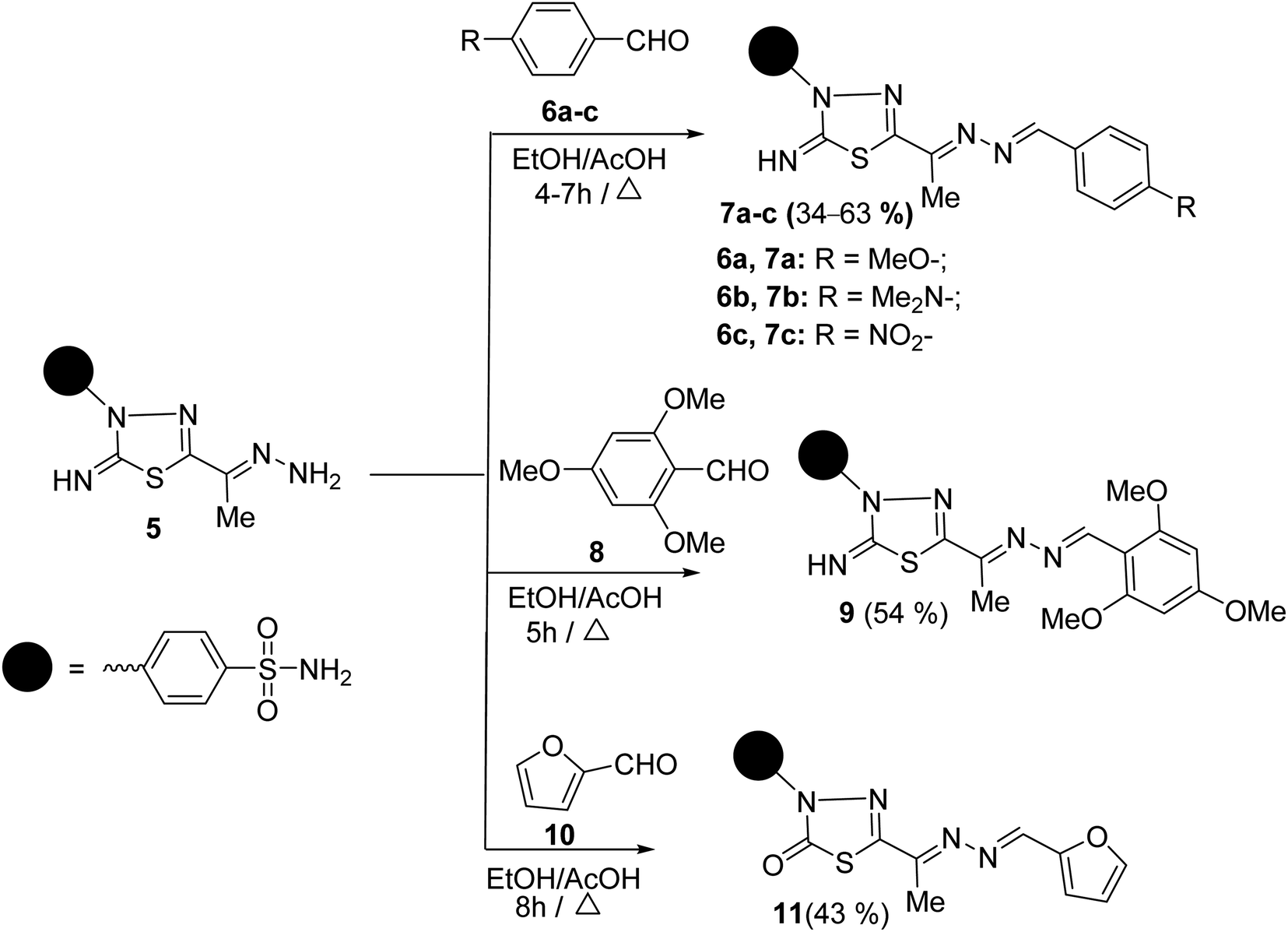 | ||
| Scheme 2 The synthesis of unsymmetrical azines 7a–c, 9, 11. | ||
A M+ at m/z 430 is also revealed in the mass spectra of 7a, which corresponds to its molecular formula (C18H18N6O3S2). The mass fragmentation pattern of compound 7a (Scheme 1 at ESI†). The M+ undergoes N–N bond cleavages to give a cation ion peak at m/z = 134 a.m.u. followed by loss of hydrogen atom to afford ion peak at m/z = 133 a.m.u. Due to the formation of p-methoxybenzonitrile. Then, due to phenyl cation, it loses further methoxy and nitrile groups to afford the base peak at m/z = 77. The parent ion peak can also undergo denitrogenation with the formation of ion peaks at m/z = 402 and 403 a.m.u. assignable to 4-(2-imino-5-(1-(4-methoxyphenyl)prop-1-en-2-yl)-1,3,4-thiadiazol-3(2H)-yl)benzenesulfonamide and its protonated form, respectively (Scheme 1 at ESI†).
Similarly, the treatment of aminohydrazone 5 with furfural 10, as heterocyclic aldehyde, in ethanol afforded 1-(furan-2-ylmethylene)-2-(1-(2-oxo-3-(4-sulfamoylphenyl)-1,3,4-thiadiazol -5-yl) ethylidene)hydrazine 11 (Scheme 2). The IR spectrum of 11 exhibited bands at 3338–3245, 1685, 1625, 1325, and 1291 cm−1, which were attributed to NH2, CO, CN, and SO2 functions, respectively. Furthermore, the MS of 11 showed a M+ at m/z = 391 and an ion peak at m/z = 393 (M+ + 2). The 1H-NMR spectrum of 11 revealed the absence of imine proton at about 9.50 ppm. It disclosed the existence of three singlet signals at δ 2.44, 7.47, 8.48 ppm assigned to CH3, NH2, and CHN protons, along with the expected signals of furan and benzene residue. Its 13C-NMR spectrum displayed important signals resonating at δ 13.68, 159.01, and 168.29 ppm characteristics for CH3, CHN, and CO carbons, respectively.
The scope of these condensation reactions was extended via the treatment of aminohydrazone 2 with heterocyclic ketones, namely, isatin 12, 1-(N,N-dimethylaminomethyl)isatin 14a,23 and 1-(N,N-diethylaminomethyl)isatin 14b (ref. 23) to afford the respective unsymmetrical azines 13, 15a and 15b (Scheme 3). The MS of 13, 15a, and 15b showed parent ion peaks at m/z = 441, 498, and 526 a.m.u. which is consistent with their molecular formulas (C18H15N7O3S2), (C21H22N8O3S2) and (C23H26N8O3S2), respectively. The IR spectrum of 13 showed bands at 3303–3257, 3115, 1717, 1607, and 1587 characteristics for NH2, NH, CO, CN, and CC functions, respectively. The 1H-NMR spectrum of 13 showed three D2O-exchangeable signals at δ 10.92, 9.54, 7.41 ppm assignable to indoline-NH, imine-NH, NH2 proton, and a singlet signal at 2.32 ppm assignable to the CH3 protons. Its 13C-NMR spectrum revealed signals at δ 163.91, 158.09, 154.71, and 13.49 ppm specific for carbonyl, thiadiazole-C2, CN, and CH3 carbons, respectively. Additionally, the structures of 15a and 15b were chemically indicated via Mannich reactions of 13 with each formalin and dimethylamine or formalin and diethylamine.
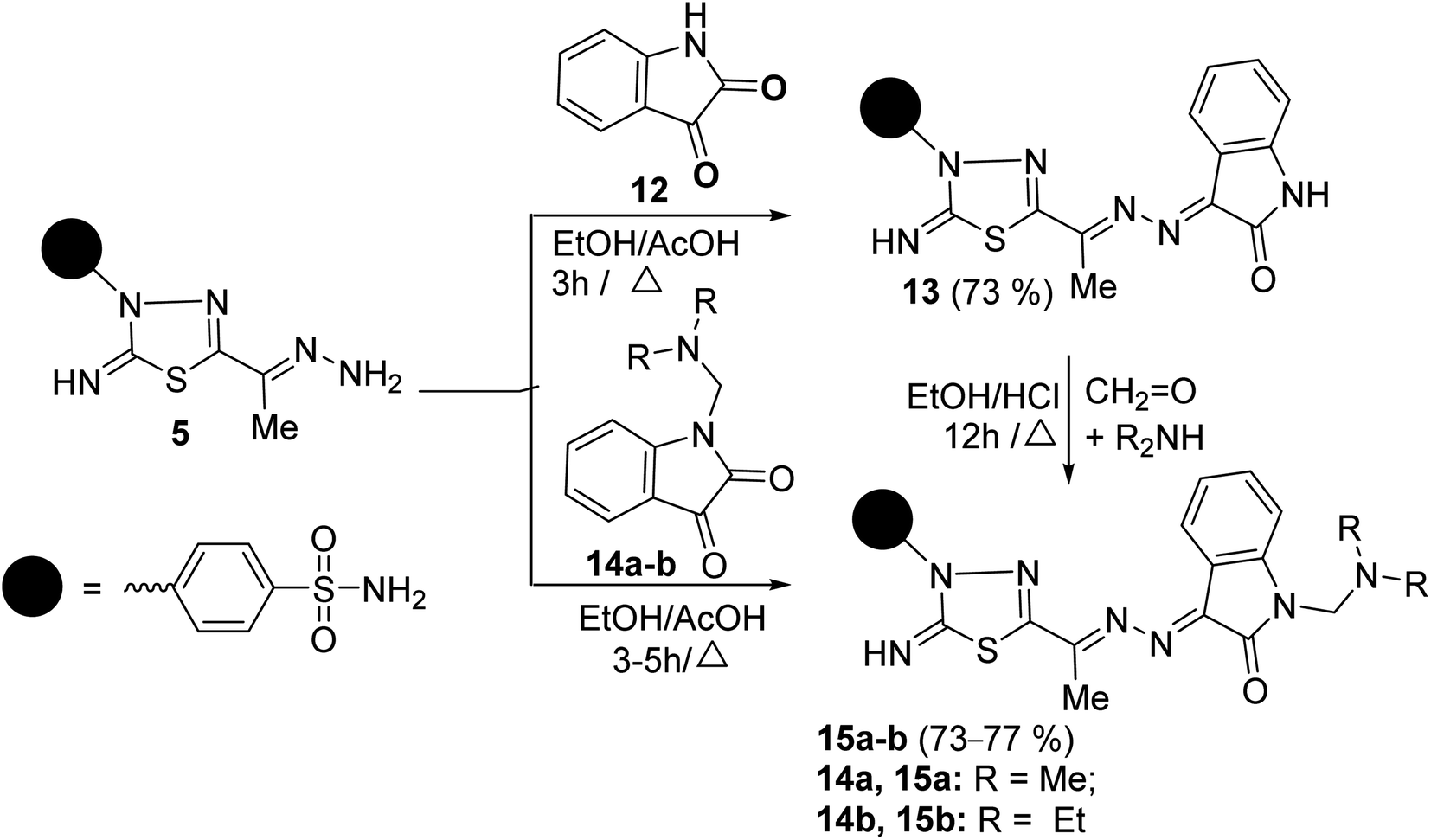 | ||
| Scheme 3 Reaction of aminohydrazone 5 with heterocyclic ketones 12, 14a–b. | ||
2.2. Quantum mechanical study
N bond where they are in the same (Z isomer) or opposite sides (E isomer), see Fig. 3. The computational outcomes using B3LYP functional combined with the 6-31G(d) basis set reveal that the E configuration is more stable than the Z form by 769 cm−1 (2.21 kcal mol−1). While adopting the E/Z configurations, we need to explore the exact conformation of –C(CH3)NNH2, phenyl sulfonamide, sulfonamide, and methyl moieties which result from free rotation about C2–C6, N4–C15, C18–S25 and C6–C7 single bonds, respectively. For this purpose, a relaxed scan of the PES was performed throughout the rotation of the dihedral angles τ N8C6C2N3, τ C16C15N4N3, τ N28S25C18C19, and τ H12C7C6C2 from 0° to 360° in steps of 10° proceeded by optimization process after every scan point. The obtained curves of the PES scan using B3LYP/6-31G(d) calculations for E and Z isomers are given at ESI (Fig. S1 and S2†). For the E isomer, the predicted PES curve from the rotation of –C(CH3)NNH2 moiety shows a global minimum when τ N8C6C2N3 reaches 180.0° in which the imine bonds (CN) are trans to each other.
 | ||
| Fig. 3 Optimized geometries and atom numbering for Z and E isomers of 5 predicted using the B3LYP/6-31G(d) calculations. | ||
In contrast, minimum energy for Z-isomer was obtained at τ N8C6C2N3 equal to 0.0° where both CN bonds are cis to each other where stabilization could be attributed to intra-molecular H-bonding interaction between amino hydrogen and thiadiazol nitrogen. Moreover, the energetically favored conformation of benzenesulfonamide moiety (–C6H4SO2NH2) concerning the thiadiazol ring was assigned at τ C16C15N4N3 of 10.0° where the phenyl ring is almost planar towards the thiadiazol ring. The predicted curve for the scan of PES throughout the rotation of sulfonamide moiety for both E and Z isomer exhibit a minimum conformation at τ N28S25C18C19 equal to 260.0° in which the amino group is perpendicular to the phenyl ring. The internal rotation of the methyl group gives rise to a minimum at τ H12C7C6C2 equal to 60.0° and 0.0° for Z and E configuration, where the methyl group is orientated in eclipsed and staggered conformation to the adjacent CN bond, respectively. In conclusion, opposite conformations of C(CH3)NNH2 and methyl moieties were obtained for E and Z isomers. In contrast to the E isomer, the global minimum of the Z isomer has the CN bond in cis orientation to the thiadiazol CN bond with the eclipsed conformation of the methyl group towards the CN bond (Fig. 3).
The measured 13C NMR spectrum for 5 displays 20 signals, double the number of carbon atoms, and reveals the presence of both Z and E isomers in the solution phase. Thus, the 1H and 13C NMR chemical shifts (δ in ppm) were calculated for both isomers using B3LYP/6-31G(d) calculations and compared to those observed experimentally. The computed 1H and 13C chemical shifts for Z and E isomers have equivalent values in the experimental spectra, as Table 1 shows that both isomers exist in the solution phase. In contrast to the E isomer, one proton of amino-hydrazone moiety (H11) for the Z isomer was predicted to resonate at 7.31 ppm and matches the observed signal at 7.33 ppm, confirming the presence of the Z isomer in the solution phase. The downfield shift for H11 accounts for the intra-molecular H bonding with adjacent thiadiazol nitrogen. The 1H NMR spectrum shows three signals at 7.36, 7.36, and 7.43 ppm corresponding to amino protons of sulfonamide moiety (H29 and H30) in excellent agreement with those calculated for both E and Z isomers, 7.34–7.41 ppm. It's worth noting that, compared to tetramethylsilane (TMS), the prediction of chemical shifts for amino protons was improved when a multi-standard technique24,25 was applied using a comparable skeleton as a reference.
| Atomb | δcalc | δexpc | Atomb | δcalc | δexpc | ||
|---|---|---|---|---|---|---|---|
| Z | E | Z | E | ||||
| a Calculations were carried out in DMSO solution by B3LYP/6-31G(d) method utilizing the IEF-PCM solvation model.b For the structures and atom numbering of Z and E isomers (Fig. 3).c The values of chemical shifts between brackets are assigned to more than one atom.d rms refers to the root mean square deviations of the theoretical values of chemical shifts from their corresponding experimental values.e The correlation coefficient (R2) between the computed and observed values of chemical shifts. | |||||||
| C2 | 149.14 | 151.26 | H10 | 5.52 | (5.67) | ||
| 155.67 | 153.01 | 5.16 | (5.67) | ||||
| C5 | 160.48 | (159.23) | H11 | 7.31 | 7.33 | ||
| 164.53 | (159.23) | 5.63 | 5.67 | ||||
| C6 | 126.49 | 126.80 | H12 | 1.95 | (2.08) | ||
| 138.02 | 139.07 | 1.76 | (2.08) | ||||
| C7 | 20.14 | 11.36 | H13 | 2.19 | (2.08) | ||
| 7.64 | 10.27 | 1.68 | (2.08) | ||||
| C15 | 144.59 | 142.89 | H14 | 2.21 | (2.08) | ||
| 145.49 | 145.89 | 2.71 | (2.08) | ||||
| C16 | 121.93 | 122.43 | H21 | 7.66 | 7.87 | ||
| 120.39 | 120.24 | 7.79 | 8.28 | ||||
| C17 | 128.93 | 127.15 | H22 | 7.73 | 8.27 | ||
| 129.00 | (131.35) | 7.70 | 7.88 | ||||
| C18 | 138.60 | 141.75 | H23 | 7.65 | 7.86 | ||
| 136.97 | 134.63 | 7.60 | 7.84 | ||||
| C19 | 129.02 | (131.35) | H24 | 8.57 | 8.35 | ||
| 129.49 | (131.35) | 8.97 | 8.36 | ||||
| C20 | 120.83 | 121.33 | H29 | 7.41 | 7.43 | ||
| 118.09 | 118.60 | 7.36 | (7.36) | ||||
| H30 | 7.38 | (7.36) | |||||
| 7.34 | 7.34 | ||||||
| H32 | 10.71 | 9.15 | |||||
| 10.82 | 9.31 | ||||||
| rmsd | 3.24 | 2.41 | 0.49 | 0.57 | |||
| R2e | 0.9988 | 0.9988 | 0.9838 | 0.9804 | |||
The predicted chemical shift for the Z isomer at 126.49 ppm belongs to the hydrazone carbon atom (C6) and better matches the 13C signal observed at 126.80 ppm. The counterpart value for the E isomer was computed at 138.02 ppm, which is consistent with the signal observed at 139.07 ppm. The experimental 13C NMR spectrum shows signals at 151.26, 141.75, 122.43 and 121.33 ppm are better correlated to the Z isomer's computed values of 149.14 (C2), 138.60 (C18), 121.93 (C16) and 120.83 (C20) ppm, respectively. Regarding the E isomers, these atoms are predicted to resonate at 155.67, 136.97, 120.39, and 118.09 ppm, respectively, which match the signals displayed at 153.01, 134.63, 120.24, and 118.60 ppm, respectively. Compared to the experimental value, the calculated chemical shifts of thiadiazol C5 (for E isomer) and methyl C7 (for Z isomer) were noticeably overestimated by 5.30 and 8.78 ppm, respectively. In sum, the computed chemical shifts of hydrogen/carbon nuclei for both Z and E isomers are well compared to those observed in the experimental 1H/13C NMR spectra, with high correlation coefficients (R2) of 0.9838/0.9988 and 0.9804/0.9988, respectively.
The E/Z configuration for the synthesized products 7a–c, 9, 11, 13 and 15a–b were theoretically explored based on the orientation around the hydrazonyl CN bonds, which results in four possible configurations, 1 (EE), 2 (ZZ), 3 (EZ) and 4 (ZE). Therefore, a full geometry optimization was carried out for each configuration followed by frequency applying B3LYP method at 6-31G(d) basis set. The ESI† provides the equilibrium geometries and computed energy difference for the suggested configurations (Fig. S3–S10/Table S1†). For all synthesized compounds, the computational outcomes revealed EE (1) isomer to be the favored configuration with the lowest energy. Fig. 4 shows the predicted equilibrium geometries for the most stable configuration (1, EE) for 7a–c, 9, 11, 13, 15a and 15b.
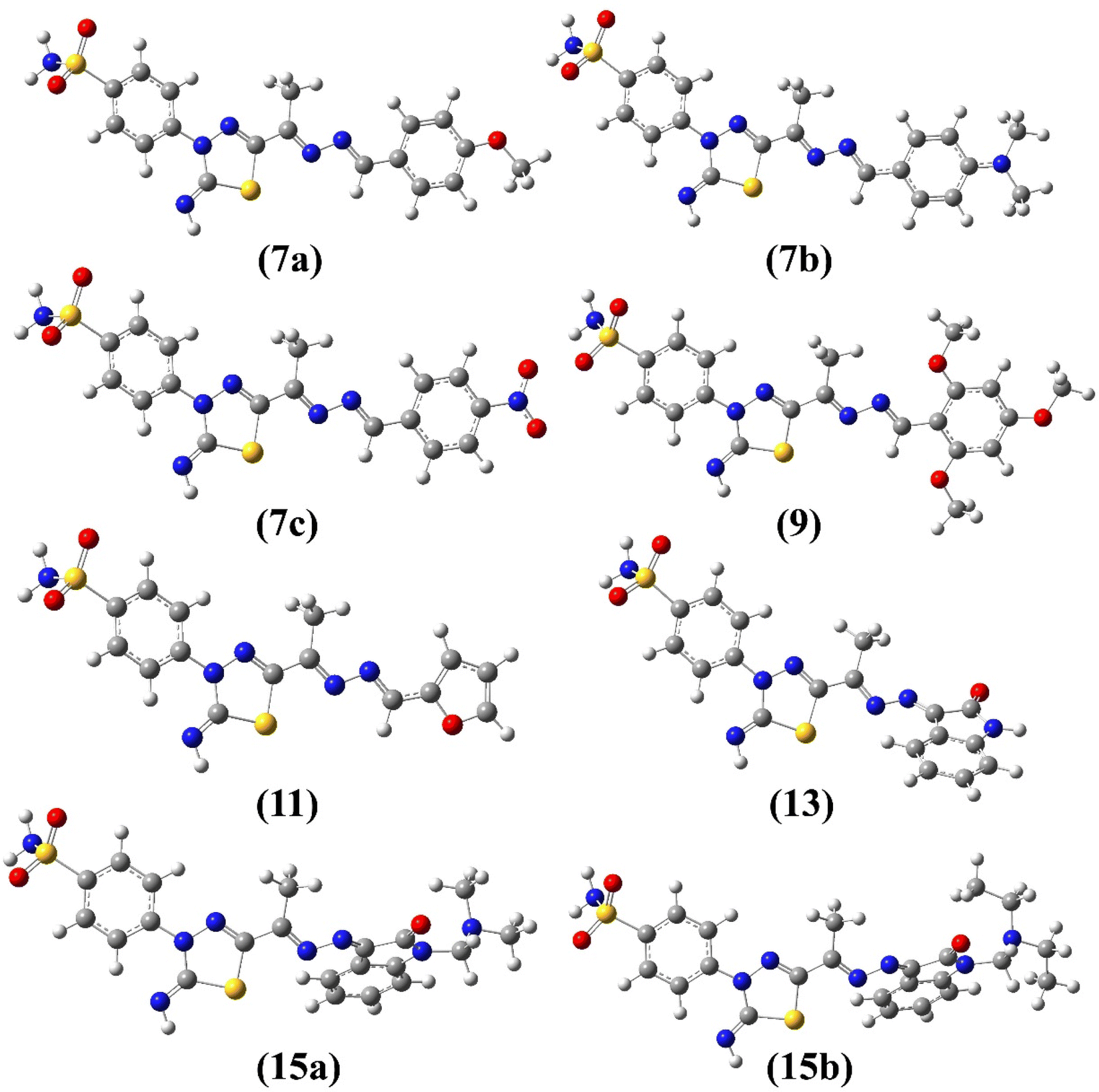 | ||
| Fig. 4 Optimized geometries of 7a–c, 9, 11, 13 and 15a–b obtained from B3LYP/6-31G(d) calculations. | ||
The impact of FMOs and their associated molecular reactivity descriptors on molecule biological reactivity has recently been considered.26,27 Herein, The FMOs were predicted for the optimized geometries of the synthesized molecules (5, 7a–c, 9, 11, 13 and 15a–b) to evaluate the reactivity and to correlate their biological activities. Subsequently, the computed energies for HOMO and LUMO were used to calculate a set of quantum chemical descriptors (Table 2), which are useful in assessing the molecule's overall reactivity.28,29
| Cpd | E | I | A | Eg | η | μ | ω | μtot | α |
|---|---|---|---|---|---|---|---|---|---|
| a Calculations were carried out using 6-31G(d) basis set.b E, total energy; I, ionization potential = −EHOMO; A, electron affinity = −ELUMO; Eg, energy gap = ELUMO–EHOMO; η, hardness = (I − A)/2; μ, chemical potential = −(I + A)/2; ω, electrophilicity index = μ2/2η (all in eV); μtot, total dipole moment in Debye; α, polarizability in a.u. | |||||||||
| 5 | −45266.30 |
5.75 | 1.64 | 4.11 | 2.06 | −3.69 | 3.32 | 7.01 | 210.19 |
| 7a | −55706.90 |
5.76 | 2.31 | 3.45 | 1.73 | −4.03 | 4.71 | 9.53 | 367.89 |
| 7b | −56236.08 |
5.36 | 2.11 | 3.24 | 1.62 | −3.74 | 4.30 | 12.07 | 407.38 |
| 7c | −58155.23 |
6.17 | 3.21 | 2.96 | 1.48 | −4.69 | 7.43 | 3.69 | 368.79 |
| 9 | −61939.41 |
5.48 | 2.00 | 3.49 | 1.74 | −3.74 | 4.01 | 11.91 | 402.22 |
| 11 | −52529.97 |
5.83 | 2.48 | 3.35 | 1.68 | −4.15 | 5.15 | 7.73 | 321.64 |
| 13 | −57148.21 |
6.08 | 2.85 | 3.23 | 1.61 | −4.46 | 6.17 | 5.25 | 339.36 |
| 15a | −61863.38 |
6.02 | 2.76 | 3.25 | 1.63 | −4.39 | 5.93 | 8.37 | 386.01 |
| 15b | −64002.88 |
6.00 | 2.76 | 3.24 | 1.62 | −4.38 | 5.92 | 8.77 | 409.20 |
| Staurosporine | −41561.54 |
4.90 | 0.79 | 4.12 | 2.06 | −2.84 | 1.96 | 7.91 | 342.39 |
| Valdecoxib | −36773.65 |
6.60 | 1.34 | 5.25 | 2.63 | −3.97 | 3.00 | 3.07 | 203.20 |
| Methazolamide | −39083.89 |
6.58 | 1.78 | 4.80 | 2.40 | −4.18 | 3.64 | 1.50 | 126.35 |
| ZRL4 | −55935.10 |
6.06 | 2.12 | 3.94 | 1.97 | −4.09 | 4.25 | 2.18 | 279.79 |
The distribution for electron density in HOMOs and LUMOs for the investigated compounds is shown in Fig. 5, along with their energy gaps predicted using the B3LYP/6-31G(d) computations. The HOMO is largely localized on the thiadiazol ring and phenyl ring of the benzenesulfonamide moiety except for 7b, where the HOMO is mostly distributed over the dimethylphenylamine moiety. The LUMO localized over the whole molecule for all synthesized molecules except the region containing the benzenesulfonamide moiety.
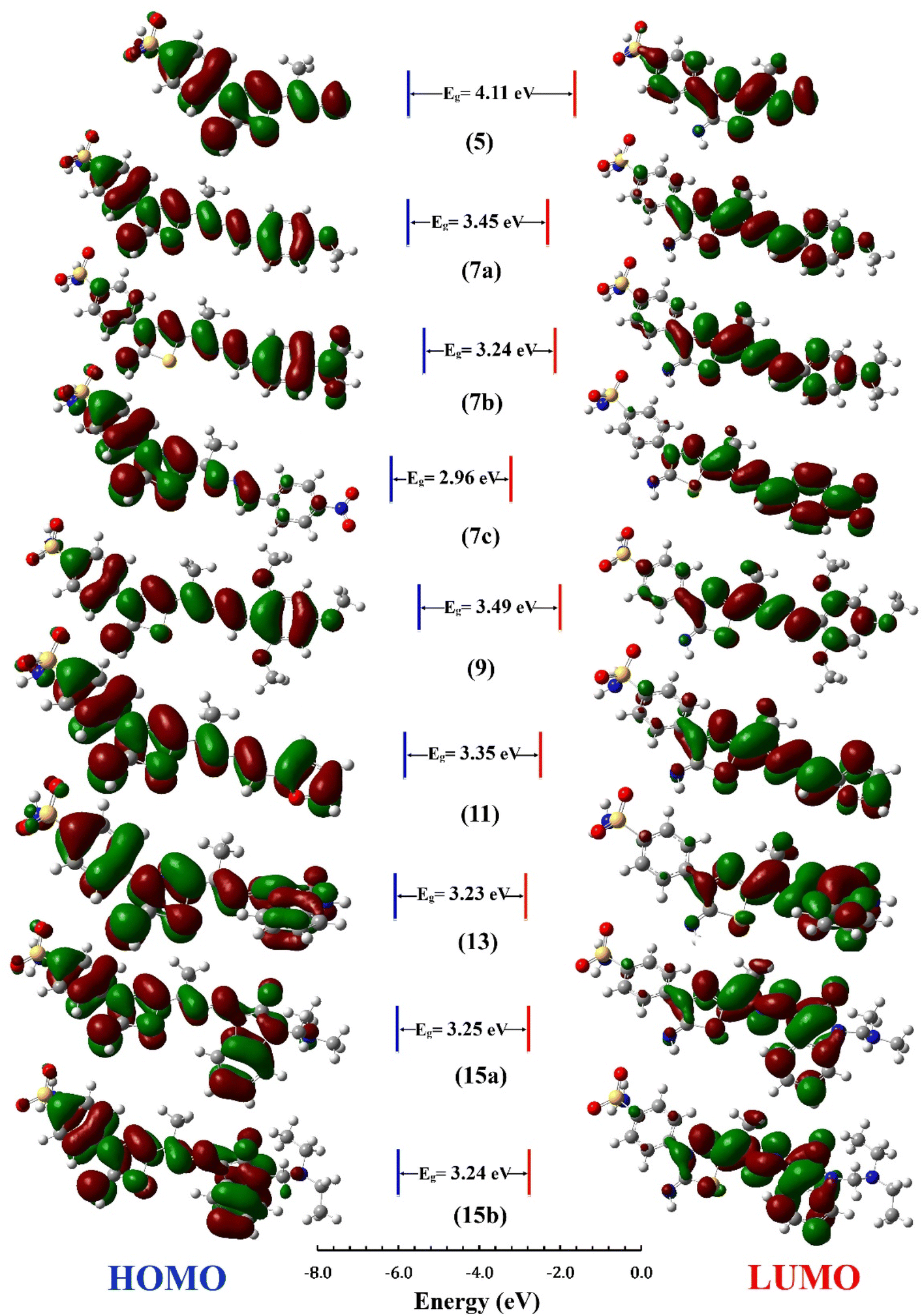 | ||
| Fig. 5 HOMO and LUMO Frontier MOs and energy gaps (Eg) for the synthesized compounds predicted using B3LYP/6-31G(d) calculations. | ||
The energy gap (EHOMO–ELUMO) is a valuable sign of a molecule's chemical reactivity and kinetic stability. A molecule with a small energy gap is more polarized and has a higher chemical reactivity and lower kinetic stability.30 Furthermore, Small energy gaps suggest that the molecule undergoes a large intramolecular charge transfer, which might affect the molecule's biological activity.31 The energy gaps calculated for the synthesized compounds ranged from 2.96 to 4.11 eV, comparable to the reported values for bioactive molecules32–34 and that computed for the standard reference drug, staurosporine (4.12 eV). For all compounds, the calculated energy gap Eg (Table 2) decreases in the order 5 (4.11 eV) > 9 > 7a > 11 > 15a > 15b = 7b > 13 > 7c (2.96 eV). These results are compatible with the observed high activities for 7c, 13, 15b, and 15a against HepG-2 and Caco2 (Table 3).
| Compound No. | IC50 (μM) | |||
|---|---|---|---|---|
| HepG-2 | Caco2 | MCF-7 | WI 38 | |
| 7a | 4.17 ± 0.20 | 3.16 ± 0.20 | 31.50 ± 1.4 | 28.20 ± 1.50 |
| 7b | 26.80 ± 1.50 | 33.60 ± 1.70 | 35.20 ± 1.6 | 49.80 ± 2.60 |
| 7c | 3.04 ± 0.20 | 3.37 ± 0.20 | 9.59 ± 0.4 | 25.40 ± 1.90 |
| 9 | 12.20 ± 0.70 | 47.20 ± 2.30 | 7.21 ± 0.3 | 25.40 ± 1.30 |
| 11 | 15.00 ± 0.80 | 8.55 ± 0.40 | 3.34 ± 0.2 | 24.50 ± 1.80 |
| 13 | 1.76 ± 0.10 | 8.99 ± 0.40 | 17.70 ± 0.8 | 11.40 ± 0.60 |
| 15a | 6.54 ± 0.40 | 2.33 ± 0.10 | 4.34 ± 0.2 | 13.40 ± 0.70 |
| 15b | 1.62 ± 0.10 | 0.78 ± 0.04 | 4.43 ± 0.2 | 16.80 ± 0.90 |
| Staurosporine | 13.60 ± 0.80 | 8.18 ± 0.70 | 6.19 ± 0.3 | 25.20 ± 1.30 |
Chemical potential (μ) is the inverse of electronegativity (χ), and it defines how much energy a molecule absorbs or releases during a chemical process. Both descriptors significantly impact a molecule's inhibitory effectiveness.35 Compound 7c, with a high χ value (of 4.69 eV), is more active than 7a and 7b with low values (4.03 and 3.74 eV), which is attributed to the presence of the nitro group in 7c and validates the observed higher activity against MCF-7 (Table 3). Also, the hardness value for 7c (1.48 eV) is lower than that calculated for 7a and 7b. It shows that it is a softer and more reactive molecule compared to 7a and 7b, in agreement with the estimated energy gap, see Table 2. Owing to the values of the electrophilicity index (ω), organic compounds were categorized as strong (ω > 1.5 eV), moderate (0.8 < ω < 1.5 eV), or weak (ω < 0.8 eV) electrophiles.36 Compounds 7c and 13 are powerful electrophiles in this study, with ω values of 7.43 and 6.17, respectively. Compound 9 has the lowest biological activity against Caco2, which might be explained by its high energy gap (3.49 eV) and poor electrophilicity (4.01 eV) between all the synthesized compounds. To support the combi-targeting technique in this study, QM descriptors were computed for three commercial drugs with similar cores, valdecoxib, methazolamide, and ZSL4 (Fig. 1, Table 2). As a result, the computed Eg and η values for the ZSL4 drug matched those predicted for compound 5 and staurosporine. Also, compounds 7a and 11 have μ values of −4.03 and −3.97 eV, respectively, and are similar to those obtained for valdecoxib and ZSL4 drugs.
The dipole moment is also considered when correlating the molecule's biological activity, which might influence the degree of interaction between drugs and the active sites of protein.37 Compounds 15a–b, for example, are more active against Caco2 and MCF-7 than 13, which may be explained by the fact that 15a–b has a higher total dipole moment (8.37–8.77 Debye) than 13 (5.25). As shown in Table 2, compounds 7b and 9 have high total dipole moments (12.07 and 11.91 Debye, respectively) and exhibit high polarizabilities (407.38 and 402.22 a.u.), indicating that they are a good candidate for non-linear optical (NLO) materials.38
| Parametera | 7a | 7b | 7c | 9 | 11 | 13 | 15a | 15b | Staurosporine |
|---|---|---|---|---|---|---|---|---|---|
| a MW, molecular weight (g mol−1); nrot, number of rotatable bonds; HBA, number of H-bond acceptors; HBD, number of H-bond donors; TPSA, topological polar surface area; %Abs, intestinal absorption (human); logPo/w, lipophilicity; logS, water solubility; GI abs, gastrointestinal absorption; logKp, skin permeation, Lipinski, rule of five for drug likeness; LD50, oral rat acute toxicity. |
|||||||||
| MW (g mol−1) | 430.50 | 443.55 | 445.48 | 490.56 | 390.44 | 441.49 | 498.58 | 526.63 | 466.53 |
| nrot | 6 | 6 | 6 | 8 | 5 | 4 | 6 | 8 | 2 |
| HBA | 8 | 7 | 9 | 10 | 8 | 8 | 9 | 9 | 4 |
| HBD | 2 | 2 | 2 | 2 | 2 | 3 | 2 | 2 | 2 |
| TPSA (Å2) | 172.40 | 166.41 | 208.99 | 190.86 | 176.31 | 192.27 | 186.72 | 186.72 | 69.45 |
| %Abs | 77.97 | 76.85 | 75.87 | 74.37 | 78.33 | 78.25 | 74.61 | 76.12 | 95.79 |
| logPo/w |
2.42 | 2.40 | 2.40 | 2.33 | 2.33 | 2.33 | 1.83 | 1.83 | 3.03 |
| logS |
−4.10 | −4.26 | −4.26 | −4.25 | −4.25 | −4.25 | −4.26 | −4.26 | −5.06 |
| GI abs | Low | Low | Low | Low | Low | Low | Low | Low | High |
| logKp (cm s−1) |
−7.17 | −7.15 | −7.15 | −7.58 | −7.58 | −7.58 | −7.81 | −7.81 | −6.85 |
| Lipinski, violation | Yes; 0 | Yes; 0 | Yes; 0 | Yes; 1 | Yes; 1 | Yes; 1 | Yes; 1 | Yes; 1 | Yes; 0 |
| AMES toxicity | No | No | Yes | No | No | No | No | No | Yes |
| LD50 (mol kg−1) | 2.14 | 2.24 | 3.32 | 2.20 | 2.18 | 2.02 | 2.13 | 2.21 | 2.46 |
| Skin sensitization | No | No | No | No | No | No | No | No | No |
The oral bioavailability was estimated using the SwissADME's bioavailability radar, which considers six physicochemical parameters: size, solubility, lipophilicity, polarity, flexibility, and saturation. The bioavailable radars for the investigated compounds in this work are given (Fig. S11 at ESI†). Accordingly, the pink zone for all examined compounds has four parameters that provide physicochemical space acceptable for oral bioavailability. However, saturation and polarity were found beyond the bioavailability radar's pink zone due to a low saturation (fraction of Csp3 < 0.25) and high polarity (TPSA > 130 Å2).
2.3. Bio-evaluation
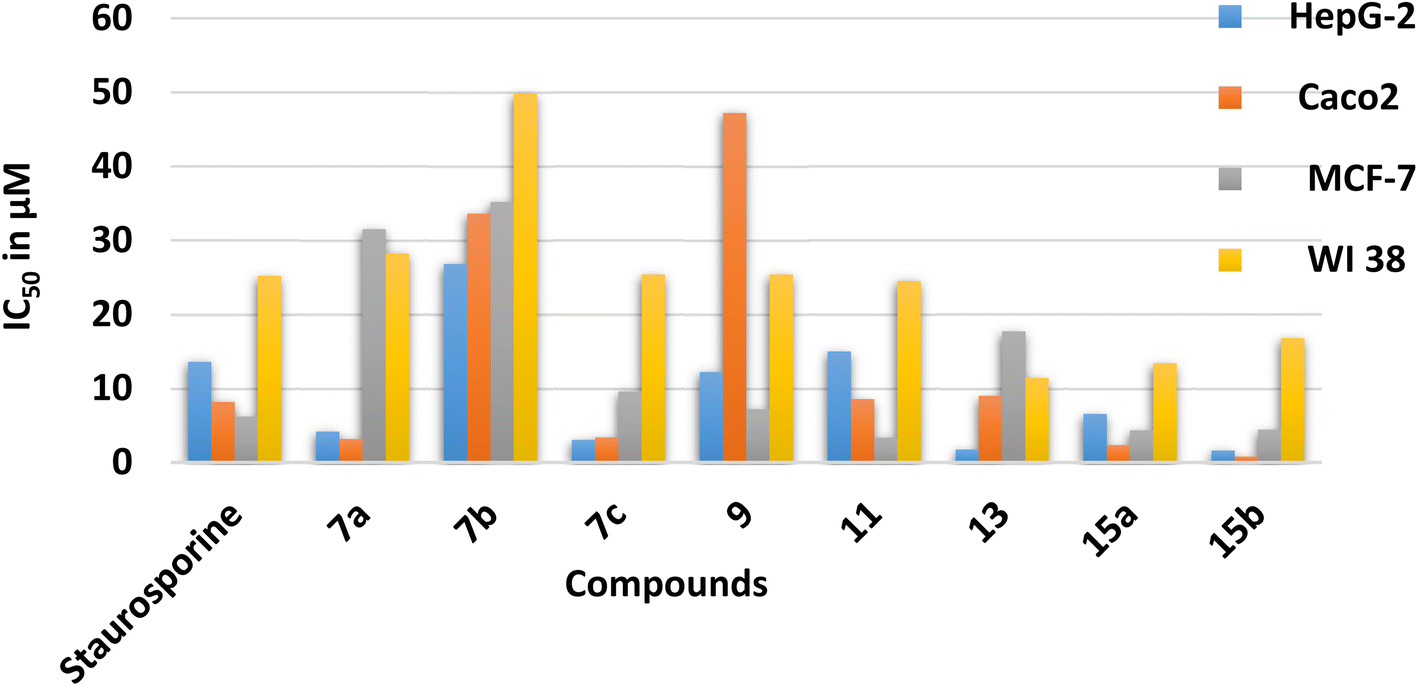 | ||
| Fig. 6 The in vitro cytotoxic evaluation of derivatives 7a–c, 9, 11, 13, 15a and 15b presented as IC50 in μM. | ||
3. Experimental section
3.1. Synthesis and spectroscopic characterization
O), 1599 (CN), 1579 (CC), 1330, 1298 (SO2); 1H-NMR (DMSO-d6): δppm = 2.50 (s, 3H, CH3), 7.45 (s, 2H, NH2), 7.92 (d, J = 9.35 Hz, 2H, Ar–H2,6), 8.18 (d, J = 9.35 Hz, 2H, Ar–H3,5), 9.62 (s, 1H, NH); 13C-NMR (DMSO-d6): δppm = 24.88 (CH3), 122.00 (2CH, Ar–C2,6), 126.54 (2CH, Ar–C3,5), 141.28 (Ar–C4), 141.48 (Ar–C1), 148.04 (thiadiazole-C5), 158.30 (thiadiazole-C2), 189.83 (CO); MS m/z (%): 298 (M+, 55.9), 297 (43.7), 241 (0.9), 218 (1.0), 198 (10.2), 182 (0.8), 181 (4.6), 176 (1.3), 172 (3.5), 169 (1.8), 148 (1.0), 134 (2.2), 120 (1.1), 106 (100), 93 (2.3), 78 (10.0), 54 (4.9). Anal. calc. For C10H10N4O3S2 (298.34): C, 40.26; H, 3.38; N, 18.78%; found: C, 40.24; H, 3.39; N, 18.77%.N), 1585 (CC), 1323, 1305 (SO2); 1H-NMR (DMSO-d6): δppm = 2.07 (s, 3H, CH3), 5.66 (s, 2H, NH2), 7.42 (s, 2H, NH2), 7.84 (d, J = 8.5 Hz, 2H, Ar–H2,6), 7.86 (d, J = 8.5 Hz, 2H, Ar–H3,5), 9.49 (s, 1H, NH); 2.05 (s, 3H, CH3), 7.32 (s, 1H, 2N-Hb), 7.33 (s, 1H, 2N-Ha), 7.36 (s, 2H, NH2), 8.26 (d, J = 8.5 Hz, 2H, Ar–H2,6), 8.34 (d, J = 8.5 Hz, 2H, Ar–H3,5), 9.49 (s, 1H, NH); 13C-NMR (DMSO-d6): δppm = 11.55 (CH3), 120.24 (2CHAr–C2,6), 126.80 (2CHAr–C3,5), 131.35 (Ar–C4), 139.75 (Ar–C1), 145.89 (CN), 151.28 (thiadiazole-C5), 159.23 (thiadiazole-C2); MS m/z (%): 312 (M+, 10.3), 270 (1.3), 251 (2.5), 227 (2.2), 205 (1.4), 193 (1.5), 176 (9.6), 158 (2.4), 145 (2.2), 129 (7.1), 111 (8.9), 88 (2.2), 65 (11.7), 57 (100); anal. calc. For C10H12N6O2S2 (312.37): C, 38.45; H, 3.87; N, 26.90%; found: C, 38.46; H, 3.86; N, 26.91%.3.1.3.1. 4-[2-Imino-5-(-1-((4-methoxybenzylidene)hydrazineylidene)ethyl)-1,3,4-thiadiazol-3(2H)-yl]benzenesulfonamide (7a). The compound was obtained by heating a solution of 4-methoxybenzaldehyde 6a (0.43 g, 0.003 mol) for 5 h under reflux. Brown powder (0.70 g, 54%); M.P. 249–252 °C; IR νmax/cm−1 = 3360, 3255 (NH2), 3110 (NH), 1604 (C
N), 1550 (CC), 1332, 1305 (SO2); 1H-NMR (DMSO-d6): δppm = 2.49 (s, 3H, CH3), 3.84 (s, 3H, OCH3), 7.07 (d, J = 9.3 Hz, 2CH, Ar–H3′,5′), 7.49 (s, 2H, NH2), 7.88 (d, J = 9.3 Hz, 2CHAr–H2′,6′), 7.97 (d, J = 2.5 Hz, 2CH, Ar–H2,6), 8.03 (d, J = 2.5 Hz, 2CHAr–H3,5), 8.63 (s, 1H, CHN), 9.42 (s, 1H, CNH); 13C-NMR (DMSO-d6): δppm = 13.46 (CH3), 55.85 (OCH3), 114.87 (2CHAr–C3′,5′), 121.32 (2CHAr–C2,6), 126.88 (2CHAr–C2′,6′), 127.34 (Ar–C1′), 131.16 (2CHAr–C3,5), 131.33 (Ar–C4), 142.91 (Ar–C1), 149.40 (Ar–C4′), 161.91 (CHN), 162.71 (CN), 162.89 (thiadiazole-C5), 168.75 (thiadiazole-C2); MS m/z (%): 430 (M+, 4.2), 429 (0.4), 417 (3.9), 404 (11.2), 386 (0.09), 357 (0.1), 333 (0.1), 318 (0.1), 299 (0.1), 279 (0.1), 264 (0.4), 247 (0.2), 225 (0.7), 200 (1.5), 177 (6.4), 160 (2.1), 147 (4.3), 115 (3.0), 77 (100), 63 (50.4), 57 (31.6); anal. calc. For C18H18N6O3S2 (430.50); C, 50.22; H, 4.21; N, 19.52%; found: C, 50.21; H, 4.22; N, 19.51%.
3.1.3.2. 4-[5-(1-((4-(Dimethylamino)benzylidene)hydrazineylidene)ethyl)-2-imino-1,3,4-thiadiazol-3(2H)-yl]benzenesulfonamide (7b). The compound was obtained by heating a solution of 4-(dimethylamino)benzaldehyde 6b (0.47 g, 0.003 mol) for 4 h under reflux. Brown powder (0.45 g, 34%); M.P. 235–237 °C; IR νmax/cm−1 = 3310, 3225 (NH2), 3116 (NH), 1601 (C
N), 1524 (CC), 1324, 1230 (SO2); 1H-NMR (DMSO-d6): δppm = 2.46 (s, 3H, CH3), 3.02 (s, 6H, 2CH3), 7.39 (s, 2H, NH2), 6.77 (d, J = 9.3 Hz, 2CH, Ar–H2′,6′), 7.73 (d, J = 9.3 Hz, 2CH, Ar–H3′,5′), 7.90 (d, J = 8.5 Hz, 2CH, Ar–H2,6), 8.04 (d, J = 8.5 Hz, 2CH, Ar–H3,5), 8.43 (s, 1H, CH), 9.38 (s, 1H, NH); 13C-NMR (DMSO-d6): δppm = 13.33 (CH3), 40.30 (2CH3), 112.08 (2CHAr–C3′,5′), 121.19 (2CHAr–C2,6), 122.02 (Ar–C1′), 126.87 (2CHAr–C3,5), 127.33 (Ar–C4), 131.31 (2CHAr–C2′,6′), 142.80 (Ar–C1), 153.32 (Ar–C4′), 158.83 (CHN), 162.66 (CN), 163.69 (thiadiazole-C5), 168.80 (thiadiazole-C2); MS m/z (%): 443 (M+, 12.9), 442 (1.4), 428 (1.1), 414 (3.0), 383 (1.3), 353 (0.9), 342 (1.1), 299 (1.0), 263 (1.0), 251 (1.9), 223 (1.0), 195 (1.0), 181 (1.3), 149 (3.6), 116 (6.2), 90 (17.8), 63 (18.4), 57 (100); anal. calc. For C19H21N7O2S2 (443.54): C, 51.45; H, 4.77; N, 22.11%; found: C, 51.46; H, 4.78; N, 22.12%.
3.1.3.3. 4-[2-Imino-5-(1-((4-nitrobenzylidene)hydrazineylidene)ethyl)-1,3,4-thiadiazol-3(2H)-yl]benzenesulfonamide (7c). The compound was obtained by heating a solution of 4-nitrobenzaldehyde 6c (0.48 g, 0.003 mol) for 7 h under reflux. Brown powder (0.84 g, 63%); M.P. 177–179 °C; IR νmax/cm−1 = 3305, 3258 (NH2), 3112 (NH), 1623 (C
N), 1516 (CC), 1307, 1291 (SO2); 1H-NMR (DMSO-d6): δppm = 2.47 (s, 3H, CH3), 7.49 (s, 2H, NH2), 7.90 (d, J = 1.7 Hz, 2CH, Ar–H2,6), 8.04 (d, J = 1.7 Hz, 2CHAr–H3,5), 8.16 (d, J = 3.4 Hz, 2CH, Ar–H2′,6′), 8.36 (d, J = 3.4 Hz, 2CHAr-H3′,5′), 8.67 (s, 1H, NCH), 9.49 (s, 1H, NH); 13C-NMR (DMSO-d6): δppm = 14.31 (CH3), 121.53 (2CHAr–C2,6), 124.73 (2CHAr–C3′,5′), 127.04 (2CHAr–C2′,6′), 130.12 (2CHAr–C3,5), 130.23 (Ar–C4), 140.05 (Ar–C1), 142.16 (Ar–C1′), 149.40 (NCH), 151.55 (Ar–C4′), 158.41 (CN), 159.60 (thiadiazole-C5), 168.70 (thiadiazole-C2); MS m/z (%): 445 (M+, 7.9), 444 (8.3), 429 (2.7), 416 (2.8), 393 (0.6), 368 (1.9), 354 (1.9), 335 (0.6), 308 (0.8), 269 (0.7), 237 (0.8), 223 (0.6), 205 (1.5), 190 (19.8), 174 (2.0), 141 (3.0), 119 (20.3), 95 (16.1), 73 (16.7), 57 (100); anal. calc. For C17H15N7O4S2 (445.47): C, 45.84; H, 3.39; N, 22.01%; found: C, 45.83; H, 3.40; N, 22.02%.
3.1.3.4. 4-[2-Imino-5-(1-((2,4,6-trimethoxybenzylidene)hydrazineylidene)ethyl)-1,3,4-thiadiazol-3(2H)-yl]benzenesulfonamide (9). The compound was obtained by heating a solution of 2,4,6-trimethoxybenzaldehyde 8 (0.62 g, 0.003 mol) for 5 h under reflux. Brown powder; (0.79 g, 54%); M.P. 189–192 °C; IR νmax/cm−1 = 3305, 3247 (NH2), 3115 (NH), 1620 (C
N), 1592 (CC), 1325, 1294 (SO2); 1H-NMR (DMSO-d6): δppm = 2.50 (s, 3H, CH3), 3.82 (s, 3H, OCH3), 3.86 (s, 6H, 2OCH3), 6.33 (s, 2H, Ar–H), 7.42 (s, 2H, NH2), 7.97 (d, J = 8.5 Hz, 2H, Ar–H2,6), 8.01 (d, J = 8.5 Hz, 2H, Ar–H3,5), 8.68 (s, 1H, CH), 10.3 (s, 1H, NH); 13C-NMR (DMSO-d6): δppm = 13.55 (CH3), 56.50 (CH3), 56.64 (CH3), 56.76 (CH3), 91.64 (2CHAr–C3′,5′), 103.76 (Ar–C1′), 122.13 (2CHAr–C2,6), 122.29 (2CHAr–C3,5), 127.34 (Ar–C4), 127.38 (Ar–C1), 142.85 (thiadiazole-C5), 143.07 (CN), 149.4 (CHN), 159.2 (Ar–C4′), 161.98 (Ar–C–O), 168.89 (thiadiazole-C2); MS m/z (%): 490 (M+, 6.9), 489 (8.3), 472 (1.9), 461 (1.9), 443 (2.3), 408 (3.5), 378 (2.7), 354 (2.2), 322 (2.6), 280 (3.2), 266 (3.3), 229 (3.0), 217 (4.6), 196 (3.6), 179 (100), 142 (4.4), 121 (32.4), 76 (26.6), 57 (41.5); anal. calc. For C20H22N6O5S2 (490.55): C, 48.98; H, 4.53; N, 17.12%; found: C, 48.99; H, 4.52; N, 17.13%.
3.1.3.5. 4-[5-(1-((Furan-2-ylmethylene)hydrazineylidene)ethyl)-2-oxo-1,3,4-thiadiazol-3(2H)-yl]benzene sulfonamide (11). The compound was obtained by heating a solution of furan-2-carbaldehyde 10 (0.30 g, 0.003 mol) for 8 h under reflux. Brown powder; (0.50 g, 43%); M.P. 222–223 °C; IR νmax/cm−1 = 3338, 3245 (NH2), 1685 (C
O), 1625 (CN), 1593 (CC), 1325, 1291 (SO2); 1H-NMR (DMSO-d6): δppm = 2.44 (s, 3H, CH3), 6.72 (d, J = 3.4 Hz, 1H, furan-H3), 6.92 (t, J = 3.4 Hz, 1H, furan-H4), 7.21 (d, J = 3.4 Hz, 1H, furan-H2), 7.47 (s, 2H, NH2), 7.98 (d, J = 5.1 Hz, 2CH, Ar–H2,6), 8.01 (d, J = 8.5 Hz, 2CH, Ar–H3,5), 8.48 (s, 1H, NCH); 13C-NMR (DMSO-d6): δppm = 13.68 (CH3), 113.48 (furan-C3), 119.59 (2CHAr–C2,6), 121.68 (furan-C4), 126.91 (2CHAr–C3,5), 139.67 (Ar–C4), 146.64 (Ar–C1), 148.87 (furan-C2), 150.35 (thiadiazole-C5), 151.53 (furan-C5), 152.54 (CN), 159.01 (CNH), 168.29 (CO); MS m/z (%): 391 (M+, 7.5), 390 (0.6), 378 (0.1), 365 (3.0), 341 (0.1), 311 (0.3), 295 (0.1), 280 (0.1), 262 (0.1), 239 (0.7), 221 (0.1), 205 (0.1), 198 (0.2), 188 (0.1), 176 (0.1), 154 (0.2), 128 (0.4), 116 (1.1), 94 (100), 78 (4.7), 65 (6.0); anal. calc. For C15H13N5O4S2 (391.42): C, 46.03; H, 3.35; N, 17.89%; found: C, 46.02; H, 3.36; N, 17.90%.
3.1.4.1. 4-[2-Imino-5-(1-((2-oxoindolin-3-ylidene)hydrazineylidene)ethyl)-1,3,4-thiadiazol-3(2H)-yl]benzenesulfonamide (13). The compound was obtained by heating a solution of indoline-2,3-dione 12 (0.47 g, 0.003 mol) for 4 h under reflux. Green powder; (0.96 g, 73%); M.P. > 300 °C; IR νmax/cm−1 = 3303, 3257 (NH2), 3115, 3019 (2NH), 1717 (C
O), 1607 (CN), 1587 (CC), 1322, 1292 (SO2); 1H-NMR (DMSO-d6): δppm = 2.32 (s, 3H, CH3), 6.90–7.39 (m, 4H, Ar–H), 7.41 (s, 2H, NH2), 7.89 (d, J = 8.5 Hz, 2H, Ar–H2,6), 8.23 (d, J = 8.5 Hz, 2H, Ar–H3,5), 9.54 (s, 1H, NH), 10.92 (s, 1H, indole-NH); 13C-NMR (DMSO-d6): δppm = 13.49 (CH3), 111.12 (2CHAr–C2,6), 116.13 (indole-C4′), 121.22 (indole-C7), 122.47 (indole-C5), 126.48 (indole-C4), 126.94 (2CHAr–C3,5), 128.19 (Ar–C4), 134.34 (indole-C6), 140.80 (indole-C3′), 141.70 (indole-C5′), 145.40 (Ar–C1), 147.30 (thaiadiazole-C5), 154.71 (CN), 158.09 (thiadiazole-C2), 163.91 (CO); MS m/z (%): 441 (M+, 11.5), 440 (2.3), 417 (0.7), 381 (0.7), 367 (1.3), 335 (0.7), 300 (1.7), 292 (0.7), 266 (0.9), 232 (0.6), 206 (2.3), 171 (2.5), 144 (9.3), 104 (100), 65 (41.8), 50 (34.8); anal. calc. For C18H15N7O3S2 (441.48): C, 48.97; H, 3.42; N, 22.21%; found: C, 48.96; H, 3.41; N, 22.22%.
3.1.4.2. 4-[5-(1-((1-((Dimethylamino)methyl)-2-oxoindolin-3-ylidene)hydrazineylidene)ethyl)-2-imino-1,3,4-thiadiazol-3(2H)-yl]benzenesulfonamide (15a). The compound was obtained by heating a solution of 1-((dimethylamino)methyl)indoline-2,3-dione 14a (0.65 g, 0.003 mol) for 3 h under reflux. Brown powder; (1.09 g, 54%); 73%; M.P. > 300 °C; IR νmax/cm−1 = 3303, 3269 (NH2), 3115 (NH), 1721 (C
O), 1609 (CN), 1590 (CC), 1320, 1304 (SO2); 1H-NMR (DMSO-d6): δppm = 2.42 (s, 3H, CH3), 2.50 (s, 6H, 2CH3), 4.36 (s, 2H, CH2–N), 6.92 (d, J = 7.6 Hz, 1CH, indole-H7), 7.06 (t, J =7.6 Hz, 1CH, indole-H5), 7.44(t, J = 7.6 Hz, 1CH, indole-H6), 7.49 (s, 2H, NH2), 7.61 (d, J = 7.6 Hz, 1CH, indole-H4), 7.90 (d, J = 8.5 Hz, 2CH, Ar–H2,6), 8.01 (d, J = 8.5 Hz, 2CH, Ar–H3,5), 10.94 (s, 1H, NH); 13C-NMR (DMSO-d6): δppm = 14.18 (CH3), 40.39 (CH3)2, 65.03 (N–CH2–N), 111.55 (2CHAr–C2,6), 116.49 (indole-C7), 121.17 (indole-C5), 123.06 (indole-C4), 127.85 (2CHAr–C3,5), 128.80 (Ar–C4), 142.00 (Ar–C1), 147.61 (thiadiazole-C5), 153.27 (CN), 154.63 (thiadiazole-C2), 164.35 (CO); MS m/z (%): 498 (M+, 2.6), 497 (3.1), 462 (2.9), 402 (2.6), 336 (3.0), 321 (2.5), 304 (2.6), 270 (3.6), 256 (3.1), 220 (3.8), 202 (2.5), 188 (6.1), 164 (4.0), 128 (7.0), 111 (16.0), 90 (40.7), 67 (31.5), 55 (100); anal. calc. For C21H22N8O3S2 (498.58): C, 50.59; H, 4.45; N, 22.48%; found: C, 50.58; H, 4.44; N, 22.47%.
3.1.4.3. 4-[5-(1-((1-((Diethylamino)methyl)-2-oxoindolin-3-ylidene)hydrazineylidene)ethyl)-2-imino-1,3,4-thiadiazol-3(2H)-yl)benzenesulfonamide (15b). The compound was obtained by heating a solution of 1-((diethylamino)methyl)indoline-2,3-dione 14b (0.74 g, 0.003 mol) for 5 h under reflux. Brown powder; (1.22 g, 77%); M.P. > 300 °C; IR νmax/cm−1 = 3305, 3264 (NH2), 3115 (NH), 1728 (C
O), 1609 (CN), 1589 (CC), 1380, 1304 (SO2); 1H-NMR (DMSO-d6): δppm = 2.03 (t, J = 7.6 Hz, 3H, CH3), 2.34 (s, 3H, CH3), 2.92 (q, J = 7.6 Hz, 2H, CH2), 4.36 (s, 2H, CH2), 6.92 (d, J = 7.6 Hz, 1CH, indole-H7), 7.06 (t, J = 7.6 Hz, 1CH, indole-H5), 7.41 (s, 2H, NH2), 7.45 (t, J =7.6 Hz, 1CH, indole-H6), 7.90 (d, J = 8.5 Hz, 2CH, Ar–H2,6), 8.01 (d, J = 8.5 Hz, 2CH, Ar–H3,5), 9.56 (s, 1H, NH); 13C-NMR (DMSO-d6): δppm = 11.52 (CH3), 14.18 (CH3), 41.61 (CH2N), 65.03 (N–CH2–N), 111.58 (2CHAr–C2,6), 116.49 (indole-C4), 121.17 (indole-C7), 123.05 (indole-C5), 126.95 (indole-C4), 127.34 (2CHAr–C3,5), 127.85 (Ar–C4), 128.74 (1CHAr–C6), 134.83 (CN–N), 142.00 (Ar–C1), 145.85 (1H-indole-C5′), 147.61 (thiadiazole-C5), 148.07 (CN), 154.60 (thiadiazole-C2), 164.29 (CO); MS m/z (%): 526 (M+, 1.4), 451 (1.9), 409 (1.7), 383 (2.5), 369 (4.7), 325 (2.0), 299 (3.5), 265 (2.9), 241 (1.4), 208 (1.8), 175 (1.9), 166 (2.2), 145 (3.0), 123 (12.7), 110 (10.3), 91 (8.5), 69 (58.8), 57 (100); anal. calc. For C23H26N8O3S2 (526.63): C, 52.46; H, 4.98; N, 21.28%; found: C, 52.45; H, 4.97; N, 21.29%.
3.2. Computational details
Gaussian 09 package software43 was used to carry out all quantum chemical calculations using the method of DFT-B3LYP12,13 combined with a standard 6-31G(d) basis set. The suggested geometries for the synthesized compounds were initially optimized using Pulay's gradient approach.44 Frequency calculations were performed to verify that the optimized geometries are actual minimums with real wavenumbers. The geometry of the E and Z forms of 5 were reoptimized in DMSO solution using the IEF-PCM solvation model,45 followed by 1H and 13C NMR chemical shift calculations applying the GIAO approach.14,15 The multi-standard approach24,25 was applied to get the theoretical chemical shifts for E and Z isomers using the isotropic magnetic shielding values (σi, ppm) for H and C atoms acquired from the Gaussian output file. In this approach, methanol and benzene were used as references to predict sp3 and sp2 hybridized Cs/C–Hs, respectively, whereas comparable skeletons were used in the case of N–H protons. Thus, benzenesulfonamide,46 acetophenonehydrazone,47 and 5-iso-propyl-3-methyl-2-imino-1,3,4-thiadiazole48 were used as references to calculate the N–H chemical shifts in the benzenesulfonamide, hydrazone and iminothiadiazoles moieties, respectively. Furthermore, the HOMO/LUMO (FMOs) and energy gaps were predicted for the optimized geometries of the synthesized compounds 7a–c, 9, 11, 13, 15a and 15b. Accordingly, molecular reactivity descriptors such as ionization potential, electron affinity, hardness, chemical potential, and electrophilicity index were calculated using the values of energies for FMOs.28,29 The synthesized compounds' pharmacokinetics, drug-like characteristics, and toxicities were evaluated using the SwissADME20 and pkCSM21 online servers.3.3. Biological evaluation
4. Conclusions
Herein, the combi-molecule strategy was used to synthesize a bundle of differently hybrid 1,3,4-thiadiazole sulfonamide derivatives and unsymmetrical azines 7a–c, 9, 11, 13, 15a and 15b started with 5-acetyl-3-N-(4-sulfamoylphenyl)-2-imino-1,3,4-thiadiazoline 4. In the solution phase, compound 5 exists as a mixture of E and Z configurations, according to the results of computed/observed 1H and 13C NMR chemical shifts. The DFT-B3LYP calculations favor the final synthesized products in the EE configuration with reference to the azomethine CN bonds. For the newly synthesized compounds, quantum chemical descriptors and drug-likeness properties were predicted and correlated to their in vitro bioactivities. Experimentally, the synthesized compounds were investigated for their anticancer effects in vitro against cancer cell lines: HepG-2, Caco-2, MCF-7, and WI-38, where they exhibited promising results.
Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
The authors thank the Deanship of Scientific Research at King Khalid University for funding this work through a research group project (Grant no. RGP.1/98/43).References
- M. Wu, M. Wang, H. Jia and P. Wu, Drug Delivery, 2022, 29, 2513–2538 CrossRef CAS PubMed.
- M. Szumilak, A. Wiktorowska-Owczarek and A. Stanczak, Molecules, 2021, 26, 2601 CrossRef CAS PubMed.
- Y. Wan, G. Fang, H. Chen, X. Deng and Z. Tang, Eur. J. Med. Chem., 2021, 226, 113837 CrossRef CAS PubMed.
- A. Ovung and J. Bhattacharyya, Biophys. Rev., 2021, 13, 259–272 CrossRef CAS.
- J. E. Lesch, The first miracle drugs: how the sulfa drugs transformed medicine, Oxford University Press, USA, 2007 Search PubMed.
- M. Mustafa and J. Y. Winum, Expert Opin. Drug Discovery, 2022, 17, 501–512 CrossRef CAS.
- A. K. Jain, S. Sharma, A. Vaidya, V. Ravichandran and R. K. Agrawal, Chem. Biol. Drug Des., 2013, 81, 557–576 CrossRef CAS PubMed.
- M. F. Khan, G. Verma, W. Akhtar, M. Shaquiquzzaman, M. Akhter, M. A. Rizvi and M. M. Alam, Arabian J. Chem., 2019, 12, 5000–5018 CrossRef CAS.
- G. H. Elgemeie, R. A. Azzam and R. E. Elsayed, Med. Chem. Res., 2019, 28, 1099–1131 CrossRef CAS.
- G. Wang, Q. Han and R. Zhu, J. Mol. Model., 2022, 28, 393 CrossRef CAS.
- S. Fortin and G. Bérubé, Expert Opin. Drug Discovery, 2013, 8, 1029–1047 CrossRef CAS.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- S. G. Patel, R. M. Vala, P. J. Patel, D. B. Upadhyay, V. Ramkumar, R. L. Gardas and H. M. Patel, RSC Adv., 2022, 12, 18806–18820 RSC.
- E. Eroglu and H. Türkmen, J. Mol. Graphics Modell., 2007, 26, 701–708 CrossRef CAS.
- D. B. Chesnut and C. G. Phung, J. Chem. Phys., 1989, 91, 6238–6245 CrossRef CAS.
- S. Bharadwaj, A. Dubey, U. Yadava, S. K. Mishra, S. G. Kang and V. D. Dwivedi, Briefings Bioinf., 2021, 22, 1361–1377 CrossRef CAS.
- A. Sheikh Ali, D. Khan, A. Naqvi, F. F. Al-Blewi, N. Rezki, M. R. Aouad and M. Hagar, ACS Omega, 2020, 6, 301–316 CrossRef.
- P. K. Singh, J. Singh, T. Medhi and A. Kumar, ACS Omega, 2022, 7, 33067–33078 CrossRef CAS PubMed.
- İ. Çapan, S. Servi, İ. Yıldırım and Y. Sert, ChemistrySelect, 2021, 6, 5838–5846 CrossRef.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef PubMed.
- D. E. V. Pires, T. L. Blundell and D. B. Ascher, J. Med. Chem., 2015, 58, 4066–4072 CrossRef CAS PubMed.
- V. T. Angelova, V. Valcheva, N. G. Vassilev, R. Buyukliev, G. Momekov, I. Dimitrov, L. Saso, M. Djukic and B. Shivachev, Bioorg. Med. Chem. Lett., 2017, 27, 223–227 CrossRef CAS PubMed.
- H. Hellmann and I. Löschmann, Chem. Ber., 1954, 87, 1684–1690 CrossRef CAS.
- A. M. Sarotti and S. C. Pellegrinet, J. Org. Chem., 2009, 74, 7254–7260 CrossRef CAS.
- A. M. Sarotti and S. C. Pellegrinet, J. Org. Chem., 2012, 77, 6059–6065 CrossRef CAS PubMed.
- S. Tighadouini, S. Radi, F. Abrigach, R. Benabbes, D. Eddike and M. Tillard, J. Chem. Inf. Model., 2019, 59, 1398–1409 CrossRef CAS.
- A. I. Khodair, M. K. Awad, J.-P. Gesson and Y. A. M. M. Elshaier, Carbohydr. Res., 2020, 487, 107894 CrossRef CAS PubMed.
- T. Koopmans, Physica, 1934, 1, 104–113 CrossRef.
- R. G. Parr, L. v. Szentpály and S. Liu, J. Am. Chem. Soc., 1999, 121, 1922–1924 CrossRef CAS.
- J. I. Aihara, J. Phys. Chem. A, 1999, 103, 7487–7495 CrossRef CAS.
- Y. N. Mabkhot, F. D. Aldawsari, S. S. Al-Showiman, A. Barakat, S. M. Soliman, M. I. Choudhary, S. Yousuf, M. S. Mubarak and T. B. Hadda, Chem. Cent. J., 2015, 9, 24 CrossRef PubMed.
- R. Srivastava, ACS Omega, 2021, 6, 24891–24901 CrossRef CAS PubMed.
- A. R. Bhat, R. S. Dongre, F. A. Almalki, M. Berredjem, M. Aissaoui, R. Touzani, T. B. Hadda and M. S. Akhter, Bioorg. Chem., 2021, 106, 104480 CrossRef CAS PubMed.
- S. K. Nagarajan, S. Babu, H. Sohn and T. Madhavan, ACS Omega, 2020, 5, 21145–21161 CrossRef CAS PubMed.
- C.-G. Zhan, J. A. Nichols and D. A. Dixon, J. Phys. Chem. A, 2003, 107, 4184–4195 CrossRef CAS.
- L. R. Domingo and P. Pérez, Org. Biomol. Chem., 2011, 9, 7168–7175 RSC.
- M. Kouza, A. Banerji, A. Kolinski, I. Buhimschi and A. Kloczkowski, Molecules, 2018, 23, 1995 CrossRef.
- M. Marinescu, A. Emandi, G. Marton, L. O. Cinteza and C. Constantinescu, Nanosci. Nanotechnol. Lett., 2015, 7, 846–854 CrossRef.
- J. G. Cumming, A. M. Davis, S. Muresan, M. Haeberlein and H. Chen, Nat. Rev. Drug Discovery, 2013, 12, 948–962 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- M. M. Hammouda, A. A. Elmaaty, M. S. Nafie, M. Abdel-Motaal, N. S. Mohamed, M. A. Tantawy, A. Belal, R. Alnajjar, W. M. Eldehna and A. A. Al-Karmalawy, Bioorg. Chem., 2022, 127, 105995 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09, (Revision B.01), Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- P. Pulay, Mol. Phys., 1969, 17, 197–204 CrossRef CAS.
- J. Tomasi, B. Mennucci and E. Cancès, J. Mol. Struct.: THEOCHEM, 1999, 464, 211–226 CrossRef CAS.
- S. L. Graham and T. H. Scholz, Synthesis, 1986, 1986, 1031–1032 CrossRef.
- T. Kleine, R. Fröhlich, B. Wibbeling and E.-U. Würthwein, J. Org. Chem., 2011, 76, 4591–4599 CrossRef CAS PubMed.
- D. Leppard and H. Sauter, J. Heterocycl. Chem., 1980, 17, 1469–1472 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra00123g |
| This journal is © The Royal Society of Chemistry 2023 |