
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCargo-loaded lipid-shielded breakable organosilica nanocages for enhanced drug delivery†
María
Sancho-Albero‡
 a,
Giada
Rosso‡
b,
Luisa
De Cola
*ac and
Valentina
Cauda
*b
a,
Giada
Rosso‡
b,
Luisa
De Cola
*ac and
Valentina
Cauda
*b
aDepartment of Biochemistry and Molecular Pharmacology, Istituto di Ricerche Farmacologiche Mario Negri IRCCS, Via Mario Negri, 2, 20156, Milan, Italy. E-mail: luisa.decola@marionegri.it
bDepartment of Applied Science and Technology, Politecnico di Torino, Corso Duca degli Abruzzi 24, 10129 Turin, Italy. E-mail: valentina.cauda@polito.it
cDepartment of Pharmaceutical Science, DISFARM, Università degli Studi di Milano, 20133, Milan, Italy
First published on 24th August 2023
Abstract
The recent nanomedicine advancements have introduced a variety of smart nanoparticles in cancer treatment and diagnostics. However, their application to the clinic is still hindered by several challenges related to their biocompatibility, elimination and biodistribution. Here we propose breakable organosilica mesoporous nanoparticles, i.e. nanocages, able to efficiently incorporate cargo molecules and be coated, with different lipid compositions, to enhance their biomimetic behaviour. We exploit the electrostatic interactions between the organosilica surface and the opposite charge of the lipid mixtures in order to obtain an efficient organosilica coverage. The lipid-coated nanocages are proved to have an incredibly high hemocompatibility, significantly increased with respect to pristine nanocages, and excellent colloidal stability and biocompatibility. The cargo-loaded and lipid-coated nanocages are tested and compared in vitro on two different cancer cell lines, demonstrating the key role played by the lipid coating in mediating the internalization of the nanocages, evaluated by the enhanced and rapid cellular uptake. The efficient intracellular delivery of the therapeutic agents is then assured by the destruction of the organosilica, due to the disulfide bridges, introduced into the silica framework, that in reducing media, like the intracellular one, are reduced to thiols causing the breaking of the nanoparticles. The possibility to image and effectively kill cancer cells demonstrates the potentiality of the lipid-coated nanocages as a powerful tool in anticancer research and as a promising smart theranostic platform.
Introduction
Within the broad literature panorama of drug-delivery nanosystems, mesoporous silica-based nanoparticles offer unique properties, such as a very high porosity (up to 1 cm3 g−1) and a huge surface area (up to 1200 m2 g−1), with very uniform and easily tunable pore sizes (ranging from 2 up to 15 nm).1,2 Silica-based nanoparticles, having a size from 20 to 200 nm, have shown excellent biocompatibility and have been broadly proposed as smart nano-sized platforms for theranostic applications3 and for carrying and releasing drugs in a site-selective, controlled, and even mechanized manner.4–6 Recently porous organosilica nanoparticles have been reported with a cage-like morphology.7–9 De Cola's group realized a breakable structure, using disulfide-bridges as redox responsive units in the silica network.10,11 These organosilica nanocages, ssOSCs, have been proved to easily degrade in reducing media, such as those typically present intracellularly, and in view of their single large cavity, they have been established to successfully deliver different cargo molecules, ranging from large biomolecules12,13 to small anticancer drug molecules.14,15 Importantly, some of us have recently reported the unique capability of 20 nm disulfide-bridged organosilica nanocages to avoid phagocytosis by hepatic macrophages,10 achieving an important step towards the possible translation of these stimuli-responsive organosilica nanocages to clinical applications. Drug delivery was also demonstrated for malignant mesothelioma and the reported in vivo experiments on the reduction of the tumor growth corroborate the possible use of such particles.16The silica surface, rich in hydroxyl groups, offers not only a highly-hydrophilic nature, but also efficacious chemical anchoring sites for functional molecules,17 such as targeting ligands,18 polymers19 and lipid bilayers20 able to improve the biodistribution and colloidal stability of the porous silica nanoparticles. In fact, the colloidal stability of a generic nanoparticle in biological media has to be strictly preserved to avoid the aggregation and degradation phenomenon,21 as it can negatively affect the biodistribution, the endocytosis process and the delivery of the entrapped active molecules to the target tissue. A strategy was indeed proposed, consisting of phospholipidic bilayers encapsulating solid-state nanoparticles, and among others also mesoporous silica.22 This approach offers several advantages: besides guaranteeing the colloidal stability of nanoparticles in biological media, it increases their biocompatibility and biomimicking features and improves the interaction with the cell membrane, facilitating the endocytosis process.23 The lipidic bilayer also offers a valid barrier to the rapid drug leakage out of the nanoparticle pores,20,24 avoiding off-target release, while assuring the drug delivery only intracellularly. Indeed, literature reports have shown the proficient use of lipid bilayer-coated nanoparticles for effective biostabilization in biological media25 and for efficient cell internalization. Moreover, in combination with mesoporous silica nanoparticles, lipidic bilayers allowed the retention of anticancer drugs, such as doxorubicin, paclitaxel, curcumin, irinotecan,26–28 enzyme inhibitors,24 and nucleic acids, such as mRNA or dsDNA.29,30
However, efficient endocytosis needs to be coupled with an effective endosomal escape to intracellularly release the therapeutic content. For this reason, some solutions were developed to overcome the endosomal membrane “barrier” and allow the therapeutic content to efficiently reach the cell sub-compartment. Towards this goal, mesoporous silica, coated by an artificial lipid bilayer, can be equipped with stimuli responsive surface modifiers, e.g. to induce an osmotic pressure inside the endosome,31 or photoactive molecules32–34 to escape from the endosomal compartments. Another successful strategy relies on the nanoparticle degradation itself, triggered by acidic pH or redox reactions.23,35
In this work, we report breakable organosilica nanocages, ssOSCs, containing disulfide groups in the silica structure, coated by lipidic bilayers. The nanocages are able to encapsulate an anticancer drug, doxorubicin, and are effectively internalized into cancer cells, functioning as an anticancer therapy. We demonstrate that these nanostructures can be covered with different lipid mixtures using electrostatic interactions, and the resulting hybrids maintain high colloidal stability and, most importantly, improve the cellular uptake and hemocompatibility. Remarkably, we observed that lipid bilayers can modify the cell interaction of the disulfide-bridged organosilica nanocages: once they are coated by a negatively charged lipid mixture, a prompt internalization is observed, guaranteeing a successful cargo release into cancer cells. Therefore, the combination of two sophisticated strategies, i.e. the breakable organosilica carrying an anticancer cargo and a biomimicking lipid bilayer, produces a smart hybrid nanoparticle with improved properties and is ready to be further tested in a preclinical context.
Results and discussion
The disulfide-breakable nanoparticles (ssOSCs) were synthesized using a sol–gel template assisted method, following a reported procedure,11 together with non-breakable nanocages (OSCs)8 for comparison. After their purification by dialysis, to eliminate the template, the nanomaterials were further functionalized with amino–propyl groups to achieve a net positive surface charge in order to promote electrostatic interactions with the lipid mixtures.Characterization of naked and functionalized nanoparticles
The obtained nanostructures were characterized with a variety of techniques to assess their morphology, size, porosity and functionalization. Transmission Electron Microscopy (TEM) images revealed the typical cage structure11 with the presence of a single pore of about 10 nm (Fig. S1A†). The size of the nanocages, estimated at around 18 nm, was in excellent agreement with the hydrodynamic diameter analysed by DLS (Fig. S1B†). As expected, the NPs possessed a negative ζ potential of −11 ± 0.5 mV. In order to corroborate the proper elimination of the surfactant employed during the synthesis procedure and to evaluate the presence of the S–S bonds that provide the NPs with breakable properties, Fourier Transform Infrared Spectroscopy (FTIR) and thermogravimetric analysis (TGA) were also performed. ssOSCs exhibited typical FTIR spectra characteristic of organosilica NPs with the Si–OH (3445 cm−1), Si–O–Si (1077 cm−1) Si–OH (947 cm−1) and C–H (805 cm−1) vibrational peaks coming from the organosilica framework. When comparing the FTIR spectra of OSCs and ssOSCs, the 2927–2868 cm−1 bands (attributed to the alkyl chain of the methylene groups of the breakable linker present in the NPs) were observed (inset of Fig. S1C†). The TGA study of the non-breakable OSCs indicates that the CTAB employed in the synthetic approach was completely eliminated. Almost 100% of the sample weight corresponded with the silica structure (Fig. S1D†). However, when comparing the TGA performed on OSCs and ssOSCs (Fig. S1E†), the number of breakable groups introduced into the silica framework was quantified (around 10% of sample weight). This is consistent with the previously published results.11,36 Nitrogen adsorption–desorption experiments were also performed to assess the porosity of the NPs. The results revealed the size of the particle's pore to be around 6–8 nm, in accordance with TEM analysis and with previous works (Fig. S1F and S1G†).11In order to create an electrostatic interaction between the ssOSCs and the phospholipids, the nanocages were functionalized with amino groups (ssOSCs–NH2) that are protonated under physiological conditions, leading to positive charges. TEM images depicted in Fig. 1A indicate that the functionalized ssOSCs preserved their morphology and size even after functionalization with amino silane (see the Experimental section for further details). DLS (Fig. 1B) revealed a hydrodynamic diameter of around 30 nm and a positive ζ potential (+23.6 ± 0.7 mV) was also obtained, corroborating the correct surface functionalization of the ssOSCs with the amino groups. Similarly, by FTIR analysis (Fig. 1C) it was possible to confirm, besides the other bands, the characteristic fingerprint of amino silane employed for the NPs functionalization, and the bands at 3378 and 1416 cm−1 were attributed to the NH2 groups. When comparing TGA of the naked and the NH2 functionalized ssOSCs (Fig. 1D), an estimate of a functionalization corresponding to 5 to 10% in weight of the amino groups was achieved.
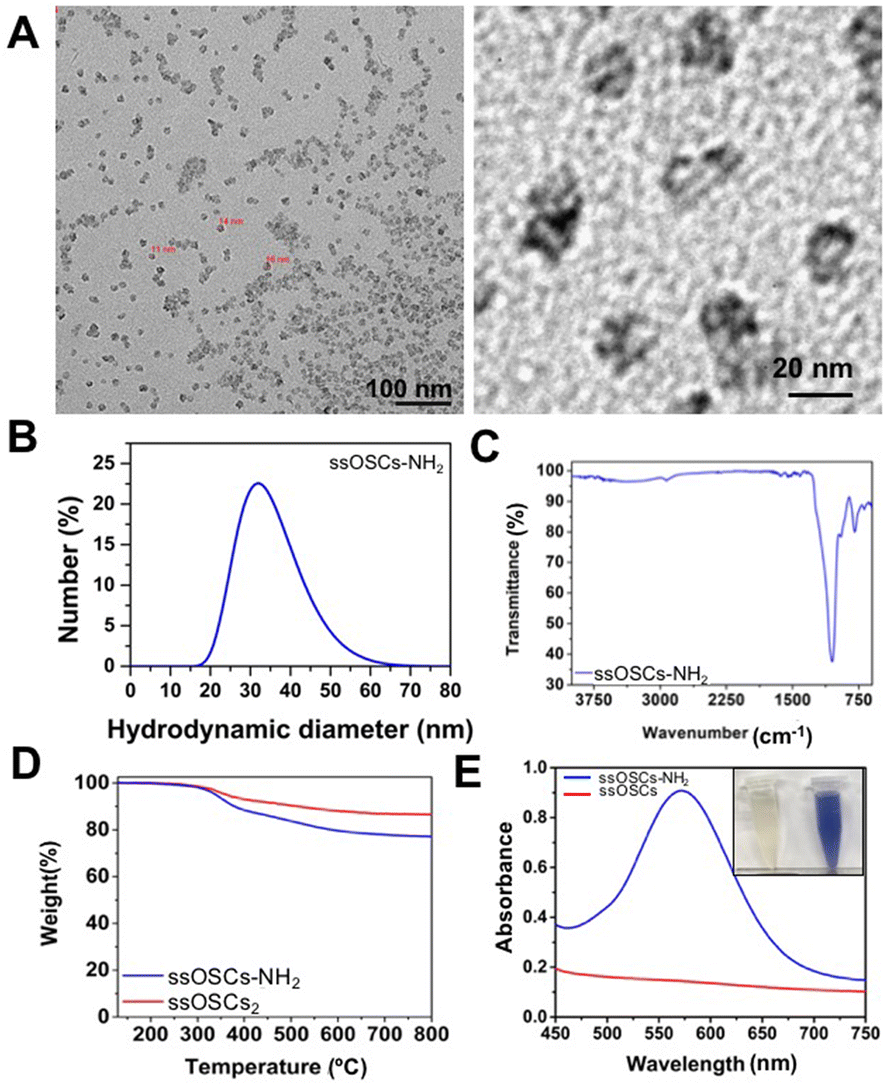 | ||
| Fig. 1 Characterization of the breakable ssOSCs–NH2. (A) Transmission electron microscopy images of the ssOSCs functionalized with NH2 groups. (B) DLS analysis, (C) FTIR spectrum, (D) TGA of ssOSCs–NH2 compared to ssOSCs. (E) Kaiser test for determining primary amines with the maximum absorption peak at 570 nm. | ||
Finally, the colorimetric Kaiser test allowed us to determine the presence of the positively-charged amine groups and roughly estimate the amount of the amine groups on the silica surface. Fig. 1E shows that the ssOSC solution remained colorless (when no free primary amines were present); in contrast, the ssOSCs–NH2 solution turned dark blue, indicating the presence of primary amines. The absorption spectra of both solutions recorded from 450 nm to 750 nm are shown in Fig. 1E. Considering the absorbance obtained at 570 nm and taking into account the density of SiO2 (2.65 g mL−1), the diameter of NPs obtained by TEM and the sample concentration, we roughly estimated the presence of 105 NH2 molecules per particle (Table S1 in the ESI†).
Lipid bilayer shell formation
Lipidic shell formation is fundamental to achieve high stability in biological media and a superior biocompatibility of the nanocages as well as to efficiently retain the adsorbed drug and achieve improved cell internalization.In order to evaluate the effect of the nanoparticle's surface functionalization and its interaction with differently charged lipids, various combinations of nanoparticles and lipid formulations were tested and characterized (Fig. 2). In particular, ssOSCs and ssOSCs–NH2 were combined with two lipid mixtures: one possessing a neutral charge and one with a negative net charge, as already reported by some of us.22 We indeed expected that the correct matching between the superficial charge of the nanoparticles and the charge manifested by the lipid mixture could promote efficient encapsulation of the nanoparticles, through favourable electrostatic interactions. All combinations of lipids and nanoparticles were thus tested by DLS, ζ potential measurements, and fluorescence microscopy to verify our hypothesis. Additionally, nanoparticles without lipid coverage and liposomes without nanoparticles were tested as controls. Interestingly, the DLS measurements in Fig. 2A evidenced a shift in the hydrodynamic diameter from the naked and unfunctionalized nanocages (26 nm) to the coated ones with a neutral lipid mixture (50 nm); the peak of these lipid-coated ssOSCs is also distant from that of neutral liposomes as the control, which have a size distribution peaking at 28 nm, approximately in the same range of pristine uncovered ssOSCs, but with a broader size distribution. Similar observations can be made for ssOSCs–NH2 combined with neutral lipids, as shown in Fig. 2B. The measurements evidenced a clear shift in the size distribution from uncoated (28 nm) to coated ssOSCs–NH2 (78 nm). In contrast, the negative lipid formulation combined with pristine ssOSCs (Fig. 2C) did not produce a narrow size distribution, but a range of sizes with two larger populations (peaks at 33 and 114 nm). This result indicates the presence of particles which could be a mixture of covered and uncovered ssOSCs and even free liposomes and suggests an unsuccessful nanocage encapsulation in the lipidic shell, probably due to electrostatic repulsions.
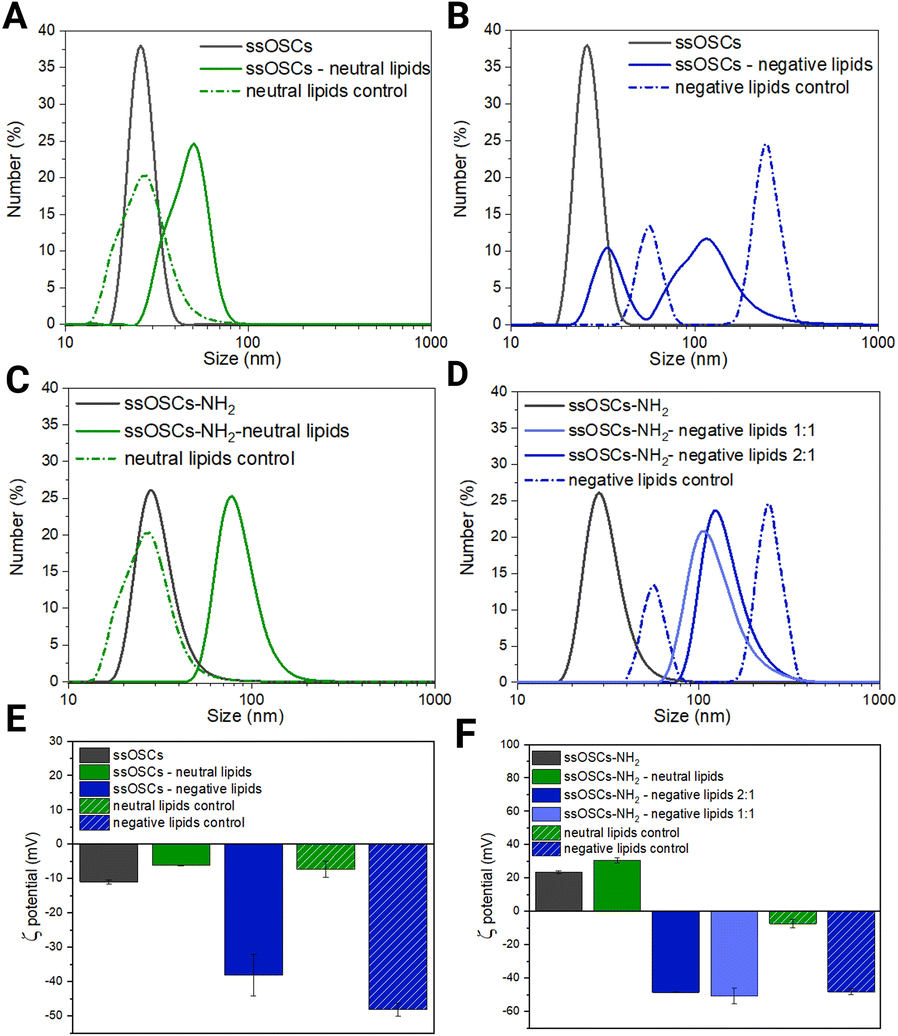 | ||
Fig. 2 Characterization of lipid coated nanoparticles. (A) DLS results of non-functionalized ssOSCs, uncovered and covered with neutral lipids, and control liposomes; (B) DLS results of non-functionalized ssOSCs, uncovered and covered with negative lipids, and control liposomes; (C) DLS results of NH2-functionalized ssOSCs uncovered and covered with neutral lipids and control liposomes; (D) DLS results of NH2-functionalized ssOSCs uncovered and covered with negative lipids and control liposomes; for negative lipids, two ssOSCs:lipids mass ratios were tested: 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:1; (E) ζ potential results of non-functionalized ssOSCs, covered with neutral and negative lipids; and (F) ζ potential results of non-functionalized ssOSCs, covered with neutral and negative lipids; for negative lipids, two ssOSCs:lipids mass ratios were tested: 2:1 and 1:1. A table of the above plotted data is available in the ESI (Table S2†). :1 and 1:1; (E) ζ potential results of non-functionalized ssOSCs, covered with neutral and negative lipids; and (F) ζ potential results of non-functionalized ssOSCs, covered with neutral and negative lipids; for negative lipids, two ssOSCs:lipids mass ratios were tested: 2:1 and 1:1. A table of the above plotted data is available in the ESI (Table S2†). | ||
The coating of ssOSCs–NH2 with negative lipids was explored for two different lipid amounts (mass ratios 2:1 and 1:1 of ssOSCs:lipids, see Fig. 2D); both lipid coatings produced a uniform and significant shift in size distribution, with respect to the pristine nanocages. The size distribution peaks were at 124 nm and 108 nm for 2:1 and 1:1 mass ratio, respectively, and were very different from the two peaks obtained by the negative control liposomes (56 and 246 nm), also characterized by the highest polydispersity index (PdI = 1, see Table S2 in the ESI†). Interestingly the different ratios do not lead to markedly different sizes suggesting, already at a low ratio, good coverage of the nanocages by the lipids. Fig. 2E and F show the ζ potential results. The value obtained for the pristine ssOSCs (Fig. 2E) was −11 ± 0.5 mV; for the control neutral liposomes, it is −7.3 ± 2.4 mV and for the negative control liposome, it is −48.2 ± 1.8 mV. Both zeta potentials of neutral and negative lipid-coated ssOSCs were slightly negative: −6.2 ± 0.12 mV and −13.7 ± 0.3 mV, respectively, with neither reaching the desired colloidal stability. On the other hand, ssOSCs–NH2 showed a positive ζ potential due to the presence of protonated amine groups on the surface and, when covered with the neutral lipid mixture, they evidenced a ζ potential of +30.7 ± 1.5 mV. The ssOSCs–NH2 coated with the negative lipid mixture showed a ζ potential of −48.6 ± 0.3 mV and −50.7 ± 4.8 mV for 2:1 and 1:1 nanocages to lipid mass ratios respectively. These data are also summarized in Table S1.† Interestingly, the addition of negatively charged lipids on the ssOSCs–NH2 sample produced a drastic shift in the ζ potential value, which was very similar to the free negative liposomes, indicating a well-formed lipid bilayer on the functionalized silica cores due to the favourable electrostatic interaction. In both cases, the absolute value of the zeta potential for the lipid coated ssOSCs–NH2 was higher than +30 mV, indicating the colloidal stability of the obtained nanoconstructs. Taken together, the ζ potential and DLS results suggest that functionalization of the nanocages is fundamental to achieve an effective, stable, homogeneous lipid bilayer coverage of the negative lipid mixture because of an optimal electrostatic interaction, with the positively charged ssOSCs–NH2. Fig. S2† presents the nanoparticle tracking analysis results of the ssOSC–NH2-negative lipid sample with a 2:1 nanoparticle to lipid ratio, which shows a mode size of 125 nm, confirming DLS data and corroborating the achievement of a quite narrow and an almost Gaussian-shaped distribution.
To further evaluate the efficacy of the self-assembly of lipids around the nanocages, a fluorescence colocalization analysis was performed. ssOSCs were covalently labelled with a red cyanine (Cy5) fluorescent dye through the coupling of the sulfo–Cy5–NHS derivative with the amino groups grafted on the nanocages. Independently, the lipids were labelled with DiO, a lipophilic dye, exhibiting green emission. Although the resolution limit dictated by Abbey's law does not permit appreciation of the core–shell structure, the observation of the spots collected from the two channels suggests the co-localization of lipids and nanocages. In Fig. 3 it is clearly visible that the ssOSC–NH2-negative lipid sample with a 2:1 nanocage to lipid ratio presented the best colocalization of the lipidic shells with the organosilica cores, since almost the totality of the spots appears white. This means that the visible objects in the far-red channel and in the green one were completely superimposed. The number of white spots was remarkably lower for the other samples, where single green or magenta spots were identified, indicating the presence of free nanoparticles or liposomes without the organosilica core. Considering the multitude of images collected in the two fluorescence channels, an estimation of the encapsulation efficacy was performed. Remarkably, the calculated colocalization percentages (reported in Tables S3 and S4, ESI†) reflected the scenario prospected by DLS and ζ potential measurements. The best results were again obtained for the NH2-functionalized nanocages covered by negative lipids, achieving a colocalization percentage above 80%. In contrast, both ssOSCs and ssOSC–NH2 covered with neutral lipids showed a moderate colocalization percentage of 65.7% and 50.1% on average, respectively. Finally, the lowest colocalization percentage (5.3%) was registered for the ssOSC-negative lipid sample, which showed the poorest size distribution among all lipid-coated samples, together with a low ζ potential, despite the charge of the lipids. Regarding the coupling between negative lipid formulation and functionalized nanocages, which was expected to be the best match, two different ssOSCs–NH2 to lipid (w/w) ratios were tested and compared: 1:1 and 2:1. While DLS (Fig. 2D) and the ζ potential measurements (Fig. 2F) showed rather similar results, the colocalization percentage was higher (92%) for the 2:1 ratio, compared to the 1:1 ratio (82%). For this reason, the nanocages to lipid ratio equal to 2:1 w/w was chosen for the subsequent experiments. Fig. 3 shows a representative set of fluorescence microscopy images (merged channels) of the lipid coated nanoparticles, whereas single-channel images are reported in Fig. S3 and S4 of the ESI.†
 | ||
| Fig. 3 Representative set of multichannel epifluorescence microscopy images of ssOSCs (top) and ssOSCs–NH2 (bottom) coated with neutral (left) and negative (right) lipids. Merged channels and their relative close-ups are here reported. The far-red channel (magenta) represents Cy5-labelled nanoparticles, and the green channel (green) represents DiO-labelled lipids. The colocalized particles are visible as white spots in the merged image. Samples were prepared with a ssOSCs/lipids mass ratio equal to 2:1. Scale bar = 10 μm; magnification = 100×. | ||
FTIR analysis of the negative lipid-coated and the naked ssOSCs–NH2 (Fig. S5†) confirmed once again the correct wrapping of the phospholipid coating around the silica core: together with the silica characteristic bands (previously described), the typical phospholipid fingerprinted peaks37,38 were observed. In particular, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (1765–1720 cm−1), PO2 (1200–1145 cm−1 and 830–740 cm−1), P–O–C (1145–970 cm−1), P–O–C + PO2 (1200–970 cm−1) and CH2 (2926–2855 cm−1) vibrational bands were clearly observed. TGA confirmed the last results and demonstrated the effective coating of the nanocages with the lipids (almost 50% of the sample weight corresponds to the lipidic fraction).
O (1765–1720 cm−1), PO2 (1200–1145 cm−1 and 830–740 cm−1), P–O–C (1145–970 cm−1), P–O–C + PO2 (1200–970 cm−1) and CH2 (2926–2855 cm−1) vibrational bands were clearly observed. TGA confirmed the last results and demonstrated the effective coating of the nanocages with the lipids (almost 50% of the sample weight corresponds to the lipidic fraction).
Hemocompatibility
To assess the role of the lipid coating in the biocompatibility of the nanoparticles, a hemocompatibility test was performed, evaluating the coagulation time of plasma, caused by a coagulating agent, in the presence of pristine and lipid-coated nanocages. The different samples were added to plasma citrate; successively, the obtained plasma and nanoparticles suspensions were treated with calcium chloride (CaCl2), which nullifies the anticoagulant effects of the citrate, enabling the plasma to clot. The coagulation time was calculated from the measured absorbance curves (Fig. S6†) for pristine and negative lipid-coated ssOSCs and ssOSCs–NH2, as described in the Experimental section. The obtained coagulation times are reported in Fig. 4. Results indicate that ssOSCs showed the shortest coagulation time and thus the lowest hemocompatibility. Interestingly, also ssOSC-negative lipids showed a similarly short coagulation time, suggesting that the lack of functionalization is detrimental to the haemocompatibility of the nanocages. Both these results were significantly different from the control sample, consisting of plasma treated with a physiological solution.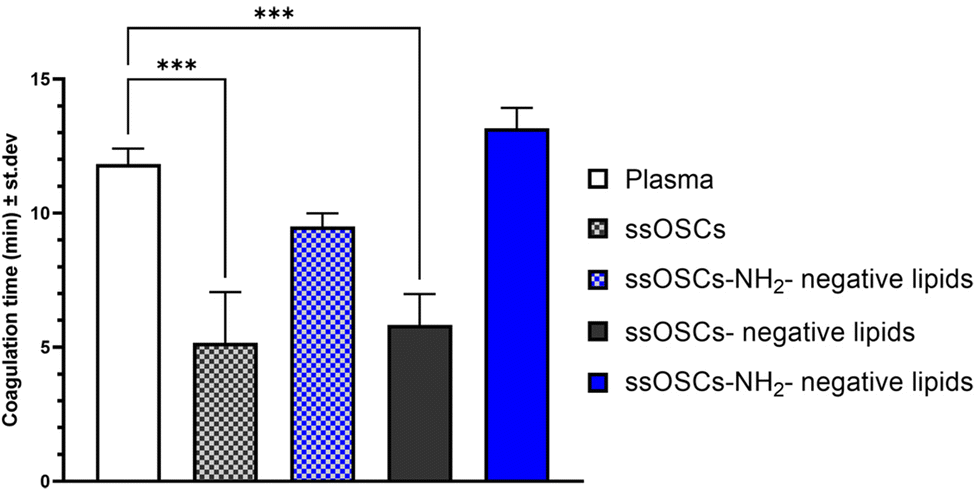 | ||
| Fig. 4 Hemocompatibility assessment through plasma recalcification assay of pristine and negative lipid-coated ssOSCs and ssOSCs–NH2, suspended in a physiological solution. The control sample (plasma) consists of plasma treated with pure physiological solution. The higher the coagulation time, the higher the hemocompatibility. Statistically significant differences were indicated as follows: ***p < 0.001. | ||
Remarkably, both ssOSCs–NH2 and ssOSCs–NH2-negative lipids showed coagulation times that were not significantly different from the control sample: the ssOSC–NH2 had an average coagulation time slightly inferior to the control and the ssOSCs–NH2-negative lipids even slightly superior. These results corroborate the data obtained with the previous characterization methods; in fact, the fast coagulation time of ssOSC-negative lipids can be interpreted as proof of an unsuccessful encapsulation of the nanocages, supporting the hypothesis that the functionalization with aminopropyl groups is fundamental. Remarkably, ssOSC–NH2-negative lipids are characterized by the highest coagulation time, confirming that lipids confer to the nanoparticles a superior hemocompatibility. Moreover, the higher coagulation time reported for lipid-coated ssOSCs–NH2 with respect to the uncoated ones is indirect proof of the optimal encapsulation achieved with these functionalized nanocages and the negatively charged lipid formulation.
Cell internalization
In order to test not only the colloidal stability of the lipid-coated nanocages, but also their improved cell internalization, confocal fluorescence microscopy measurements were performed in two different cell lines. The images reported in Fig. S7† show the cellular uptake of ssOSCs, ssOSCs–NH2 and ssOSCs–NH2-negative lipids (0.1 mg mL−1) after 8 h, 24 h and 48 h in contact with two cancer cell lines of melanoma (B16-BL6) and prostate carcinoma (PC-3). While almost no internalization was observed for a short time for the pristine and amine-functionalized nanocages, a more prominent internalization was achieved when nanocages were coated by the negative lipid formulation (mass ratio 2:1 ssOSCs:lipids). The internalization dynamics of the ssOSCs–NH2 and ssOSCs–NH2-negative lipids were also evaluated, using Cy5-labelled NPs, by flow cytometry (Fig. 5). The in vitro uptake of the lipid coated ssOSCs–NH2 was found to be higher than that of uncoated NPs. This trend was clearly observed for all the tested incubation times (2, 4, 6 and 24 h). Moreover, flow cytometry results indicate time-dependent internalization kinetics. These results are in agreement with the previously published works indicating that lipid shell increases the colloidal stability of nanomaterials and favors their entrance into the intracellular compartments compared to naked NPs.25,39,40
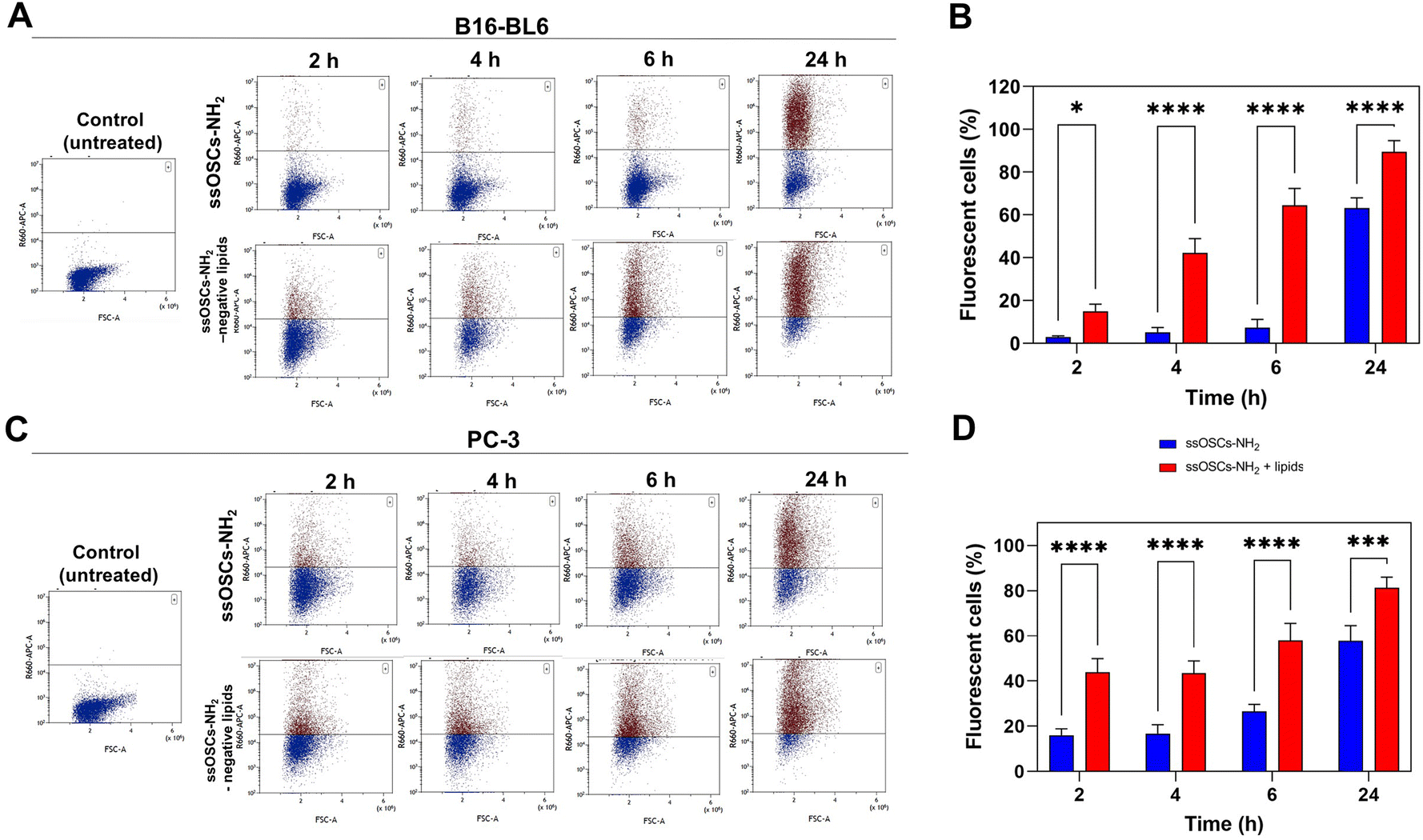 | ||
| Fig. 5 Uptake of both naked and lipid-coated ssOSCs@NH2 in B16-BL6 and PC3 cells. (A) Cy5 fluorescence histogram plots of the analysed B16-BL6 cells at the different time points incubated with ssOCs–NH2 and ssOSCs–NH2-negative lipids (0.1 mg mL−1). (B) Flow cytometry-based kinetics of the internalization process in B16-BL6 cells. (C) Cy5 fluorescence histogram plots of the analysed B16-BL6 cells at the different time points incubated with ssOCs–NH2 and ssOSCs–NH2-negative lipids (0.1 mg mL−1). (D) Flow cytometry-based kinetics of the internalization process in PC3 cells. *p < 0.05; **p < 0.01; ***p < 0.0001 and ****p < 0.00001. | ||
Calcein encapsulation: role of lipids in the cellular internalization of ssOSCs@calcein
In order to evaluate the potential of lipid–silica ssOSCs as delivery systems, doxorubicin and calcein molecules were encapsulated within the nanocapsules. First, calcein molecules were entrapped with the aid of CaCl2 (see the Experimental section) in the internal cavity of the nanocages by impregnation in order to use them as imaging agents by fluorescence microscopy. TEM and DLS of the ssOSCs loaded calcein (ssOSCs@CAL) display the same morphology and a hydrodynamic diameter comparable with the empty ssOSCs (Fig. S8A and S8B†), with a ζ potential of −17.2 ± 3.2 (see Table S2 of the ESI†). The FTIR analysis in Fig. S8C† also demonstrated the presence of the calcein peaks (red line) together with the silica bands previously described (blue line). Finally, the UV-VIS absorbance spectrum was recorded, demonstrating the presence of the calcein maximum around λ = 480 nm (Fig. S8D†). After the encapsulation of the dye molecules, the ssOSCs were functionalized with positive amino–silane (see the Experimental section). After the reaction, they exhibited a positive ζ potential (+9.2 ± 2.8 mV), useful for being further coupled with the negative lipid formulation. FTIR analysis of ssOSCs@CAL–NH2-negative lipids demonstrated the presence of the characteristic bands coming from the silica structure, from the calcein molecule and from the lipids on their surface (Fig. S9A†). The final nanomaterial, ssOSCs@CAL–NH2-negative lipids, showed a negative ζ potential (−18.8 ± 3.4 mV), and DLS analysis showed a diameter around 130 nm (Fig. S9B†). To corroborate the internalization of the negative lipid-covered and calcein-loaded ssOSCs, confocal microscopy was performed in two different cell lines: (1) PC3 prostate cancer line and (2) B16-BL6 melanoma cells (Fig. 6). In particular, ssOSCs and ssOSCs–NH2-negative lipids loaded with calcein were incubated with the different cell lines at 100 μg mL−1 and for 8, 24 and 48 h. The actin filaments of the cells can be observed in grey (being labelled with phalloidin-Alexa564), the ssOSCs were labelled with Cy5 (red), nuclei were coloured with Hoechst (blue) and calcein was observed upon excitation at 488 nm (green). It was demonstrated again how the ssOSCs@CAL–NH2-negative lipids were successfully and rapidly internalized in both cancer cell lines (PC3 and B16-BL6) in a time-dependent manner. In particular, at the shortest time-point (8 h) a significantly high accumulation of ssOSCs was already observed in the cytoplasm of PC3 cells when they were coated with the lipid formulation. This internalization increases even further after 24 hours. These results are in accordance with previous data shown in Fig. 5 and S7.† Furthermore, the release of calcein can be clearly observed (Fig. 6 and supporting videos of the 3D confocal images): pixels in orange indicate the colocalization of the ssOSCs and calcein. In contrast, the green spots correspond to the released calcein inside the cells after the breakage of the ssOSCs as a result of the reduction of the disulfide groups, as clearly observable within the two cancer cell lines. These data demonstrate their efficient release from the NPs and successful migration and spreading in the cell interior.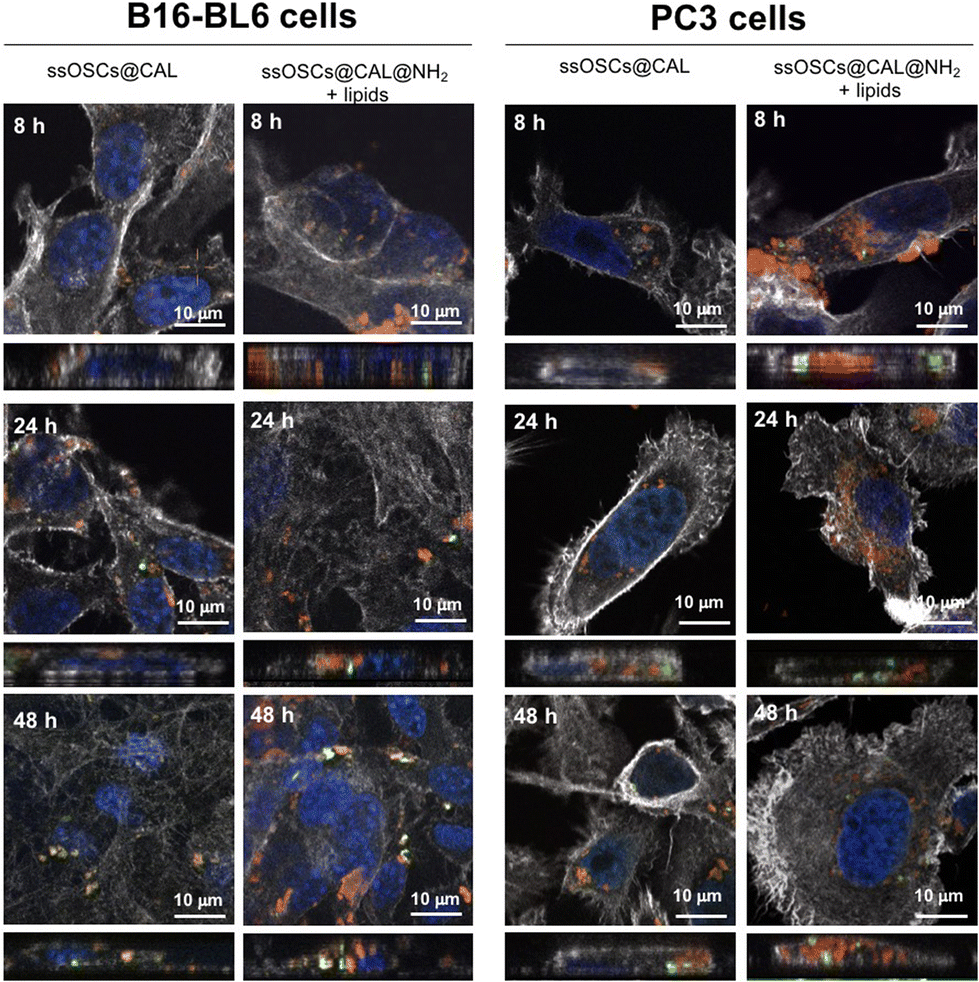 | ||
| Fig. 6 Confocal microscopy of B16-BL6 melanoma cells and PC3 prostate cancer cells incubated with ssOSCs@calcein·NH2-negative lipids during 8, 24, and 48 h. Grey: the actin filaments of the cells labelled with phalloidin-Alexa564; red: ssOSCs labelled with Cy5; blue: nuclei colored with Hoechst; and green: calcein. Above every fluorescence image, the lateral projected view is reported retrieved from the z-stacking and clearly proving the presence or the absence of internalized nanoparticles. The scale bar is 10 μm. | ||
Doxorubicin encapsulation: release kinetics, biocompatibility and therapeutic efficacy
Doxorubicin was also encapsulated into the nanocages (ssOSCs@DOX) following a conventional impregnation protocol. The loaded nanomaterials were characterized by DLS, ζ potential measurements, FTIR and absorption spectroscopy. The TEM images confirmed that even after the doxorubicin encapsulation, the NPs were still monodispersed and with a diameter of around 20 nm (Fig. 7A).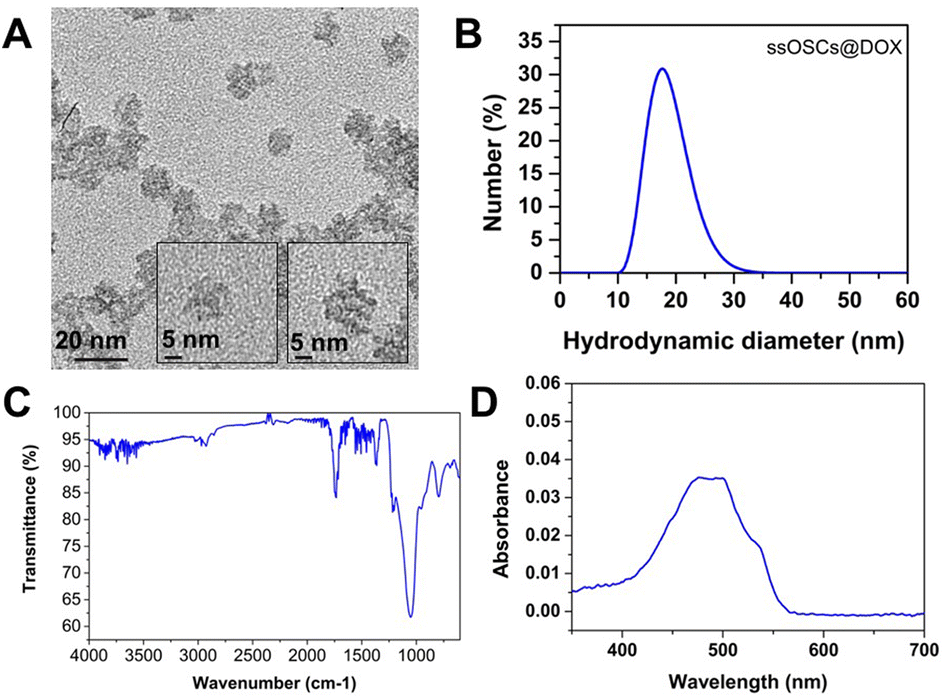 | ||
| Fig. 7 Characterization of ssOSCs@DOX. (A) TEM images, (B) DLS analysis, (C) FTIR and (D) absorption spectrum. | ||
The DLS results and ζ potential (−8.3 ± 1.3 mV) of the ssOSCs@DOX (Fig. 7B) corroborated the TEM findings, confirming that doxorubicin was encapsulated inside the NP pores rather than being attached to the surface. In the FTIR spectra of Fig. 7C, the characteristic peaks of the organosilica NPs were observed together with the main bands of doxorubicin (the characteristic sharp peak at 1727 cm−1 is related to the carbonyl (CO) of the pure doxorubicin). Finally, the absorption spectrum of the ssOSCs@DOX was also recorded showing maximum peaks at 470 and 496 nm (Fig. 7D). The ssOSCs@DOX were then functionalized with amino–silane and further covered with the negative lipids, as previously described for the calcein loaded samples. Fig. S10A† includes the FTIR spectra of ssOSCs@DOX–NH2-negative lipids. It is possible to observe the presence of the characteristic bands coming from the organosilica groups, the disulfide breakable bonds, the doxorubicin fingerprint and the lipid bands. The hydrodynamic diameter obtained by DLS was around 100 nm (Fig. S10B†) and the ζ potential was −29.2 ± 2.2 mV, demonstrating again the successful coating of the silica core with the lipidic shell.
The breakable properties of the nanocages and the effect of the lipidic shell on the release kinetics were evaluated for the doxorubicin-loaded ssOSCs and conducted by mimicking the physiological conditions in cancer cells, and in particular the glutathione concentration. The reduction of the disulfide groups in the ssOSCs was achieved by adding a solution of glutathione (10 mM) to the particles suspended in a PBS solution at a final concentration of 0.1 mg mL−1 incubated at 37° C (pH = 7.4). Then, centrifugation of each vial led to the formation of a pellet with a supernatant that was collected at different time points (1, 3, 5, 7 and 24 h). In the same volume of supernatant, we expected to see different amounts of the released drugs depending on the time. Fig. S11A and S11B† show the absorption spectra of the supernatant of naked and lipid-coated ssOSCs@DOX–NH2, respectively. A time-dependent increment in the doxorubicin absorption signal intensity in the supernatant was observed only in the presence of the reducing agent (GSH) for both naked and lipid-coated NPs. These results confirmed that the degradation of the NPs was triggered by the reduction of the disulfide bonds present in the silica framework of the ssOSCs. However, differences were observed between naked and lipid-coated ssOSCs@DOX–NH2 (Fig. S11C†): lipid-coated ssOSCs exhibited a slower and smaller amount of dox release compared with the naked NPs. In particular within the first hour without the lipidic coverage the naked nanoparticles released more than 50% of the entrapped DOX. Data are in agreement with previously published works, demonstrating that the lipidic bilayer offers a valid shell to (1) retain the encapsulated molecule and (2) prevent the rapid drug leakage and release from the NP pores, maximizing the drug accumulation in the intracellular compartments.20,24,26
Finally, the tolerability of the tested cells (B16-BL6 and PC3 cells) to the exposure of ssOSCs@DOX (both lipid-coated and naked NPs) was determined by incubating increasing quantities of NPs (15, 30, 60 and 120 μg mL−1) for 2, 4, 24 and 48 h (Fig. 8). Unloaded ssOSCs were also tested as a negative control to corroborate the absence of toxicity for the carriers in the absence of the anticancer drug. The cell viability assays revealed that the naked and empty ssOSCs induce minimal effects on cell toxicity, even at the highest dose during the last time point tested (Fig. 8A and B). The lipid-coated and empty ssOSCs produced a minimal decrease in cell proliferation, probably attributed to their higher internalization level inside the cells (yet confirmed by confocal microscopy and flow cytometry). Remarkably, in the two tested cell lines, both ssOSCs@DOX–NH2 and lipid-coated ssOSCs@DOX–NH2 induced significant mortality. More interestingly, Fig. 8C and D demonstrate that when the DOX-loaded nanocages were coated with the lipid layer, the proliferation was significantly reduced compared with the DOX-loaded naked ones in the two cell lines. This trend, in the decrease of the cell viability, when treating both cell lines with the ssOSCs@DOX–NH2-negative lipids (120 μg mL−1), can be observed after 4 h and 24 h of incubation. The viability was compared to the cultures treated with the naked ssOSCs loaded with the same amount of DOX at the same time points. Indeed, statistically significant differences were obtained when comparing the cell viability of PC3 cells treated with naked or lipid coated ssOSCs@DOX after 24 h. These findings demonstrate how the presence of the lipid layer around the NPs favour their internalization toward the cell cytoplasm and does not hinder the cytotoxic drug release resulting in the highest efficiency of the delivered drug.
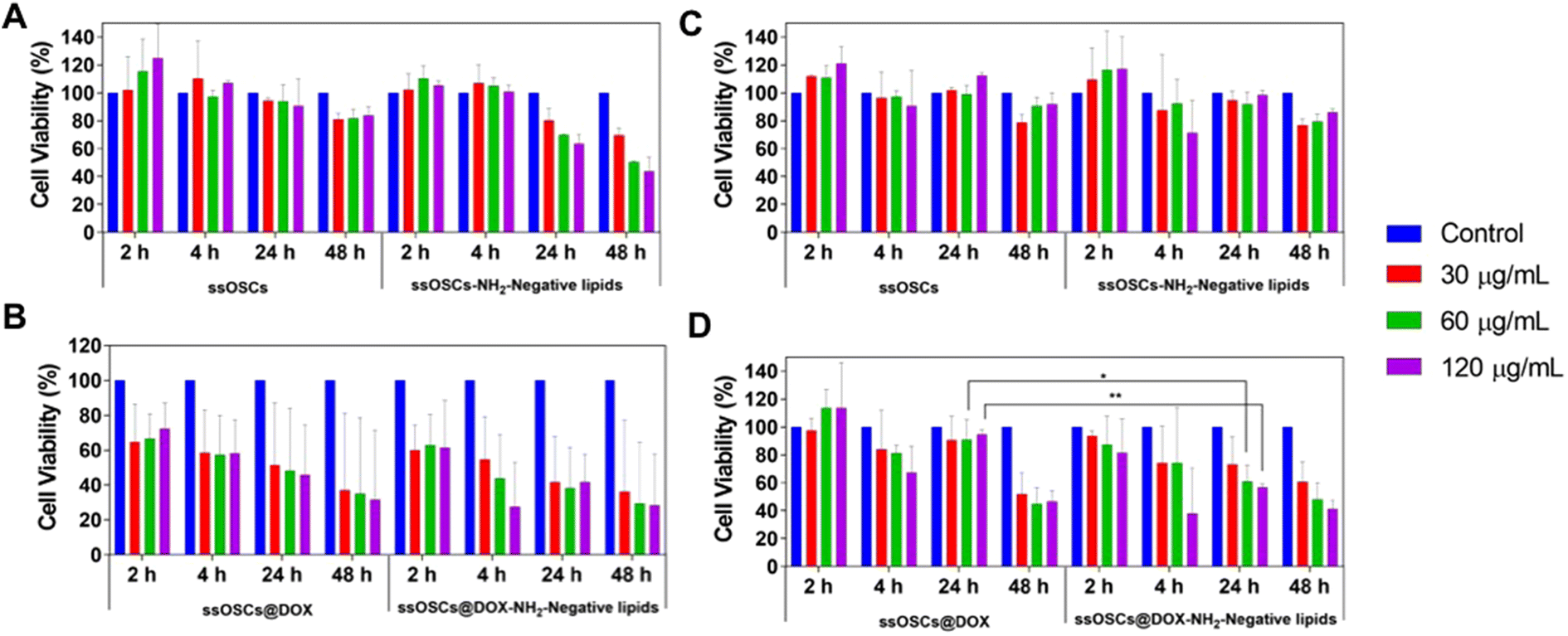 | ||
| Fig. 8 Metabolic cytotoxicity of empty ssOSCs and lipid-covered ssOSCs on (A) B16-BL6 cells and (B) PC3 cells and of DOX-loaded (both lipid-coated and naked ssOSCs@DOX) on (C) B16-BL6 melanoma cells and (D) PC3 prostate cancer cells. Statistically significant differences were indicated as follows: *p < 0.05 and **p < 0.01. | ||
Experimental
Synthesis and functionalization of nanoparticles
All the materials were purchased from Merck and used as received. Sulfo–Cy5–NHS ester was acquired from Lumiprobe. Phalloidin-Alexa-568 and Hoechst 3342 were purchased from Life Technologies, Invitrogen.A previously published strategy was employed for the synthesis of the breakable ssOSCs. In summary, 408 mg (1.12 mmol) of cetyltrimethylammonium (CTAB) was dissolved in 50 mL of deionized water and magnetically stirred for 30 min at 250 rpm at 50 °C. Then, an ammonia aqueous solution (12.5 μL) was added and the stirring speed was increased to 750 rpm. Subsequently, 448.1 μL (2 mmol) of tetraethyl orthosilicate (TEOS) and 102.8 μL (0.223 mmol) of bis[3-(triethoxysilyl)propyl]disulphide (BTDS) were mixed and added to the previous solution and stirred at 750 rpm, for 20 h at 50 °C. Subsequently, APTES (87.2 μL) was added to the stirring solution and the reaction mixture was stirred for 14 h at 50 °C. The particle dispersion was purified by dialysis against an EtOH/H2O/AcOH (1:1:0.007 v/v/v) mixture for 24 h and finally against an EtOH/H2O (1:1 v/v) mixture for another 24 h whereby the dialysis solution was replaced every 2 h, when possible.
For the colocalization study and the internalization experiments, the ssOSCs–NH2 were labelled with the sulfo–Cy5–NHS probe. First, a sulfo–Cy5–silane was prepared by suspending a solution of sulfo–Cy5–NHS ester (0.5 mg, 0.643 μmol) in dry dimethylsulfoxide (DMSO). Afterwards, APTES (4.5 μL, 0.142 M in dry DMSO) was added and the reaction mixture was allowed to stir for 30 min in the dark.
The obtained ssOSCs were purified by dialysis against an EtOH/H2O/AcOH (1/1/0.007 v/v/v) mixture for 72 h, then against EtOH/H2O (1/1 v/v) for 24 h and finally against dH2O for 24 h. The dialysis solutions were replaced every 12 h.
The resulting naked and functionalized ssOSCs were characterized by a broad battery of physicochemical techniques. The dimensions and morphology of the NPs were evaluated by transmission electron microscopy (TEM) using an Energy Filter Transmission Electron Microscope (EFTEM, Zeiss Libra 120). For TEM observations, samples were deposited onto a TEM copper grid. The size distribution and the surface charge of the nanoparticles were obtained employing a dynamic light scattering (DLS) instrument. The DLS measurements were performed on dispersions of ssOSCs in dH2O (pH 7) using Zetasizer Ultra equipment equipped with Multi-Angle Dynamic Light Scattering (MALDLS®) technology from Malvern Panalytical and using the software ZS Xplorer. Fourier Transform Infra-Red (FTIR) spectra were recorded using a Shimadzu IRAffinity-1 spectrometer. The transmittance spectrum was collected using a spectral resolution of 1 cm−1 accumulating 64 scans from 600 to 4000 cm−1. Thermogravimetric assay (TGA) was conducted on a Netzsch model STA 449 fi Jupiter instrument. The samples (0.5–3 mg) were maintained at 100 °C for 30 minutes for stabilization and then heated from 130 to 800 °C at a speed of 10 °C min−1, before being held at this temperature for a further 30 minutes at the end of the measurement. The analysis was carried out under a 20 mL min−1 airflow. Nitrogen physisorption isotherms were obtained to evaluate the porosity of the ssOSCs with a Micromeritics ASAP-2020 physisorption instrument. The particle sample was degassed at 100 °C for 12 h. The N2 adsorption/desorption measurement was performed at −196 °C. The surface areas were calculated using the Brunauer–Emmett–Teller (BET) method in the relative pressure range (p/p0) of 0.06–0.3. The pore size distributions and pore volumes were calculated using the classical method on the adsorption branch using a cylindrical model. The total pore volume was calculated to be P/P0 0.99. The emission spectra of Cy5 functionalized NPs were recorded on a Horiba Jobin–Yvon IBH FL-322 Fluorolog 3 spectrometer equipped with a 450 W xenon arc lamp, double grating excitation, emission monochromators (2.1 nm mm−1 of dispersion; 1200 grooves per mm) and a TBX-04 single photon-counting detector. The samples were excited by a wave laser at λex = 633 nm. The emission of the samples was collected in the range from 640 to 820 nm (Fig. S12†). The presence of primary amines in the surface of the ssOSCs was assessed using the Kaiser test (Sigma Aldrich) and by measuring the absorbance in a Shimadzu UV-3600 spectrophotometer double-beam UV-VIS-NIR spectrometer with quartz cuvettes with a path length of 10 mm.
Drug encapsulation
Doxorubicin and calcein molecules were loaded inside the inner cavity of the ssOSCs before their functionalization with the APTES and following an impregnation strategy. For the encapsulation of doxorubicin, 20 mg of naked ssOSCs was suspended in 5 mL of EtOH. In parallel, doxorubicin (20 mg) was also dissolved in 5 mL of EtOH. Both solutions were sonicated for 10 min separately. Then, they were mixed and briefly sonicated for a further 20 min and stirred for 12 h at room temperature. Subsequently, to force the encapsulation, the vacuum was exploited and the sample was placed in a rotavapor for a few minutes until the solvent was completely evaporated. Then, the NPs were resuspended in toluene (2 mL), 150 μL of 2,2-dimethoxy-1,6-diaza-2-silacyclooctane and 1 μL of NH4OH (28%) were added and the solution was left for 12 h in continuous agitation at room temperature. Then, the loaded NPs were washed with water five times to eliminate the non-encapsulated drug.In the case of calcein, a solution of CaCl2 2 M in ethanol was employed as a solvent for the encapsulation approach. Briefly, 20 mg of naked ssOSCs was dissolved in 5 mL of CaCl2 2 M (in ethanol). Meanwhile, 20 mg of calcein molecules were also dissolved in 5 mL of CaCl2 2 M (in ethanol). Both solutions were vigorously vortexed and sonicated for 10 min separately until they were completely dissolved. Then, they were mixed and briefly sonicated for a further 20 min and stirred for 12 h at room temperature. Successively, NPs were functionalized with cyclic silane, as previously mentioned, and they were finally washed several times to discard the non-encapsulated calcein.
Before the lipid coating, ssOSCs@DOX and ssOSCs@CAL were covered with a positive cyclic silane and stored at 4 °C until use. In particular, 10% of 2,2-dimethoxy-1,6-diaza-2-silacyclooctane was incubated overnight at room temperature with the loaded NPs using toluene as the solvent and in the presence of NH3 28%. The positive NPs were washed three times with water and stored at 4 °C until further use. The loaded ssOSCs were characterized as previously described by DLS, ζ potential measurements, TEM and FTIR. Moreover, the absorption of the ssOSCs@DOX and ssOSCs@CAL was measured on a Shimadzu UV-3600 spectrophotometer double-beam UV-VIS-NIR spectrometer and baseline corrected. Quartz cuvettes with a path length of 10 mm were used.
Lipid shell formation
In order to confer to ssOSCs a higher biocompatibility and stability in biological media and to seal the therapeutic compound inside the nanocage, they were covered by a lipidic bilayer, following a solvent exchange method, as reported in a previous work.20 This technique consists in preparing a formulation of different phospholipids and cholesterol, collecting them from their stock chloroform solution, and rehydrating them with a solution of 40%v ethanol and 60%v water. The organosilica nanoparticles are then mixed with the lipid solution and water is then added in excess. The rapid increase of water content is crucial for this method, as it drives the self-assembly of the phospholipids on the surface of the nanoparticles.The functionalized organosilica nanocages (ssOSCs–NH2) synthesized in this work show a positive ζ potential; therefore a negatively charged phospholipid mixture was ad hoc designed to efficiently exploit electrostatic interactions driving the lipid self-assembly on the nanocages surface, as recently reported.40 The phospholipids were purchased from Avanti Polar Lipids: DOPA (1,2-dioleoyl-sn-glycero-3-phosphate) as a negatively charged lipid, DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine dissolved in chloroform, by Avanti Polar Lipids) as a neutral one, and DSPE–PEG(2000)–amine (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000]. The phospholipids were mixed with cholesterol (from Sigma Aldrich) in chloroform, in a molar ratio of DOPA/DOPC/DSPE–PEG(2000)–amine/cholesterol equal to 50/10/1.5/38.5, and dried under vacuum overnight in glass vials. The obtained lipid film was then rehydrated with a 2:3 volume ratio of ethanol/bd water solution, obtaining a final concentration of 3 mg mL−1. As a control, a non-charged lipid mixture was prepared, substituting the DOPA fraction with DOPC, thus having DOPC/DSPE–PEG(2000)–amine/cholesterol with a 60/1.5/38.5 molar ratio.
ssOSCs or ssOSCs–NH2, dispersed in water, were sonicated for 3 minutes at 59 kHz using a sonication bath (Branson 3800 CPXH, Branson Ultrasonics Corporation); subsequently, ethanol was added to the nanoparticle dispersion to reach a volume ratio of 2:3 ethanol to water. Then, the lipid solution was added to the nanoparticle dispersion at a weight ratio of 2:1 (preferred option) or a 1:1 nanoparticle to lipid ratio. Thereafter, the mixture was sonicated for 3 minutes to increase homogeneity. Finally, to self-assemble the lipid bilayer on the nanoparticle surface, an excess of deionized water was added to the mixture, until reaching a 1:9 ethanol to water volume ratio. The final suspension was then sonicated for 5 minutes to obtain better size uniformity and to help the formation of a homogeneous self-assembled lipid bilayer encasing the nanoparticles. Similarly, control liposomes were obtained: the lipid mixture in 2:3 ethanol to water solution was sonicated for 3 minutes; successively, water was added achieving a 1:9 ethanol to water volume ratio and 5 minutes sonication was eventually performed for homogeneity.
To characterize the size distribution and colloidal stability of the coated nanoparticles, DLS and Z-potential measurements (Zetasizer Nano ZS90 from Malvern Instruments) and Nanoparticles Tracking Analysis (NTA, NanoSight NS300 from Malvern Panalytical) were performed in deionized water and at room temperature. All the measurements were conducted 3 times and then averaged. In the case of NTA, for each sample, three 60-second videos of the samples flowing through the instrument chamber were recorded and analyzed with the NTA 3.4 software from Malvern Panalytical.
In order to prove the correct formation of the lipid layer around the nanocages, fluorescence microscopy colocalization analyses were performed. In particular, Cyanin 5-labelled nanocages (ssOSCs–NH2–Cy5) were obtained in a one-pot synthetic approach. Lipids, after nanocages coating, were labelled with DiO (DiOC18(3) (3,3′-dioctadecyloxacarbocyanine perchlorate)) from ThermoFisher Scientific, adding 0.5 μL of 10 μg mL−1 DiO solution in DMSO for each mg of lipid-coated nanocages. The nanoconstructs were then incubated at 37 °C for 30 minutes in shaking mode (200 rpm). After the incubation, 5 μL of lipid-coated nanocage dispersion was deposited on a glass slide, coated by a cover glass slip and analyzed through an inverted wide-field fluorescence microscope (Eclipse TiE from Nikon) equipped with a 100× oil-immersion PlanAPO objective. At least 10 multichannel images from different regions were taken for each sample and analyzed with the colocalization tool of the NIS software (NIS-Elements AR 4.5, Nikon). In particular, the objects in the far-red channel (representing the nanocages), the ones in the green channel (representing the lipids) and the colocalized spots in the merged image were counted and the colocalization percentage was calculated following the formula:
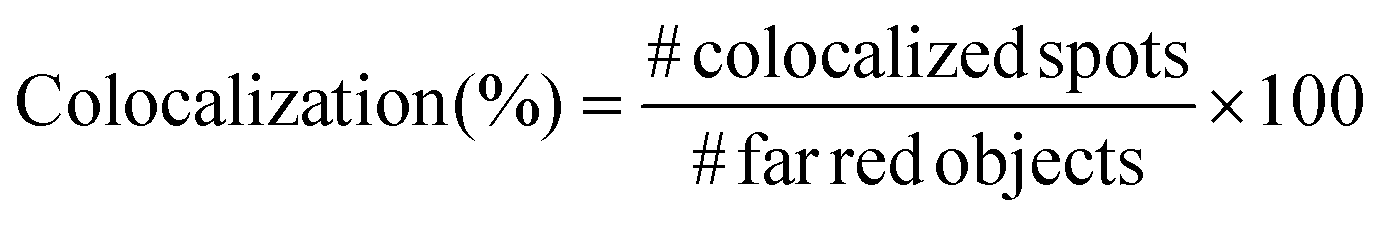
For each sample, the colocalization percentage was calculated for 10 images and then the results were averaged.
Hemocompatibility test
In order to evaluate the hemocompatibility of pristine and negative lipid-coated nanocages, their capability to coagulate plasma was evaluated through a plasma recalcification test, as previously reported.24,41 ssOSCs and ssOSCs–NH2 were coated with negative lipids, as described above, under sterile conditions. Pristine and coated nanocages were resuspended in 0.1 μm filtered physiological solution (0.9% NaCl w/w water solution), obtaining a total of 4 samples with a final concentration equal to 450 μg mL−1. A pure physiological solution was used as the control sample. 6 wells per sample of a 96-well plate were filled with 75 μL of human citrated plasma (Human Recovered Plasma Pooled-frozen – NaCitrate from ZenBio) pre-heated at 37 °C, and 75 μL of the control sample or nanocage samples was added in each well. Then, after 5 minutes of incubation at 37 °C, 150 μL of 25 mM calcium chloride was quickly added into 3 wells for each sample, thus inducing the coagulation of plasma. Immediately after, the plate was inserted in a 37 °C pre-heated UV-Vis spectrometer and the absorbance at 405 nm was measured periodically. In particular, the protocol consists of a measurement every 30 s for 45 min, with incubation of the plate at 37 °C. The chosen wavelength demonstrates the changes of turbidity in the solutions treated with calcium chloride which is relative to the formation of fibrins during the recalcification of plasma, as a consequence of the presence of calcium cations (Ca2+). The periodical measure of absorbance produced sigmoid curves in which the plateau represents the coagulation of plasma (Fig. S6†). For each sample, the central absorbance point (ac) was calculated as the mean point between the maximum and the minimum of the absorbance (a):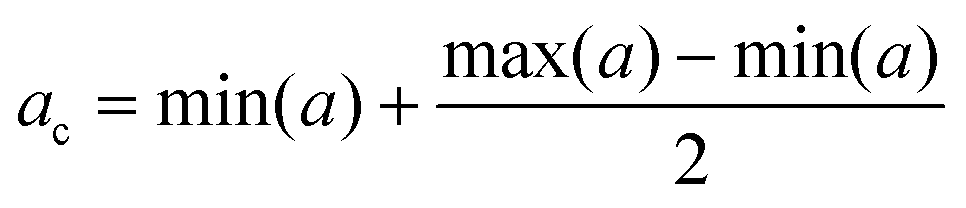
The point corresponding to ac on the t axis was individuated as the coagulation time (tc) and used to compare the samples. Three independent experiments were carried out and the resulting coagulation times were averaged.
Naked and lipid-coated ssOSCs’ drug release kinetics
The breakability of the ssOSCs@DOX–NH2 and ssOSCs@DOX–NH2 + lipids was assessed using reduced glutathione (GSH) as a reducing agent. The loaded particles were suspended in a PBS solution of reduced GSH (10 mM) at a final concentration of 0.1 mg mL−1 and incubated at 37 °C in PBS (pH = 7.4). A control experiment was also carried out in parallel but in the absence of GSH. Aliquots were taken at different time points (1, 3, 5, 7 and 24 h) and analyzed by absorption spectroscopy. Before the measurements, the aliquots were centrifuged to allow the sedimentation of the intact particles; hence the supernatant fraction was collected and its absorbance was recorded using a Shimadzu UV-3600 spectrophotometer double-beam UV-VIS-NIR spectrometer and the baseline was corrected using a UV-VIS-NIR spectrometer. Quartz cuvettes with a path length of 10 mm were used.Cell lines
PC3 cells were kindly provided by Prof. Esther Nolte-‘t Hoen's research group. B16-BL6 cells were provided by the Department of Biochemistry and Molecular Pharmacology and by the Department of Oncology from Mario Negri Institute for Pharmacological Research, respectively. PC3 cells were grown in Dulbecco's modified Eagle's medium (DMEM, Biowest, France) supplemented with 10% fetal bovine serum (FBS, GIBCO, USA), 1% penicillin/streptomycin (Biowest, France) and 1% L-glutamine (Euroclone, Italy) and maintained at 37 °C in a 3.5% CO2-humidified atmosphere under normoxic conditions. B16-BL6 melanoma cells were grown in a Minimum Essential Medium (MEM, Euroclone, Italy) supplemented with 10% FBS (GIBCO, USA), 1% penicillin/streptomycin (Biowest, France), 1% L-glutamine (Euroclone, Italy), 1% non-essential amino acids (Euroclone, Italy), 1% sodium pyruvate (Euroclone, Italy) and 2% of vitamin solution (Euroclone, Italy). They were maintained at 37 °C in a 3.5% CO2-humidified atmosphere under normoxic conditions.Cytotoxicity experiments
To determine the cytotoxic and antiproliferative properties of the doxorubicin loaded breakable nanocages, the MTT Cell Viability assay (ThermoFisher Scientific) was employed following the manufacturer's instructions. For carrying out the test, the two cell lines were seeded at a density of 4 × 103 cells per well (into a 96-well plate) in 100 μL of the previously mentioned medium. After incubation at 37 °C for 24 h, the medium was changed to 100 μL of freshly prepared medium enriched with ssOSCs@DOX or with ssOSCs@DOX–NH2-negative lipids (15, 30, 60, and 125 μg mL−1) and the cells were cultured for another 2, 4, 24 and 48 h. At these different time-points, cells were washed with PBS and then treated for the MTT test. Briefly, a tetrazolium solution (20 μL of 5 mg mL−1) was added to each well and incubated for 4 h. The medium was replaced with acidified isopropanol (0.04 M HCl) to dissolve the purple precipitate and the absorbance intensity was measured at 570 nm using a plate reader (Infinite M200, Tecan).Internalization study by confocal microscopy
The internalization of the NPs and the NPs covered with the lipids was evaluated by confocal microscopy. B16-BL6 melanoma cells and PC3 prostate cancer cells were seeded at a density of 3 × 104 cells onto 20 mm coverslips (in a 24-well plate) and cultured for 24 h. In order to visualize the ssOSCs they were labelled with sulfo–Cy5–NHS as previously described.29 Then, ssOSCs, ssOSCs–NH2 and ssOSCs–NH2-negative lipids (0.1 mg mL−1) were resuspended in DMEM and were added to the cells at 2, 4, 8, 24 and 48 h. Afterwards, cells were fixed with 4% para-formaldehyde (PFA) and finally washed three times with PBS. In order to label the cytoplasm actin, cells were stained with phalloidin-Alexa568 (Invitrogen, USA). Hoechst 33342 was used to observe the nuclei. Samples were observed by confocal microscopy (Nikon A1 confocal scan unit with a 100 × 1.49 NA oil immersion objective managed by NIS elements software). The uptake of naked and lipid-coated ssOSCs@NH2 was also assessed and quantified by flow cytometry. Briefly, 3 × 104 cells were seeded onto a 24-well plate. After 24 h, the cells were treated with Cy5-labelled ssOSCs–NH2 or ssOSCs–NH2-negative lipids (0.1 mg mL−1) for 2, 4, 6 and 24 h. Then, they were collected and analyzed by flow cytometry using a Cytoflex LX instrument (Beckman Coulter), and the data were analyzed using the Kaluza Sofware (Beckman Coulter). Non-treated cells were used to set the gate and the percentage of positive cells and determine the nanoparticle internalization. For the detection of Cy5-labeled NPs, an excitation wavelength of 633 nm was employed. The emitted signal was collected with BP 610/10 filers. All the samples were analyzed acquiring at least 10000 events.
Finally, the release of calcein loaded within the lipid-coated NPs (ssOSCs@CAL–NH2-negative lipids) was also assessed by confocal microscopy. Cells were incubated for 8, 24 and 48 h with the calcein-loaded and lipid-coated ssOSCs (0.05 mg mL−1). Calcein was observed exciting the sample at λ = 488 nm, cell cytoplasm was stained with phalloidin-Alexa568 (λ = 568 nm), nuclei were labelled with Hoechst (λ = 405 nm) and NPs were visualized upon excitation at λ = 633 nm, thanks to Cy5.
Statistical analysis
All the data are indicated as mean ± SD. Statistical analysis of the in vitro data and the significant differences among the means were analyzed by two-way analysis of variance (ANOVA) for multiple comparisons by Dunnett's multiple comparisons test (GraphPad Software). Statistically significant differences were expressed as follows: *p < 0.05; **p < 0.01; ***p < 0.0001 and ****p < 0.00001.Conclusions
In the present work, we have developed a biomimetic nanocarrier using a breakable organosilica nanocage, functionalized with amines and decorated with a lipid bilayer. The nanohybrid was fully characterized to assess the best combination of lipids and surface charges. Finally, the nanocarriers have been filled with dyes or doxorubicin to test their abilities for drug release and evaluate cell internalization in two different cell lines. Such lipid-coated nanoconstructs were proved to have an incredibly high hemocompatibility, significantly increased with respect to pristine nanocages.Furthermore, an enhanced cellular uptake, which proves the key role of the lipid coating in mediating the internalization of the nanoparticles, was observed. The efficient intracellular delivery of the therapeutic agent effectively affected cancer cells, demonstrating the potentiality of the lipid-coated nanocages as a powerful tool in anticancer research.
Author contributions
MSA and GR equally contributed to the present work, performing all the experiments. LDC and VC conceptualized and ideated the experiments and performed the supervision. The manuscript has been written and reviewed by all authors.Conflicts of interest
All authors declare to be co-inventors of patent application no. 102023000015396 of 21st July 2023, describing a method to coat nanoparticles with lipid bilayers.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement no. 964386 (FET Open Mimic-Key project). M. S-A is thankful for the financial support from Mario Negri Young Investigator Grant ExAC project (8702) and support from FIRC-AIRC fellowship for Italy (26907-2021). Thanks to Dr Xuequan Zou for helping us with amine group estimation on the surface of NPs. We thank Mario Negri Institute for Pharmacological Research and Dr Victor Sebastián from the University of Zaragoza and ICTS LMA for the TEM images. We are grateful to the support of Mrs Marzia Conte, MD Eng., who is also a co-inventor with the authors of the patent application no. 102023000015396 of 21st July 2023, for the development of both negative and neutral lipid formulations adopted in this work.References
- M. Colilla, B. González and M. Vallet-Regí, Mesoporous silica nanoparticles for the design of smart delivery nanodevices, Biomater. Sci., 2013, 1, 114–134 RSC.
- Z. Li, J. C. Barnes, A. Bosoy, J. F. Stoddart and J. I. Zink, Mesoporous silica nanoparticles in biomedical applications, Chem. Soc. Rev., 2012, 41, 2590–2605 RSC.
- C. Argyo, V. Weiss, C. Bräuchle and T. Bein, Multifunctional Mesoporous Silica Nanoparticles as a Universal Platform for Drug Delivery, Chem. Mater., 2014, 26, 435–451 CrossRef CAS.
- M. W. Ambrogio, C. R. Thomas, Y.-L. Zhao, J. I. Zink and J. F. Stoddart, Mechanized Silica Nanoparticles: A New Frontier in Theranostic Nanomedicine, Acc. Chem. Res., 2011, 44, 903–913 CrossRef CAS PubMed.
- F. Chen, et al., Increasing the Efficacy of Stem Cell Therapy via Triple-Function Inorganic Nanoparticles, ACS Nano, 2019, 13, 6605–6617 CrossRef CAS PubMed.
- R. Juthani, et al., Ultrasmall Core-Shell Silica Nanoparticles for Precision Drug Delivery in a High-Grade Malignant Brain Tumor Model, Clin. Cancer Res., 2020, 26, 147–158 CrossRef CAS PubMed.
- K. Ma, H. Sai and U. Wiesner, Ultrasmall Sub-10 nm Near-Infrared Fluorescent Mesoporous Silica Nanoparticles, J. Am. Chem. Soc., 2012, 134, 13180–13183 CrossRef CAS PubMed.
- T. Aubert, J.-Y. Huang, K. Ma, T. Hanrath and U. Wiesner, Porous cage-derived nanomaterial inks for direct and internal three-dimensional printing, Nat. Commun., 2020, 11, 4695 CrossRef CAS PubMed.
- K. Ma, et al., Self-assembly of highly symmetrical, ultrasmall inorganic cages directed by surfactant micelles, Nature, 2018, 558, 577–580 CrossRef CAS PubMed.
- L. Talamini, et al., Organosilica Cages Target Hepatic Sinusoidal Endothelial Cells Avoiding Macrophage Filtering, ACS Nano, 2021, 15, 9701–9716 CrossRef CAS PubMed.
- P. Picchetti, et al., Smart Nanocages as a Tool for Controlling Supramolecular Aggregation, J. Am. Chem. Soc., 2021, 143(20), 7681–7687 CrossRef CAS PubMed.
- M. Wu, et al., Large-pore ultrasmall mesoporous organosilica nanoparticles: micelle/precursor co-templating assembly and nuclear-targeted gene delivery, Adv. Mater., 2015, 27, 215–222 CrossRef CAS PubMed.
- Y. Yang, et al., Structure-Dependent and Glutathione-Responsive Biodegradable Dendritic Mesoporous Organosilica Nanoparticles for Safe Protein Delivery, Chem. Mater., 2016, 28, 9008–9016 CrossRef CAS.
- S. P. Hadipour Moghaddam, M. Yazdimamaghani and H. Ghandehari, Glutathione-sensitive hollow mesoporous silica nanoparticles for controlled drug delivery, J. Controlled Release, 2018, 282, 62–75 CrossRef CAS PubMed.
- N. X. D. Mai, et al., Biodegradable Periodic Mesoporous Organosilica (BPMO) Loaded with Daunorubicin: A Promising Nanoparticle-Based Anticancer Drug, ChemMedChem, 2020, 15, 593–599 CrossRef CAS PubMed.
- M. Sancho-Albero, et al., Enhancing Pt(IV) Complexes’ Anticancer Activity Upon Encapsulation in Stimuli Responsive Nanocages, Adv. Healthcare Mater., 2023, 12(17), e2202932, DOI:10.1002/adhm.202202932.
- V. Cauda, A. Schlossbauer, J. Kecht, A. Zürner and T. Bein, Multiple Core–Shell Functionalized Colloidal Mesoporous Silica Nanoparticles, J. Am. Chem. Soc., 2009, 131, 11361–11370 CrossRef CAS PubMed.
- S. Barui and V. Cauda, Multimodal Decorations of Mesoporous Silica Nanoparticles for Improved Cancer Therapy, Pharmaceutics, 2020, 12(6), 527 CrossRef CAS PubMed.
- V. Cauda, C. Argyo and T. Bein, Impact of different PEGylation patterns on the long-term bio-stability of colloidal mesoporous silica nanoparticles, J. Mater. Chem., 2010, 20, 8693–8699 RSC.
- V. Cauda, et al., Colchicine-Loaded Lipid Bilayer-Coated 50 nm Mesoporous Nanoparticles Efficiently Induce Microtubule Depolymerization upon Cell Uptake, Nano Lett., 2010, 10, 2484–2492 CrossRef CAS PubMed.
- V. Cauda, A. Schlossbauer and T. Bein, Bio-degradation study of colloidal mesoporous silica nanoparticles: Effect of surface functionalization with organo-silanes and poly(ethylene glycol), Microporous Mesoporous Mater., 2010, 132, 60–71 CrossRef CAS.
- M. Percivalle, et al., Artificial and Naturally Derived Phospholipidic Bilayers as Smart Coatings of Solid-State Nanoparticles: Current Works and Perspectives in Cancer Therapy, Int. J. Mol. Sci., 2022, 23, 15815 CrossRef PubMed.
- E. Ploetz, et al., Metal-Organic Framework Nanoparticles Induce Pyroptosis in Cells Controlled by the Extracellular pH, Adv. Mater., 2020, 32, e1907267 CrossRef PubMed.
- V. Cauda, et al., Biomimetic mesoporous vectors enabling the efficient inhibition of wild-type isocitrate dehydrogenase in multiple myeloma cells, Microporous Mesoporous Mater., 2021, 325, 111320 CrossRef CAS.
- B. Dumontel, et al., Enhanced biostability and cellular uptake of zinc oxide nanocrystals shielded with a phospholipid bilayer, J. Mater. Chem. B, 2017, 5, 8799–8813 RSC.
- J. Lin, et al., PEGylated Lipid bilayer coated mesoporous silica nanoparticles for co-delivery of paclitaxel and curcumin: Design, characterization and its cytotoxic effect, Int. J. Pharm., 2018, 536, 272–282 CrossRef CAS PubMed.
- Y. Qiu, et al., Lipid-coated hollow mesoporous silica nanospheres for co-delivery of doxorubicin and paclitaxel: Preparation, sustained release, cellular uptake and pharmacokinetics, Mater. Sci. Eng., C, 2017, 71, 835–843 CrossRef CAS PubMed.
- X. Liu, et al., Irinotecan Delivery by Lipid-Coated Mesoporous Silica Nanoparticles Shows Improved Efficacy and Safety over Liposomes for Pancreatic Cancer, ACS Nano, 2016, 10, 2702–2715 CrossRef CAS PubMed.
- W. Yuan, X. He and R. J. Lee, Delivery of miR-375 and doxorubicin hydrochloride by lipid-coated hollow mesoporous silica nanoparticles to overcome multiple drug resistance in hepatocellular carcinoma, Int. J. Nanomed., 2017, 12, 5271–5287 CrossRef PubMed.
- K. S. Butler, et al., Protocells: Modular Mesoporous Silica Nanoparticle-Supported Lipid Bilayers for Drug Delivery, Small, 2016, 12, 2173–2185 CrossRef CAS PubMed.
- C. E. Ashley, et al., Delivery of Small Interfering RNA by Peptide-Targeted Mesoporous Silica Nanoparticle-Supported Lipid Bilayers, ACS Nano, 2012, 6, 2174–2188 CrossRef CAS PubMed.
- A. Schlossbauer, et al., Cascaded photoinduced drug delivery to cells from multifunctional core-shell mesoporous silica, Adv. Healthcare Mater., 2012, 1, 316–320 CrossRef CAS PubMed.
- Y. Wang, et al., Endosomolytic and Tumor-Penetrating Mesoporous Silica Nanoparticles for siRNA/miRNA Combination Cancer Therapy, ACS Appl. Mater. Interfaces, 2020, 12, 4308–4322 CrossRef CAS PubMed.
- A. M. Sauer, et al., Role of Endosomal Escape for Disulfide-Based Drug Delivery from Colloidal Mesoporous Silica Evaluated by Live-Cell Imaging, Nano Lett., 2010, 10, 3684–3691 CrossRef CAS PubMed.
- Y. Xu, et al., pH and Redox Dual-Responsive MSN-S-S-CS as a Drug Delivery System in Cancer Therapy, Materials, 2020, 13, 1279 CrossRef CAS PubMed.
- L. Talamini, et al., Organosilica, ACS Nano, 2021, 15, 9701–9716 CrossRef CAS PubMed.
- J. M. Nzai and A. Proctor, Determination of Phospholipids in Vegetable Oil by Fourier Transform Infrared Spectroscopy, J. Am. Oil Chem. Soc., 1998, 75, 1281–1289 CrossRef CAS.
- X. Chen, et al., The Stability, Sustained Release and Cellular Antioxidant Activity of Curcumin Nanoliposomes, Molecules, 2015, 20, 14293–14311 CrossRef CAS PubMed.
- Y. Yang, et al., Lipid coated mesoporous silica nanoparticles as photosensitive drug carriers, Phys. Chem. Chem. Phys., 2010, 12, 4418–4422 RSC.
- M. Conte, M. Carofiglio, G. Rosso and V. Cauda, Lipidic Formulations Inspired by COVID Vaccines as Smart Coatings to Enhance Nanoparticle-Based Cancer Therapy, Nanomaterials, 2023, 13(15), 2250 CrossRef CAS PubMed.
- L. Bircher, et al., Characterization of carbon-coated magnetic nanoparticles using clinical blood coagulation assays: effect of PEG-functionalization and comparison to silica nanoparticles, J. Mater. Chem. B, 2014, 2, 3753–3758 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Estimation of amine groups on the NP surface, DLS and Z-potential, nanoparticle tracking analysis, fluorescence microscopy co-localization results, additional characterization of nanocages and lipid-coated nanocages, haemocompatibility assays, additional characterization of nanocages loaded with calcein and doxorubicin, and videos of 3D reconstruction of internalization of lipid-coated nanocages within cells. See DOI: https://doi.org/10.1039/d3nr02155f |
| ‡ These authors have equally contributed to the work. |
| This journal is © The Royal Society of Chemistry 2023 |