
Non-thermal plasma assisted non-oxidative methane liquefaction for fuel production at near ambient conditions†
Shijun
Meng
,
Wenping
Li
,
Zhaofei
Li
and
Hua
Song
 *
*
Department of Chemical and Petroleum Engineering, University of Calgary, 2500 University Dr. NW, Calgary, Alberta T2N 1N4, Canada. E-mail: sonh@ucalgary.ca
First published on 6th July 2023
Abstract
The utilization of natural gas, one of the most important energy sources, has a series of limitations because of its inert characteristics. In this study, the non-thermal plasma assisted non-oxidative methane liquefaction process at near ambient conditions (<100 °C and atmospheric pressure) is reported. As the primary component of natural gas, methane has been successfully transformed into gasoline-range chemicals under mild conditions. The engagement of a mesoporous silica catalyst, SBA-15, can enhance the methane conversion, liquid selectivity, and quality of the collected liquid product. Because of the possible generation of micro-discharge within the mesopores, the methane conversion increased from 38.4% to 43.3% over SBA-15. This catalyst could also greatly mitigate the coke formation, where the coke selectivity decreased from 8.0 C% to 2.6 C%. As the main target of this study, the liquid product with high content of desired iso-paraffins was collected. The loading of the SBA-15 enhanced liquid selectivity (increased from 39.0 C% to 45.8 C%) and the quality of liquid product, where less undesired side-products such as aromatics and olefins (total content decreased from 23.6 C% to 9.6 C% in collected liquid) and more paraffins (elevated from 76.5 C% to 90.4 C%) were collected. The effects of the dielectric constant and acidity of the packing material were also investigated. Several 4-hour long-term experiments were performed to learn the deactivation mechanism. It was proved that deactivation was mainly caused by coke generated between the working electrode and the quartz tube. This study demonstrates a promising process for natural gas utilization at mild conditions.
1. Introduction
Nowadays, natural gas is still considered as one of the most fundamentally important energy resources, thanks to its abundant reserve and low price.1,2 The main applications of natural gas include heating, cooking, and power generation. The discovery of substantial shale reserves has sparked researchers' curiosity in transforming natural gas into more valuable liquid fuels or chemicals with high economic value. Unfortunately, the inert characteristics of methane (the primary component of natural gas) due to its highly symmetric molecular structure brings great difficulties to the chemical transformation of natural gas. Traditional thermo-catalytic processes are able to convert methane into syngas,3 C2 hydrocarbons,4 C1–C2 oxygenates (e.g., formaldehyde),5 and aromatics.6 However, even at an elevated temperature (>400 °C), these processes usually end up with low methane conversion,7 while the high reaction temperature may also cause severe coke formation and poor selectivity of desired products.8,9To utilize methane at a mild condition with boosted conversion, several novel techniques have been well-investigated in the last few decades, including plasma-catalysis,10,11 electro-catalysis,12,13 and photo-catalysis.14,15 Out of all these processes, non-thermal plasma (NTP) is considered as the most promising one in consideration of its high methane conversion, and more importantly, it is considered as a promising technique that could be potentially scaled up for industrial applications.16,17 Plasma is composed of multiple energetic species, such as ions, electrons, and free radicals. The collision between methane and these highly energetic species can break the stable structure of methane. Depending on its operating temperature, plasma can be briefly classified as thermal plasma and non-thermal plasma (NTP). Thanks to its non-equilibrium nature, NTP could operate at as low as room temperature while keeping a very high electron temperature in the range of 104 to 105 K.18 Therefore, NTP can trigger thermodynamically unfavorable chemical reactions under mild conditions.
Over the last few decades, significant effort has been made to convert methane and other light hydrocarbons with the assistance of NTP, including methane reforming to produce syngas,19,20 partial oxidation of methane for methanol production,21,22 and non-oxidative activation of methane to higher hydrocarbons.23–30 Unfortunately, to our best knowledge, the majority of studies focused on the production of C2 hydrocarbons, while the directly non-oxidative methane liquefaction has not been well-studied. The NTP-assisted liquefaction of simulated natural gas has been reported previously, where propane was co-fed with methane as the model compound of higher hydrocarbons contained in the natural gas. C6–C9 hydrocarbons were detected in the liquid product, and the synergistic interaction between catalyst and non-thermal plasma on the liquid yield was witnessed.31 However, the addition of propane might be questionable because propane can produce longer radicles to make liquid production easier.
Therefore, we report the NTP-assisted non-oxidative liquefaction of pure methane in this study. Mesoporous silica catalysts have been evaluated, considering that the potential generation of micro-discharge inside of the mesopore might enhance methane activation.32–35 The effect of the dielectric constant and surface acidity of the charged catalyst was also studied. Several long-term experiments were performed to assess the stability of the plasma-catalysis reaction, and the deactivation mechanism has been discussed.
2. Experimental
2.1 Catalyst preparation
The SBA-15 powder was synthesized by hydrothermal method. 27.84 g P123, 496 g H2O, and 36 g concentrated HCl were added into a beaker simultaneously. After P123 was completely dissolved, 60 g TEOS was added dropwise into the beaker. The mixture was stirred at 45 °C for 24 h and then transferred to a Teflon-lined hydrothermal autoclave, where the hydrothermal synthesis was performed at 100 °C for 24 h. The resulting powder was then recovered by centrifugation, followed by being washed with deionized water (DI water) at least 3 times. The resulting paste was dried at 110 °C for 4 h and calcined under air at 550 °C for 4 h in a muffle oven (Vulcan 3-550PD) to obtain SBA-15 powder. The FDU-12 powder was provided by ACS material.HZSM-5 powder was obtained by calcining ammonium ZSM-5 zeolite powder with SiO2/Al2O3 ratio of 23, 80, and 280 (Zeolyst CBV 2314, 8014, and 28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 014) at 600 °C for 5 h under air. K-doped UZSM-5(80) was synthesized by hydrothermal method. Al(NO3)3·9H2O (98%, Alfa Aesar) was dissolved in tetrapropylammonium hydroxide (TPAOH, 1.0 M) and stirred at 360 rpm for 2 h at ambient temperature. The TPAOH provided by Sigma-Aldrich contains 0.49 wt% of potassium. Then, tetraethyl orthosilicate (TEOS, 98%, Sigma-Aldrich) was added dropwise into the mixture. The molar ratio of these chemicals is Al2O3:80SiO2:21TPAOH:943H2O. The gel was obtained after the mixture was kept stirring at room temperature for at least 2 h. The gel was then transferred to a Teflon-lined autoclave and kept at 170 °C for 3 days. The UZSM-5 powder was recovered by centrifugation and washed with DI water at least 3 times. The resulting paste was dried at 110 °C for 12 h, ramped to 300 °C at 5 °C min−1, and then calcined in the air at 600 °C for 6 h. The collected powder was labelled as UZSM-5(80).
014) at 600 °C for 5 h under air. K-doped UZSM-5(80) was synthesized by hydrothermal method. Al(NO3)3·9H2O (98%, Alfa Aesar) was dissolved in tetrapropylammonium hydroxide (TPAOH, 1.0 M) and stirred at 360 rpm for 2 h at ambient temperature. The TPAOH provided by Sigma-Aldrich contains 0.49 wt% of potassium. Then, tetraethyl orthosilicate (TEOS, 98%, Sigma-Aldrich) was added dropwise into the mixture. The molar ratio of these chemicals is Al2O3:80SiO2:21TPAOH:943H2O. The gel was obtained after the mixture was kept stirring at room temperature for at least 2 h. The gel was then transferred to a Teflon-lined autoclave and kept at 170 °C for 3 days. The UZSM-5 powder was recovered by centrifugation and washed with DI water at least 3 times. The resulting paste was dried at 110 °C for 12 h, ramped to 300 °C at 5 °C min−1, and then calcined in the air at 600 °C for 6 h. The collected powder was labelled as UZSM-5(80).
To provide enough void space in the NTP reactor for easing plasma generation, the as-prepared powder catalyst was shaped into pellet form. The powder catalyst (SBA-15, FDU-12, HZSM-5, and UZSM-5) was mixed with LUDOX AM-30 colloidal silica, citric acid, deionized water, and methylcellulose with a certain ratio. Then, the resulting paste was extruded into pellet form using a JW50 extruder (provided by Sino-Green Hi-Tech Co. Ltd). The shaped catalyst was then dried at 110 °C for 4 h and calcined at 550 °C for 4 h under the air. The resulting catalyst was cut into cylindrical pellets with a length of 5–10 mm and a diameter of 2 mm.
2.2 Performance evaluation
As shown in Fig. S1,† a homemade dielectric barrier discharge (DBD) reactor was employed as described in our previous study.36 The DBD reactor was comprised of a quartz tube with an outer and inner diameter of 25.4 and 20.3 mm, respectively. A coaxial inner electrode (as known as the working electrode) made of stainless steel with a diameter of 14.3 mm and a copper coil worked as the outer electrode (as known as the grounding electrode). The outer electrode was finely grounded. The distance between the inner electrode and the inner wall of the quartz tube was 3 mm and the length of the outer electrode was 8 cm, which was also the length of the plasma zone. A G2000 plasma generator manufactured by HVP applied AC high voltage to the inner electrode to generate plasma. The applied power was controlled at 21 W for all experiments, while the applied voltage varied from 12–15 kV (peak-to-peak).The reactions were performed with methane (>99.9%, provided by Praxair) with a flow rate of 10 sccm. Argon (>99.9%, Praxair) was also used to assist plasma generation with a flow rate of 40 sccm. To evaluate the catalytic performance under plasma, ∼13 cm3 of shaped catalyst pellet was packed in the plasma zone, with the quartz wool placed underneath to hold the catalyst bed.
The gas mixture (methane and argon) was introduced into the reactor from the top. Methane was converted when flowing through the plasma zone. After flowed out from the bottom of the reactor, the produced hydrogen and heavier hydrocarbons passed through a liquid trap filled with 10 mL TCE (1,1,2,2-tetrachloroethane), which was placed at the end of the reactor to collect the liquid product. The remaining gas product entered into a connected micro-GC to analyze its composition. The flow rate of the gas product was determined by a soap bubble flow meter. After 40 min reaction, the TCE in the liquid trap was used to rinse the whole reactor and spent catalyst to collect the remaining liquid product. A Gas Chromatography-Mass Spectrometer (GC-MS) was applied to analyze the composition of extracted liquid product. The solid product was collected from the spent working electrode and spent catalyst. The amount of solid product over the spent catalyst was quantified based on TGA analysis.
A four-channel micro-GC (Agilent 490) with thermal conductivity detectors was used to analyze the composition of gas products (H2, CH4, C2H6, C2H4, C2H2, C3H8, C3H6, C4H10, and C4H8). A 10 m molecular sieve 5A column served as the first channel, which can accurately quantify H2 and CH4. C2 hydrocarbons (C2H2, C2H4, and C2H6) were analyzed in a 10 m PPU column that worked as the second channel. C3–C4 species can be analyzed in the third and fourth channels equipped with a 10 m alumina column and an 8 m CP-Sil 5 CB column, respectively. Ar was applied as the carrier gas for the first channel, and He was used for the rest channels. The collected data was used to calculate the composition of the gas species and the methane conversion.
A pre-calibrated Gas Chromatography-Mass Spectrometry (GC-MS, PerkinElmer GC Claus 680 and MS Clarus SQ 8 T) was applied to analyze the composition of the liquid product, and an HP-PONA column provided by Agilent was equipped. The temperature of GC oven was set at 35 °C and hold for 15 min, ramp to 70 °C at 1.5 °C min−1, heat to 150 °C at 3 °C min−1 and hold for 15 min, rise to 250 °C at 5 °C min−1 and hold for 20 min. A split ratio of 20 was used for the GC-MS analysis. The mass detector was set to scan the m/z range from 10 to 400. Identification of the species was performed by comparing the mass spectra obtained with the standard spectra installed in the system's database (NIST). The data collected from GC-MS were used to quantify the amount of each species in the liquid product.
A Simultaneous Thermal Analyzer (PerkinElmer STA 6000) was used to quantitatively analyze the coke over the spent catalyst. The catalyst samples were set at 50 °C and hold for 5 min, then rise to 800 °C at 20 °C min−1 with 50 sccm of air flow. The coke on the spent electrode was removed with sandpaper. The mass difference between clean and spent sandpaper was the mass of coke over the electrode.
A UV/vis spectrometer (AvaSpec-Uls4096CL-EVO) was used to collect optical emission spectra (OES). The slit width of the spectrometer was 10 μm, while the grating was set at 300 lines per mm and the resolution is around 0.6 nm.
2.3 Characterizations
The Micromeritics ASAP 2020 Plus was used to conduct N2 adsorption analysis of catalysts. Before the analysis was performed, the sample was degassed at 350 °C for 4 hours with a temperature ramping rate of 10 °C min−1 and a vacuum level of 10 mmHg. Then, the N2-adsorption test was conducted in liquid nitrogen to collect a 28-point adsorption isotherm. The BET method was used to calculate the total surface area, and the total pore volume was determined at 0.995 relative pressure. The BJH method was used to quantify the pore size distribution.Ammonia-Temperature Programmed Desorption (NH3-TPD) experiments were carried out on the Micromeritics AutoChem II 2920, while ammonia was used as a probe to quantify the concentration of strong and weak acid sites on the catalyst. Prior to measurements, ∼200 mg of fresh sample was pre-treated with pure He at 120 °C for 1 h to remove physically adsorbed moisture over the catalyst surface. Ammonia adsorption was performed using a flow of 10% NH3/He (provided by Praxair) for 30 min at 120 °C with a flow rate of 50 sccm. After flowing He for 30 min at 120 °C to remove any physically adsorbed NH3, temperature-programmed desorption was carried out by ramping to 800 °C at 10 °C min−1 and holding of 3 min under He with the rate of 50 sccm. The amount of desorbed NH3 was determined using a well-calibrated thermal conductivity detector (TCD), and the acquired profiles were fitted and deconvoluted for acid site quantification.
SEM (Scanning Electron Microscopy) analysis was conducted with a Quanta FEG 250 Field Emission SEM system. The images of coke were collected using secondary electrons and a primary electron of 1 keV under high vacuum conditions.
3. Results and discussions
3.1 The effect of mesoporous materials
As shown in Fig. 1, methane can be effectively activated by non-thermal plasma even without the involvement of a catalyst, and activated species have been detected in optical emission spectrum (Fig. S2†). Loading mesoporous silica materials, such as SBA-15 and FDU-12, could improve the methane conversion by the possible generation of micro-discharge within the catalyst mesopores, which have been experimentally and theoretically witnessed in other studies.32–35,37 It is also reported that the pore size of the catalyst can greatly change the intensity of generated micro-discharge. As listed in Table 1, N2-physisorption results confirmed that the mesoporous catalysts tested in this study have an average pore size of around 8 nm, which agreed well with the most ideal pore size (around 10 nm) for micro-discharge generation.35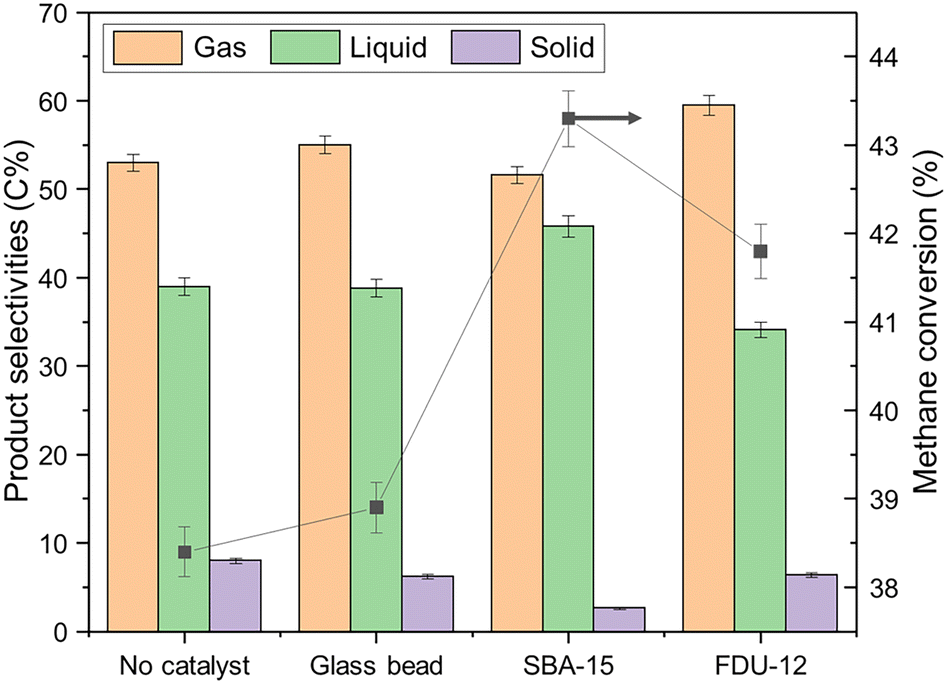 | ||
| Fig. 1 Methane conversion and product selectivities of glass bead, SBA-15, FDU-12, and control runs. | ||
| Material | Surface area (m2 g−1) | Pore volume (cm3 g−1) | Average pore size (nm) |
|---|---|---|---|
| FDU-12 powder | 397 | 0.4 | 8.0 |
| SBA-15 powder | 864 | 1.2 | 7.9 |
| SBA-15 pellet | 344 | 0.5 | 7.5 |
Compared with FDU-12, SBA-15 exhibited a better performance in terms of methane conversion, which was increased from 38.4% to 43.3% when compared with the control experiment. As shown in Table S1,† an energy efficiency of 0.140 mmol CH4 converted per kJ has been achieved over SBA-15, which is relatively high compared with the reported data (0.005–0.145 mmol kJ−1).
For all experiments presented in this study, H2 was the main component in the gas product with a concentration of ∼75 vol%, excluding the unreacted methane. More than half of the carbon in the converted methane was used to synthesize the gas products listed in Fig. 1. In addition to H2, the gas product mainly consisted of C2–C4 paraffins, with a certain amount of olefins, as detailed in Table 2.
| Composition (C%) | No catalyst | Glass bead | SBA-15 |
|---|---|---|---|
| C2H6 | 44.4 | 44.1 | 44.5 |
| C2H4 | 5.0 | 4.8 | 4.2 |
| C2H2 | 7.5 | 7.6 | 6.7 |
| C3H8 | 22.8 | 23.6 | 25.1 |
| C3H6 | 5.6 | 5.5 | 5.3 |
| C4H10 | 12.4 | 12.6 | 12.6 |
| C4H8 | 2.3 | 1.9 | 1.6 |
As mentioned earlier, the production of C2–C4 hydrocarbons from NTP-assisted methane conversion was well-studied. However, to the best of our knowledge, the direct synthesis of liquid products from non-oxidative NTP-assisted methane conversion has not been reported. As listed in Fig. 1 and Table 3, ∼40% of carbon in activated methane was transformed into a liquid product composed of paraffins, olefins, and mono-aromatics, while the rest ended up with coke as an undesired side-product. Other than high methane conversion, SBA-15 also demonstrated outstanding performance in producing a larger amount of liquid product with upgraded quality. Due to the reduction in unfavorable gas and solid product, the packing of the SBA-15 catalyst boosted the liquid selectivity from 39.0 C% to 45.8 C%. In the meanwhile, less problematic aromatics and olefins and more desired C4–C9 paraffins were collected over SBA-15, as detailed in Table 3 and Fig. S3.† The liquid fuel with high iso-paraffin content was regarded as high-quality motor fuel, which can provide better engine-combustion characteristics. More than 90 C% of liquid products collected from the SBA-15 run were paraffins, while more than 80 C% of produced paraffins were iso-paraffins.
| Composition (C%) | No catalyst | Glass bead | SBA-15 |
|---|---|---|---|
| Aromatics | 8.4 | 3.8 | 3.6 |
| Olefins | 15.2 | 13.6 | 6.0 |
| N-paraffins | 9.1 | 7.2 | 15.7 |
| Iso-paraffins | 67.4 | 75.4 | 74.7 |
| Total paraffins | 76.5 | 82.6 | 90.4 |
Coke formation brings a series of problems to plasma catalytic reactions, such as blocking active sites over a catalyst, causing catalyst deactivation, and preventing normal plasma generation. SBA-15 was reported as a promising material with high coke resistance in methane-related catalytic reactions.38 Similar coke control performance was also witnessed in this study. As detailed in Fig. 1, the SBA-15 packing could greatly mitigate coke formation, where the solid selectivity decreased from 8.0 C% to 2.6 C%. By conducting TGA analysis for spent SBA-15 (Fig. S4a†), there is no obvious mass loss peak during high-temperature treatment under air, suggesting the amount of coke over spent SBA-15 was negligible.
3.2 The effect of dielectric constant
The dielectric constant is defined as the ratio of the electric permittivity of a substance to the electric permittivity of a vacuum, which plays an important role in the plasma-catalysis hybrid system.39,40 With high dielectric constant material charged, a higher methane conversion is reported in plasma-assisted dry reforming of methane.41,42 Therefore, to test the effect of the dielectric constant in this study, the packing materials with various dielectric constants were evaluated. In order to eliminate the influence of morphology and porous structure, solid beads with similar diameters and very limited porous structure were selected, including borosilicate glass bead, Al2O3 bead, and ZrO2 bead, with dielectric constants ranging from 6 (glass),43 9 (alumina),44 and 25 (zirconia).45As shown in Table 4, the packing of glass bead exhibited insignificant differences in conversion and selectivities when the empty run (i.e., the test conducted over an empty reactor without packing) was referred, while the experiment loaded with Al2O3 bead produced slightly less liquid with a lightly decreased methane conversion, suggesting that a packing material with a low dielectric constant (<10) can not greatly affect the plasma-catalytic performance. On the other side, as a material with a higher dielectric constant, ZrO2 packing notably improved methane conversion from 38.4% to 44.4%. In the meanwhile, an exaggerated coke generation was recorded, while the solid selectivity was almost doubled when compared to the glass bead run. The possible explanation is, a material with a high dielectric constant can trigger the generation of stronger discharge,46,47 which leads to the enhanced conversion of reactant and the production of over-reacted coke. In general, the trade-off between high methane conversion and desired liquid selectivity (or low coke formation) is inevitable when evaluating materials with varying dielectric constants.
| Catalyst | Dielectric constant | Methane conversion (%) | Selectivities (C%) | ||
|---|---|---|---|---|---|
| Gas | Liquid | Solid | |||
| No catalyst | N/A | 38.4 | 53.0 | 39.0 | 8.0 |
| Glass bead | ∼6 | 38.9 | 55.0 | 38.8 | 6.2 |
| Al2O3 bead | ∼9 | 35.2 | 58.4 | 33.3 | 8.3 |
| ZrO2 bead | ∼25 | 44.4 | 47.7 | 40.2 | 12.1 |
3.3 The effect of catalyst acidity
Because of its adjustable acidity, HZSM-5 was selected as the material to study the effect of catalyst acidity on NTP-assisted methane liquefaction. Therefore, several HZSM-5 based catalysts with different silica-to-alumina ratios (SiO2/Al2O3 = 23, 80, and 280) were evaluated. The surface acidity of these catalysts was quantitively analyzed by NH3-TPD analysis. As shown in Table 5, the HZSM-5 with a lower SiO2/Al2O3 ratio kept a higher density of acidic sites. As detailed in Fig. 2, the loading of HZSM-5 catalysts showed an unnoticeable effect on methane conversion, probably attributing to its smaller pore diameter (0.5–0.6 nm) compared with the aforementioned mesoporous catalysts.48 Unfortunately, the introduction of the catalyst with strong acidity dramatically accelerated the coke formation (Fig. 2 and S4b–d†), while a clear trend between the catalyst acidity and the solid selectivity was witnessed. However, even for the catalyst with the weakest acidity among all tested HZSM-5 catalysts, more coke was produced from HZSM-5(280) run compared with that from the no catalyst control experiment.| Catalysts | The densities of different types of acidic sites (μmol NH3 g−1) | ||
|---|---|---|---|
| Weak (<400 °C) | Strong (>400 °C) | Total | |
| HZSM-5 (23) | 706 | 497 | 1203 |
| HZSM-5 (80) | 247 | 168 | 415 |
| HZSM-5 (280) | 94 | 17 | 111 |
| UZSM-5 (80) | 44 | 0 | 44 |
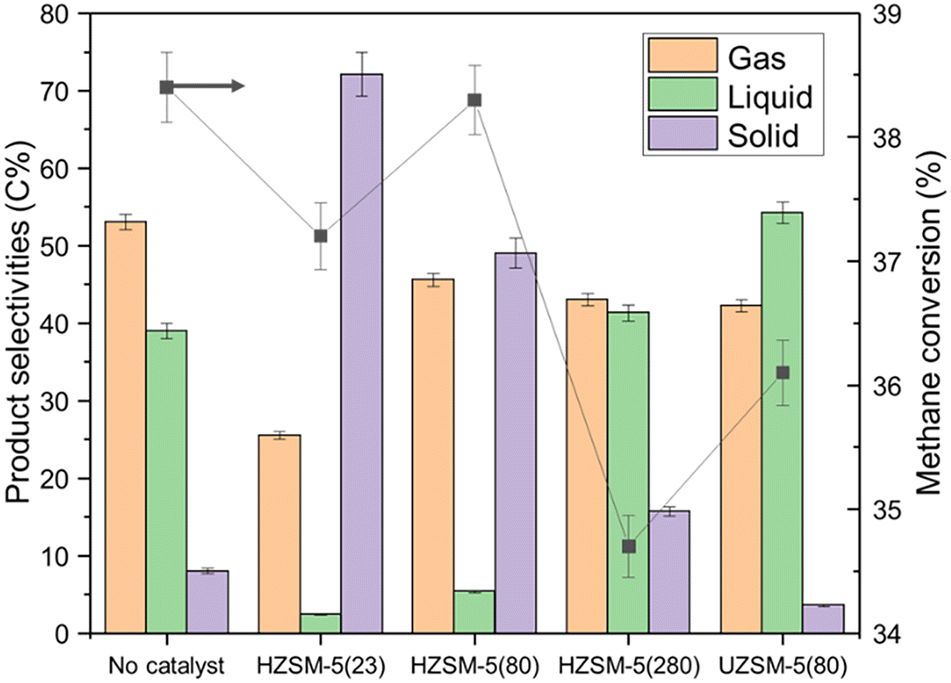 | ||
| Fig. 2 The methane conversion and product selectivities of UZSM-5, and HZSM-5 catalysts with various SiO2/Al2O3 ratios. | ||
To evaluate the performance of the catalyst with even weaker acidity, a K-doped UZSM-5(80) was prepared by hydrothermal synthesis. In our previous study,49 the weaker acidity of UZSM-5(80) was confirmed, and it exhibited excellent performance in controlling coke formation. The acidity properties of UZSM-5(80) were also qualified. As presented in Table 5, UZSM-5(80) exhibited a very low concentration of acidic sites, and the strength of its acidic sites was weaker than other HZSM-5 catalysts. As shown in Fig. 2, compared with no catalyst control experiment, the packing of UZSM-5(80) could decrease solid selectivity from 8.0 C% to 3.6 C%. However, similar to the other ZSM-5 zeolites, UZSM-5 did not improve the methane conversion due to the limited pore diameter. Therefore, SBA-15 is the most promising catalyst among all tested materials, which exhibited high methane conversion and low coke selectivity simultaneously. In the next section, SBA-15 was thus selected as the material to study the deactivation mechanism in prolonged runs.
3.4 Deactivation studies
A stable methane conversion was observed for all previously mentioned experiments during a 40-minute period. To study the stability of NTP-assisted methane liquefaction, several experiments were conducted over SBA-15, which were extended to 4 hours. However, as shown in Fig. S5,† although the methane conversion was stable during the first hour with fresh SBA-15 charged, an obvious decrease in methane conversion was observed subsequently. The picture of the spent reactor was shown in Fig. 3a. The picture clearly showed the generation of coke between the working electrode and quartz tube, which was referred to as “gas gap coke”. This type of coke was initially generated over the surface of the working electrode, which kept growing radially toward the quartz tube. Once the coke bridged the working electrode and the quartz tube, the coke could continuously grow over the inner surface of the quartz tube. The formation of gas gap coke can inhibit the generation of normally homogenous plasma. Similar to carbon nanotube and graphene, carbon materials were regarded as semi-conductor with excellent electrical conductivity.50,51 Therefore, it may work as the extension of the working electrode to reduce the discharge gap.52,53 With the reduced gap, gas gap coke created a shortcut for plasma generation, and the plasma was only generated at the tip of gas gap coke, which greatly weakened the intensity of plasma discharge. The gas gap coke was collected and sent for SEM analysis. As shown in Fig. 3b–e, the collected coke has a complex morphology, which is likely due to its exposure to intense plasma discharge during its growth. | ||
| Fig. 3 Pictures of the coke generated between the electrode and the quartz tube. a) the picture of the spent reactor; b–e) the SEM images of the coke. | ||
To further confirm that the gas gap coke was the primary cause of performance loss, another 4-hour reaction was performed using the spent reactor and reloaded spent SBA-15, while only the gas gap coke was removed. As presented in Fig. S5,† after removing the gas gap coke, the methane conversion can be recovered back to the initial performance where a clean reactor and fresh SBA-15 catalyst were engaged. However, a decrease in conversion was observed again after less than 1 hour of stability, as the generation of gas gap coke was unavoidable with the current experimental setup. In general, the results have confirmed that the generation of gas gap coke was the main reason for performance loss during long-term testing. To prevent its formation, one potential solution is coupling fluidized bed operation with a non-thermal plasma reactor. The generated coke could be collided and destroyed by the fluidized catalyst, and its stable generation might be effectively avoidable.
4. Conclusion
The direct liquid formation from non-oxidative NTP-assisted methane activation was reported. SBA-15 exhibited the best performance regarding methane conversion, liquid selectivity, and the enhanced quality of produced liquid product. Over SBA-15, the liquid product mainly composed of C4–C9 iso-paraffins was collected, which agreed well with the main component of gasoline. The loading of mesoporous silica catalyst can enhance the methane conversion, due to the possible generation of micro-discharge inside of the mesopores. The effect of dielectric constant and catalyst acidity was also studied. The results indicated that the packing materials with low dielectric constant had an insignificant catalytic performance. A higher methane conversion was observed over the material with a higher dielectric constant, while more coke was produced. A clear trend between the solid yield and acidity was witnessed. More coke was collected when a catalyst with stronger acidity was packed. By conducting several extended reactions, it has been confirmed that the formation of gas gap coke was the primary cause of performance loss during long-term testing. Overall, this proof-of-concept study highlights the non-oxidative methane liquefaction for fuel production in an NTP reactor. The cheap natural gas can be directly converted into gasoline range chemicals at mild conditions, which provides a novel process to the current petrochemical industry.Author contributions
Shijun Meng: conceptualization, investigation, writing – original draft, Wenping Li: validation, writing – review & editing, Zhaofei Li: resources, writing – review & editing, Hua Song: conceptualization, supervision, project administration, writing – review & editing.Conflicts of interest
The authors declare no competing financial interests.References
- J. Du, J. Zheng, Y. Liang, B. Wang, J. J. Klemeš, X. Lu, R. Tu, Q. Liao, N. Xu and Y. Xia, Energy, 2023, 263, 125976 CrossRef.
- G.-F. Fa, Z.-Q. Wang, F. Gao, Z.-S. Wang and Z.-Y. Li, Global Natural Gas Production and Development Status, Supply and Demand Situation Analysis and Prospects, Proceedings of the International Field Exploration and Development Conference 2021, 2022, pp. 574–585 Search PubMed.
- F. Sharifianjazi, A. Esmaeilkhanian, L. Bazli, S. Eskandarinezhad, S. Khaksar, P. Shafiee, M. Yusuf, B. Abdullah, P. Salahshour and F. Sadeghi, Int. J. Hydrogen Energy, 2022, 47, 42213–42233 CrossRef CAS.
- A. Maitra, Appl. Catal., A, 1993, 104, 11–59 CrossRef CAS.
- A. De Vekki and S. Marakaev, Russ. J. Appl. Chem., 2009, 82, 521–536 CrossRef.
- X. Guo, G. Fang, G. Li, H. Ma, H. Fan, L. Yu and D. Tan, Science, 2014, 344, 616–619 CrossRef CAS PubMed.
- Y. Li, P. He, Z. Li, H. Xu, J. Jarvis, S. Meng and H. Song, Fuel, 2021, 289, 119864 CrossRef CAS.
- J. J. Spivey and G. Hutchings, Chem. Soc. Rev., 2014, 43, 792–803 RSC.
- U. Zavyalova, M. Holena, R. Schlögl and M. Baerns, ChemCatChem, 2011, 3, 1935–1947 CrossRef CAS.
- P. Chawdhury, S. B. Rawool, M. U. Rao and C. Subrahmanyam, Chem. Eng. Sci., 2022, 258, 117779 CrossRef CAS.
- Y. Diao, X. Zhang, Y. Liu, B. Chen, G. Wu and C. Shi, Appl. Catal., A, 2022, 301, 120779 CrossRef CAS.
- H. Jiang, L. Zhang, Z. Han, Y. Tang, Y. Sun, P. Wan, Y. Chen, M. D. Argyle and M. Fan, Green Energy Environ., 2022, 7, 1132–1142 CrossRef CAS.
- Q. Lu, Y. Hou, S. R. Laraib, O. Khalifa, K. Li, W.-L. Xie, M.-S. Cui and Y.-P. Yang, Fuel Process. Technol., 2019, 192, 57–64 CrossRef CAS.
- X. Cai, S. Fang and Y. H. Hu, J. Mater. Chem. A, 2021, 9, 10796–10802 RSC.
- Y. Zhu, S. Chen, S. Fang, Z. Li, C. Wang and Y. H. Hu, J. Phys. Chem. Lett., 2021, 12, 7459–7465 CrossRef CAS PubMed.
- W.-C. Chung and M.-B. Chang, Renewable Sustainable Energy Rev., 2016, 62, 13–31 CrossRef CAS.
- M. F. Mustafa, Y. Abbas, M. Ajmal, A. Afreen, Z. Ahmed, C. Liu and W. Lu, J. Cleaner Prod., 2022, 379, 134504 CrossRef CAS.
- R. Hippler, H. Kersten, M. Schmidt and K. H. Schoenbach, Low Temperature Plasmas: Fundamentals, Technologies and Techniques, ed. R. Hippler, et al., Wiley, Berlin, 2nd edn, 2008, vol. 787, ISBN: 978-3-527-40673-9 Search PubMed.
- R. Vakili, R. Gholami, C. E. Stere, S. Chansai, H. Chen, S. M. Holmes, Y. Jiao, C. Hardacre and X. Fan, Appl. Catal., A, 2020, 260, 118195 CrossRef CAS.
- X. Gao, Z. Lin, T. Li, L. Huang, J. Zhang, S. Askari, N. Dewangan, A. Jangam and S. Kawi, Catalysts, 2021, 11, 455 CrossRef CAS.
- P. Chawdhury, D. Kumar and C. Subrahmanyam, Chem. Eng. J., 2019, 372, 638–647 CrossRef CAS.
- P. Fathollahi, M. Farahani, R. H. Rad, M. R. Khani, A. Asadi, M. Shafiei and B. Shokri, J. Electrost., 2021, 112, 103594 CrossRef CAS.
- M. Ghanbari, M. Binazadeh, S. Zafarnak, H. Taghvaei and M. R. Rahimpour, Int. J. Hydrogen Energy, 2020, 45, 13899–13910 CrossRef CAS.
- R. Snoeckx, A. Rabinovich, D. Dobrynin, A. Bogaerts and A. Fridman, Plasma Processes Polym., 2017, 14, 1600115 CrossRef.
- W. Cho, Y. C. Kim and S.-S. Kim, J. Ind. Eng. Chem., 2010, 16, 20–26 CrossRef CAS.
- J. Lü and Z. Li, J. Nat. Gas Chem., 2010, 19, 375–379 CrossRef.
- H. Miao and C. Jierong, Plasma Sci. Technol., 2007, 9, 269 CrossRef.
- W. Cho, Y. Baek, Y. C. Kim and M. Anpo, Res. Chem. Intermed., 2002, 28, 343–357 CrossRef CAS.
- W. Cho, Y. Baek, D. Park, Y. C. Kim and M. Anpo, Res. Chem. Intermed., 1998, 24, 55–66 CrossRef CAS.
- B. Wang and G. Xu, Sci. China, Ser. B: Chem., 2002, 45, 299–310 CrossRef CAS.
- S. Meng, A. Wang, P. He and H. Song, J. Phys. Chem. Lett., 2020, 11, 3877–3881 CrossRef CAS PubMed.
- F. Holzer, U. Roland and F. D. Kopinke, Appl. Catal., B, 2002, 38, 163–181 CrossRef CAS.
- U. Roland, F. Holzer and F. D. Kopinke, Appl. Catal., B, 2005, 58, 217–226 CrossRef CAS.
- K. Hensel, S. Katsura and A. Mizuno, IEEE Trans. Plasma Sci., 2005, 33, 574–575 CAS.
- Y. Zhang, H. Wang, Y. Zhang and A. Bogaerts, Plasma Sources Sci. Technol., 2017, 26, 054002 CrossRef.
- A. G. Wang, J. H. Harrhy, S. J. Meng, P. He, L. J. Liu and H. Song, Energy Convers. Manage., 2019, 191, 93–101 CrossRef CAS.
- H. Wang, J. Han, Z. Bo, L. Qin, Y. Wang and F. Yu, Mol. Catal., 2019, 475, 110486 CrossRef CAS.
- B. Erdogan, H. Arbag and N. Yasyerli, Int. J. Hydrogen Energy, 2018, 43, 1396–1405 CrossRef CAS.
- L. I. U. Jin, Z. H. U. Xinbo, H. U. Xueli and T. U. Xin, Plasma Sci. Technol., 2022, 24, 025503 CrossRef.
- H. Song, F. Hu, Y. Peng, K. Li, S. Bai and J. Li, Chem. Eng. J., 2018, 347, 447–454 CrossRef CAS.
- W.-C. Chung, K.-L. Pan, H.-M. Lee and M.-B. Chang, Energy Fuels, 2014, 28, 7621–7631 CrossRef CAS.
- A. H. Khoja, M. Tahir and N. A. S. Amin, Energy Convers. Manage., 2017, 144, 262–274 CrossRef CAS.
- Z. Wang, Y. Hu, H. Lu and F. Yu, J. Non-Cryst. Solids, 2008, 354, 1128–1132 CrossRef CAS.
- K. Z. Rajab, M. Naftaly, E. H. Linfield, J. C. Nino, D. Arenas, D. Tanner, R. Mittra and M. Lanagan, J. Microelectron. Electron. Packag., 2008, 5, 2–7 CrossRef CAS.
- M.-T. Wang, T.-H. Wang and J. Y.-M. Lee, Microelectron. Reliab., 2005, 45, 969–972 CrossRef CAS.
- K. Van Laer and A. Bogaerts, Plasma Sources Sci. Technol., 2017, 26, 085007 CrossRef.
- K. Van Laer and A. Bogaerts, Plasma Processes Polym., 2017, 14, 1600129 CrossRef.
- C. Hu, H. Zhang, S. Wu and R. Xiao, Energy Convers. Manage., 2020, 210, 112678 CrossRef CAS.
- S. Meng, W. Li, H. Xu, Z. Li, Y. Li, J. Jarvis and H. Song, Appl. Catal., A, 2021, 297, 120459 CrossRef CAS.
- M. Raji, N. Zari, A. El Kacem Qaiss and R. Bouhfid, Functionalized graphene nanocomposites and their derivatives, 2019, pp. 245–263 Search PubMed.
- T. W. Ebbesen, H. J. Lezec, H. Hiura, J. W. Bennett, H. F. Ghaemi and T. Thio, Nature, 1996, 382, 54–56 CrossRef CAS.
- A. Jahanmiri, M. R. Rahimpour, M. M. Shirazi, N. Hooshmand and H. Taghvaei, Chem. Eng. J., 2012, 191, 416–425 CrossRef CAS.
- H. Taghvaei, M. M. Shirazi, N. Hooshmand, M. R. Rahimpour and A. Jahanmiri, Appl. Energy, 2012, 98, 3–10 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy00835e |
| This journal is © The Royal Society of Chemistry 2023 |