
High performance, recyclable and sustainable by design natural polyphenol-based epoxy polyester thermosets†
Roxana
Dinu
a,
Anastasiia
Pidvoronia
a,
Ugo
Lafont
b,
Olivier
Damiano
c and
Alice
Mija
*a
aUniversité Côte d'Azur, Institut de Chimie de Nice, France. E-mail: Alice.Mija@univ-cotedazur.fr
bESA, ESTEC, Keplerlaan 1, PO Box 299, NL-2200 AG Noordwijk, The Netherlands
cThales Alenia Space, 5 Allée des Gabians, 06156 Cannes la Bocca, France
First published on 13th February 2023
Abstract
Renewable flavonoid and phlorotannin extracts were used as building blocks to synthesize biobased tris-epoxy monomers and further to design and develop sustainable thermosetting resins. The triglycidyl ethers of phloroglucinol and naringenin were combined and crosslinked with a series of anhydrides, four of them being bio-based. The thermosetting resins’ reactivity has been studied, as well as the influence of the chemical structure of anhydride crosslinkers allowing us to obtain highly crosslinked networks with high performance. The designed thermosets show high glass transition values (134–199 °C), high storage moduli at 25 °C (2.7–3.5 GPa), and high crosslinking densities and gel content (>99%). These polyaromatic thermosets are characterized by high mechanical strength and toughness (Young's moduli ∼1.26–1.68 GPa), and they proved to be high heat-resistant materials (LOI ∼28–33%). The sustainability aspect could be related to a particularly important feature of these thermosets, their ability to be chemically recycled by bond exchange reactions. Therefore, the polyaromatic thermosets developed in this study demonstrate important characteristics recommending them as environmentally friendly alternatives to petroleum-based epoxy resins used in various industries such as civil, automotive, marine, aerospace, and space.
Introduction
The continuously increasing price of oil and its volatile availability, as well as the global political trends regarding mandatory CO2 emission regulations in Europe, are putting pressure on researchers and industries to develop a sustainable economy.1,2 If we look at polymer applications, the majority of the most used ones (polyolefins for everyday use: PE, PP) or the most performing ones (high industry grade) were discovered almost a hundred years ago. This is also the case for the epoxy resins discovered by P. Castan in 1940, one of the most commercially used thermosets with excellent properties and a myriad of applications.3–5 These resins have small flaws such as their brittleness and recently, they have shown some negative impacts on the environment due to the bisphenol A used in their synthesis, which is reported to be an endocrine disruptor,6 classified as carcinogenic, mutagenic and reprotoxic (CMR), and thus prohibited in many countries.7 Despite the fact that there are numerous studies reporting the use of renewable resources (vegetable oils, tannins, lignin, catechins, terpenes, cardanol and rosins) in the development of epoxy polymers, at the industrial level, only epoxidized vegetable oils and modified cardanol are commercially available.8–10 Bio-based epoxy polymers with aromatic units, however, have shown high performances and are therefore the main sustainable candidates for the disposal of DGEBA-based polymers.11–13 The intensive use of thermoset resins increased environmental concerns, as the common thermosets generally do not have the recycling and reforming characteristics of thermoplastics.14The main goal of this work was to develop high performance materials with a high content of aromatic units and high functionality, using natural and renewable polyphenolic synthons such as naringenin and phloroglucinol. Naringenin belongs to a subclass of natural flavonoids named flavanones, mainly found in the peel and pulp of vegetables, citrus fruits, cherries, tomatoes, etc.15 The pharmacological and curative activities of naringenin on human health have been intensively studied showing a multitude of beneficial effects, such as anti-carcinogenic, anti-estrogenic, anti-atherosclerotic, anti-diabetic, anti-viral, anti-bacterial, etc.16–18 Complementary to its applicability in the medical field, naringenin is also used in perfumery and cosmetics. In the domain of materials, there are only very few studies reporting the use of naringenin.19–21 Very recently, Du et al.19 have developed bio-epoxy resins based on daidzein, naringenin, and luteolin. To our knowledge, this is the single study showing the synthesis of materials with high Tg and thermal stabilities starting from flavonoids. The mechanical properties (Tg = 151 °C), thermal stability (T5% = 302 °C), and fire retardancy (LOI = 22.7%) of the produced bio-based epoxy resins were characterized and compared with those of DGEBA, demonstrating that the properties of the developed resins were significantly superior to those of the reference. Latos-Brozio et al.20 crosslinked naringenin with glycerol diglycidyl ether (GDE), with the obtained poly(naringenin) exhibiting good oxidation resistance as well as a thermal stability of about 100 °C (T5%). The antimicrobial properties of these materials were demonstrated. Due to these characteristics, as well as the ability to reduce free radicals and Cu2+ ions, the authors showed that poly(naringenin) can be used in the development of environmentally friendly materials or packaging with antimicrobial properties. A naringenin-based thermoset with modular recycling capabilities as well as shape memory behaviour was developed by Oh et al.21 by naringenin epoxidation followed by its copolymerization with poly(ethylene glycol)-diacid. The obtained material showed an elastic modulus of about 40 MPa, a value close to that of articular cartilage or rubber. Also, in addition to selectively absorbing water by swelling >400%, it rapidly restored its initial shape (in response to heat and water) after physical deformation and was chemically recycled by transesterification. Although naringenin has started to gain interest in the field of polymeric materials, to date, this natural compound has not yet been combined with any other natural polyphenol to produce recyclable high functional performance grade materials.
Phloroglucinol (1,3,5-trihydroxybenzene) is the main component in polyphenolic compounds such as phlorotannins22 extracted from non-vascular plant tannins such as marine brown algae (Ecklonia Cava) or the bark of fruits, but can also be chemically synthesized from benzene.23 Over time, phloroglucinol has been studied in the medical field as a bioactive compound.24,25 Recently, phloroglucinol was used to generate monomers in various applications as fire retardant additives,26 coatings,27 thermosetting materials,28–30 absorbents31,32 and electrode33 materials.
In this study, the two natural and renewable polyphenolics, phloroglucinol and naringenin, one monoaromatic and the other polyaromatic, were selected and glycidylated to obtain bio-based aromatic tris-epoxy monomers. These monomers were then combined to design recyclable and highly functional thermosets with a high content of aromatic moieties. These networks were designed to exhibit high performances suitable for industries such as construction, automotive, marine and space. For the polyester thermosetting resin design, different ratios of monoaromatic/polyaromatic monomers were reacted with five anhydrides, four of them being bio-based. The formulations’ reactivity was studied by differential scanning calorimetry (DSC) and in situ Fourier-transform infrared spectroscopy (FT-IR). Then, the physico-chemical and thermo-mechanical properties of the new thermosets were evaluated by DSC, FT-IR, thermogravimetric analysis (TGA), dynamic mechanical analysis (DMA), and tensile tests. The chemical recycling of these polyester thermosets was the purpose of this study, through their depolymerization. These polyester thermosets were subjected to dynamic bond exchange reactions (BER) in the presence of 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD)-ethylene glycol (EG) solution.
Experimental
Materials
Phloroglucinol, naringenin, epichlorohydrin, benzyltriethylammonium bromide (TBAB, 99%), sodium hydroxide, dichloromethane, maleic anhydride (MA), succinic anhydride (SA), 1,2,4-benzene tricarboxylic anhydride (TA), cis-1,2,3,6-tetrahydrophthalic anhydride (THPA), phthalic anhydride (PA), N,N-dimethylbenzylamine (BDMA) and all the solvents used in this study were purchased from Merck and were used as received, without any prior purification or modification.The two epoxy monomers, derived from phloroglucinol and naringenin, were laboratory-synthesized by glycidylation of the natural polyphenols following the patented methods.34–36 The triglycidyl ether of phloroglucinol (TGPh) is a brown viscous liquid with an average functionality of 2.8, a molar mass of 301.6 g mol−1 (theoretical mass = 294.3 g mol−1) and an EEW (epoxy equivalent weight) of ∼99.51 g per equiv. TGPh:1H NMR (400 MHz, CDCl3) δ 6.07 (s, 3H), 4.12 (dd, J = 11.1, 3.1 Hz, 3H), 3.82 (dd, J = 11.1, 5.8 Hz, 3H), 3.26 (ddt, J = 5.7, 4.2, 2.8 Hz, 3H), 2.82 (dd, J = 4.9, 4.1 Hz, 3H), 2.67 (dd, J = 4.9, 2.7 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 160.31, 94.72, 94.71, 68.87, 68.85, 68.83, 50.01, 44.67. TGEN is a yellow viscous liquid with an average functionality of 3.6 and a molar mass of 448.99 g mol−1 (theoretical mass = 440.44 g mol−1) and an EEW of 148.54 g per equiv. TGEN:1H NMR (400 MHz, CDCl3) δ 7.45–7.38 (m, 2H), 6.90–6.84 (m, 2H), 6.19 (d, J = 1.0 Hz, 2H), 5.23 (s, 1H), 4.20 (tdd, J = 10.3, 4.9, 2.9 Hz, 3H), 3.94–3.85 (m, 6H), 3.66–3.60 (m, 1H), 3.29 (td, J = 6.2, 2.7 Hz, 5H), 3.17 (tdd, J = 5.4, 3.9, 2.5 Hz, 7H), 2.73–2.65 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 193.35, 160.98, 160.28, 157.68, 130.44, 130.12, 114.92 (d, J = 9.8 Hz), 94.80, 93.70, 92.21, 68.81, 51.27, 46.94, 45.00. The molecular structures of the compounds used in the thermosetting resin formulations are displayed in Fig. 1.
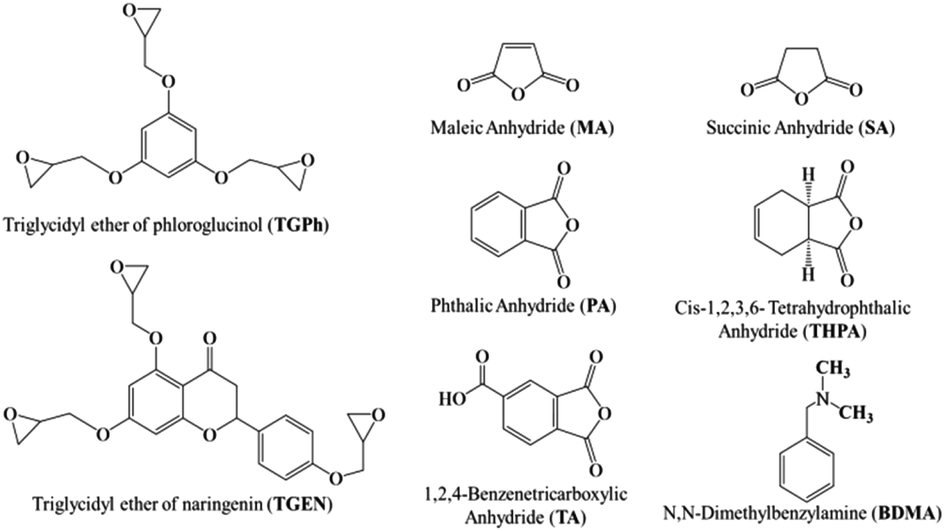 | ||
| Fig. 1 Chemical structures of the compounds used in the design of thermosetting resins. | ||
Preparation of epoxy resins
The two-laboratory synthesized triglycidyl ethers of phloroglucinol (TGPh) and naringenin (TGEN) were combined in different mass ratios: 75/25, 50/50 and 25/75, thus corresponding to the TGPh/TGEN molar fractions of 0.8/0.2, 0.6/0.4 and 0.25/0.75, respectively. A stoichiometric ratio between epoxy and anhydride groups was used. Then, the BDMA initiator was added at room temperature in different mass proportions (0.5 wt%, 1 wt%, and 2 wt%) in order to determine its optimum amount. The mixtures were poured into silicone molds and crosslinked in an oven, following a curing protocol at 80 °C for 1 h and another hour at 150 °C. The crosslinking of thermosets was completed by post-curing at 180 °C for 2 h. After the completion of the polymerization reaction, the thermoset materials were analysed again by dynamic DSC, confirming the complete crosslinking in the absence of residual heat.Experimental techniques
where E′ is the storage modulus in the rubbery plateau, R is the gas constant, and T is the absolute temperature. Then, the mass of the segments between the crosslinking bridges (Mc) was determined using the equation:38
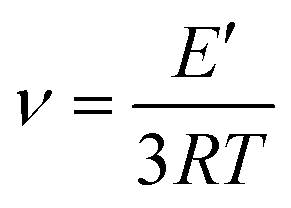
where ρ is the sample density, E′ is the storage modulus in the rubbery region, R is the gas constant, and T is the absolute temperature.
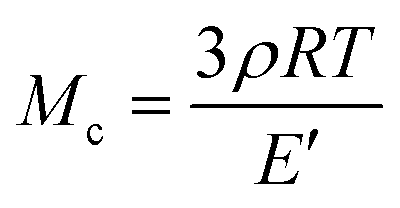
| B = 1/εbE′ |
| Ts = 0.49[T5% + 0.6(T30% − T5%)] |
Using the carbon content resulting from the TGA analyses under nitrogen at 850 °C (Cy850), the flame retardant properties were investigated by calculating the limiting oxygen index (LOI):43,44
| LOI = 17.5 + 0.4 × Cy850. |
where W0 is the initial mass of the specimen and Wf is its dried mass after THF immersion.
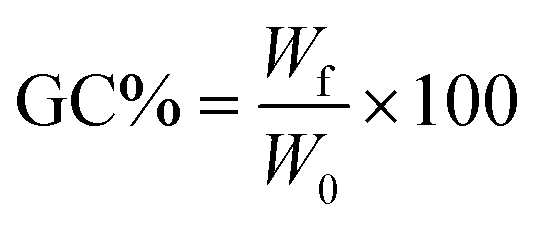
Results and discussion
Polyaromatic network design
In a preliminary test, the polymerization behaviour of formulations containing a variation of monoaromatic/polyaromatic TGPh/TGEN epoxy monomers 25/75, 50/50, and 75/25 was studied. The dynamic DSC thermograms obtained during the crosslinking of formulations with the selected anhydrides in the presence of 1 wt% BDMA are presented in Fig. S1, ESI,† and the main results are reported in Table S2, ESI.† As can be seen, for all TGPh/TGEN ratios, the crosslinking reactions with epoxy/anhydride, initiated by 1 wt% BDMA, exhibited a single well-defined exothermal peak. The highest values of reaction enthalpies (ΔH ∼221–364 J g−1) were measured for the formulations with the greatest content of monoaromatic epoxy, TGPh75/TGEN25//anhydride, followed closely by the formulation TGPh50/TGEN50//anhydride (ΔH 215–360 J g−1). Although the formulation TGPh75/TGEN25//anhydride showed the highest reactivity, if we look at the final material properties, the TGPh50/TGEN50//anhydride thermosets are characterized by the greatest values of glass transition (Tg-DSC, Table S3, ESI†). Therefore, the combination TGPh50/TGEN50 was selected for further use in this study, for crosslinking with the selected anhydrides.The initiator percentage was varied to determine the optimal amount it requires to reach the ultimate conversion, using 0.5 wt%, 1 wt% and 2 wt% BDMA. The evolution of heat flows with temperature is given in Fig. 2(a) and Fig. S2, ESI,† while ΔH, Tmax, and the reaction interval are listed in Table 1. In accordance with Fig. 2(a) and Fig. S2, ESI,† the reaction between the mixture of monoaromatic and polyaromatic tris-epoxies and anhydride appears as a narrow and well-defined exothermic event for all the systems, but the presence of a small secondary exotherm can also be observed at high temperature (>150 °C) for the systems with 2 wt% BDMA. In terms of both formulations’ reactivities (Table 1) and thermosets’ glass transition values, (Table S4, ESI†) the optimal results were obtained using 0.5 wt% initiator. These results can be explained by an accelerated crosslinking that occurred when using a higher initiator concentration, leading to a network structure disruption and a reduction in the crosslink density, thus affecting the final thermo-mechanical properties.46,47
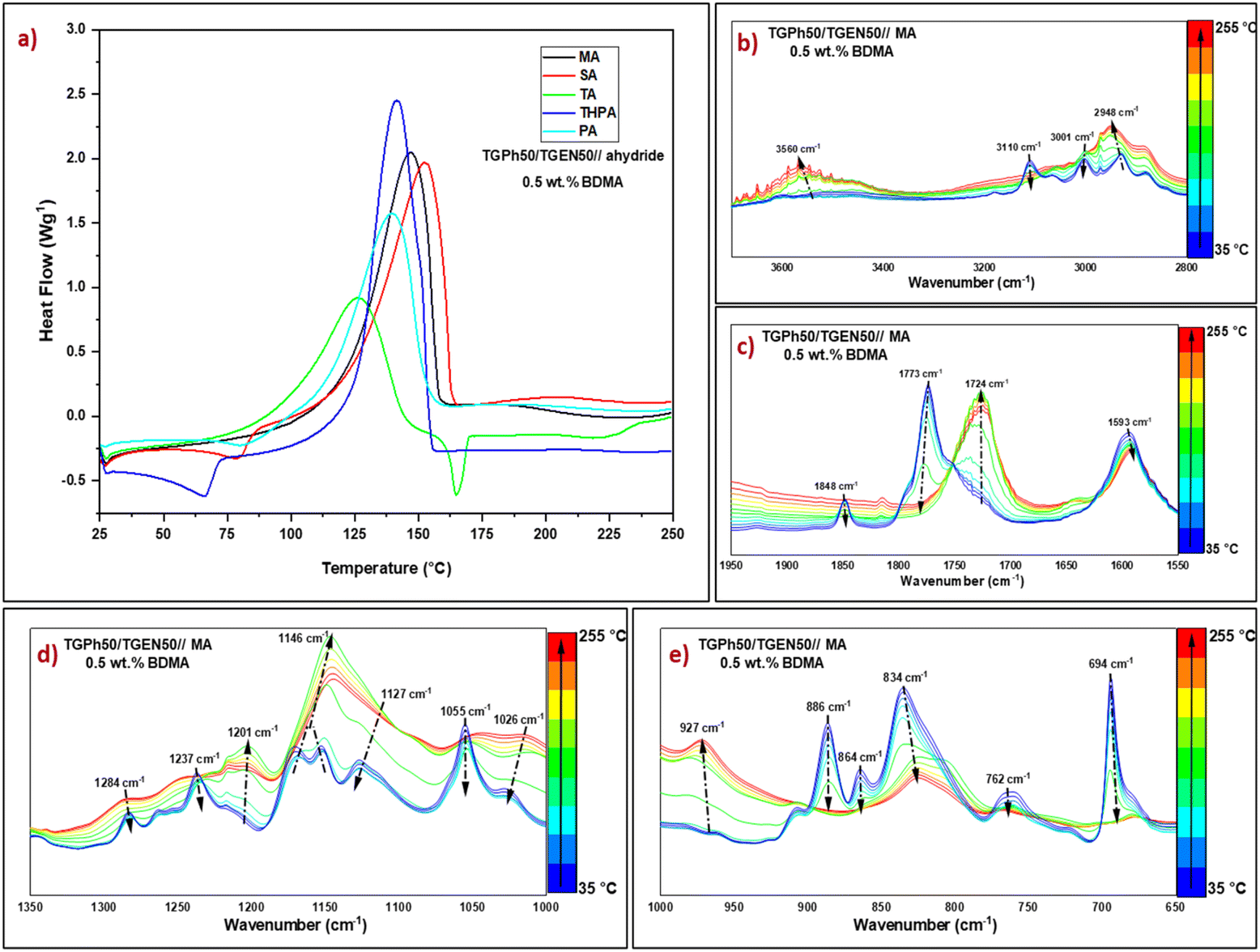 | ||
| Fig. 2 (a) Dynamic DSC thermograms obtained during the crosslinking of TGPh50/TGEN50//anhydride formulations initiated by 0.5 wt% BDMA, and the temperature-dependent FT-IR spectra of the TGPh50/TGEN50//MA system during curing in the regions (b) 3800–2800 cm−1, (c)1950–1550 cm−1, (d) 1350–1000 cm−1, and (e) 1000–650 cm−1. | ||

|
As a comparison, formulations developed with individual monomers TGPh or TGEN have also been prepared with the corresponding anhydrides and 0.5 wt% BDMA (Fig. S3, Tables S5 and S6, ESI†). Therefore, we can observe that the TGPh50/TGEN50 combination of the two monomers gives systems with superior reactivity, and then materials with higher glass transitions, compared with the case of the curing of individual monomers and their associated thermosets.
From Table 1 and Fig. 2(a) we can highlight that TGPh50/TGEN50//anhydride formulations initiated with 0.5 wt% BDMA have similar reactivities, except for the mixture crosslinked with TA anhydride. Due to its distinct structure, in the presence of a carboxylic function, TA has a superior melting temperature (Tm = 163–166 °C) compared to the other anhydrides (Table S1, ESI†), and it may also be used in epoxy–carboxylic acid addition reactions. The maximum crosslinking reaction temperature varies between 140 and 153 °C, except for the TGPh50/TGEN50//TA system with a curing Tmax of ∼126 °C. The onset curing temperatures are quite low, varying between 46 and 81 °C, depending on the hardener. We can notice that the enthalpy of polymerization for the TGPh50/TGEN50//TA formulation is almost half the value measured for the other systems, i.e., 238 J g−1vs. 487 J g−1 (TGPh50/TGEN50//SA). This result can be explained by (i) TA can liberate a proton to the anionic chains in growth therefore it will combine with the polymerization active species, with the formation of –OH or –COOH functions (ii) H bonding implying the –COOH functions from TA, hindering the epoxy-anhydride polymerization, and (iii) epoxide functions consumed by addition to carboxylic groups. Interestingly, maleic anhydride has a smaller enthalpy of reaction towards TGPh50/TGEN50 compared with succinic anhydride: 392 J g−1vs. 487 J g−1.
Phthalic vs. tetrahydrophthalic anhydride has the same configuration with enthalpies of polymerization with TGPh50/TGEN50 of 352 J g−1vs. 428 J g−1. These results suggest that the anhydride's unsaturation plays a role in the epoxy–anhydride polymerization mechanism. To investigate the structural changes, FTIR analyses in a dynamic heating program similar to that applied in DSC were realized on freshly prepared TGPh50/TGEN50//anhydride formulations (Fig. S7, ESI†). As illustrated for TGPh50/TGEN50//MA (Fig. S7a† and Fig. 2c, ESI†), in the region 1950–1550 cm−1 (characteristic area for unreacted anhydride), the thermal program provoked modifications in the characteristic peaks of the symmetric and antisymmetric C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations of the anhydride at 1848 cm−1 and 1773 cm−1. These peaks completely disappeared at the end of thermal crosslinking, with the area being dominated by a sharp peak centred at 1724 cm−1, linked to the formation of ester bonds during polymerization48 (Fig. 2(c)), indicating the full participation and consumption of anhydrides in the curing process.49,50 Likewise, esterification reactions are very well highlighted by the increase in the intensity of the peak at 1146 cm−1 characteristic of the C–O stretching vibration of the ester group formed as a result of copolymerization of epoxy–anhydride, and by the almost total decrease of ether peaks present in epoxy monomers at 1284–1237 cm−1 (C–O–benzene ring symmetrical st. vibration) and 1127–1055 cm−1 (C–O–benzene ring asymmetrical st. vibration) (Fig. 2(d)).51
O stretching vibrations of the anhydride at 1848 cm−1 and 1773 cm−1. These peaks completely disappeared at the end of thermal crosslinking, with the area being dominated by a sharp peak centred at 1724 cm−1, linked to the formation of ester bonds during polymerization48 (Fig. 2(c)), indicating the full participation and consumption of anhydrides in the curing process.49,50 Likewise, esterification reactions are very well highlighted by the increase in the intensity of the peak at 1146 cm−1 characteristic of the C–O stretching vibration of the ester group formed as a result of copolymerization of epoxy–anhydride, and by the almost total decrease of ether peaks present in epoxy monomers at 1284–1237 cm−1 (C–O–benzene ring symmetrical st. vibration) and 1127–1055 cm−1 (C–O–benzene ring asymmetrical st. vibration) (Fig. 2(d)).51
The epoxy groups’ breathing vibration at 1237 cm−1 disappeared together with the peaks at 886 cm−1, 864 cm−1 and 762 cm−1, showing their involvement in polymerization (Fig. 2(d) and (e)). At the same time, we can notice some changes affecting the MA's unsaturation: the diminution until disappearance of the C–H vibration at 3110 cm−1 and that of CC at 694 cm−1 together with a decrease in CC vibration at 1593 cm−1. With increasing temperature a new band surprisingly appeared at 3560 cm−1 (Fig. S7, ESI,†Fig. 2(b)), which can be due to a non-catalyzed anhydride–epoxy reaction with the formation of monoacid dangling chains, or due to the conversion of alkoxide ions into –OH groups due to the presence of some protons from residual unreacted –OH from phloroglucinol or naringenin or from traces of moisture (Scheme S1, ESI†).48,52
The tertiary amine catalysed crosslinking mechanisms are complex, as many factors play a significant role in their evolution, besides the main epoxy–anhydride chemical reactions. The corroboration of reactivity aspects (DSC) with the evolution of the functional groups (FTIR) allowed us to observe that the unsaturation of anhydrides affects the reactivity and we could also observe that MA unsaturation disappeared from the FTIR spectrum of its TGPh50/TGEN50//MA thermoset. This is not the case in THPA unsaturation that remains constant in the thermoset FTIR spectrum at 664 cm−1 (Fig. S7c, ESI†). We can suppose that under the action of BDMA, the initiation step is described by the nucleophilic attack of the tertiary amine either on the anhydride group, generating a zwitterionic carboxylate anion, or on the oxirane ring, forming a zwitterionic alkoxide anion. Then, an anionic copolymerization reaction takes place as an alternative addition reaction of anhydride and/or epoxy groups, resulting in a polyester network. In parallel, as we observed the formation of –OH functions in the FTIR spectrum, we can suppose that a step growth polymerization can occur probably (Scheme S1, ESI†).
Polyaromatic network performance characterisation
The viscoelastic response of the designed thermosets, specifically their molecular relaxations during a dynamic temperature ramp was first investigated by DMA. The storage modulus (E′) and the internal friction factor (tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ) of thermosets as a function of temperature are plotted in Fig. 3, while significant parameters such as storage moduli in the glassy state (at 25 °C) and the rubbery plateau (at 250 °C), and the maximum values of the damping factor of each sample are depicted in Table 2.
δ) of thermosets as a function of temperature are plotted in Fig. 3, while significant parameters such as storage moduli in the glassy state (at 25 °C) and the rubbery plateau (at 250 °C), and the maximum values of the damping factor of each sample are depicted in Table 2.
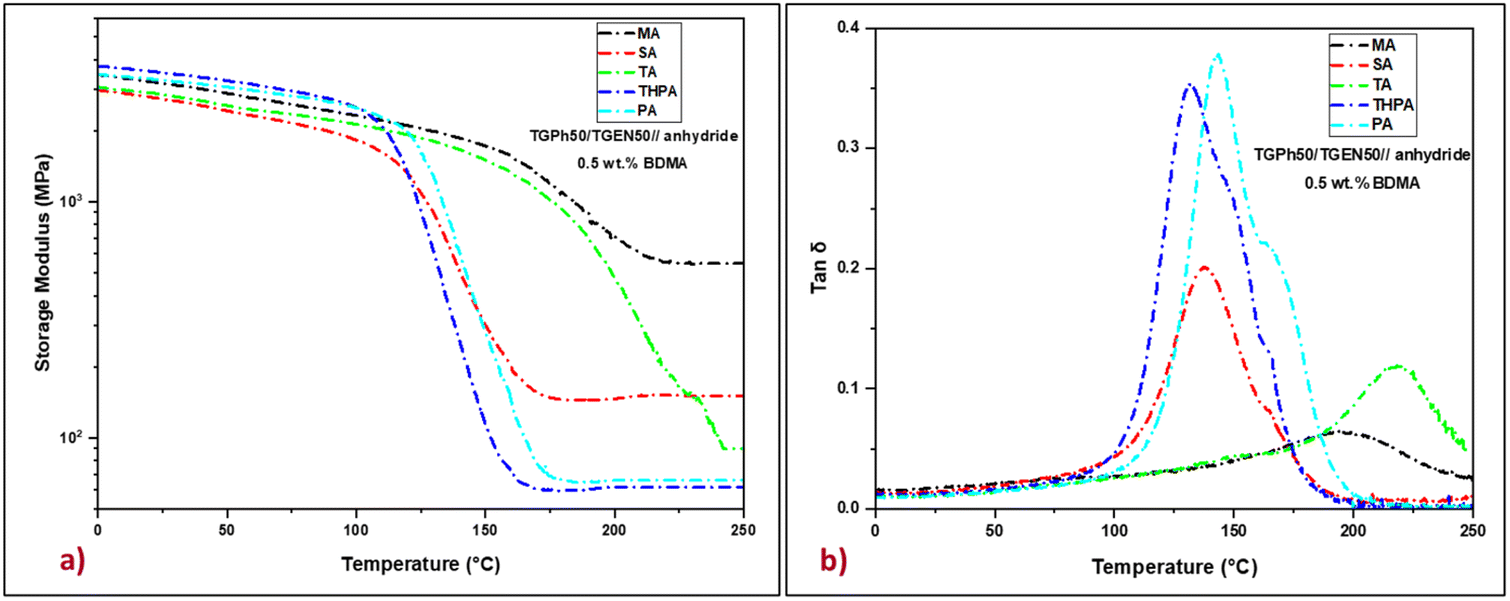 | ||
| Fig. 3 Storage moduli (a) and tanδ (b) curves of the TGPh50/TGEN50//anhydride thermosets. | ||
| Thermosets | Density [g cm−3] | Shore D [SD] | E′ at 25 °C [GPa] | E′ at 250 °C [MPa] | tanδ [°C] |
T g-DSC [°C] | ν [mmol cm−3] | M c [g mol−1] | GC [%] |
|---|---|---|---|---|---|---|---|---|---|
| TGPh50/TGEN50//MA | 1.20 | 90.5 | 3.2 | 546 | 197 | 180 | 40.5 | 28 | 99.87 |
| TGPh50/TGEN50//SA | 1.25 | 88.2 | 2.7 | 151 | 138 | 138 | 12.5 | 100 | 99.88 |
| TGPh50/TGEN50//TA | 1.31 | 92.3 | 2.9 | 90 | 220 | 199 | 6.4 | 204 | 99.24 |
| TGPh50/TGEN50//THPA | 1.25 | 95.5 | 3.5 | 62 | 133 | 134 | 5.2 | 240 | 99.93 |
| TGPh50/TGEN50//PA | 1.26 | 93 | 3.3 | 66 | 144 | 149 | 5.4 | 232 | 99.96 |
In the glassy region, the materials show storage moduli that vary between 2.7 GPa for the TGPh50/TGEN50//SA resin and 3.5 GPa for the TGPh50/TGEN50//THPA thermoset, values that prove the stiffness of the designed thermosets. During heating, the storage moduli decrease until reaching the rubbery plateau where they are constant, indicating a complete and stable high cross-linking (GC > 99.8%, Table 2). In the elastic region, the E′ values are substantially affected by the nature of the anhydride, fluctuating from 62 MPa for the TGPh50/TGEN50//THPA resin to about 550 MPa for the TGPh50/TGEN50//MA material. This very high value of the storage modulus in the rubbery state for the TGPh50/TGEN50//MA thermoset, around 8.9 times higher than that of TGPh50/TGEN50//THPA, indicates the higher stiffness of this material, a sign of a tighter network structure by a high density of the crosslinks.
To confirm this presumption we calculated the crosslink density (ν) of the thermoset materials based on rubber elasticity theory.37,53 To estimate a realistic value of the crosslink density using this theory, we applied the equation considering the temperature ∼tanδ + 70 °C (ref. 54 and 55) due to the quite high values of tanδ for some systems. In general, the crosslink density value is consistent with the glass transition values, which is not the case for all materials designed in this study. For example, the TGPh50/TGEN50//TA resin, which has the highest tanδ (220 °C), has a crosslinking density value of 6.4 mmol cm−3, which is 6 times lower than that of TGPh50/TGEN50//MA (tanδ = 180 °C, ν = 40.5 mmol cm−3, Table 2). If we compare again the thermosets designed with similar MA vs. SA anhydrides, we should again highlight the unusual results obtained with MA: a 1.5 times higher tanδ (180 °C vs. 138 °C) and 3.24 times higher crosslink density (ν = 40.5 mmol cm−3vs. ν = 12.5 mmol cm−3). In the same trend follows the amplitude of tan δ peaks ∼0.05 for TGPh50/TGEN50//MA < 0.1 for TGPh50/TGEN50//TA < 0.18 for TGPh50/TGEN50//SA < 0.33 for TGPh50/TGEN50//THPA < 0.37 for TGPh50/TGEN50//PA. Thus, the denser networks are those based on MA and TA. These results are in agreement with the previous studies concerning the differences in reactivity by DSC and the unexpected structural observation by FTIR showing the disappearance of unsaturation in the MA fragment structure, sign that extra-linkages were formed by the participation of this unsaturation to new covalent bonds with the network structure.
The mass between crosslinks (Mc) was also calculated, based on the Tobolsky theory.38 In the case of the material with the highest crosslink density, i.e. TGPh50/TGEN50//MA, the Mc has the smallest value, around 28 g mol−1. Meanwhile the TGPh50/TGEN50//SA thermoset that should have a similar value of Mc, since the distance between the crosslinks should be the same, has a 3.6 times higher Mc of ∼100 g mol−1 (Table 2).
A denser and packed network was therefore obtained with the MA crosslinker, a sign of participation of its unsaturation to extra-linkages inside the polyaromatic network. If we compare TGPh50/TGEN50//TA and TGPh50/TGEN50//PA materials, we can also observe that the first thermoset has a crosslink density ∼1.18 times higher (6.4 mmol cm−3vs. 5.4 mmol cm−3) while its average mass of the segments between crosslinks is shorter (240 g mol−1vs. 232 g mol−1). These results show that the –COOH group from TA contributes through carboxylic acid–epoxy reactions to the network connectivity.
In strong correlation with these network parameters, ν and Mc, the internal friction factor (the so-called damping factor, tanδ) values show that the stiffest thermosets are those provided by the anhydrides that give the extra-crosslinking, i.e. TA (tanδ = 220 °C) and MA (tanδ = 197 °C) anhydrides, followed by PA, SA and THPA with tanδ ranging between 144 and 133 °C. The glass transition values determined by DSC are in good agreement with the tanδ values (Fig. S4, ESI† and Table 2). These variations in mechanical properties can be attributed to the differences in the chemical structure of the five thermoset resins, whereby, with increasing crosslink density, the motion of the polymer chains became more problematic causing an increase in both E′ in the rubbery plateau and the damping factor.
According to DMA, the achieved thermo-mechanical characteristics confirmed that the designed thermosets have thermomechanical characteristics suitable for high-end industries.56,57 In accordance with the MatWeb database, these materials exhibit glass transition values comparable to commercial resins: Park Aerospace Nelco® N4000-29: Tg = 175–185 °C, Solvay MTM® 71: Tg = 128–155 °C, Dow VORAFORCE™ TW 103/TW 165/TC 3000: Tg = 140–150 °C, etc.
The hardness of the designed thermosets was validated by the Shore D tests, with the achieved values (Table 2) being specific for extra-hard materials and similar to those currently used in industry (Epoxy Technology EPO-TEK® H77 = 90 SD, Resinlab® EP1330 = 90 SD).
The tensile properties of the five bio-based thermosets were investigated by tensile tests (Fig. 4 and Table 3). Based on the Young's modulus values (Table 3) we can evaluate that the designed polyaromatic thermosets show significant stiffness. In addition, varying the nature of the crosslinking agent led to the development of materials with different networks exhibiting variable mechanical properties. The highest value of the Young's modulus was obtained for the TGPh50/TGEN50//TA material (1.68 GPa), closely followed by TGPh50/TGEN50//MA (1.66 GPa). These results are in good agreement with the previous discussions about the effect of the extra-linkages developed when using TA or MA crosslinkers on network parameters (v and Mc) and thermomechanical properties (tanδ, E′). Moreover, the higher toughness thermosets are TGPh50/TGEN50//SA (1.29 GPa) and TGPh50/TGEN50//THPA (1.26 GPa), with the difference between the five thermosets being very small (∼0.2–0.4 GPa). As far as elongation at break is concerned, it varies oppositely according to the nature of the anhydrides, with the highest being shown by materials with THPA (8.2%) and the lowest by those with TA (2.13%).
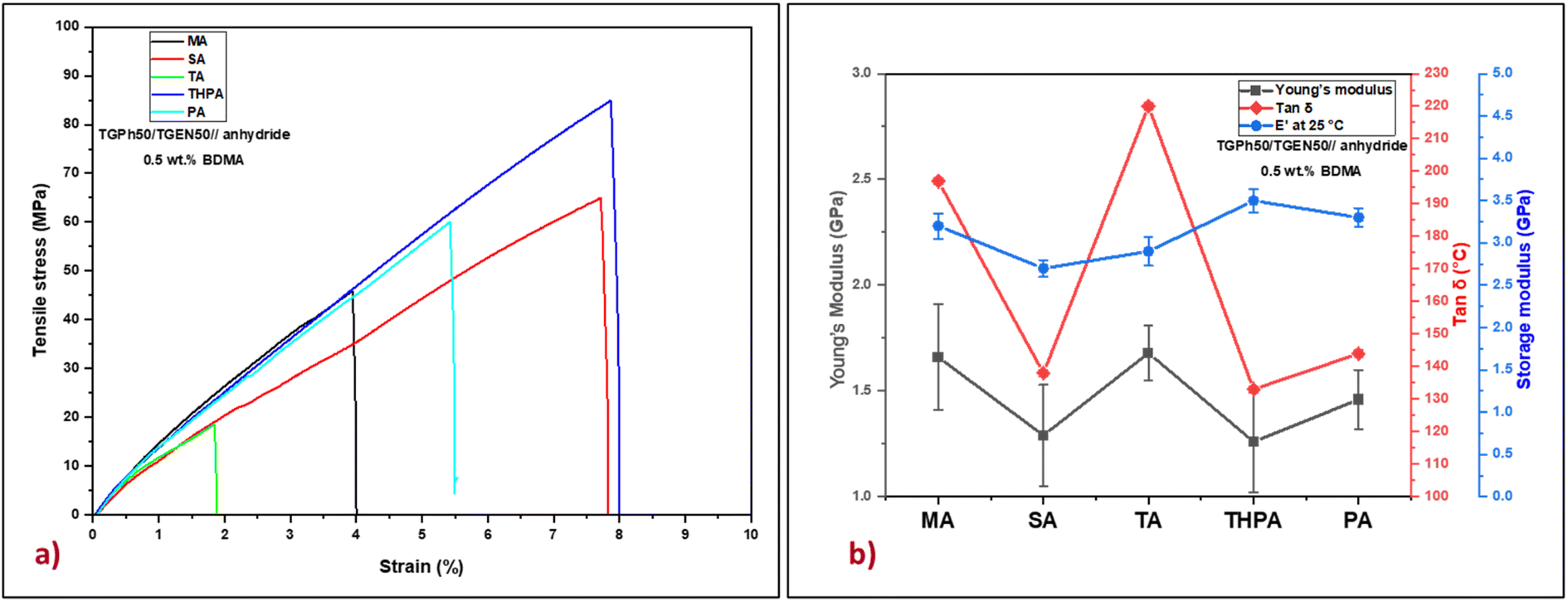 | ||
| Fig. 4 (a) Representative stress–strain curves and (b) Young's modulus, tanδ and storage modulus (E′) at 25 °C for the designed thermosets. | ||
| Thermosets | Young's modulus [GPa] | Tensile stress [MPa] | Elongation at break [%] | Specific modulus – E/ρ –[106 m2 s−2] | Specific strength – σ/ρ – [kN m−1 kg−1] | Specific length – σ/ρ g – [km] | B [% Pa/1010] | Energy at break [J] |
|---|---|---|---|---|---|---|---|---|
| TGPh50/TGEN50//MA | 1.66 ± 0.25 | 43 ± 6.3 | 4.57 ± 1.05 | 1.38 | 36 | 3.66 | 0.068 | 0.5 ± 0.22 |
| TGPh50/TGEN50//SA | 1.29 ± 0.24 | 62 ± 13.5 | 7.25 ± 1.82 | 1.04 | 50 | 5.07 | 0.051 | 1.4 ± 0.57 |
| TGPh50/TGEN50//TA | 1.68 ± 0.13 | 18 ± 2.5 | 2.13 ± 0.47 | 1.29 | 14 | 1.41 | 0.162 | 0.1 ± 0.04 |
| TGPh50/TGEN50//THPA | 1.26 ± 0.24 | 80 ± 11.4 | 8.20 ± 1.20 | 1.01 | 64 | 6.52 | 0.035 | 1.8 ± 0.53 |
| TGPh50/TGEN50//PA | 1.46 ± 0.14 | 65 ± 19 | 6.42 ± 1.76 | 1.16 | 52 | 5.27 | 0.047 | 1.2 ± 0.63 |
The stiffness of polymeric materials can also be characterized by a significant mechanical parameter called brittleness which was described by Brostow et al.39–41 As presented in Table 3, very low brittleness values were obtained: 0.035–0.068% Pa/1010 (except for the TGPh50/TGEN50//TA thermoset with B = 0.162% Pa/1010), which confirmed once again the strength of the developed thermosets. Additional important mechanical parameters such as specific modulus, specific strength, and specific length were calculated and are reported in Table 3. The values of these parameters define the maximum strength of a material at a minimum weight, thus aiming to reduce the weight of the final element without affecting its mechanical properties. In the case of the materials developed in this study the specific modulus varies between 1.01 and 1.36 106 m2 s−2, and the specific strength is between 14 and 64 kN m−1 kg−1, values comparable to those of commercial resins (Hexcel HexPly 108 – Sp. strength = 46.8 kN m−1 kg−1, Sp. modulus = 2.81 × 106 m2 s−2, and Sp. length = 4.78 km; Hexcel HexPly EH25 – Sp. strength = 56.4 kN m−1 kg−1, Sp. modulus = 2.78 × 106 m2 s−2, and Sp. length = 5.75 km) used in high level fields such as automotive, naval, aerospace and space.
The thermal stability of the designed polyaromatic thermosets was analyzed under both air and nitrogen atmospheres (Fig. S8, ESI†) and the related parameters T5%, Tdmax and Cy800 are summarized in Table S7 ESI.† The thermal behaviour of the designed resins under an oxidative atmosphere is divided into two main stages (except for the TA-based resin which shows a small degradation step before 300 °C). The first thermal decomposition stage occurs between 300 and 450 °C, with a maximum degradation around 360–380 °C and a mass loss of about 52–65%, associated with the thermolysis of the polymeric network in which the breaking and degradation of OC–O and C–O–C groups take place.42,49 In the second thermal degradation step, between 530 and 930 °C, the mass loss is slightly lower, ∼35–48%, and is generally characteristic of the thermo-oxidative degradation of the polymer structures in which the carbonization and oxidation of the aromatic rings take place.42,49 As can be seen in Table S7, ESI,† the polyaromatic thermosets have T5% values around 335–355 °C, similar to30,58,59 or even higher50 than those reported in the literature.
The statistical heat resistance index, Ts, a parameter describing the physical heat tolerance limit temperature42,60 shows similar values under both air and nitrogen atmospheres of ∼170–183 °C (Table S7, ESI†), therefore the thermosets can be framed in the high heat-resistant material category. The char yield at 850 °C is significantly high, varying between 26 and 38% depending on the thermoset (Table S7, ESI†). This char can function as a flame retardant by forming a barrier at the polymer–air interface that will reduce the heat conduction and slow down the combustion process. Therefore, higher the amount of carbon residue, more thermally stable is the material. Table S7, ESI† gives the values obtained for LOI parameters in the range between 28 and 32%. The materials with LOI > 26% are classified as self-extinguishing materials.61
Thermoset recycling through a chemical decomposition procedure
Fig. 5 displays the evolution of the five TGPh50/TGEN50//anhydride thermosets from the initial immersion until their total transesterification in the TBD-EG solution. | ||
| Fig. 5 Physical appearance of the thermosets during immersion in TBD-EG solution at 170 °C: (a) t = 0 min; (b) t = 60 min; (c) end of dissolution at t = 70 min for MA & SA systems, at t = 150 min for THPA & PA systems and t = 12 h 30 min for the TA system. | ||
As can be seen, the sample decomposition is quite rapid and occurs through erosion of the sample surface. Immediately after the samples were immersed in the liquid medium, they initially swelled and then began to decrease in weight, maintaining their initial geometry. Under the influence of temperature, the reactive hydroxyl groups present in the EG-catalyzed molecules diffuse into the network and swell the thermoset structure. Then, once in the polymer network, the hydroxyls attack the ester linkages breaking the network into polymeric fragments.62–65 The TGPh50/TGEN50//MA and TGPh50/TGEN50//SA thermosets were completely decomposed after 70 min at 170 °C (and normal pressure), while TGPh50/TGEN50//THPA and TGPh50/TGEN50//PA were completely dissolved in 150 min. For the formulation developed with TA, the erosion process was much slower, taking around 12 hours and 30 minutes of holding in the BER solution at 170 °C. The transesterification of the polymeric networks was also investigated using EG without a catalyst (Fig. S9, ESI†), but since EG is a weak reactant, it cannot cleave the ester bonds without the action of a catalyst.
Conclusions
Starting from two natural and bio-renewable molecules were designed new sustainable and eco-friendly bio-based thermosets with excellent physico-chemical and thermo-mechanical performances, as well as with chemical recycling abilities. The highly crosslinked polyaromatic thermosets (GC > 99%) exhibited high Tg values from 134 to over 199 °C, high storage moduli of 2.7 and 3.5 GPa (room temperature) while Young's moduli ranged from 1.26 to 1.68 GPa and their elongation at break ranged from ∼2 to 8%. The excellent stiffness of the thermosets is also accompanied by a high thermal stability (T5% = 335–355 °C) and good flame-retardancy properties without the addition of any supplementary additive (LOI ∼ 28–33%). The five polyester based thermosets were completely depolymerized in a TBD-EG/mixture, in a quite short time (60–150 min), at a reasonable temperature (170 °C). This capability of the designed materials facilitates their use as polymer matrices in the development of composites, where the fibers can be recovered and reused in a new generation of materials.Author contributions
Conceptualization: R.D. and A.M.; methodology: R.D. and A. M.; epoxy monomer synthesis and investigation: A.P. and A. M.; thermoset development and investigation: R.D. and A. M.; data curation: R.D.; writing—original draft preparation: R.D. and A.M.; writing—review and editing: R.D., U.L., O.D. and A.M.; visualization: R.D., U.L., O.D. and A.M. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work received financial support from the French Government, managed by the National Research Agency under the “Investissements d'Avenir UCAJEDI” project with Reference No. ANR-15-IDEX-01 and from the European Space Agency under ESA Contract 4000134653. A. P. is grateful to UCA JEDI IDEX including the Advanced Research Programme for a grant.References
- Z. Wang, M. S. Ganewatta and C. Tang, Prog. Polym. Sci., 2020, 101, 101197 CrossRef CAS.
- A. Llevot and M. A. R. Meier, Green Chem., 2016, 18, 4800–4803 RSC.
- H. Sukanto, W. W. Raharjo, D. Ariawan, J. Triyono and M. Kaavesina, Open Eng., 2021, 11, 797–814 CrossRef CAS.
- J. C. Capricho, B. Fox and N. Hameed, Polym. Rev., 2020, 60, 1–41 CrossRef CAS.
- H. Lee and K. Neville, Handbook of Epoxy Resins, McGraw-Hill, New York, 1967 Search PubMed.
- J. R. Rochester, Reprod. Toxicol., 2013, 42, 132–155 CrossRef CAS PubMed.
- J. Moreman, O. Lee, M. Trznadel, A. David, T. Kudoh and C. R. Tyler, Environ. Sci. Technol., 2017, 51, 12796–12805 CrossRef CAS PubMed.
- J. Liu, S. Wang, Y. Peng, J. Zhu, W. Zhao and X. Liu, Prog. Polym. Sci., 2021, 113, 101353 CrossRef CAS.
- G. M. Roudsari, A. K. Mohanty and M. Misra, ACS Sustainable Chem. Eng., 2017, 5, 9528–9541 CrossRef.
- F. A. M. M. Gonçalves, M. Santos, T. Cernadas, P. Ferreira and P. Alves, Int. Mater. Rev., 2022, 67, 119–149 CrossRef.
- E. A. Baroncini, K. S. Yadav, G. R. Palmese and J. F. Stanzione, J. Appl. Polym. Sci., 2016, 133(45), 44103 CrossRef.
- F. Ng, G. Couture, C. Philippe, B. Boutevin and S. Caillol, Molecules, 2017, 22, 149 CrossRef PubMed.
- J. Wan, J. Zhao, X. Zhang, H. Fan, J. Zhang, D. Hu, P. Jin and D.-Y. Wang, Prog. Polym. Sci., 2020, 108, 101287 CrossRef CAS.
- Bio-Based Epoxy Polymers, Blends, and Composites - Synthesis, Properties, Characterization, and Applications, ed. J. Parameswaranpillai, S. M. Rangappa, S. Siengchin and S. Jose, WILEY-VCH GmbH, Weinheim, 2021 Search PubMed.
- M. F. ur Rehman, A. I. Batool, R. Qadir and M. Aslam, in A Centum of Valuable Plant Bioactives, Elsevier, 2021, pp. 403–444 Search PubMed.
- A. Stasiłowicz-Krzemień, M. Gołębiewski, A. Płazińska, W. Płaziński, A. Miklaszewski, M. Żarowski, Z. Adamska-Jernaś and J. Cielecka-Piontek, Int. J. Mol. Sci., 2022, 23, 755 CrossRef PubMed.
- J. Stabrauskiene, D. M. Kopustinskiene, R. Lazauskas and J. Bernatoniene, Biomedicines, 2022, 10, 1686 CrossRef CAS PubMed.
- J. Kaur, M. Vyas, J. Singh, R. Prasad and J. Gupta, Phyton, 2020, 89, 795–803 Search PubMed.
- Y. Du, G. Zhao, G. Shi, Y. Wang, W. Li and S. Ren, Eur. Polym. J., 2022, 162(17), 110898 CrossRef CAS.
- M. Latos-Brozio, A. Masek and M. Piotrowska, Materials (Basel), 2021, 14(9), 2142 CrossRef CAS PubMed.
- Y. Oh, K. M. Lee, D. Jung, J. A. Chae, H. J. Kim, M. Chang, J. J. Park and H. Kim, ACS Macro Lett., 2019, 8, 239–244 CrossRef CAS PubMed.
- Y. Li, X. Fu, D. Duan, X. Liu, J. Xu and X. Gao, Mar. Drugs, 2017, 15(2), 49 CrossRef PubMed.
- M. T. H. Chowdhury, I. Bangoura, J. Y. Kang, N. G. Park, D. H. Ahn and Y. K. Hong, Fish. Aquat. Sci., 2011, 14, 198–204 CAS.
- C. Park, H. J. Cha, S. H. Hong, G. Y. Kim, S. Kim, H. S. Kim, B. W. Kim, Y. J. Jeon and Y. H. Choi, Mar. Drugs, 2019, 17, 1–16 Search PubMed.
- P. Bhanja, S. Mishra, K. Manna, K. Das Saha and A. Bhaumik, ACS Omega, 2018, 3(1), 529–535 CrossRef CAS PubMed.
- R. Ménard, C. Negrell, M. Fache, L. Ferry, R. Sonnier and G. David, RSC Adv., 2015, 5, 70856–70867 RSC.
- F. Ng, L. Bonnet, G. David and S. Caillol, Prog. Org. Coat., 2017, 109, 1–8 CrossRef CAS.
- D. Santiago, D. Guzmán, F. Ferrando, À. Serra and S. De la Flor, Polymers, 2020, 12, 1–14 CrossRef PubMed.
- D. Guzmán, D. Santiago, À. Serra and F. Ferrando, Polymers (Basel), 2020, 12(2), 337 CrossRef PubMed.
- A. Genua, S. Montes, I. Azcune, A. Rekondo, S. Malburet, B. Daydé-Cazals and A. Graillot, Polymers, 2020, 12, 1–14 CrossRef PubMed.
- P. K. Tripathi, L. Gan, M. Liu, X. Ma, Y. Zhao, D. Zhu, Z. Xu, L. Chen and N. N. Rao, Mater. Lett., 2014, 120, 108–110 CrossRef CAS.
- B. Wang, Q. Zhang, G. Xiong, F. Ding, Y. He, B. Ren, L. You, X. Fan, C. Hardacre and Y. Sun, Chem. Eng. J., 2019, 366, 404–414 CrossRef CAS.
- M. Gao, J. Fu, M. Wang, K. Wang, S. Wang, Z. Wang, Z. Chen and Q. Xu, J. Colloid Interface Sci., 2018, 524, 165–176 CrossRef CAS PubMed.
- R. W. H. Tess and O. Calif, Process for Manufacture of Glycidyl Ethers of Polyhydric Phenols, US2879259A, 1959.
- W. E. S. Claire, Process for Preparing Epoxyalkyl Aryl Ethers, US2892849A, 1959.
- N. H. Reinking, Preparation of Monomeric Glycidyl Polyethers of Polyhydric Phenols, US2943096A, 1960.
- P. J. Flory, Principles of Polymer Chemistry, Cornell University Press, Ithaca, New York, 1953 Search PubMed.
- A. V. Tobolsky, Properties and structure of polymers, Wiley, New York, 1960 Search PubMed.
- W. Brostow, H. E. Hagg Lobland and M. Narkis, J. Mater. Res., 2006, 21, 2422–2428 CrossRef CAS.
- W. Brostow and H. E. Hagg Lobland, J. Mater. Sci., 2010, 45, 242–250 CrossRef CAS.
- W. Brostow, H. E. Hagg Lobland and M. Narkis, Polym. Bull., 2011, 67, 1697–1707 CrossRef CAS.
- F. Mustata and N. Tudorachi, Ind. Crops Prod., 2021, 159, 113087 CrossRef CAS.
- D. W. van Krevelen, Polymer, 1975, 16, 615–620 CrossRef CAS.
- A. Kumar and J. T'Ien, Int. J. Spray Combust. Dyn., 2012, 4, 299–322 CrossRef CAS.
- Determination of Gel Content and Swell Ratio of Crosslinked Ethylene, Plastics, 2016.
- A. I. Barabanova, B. V. Lokshin, E. P. Kharitonova, E. S. Afanasyev, A. A. Askadskii and O. E. Philippova, Polymer (Guildf), 2019, 178, 121590 CrossRef CAS.
- M. Giebler, C. Sperling, S. Kaiser, I. Duretek and S. Schlögl, Polymers, 2020, 12, 1148 CrossRef CAS PubMed.
- M. K. Antoon and J. L. Koenig, J. Polym. Sci., Polym. Chem. Ed., 1981, 19, 549–570 CrossRef CAS.
- Y. Liu, Z. Du, C. Zhang, C. Li and H. Li, J. Appl. Polym. Sci., 2007, 103, 2041–2048 CrossRef CAS.
- Y. Du, G. Zhao, G. Shi, Y. Wang, W. Li and S. Ren, Eur. Polym. J., 2022, 162, 110898 CrossRef CAS.
- Y. Yang, G. Xian, H. Li and L. Sui, Polym. Degrad. Stab., 2015, 118, 111–119 CrossRef CAS.
- E. Duemichen, M. Javdanitehran, M. Erdmann, V. Trappe, H. Sturm, U. Braun and G. Ziegmann, Thermochim. Acta, 2015, 616, 49–60 CrossRef CAS.
- L. W. Hill, Prog. Org. Coat., 1997, 31, 235–243 CrossRef CAS.
- A. Rios de Anda, P. Sotta, T. Modjinou, V. Langlois, D. Versace and E. Renard, Macromolecules, 2020, 53, 2187–2197 CrossRef CAS.
- F. Hübner, E. Szpoganicz, M. Demleitner, J. Kuhnigk, V. Altstädt and A. Rios De Anda, ACS Appl. Polym. Mater., 2020, 2, 4779–4789 CrossRef.
- M. Biron, Thermosets and Composites Material Selection, Applications, Manufacturing and Cost Analysis, Elsevier, 2nd edn, 2013 Search PubMed.
- Q. Guo, Thermosets - Structure, Properties, and Applications, Elsevier, 2nd edn, 2017 Search PubMed.
- A. Marotta, N. Faggio, V. Ambrogi, P. Cerruti, G. Gentile and A. Mija, Biomacromolecules, 2019, 20, 3831–3841 CrossRef CAS PubMed.
- C. François, S. Pourchet, G. Boni, S. Rautiainen, J. Samec, L. Fournier, C. Robert, C. M. Thomas, S. Fontaine, Y. Gaillard, V. Placet and L. Plasseraud, C. R. Chim., 2017, 20, 1006–1016 CrossRef.
- S. Ma, X. Liu, L. Fan, Y. Jiang, L. Cao, Z. Tang and J. Zhu, ChemSusChem, 2014, 7, 555–562 CrossRef CAS PubMed.
- T. Weiss, H. Schuster, J. Müller, K.-H. Schaller, H. Drexler, J. Angerer and H. U. Käfferlein, Ann. Occup. Hyg., 2011, 55(8), 886–892 CAS.
- X. Kuang, Q. Shi, Y. Zhou, Z. Zhao, T. Wang and H. J. Qi, RSC Adv., 2018, 8, 1493–1502 RSC.
- X. Kuang, Y. Zhou, Q. Shi, T. Wang and H. J. Qi, ACS Sustainable Chem. Eng., 2018, 6, 9189–9197 CrossRef CAS.
- Q. Mu, L. An, Z. Hu and X. Kuang, Polym. Degrad. Stab., 2022, 199, 109895 CrossRef CAS.
- X. Shi, C. Luo, H. Lu and K. Yu, Polym. Eng. Sci., 2019, 59, E111–E119 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc04414e |
| This journal is © The Royal Society of Chemistry 2023 |