
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA super-resolution and transmission electron microscopy correlative approach to study intracellular trafficking of nanoparticles†
Teodora
Andrian
 a,
Yolanda
Muela
b,
Lidia
Delgado
b,
Lorenzo
Albertazzi
*ac and
Silvia
Pujals
*d
a,
Yolanda
Muela
b,
Lidia
Delgado
b,
Lorenzo
Albertazzi
*ac and
Silvia
Pujals
*d
aInstitute for Bioengineering of Catalonia (IBEC), Carrer Baldiri Reixac 15-21, 08024 Barcelona, Spain. E-mail: l.albertazzi@tue.nl
bElectron Cryomicroscopy Unit, Centres Científics i Tecnològics de la Universitat de Barcelona (CCiTUB), Carrer Baldiri i Reixac 10-12, 08028 Barcelona, Spain
cDepartment of Biomedical Engineering, Institute for Complex Molecular Systems (ICMS), Eindhoven University of Technology, 5612 AZ Eindhoven, The Netherlands
dDepartment of Biological Chemistry, Institute for Advanced Chemistry of Catalonia (IQAC-CSIC), Carrer Jordi Girona 18-26, 08034 Barcelona, Spain. E-mail: silvia.pujals@iqac.csic.es
First published on 14th August 2023
Abstract
Nanoparticles (NPs) are used to encapsulate therapeutic cargos and deliver them specifically to the target site. The intracellular trafficking of NPs dictates the NP-cargo distribution within different cellular compartments, and thus governs their efficacy and safety. Knowledge in this field is crucial to understand their biological fate and improve their rational design. However, there is a lack of methods that allow precise localization and quantification of individual NPs within distinct cellular compartments simultaneously. Here, we address this issue by proposing a correlative light and electron microscopy (CLEM) method combining direct stochastic optical reconstruction microscopy (dSTORM) and transmission electron microscopy (TEM). We aim at combining the advantages of both techniques to precisely address NP localization in the context of the cell ultrastructure. Individual fluorescently-labelled poly(lactide-co-glycolide)-poly(ethylene glycol) (PLGA-PEG) NPs were directly visualized by dSTORM and assigned to cellular compartments by TEM. We first tracked NPs along the endo-lysosomal pathway at different time points, then demonstrated the effect of chloroquine on their intracellular distribution (i.e. endosomal escape). The proposed protocol can be applied to fluorescently labelled NPs and/or cargo, including those not detectable by TEM alone. Our studies are of great relevance to obtain important information on NP trafficking, and crucial for the design of more complex nanomaterials aimed at cytoplasmic/nucleic drug delivery.
1. Introduction
Nanoparticles (NPs) are used in medicine to encapsulate therapeutic molecules (e.g. chemotherapeutics, protein inhibitors and nucleic acids) in order to improve their target selectivity,1,2 solubility,3,4 and to reduce their toxicity.5–7 Yet, although several NP formulations have been successfully marketed, achieving efficient intracellular delivery remains a significant bottleneck in nanomedicine.8–11 Following cellular uptake, the next critical stage in NP delivery is intracellular trafficking, which determines the NP-cargo distribution within cellular compartments and hence the therapeutic efficacy and safety.12,13 For example, a pre-requisite for RNA and DNA delivery is for the molecules to reach the cytoplasm or nucleus intact to perform their therapeutic function – hence NPs carrying such degradation-prone molecules must overcome several cellular barriers, including trafficking to acidic endosomes and lysosomes that can lead to NP disassembly, cargo degradation and loss of efficacy.14–16 In order to improve the therapeutic efficacy of nanomaterials it is thus essential to understand their biological interaction with cells, including intracellular trafficking.17–19However, considerable uncertainties still remain about the exact intracellular pathways taken by nanocarriers, and this largely stems from a lack of reliable methods to visualize and quantify with enough resolution NPs at a single particle level within the ultracellular environment.16,19,20 NP uptake and intracellular trafficking are typically assessed using flow cytometry, fluorescence microscopy (FM) and/or transmission electron microscopy (TEM).19–22 Notably, flow cytometry measures relative fluorescence intensity and not individual NPs and does not distinguish the NPs location (e.g. membrane-bound, endosomal, cytosolic). FM lacks the adequate resolution to resolve individual NP, and objects that are found closer together than the resolution of the microscope (≈250 nm) can falsely appear co-localized.22 It is also limited by the number of targets that can be labelled and lacks ultrastructural information. An upgrade to classical FM are super-resolution microscopy (SRM) techniques that allow localization of single molecules and single NPs down to tens of nanometres.23 For example, stochastic optical reconstruction microscopy (STORM) is based on the localization of individual, photoswitchable fluorophores, offering a resolution of ≈20 nm, and has been previously applied for the intracellular trafficking of several types of nanomaterials.24–29 Despite this achievement, it still lacks cellular ultrastructural information, and can typically be used to image co-localization in only two simultaneous targets. In contrast, TEM offers a powerful approach to study intracellular trafficking pathways, due to its excellent resolution (<1 nm) and ultracellular detail, but it only offers analysis on grayscale images and is restricted to nanomaterials with adequate atomic contrast (e.g. metallic NPs).30–32
To overcome the limitations of FM and EM, a group of techniques that combine both microscopies through detailed images of the same field-of-view (FOV) have been developed, namely correlative light and electron microscopy (CLEM).33,34 Despite the potential of combining two very powerful techniques, only a handful of articles demonstrate the applicability of CLEM on intracellular trafficking of nanomaterials.35,36 Whilst these studies clearly demonstrate the power of CLEM in understanding NP intracellular metabolism, there is still a resolution gap between the FM and EM techniques used, preventing the assignment of single particles to specific compartments.
To bridge the resolution gap between FM and EM, the correlation of SRM-EM has been established, albeit not in the nanomedicine field, substantially improving image quality.33,34,37 Various combinations of SRM-EM approaches have been used in biology to image intracellular fluorescent proteins,38 track intracellular pathogens39,40 and to study budding of influenza viruses from infected cells.41 Recently, we have developed a DNA-PAINT-TEM correlative approach to study the relationship between NP functional ligand numbers and size at a single particle level.42 However, despite the potential of CLEM in this field, and to the best of our knowledge, SRM-TEM correlative approaches have not yet been applied for the intracellular trafficking of NPs. At a cellular level, SRM-TEM correlation would offer a means to specifically label a variety of nanomaterials including their cargo, and to track them within specific cellular compartments with nanoscale precision.
To address this issue, we propose a dSTORM-TEM correlative protocol to track fluorescently labelled polymeric poly(lactide-co-glycolide)-poly(ethylene glycol) (PLGA-PEG) NPs at a single particle level within different cellular compartments. Polymeric NPs were chosen as a standard NP formulation for this protocol due to their well-studied properties; however they also show great promise for targeted drug delivery systems due to their biocompatibility, biodegradability, design flexibility and safety.43–46 Notably, these NPs are undetectable by TEM alone in the cellular milieu due to their low contrast, highlighting the relevance of this correlative approach for imaging a wide range of nanomaterials.
We first describe our adapted dSTORM-TEM correlative method and demonstrate that the superior resolution of dSTORM, as opposed to low-resolution FM, offers localization of single NPs within specific cell compartments in TEM. First, we use the proposed technique to track NPs within different compartments (early and late endosomes, lysosomes and non-endosomal compartments) at different time points. Second, we study the effect of the lysomotropic agent chloroquine on the NP intracellular distribution, with nanometric resolution. We envision that this technique will be applicable to answer various questions on intracellular trafficking and endosomal escape, and to study more complex nanomaterials such as polyplexes for nucleic acid delivery, overall offering unique information that we are currently lacking on the biological fate of nanomaterials.
2. Results and discussion
2.1. dSTORM-TEM correlative method
To track NPs within different cellular compartments at a single particle level, we developed a dSTORM-TEM correlative approach that benefits from nanometer-scale resolution in both techniques. The method described here (Fig. 1) is an adaptation of the confocal microscopy – TEM correlative protocol used by Katheder et al. to assess autophagy in Drosophila tumour growth47 and based on previous findings that demonstrate that fluorescent signal is maintained and ultrastructure is preserved in cells which have been high-pressure frozen, freeze-substituted and embedded in lowicryl resin sections.48–50 Other more classical protocols were attempted without high-pressure freezing (HPF) and freeze-substitution (FS), using (a) an epoxy resin or (b) LR white acrylic resin (Fig. S1†). The three different protocols shared some common aspects, for example that dSTORM imaging was carried out directly on the thin sections and both FM and EM were done at RT. We chose to carry out dSTORM on ultrathin sections (after resin embedding) as it reduces the risk of morphological changes between the two microscopies.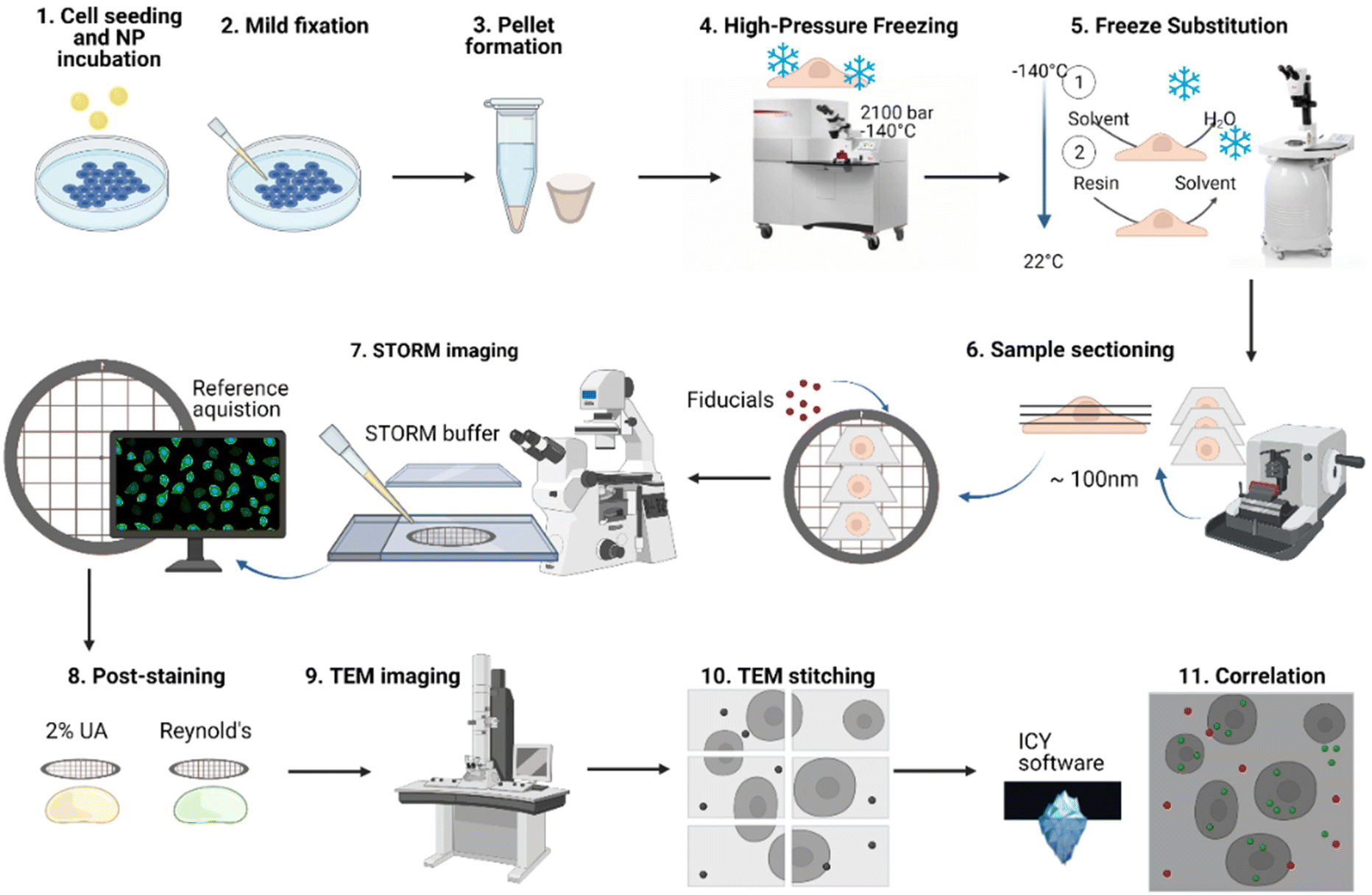 | ||
| Fig. 1 Overview of dSTORM-TEM correlative protocol. After the formulation of PLGA-PEG DiI loaded NPs, the NPs were incubated with HeLa cells for specific time periods, and mildly fixed. Then, after scraping, the cells were centrifuged and the resulting pellets were high-pressure frozen and freeze substituted, then sectioned using an ultramicrotome to obtain ultrathin sections (70–100 nm). The ultrathin sections were picked up on a TEM grid and fiducials markers attached to aid in correlation. The grids were first imaged by dSTORM microscopy using a dSTORM buffer, and reference images of the region of interest (ROI) were acquired. The sections were then post-stained using the heavy metals 2% uranyl acetate and 3% lead citrate (Reynolds). Then, the ROI was found in TEM and imaged by sequential imaging, followed by stitching of the sequential images. Finally, the respective dSTORM and TEM images of the same ROI were overlapped using the open-source software ICY (ec-CLEM) by using the fiducial markers in both images. | ||
However, for 2 out of the 3 protocols, we had the issue of fluorescence quenching due to harsh fixation steps and the use of osmium tetroxide. For the first protocol, RT pathway using an epoxy resin, the ultrastructure preservation was poor (Fig. S1B†), probably due to using milder fixation protocols, and the fluorescence signal quenched in the process as can happen even at low osmium tetroxide concentrations and upon epon resin embedding. The second protocol consisted of a softer hydrophilic acrylic resin, namely LR white, that has been previously used for both EM and LM. Whilst the ultrastructure preservation was slightly better than with the previous resin, the fluorescence signal was once again quenched which came as a surprise since no osmium was used but could have been caused by the high temperatures used during resin polymerization.
The third and final protocol we attempted was using Lowicryl HM20 resin together with high-pressure freezing (HPF) and freeze substitution (FS) (Fig. S1A, S1D and Fig. S2†). This combines a great structural preservation with the advantage of imaging with high numerical aperture lenses at RT. We hoped that aside from the ultrastructural preservation, the reduction in chemical fixatives used, avoidance of both osmium tetroxide and high temperatures would also prevent fluorescence quenching.
Briefly, PLGA-PEG DiI loaded NPs were formulated via the nanoprecipitation method42,51,52 and characterized using TEM for diameter and morphology (Fig. S3†), dynamic light scattering (DLS) for the hydrodynamic radius, (Fig. S4†) and zeta potential for the surface charge (Fig. S4†). HeLa cells were then incubated with the NPs for specific time points. The cells were then mildly chemically fixed as this aids in cross-linking cellular structures and preserving membrane structures, without quenching fluorescent signal.47 The cells were then pelleted and high-pressure frozen (HPF), ensuring vitrification at high pressures without the formation of ice crystals. The samples were then processed by freeze-substitution (FS; Table S1†). During this process, cells were further stained and fixed and resin embedded at low temperatures which were gradually increased to ambient temperature. As a fixing and staining agent uranyl acetate (0.1% UA) was used, since strong fixatives such as osmium tetroxide can strongly quench fluorescence even at low concentrations.41,53 A lowicryl HM20 resin was chosen, as it is designed for low-temperatures and due to its low viscosity, it can allow penetration of dSTORM imaging buffers into the resin-embedded samples. The resin-embedded cells were then sectioned using an ultramicrotome to obtain ultrathin sections (70–100 nm) that were then picked up on TEM grids. These specific grids were used since formvar layer allows an easier pick up of ultrathin sections, whilst the nickel grid does not react with any chemicals in the dSTORM buffer. Please note that standard copper grids reacted with the thiol-containing STORM buffer and were unsuitable for imaging. 100 nm Tetraspeck beads were then attached to the ultrathin sections and used as fiducial markers, due to their visibility in both microscopes. Next, the grid was sandwiched between a coverslip and a glass-slide with the dSTORM buffer and sealed. The sample was first imaged by dSTORM, as subsequent staining steps for TEM quench fluorescence, and reference images were acquired showing the location of the region of interest (ROI) on the grid. This step was crucial, as finding the exact same area during TEM imaging was not an easy task. Grids with a guide (a number 1 marked on the outer part of the grid) were used to ease this step. To enhance the contrast, the grids were post-stained after dSTORM imaging with 2% UA and 3% lead citrate (Reynolds), then later imaged by TEM. This post-staining was a key to avoid fluorescence quenching in dSTORM imaging. Due to its superior resolution, the field-of-view (FOV) is smaller than that in dSTORM, thus consecutive images were acquired to cover the required FOV, and later stitched automatically in Adobe Photoshop. The dSTORM-TEM image correlation was achieved using the open access eC-CLEM software package.54 Fine correlation and high location accuracy was achieved by using a high number of fiducial markers on the grid. For extra protocol details see the Experimental section.
2.2. NP intracellular trafficking using dSTORM-TEM correlation
An example correlative image using both low-resolution FM and dSTORM images is shown in Fig. 2. The TEM canvas of the cells is appreciated in Fig. 2A, with the respective PLGA-PEG NP fluorescence visible in green (Fig. 2B) and fiducial markers in red (Fig. 2C). The resulting low-resolution FM-TEM overlap of these images is shown in Fig. 2D. Whilst the low-resolution gives a clearer representation of the NP signal at larger FOVs (Fig. 2D), the superior resolution of dSTORM is better appreciated at greater magnifications (Fig. 2E and F). Due to the greater match in resolutions between dSTORM and TEM, single NPs can be accurately assigned to specific cellular compartments. The ultrastructure in TEM was very well maintained even after using high laser power by dSTORM (Fig. S2†), allowing the morphological detection of various cellular organelles, including early endosomes (EEs), late endosomes (LEs), lysosomes, mitochondria, and nuclei.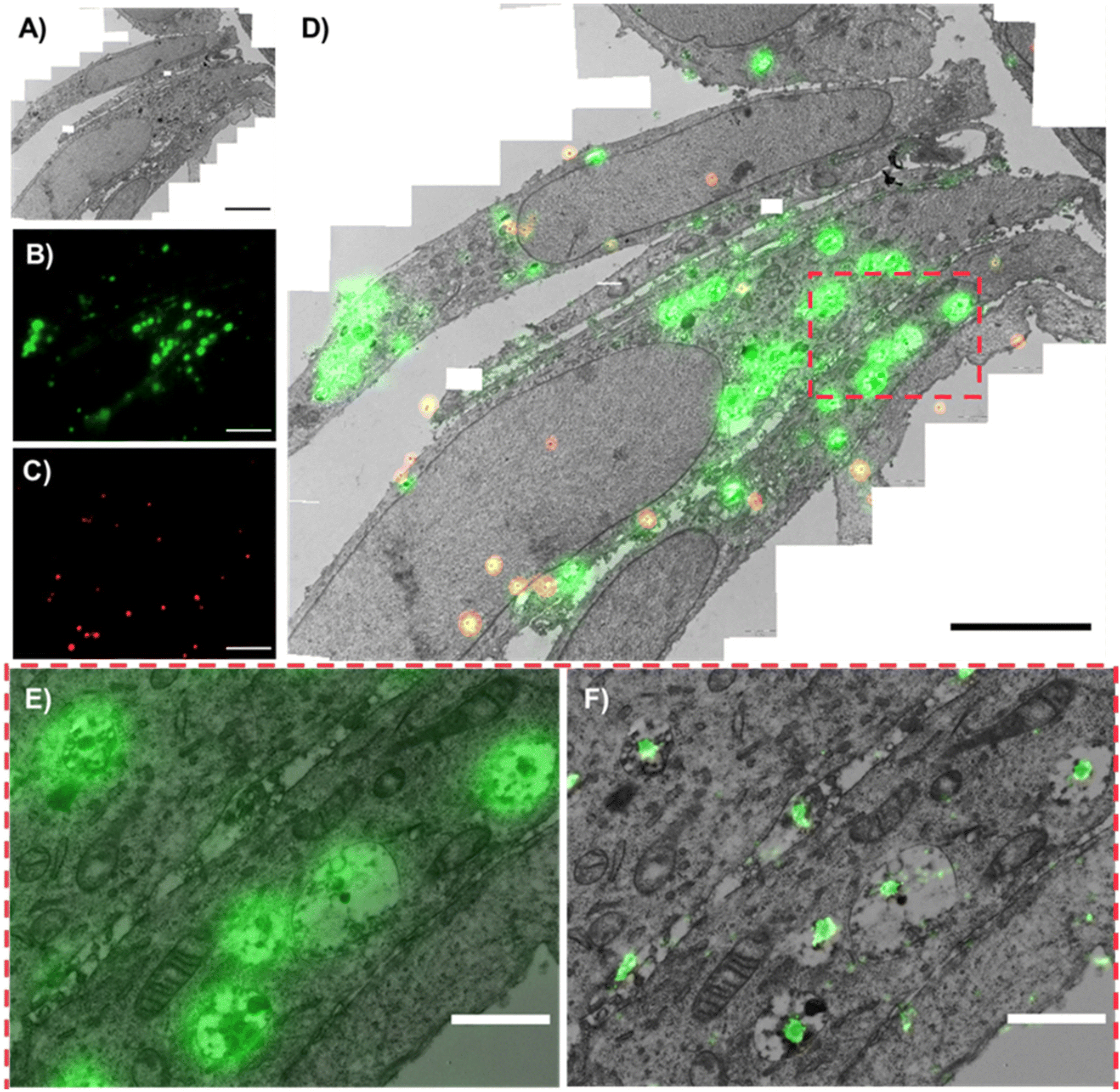 | ||
| Fig. 2 Correlative low-resolution FM/dSTORM and TEM images of PLGA-PEG DiI loaded NPs internalized in HeLa cells. Large view images of the ROI of (A) the stitched TEM image, (B) PLGA-PEG NPs (DiI) signal and (C) fiducial markers both in low-resolution FM, and (D) correlative low-res FM-TEM image (scale bars A–D = 5 μm). Magnified views of the boxed region in (D), where (E) low-resolution FM-TEM correlated image of PLGA-PEG NPs and (F) dSTORM-TEM correlated image of PLGA-PEG NPs, highlighting the difference in resolution between the two FM techniques (scale bars E and F = 1 μm). | ||
Markedly, the mild chemical fixation protocol (4% PFA and 0.1% GA) prior to HPF and FS did not lead to background autofluorescence (Fig. S5†).
Additionally, our protocol did not rely on the use of osmium tetroxide (OsO4) as a contrast enhancer/fixative, whilst the use of low concentration of UA (0.1%), resin and UV polymerization treatments maintained an adequate fluorescence signal viable for further measurements and did not affect cellular ultrastructure. Using the dSTORM-TEM images, we were able to assign single NPs within specific cellular compartments and track their cellular voyage at different time points.
The accuracy error in correlation was calculated automatically using the eC-CLEM plug-in in ICY and ranged between 40–60 nm depending on the number of fiducial markers in the FOV. For this image, 20 fiducial markers were present, and the accuracy was of 52 nm (Fig. S6†), in line with other values reported using super-resolution CLEM and fiducial markers of similar size.55 Briefly, if the registration error of the fiducials was greater than the predicted registration error, a non-rigid transformation was applied (warping), which is useful if any deformations have occurred during preparation steps such as chemical fixation/dehydration.54 Notably, correlation between two techniques can be limited by (i) the limit of resolution of the least resolved technique and (ii) the correlation accuracy between the two techniques. Since the correlation accuracy error is slightly greater than the resolution limit of dSTORM (approx. 20 nm),56 dSTORM-TEM provides a more accurate assignment of single NPs to specific organelles as opposed to correlation with low-resFM (approx. 250 nm). This can be clearly visualized in Fig. 2E and F.
NPs are typically taken up via endocytosis, a mechanism that can be further characterised as caveolae- and clathrin mediated endocytosis, phagocytosis, macropinocytosis and pinocytosis.57 After endocytosis, NPs are engulfed by endocytoic vesicles that fuse to form early endosomes (EEs), and then mature to late endosomes (LEs) and lysosomes12,58–61 (Fig. 3A). This intracellular pathway is thought to be an important limiting step for various formulations that require cytosolic/nuclear delivery of cargo such as chemotherapeutics/RNA/DNA. The lack of methods that allow single NP tracking within the ultrastructure of the cell prevents gaining important insights into their intracellular trafficking and developing more effective nanomaterials. Although in this work we use standard non-functionalized PLGA-PEG NPs to demonstrate the capability of super-resolution CLEM, the protocol is extendable to other fluorescently labelled nanomaterials and/or their cargo.
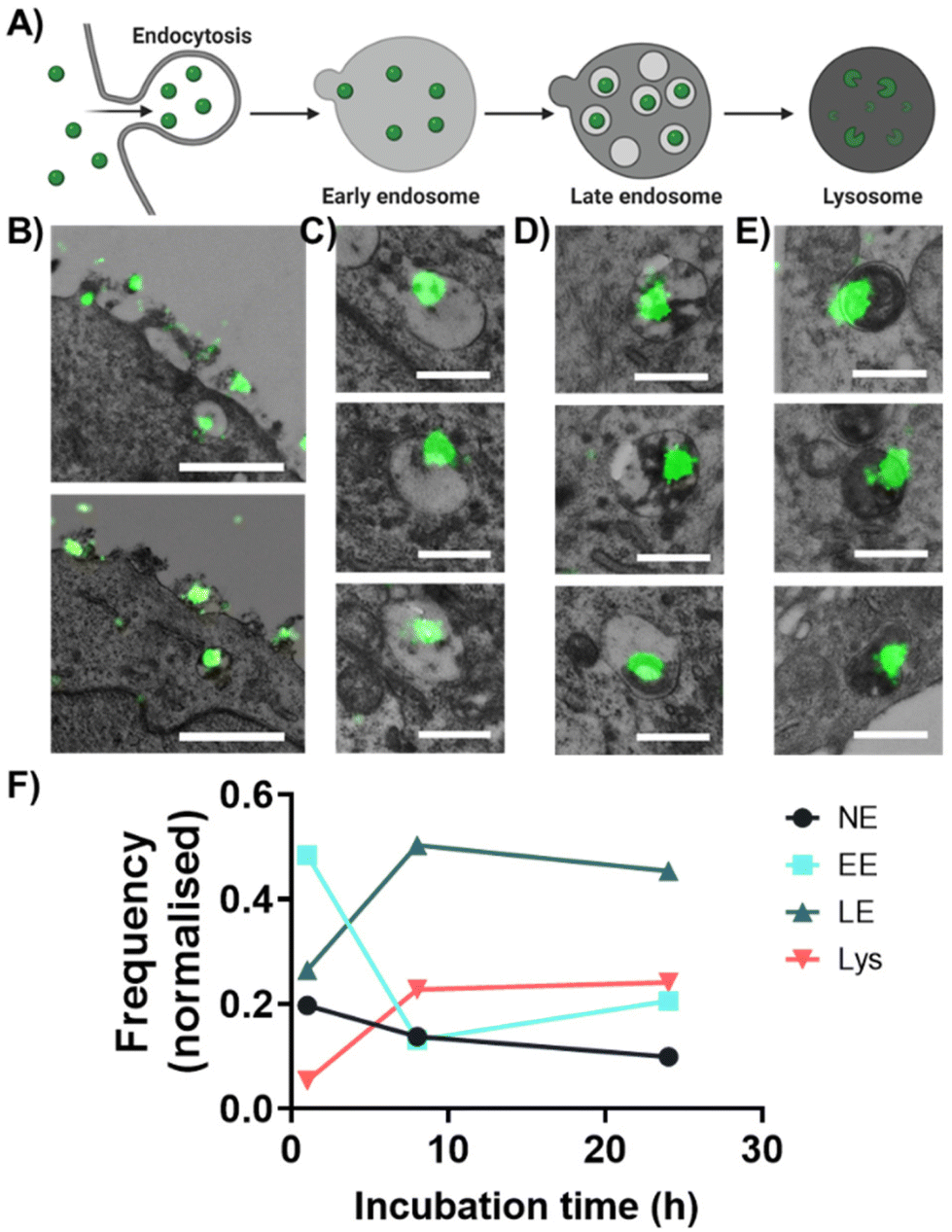 | ||
| Fig. 3 dSTORM-TEM correlation for intracellular trafficking of PLGA-PEG NPs. Scheme of (A) endo-lysosomal pathway consists of NP endocytosis, and trafficking by early endosomes (EE) to late endosomes (LE) and to lysosomes in HeLa cells. (B) PLGA-PEG DiI loaded NPs detected by dSTORM and localized within different cellular compartments by TEM: (B) during endocytosis, (C) in EE, (D) in LE and (E) in lysosomes. (F) Time course of quantitative dSTORM-TEM analysis of PLGA-PEG NPs progressing through EE, LE, lysosomes and non-endosomal compartments (NE). Normalization was done using the total number of NPs at each time point. At 1 h N = 223, at 8 h N = 145 and at 24 h N = 141. Scale bar B = 1 μm, C–E = 500 nm. | ||
For this purpose, we used the proposed dSTORM-TEM protocol to explore the intracellular fate of PLGA-PEG NPs, by localizing single NPs using dSTORM signal and assigning them to specific cellular compartment using the morphological information from TEM. To do this, we first incubated HeLa cells with NPs for different periods of time (1 h, 8 h and 24 h) and treated the cells as described in the previous section. A 4 h pulse was done for the 8 h and 24 h time-points and then NPs were chased for the remaining respective time. We first correlated the low-resolution FM images with the respective TEM images as this allows a clearer correlation at a large FOV (Fig. S7†). Notably, at 1 h NPs were still found attached to/surrounding the cell membrane, which reduced with increasing incubation times. Punctate intracellular signal is seen at all time points (Fig. S7†), indicating NP uptake into endo-lysosomal compartments had started prior to 1 h, agreeing with previous results under similar conditions.62–64
We then correlated the dSTORM and TEM images, since the increase in resolution from dSTORM allowed precise tracking of individual NPs within the cellular milieu. NPs were imaged during their cell uptake (Fig. 3B, left panel), to their distribution in EE (Fig. 3C), LE (Fig. 3D) and lysosomes (Fig. 3E). The NPs were assigned to different compartments by morphological comparison between different vesicles as previously described by Fermie et al. using a CLEM approach.65 EE typically have a diameter between 100–500 nm with an electron-lucent lumen and loosely packed intraluminal vesicles (ILVs); LE have a diameter between 250–1000 nm with densely packed ILVs; lysosomes have diameters between 200 to >1000 nm with an electron-dense lumen and membrane whorls. For extra correlative images at 1 h, 8 h and 24 h see Fig. S8, S9 and S10,† respectively. Occasionally we were also able to detect non-endosomal NPs (NE), i.e. found inside the cell but not in any of the three compartments/nucleus (Fig. S11†). Notably, polymeric nanoparticles, such as PLGA-PEG, may be slightly visible at the cell membrane but not at all visible within the cellular milieu by TEM alone, showing the suitability of this method to a wide range of nanomaterials.
For the quantification, NP clusters were counted in each compartment and normalized based on the total number of NPs (Fig. 3F). At 1 h, most NPs were found within the EE (∼48%), followed by LE (∼26%) and NE (∼20%), with the minority in lysosomes (∼5%). This suggests that PLGA-PEG NPs were taken up by cells via endocytosis and then transported through the classic endocytic pathway, as has been previously reported.61,66 As the incubation time increased to 8 h, the population of NPs in EE decreased to ∼13%, whilst it increased in LE to ∼50% and in lysosomes to ∼23%, suggesting that NPs have progressed along the endo-lysosomal pathway. At 24 h the distribution of NPs within the specific compartments remained similar to that at 8 h. This could be explained by the fact that at 8 h more NPs were still found at the membrane compared to 24 h (Fig. S7 and S12†), suggesting that between 8–24 h NPs were still being endocytosed and transported through the endocytic pathways, agreeing with previous results.59
As previously stated, NPs were also found in NE compartments (Fig. S11†), albeit in small amounts. Since PLGA-PEG NPs are not expected to achieve endosomal escape, we believe these results could be due to broken organelles releasing NPs into the cytoplasm,67 as partially broken endo-lysosomal compartments containing NPs were seen by dSTORM-TEM (Fig. S13†). However, these results could also indicate partial degradation of NPs in the lysosomes and release of the dye in the cytosol. Although in this experiment we were not interested in studying endosomal escape, being able to localize and quantify NPs outside of endo-lysosomal compartments indicates that our technique is well-tailored to study this process as will be demonstrated later.
Notably, a 72 h time point was also carried out, which showed no fluorescence signal by CLEM (Fig. S14†), as opposed to the other time points. To confirm NP internalization at these various time points, a flow-cytometry assay was carried out (Fig. S15†). As expected, the lowest internalization is seen at 1 h, followed by a significant increase at 8 h. At 24 h there was a decrease in the number of internalized NPs, followed by a further decrease at 72 h, comparable to the uptake at 1 h. These results agree with the decrease in fluorescence seen at 72 h in Fig. S13† and could be due to a release of NPs from the cells through the recycling endosome system and/or through the exocytosis pathway as demonstrated previously for PLGA NPs61,66 and other nanomaterials.57,68–70 Since these NPs reach the lysosomes, it is also possible that they are degraded, as previously shown for PLGA NPs.61 Another possible reason is that since HeLa cells have a doubling time of roughly 33–35 h,71 after this time intracellular NP load is expected to decrease as a result of cell division.72 Finally, CLEM is still a low-throughput technique, meaning that if the NP load is low or has been diluted/degraded, there is a high chance that NP presence will not be detected. Further systematic analysis of longer time points would be required to fully understand these observations.
Using the information from dSTORM-TEM correlative images, we were able to assign and quantify the distribution of NPs among different cellular compartments at a given time point. This is not possible with the classical confocal colocalization analysis that are generally limited to two structures at a time and suffer of resolution issues (structures in proximity tend to colocalize due to lack of resolving power). Overall, these results demonstrate the applicability of dSTORM-TEM correlative imaging in tracking individual nanomaterials and/or their cargo intracellularly, for a better understanding of their biological fates. However, there is a clear need to develop more high-throughput protocols that would result in more user-friendly technique, more significant statistics and the ability to catch rare events in the act.
2.3 The effect of chloroquine on NP intracellular distribution using dSTORM-TEM
Our previous results showed that PLGA-PEG NPs were mainly entrapped within the endolysosomal pathway. Endosomal entrapment is a process that typically happens after endocytosis, whereby the carrier–drug ensemble is entrapped and degraded in endo-lysosomal compartments, and thus presents a key bottleneck in ensuring effective therapeutic delivery using nanocarriers.11,19,20,73 As previously mentioned, current ensemble techniques used in this field prevent the thorough understanding of endosomal escape mechanisms. We therefore wanted to demonstrate the applicability of our proposed dSTORM-TEM correlative protocol on studying endosomal escape. To promote the escape of PLGA-PEG NPs from endo-lysosomal compartments, we used the lysomotropic endosomal escape enhancing compound chloroquine74,75 (Fig. 4). Cells were incubated with 100 μM chloroquine for 4 h, followed by a 4 h NP pulse and a 4 h NP chase (total 8 h) (+CQ), and compared with an identical sample without chloroquine treatment (−CQ). The ultrathin sections were prepared in accordance to the described CLEM protocol. After imaging by dSTORM-TEM (Fig. 4A and B), NP clusters were counted in each compartment and normalized according to the total number of NPs (Fig. 4C).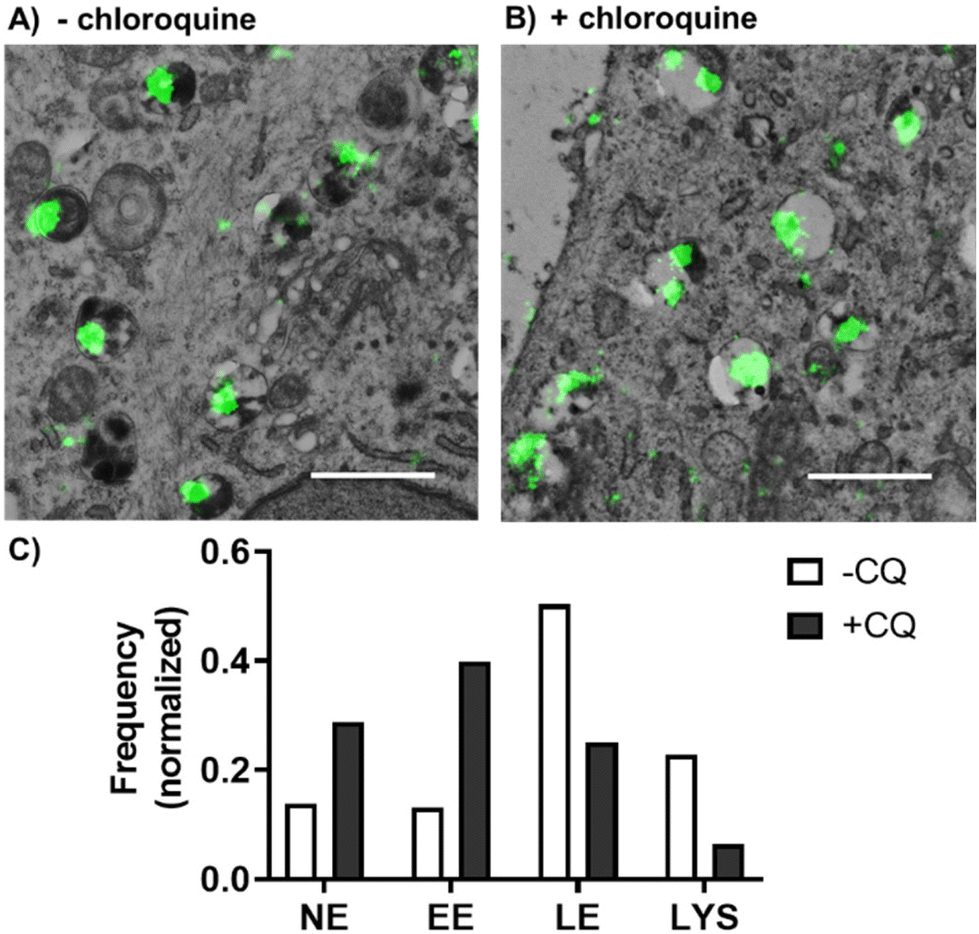 | ||
| Fig. 4 dSTORM-TEM correlation for studying the effect of chloroquine on PLGA-PEG NP intracellular trafficking. PLGA-PEG DiI loaded NPs were detected by dSTORM and localized within different cellular compartments using TEM. dSTORM-TEM correlative images of (A) NPs incubated with HeLa cells for 8 h, and (B) HeLa cells treated with 100 μM chloroquine for 4 h prior to 8 h NP incubation. A 4 h NP pulse and a 4 h NP chase were carried out for both samples. Scale bars = 1000 nm. (C) Quantitative analysis of NP distribution within different cellular compartments without (−CQ) and with chloroquine treatment (+CQ). Normalization was done using the total number of NPs at each condition: −CQ N = 145, +CQ N = 236. | ||
For the cells not treated with chloroquine (−CQ), PLGA-PEG NPs were found mainly in LE and lysosomes, with a minority found in EE and NE compartments. In contrast, chloroquine treatment (+CQ) caused a 4-fold and a 2-fold decrease in NPs found in lysosomes and LE, respectively, whilst causing a 3-fold and a 2-fold increase in NPs found in EE and NE compartments, respectively (Fig. 4C). For extra dSTORM-TEM images see Fig. S16.† These results agree with the documented behaviour of chloroquine as a damage-inducing agent for vesicles in the late stages of the endo-lysosomal pathway (low pH compartments), leading to less NP found in LE and lysosomes, but more within the EE and cytoplasm.
Endosomal entrapment is a recognised bottleneck in drug delivery and for this increasing research is aimed at designing formulations with endosomal escape in mind. However, whilst the arsenal of nanomaterials is increasing, there is still a tendency in the field to use qualitative and inconsistent techniques (e.g. co-localization studies using confocal microscopy and transfection efficiency) to study their endosomal escape properties. To be able to design more successful nanocarriers, we must be able to track their intracellular pathways using more quantitative and direct techniques, and this work highlights the relevance of dSTORM-TEM in achieving this quest.
3. Experimental
3.1. Nanoparticle formulation
PLGA-PEG NPs were formulated via the precipitation-solvent evaporation (nanoprecipitation) method according to literature1 and to our previously reported data.2–4 PLGA (PLGA, 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 LA:GA, Mw 25–35 kDa; PolySciTech, Akina) and PLGA-PEG (PLGA LA:GA 50:50, Mw. 30 kDa and PEG 5 kDa; PolySciTech, Akina) were dissolved in the solvent acetonitrile (ACN, Chem-Lab, HPLC grade, Sigma Aldrich) to give stock solutions. To ensure maximum dissolution, the solutions were maintained at R.T. for 2 h, with 1 min vortex every 30 min, followed by a final 10 min bath sonication. Fresh milliQ water was filtered with a 0.22 μm sterile filter (Merck Milipore, Millex GP) and used as the antisolvent phase. 1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI, lipophilic cationic carbocyanine dye; Sigma Aldrich) was weighed and dissolved in ACN to a concentration of 1.1 mM and used as a stock solution. PLGA and PLGA-PEG stock solutions were mixed to give a 15:85 (wt:wt) ratio, respectively, and DiI stock solution was added to give a final concentration of 30 μM.
:50 LA:GA, Mw 25–35 kDa; PolySciTech, Akina) and PLGA-PEG (PLGA LA:GA 50:50, Mw. 30 kDa and PEG 5 kDa; PolySciTech, Akina) were dissolved in the solvent acetonitrile (ACN, Chem-Lab, HPLC grade, Sigma Aldrich) to give stock solutions. To ensure maximum dissolution, the solutions were maintained at R.T. for 2 h, with 1 min vortex every 30 min, followed by a final 10 min bath sonication. Fresh milliQ water was filtered with a 0.22 μm sterile filter (Merck Milipore, Millex GP) and used as the antisolvent phase. 1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI, lipophilic cationic carbocyanine dye; Sigma Aldrich) was weighed and dissolved in ACN to a concentration of 1.1 mM and used as a stock solution. PLGA and PLGA-PEG stock solutions were mixed to give a 15:85 (wt:wt) ratio, respectively, and DiI stock solution was added to give a final concentration of 30 μM.
4 mL of the anti-solvent phase (water) was added to a glass vial (5 mL) with a magnetic stirring bar (VWR, 8 × 3 mm) and placed on a magnetic stirring plate (IKA3050009 Big Squid White) at 200–300 rpm. Maintaining a 10:1 ratio between antisolvent and solvent, 400 μL of the solvent phase (polymeric solution) was added dropwise to the stirring antisolvent phase using a 20–200 μL pipette. The final NP suspension was allowed to stir for 5 h to enhance solvent evaporation. The capped glass vial was then kept in the fridge at 4–8 °C until further use.
3.2. Cell plating and nanoparticle incubation
HeLa cells (ATCC) were seeded on a 92 × 17 mm cell culture dish (NuncTM, Fisher Scientific) and incubated for 24 h in Dulbecco's modified Eagle's medium (DMEM, with L-glutamine 10 mM, 4.5 g L−1D-glucose and pyruvate, Gibco) supplemented with FBS 5% (Gibco) and penicillin/streptomycin 1% (Biowest), at 37 °C and 5% CO2, to full confluency. After the incubation, the medium was removed and the cells washed thrice with warm PBS (1×, pH 7; Thermo Fisher). Then PLGA-PEG NPs diluted to 1.5 mg/5 mL in OptiMEMTM (Thermo Fisher) were added to the cell plate and incubated for either 1 h, 8 h, 24 h or 72 h at 37 °C and 5% CO2. For the latter 3 time points, the NP solution was removed after 4 h and washed thrice with PBS (at 37 °C), then replaced with medium for the remaining incubation time.Next, the cells were first fixed for 30 min at R.T. by adding an equal volume of fixative 1 (4% paraformaldehyde, 0.1% glutaraldehyde, in 0.1 M PHEM (240 mM PIPES, 100 mM HEPES, 8 mM MgCl2, 40 mM EGTA; pH 6.9)), in an equal volume of the FDMEM found in the cell culture dish. Then, fixative solution was discarded and replaced with 5 mL fixative 1 for another 30 min at R.T. Then, the solution was removed and replaced with 1.5 mL of fixative 2 (2% paraformaldehyde in 0.1 M PHEM) for 5 min at R.T., ensuring that the cell dish is evenly covered. The cells were then scraped off (whilst in fixative 2) using a cell scraper and transferred to a 1.5 mL Eppendorf. These were then pelleted using a centrifuge (Eppendorf 5804 R) at 2500 rpm (1200 rcf) for 10 min at R.T. The supernatant was replaced with 1 mL fixative 2, then centrifuged again. The pellet was kept in fixative 2 at 4 °C and covered from light until next steps.
3.3. High-pressure freezing, freeze substitution, embedding and ultramicrotomy
The following protocol is an adaptation of the protocol used by Katheder et al.,47 with some modifications. Briefly, pellets were suspended in 20% dextran (in PBS), pipetted to 200 μm-depth planchettes and high-pressure frozen (Leica HPM100). The freeze substitution and resin embedding steps were carried out using a temperature-controlled AFS2 (Leica). For full details on freeze substitution protocol see Table S1.† Briefly, sample carriers were transferred to 4 mL freeze substituent (0.1% (w/v) uranyl acetate in acetone) inside the pre-cooled AFS2 chamber, and the freeze substitution steps occurred from −140 °C to −45 °C (steps 1–5). Then, the samples were washed 4 times in acetone at −45 °C (steps 6–9), followed by infiltration with increasing concentrations of Lowicryl HM20 resin (25%, 50%, 75%, and 100%; steps 10–14; Electron Microscopy Sciences). Finally, two last steps of HM20 infiltration take place, whilst the temperature is increased slowly from −48 °C to 22 °C (steps 15–16). UV-polymerization occurred in steps 14–16.Two semithin sections (500 nm) were obtained for each sample on an Ultracut UC7 ultramicrotome (Leica). One was stained using methylene blue, for a first visual check-up of cell morphology. The second semithin section was stained using 20 μg mL−1 Hoechst (Sigma-Aldrich) in PBS for 10 min at R.T. and imaged using a Nikon N-STORM microscope to check for fluorescence. Then, ultrathin sections (70–100 nm) were obtained and collected on a 200-mesh formvar film-coated hexagonal nickel grids.
3.4. Preparation for STORM imaging
As per Fig. S17,† the grids were first placed on top of 40 μL drops of nuclei stain Hoechst 20 μg μL−1 (in PBS) for 10 min at R.T. using a fine tweezer. The grids were then washed in 40 μL ×3 consecutive milliQ water drops (1 min each). Next, for addition of fiducial markers, the grids were placed on a 40 μL drop of Tetraspeck (0.1 μm diameter, ∼1.8 × 1011 particles per mL in 2 mM sodium azide; Invitrogen) diluted ×500 in PBS for 10 min, then washed through a drop of milliQ water.A 40 μL drop of GLOX imaging buffer was placed on top of a glass microscopy slide (FisherBrand), containing (a) 100 mM mercaptoethylamine (MEA), (b) an oxygen scavenger system consisting of 5% glucose (wt/vol), 0.5 mg mL−1 glucose oxidase (Sigma-Aldrich) and 40 mg mL−1 catalase (Sigma), (c) 15% glycerol, in PBS at pH 8.5. This was covered with a glass coverslip (Corning Cover Glass, thickness 1½, 22 × 22 mm). The imaging chamber was then sealed using transparent nail polish to prevent buffer evaporation (see Fig. S16†).
3.5. STORM imaging
First, several reference images were taken as seen in Fig. S18:† (1) the EPI lamp on DAPI filter was used to take a 13 × 13 stitch (1024 × 1024) of the distribution of the ultrathin sections on the grid, using the NIS-Elements stitch function. (2) The second reference image was taken using the same lamp and filter at 1024 × 1024 inside the grid hexagon of interest, showing the cell nuclei, then (3) one image was acquired at 256 × 256, which gives the exact FOV that will be imaged by STORM. Then, using the Nikon N-STORM system configured for highly inclined and laminated optical sheet (HILO) imaging, a low-resolution image of NPs was taken using the 561 nm (80 mW) laser, and a low-resolution image of the fiducial markers using the 647 (160 mW) laser, both at 2% laser power and 300 ms. These latter two images are used for correlation of low-resCLEM images.NPs were imaged by STORM by illuminating the sample with the 561 nm (80 mW) laser at 100% power, and the fiducial markers with the 647 nm (160 mW) laser at 5% power. Fluorescence was collected using a Nikon 100×, 1.49 NA oil immersion objective and passed through a quadband pass dichroic filter (97335 Nikon). Images were acquired onto a 256 × 256-pixel region (pixel size 0.16 μm) of a Hamamatsu ORCA Flash 4.0 camera at 20 ms integration time. For the 561 channel 10000 frames were acquired, and for the fiducial markers one frame was acquired every 100 frames in the 647 channel. Single molecules localization movies were analyzed with NIS element Nikon software. Data was analyzed with NIS Elements (Nikon) and ImageJ. No filters were applied to the STORM images presented in the article.
3.6. Preparation for TEM imaging
To remove the grid from the microscopy chamber without ripping/damaging the grid, it is important to be as gentle as possible whilst doing the following steps. First, tissue embedded in acetone was used to gently remove the nail polish around the edges of the coverslip, trying to not move the coverslip whilst doing this. Then, as seen in Fig. S19,† the microscopy chamber was placed in a Petri dish filled with milliQ water, and the coverslip was gently moved left and right, until detached from the microscopy slide, then with a tweezer, gently lifted. Next, the grid was picked up using a tweezer and washed to remove buffer salts on 5 × 40 μL milliQ water drops for 1 min each. The grid was allowed to dry for at least 15 min, and then stained.For staining of the ultrathin sections, the grid was first placed on a 20 μL drop of 2% uranyl acetate (in milliQ water) for 30 min at R.T. Then, the grid was removed using a tweezer and rinsed thoroughly in 200 mL milliQ water using a jet of water. If the grid dropped in the beaker, it was gently removed and washed with another 200 mL of milliQ water. The grid was then allowed to dry for at least 15 minutes. To prevent precipitation of lead in Reynolds’ solution (prepared in house) by exposure to atmospheric CO2, a pellet of NaOH was placed underneath a small Petri dish, to exclude atmospheric CO2. Then, a 20 μL drop of 3% lead citrate (Reynolds’) was placed quickly underneath the Petri dish, next to the NaOH pellet. The grid was then stained for 1 min and washed thoroughly using a 200 mL jet of milliQ water. The grid was then allowed to dry inside a desiccator for at least 2 h before imaging in TEM.
3.7. TEM imaging
The grids were imaged using a Jeol 1010 (Gatan, Japan) from the Electron Cryomicroscopy Unit from the CCiTUB, equipped with a tungsten cathode. Images were acquired at 80 kV with a CCD Megaview 1k × 1k and the Analysis 3.2 software. First, the ROI was located using a low-magnification function and the reference images acquired previously. Then, sequential images were taken of the ROI at a magnification of ×15000, which were later stitched using the ‘photomerge’ function in Adobe Photoshop CS9. Images were also taken at greater magnifications such as ×30000 to highlight in better resolution structures of interest.
3.8. Correlation and data analysis
The low-resolution/STORM and TEM images were first pre-processed to enhance contrast and brightness, and overlay was achieved using the open access eC-CLEM plugin using the ICY bioimage analysis platform. To do this, fiducial markers visible in both images were selected, and the program would achieve the correlation. Typically, a non-rigid (warping) transformation would be carried out if the registration error of the fiducials was greater than the predicted registration error. Briefly, as seen in Fig. S20,† around 20–30 fiducial markers were localized in both images, and a non-rigid transformation was applied. The correlation accuracy (in nm) was calculated and was between 40–60 nm when using 20–30 fiducial markers. Note that the greater the number of fiducials per image, the greater the accuracy and vice versa.STORM images were reconstructed using the NIS Elements software in Nikon using a 2D Gaussian fitting and a threshold of 180 for both channels. The threshold is the difference between the photon number in the peak pixels and the background pixels. The trace length parameter was set to 5 to avoid different molecules blinking consecutively, meaning that molecules identified in 5 consecutive frames would be counted as 1, or removed if more than 5 frames.
For NP quantification, STORM signal with 50–300 nm diameter and 10 or more localizations was considered a true NP. If signal of 2 or more NPs was clearly distinguishable as single NPs, then these were counted as independent NPs. Larger signal (200–300 nm diameter) was considered a single NP. As opposed to TEM, if NPs are closer to each other than the resolution limit of STORM (∼20 nm) they are not distinguishable as individual NPs, and thus a limitation of this method is that the number of NPs may be understated.
4. Conclusions
In this work, we have introduced an efficient dSTORM-TEM correlative protocol to study the intracellular pathway of PLGA-PEG NPs in HeLa cells, that allows the localization of single NPs (dSTORM) within cellular compartments (TEM) with nanometric resolution in both microscopies, whilst maintaining NP fluorescence and preserving cellular ultrastructure.Notably, our method performs the heavy metal staining steps after dSTORM imaging, thus preserving the fluorescence in the sample.
PLGA-PEG NPs were localized at increasing incubation times in different cellular endo-lysosomal and in non-endosomal compartments. We showed that at early time points, NPs were found mainly in EE, whilst at later time points in LE and lysosomes. After 72 h however we were unable to image NPs by CLEM due to a loss of fluorescence, which we attributed to various already proven factors, including exocytosis, cell division and/or particle degradation. Since PLGA-PEG NPs were entrapped within the endo-lysosomal pathway, we then studied their endosomal escape upon incubation with the lysomotropic agent chloroquine. Using our dSTORM-TEM protocol we quantified the cellular distribution of NPs, and showed a decrease of NP localization within low pH compartments (LE and lysosomes), and an increase in EE and NE compartments (i.e. endosomal escape).
Importantly, the proposed dSTORM-TEM protocol can image and localize NPs that are not detectable by TEM alone and thus we envision that it could be applied to track many fluorescently labelled nanomaterials and/or their cargo within the ultracellular environment. Particularly, limiting steps in intracellular delivery such as endosomal entrapment could be studied for more complex nanomaterials such as polyplexes or lipoplexes that require nucleic acid delivery to the nucleus. We believe that advanced microscopy techniques such as CLEM are of great relevance in obtaining complex information that we are currently lacking and could facilitate the rational and efficient design of nanomaterials for drug delivery.
Although the focus of this work is on tracking NPs to understand their fate inside the cell, the applicability of this method expands beyond nanomaterial research and could be applied to other fields in biology, such as the study of virus or extracellular vesicles trafficking, immunology or neuroscience, among others, when labelled with dSTORM suitable dyes (by using genetic tags or click-chemistry). Even though commercial systems for integrated microscopy have been developed,34 they are still a long way from being incorporated into general laboratories. Our protocol requires only equipment typically found within light and electron microscopy facilities, thus allows a wider implementation by the scientific community. Yet, for correlative techniques to be picked up as standard techniques in nanomaterial characterization and demonstrate their worth, more automated, and high-throughput imaging protocols will be required. We believe that a synergistic effort from various scientific disciplines could soon make this a reality.
Author contributions
T. A., L. A. and S. P. conceived the idea and designed the experiments. T. A. performed nanoparticle formulation, cell plating and nanoparticle incubation, preparation for STORM imaging, preparation for TEM imaging and correlation and data analysis. Y. M. and L. D. performed high-pressure freezing, freeze substitution, embedding and ultramicrotomy. T. A. and S. P. performed STORM imaging and TEM imaging. L. A. and S. P. directed and oversaw the project. T. A. wrote the initial manuscript draft. All authors discussed results and provided suggestions for the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The project that gave rise to these results received the support of a fellowship from “La Caixa” Foundation (ID 100010434). The fellowship code is LCF/BQ/DI18/11660039. This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 713673. S. P. and L. A. acknowledge the financial support by the Spanish Ministry of Science and Innovation (PID2019-109450RB-I00/AEI/10.13039/501100011033) and “la Caixa” Foundation (ID 100010434). The authors would like thank Larry Fitzpatrick and Laura Woythe for their input in proof-reading the manuscript.References
- B. Bahrami, Nanoparticles and Targeted Drug Delivery in Cancer Therapy, Immunol. Lett., 2017, 190, 64–83 CrossRef CAS PubMed.
- S. T. Jahan, S. M. A. Sadat, M. Walliser and A. Haddadi, Targeted Therapeutic Nanoparticles: An Immense Promise to Fight against Cancer, J. Drug Delivery, 2017, 2017, 9090325 Search PubMed.
- S. Kumar, N. Dilbaghi, R. Saharan and G. Bhanjana, Nanotechnology as Emerging Tool for Enhancing Solubility of Poorly Water-Soluble Drugs, BioNanoSci., 2012, 2(4), 227–250, DOI:10.1007/s12668-012-0060-7.
- S. Kalepu and V. Nekkanti, Improved Delivery of Poorly Soluble Compounds Using Nanoparticle Technology: A Review, Drug Delivery Transl. Res., 2016, 6(3), 319–332 CrossRef CAS PubMed.
- J. Wu, The Enhanced Permeability and Retention (EPR) Effect: The Significance of the Concept and Methods to Enhance Its Application, J. Pers. Med., 2021, 11(8), 771 CrossRef PubMed.
- H. Wang, J. Yu, X. Lu and X. He, Nanoparticle Systems Reduce Systemic Toxicity in Cancer Treatment, Nanomedicine, 2016, 11(2), 103–106, DOI:10.2217/nnm.15.166.
- T. M. Allen, Ligand-Targeted Therapeutics in Anticancer Therapy, Nat. Rev. Cancer, 2002, 2, 750–763, DOI:10.1038/nrc903.
- S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, J. Audet, H. F. Dvorak and W. C. W. Chan, Analysis of Nanoparticle Delivery to Tumours, Nat. Rev. Mater., 2016, 1(5), 16014, DOI:10.1038/natrevmats.2016.14.
- S. Patel, J. Kim, M. Herrera, A. Mukherjee, A. V. Kabanov and G. Sahay, Brief Update on Endocytosis of Nanomedicines, Adv. Drug Delivery Rev., 2019, 144, 90–111, DOI:10.1016/j.addr.2019.08.004.
- C. Battistella and H.-A. Klok, Controlling and Monitoring Intracellular Delivery of Anticancer Polymer Nanomedicines, Macromol. Biosci., 2017, 17(10), 1700022, DOI:10.1002/mabi.201700022.
- D. Rosenblum, N. Joshi, W. Tao, J. M. Karp and D. Peer, Progress and Challenges towards Targeted Delivery of Cancer Therapeutics, Nat. Commun., 2018, 9(1), 1410, DOI:10.1038/s41467-018-03705-y.
- P. Foroozandeh, Insight into Cellular Uptake and Intracellular Trafficking of Nanoparticles, Nanoscale Res. Lett., 2018, 13, 339 CrossRef PubMed.
- A. Albanese, P. S. Tang and W. C. W. Chan, The Effect of Nanoparticle Size, Shape, and Surface Chemistry on Biological Systems, Annu. Rev. Biomed. Eng., 2012, 14(1), 1–16, DOI:10.1146/annurev-bioeng-071811-150124.
- L. I. Selby, C. M. Cortez-Jugo, G. K. Such and A. P. R. Johnston, Nanoescapology: Progress toward Understanding the Endosomal Escape of Polymeric Nanoparticles: Endosomal Escape of Polymeric Nanoparticles, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2017, 9(5), e1452, DOI:10.1002/wnan.1452.
- I. M. S. Degors, C. Wang, Z. U. Rehman and I. S. Zuhorn, Carriers Break Barriers in Drug Delivery: Endocytosis and Endosomal Escape of Gene Delivery Vectors, Acc. Chem. Res., 2019, 52(7), 1750–1760, DOI:10.1021/acs.accounts.9b00177.
- M. P. Stewart, A. Lorenz, J. Dahlman and G. Sahay, Challenges in Carrier-Mediated Intracellular Delivery: Moving beyond Endosomal Barriers: Challenges in Carrier-Mediated Intracellular Delivery, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2016, 8(3), 465–478, DOI:10.1002/wnan.1377.
- T.-G. Iversen, Endocytosis and Intracellular Transport of Nanoparticles: Present Knowledge and Need for Future Studies, Nano Today, 2011, 6, 176–185 CrossRef CAS.
- L. Y. T. Chou, K. Ming and W. C. W. Chan, Strategies for the Intracellular Delivery of Nanoparticles, Chem. Soc. Rev., 2011, 40(1), 233–245 RSC.
- T. Andrian, R. Riera, S. Pujals and L. Albertazzi, Nanoscopy for Endosomal Escape Quantification, Nanoscale Adv., 2021, 3(1), 10–23, 10.1039/D0NA00454E.
- S. A. Smith, L. I. Selby, A. P. R. Johnston and G. K. Such, The Endosomal Escape of Nanoparticles: Towards More Efficient Cellular Delivery, Bioconjugate Chem., 2018, 30, DOI:10.1021/acs.bioconjchem.8b00732.
- L. M. P. Vermeulen, T. Brans, S. C. De Smedt, K. Remaut and K. Braeckmans, Methodologies to Investigate Intracellular Barriers for Nucleic Acid Delivery in Non-Viral Gene Therapy, Nano Today, 2018, 21, 74–90, DOI:10.1016/j.nantod.2018.06.007.
- T. F. Martens, K. Remaut, J. Demeester, S. C. De Smedt and K. Braeckmans, Intracellular Delivery of Nanomaterials: How to Catch Endosomal Escape in the Act, Nano Today, 2014, 9(3), 344–364, DOI:10.1016/j.nantod.2014.04.011.
- S. Pujals, N. Feiner-Gracia, P. Delcanale, I. Voets and L. Albertazzi, Super-Resolution Microscopy as a Powerful Tool to Study Complex Synthetic Materials, Nat. Rev. Chem., 2019, 3(2), 68–84, DOI:10.1038/s41570-018-0070-2.
- D. van der Zwaag, N. Vanparijs, S. Wijnands, R. De Rycke, B. G. De Geest and L. Albertazzi, Super Resolution Imaging of Nanoparticles Cellular Uptake and Trafficking, ACS Appl. Mater. Interfaces, 2016, 8(10), 6391–6399, DOI:10.1021/acsami.6b00811.
- S. K. Chakkarapani, Quantifying Intracellular Trafficking of Silica-Coated Magnetic Nanoparticles in Live Single Cells by Site-Specific Direct Stochastic Optical Reconstruction Microscopy, J. Nanobiotechnol., 2021, 19(1), 398 CrossRef CAS PubMed.
- M. Wojnilowicz, A. Glab, A. Bertucci, F. Caruso and F. Cavalieri, Super-Resolution Imaging of Proton Sponge-Triggered Rupture of Endosomes and Cytosolic Release of Small Interfering RNA, ACS Nano, 2019, 13(1), 187–202, DOI:10.1021/acsnano.8b05151.
- R. Riera, N. Feiner-Gracia, C. Fornaguera, A. Cascante, S. Borrós and L. Albertazzi, Tracking the DNA Complexation State of PBAE Polyplexes in Cells with Super Resolution Microscopy, Nanoscale, 2019, 11(38), 17869–17877 RSC.
- R. Riera, J. Tauler, N. Feiner-Gracia, S. Borrós, C. Fornaguera and L. Albertazzi, Complex PBAE Nanoparticle Cell Trafficking: Tracking Both Position and Composition Using Super Resolution Microscopy, ChemMedChem, 2022, 17(13), e202100633 CrossRef CAS PubMed.
- L. Woythe, P. Madhikar, N. Feiner-Gracia, C. Storm and L. Albertazzi, A Single-Molecule View at Nanoparticle Targeting Selectivity: Correlating Ligand Functionality and Cell Receptor Density, ACS Nano, 2022, 16(3), 3785–3796, DOI:10.1021/acsnano.1c08277.
- M. Costanzo, F. Carton, A. Marengo, G. Berlier, B. Stella, S. Arpicco and M. Malatesta, Fluorescence and Electron Microscopy to Visualize the Intracellular Fate of Nanoparticles for Drug Delivery, Eur. J. Histochem., 2016, 60(2), 2640, DOI:10.4081/ejh.2016.2640.
- M. Malatesta, Transmission Electron Microscopy as a Powerful Tool to Investigate the Interaction of Nanoparticles with Subcellular Structures, Int. J. Mol. Sci., 2021, 17 Search PubMed.
- W. Jiang, B. Y. S. Kim, J. T. Rutka and W. C. W. Chan, Nanoparticle-Mediated Cellular Response Is Size-Dependent, Nat. Nanotechnol., 2008, 3, 6 CrossRef PubMed.
- T. Ando, S. P. Bhamidimarri, N. Brending, H. Colin-York, L. Collinson, N. De Jonge, P. J. de Pablo, E. Debroye, C. Eggeling, C. Franck, M. Fritzsche, H. Gerritsen, B. N. G. Giepmans, K. Grunewald, J. Hofkens, J. P. Hoogenboom, K. P. F. Janssen, R. Kaufmann, J. Klumperman, N. Kurniawan, J. Kusch, N. Liv, V. Parekh, D. B. Peckys, F. Rehfeldt, D. C. Reutens, M. B. J. Roeffaers, T. Salditt, I. A. T. Schaap, U. S. Schwarz, P. Verkade, M. W. Vogel, R. Wagner, M. Winterhalter, H. Yuan and G. Zifarelli, The 2018 Correlative Microscopy Techniques Roadmap, J. Phys. D: Appl. Phys., 2018, 51(44), 443001, DOI:10.1088/1361-6463/aad055.
- P. de Boer, J. P. Hoogenboom and B. N. G. Giepmans, Correlated Light and Electron Microscopy: Ultrastructure Lights Up!, Nat. Methods, 2015, 12(6), 503–513, DOI:10.1038/nmeth.3400.
- S. Han, M. Raabe, L. Hodgson, J. Mantell, P. Verkade, T. Lasser, K. Landfester, T. Weil and I. Lieberwirth, High-Contrast Imaging of Nanodiamonds in Cells by Energy Filtered and Correlative Light-Electron Microscopy: Toward a Quantitative Nanoparticle-Cell Analysis, Nano Lett., 2019, 19(3), 2178–2185, DOI:10.1021/acs.nanolett.9b00752.
- M. Frey, S. Han, H. Halim, A. Kaltbeitzel, A. Riedinger, K. Landfester and I. Lieberwirth, Nanocarriers Made of Proteins: Intracellular Visualization of a Smart Biodegradable Drug Delivery System, Small, 2022, 2106094, DOI:10.1002/smll.202106094.
- I. M. Dobbie, Bridging the Resolution Gap: Correlative Super-Resolution Imaging, Nat. Rev. Microbiol., 2019, 17(6), 337–337, DOI:10.1038/s41579-019-0203-8.
- E. Betzig, G. H. Patterson, R. Sougrat, O. W. Lindwasser, S. Olenych, J. S. Bonifacino, M. W. Davidson, J. Lippincott-Schwartz and H. F. Hess, Imaging Intracellular Fluorescent Proteins at Nanometer Resolution, Science, 2006, 313(5793), 1642–1645, DOI:10.1126/science.1127344.
- D. M. van Elsland, S. Pujals, T. Bakkum, E. Bos, N. Oikonomeas-Koppasis, I. Berlin, J. Neefjes, A. H. Meijer, A. J. Koster, L. Albertazzi and S. I. van Kasteren, Ultrastructural Imaging of Salmonella -Host Interactions Using Super-Resolution Correlative Light-Electron Microscopy of Bioorthogonal Pathogens, ChemBioChem, 2018, 19(16), 1766–1770, DOI:10.1002/cbic.201800230.
- T. Andrian, T. Bakkum, D. M. van Elsland, E. Bos, A. J. Koster, L. Albertazzi, S. I. van Kasteren and S. Pujals, Super-Resolution Correlative Light-Electron Microscopy Using a Click-Chemistry Approach for Studying Intracellular Trafficking, in Methods in Cell Biology, Elsevier, 2021, vol. 162, pp. 303–331. DOI:10.1016/bs.mcb.2020.09.001.
- D. Kim, T. J. Deerinck, Y. M. Sigal, H. P. Babcock, M. H. Ellisman and X. Zhuang, Correlative Stochastic Optical Reconstruction Microscopy and Electron Microscopy, PLoS One, 2015, 10(4), e0124581, DOI:10.1371/journal.pone.0124581.
- T. Andrian, P. Delcanale, S. Pujals and L. Albertazzi, Correlating Super-Resolution Microscopy and Transmission Electron Microscopy Reveals Multiparametric Heterogeneity in Nanoparticles, Nano Lett., 2021, 21(12), 5360–5368, DOI:10.1021/acs.nanolett.1c01666.
- D. Bobo, K. J. Robinson, J. Islam, K. J. Thurecht and S. R. Corrie, Nanoparticle-Based Medicines: A Review of FDA-Approved Materials and Clinical Trials to Date, Pharm. Res., 2016, 33(10), 2373–2387, DOI:10.1007/s11095-016-1958-5.
- C. L. Ventola, Progress in Nanomedicine: Approved and Investigational Nanodrugs, Pharm. Ther., 2017, 42(12), 742–755 Search PubMed.
- B. Li, Q. Li, J. Mo and H. Dai, Drug-Loaded Polymeric Nanoparticles for Cancer Stem Cell Targeting, Front. Pharmacol., 2017, 8, DOI:10.3389/fphar.2017.00051.
- E. Pérez-Herrero and A. Fernández-Medarde, Advanced Targeted Therapies in Cancer: Drug Nanocarriers, the Future of Chemotherapy, Eur. J. Pharm. Biopharm., 2015, 93, 52–79, DOI:10.1016/j.ejpb.2015.03.018.
- N. S. Katheder, R. Khezri, F. O'Farrell, S. W. Schultz, A. Jain, M. M. Rahman, K. O. Schink, T. A. Theodossiou, T. Johansen, G. Juhász, D. Bilder, A. Brech, H. Stenmark and T. E. Rusten, Microenvironmental Autophagy Promotes Tumour Growth, Nature, 2017, 541(7637), 417–420, DOI:10.1038/nature20815.
- W. Kukulski, M. Schorb, S. Welsch, A. Picco, M. Kaksonen and J. A. G. Briggs, Precise, Correlated Fluorescence Microscopy and Electron Tomography of Lowicryl Sections Using Fluorescent Fiducial Markers, in Methods in Cell Biology, Elsevier, 2012, vol. 111, pp. 235–257. DOI:10.1016/B978-0-12-416026-2.00013-3.
- S. J. Nixon, R. I. Webb, M. Floetenmeyer, N. Schieber, H. P. Lo and R. G. Parton, A Single Method for Cryofixation and Correlative Light, Electron Microscopy and Tomography of Zebrafish Embryos, Traffic, 2009, 10(2), 131–136, DOI:10.1111/j.1600-0854.2008.00859.x.
- W. Kukulski, M. Schorb, S. Welsch, A. Picco, M. Kaksonen and J. A. G. Briggs, Correlated Fluorescence and 3D Electron Microscopy with High Sensitivity and Spatial Precision, J. Cell Biol., 2011, 192(1), 111–119, DOI:10.1083/jcb.201009037.
- J. M. Barichello, M. Morishita, K. Takayama and T. Nagai, Encapsulation of Hydrophilic and Lipophilic Drugs in PLGA Nanoparticles by the Nanoprecipitation Method, Drug Dev. Ind. Pharm., 1999, 25(4), 471–476, DOI:10.1081/DDC-100102197.
- T. Andrian, S. Pujals and L. Albertazzi, Quantifying the Effect of PEG Architecture on Nanoparticle Ligand Availability Using DNA-PAINT, Nanoscale Adv., 2021, 3(24), 6876–6881, 10.1039/D1NA00696G.
- S. Watanabe, A. Punge, G. Hollopeter, K. I. Willig, R. J. Hobson, M. W. Davis, S. W. Hell and E. M. Jorgensen, Protein Localization in Electron Micrographs Using Fluorescence Nanoscopy, Nat. Methods, 2011, 8(1), 80–84, DOI:10.1038/nmeth.1537.
- P. Paul-Gilloteaux, X. Heiligenstein, M. Belle, M.-C. Domart, B. Larijani, L. Collinson, G. Raposo and J. Salamero, EC-CLEM: Flexible Multidimensional Registration Software for Correlative Microscopies, Nat. Methods, 2017, 14(2), 102–103, DOI:10.1038/nmeth.4170.
- Z. Fu, D. Peng, M. Zhang, F. Xue, R. Zhang, W. He, T. Xu and P. Xu, MEosEM Withstands Osmium Staining and Epon Embedding for Super-Resolution CLEM, Nat. Methods, 2020, 17(1), 55–58, DOI:10.1038/s41592-019-0613-6.
- M. J. Rust, M. Bates and X. Zhuang, Sub-Diffraction-Limit Imaging by Stochastic Optical Reconstruction Microscopy (STORM), Nat. Methods, 2006, 3(10), 793–796, DOI:10.1038/nmeth929.
- J. H. Park and N. Oh, Endocytosis and Exocytosis of Nanoparticles in Mammalian Cells, Int. J. Nanomed., 2014, 51, DOI:10.2147/IJN.S26592.
- C. Fornaguera, C. Castells-Sala and S. Borrós, Unraveling Polymeric Nanoparticles Cell Uptake Pathways: Two Decades Working to Understand Nanoparticles Journey to Improve Gene Therapy, in Cell Biology and Translational Medicine, Volume 9, ed. K. Turksen, Advances in Experimental Medicine and Biology, Springer International Publishing, Cham, 2019, vol. 1288, pp. 117–138. DOI:10.1007/5584_2019_467.
- P. Sandin, L. W. Fitzpatrick, J. C. Simpson and K. A. Dawson, High-Speed Imaging of Rab Family Small GTPases Reveals Rare Events in Nanoparticle Trafficking in Living Cells, ACS Nano, 2012, 6(2), 1513–1521, DOI:10.1021/nn204448x.
- J. Huotari and A. Helenius, Endosome Maturation: Endosome Maturation, EMBO J., 2011, 30(17), 3481–3500, DOI:10.1038/emboj.2011.286.
- J. Zhang, D. Chang, Y. Yang, X. Zhang, W. Tao, L. Jiang, X. Liang, H. Tsai, L. Huang and L. Mei, Systematic Investigation on the Intracellular Trafficking Network of Polymeric Nanoparticles, Nanoscale, 2017, 9(9), 3269–3282, 10.1039/C7NR00532F.
- L. B. Sims, L. T. Curtis, H. B. Frieboes and J. M. Steinbach-Rankins, Enhanced Uptake and Transport of PLGA-Modified Nanoparticles in Cervical Cancer, J. Nanobiotechnol., 2016, 14(1), 33, DOI:10.1186/s12951-016-0185-x.
- C.-H. Chu, Y.-C. Wang, H.-Y. Huang, L.-C. Wu and C.-S. Yang, Ultrafine PEG-Coated Poly(Lactic- Co -Glycolic Acid) Nanoparticles Formulated by Hydrophobic Surfactant-Assisted One-Pot Synthesis for Biomedical Applications, Nanotechnology, 2011, 22(18), 185601, DOI:10.1088/0957-4484/22/18/185601.
- F. Danhier, N. Lecouturier, B. Vroman, C. Jérôme, J. Marchand-Brynaert, O. Feron and V. Préat, Paclitaxel-Loaded PEGylated PLGA-Based Nanoparticles: In Vitro and in Vivo Evaluation, J. Controlled Release, 2009, 133(1), 11–17, DOI:10.1016/j.jconrel.2008.09.086.
- J. Fermie, N. Liv, C. ten Brink, E. G. van Donselaar, W. H. Müller, N. L. Schieber, Y. Schwab, H. C. Gerritsen and J. Klumperman, Single Organelle Dynamics Linked to 3D Structure by Correlative Live-Cell Imaging and 3D Electron Microscopy, Traffic, 2018, 19(5), 354–369, DOI:10.1111/tra.12557.
- J. Panyam and V. Labhasetwar, Dynamics of Endocytosis and Exocytosis of Poly(D,L-Lactide-Co-Glycolide) Nanoparticles in Vascular Smooth Muscle Cells, Pharm. Res., 2003, 20(2), 212–220 CrossRef CAS PubMed.
- Z. Chu, Y. Huang, Q. Tao and Q. Li, Cellular Uptake, Evolution, and Excretion of Silica Nanoparticles in Human Cells, Nanoscale, 2011, 3(8), 3291, 10.1039/c1nr10499c.
- R. Sakhtianchi, R. F. Minchin, K.-B. Lee, A. M. Alkilany, V. Serpooshan and M. Mahmoudi, Exocytosis of Nanoparticles from Cells: Role in Cellular Retention and Toxicity, Adv. Colloid Interface Sci., 2013, 201–202, 18–29, DOI:10.1016/j.cis.2013.10.013.
- S. Behzadi, V. Serpooshan, W. Tao, M. A. Hamaly, M. Y. Alkawareek, E. C. Dreaden, D. Brown, A. M. Alkilany, O. C. Farokhzad and M. Mahmoudi, Cellular Uptake of Nanoparticles: Journey inside the Cell, Chem. Soc. Rev., 2017, 46(14), 4218–4244, 10.1039/C6CS00636A.
- B. D. Chithrani and W. C. W. Chan, Elucidating the Mechanism of Cellular Uptake and Removal of Protein-Coated Gold Nanoparticles of Different Sizes and Shapes, Nano Lett., 2007, 7(6), 1542–1550, DOI:10.1021/nl070363y.
- S. Sato, A. Rancourt, Y. Sato and M. S. Satoh, Single-Cell Lineage Tracking Analysis Reveals That an Established Cell Line Comprises Putative Cancer Stem Cells and Their Heterogeneous Progeny, Sci. Rep., 2016, 6(1), 23328, DOI:10.1038/srep23328.
- J. A. Kim, C. Åberg, A. Salvati and K. A. Dawson, Role of Cell Cycle on the Cellular Uptake and Dilution of Nanoparticles in a Cell Population, Nat. Nanotechnol., 2012, 7(1), 62–68, DOI:10.1038/nnano.2011.191.
- T. Bus, A. Traeger and U. S. Schubert, The Great Escape: How Cationic Polyplexes Overcome the Endosomal Barrier, J. Mater. Chem. B, 2018, 6(43), 6904–6918, 10.1039/C8TB00967H.
- P. Lönn, A. D. Kacsinta, X.-S. Cui, A. S. Hamil, M. Kaulich, K. Gogoi and S. F. Dowdy, Enhancing Endosomal Escape for Intracellular Delivery of Macromolecular Biologic Therapeutics, Sci. Rep., 2016, 6(1), 32301, DOI:10.1038/srep32301.
- A. K. Varkouhi, M. Scholte, G. Storm and H. J. Haisma, Endosomal Escape Pathways for Delivery of Biologicals, J. Controlled Release, 2011, 151(3), 220–228, DOI:10.1016/j.jconrel.2010.11.004.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr02838k |
| This journal is © The Royal Society of Chemistry 2023 |