
Organophotocatalytic silyl transfer of silylboranes enabled by methanol association: a versatile strategy for C–Si bond construction†
Yi
Wan
a,
Yumo
Zhao
a,
Jiajie
Zhu
a,
Qiyang
Yuan
a,
Wei
Wang
 *b and
Yongqiang
Zhang
*a
*b and
Yongqiang
Zhang
*a
aShanghai Frontiers Science Center of Optogenetic Techniques for Cell Metabolism, Shanghai Key Laboratory of New Drug Design, and School of Pharmacy, East China University of Science and Technology, Shanghai 200237, P. R. China. E-mail: yongqiangzhang@ecust.edu.cn
bDepartment of Pharmacology and Toxicology and BIO5 Institute, University of Arizona, Tucson, AZ 85721-0207, USA. E-mail: wwang@pharmacy.arizona.edu
First published on 8th December 2022
Abstract
Development of a mild, robust and metal-free catalytic approach for silyl transfer of silylboranes is critical for the advancement of modern organosilicon chemistry given their powerful capacity in the construction of C–Si bonds. Herein, we wish to disclose a visible light-induced organophotocatalytic strategy, where methanol association with boron atoms enables the photocatalytic generation of silyl radicals. Notably, the protocol is capable of accommodating a wide range of radial acceptors through slightly tuning the reaction conditions, which allows the formation of various types of C–Si bonds. The preparative power of the transformations has been further highlighted in a number of complex settings, including late-stage functionalization and radical cascade reactions. Furthermore, this technology could be extended to the construction of C–B, C–S and C–Sn bonds, thus offering a versatile platform for bond activation and connection of main group elements. The green aspect of the reaction has also been demonstrated by using Brij-30/water as a reaction solvent or sunlight as an alternative energy source.
Introduction
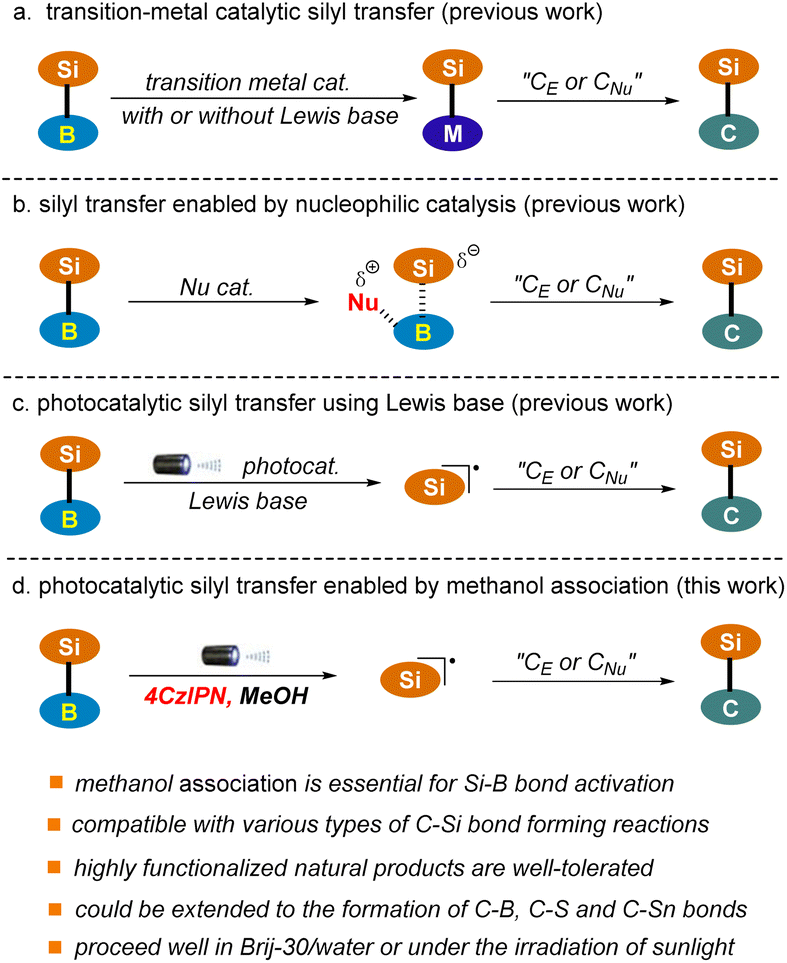
Silicon-containing compounds have gained increasing attention from the synthetic community owing to their broad range of applications in the manufacture of organic optoelectronic materials and in the pharmaceutical sector.1–5 In addition, they are emerging as some of the most versatile organometallic species for complex molecule synthesis owing to the high natural abundance and low toxicity of silicon.6–8The construction of C–Si bonds occupies the central position in the synthesis of this class of valuable targets. Various organic transformations for C–Si bond construction are available. Among them, catalytic silyl transfer from silylboranes to reactive carbon nucleophiles or electrophiles represents one of the most attractive strategies given the ready accessibility and versatile reactivity of these reagents.9–15 However, most of the classic procedures rely heavily on the use of expensive and toxic transition-metal catalysts (Pd, Au, Rh, Cu), which enable the chemo-selective cleavage of the Si–B bond via oxidative addition or transmetalation and the subsequent introduction of the silyl group into a carbon skeleton with high levels of regiocontrol (Scheme 1a).16–25 This drawback has significantly restricted the industrial application of these methods, particularly in the pharmaceutical sector. Recently, improved protocols utilizing cheap and relatively low-toxicity nickel and iron catalysts have been established.26–34 Nonetheless, stoichiometric amounts of strong Lewis bases or high temperatures are often required to facilitate the transformations.
 | ||
| Scheme 1 The construction of the C–Si bond via catalytic silyl transfer of silylboranes. | ||
Nucleophilic catalysis, particularly that employing an organocatalyst, which enables the in situ generation of reactive silicon species under metal-free conditions, has emerged as a promising alternative to the classic transition-metal catalyzed approach (Scheme 1b).35–39 For example, the Hoveyda group has reported the silyl transfer of silylboranes to α, β-unsaturated carbonyls using N-heterocyclic carbene (NHC) as the catalyst. Various pyridines, such as 4-cyanopyridine and 2-benzyl pyridine, as well as tributylphosphane (PBu3), have also been identified as suitable nucleophilic catalysts. Nonetheless, this strategy has only been applied to the addition of silylboranes to C–C or C–O multiple bonds. Furthermore, these reactions typically proceed with the assistance of a strong Lewis base, such as DBU, or at high temperature (50 °C–135 °C).
Visible light-induced photoredox catalysis has emerged as a powerful tool for the development of new and valuable transformations in organic synthesis.40–43 Recently, this strategy has been extended to silyl transfer of silylboranes using a Lewis base as the promoter, which allows the in situ generation of silyl radicals and the subsequent addition to C–C multiple bonds under mild conditions (Scheme 1c).44,45 For example, the Poisson group has reported a copper-photocatalyzed hydrosilylation of alkynes and alkenes using a stoichiometric amount of K2CO3.44 Very recently, organic photoredox-catalyzed hydrosilylation and acylsilylation of alkynes have been developed by the Ohmiya group, where a catalytic amount of DMAP is employed, enabling the generation of products in low to moderate yields.45 Herein, we wish to report a visible light-induced organophotocatalytic silyl transfer strategy of silylboranes in which the Si–B bond is pre-activated via methanol association with a more Lewis acidic boron site (Scheme 1d). The protocol is mild, robust, metal-free, and most importantly capable of accommodating a wide range of carbon nucleophiles and electrophiles upon slightly tuning the reaction conditions, which allows the facile construction of various types of C–Si bonds ranging from C(alkyl)–Si, C(aryl)–Si, C(alkenyl)–Si to C(alkynyl)–Si. The capacity of the transformations has also been demonstrated in a number of complex settings, including the late-stage functionalization of plant terpenoids and radical cascade reactions. Notably, this technology could be extended to the construction of C–B, C–S and C–Sn bonds, thus providing a versatile platform for bond activation and connection of main group elements. Furthermore, the reaction proceeded well in water with the assistance of surfactant Brij-30, where water might function as an alternative activator of the Si–B bond. Sunlight could also be used as an energy source. These results further highlight the great potential of the reaction in green synthesis.
Results and discussion
Our group has been interested in the development of visible light-induced photocatalytic protocols for C–Si bond construction and has reported an elegant hydrosilylation approach for the synthesis of β-silyl-α-amino acids where a quinuclidine-catalyzed hydrogen atom transfer (HAT) of hydrosilanes enabled by photocatalytic oxidation has been employed to generate silyl radicals.46,47 In connection with our continued interest in this field, we envisioned that this dual-catalytic strategy could be extended to silyl transfer of silylboranes via an alternative boron atom transfer (BAT). The successful implementation of this new tactic would enable the efficient construction of C–Si bonds while avoiding the use of readily oxidized and combustible hydrosilanes as silyl radical sources.To test the feasibility of this hypothesis, we first examined the hydrosilylation of alkenes since they are among the most abundant chemical feedstock. The initial investigation of the reaction started with subjecting the readily accessible bis-N-Boc-protected dehydroalanine benzyl ester 2a as a representative alkene and the cheap and easy-to-handle PhMe2Si-BPin 1a as a model silylborane to the reaction conditions established in our previous work using 4CzIPN (3 mol%) and quinuclidine (0.5 equivalent) as the catalysts under the irradiation of 24 W blue-light emitting diodes (LEDs).46 It is noted that a solvent mixture of MeCN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O = 10:1 was employed and H2O was expected to function as a proton donor to facilitate the reaction.
:H2O = 10:1 was employed and H2O was expected to function as a proton donor to facilitate the reaction.
To our delight, product 3a, an N-protected β-silyl-α-amino acid ester, was obtained in a moderate yield (64% yield, entry 1, Table 1). Surprisingly, a significant amount of product 3a was also detected in the absence of quinuclidine, which was expected to function as a BAT catalyst (52% yield, entry 2, Table 1). The increase of the ratio of water in the solvent mixture from 1:10 to 1:1 led to a higher yield (60% yield, entry 3, Table 1), while dramatically decreased reaction efficiency was observed by roughly excluding water from the reaction solvent (25% yield, entry 4, Table 1). Based on these results, as well as the reported procedures of photocatalytic C–B bond cleavage,48 it was reasonable to conclude that quinuclidine might contribute to the generation of silyl radicals through the formation of a redox-active complex with 1avia an N → B coordination, and water, particularly a large amount of water, could also facilitate the formation of the redox-active complex as an alternative nucleophile. However, the quinuclidine-mediated BAT mechanism cannot be completely excluded. Hence, various cheap and readily available alcohol solvents with enhanced nucleophilicity were screened and sterically less hindered methanol was more favorable (30%–82% yields, entries 5–9, Table 1). Nevertheless, the use of only methanol as the reaction solvent resulted in a low yield (34% yield, entry 10, Table 1). The screening of photocatalysts revealed that 4CzIPN (3 mol%) was more favorable (Table S1†). Further extensive screening of the reaction solvent and time (Table S2†) identified the optimal conditions as follows: under the irradiation of 24 W blue LEDs and in the presence of 3 mol% 4CzIPN, the reaction of 1a with 1.5 equiv. of 2a was conducted in a solvent mixture of MeCN:MeOH = 1:1 for 18 h at room temperature under N2. Under these reaction conditions, 3a was produced in an excellent yield (entry 5, 82% yield, Table 1).
|
|
||
|---|---|---|
| Entry | Solvent | Yieldb |
| a Reaction conditions: 24 W blue LEDs, 1a (0.3 mmol, 1.5 equiv.), 2a (0.2 mmol, 1.0 equiv.), 4CzIPN (0.006 mmol, 3 mol%), solvent (4 mL), N2, rt, unless otherwise noted. b Yields were determined by 1H NMR using mesitylene as an internal standard. c 0.5 equiv. of quinuclidine was employed. d Performed in darkness. e Performed in the absence of a photocatalyst. f Performed under air. g Performed in the presence of 1 equiv. of TEMPO. | ||
| 1 | MeCN:H2O = 10:1 |
64%c |
| 2 | MeCN:H2O = 10:1 |
52% |
| 3 | MeCN:H2O = 1:1 |
60% |
| 4 | MeCN only | 25% |
| 5 |
MeCN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :MeOH = 1:1 :MeOH = 1:1
|
82% |
| 6 | MeCN:EtOH = 1:1 |
77% |
| 7 | MeCN:iPrOH = 1:1 |
50% |
| 8 | MeCN:tBuOH = 1:1 |
30% |
| 9 | MeCN:Glycol = 1:1 |
33% |
| 10 | MeOH only | 34% |
| 11 | MeCN:MeOH = 1:1 |
n.dd |
| 12 | MeCN:MeOH = 1:1 |
n.de |
| 13 | MeCN:MeOH = 1:1 |
Tracef |
| 14 | MeCN:MeOH = 1:1 |
Traceg |
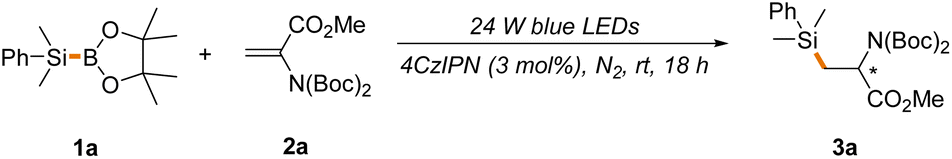
Control experiments established the importance of both visible light and the photocatalyst, as no product was observed in the absence of these two reaction promoters (entries 11 and 12, Table 1). Furthermore, the inhibition of reactivity was observed in the presence of molecular oxygen and TEMPO (entries 13 and 14, Table 1), suggesting the radical nature of the reaction.
Having identified the optimal reaction conditions (entry 6, Table 1), the scope with regard to silylboranes was first evaluated (Scheme 2a). As demonstrated, the replacement of the phenyl of 1a with alkyl, cycloalkyl or benzyl led to a slight decrease of reaction efficiency (3b–3f, 62%–78% yields), likely owing to the decreased stability of the silyl radical intermediate. Interestingly, when deuterated methanol was used in place of methanol, the reaction proceeded smoothly to give product 3a′ in a comparable yield and with a high incorporation of deuterium (79% yield, 92%D), thereby offering a mild and efficient deuteration protocol for the selective deuterium labelling of organic molecules. Meanwhile, this result further indicates that methanol functions as a proton donor in the process. The incorporation of trimethylsilyl into the carbon skeleton via photocatalytic C–Si bond formation remains elusive given that the present procedures rely heavily on the use of hydrosilanes as silyl radical sources and Me3Si-H is in the gaseous state at room temperature under normal atmospheric pressure.49–51 However, Me3Si-BPin, which is easy-to-handle and readily accessible,52 was amendable to our newly established protocol (3b, 72% yield), providing a facile photocatalytic approach for trimethylsilyl introduction. Various alkene substrates were then evaluated. As depicted in Scheme 2b, the simple benzyl acrylate displayed higher reaction efficiency (4a, 84% yield) and various other types of electron-deficient alkenes such as acrylonitrile, vinyl sulfone, acrylamides, ethyl vinyl ketone and vinyl pyridine were applicable, affording hydrosilylation products in good yields (4b–4g, 60%–76% yields). The position of vinyl on pyridine has little effect on the reaction outcome (4f–4g, 72%–76% yields). The photocatalytic hydrosilylation of acrolein has not been well-established, which might be attributed to the use of reductive hydrosilanes as silyl donors or other aldehyde-sensitive reaction conditions in present protocols. However, this mild and robust protocol was well compatible with acrolein (4h, 70% yield). Remarkably, vinyl sulfonyl fluoride was proved to be an effective reactant (4i, 76% yield), enabling the construction of diverse sulfonyl fluoride-containing building blocks for drug discovery in combination with C–Si bond-based organic transformations.53 As expected, various electron-deficient styrene substrates were also viable substrates (4j–4m, 65%–82% yields) and a variety of functional groups, such as borate, ester and chlorine, were well tolerated, providing the handles for further synthetic elaboration. Acrylates with different steric and electronic properties were suitable substrates (4n–4r, 72%–85% yields). Interestingly, oxetane and azetidine-derived methyl acrylates with a highly strained exocyclic double bond were also tolerated, although with inferior reaction efficiency (4s–4t, 30%–36% yields) and the silyl group could be transformed into the hydroxyl group under the standard conditions (4s′–4t′, 65%–70% yields), offering a powerful tool to access various oxetane and azetidine-derived functionalities for drug discovery given the prevalence of these structures in drug molecules.54,55 It is noted that the challenging Si-substituted quaternary carbon center was constructed in the reaction. However, piperidine-derived methyl acrylate displayed poor reaction efficiency (4u, Scheme 2b). A radical addition pathway driven by ring strain release might help to explain these results. Furthermore, cyclic enone and lactone were all hydrosilylated in moderate yields (4v–4w, 42%–58%). To demonstrate the synthetic utility of this new reaction, we evaluated the late-stage functionalization of three plant terpenoids, including parthenolide, oridonin, and andrographolide (Scheme 2c). Notably, the reactions proceeded smoothly, yielding the target compounds 5a–5c in 55%–84% yields with excellent chemo- and regio-selectivity, further highlighting the benign nature and excellent functional group compatibility of the reaction. Experiments probing the alkyne substrates under the standard conditions illustrated that 4-ethynylbenzonitrile 6a and 4-ethynylbenzoate 6b could also undergo hydrosilylation to give mono-silylation or bis-silylation products depending on the amount of 1a employed in the reaction (mono-silylation products 7a and 7b, 75%–80% yields, bis-silylation products 7a′–7b′, 60%–66% yields, Scheme 2d).
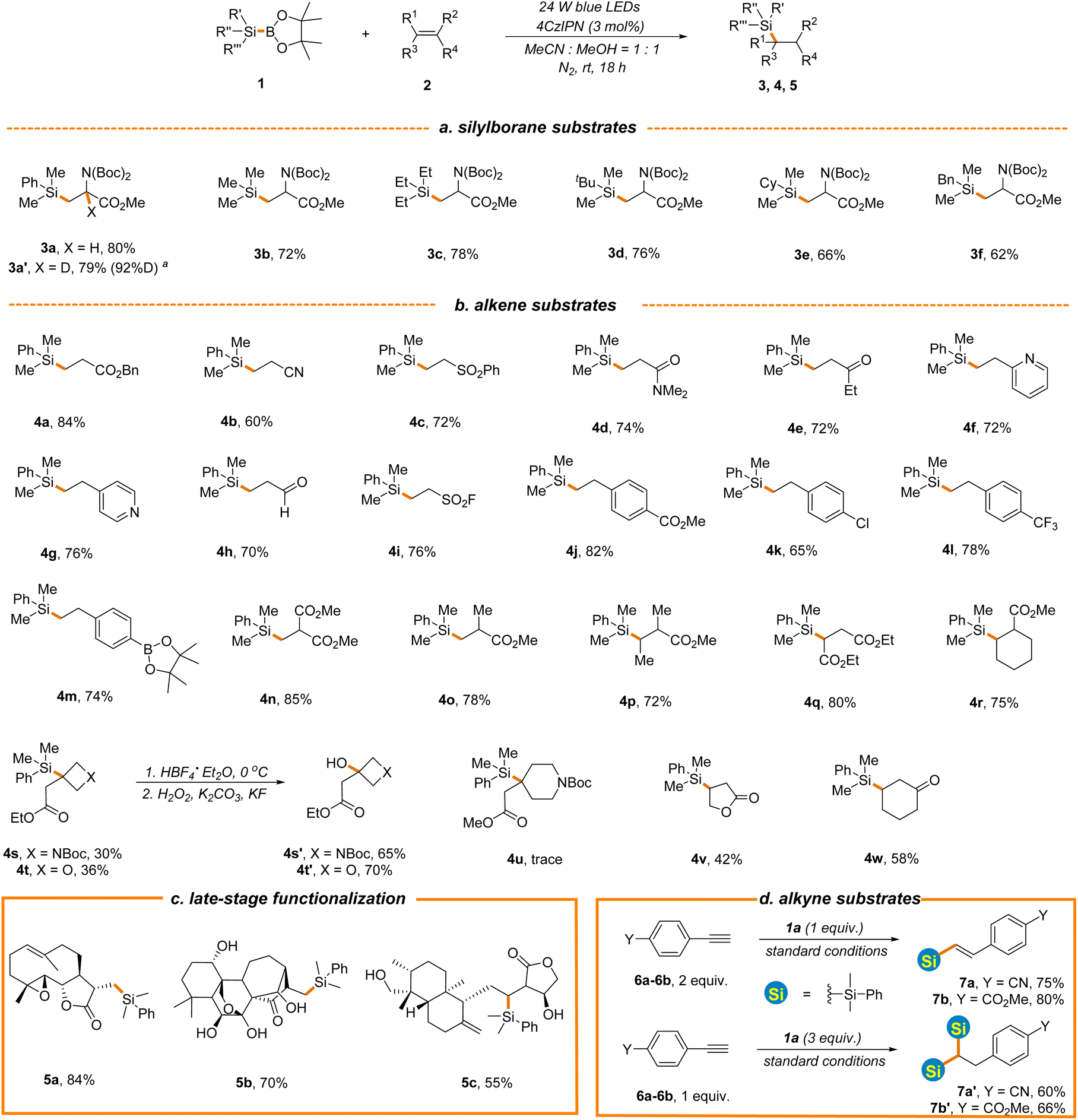 | ||
| Scheme 2 The substrate scope of alkene hydrosilylation. See general procedure A in the ESI for details.†aCD3OD was employed instead of CH3OH. | ||
Having established the scope of the hydrosilylation reaction, we performed a study to further probe and verify the reaction mechanism. The formation of the redox-active complex between methanol and silylborane 1a was further validated by 1H NMR, 11B NMR and 29Si NMR analysis (Fig. S1†). Stern–Volmer quenching experiments revealed that silylborane 1a could efficiently quench the emission of excited 4CzIPN using anhydrous MeCN/MeOH (1:1) as the solvent, while no quenching effect was observed using anhydrous MeCN as the solvent (Fig. S2A and S2B†). Furthermore, 2a did not quench the excited state of the photocatalyst (Fig. S2C†). Based on these observations, as well as the fact that methanol serves as a proton donor to facilitate the reaction and the established mechanism for photocatalytic C–B bond cleavage,48 a plausible reaction mechanism is proposed (Scheme 3). The catalytic cycle starts with the in situ formation of redox-active complex I between methanol and silylborane 1avia O → B coordination. The photocatalyst in a photoexcited state (PC*) serves as an oxidant, undergoing a single-electron (SE) redox process with I to give the reduced PC˙− species and silyl radical II, accompanied by the production of 2-methoxy-4,4,5-trimethyl-1,3,2-dioxaborolane III as a by-product. The addition of Si-radical II to electron-deficient alkenes followed by photoreduction and protonation gives the products and reconstitutes the ground state of 4CzIPN.
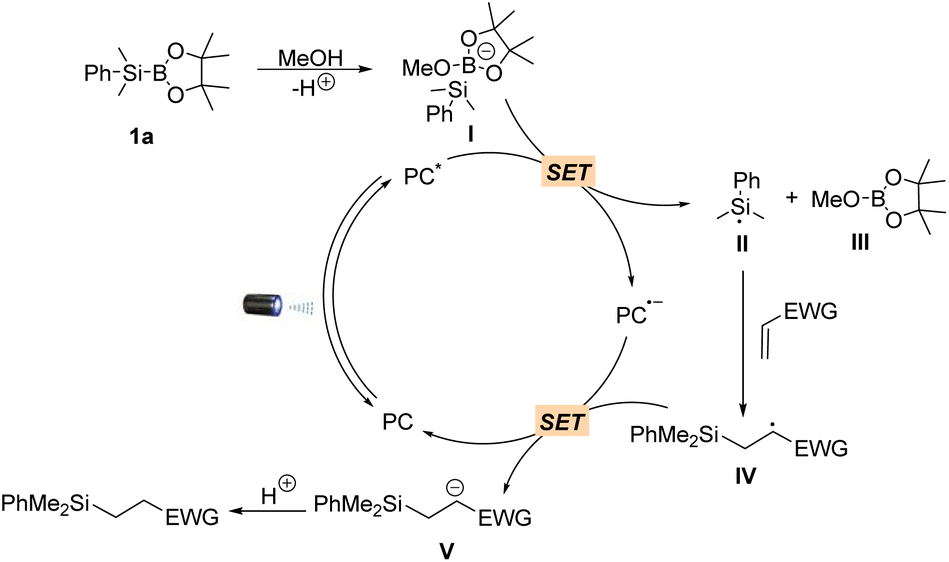 | ||
| Scheme 3 Proposed mechanism. | ||
To further probe the synthetic utility of this strategy in the construction of other types of C–Si bonds, we next attempted to extend this new protocol to electron-rich alkenes by the introduction of iPr3SiSH as a polarity-reversal catalyst.49 As expected, a broad range of aliphatic terminal alkenes were found to be good substrates, delivering the corresponding silane products in moderate to excellent yields (9a–9e, 54%–80% yields, Scheme 4a). It is worth mentioning that both vinyl silane and allyl silane could be applied to generate valuable bimetallic products 9b and 9e, which could be engaged in an array of complexity-generating processes. It should be noted that electron-rich styrene was not applicable (data not shown here), which is consistent with the photocatalytic hydrosilylation using hydrosilanes reported by Wu's group.49 The radical substitution of protonated heteroarenes, the Minisci reaction, is a well-known versatile method for direct C–H functionalization of N-heteroarenes.56 Our group has been committed to the development of mild and efficient Minisci reaction for the rapid diversification of N-heteroarenes and has reported a photocatalytic Minisci-type C–H silylation reaction using trialkylhydrosilanes, where bulky and inert tBuMe2SiH and iPr3SiH were identified as suitable substrates.46,57,58 Nonetheless, the use of stoichiometric amounts of explosive peroxide as a terminal oxidant in the process causes safety concerns and limits the reaction scale. Gratifyingly, this strategy could be extended to Minisci-type C–H silylation using tBuMe2SiBPin and iPr3SiBPin as alternative silyl radical sources with the assistance of trifluoroacetic acid (TFA) in the absence of any terminal oxidants, giving the products 11a–11k in moderate yields (48%–72% yields, Scheme 4b). As expected, the reactions typically occur at the most electrophilic sites and exhibit excellent regio-selectivity. Furthermore, silyl radicals generated via this organophotocatalytic strategy could also be captured by various ethynylbenziodoxolones (EBXs), enabling the facile construction of a variety of alkynylsilanes (13a–13e, 56%–82% yields, Scheme 4c), which could participate in various coupling reactions for complex molecule synthesis.
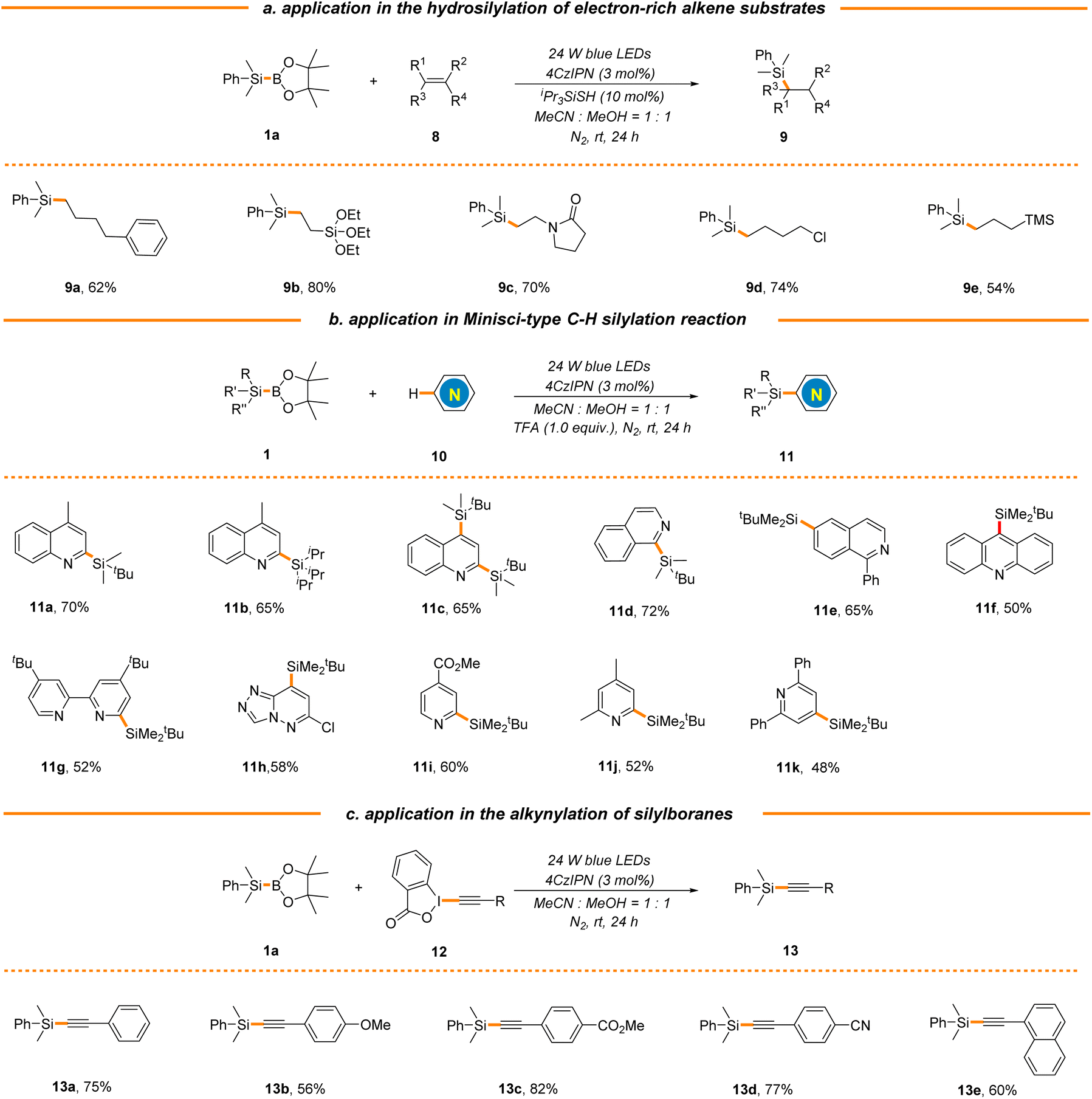 | ||
| Scheme 4 Application in the construction of other types of C–Si bonds. See general procedures B–D in the ESI for details.† | ||
We next attempted to probe the compatibility of our strategy with the photocatalytic radical cascade reaction, which presents a powerful tool to rapidly generate molecular complexity.59–61 As demonstrated, the cross-coupling of the Baylis–Hillman acetate adduct with silylborane 1a proceeded well via a radical–polar crossover pathway to give product 14a in excellent yield and stereoselectivity (85% yield, Scheme 5a), thus providing an efficient approach for the synthesis of highly functionalized allylsilanes with versatile reactivity.62 A pharmaceutically relevant indolone structure 15a could also be readily accessed via a Michael addition/radical cyclization cascade reaction in a moderate yield (67% yield, Scheme 5b). Furthermore, the current protocol could be extended to the construction of C–B and C–Sn bonds, where the transfer of boryl and stannanyl from readily available bimetallic reagents (B–B, Si–Sn or Sn–Sn) to 2a enabled the facile synthesis of B- and Sn-containing α-amino acids 17a, 19a and 21a in 75%–80% yields (Scheme 5c and d). It should be noted that stannanyl transfer is more favorable for stannosilane, which might be attributed to the stronger association of the oxygen atom in methanol with the silicon atom in stannosilane. Along this line, thiophenyl transfer was also preferred for silathiane, leading to the formation of S-containing α-amino acids in a high yield (23a, 80% yield, Scheme 5e).
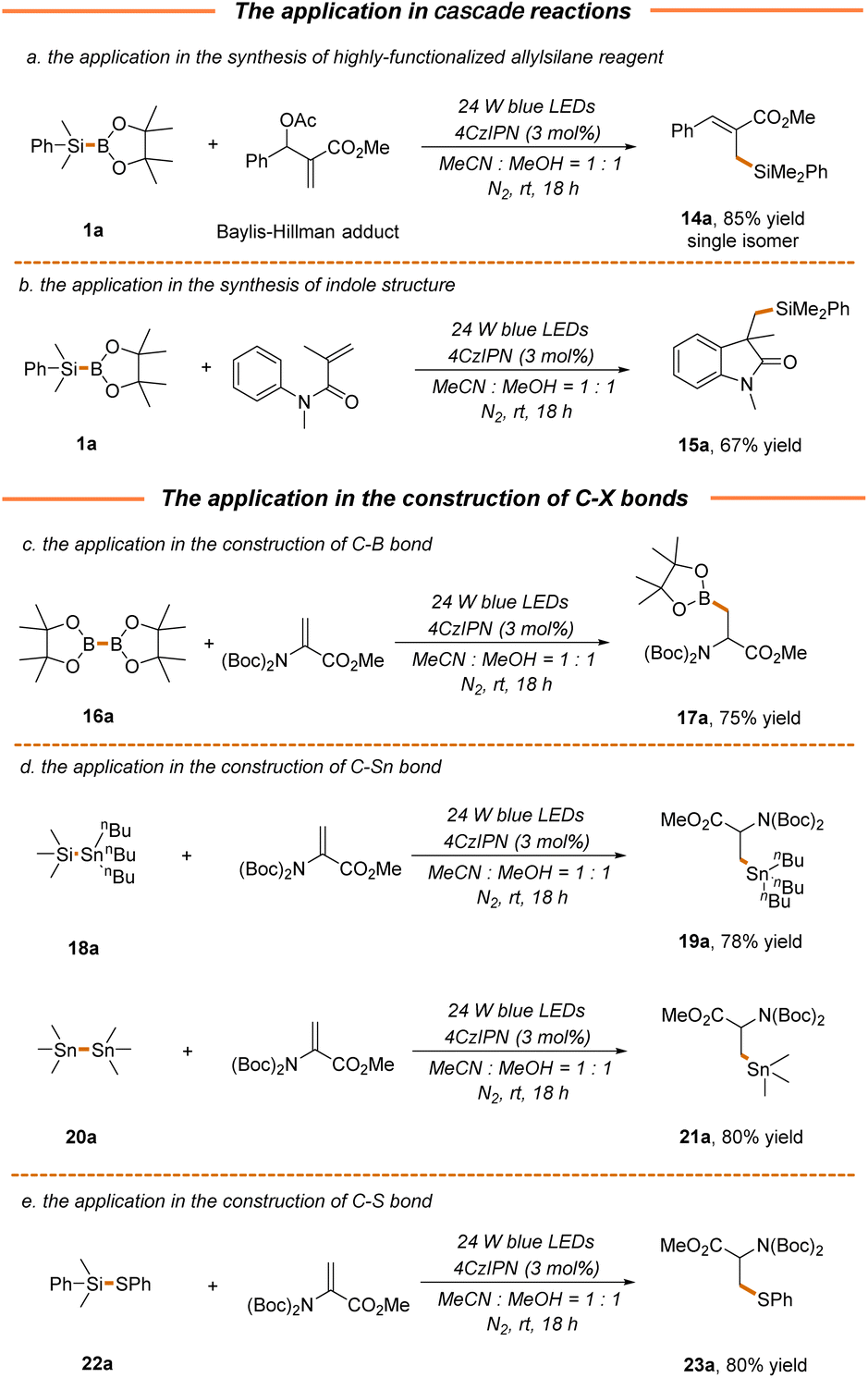 | ||
| Scheme 5 Further application. | ||
Finally, we continued to investigate the reaction in terms of the principles of green chemistry (Scheme 6). As expected, the reaction proceeded well under sunlight, a clean and sustainable energy source, although with inferior reaction efficiency (Scheme 6a). Alternatively, a more safe solvent system (ethyl acetate:ethanol = 1:1)63 could be used to facilitate the transformation (Scheme 6b). Remarkably, water was also proved to be an effective solvent with the assistance of 2 wt% surfactant Brij-30 (3,6,9,12-tetraoxatetracosan-1-ol), further highlighting the potential of the reaction in green synthesis (Scheme 6c). It is noted that ethanol and water might function as alternative activators of Si–B bonds in these two reactions. It is undeniable that the atom economy of the reaction is moderate since stoichiometric amounts of by-products such as boric acid and borate are concurrently produced in the process. Nonetheless, the uncovering of the underlying mechanism of the reaction offers the possibility of developing a highly atom economical silylboration reaction of alkenes and alkynes, which is ongoing in our laboratory.
 | ||
| Scheme 6 Investigation of the reaction in terms of the principles of green chemistry. | ||
Conclusion
In summary, visible light-induced organophotocatalytic silyl transfer of silylboranes has been established for the construction of various types of C–Si bonds, where methanol association enabled the photocatalytic generation of silyl radicals. The hydrosilylation of a broad spectrum of electron-deficient alkenes and alkynes, particularly the complex alkene-containing plant terpenoids, has been demonstrated. The challenging acrolein and some pharmaceutically relevant alkenes, such as alkenyl sulfonyl fluorides and oxetane/azetidine-derived methyl acrylates, were suitable substrates. Remarkably, trimethylsilyl could be facilely introduced given that Me3Si-BPin is readily accessible, easy-to-handle and highly reactive. Slight tuning of the reaction conditions enables the extension of this powerful strategy to the hydrosilylation of electron-rich alkenes, Minisci-type C–H silylation and alkynylation of silylboranes. It is worth mentioning that the Minisci-type C–H silylation could proceed in the absence of any terminal oxidants. Furthermore, the preparative power was further highlighted by the application in radical cascade reactions and the construction of C–B, C–Sn and C–S bonds. The reaction could also proceed in water with the assistance of surfactant Brij-30. Clean and sustainable sunlight could also be used as an alternative energy source. Overall, this promising synthetic strategy significantly expands the boundaries of Si–B chemistry and offers a versatile platform for the development of new and green main group chemistry.Conflicts of interest
There is no conflict of interest to report.Acknowledgements
The financial support from the program of the National Natural Science Foundation of China (22171080, 21871086, Y.-Q. Z.; 21738002, W. W.) is gratefully acknowledged.References
- E. A. Marro and R. S. Klausen, Chem. Mater., 2019, 31, 2202–2211 CrossRef CAS.
- Z. Zhao, B. He and B.-Z. Tang, Chem. Sci., 2015, 6, 5347–5365 RSC.
- R. Pietschnig and S. Spirk, Coord. Chem. Rev., 2016, 323, 87–106 CrossRef CAS.
- E. Rémond, C. Martin, J. Martinez and F. Cavelier, Chem. Rev., 2016, 116, 11654–11684 CrossRef PubMed.
- R. Ramesh and D. S. Reddy, J. Med. Chem., 2018, 61, 3779–3798 CrossRef CAS PubMed.
- T. Komiyama, Y. Minami and T. Hiyama, ACS Catal., 2017, 7, 631–651 CrossRef CAS.
- S. E. Denmark and A. Ambrosi, Org. Process Res. Dev., 2015, 19, 982–994 CrossRef CAS PubMed.
- F. Zhao, S. Zhang and Z. Xi, Chem. Commun., 2011, 47, 4348–4357 RSC.
- M. Oestreich, E. Hartmann and M. Mewald, Chem. Rev., 2013, 113, 402–441 CrossRef CAS PubMed.
- J.-J. Feng, W. Mao, L. Zhang and M. Oestreich, Chem. Soc. Rev., 2021, 50, 2010–2073 RSC.
- C. Moberg, Synthesis, 2020, 52, 3129–3139 CrossRef CAS.
- W. Xue and M. Oestreich, ACS Cent. Sci., 2020, 6, 1070–1081 CrossRef CAS PubMed.
- J. R. Wilkinson, C. E. Nuyen, T. S. Carpenter and S. R. Harruff, ACS Catal., 2019, 9, 8961–8979 CrossRef CAS.
- M. B. Ansell, O. Navarro and J. Spencer, Coord. Chem. Rev., 2017, 336, 54–77 CrossRef CAS.
- A. B. Cuenca, R. Shishido, H. Ito and E. Fernández, Chem. Soc. Rev., 2017, 46, 415–430 RSC.
- C.-Q. Wang, Y. Li and C.-B. Feng, Cell Rep. Phys. Sci., 2021, 2, 100461 CrossRef CAS.
- T. Ahmad, Q. Li, S.-Q. Qiu, J.-L. Xu, Y.-H. Xu and T.-P. Loh, Org. Biomol. Chem., 2019, 17, 6122–6126 RSC.
- F. Bartoccini, S. Bartolucci, S. Lucarini and G. Piersanti, Eur. J. Org. Chem., 2015, 3352–3360 CrossRef CAS.
- W. Mao and M. Oestreich, Org. Lett., 2020, 22, 8096–8100 CrossRef CAS PubMed.
- L. Zhang and M. Oestreich, ACS Catal., 2021, 11, 3516–3522 CrossRef CAS.
- T. Kitanosono, L. Zhu, C. Liu, P. Xu and S. Kobayashi, J. Am. Chem. Soc., 2015, 137, 15422–15425 CrossRef CAS PubMed.
- H. Guo, X. Chen, C. Zhao and W. He, Chem. Commun., 2015, 51, 17410–17412 RSC.
- R. Shintani, H. Kurata and K. Nozaki, J. Org. Chem., 2016, 81, 3065–3069 CrossRef CAS PubMed.
- W. Xue and M. Oestreich, Angew. Chem., Int. Ed., 2017, 56, 11649–11652 CrossRef CAS PubMed.
- W. Xue, Z.-W. Qu, S. Grimme and M. Oestreich, J. Am. Chem. Soc., 2016, 138, 14222–14225 CrossRef CAS PubMed.
- W. Srimontree, L. Guo and M. Rueping, Chem. – Eur. J., 2020, 26, 423–427 CrossRef CAS PubMed.
- R. J. Somerville and L. V. A. Hale, J. Am. Chem. Soc., 2018, 140, 8771–8780 CrossRef CAS PubMed.
- C. Zarate and R. Martin, J. Am. Chem. Soc., 2014, 136, 2236–2239 CrossRef CAS PubMed.
- J. Jia, X. Zeng, Z. Liu, L. Zhao, C.-Y. He, X.-F. Li and Z. Feng, Org. Lett., 2020, 22, 2816–2821 CrossRef CAS PubMed.
- J. Zhang, Y. Zhang, S. Geng, S. Chen, Z. Liu, X. Zeng, Y. He and Z. Feng, Org. Lett., 2020, 22, 2669–2674 CrossRef CAS PubMed.
- L. Guo, A. Chatupheeraphat and M. Rueping, Angew. Chem., Int. Ed., 2016, 55, 11810–11813 CrossRef CAS PubMed.
- X. Wang, Z. Wang and Y. Nishihara, Chem. Commun., 2019, 55, 10507–10510 RSC.
- C. Zarate, M. Nakajima and R. Martin, J. Am. Chem. Soc., 2017, 139, 1191–1197 CrossRef CAS PubMed.
- B. Cui, S. Jia, E. Tokunaga and N. Shibata, Nat. Commun., 2018, 9, 4393 CrossRef PubMed.
- K. Nagao, H. Ohmiya and M. Sawamura, Org. Lett., 2015, 17, 1304–1307 CrossRef CAS PubMed.
- Y. Morimasa, K. Kabasawa, T. Ohmura and M. Suginome, Asian J. Org. Chem., 2019, 8, 1092–1096 CrossRef CAS.
- C.-R. Jiang, C.-L. Zhao, H.-F. Guo and W. He, Chem. Commun., 2016, 52, 7862–7865 RSC.
- J. M. O'Brien and A. H. Hoveyda, J. Am. Chem. Soc., 2011, 133, 7712–7715 CrossRef PubMed.
- H. Wu, J. M. Garcia, F. Haeffner, S. Radomkit, A. R. Zhugralin and A. H. Hoveyda, J. Am. Chem. Soc., 2015, 137, 10585–10602 CrossRef CAS PubMed.
- M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed.
- C.-S. Wang, P. H. Dixneuf and J.-F. Soulé, Chem. Rev., 2018, 118, 7532–7585 CrossRef CAS PubMed.
- K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–11007 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- M. Zhong, X. Pannecoucke, P. Jubault and T. Poisson, Chem. – Eur. J., 2021, 27, 11818–11822 CrossRef CAS PubMed.
- N. Takemura, Y. Sumida and H. Ohmiya, ACS Catal., 2020, 12, 7804–7810 CrossRef.
- S.-H. Liu, P. Pan, H.-Q. Fan, H. Li, W. Wang and Y.-Q. Zhang, Chem. Sci., 2019, 10, 3817–3825 RSC.
- Y. Wan, J.-J. Zhu, Q.-Y. Yuan, W. Wang and Y.-Q. Zhang, Org. Lett., 2021, 23, 1406–1410 CrossRef CAS PubMed.
- F. Lima, U. K. Sharma, L. Grunenberg, D. Saha, S. Johannsen, J. Sedelmeier, E. V. Van der Eycken and S. V. Ley, Angew. Chem., Int. Ed., 2017, 56, 15136–15140 CrossRef CAS PubMed.
- R. Zhou, Y. Y. Goh, H. Liu, H. Tao, L. Li and J. Wu, Angew. Chem., Int. Ed., 2017, 56, 16621–16625 CrossRef CAS PubMed.
- J. Hou, A. Ee, H. Cao, H.-W. Ong, J.-H. Xu and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 17220–17224 CrossRef CAS PubMed.
- J. Zhu, W.-C. Cui, S. Wang and Z.-J. Yao, J. Org. Chem., 2018, 83, 14600–14609 CrossRef CAS PubMed.
- S. Kamio, T. Imagawa, M. Nakamoto, M. Oestreich and H. Yoshida, Synthesis, 2021, 53, 4678–4681 CrossRef CAS.
- O. O. Grygorenko, D. M. Volochnyuk and B. V. Vashchenko, Eur. J. Org. Chem., 2021, 6478–6510 CrossRef CAS.
- G. Wuitschik, E. M. Carreira, B. Wagner, H. Fischer, I. Parrilla, F. Schuler, M. Rogers-Evans and K. Müller, J. Med. Chem., 2010, 53, 3227–3246 CrossRef CAS PubMed.
- M. R. Bauer, P. Di Fruscia, S. C. C. Lucas, I. N. Michaelides, J. E. Nelson, R. I. Storer and B. C. Whitehurst, RSC Med. Chem., 2021, 12, 448–471 RSC.
- R. S. J. Proctor and R. J. Phipps, Angew. Chem., Int. Ed., 2019, 58, 13666–13699 CrossRef CAS PubMed.
- Y.-Q. Zhang, K. B. Teuscher and H.-T. Ji, Chem. Sci., 2016, 7, 2111–2118 RSC.
- S.-H. Liu, A.-X. Liu, Y.-Q. Zhang and W. Wang, Chem. Sci., 2017, 8, 4044–4050 RSC.
- L. Pitzer, J. L. Schwarz and F. Glorius, Chem. Sci., 2019, 10, 8285–8291 RSC.
- G.-Q. Xu and P.-F. Xu, Chem. Commun., 2021, 57, 12914–12935 RSC.
- J. Xuan and A. Studer, Chem. Soc. Rev., 2017, 46, 4329–4346 RSC.
- G. W. Kabalka, B. Venkataiah and G. Dong, Organometallics, 2005, 24, 762–764 CrossRef CAS.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehadad and P. J. Dunne, Green Chem., 2016, 18, 288–296 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc03577d |
| This journal is © The Royal Society of Chemistry 2023 |