
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ranking the accelerated weathering of plastic polymers†
Maryam
Hoseini
ab,
Jess
Stead
a and
Tom
Bond
 *a
*a
aSchool of Sustainability, Civil and Environmental Engineering, University of Surrey, Stag Hill, Guildford GU2 7XH, UK. E-mail: t.bond@surrey.ac.uk
bDepartment of Chemical and Biological Engineering, University of Sheffield, Western Bank, Sheffield S10 2TN, UK
First published on 19th October 2023
Abstract
The timespans over which different plastics degrade in the environment are poorly understood. This study aimed to rank the degradation speed of five widespread plastic polymers–low density polyethylene (LDPE), polypropylene (PP), polystyrene (PS), polylactic acid (PLA) and polyethylene terephthalate (PET)–in terms of their physicochemical properties. Five of the six samples were plastic films with identical dimensions, which allowed the influence of morphology to be excluded, with a polyethylene carrier bag (PEB) tested for comparison. An accelerated weathering chamber was used to photochemically degrade samples over 41 days, with degradation monitored via mass loss and changes to carbonyl index, crystallinity and contact angle. The mass loss ranking was PP ≫ LDPE > PEB > PS > PLA > PET. Estimates of the time needed for complete degradation ranged from 0.27 years for PP to 1179 years for PET. Therefore, mass loss in PP proceeded more rapidly than the other polymers, which was unexpected based on previous literature and is plausibly explained by the presence of an unlisted additive which accelerated degradation. Increases in carbonyl index proceeded more rapidly in PP and LDPE than the other polymers tested. However, changes in contact angle and crystallinity did not correspond to the mass loss ranking. Therefore, monitoring the carbonyl index during accelerated weathering trials can indicate which polymers will fragment more quickly. However, alternative approaches are needed to simulate conditions where photooxidation reactions are negligible, such as the ocean floor.
Environmental significanceUnderstanding how quickly different plastics degrade across various environmental compartments is essential to understanding the risks posed by plastic litter. Photodegradation is typically the most important degradation pathway in the presence of sunlight and is often simulated using accelerating weathering trials. This study investigated links between mass loss and physicochemical properties during accelerated weathering of widespread plastic polymers. Mass loss and increases in carbonyl index proceeded more rapidly in polypropylene (PP) and low-density polyethylene (LDPE) than other polymers tested. However, contact angle and crystallinity did not relate to mass loss. Therefore, monitoring the carbonyl index can indicate which plastics will fragment more quickly. However, alternative approaches are needed to simulate conditions where sunlight is negligible, such as the ocean floor. |
1. Introduction
Plastics have considerable societal benefits,1 as they are low-cost, easily formable, durable and many are bioinert. They are used across a wide range of sectors, though the highest demand (40.5% in 2020 in the EU27+3 (ref. 2)) relates to packaging.The global annual demand for plastics has reached 367 Mt annually,2 while estimates of mismanaged global plastic waste which accumulated in the environment in 2015 ranged from 60–99 Mt.3
The longevity of plastics poses an environmental risk: most of the plastics ever been produced remain in the environment in one form or another.4 Environmental plastics are eventually fragmented into smaller pieces,5–8i.e., microplastics and nanoplastics, which are typically defined as small pieces of plastic debris in the size range from 25 μm to 5 mm and from 1 to 1000 nm respectively.9 Microplastics and nanoplastics can be transported over large distances in freshwater, the marine environment, and atmosphere and have been detected in some of the most remote regions of Earth.8,10 Since they are bioavailable to a larger range of biota, including humans,11 across multiple trophic levels, there is concern about the toxicological risk they pose to environmental and public health.12 Understanding the environmental degradation of plastics is essential to understanding the risks posed by plastic litter, and how this varies depending on the polymer in question. Environmental degradation can release hazardous chemicals added to plastics, such as flame-retardants, stabilizers or plasticizers,13,14 and enhance the sorption of hazardous pollutants such as polycyclic aromatic hydrocarbons (PAHs), e.g., Udenby et al.15
Several weathering processes affect environmental plastics: biodegradation, photodegradation, thermo-oxidative degradation, thermal degradation, hydrolysis16 and mechanical degradation (abrasion). Polymer degradation can be defined as depolymerisation, overall mass loss, or complete mineralization to CO2 and H2O and involves both physical (related to the bulk structure) and chemical (molecular level) changes.6 Environmental plastics will be exposed to one or more degradation mechanisms,17 depending on the relevant environmental compartment. While literature is dominated by plastic litter in the marine environment, a recent study estimated that the amount accumulated in 2015 in soil was actually slightly higher than in the ocean, with urban soils accounting for 33% of modelled total environmental plastic.10 Photodegradation is typically the most important pathway, at least in the presence of sunlight.5,18–21 It acts to weaken and embrittle plastic litter and typically precedes and fragmentation by mechanical forces, e.g., abrasion with sand or rocks, wave action and swelling–deswelling, and eventually biodegradation.5,7,8,22 Conversely, in certain environmental compartments, for example, subtropical saltmarshes where biofilm formation limits transmittance of sunlight, biodegradation and mechanical abrasion can be more prevalent than photodegradation.23
The mechanism and speed of photodegradation depends on the polymer dimensions and other physicochemical properties, as well as the presence of additives.5,6,8 Polyethylene (PE) is relatively resistant to photodegradation due to a lack of chromophores in its polymer backbone (Table 1), though impurities or defects may act as chromophores to initiate photodegradation.7,18 Various types of PE are in widespread use, differentiated by their density;8 combined they represented 30.3% of polymer demand by resin type in the EU27 + 3 in 2020.2 Polypropylene (PP, 19.7% of polymer demand by resin type (Table 1)2) follows a similar degradation pathway to PE, involving free radicals and chain scission.18 Polystyrene (PS, 6.1% of polymer demand for PS and expanded PS combined (Table 1)2) has phenyl rings, which are susceptible to photodegradation (Table 1) but not biodegradation.7,20 Polyethylene terephthalate (PET, 8.4% of polymer demand (Table 1)2) contains ester bonds which are susceptible to cleavage during photodegradation and hydrolysis in the natural environment.18,24 Polylactic acid (PLA) is a bioplastic, i.e. one produced from renewable biomass substrate, rather than petroleum, which is becoming increasingly popular due to its ability to degrade under industrial composting conditions.25 It exhibits similar degradation behaviour to PET.6
| Sample code | Material | EU plastic demand2 | Chemical structure |
|---|---|---|---|
| a For LDPE and LLDPE combined. b For PS and expanded PS combined. | |||
| LDPE | Low density polyethylene | 17.4%a |
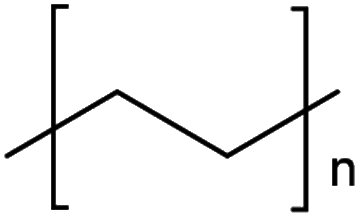
|
| PEB | Polyethylene carrier bag | — | As above |
| PP | Polypropylene | 19.7% |
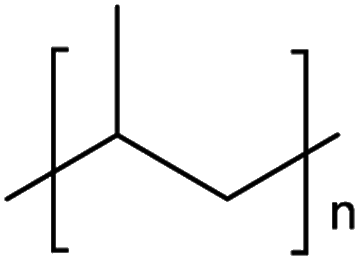
|
| PS | Polystyrene | 6.1%b |
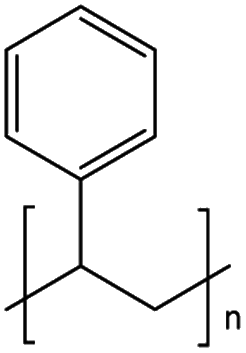
|
| PET | Polyethylene terephthalate | 8.4% |
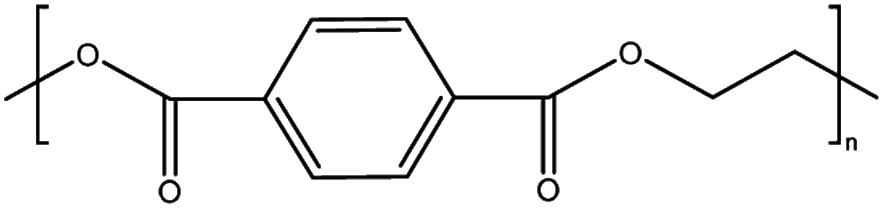
|
| PLA | Polylactic acid | — |
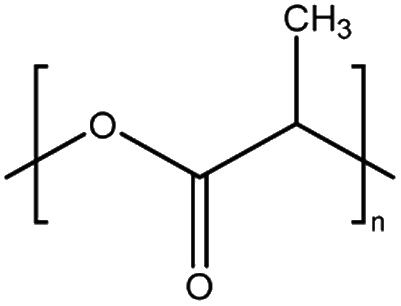
|
While degradation mechanisms for commonly-used polymers (such as those in Table 1) are well-described, e.g., ref. 7 the timespans over which degradation takes place are poorly understood and unpredictable.6,26 Thus, many unknowns remain around the speed of degradation and persistence of plastic debris in different environmental compartments.5 Min et al.26 used a theoretical approach to predict the degradation of a range of polymers in the marine environment, which highlighted the importance of molecular properties including crystallinity and hydrophobicity. Chamas et al.6 highlighted the dependence of degradation speeds on plastic shape. Since degradation is essentially a surface phenomenon, the rate of mass loss is typically proportional to the surface area of the plastic particle.6 Thus, a HDPE film was predicted to degrade 260 times faster than a fibre of the same mass and crystallinity.6
Because environmental plastic degradation is highly uncertain, potentially occurring over decades, centuries or even millennia, accelerated weathering trials are often used to deliver realistic experimental timeframes, e.g., ref. 24 such methodologies combine exposure to UV radiation, heat and moisture under more extreme conditions than experienced in the environment and were primarily developed to assess material suitability for outdoor applications.27 This work aimed to resolve some of the uncertainties surrounding how long plastics degrade under accelerated weathering conditions. The degradation speeds of six widespread plastics were ranked by monitoring polymer physicochemical properties. An accelerated weathering chamber was used to photochemically degrade samples in this presence of humidity, i.e., under conditions representative of the terrestrial environment. Five of the samples tested were plastic films of identical dimensions, which allowed us to exclude the influence of morphology on degradation speed. This approach was selected to allow us to test the hypothesis that degradation speed, at least under controlled conditions, is linked to polymer physicochemical properties.
2. Methods
2.1. Materials
Polymers were purchased from Goodfellow (United Kingdom) as 0.05 mm thick films, except for the polyethylene carrier bag (PEB), which was purchased from a UK supermarket (Sainsbury's) (Table 1). The samples prepared from plastic films had identical dimensions, which allowed us to exclude the influence of morphology on degradation speed. Together, the resins from which these films were produced account for over half of EU plastics demand2 (Table 1) and they comprise what are typically the four commonest polymers on shorelines and in surface waters: PE, PP, PET and PS.8 Smaller size fractions of environmental plastics (<1 mm) are more difficult to isolate and analyse than larger particles, but their abundance follows a power-law increase with decreasing particle size.28 For comparison, PLA and PEB and samples were also tested. Based on EU plastics demand for resin types, polyvinyl chloride (PVC) and polyurethane are respectively the third and fifth most abundant polymers, however, they were not selected as they are scarce in environmental samples.8 All the films were ordered on the understanding that they did not contain additives, as none were listed in material safety data sheets, with the intention of investigating the impact of physicochemical properties on degradation speed. A deep understanding of the accelerated weathering of commonly-used polymer samples of similar dimensions without additives should arguably be a prerequisite to studying degradation in more complex systems (i.e., samples of variable dimensions with additives present exposed to representative environmental conditions).2.2. Accelerated weathering experiments
Before handling plastic samples, disposable nitrile gloves and a cotton lab coat were put on and work surfaces were cleaned with an ethanol solution. Plastic samples were handled as little as possible, and only with metal tongs or scissors. Samples were cut into rectangles with an exposure area of 7.5 × 15 cm2 before being put into the weathering chamber. After being removed from the weathering chamber samples were stored for 24 h in carboard boxes before being analysed. Accelerated weathering was conducted in a QUV tester (Q-Lab, USA), the spectral irradiation of which was calibrated by the manufacturer shortly before the weathering trial commenced. Samples were exposed to repeated cycles of eight hours' UVA irradiance (0.76 W m−2 and UVA-340 nm, which simulates sunlight in the critical short wavelength region from 365 nm down to the solar cutoff of 295 nm, with a peak emission at 340 nm) at 60 °C, followed by four hours' condensation, with humidity created by an open water bath, following Cycle K of ISO 4892-3 (ref. 29) (Table ESI-1 of the ESI†), which specifies standard methods for simulating the weathering of materials are exposed to solar radiation. Separate samples were collected for characterization nine times over a period of 41 days (Table ESI-2†). The individual sample taken for each plastic type at each timepoint was not returned to the weathering chamber. Sample masses were based on three measurements in the 24 h period after removal from the weathering chamber, after a preliminary trial demonstrated that masses of triplicate samples did not vary by more than repeated measurements of the same sample. For differential scanning calorimetry (DSC) and contact angle measurements, three different locations or subsamples were analysed from within the whole sample, and four locations were analysed using Fourier-transform infrared (FTIR) spectroscopy. Results given in the paper are mean values of these replicate measurements, with error values based on their standard deviation. Mass loss during weathering was recorded at each time interval, as follows: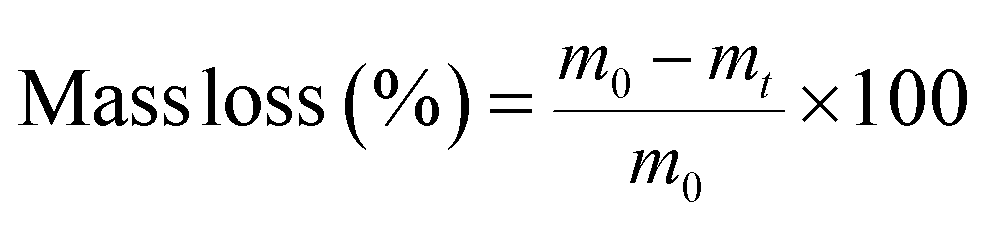 | (1) |
2.3. Characterization
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) peak between 1850 and 1650 cm−1 and the methylene (CH2) peak between 1500 and 1420 cm−1:32
O) peak between 1850 and 1650 cm−1 and the methylene (CH2) peak between 1500 and 1420 cm−1:32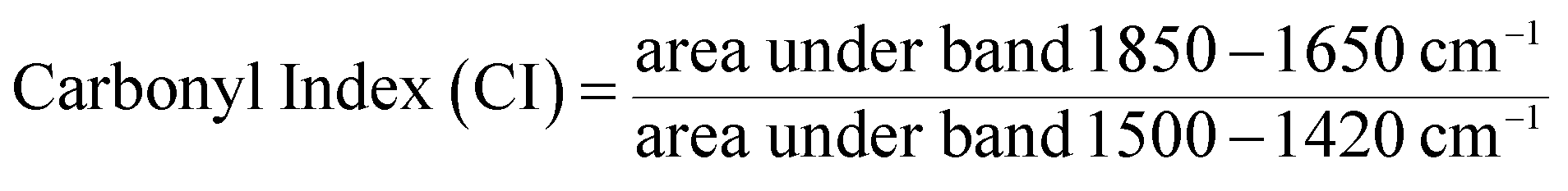 | (2) |
Peak areas were calculated using SpectraGryph 1.2 spectroscopy software.
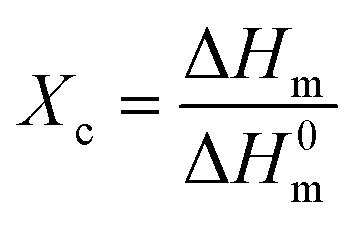 | (3) |
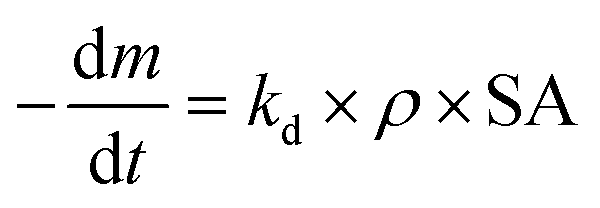 | (4) |
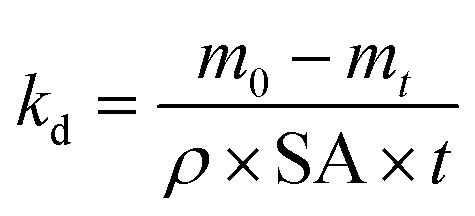 | (5) |
Chamas and co-workers subsequently derived an expression for the time needed for complete degradation of the initial polymer piece (td) for a single specific surface degradation rate, assuming constant density and surface area.6 Degradation in this context refers to overall mass loss from the initial polymer piece, which does not necessarily change the total amount of plastic present, as mineralisation is not an implied end result.6
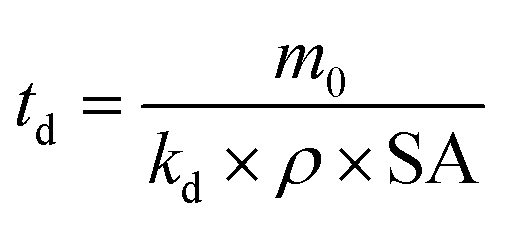 | (6) |
Eqn (6) was modified in the current work to account for the two-phase degradation observed experimentally during the study, which comprised of an initialisation and an acceleration phase:
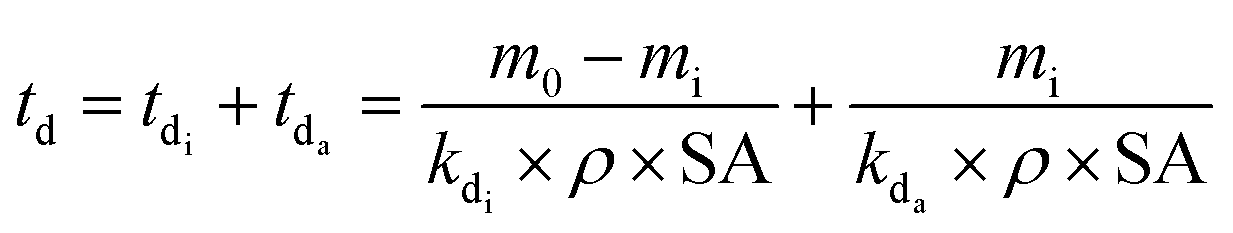 | (7) |
3. Results
3.1. Mass loss and bulk polymer degradation
In this study, mass loss was defined as overall mass loss from the initial polymer piece, as calculated by the mass difference between day 0 and the corresponding time interval (Fig. 1a). The mass loss of PP film was more noticeable than for the other plastics, especially after 16 days of accelerated weathering, reaching a mass loss of 36.8% after 41 days. During the first 15 days of weathering the mass loss of all polymers, including PP, was negligible (Fig. 1a). LDPE showed the second highest mass loss during the weathering trial, 5.8%, while PEB and PS lost respectively 3.7 and 1.1% of their mass (Fig. 1a). The mass loss of the PLA and PET films was less than the other polymers, reaching only 0.14% and 0.01%, respectively after 41 days of accelerated weathering. Thus, the overall order of mass loss was PP ≫ LDPE > PEB > PS > PLA > PET.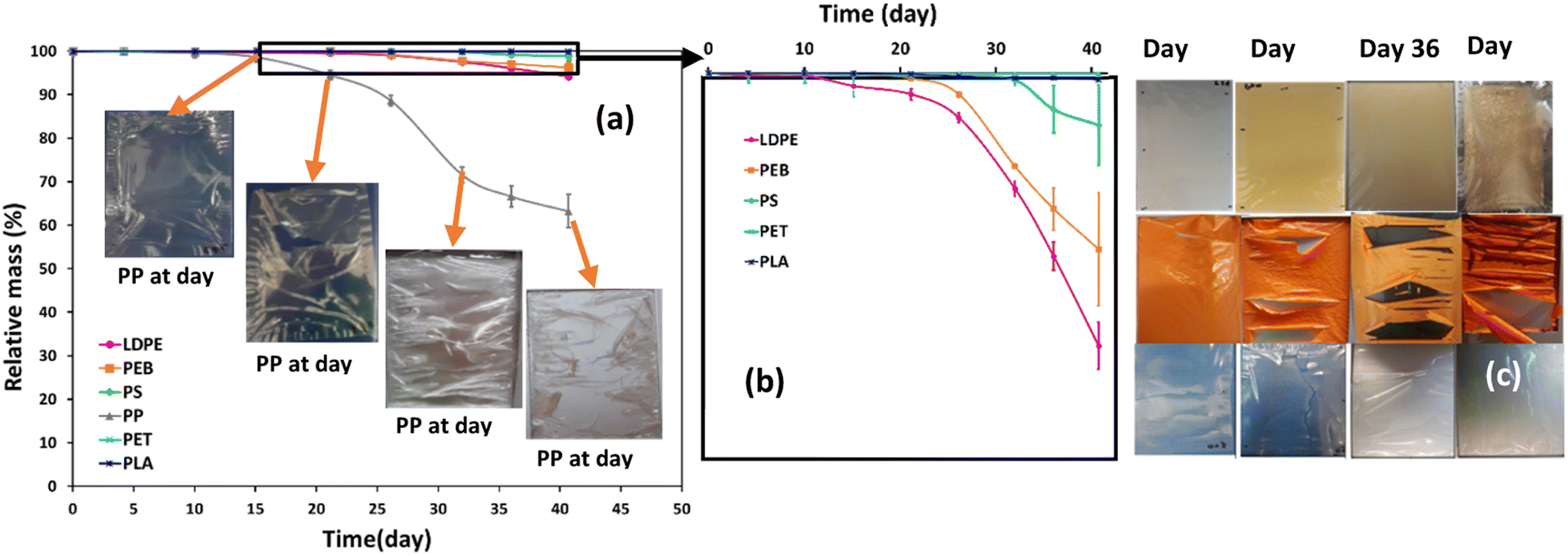 | ||
| Fig. 1 Temporal mass loss for six selected polymers during accelerated weathering. Average quantities ± standard deviations were obtained by weighing each sample three times 24 h after removal from the weathering chamber (n = 3). The photos in (a) show PP at selected time intervals, while (b) magnifies data for all polymers except PP. The top, middle and bottom rows of photos in (c) show PS, PEB and LDPE respectively. | ||
Photos of weathered samples revealed subtle physical differences caused by degradation. After 32 days of weathering, the PP and PEB samples, which experienced higher mass loss than the other polymers, exhibited an obvious curl, while there was a slight curl in the LDPE film surface (Fig. 1c). Physical changes in PS film related to changing colour and brittleness (Fig. 1c). Discoloration had started by 16 days of weathering and then rapidly increased over time. It is established that PS exhibits yellowing during photodegradation, caused by the formation of conjugated double bonds40,41 and is considered more susceptible to weathering than PP or PE in the presence of UV-irradiation.7 After 22 days of exposure, obvious cracks and imperfections appeared on the PP film surface, and small cracks on the PEB film surface, whereas the surface of the PE film was little changed. These can be ascribed to the enhanced diffusion of water out from the bulk phase of the PP and PEB films, which generated increased surface area, subsequently leading to enhanced oxidative degradation.42,43
3.2. Crystallinity
Crystallinity increased over time for all plastics, except PET (Fig. 2a). Data variability, based on the standard deviation of triplicate measurements, was ≤4% in all cases. The highest increases were observed for LDPE and PEB, in both cases 16% over 41 days (Fig. 2a). For PEB there was a particularly marked increase in crystallinity, from 37 ± 2% to 42 ± 1% between days 10 and 15 and by day 21 cracks were beginning to form (Fig. 2b). Overall increases for PP and PLA between the start and end of the trial were 10% and 4%, respectively (Fig. 2a). PET is resistant to thermal oxidation, despite the presence of bonds involving heteroatoms that are readily hydrolysed,44,45 which translates into uniform crystallinity over the weathering trial. Crystallinity fluctuated between 37 ± 2% and 39 ± 3% over the trial, with no overall crystallinity change after 41 days (Fig. 2a).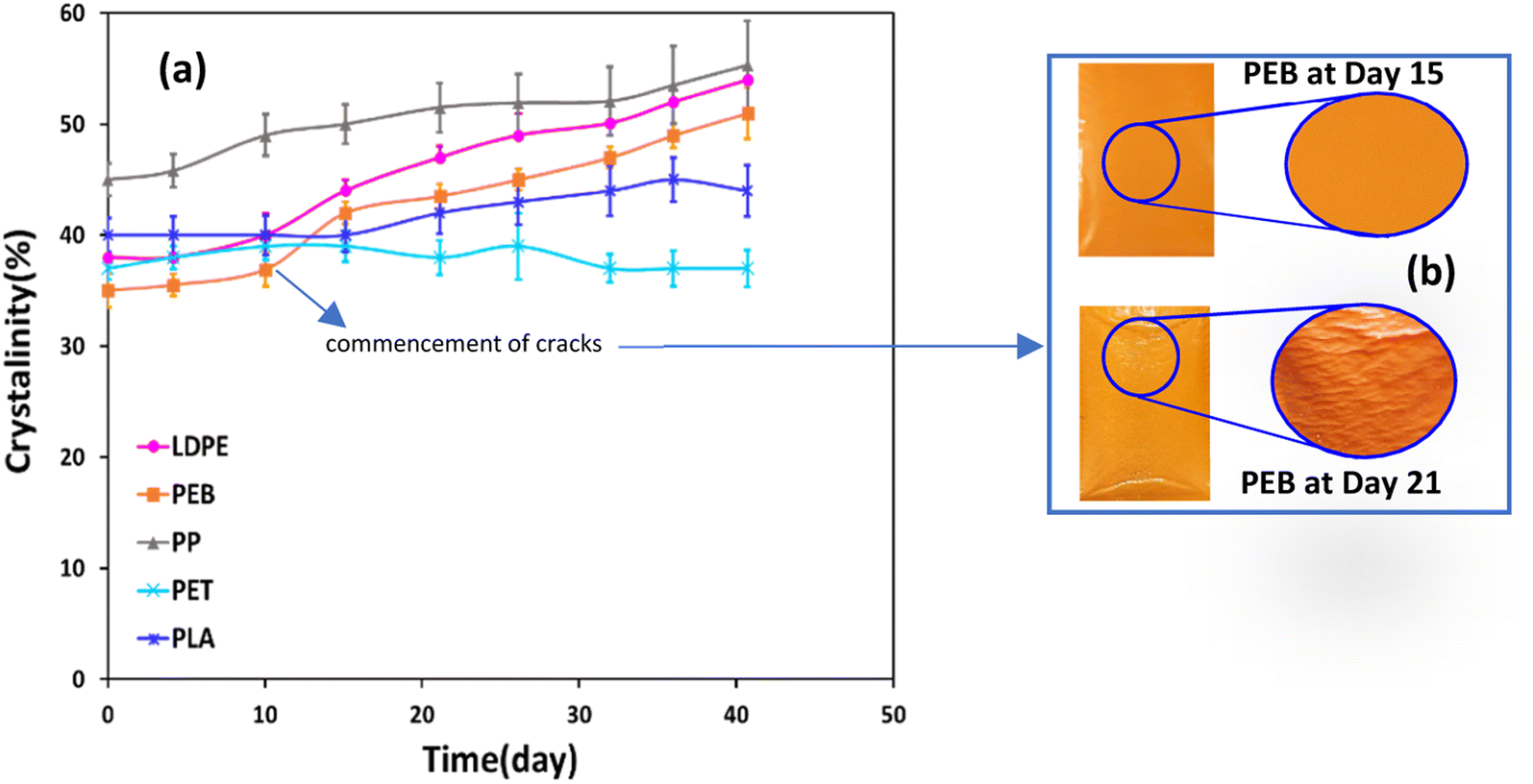 | ||
| Fig. 2 Crystallinity of selected plastics during accelerated weathering (a) and photos showing PEB after days 15 and 21 (b). DSC parameters are expressed as average quantities ± standard deviations from analysing three different subsamples for each sample (n = 3). | ||
Conversely, the higher increases in crystallinity observed for the LDPE, PEB and PP can be attributed to degradation of the amorphous polymer zones.6,31,46 Plastics such as PE and PP are semi-crystalline materials comprised of micro-scale hard crystallites embedded in a soft amorphous matrix.46 Photo-oxidation takes place almost exclusively in the amorphous zone of plastic polymers.5 On the basis of this, it was expected that PP, which had the highest initial crystallinity of all samples tested of 45 ± 2% (Fig. 2a), would be less degradable, whereas the converse was the case, at least in terms of mass loss. In heavily degraded samples, polymer chain scission and crosslinking reactions are more pronounced47,48 eventually leading to increased brittleness and surface cracking (Fig. 1c).46,49 For instance, for PEB, cracks were observable after 21 days' weathering (Fig. 2b) following a pronounced increase in crystallinity, from 37 ± 2% to 42 ± 1%, between days 10 and 15 of exposure. Regarding PS, over the experimental temperature range (from −10 to 180 °C), the glass transition temperature was observed at ∼103 °C, but there was no evidence of melting (Fig. ESI-2†). Therefore, we were not being able to measure crystallinity for PS samples, suggesting a largely amorphous structure. The glass transition temperature of PS decreased by 4.5 °C after day 26 of weathering (Fig. ESI-2†). Such a decrease is consistent with decreased polymer chain length.6,50 From 32 to 41 days of weathering the glass transition temperature increased by 2.5 °C (Fig. ESI-2†), meaning that after 41 days of exposure, the glass transition temperature had overall decreased by 2 °C, indicative of degradation due to chemical and/or thermal oxidation. DSC plots for the remaining polymers are also shown in Fig. ESI-3† LDPE, PEB, PP and PLA all showed strong positive linear correlations (r ≥ 0.81, see Table ESI-3†) between crystallinity and mass loss.
3.3. Contact angle
For all samples, contact angle decreased with weathering, due to increased hydrophilicity of the polymer surface (Fig. 3a). The most pronounced reductions in contact angle after 41 days of accelerated weathering were observed for PS, PP, PEB and LDPE: 39 ± 4%, 36 ± 9%, 34 ± 4% and 24 ± 3% relative to initial contact angle values, respectively (Fig. 3a). Hydrophilic surfaces have higher surface energies and wettability, causing lower contact angles than for more hydrophobic surfaces.51,52 This can be ascribed to the formation of polar functional groups, such as –OH and CO, due to oxidative degradation reactions,7 as illustrated for PP in Fig. 3c. Thus, in general it is expected that polymer weathering results in decreased contact angles, e.g., ref. 52 photos displaying contact angle droplets after 0, 21 and 41 days of the trial are shown in Fig. 3b for PP and for the other polymers in Fig. ESI-4† initial, i.e., before weathering, contact angles for PET and PLA were 71° and 74° (Fig. 3a). Conversely, contact angles for the other polymers were higher and ranged from 94–101°. The structures of PET and PLA already contain carbonyl groups which means they have more hydrophilic surfaces than the other plastics tested (Table 1). LDPE, PEB, PP and PLA all showed strong negative linear correlations (r ≤ −0.82, Table ESI-3†) between contact angle and mass loss. Relative reductions in contact angle for PLA and PET were respectively 12 ± 3% and 8 ± 2%, less than for the other polymers. This pattern follows the same trend as their mass loss relative to the other polymers, re-emphasising their slower degradation and overall similar chemical behaviour. The degradation of PET under landfill/compost/soil conditions was estimated to be unmeasurably slow,6 while the degradation of both PET and PLA in seawater is slower than expected on the basis of their chemical functionality.26 Further, PLA cups degraded more slowly than six other types of conventional, bio-based and biodegradable plastics (including HDPE, PS and recycled PET) over 32 weeks in a saltmarsh.53
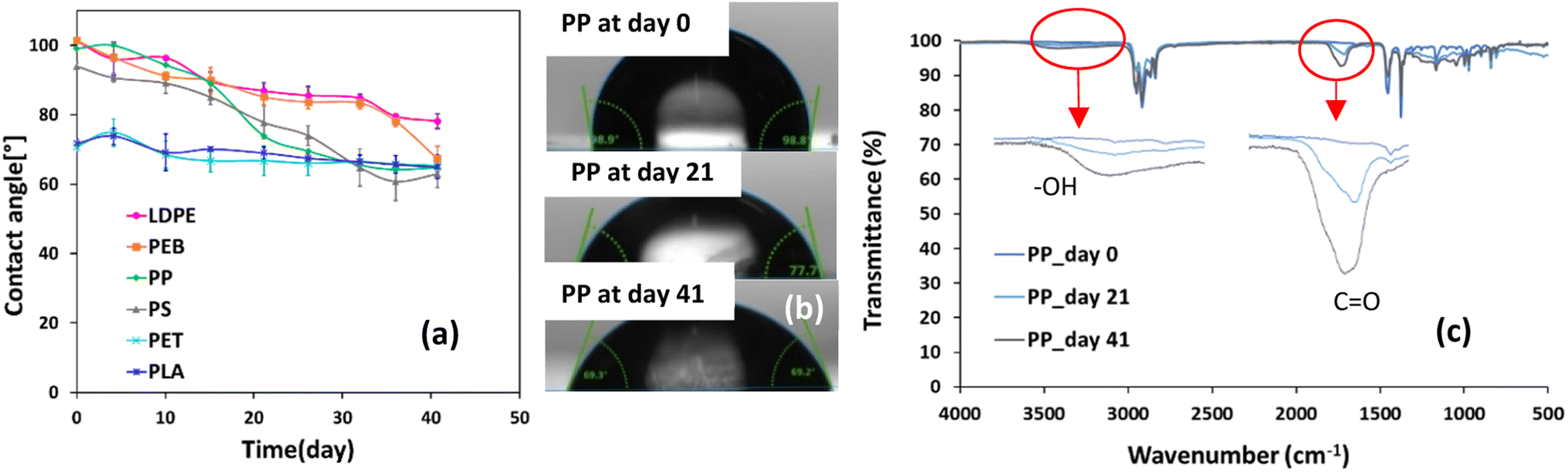 | ||
| Fig. 3 (a) Changes in contact angle over 41 days of accelerated weathering. Contact angle values are expressed as average quantities ± standard deviations from analysing three different subsamples for each sample (n = 3), (b) photos of contact angle droplets produced by PP after days 0, 21 and 41 of the trial and (c) FTIR spectra for PP after the same time periods. | ||
3.4. Carbonyl index
Increased carbonyl index over time was observed for all samples, except PET and PLA (Fig. 4). All polymers except for PS demonstrated strong positive linear correlations (r ≥ 0.8, Table ESI-3†) between carbonyl index and mass loss data. The appearance of carbonyl groups during weathering is indicative of photochemical oxidation and moreover demonstrates the polymers are photolabile and susceptible to further degradation.5,45 For PP, there was a marked increase in carbonyl index between 27 and 36 days of weathering (Fig. 4). LDPE and PEB showed a similar trend, though the increases were less pronounced than for PP. In general, these data agree with previous work describing increases in the carbonyl index during photodegradation of PP and polyethylene.31,32,48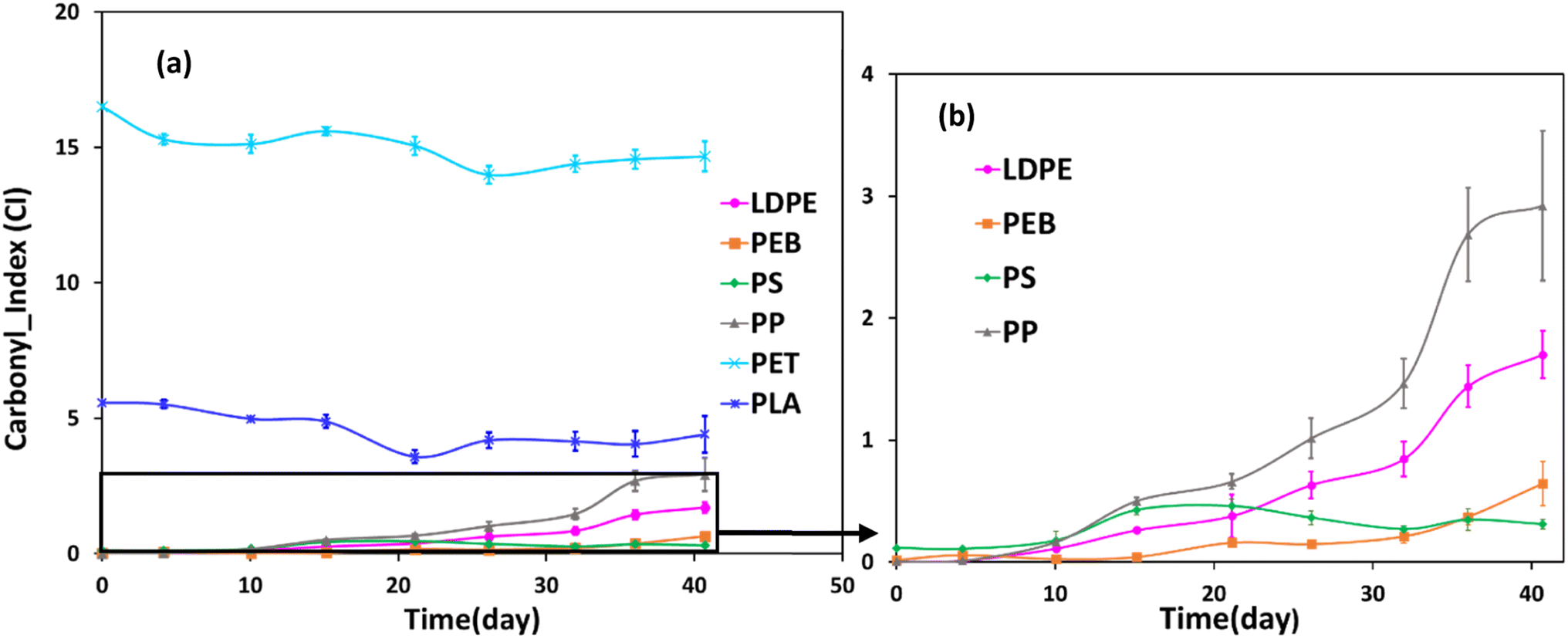 | ||
| Fig. 4 (a) Temporal change in carbonyl index during accelerated weathering for different polymer samples. Carbonyl index values are expressed as average quantities ± standard deviations of three different subsamples for each sample (n = 3). (b) Magnifies the carbonyl index for selected polymers. | ||
Regarding PET and PLA, since the FTIR spectra of unweathered samples already exhibited strong bands in the carbonyl region (Fig. ESI-1†), new bands could not be easily detected because they potentially would overlap with existing peaks. Both polymers showed a slight decrease, followed by slight increase, in carbonyl index over the accelerated weathered trial, with overall no notable change (Fig. 4). Nonetheless, FTIR spectra of PET and PLA showed some changes in both the carbonyl group (1712 cm−1) and hydroxyl group (3500 cm−1), which indicates photochemical oxidation reactions were occurring, since both these functionalities are known to be generated during the oxidative reactions during polymer degradation, e.g., ref. 7 FTIR spectra for the other polymers during the accelerated weathering trial are shown in Fig. 3c and ESI-1.†
4. Discussion: ranking polymer degradation
In this study it was hypothesized that the speed of mass loss, at least for constant surface area, can be explained by polymer physicochemical properties. However, this was found to be only partly the case. PP, followed by LDPE, were the polymers with fastest mass loss and did show a more dramatic increases in carbonyl index than the other polymers tested (Fig. 4). The underlying molecular basis for why these parameters proceeded more rapidly in PP and LDPE than the other polymers tested is obscure. The presence of tertiary carbons in PP makes photochemical degradation more rapid than for PE.7 Furthermore, Song et al.22 reported that less energy is required to break chemical bonds present in PP than PE and expanded polystyrene (EPS). Nonetheless, both PET and polystyrene contain chemical functionalities which absorb UV irradiation (i.e. chromophores), which theoretically should make them more susceptible to photodegradation than either PP or PE.7 These data can be explained by the occurrence of irregularities, impurities or additives in the plastic surface, with unpredictable distribution, and which are not captured by physicochemical characterisation, being key to initiating degradation, as literature indicates, e.g. ref. 7.Changes in contact angle and crystallinity did not correspond to the mass loss ranking. Crystallinity in particular is said to be an important property with respect to polymer degradation.5,6,26 However, PP had the highest initial crystallinity, 45%, of the six samples tested (Fig. 2), which would be expected to reduce degradation speed.
While all films used in the study were purchased on the understanding they were additive free, the supplier subsequently indicated that commercially available PP invariably contains unlisted antioxidant/s designed to prevent degradation during processing (personal communication with Goodfellow), since PP without additives is inherently unstable and will degrade in the presence of air.54 This makes it problematic to obtain additive free PP. The identity of additives present in the PP film was unknown to the supplier, however, phenolic compounds are typically used for this purpose.54 None of the other films contained any UV-stabilisers, colourants or other additives that we are aware of. Thus, the most plausible explanation for the observed mass loss ranking is that the PP sample used contained an unlisted antioxidant additive (see Section 2.1), which acted as a chromophore to initiate and stimulate photochemical weathering. A similar example was recently reported, where Irgafos 168, a widespread phosphite polymer additive which also contains tert-butylphenyl groups55 was found to increase PP degradation by a factor of four after an initialisation phase.56 Similarly, the addition of three low molecular-weight additives – benzophenone, anthraquinone and benzoyl peroxide – accelerated the photodegradation of polystyrene.41 The sample of PP tested in the current study likely contained Irgafos 168 or another additive with a similar impact on degradation.
It is interesting to compare the ranking of polymer mass loss obtained in the current study with relevant literature (Table 2). During accelerated weathering in air then mechanical abrasion by sand, particle release was in the order expanded PS pellet > PP pellet > PE pellet,22 whereas in demineralised water, a PS coffee-cup lid or PLA beverage cup released more particles at two of the three size ranges measured than other plastics studied (PP, PE and PET, Table 2 (ref. 57)). Meanwhile, Chamas et al.6 predicted theoretical mass loss based on 25 references and found that either a LDPE bag or PET water bottle were the fastest degrading plastic, depending on the environmental compartment. Overall, it is notable that there is no consensus in literature regarding the ranking of polymer degradation, even between accelerated weathering studies (e.g., Table 2). However, this is perhaps not surprising given that (i) most other studies do not use samples of standardised dimensions (ii) experimental weathering conditions vary and (iii) additives are typically unknown and can have contradictory effects on degradation speed.
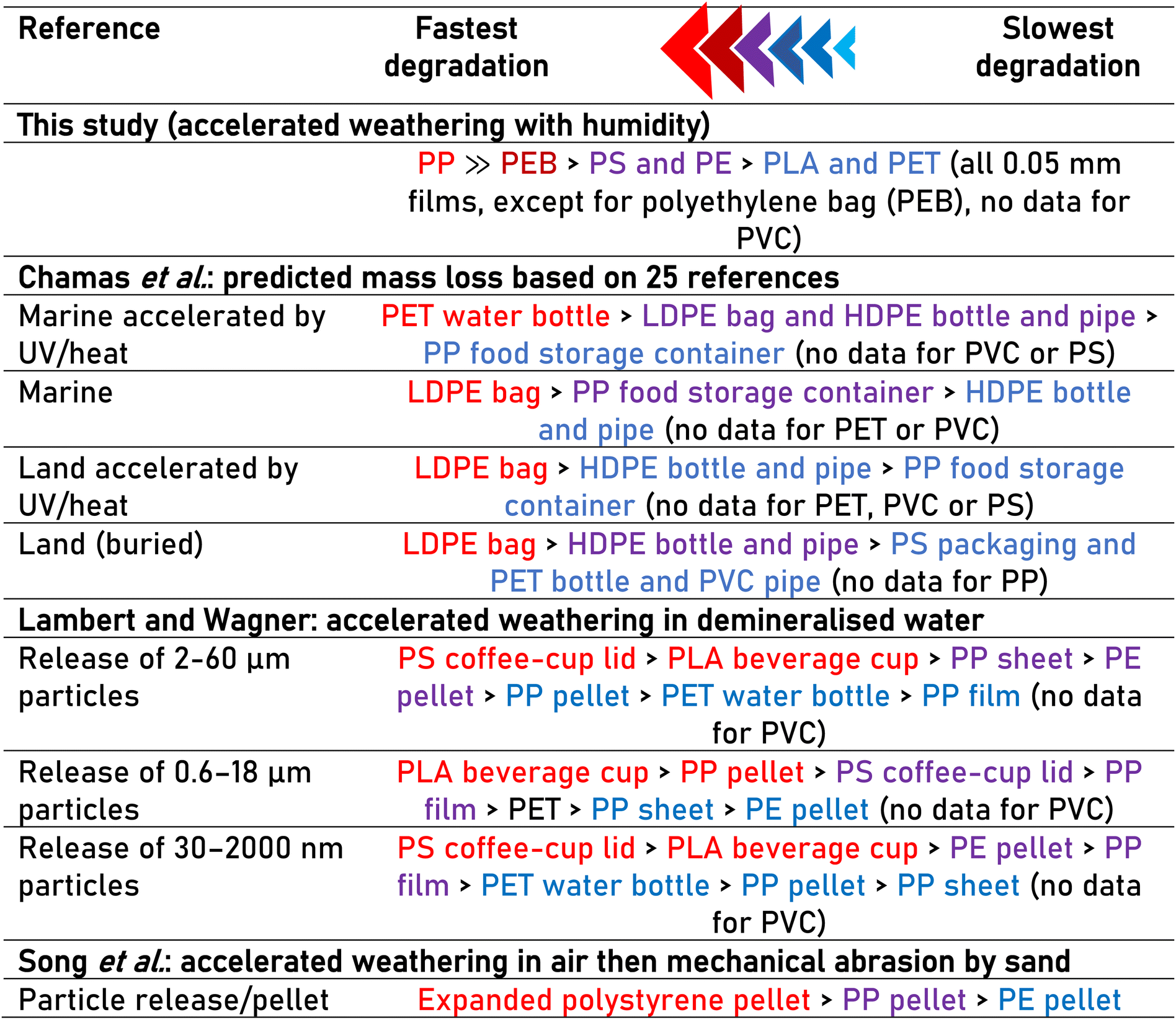
|
The specific surface degradation rate (SSDR or kd) was introduced by Chamas et al.6 as a standardised metric for quantifying the environmental degradation of plastics, which can be used to calculate the time needed for complete degradation (td, see eqn (5)–(7)). In this study SSDR values (Fig. 5) were calculated from experimental mass loss data (eqn (5)). Chamas et al.6 suggested that for thin plastic films, of the type used in this study (0.05 mm thick), surface area and specific surface degradation rate can be regarded as constant during environmental degradation.
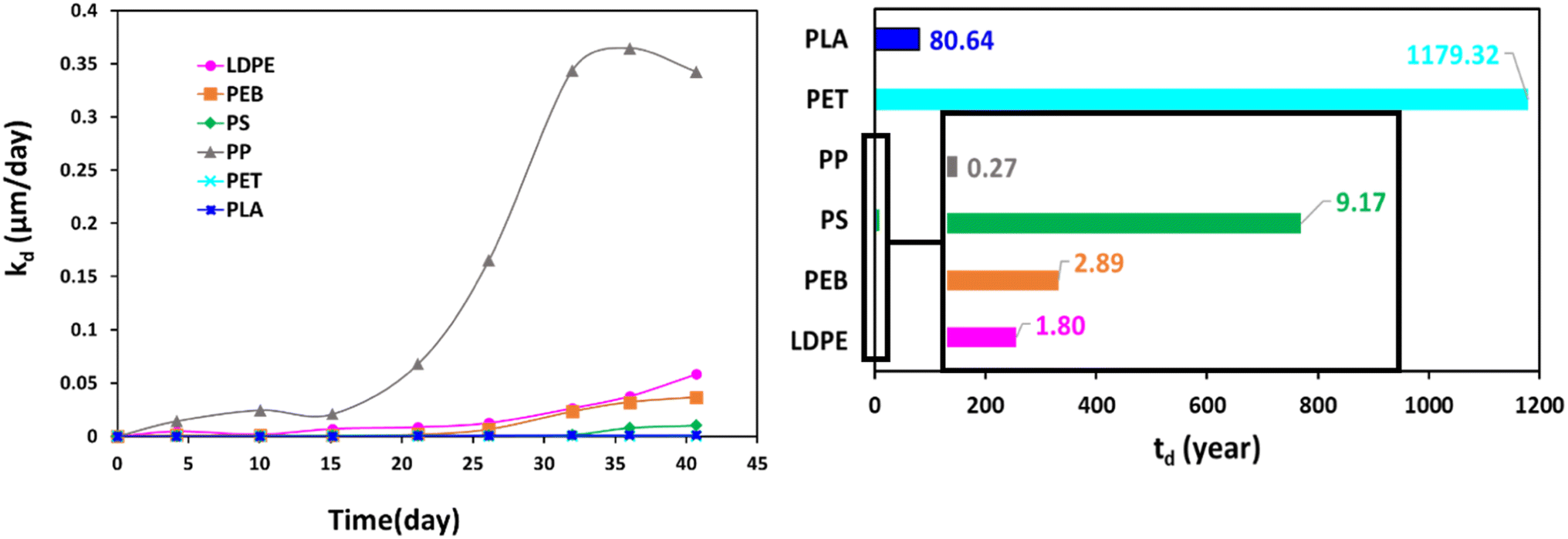 | ||
| Fig. 5 Temporal changes in specific surface degradation rate (SSDR or kd, left) and the time required for total degradation (td, right) for studied polymers. | ||
Since Fig. 1 demonstrates that the slow initialisation phase lasted until 15 days, this value was taken as the start of the acceleration phase, and td values were calculated using the final time interval of the weathering trial, when mass loss of the acceleration phase was essentially linear (eqn (7), Fig. 1).
Fig. 5 shows that specific surface degradation rates were temporally variable over the timespan of the trial, rather than uniform. Moreover, the assumption of constant surface area is not appropriate once samples enter the acceleration phase, with estimates of td values likely to be overestimates. It should also be remembered that accelerated weathering utilises more extreme conditions than typical of real-life, and the degradation reported here is faster than would be experienced by environmental plastics. Thus, calculated td values (Fig. 5) are associated with a relatively high error and are not intended to replicate environmental degradation. Nonetheless, they are helpful for ranking polymer degradation providing and approximate estimates of the time required for complete degradation, which can be refined in future work, for example, by modifying for situations where assumptions of constant surface area and/or density do not apply. Given its dependence on mass loss data, the ranking of td follows (Fig. 5). Values for td ranged from 0.27 years for PP, the fastest degrading polymer, to 1179 years for PET, the slowest degrading polymer (Fig. 5).
In the current study, experimental conditions were eight hours of UV irradiation, followed by four hours of moisture, similar to terrestrial locations with heavy rainfall, e.g., the tropics. However, it is considered unwise to make a quantitative correlation with a real-world scenario without first performing validation using outdoor weathering under relevant conditions.58 Conversely, for submerged plastic particles, such as in sediment or the ocean floor, or those covered in biofilm, photochemical reactions will be negligible and parameters which monitor them (e.g., carbonyl index) are not expected to relate to mass loss. Thus, simulated degradation trials should look beyond accelerated weathering methodologies in such circumstances. For instance, long-term trials under representative environmental conditions, which account for degradation pathways other than photochemical oxidation, e.g., biodegradation. Overall, this study provides baseline information about the accelerated weathering of polymer films under standardised conditions, i.e., samples of the same dimensions and without additives where practicable. More research is required to properly understand the degradation behaviour of commercial plastics containing a representative range of additives, including determining the identity and concentration of additives present. Moreover, the current work illustrates the perils of trying to predict the mass loss of polymers based on polymer physicochemical properties. In most cases the additives present in a plastic product are unknown, as they are unlisted by the manufacturer (and potentially vary between production sites or even batches) and can either accelerate41 or suppress54 polymer degradation. This is likely to make theoretical predictions of environmental polymer degradation inaccurate. Chamas et al.6 admit that extrapolations or predictions of this type “are fraught with uncertainty”. Greater transparency from polymer manufacturers regarding the type and amount of additives present in their products will help to improve the accuracy of future predictions of environmental plastic degradation. A similar point has been made previously about the need for increased transparency regarding chemical identities in polymer regulations.59 In the absence of specific information about the identity of additives present in plastics, experimental weathering trials, rather than theoretical predictions, are arguably required to accurately monitor degradation speed.
5. Conclusions
This study investigated relationships between the mass loss of widespread plastic polymers and their physicochemical properties during accelerated weathering. The key contributions to knowledge are as follows:• The overall order of mass loss was PP ≫ LDPE > PEB > PS > PLA > PET. Using experimental data to extrapolate the time needed for total mass loss from the initial polymer piece provided estimates from 0.27 years for PP to 1179 years for PET under accelerated weathering conditions.
• Mass loss and increases in carbonyl index proceeded more rapidly in PP and LDPE than the other polymers tested. However, changes in contact angle and crystallinity did not correspond to mass loss. As such, the carbonyl index is an indicator of which polymers will experience mass loss more rapidly.
• To the best of our knowledge, none of the films contained additives, except for PP, which contained an unlisted antioxidant additive believed to have accelerated mass loss. Therefore, this study illustrates the perils of making theoretical predictions of mass loss based on polymer physicochemical properties.
Author contributions
Maryam Hoseini: methodology, investigation, writing – original draft, writing – reviewing and editing. Jessica Stead: writing – original draft preparation, writing – reviewing and editing. Tom Bond: conceptualization, supervision, funding acquisition, writing – original draft, writing – reviewing and editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Financial support from the Engineering and Physical Sciences Research Council (EPSRC), as part of project EP/S029427/1 on Predicting the Polymer-specific Fate of Aquatic Plastic Litter is gratefully acknowledged.References
- A. L. Andrady and M. A. Neal, Philos. Trans. R. Soc., B, 2009, 364, 1977–1984 CrossRef CAS PubMed.
- PlasticsEurope, Plastics-the Facts 2021 an Analysis of European Plastics Production, Demand and Waste Data, 2021 Search PubMed.
- L. Lebreton and A. Andrady, Palgrave Commun., 2019,(5), 1–11 CAS.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e170078 Search PubMed.
- A. L. Andrady, P. W. Barnes, J. F. Bornman, T. Gouin, S. Madronich, C. C. White, R. G. Zepp and M. A. K. Jansen, Sci. Total Environ., 2022, 851, 158022 CrossRef CAS PubMed.
- A. Chamas, H. Moon, J. Zheng, Y. Qiu, T. Tabassum, J. H. Jang, M. Abu-Omar, S. L. Scott and S. Suh, ACS Sustain. Chem. Eng., 2020, 8, 3494–3511 CrossRef CAS.
- B. Gewert, M. M. Plassmann and M. Macleod, Environ. Sci.: Processes Impacts, 2015, 17, 1513–1521 RSC.
- T. Bond, V. Ferrandiz-Mas, M. Felipe-Sotelo and E. van Sebille, Crit. Rev. Environ. Sci. Technol., 2018 DOI:10.1080/10643389.2018.1483155.
- J. Gigault, A. ter Halle, M. Baudrimont, P. Y. Pascal, F. Gauffre, T. L. Phi, H. El Hadri, B. Grassl and S. Reynaud, Environ. Pollut., 2018, 235, 1030–1034 CrossRef CAS PubMed.
- M. Hoseini and T. Bond, Environ. Pollut., 2022, 300, 118966 CrossRef CAS PubMed.
- H. A. Leslie, M. J. M. van Velzen, S. H. Brandsma, A. D. Vethaak, J. J. Garcia-Vallejo and M. H. Lamoree, Environ. Int., 2022, 163, 107199 CrossRef CAS PubMed.
- B. Jiang, A. E. Kauffman, L. Li, W. McFee, B. Cai, J. Weinstein, J. R. Lead, S. Chatterjee, G. I. Scott and S. Xiao, Environ. Health Prev. Med., 2020, 251(25), 1–15 Search PubMed.
- B. Gewert, M. Macleod and M. Breitholtz, Biol. Bull., 2021, 240, 191–199 CrossRef CAS PubMed.
- E. Fries and R. Sühring, Environ. Pollut., 2023, 335, 122263 CrossRef CAS PubMed.
- F. A. O. Udenby, H. Almuhtaram, M. J. McKie and R. C. Andrews, Chemosphere, 2022, 307, 135585 CrossRef CAS PubMed.
- A. L. Andrady, Mar. Pollut. Bull., 2011, 62(8), 1596–1605 CrossRef CAS PubMed.
- O. S. Alimi, D. Claveau-Mallet, R. S. Kurusu, M. Lapointe, S. Bayen and N. Tufenkji, J. Hazard. Mater., 2022, 423, 126955 CrossRef CAS PubMed.
- K. Zhang, A. H. Hamidian, A. Tubić, Y. Zhang, J. K. H. Fang, C. Wu and P. K. S. Lam, Environ. Pollut., 2021, 274, 116554 CrossRef CAS PubMed.
- M. Masry, S. Rossignol, J. L. Gardette, S. Therias, P. O. Bussière and P. Wong-Wah-Chung, Mar. Pollut. Bull., 2021, 171, 112701 CrossRef CAS PubMed.
- P. Liu, X. Zhan, X. Wu, J. Li, H. Wang and S. Gao, Chemosphere, 2020, 242, 125193 CrossRef CAS PubMed.
- V. Albergamo, W. Wohlleben and D. L. Plata, Environ. Sci.: Processes Impacts, 2023, 25, 432–444 RSC.
- Y. K. Song, S. H. Hong, M. Jang, G. M. Han, S. W. Jung and W. J. Shim, Environ. Sci. Technol., 2017, 51, 4368–4376 CrossRef CAS PubMed.
- J. E. Weinstein, B. K. Crocker and A. D. Gray, Environ. Toxicol. Chem., 2016, 35, 1632–1640 CrossRef CAS PubMed.
- T. Sang, C. J. Wallis, G. Hill and G. J. P. Britovsek, Eur. Polym. J., 2020, 136, 109873 CrossRef CAS.
- K. Amulya, R. Katakojwala, S. Ramakrishna and S. Venkata Mohan, Compos., Part C: Open Access, 2021, 4, 100111 CAS.
- K. Min, J. D. Cuiffi and R. T. Mathers, Nat. Commun., 2020 DOI:10.1038/s41467-020-14538-z.
- W. Q. Meeker, L. A. Escobar and V. Chan, in Service Life Prediction: Methodology and Metrologies, ACS Symposium Series, 2002, pp. 396–413 Search PubMed.
- G. Erni-Cassola, M. I. Gibson, R. C. Thompson and J. A. Christie-Oleza, Environ. Sci. Technol., 2017, 51, 13641–13648 CrossRef CAS PubMed.
- ISO, ISO 4892-3:2016 Plastics — Methods of Exposure to Laboratory Light Sources — Part 3: Fluorescent UV Lamps, https://www.iso.org/standard/67793.html, accessed 2 March 2023 Search PubMed.
- E. Syranidou, K. Karkanorachaki, D. Barouta, E. Papadaki, D. Moschovas, A. Avgeropoulos and N. Kalogerakis, Environ. Sci. Technol., 2023, 57, 8130–8138 CrossRef CAS PubMed.
- C. Rouillon, P. O. Bussiere, E. Desnoux, S. Collin, C. Vial, S. Therias and J. L. Gardette, Polym. Degrad. Stab., 2016, 128, 200–208 CrossRef CAS.
- J. Almond, P. Sugumaar, M. N. Wenzel, G. Hill and C. Wallis, e-Polym., 2020, 20, 369–381 CrossRef CAS.
- A. Turner and L. Holmes, Mar. Pollut. Bull., 2011, 62, 377–381 CrossRef CAS PubMed.
- C. Badji, J. Beigbeder, H. Garay, A. Bergeret, J. C. Bénézet and V. Desauziers, Polym. Degrad. Stab., 2018, 148, 117–131 CrossRef CAS.
- S. M. Davachi and B. Kaffashi, Int. J. Polym. Mater. Polym. Biomater., 2015, 64, 497–508 CrossRef CAS.
- R. L. Blaine, Determination of Polymer Crystallinity by DSC, TA Instruments, New Castle DE, USA, 2011 Search PubMed.
- C. M. Weikart and H. K. Yasuda, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3028–3042 CrossRef CAS.
- N. W. F. Kossen and P. M. Heertjes, Chem. Eng. Sci., 1965, 20, 593–599 CrossRef CAS.
- A. Diana, M. Castillo, D. Brutin and T. Steinberg, Microgravity Sci. Technol., 2012, 24, 195–202 CrossRef CAS.
- B. G. Rånby and J. F. Rabek, Photodegradation, Photo-oxidation and Photostabilization of Polymers: Principles and Applications, John Wiley & Sons Ltd, 1975, p. 590 Search PubMed.
- H. Kaczmarek, A. Kamińska, M. Światek and S. Sanyal, Eur. Polym. J., 2000, 36, 1167–1173 CrossRef CAS.
- B. P. Chang, A. K. Mohanty and M. Misra, RSC Adv., 2020, 10, 17955–17999 RSC.
- R. S. C. Woo, Y. Chen, H. Zhu, J. Li, J. K. Kim and C. K. Y. Leung, Compos. Sci. Technol., 2007, 67, 3448–3456 CrossRef CAS.
- R. J. Müller, I. Kleeberg and W. D. Deckwer, J. Biotechnol., 2001, 86, 87–95 CrossRef PubMed.
- K. N. Fotopoulou, H. K. Karapanagioti, K. N. Fotopoulou, H. K. Karapanagioti and H. Takada, in Hazardous Chemicals Associated with Plastics in the Marine Environment, 2017, pp. 71–92 Search PubMed.
- A. Beltrán-Sanahuja, N. Casado-Coy, L. Simó-Cabrera and C. Sanz-Lázaro, Environ. Pollut., 2020, 259, 113836 CrossRef PubMed.
- J. Pabiot and J. Verdu, Polym. Eng. Sci., 1981, 21, 32–38 CrossRef CAS.
- S. G. Hirsch, B. Barel, D. Shpasser, E. Segal and O. M. Gazit, Polym. Test., 2017, 64, 194–199 CrossRef CAS.
- Y. C. Hsu, M. P. Weir, R. W. Truss, C. J. Garvey, T. M. Nicholson and P. J. Halley, Polymer, 2012, 53, 2385–2393 CrossRef CAS.
- D. J. Plazek and K. L. Ngai, in Physical Properties of Polymers Handbook, ed. J. E. Mark, Springer, 2007, pp. 187–215 Search PubMed.
- C. G. Jothi Prakash and R. Prasanth, J. Mater. Sci., 2020, 561, 108–135 Search PubMed.
- M. Van Melkebeke, C. Janssen and S. De Meester, Environ. Sci. Technol., 2020, 54, 8668–8680 CrossRef CAS PubMed.
- J. E. Weinstein, J. L. Dekle, R. R. Leads and R. A. Hunter, Mar. Pollut. Bull., 2020, 160, 111518 CrossRef CAS PubMed.
- SpecialChem, Antioxidants Stabilizers Selection for Polyolefins (PP, PE), https://polymer-additives.specialchem.com/selection-guide/antioxidants-stabilizers-selection-for-polyolefins-pp-pe, accessed 20 June 2022 Search PubMed.
- J. N. Hahladakis, C. A. Velis, R. Weber, E. Iacovidou and P. Purnell, J. Hazard. Mater., 2018, 344, 179–199 CrossRef CAS PubMed.
- A. Mauel, N. Meides, T. Menzel, H. Ruckdäschel, P. Strohriegl and J. Senker, in MICRO 2022 Conference, Lanzarote, 2022 Search PubMed.
- S. Lambert and M. Wagner, Chemosphere, 2016, 161, 510–517 CrossRef CAS PubMed.
- M. Crewdson, Outdoor Weathering Must Verify Accelerated Testing, Homestead, FL, USA, 2008 Search PubMed.
- K. J. Groh, H. P. H. Arp, M. MacLeod and Z. Wang, Environ. Sci.: Processes Impacts, 2023, 25, 10–25 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3em00295k |
| This journal is © The Royal Society of Chemistry 2023 |