
An in silico predictive method to select multi-monomer combinations for peptide imprinting†
Soumya
Rajpal
 ab and
Boris
Mizaikoff
*ac
ab and
Boris
Mizaikoff
*ac
aInstitute of Analytical and Bioanalytical Chemistry, Ulm University, Albert-Einstein-Allee 11, 89081 Ulm, Germany. E-mail: boris.mizaikoff@uni-ulm.de
bDepartment of Biochemical Engineering and Biotechnology, Indian Institute of Technology Delhi, New Delhi 110016, India
cHahn-Schickard, Sedanstraße 14, 89077 Ulm, Germany
First published on 3rd May 2022
Abstract
In silico methods enable optimizing artificial receptors such that constructive mimics of natural antibodies can be envisaged. The introduction of combinatorial synthesis strategies via multi-monomer combinations has improved the performance of molecularly imprinted polymers (MIP) significantly. However, it remains experimentally challenging to screen thousands of combinations resulting from a large library of monomers. The present study introduces a molecular mechanics based multi-monomer simultaneous docking approach (MMSD) to computationally screen monomer combinations according to their potential, facilitating selective molecular imprints. Thereby, the diversity of multipoint interactions realizable with a peptide surface is efficiently explored yielding how individual monomer binding capacities constructively or adversely add up when docked together. Additionally, spatially distributed molecular models were mapped for analyzing intermolecular H-bonding and hydrophobic interactions resulting from single monomer docking, as well as bi- and tri-monomer simultaneous docking. A direct impact of complex formation on the binding capacity of the resulting MIPs has been observed. In a first small-scale study, the predictive potential of the MMSD approach was validated via experimentally applied polymer combinations for peptide imprinting via the scoring functions established during the screening process. MMSD clearly enables rational design of MIPs for synthesizing more sensitive and selective artificial receptor materials especially for peptide and protein-epitope templates.
Introduction
Molecular imprinting involves the induction of specific recognition moieties within homogenous polymer matrices. The first step combines one or several functional monomer species with a target template in a pre-polymerization solution. This is followed by a polymerization reaction and subsequent removal of the template, which creates selective binding sites complementary in function – and to a lesser extent in shape and size - to the templating species.1 Decades of research has eventually led to a plethora of molecularly imprinted polymers (MIPs), also termed “artificial receptors” for small molecules, proteins, nucleic acids and even entire cells.2–8The ubiquitous nature of protein-macromolecular interactions is of particular interest and presents a challenge for tailoring high-affinity MIPs. From the biological perspective, there are more than 20 amino acids that uniquely arrange in all possible combinations around a specific epitope to form a strong antigen-antibody complex with affinities in the nanomolar regime. In order to constructively mimic this behavior, the employment of multiple functionalities provided by complementary types of monomers that enable matching to different regions of the template has become a common thread in rational MIP design.9 Furthermore, an epitope imprinting strategy involving certain sequential or conformational epitopes as templates rather than entire proteins closely resembles the biological synthesis and reduces the complexity of MIP-based protein recognition.3,10–16
While genetics encode the specificity in natural antibodies, the specific design of their artificial counterparts can be deciphered and fundamentally understood via computational methods. A combination of quantum mechanics/molecular mechanics, and molecular dynamics may enable modelling of multi-component systems.17–20 Quantum mechanics (QM) are based on ab initio, semi-empirical and/or functional density (DFT) approaches to elucidate the electronic structure of the system in a pre-polymerization mixture.21 The nature and extent of the interactions in this monomer-template complex directly impacts the quality and quantity of MIP recognition sites that are available for re-binding the target species. Subsequent molecular dynamics simulations may then predict the effect of solvent, time of polymerization and other environmental conditions during MIP synthesis.22–25 For example, Molinelli et al. employed AMBER for molecular modelling of non-covalent interactions between 2,4-dichlorophenoxyacetic acid (2,4-D) (template) and a functional monomer 4-vinylpyridine realizable in explicit solvents like chloroform and water.26 The characteristic nature of interactions in the solvent concurred well with experimental spectroscopic studies of pre-polymerization solutions. In more complex systems involving species such as proteins, QM studies are not considered the preferred option due to the time and computational expense involved. Alternatively, molecular mechanics studies allow for docking and faster modelling of the pre-polymerization complex to rationalize the selection of monomers and cross-linkers suitable for the imprinting process.21 Sullivan et al. employed screening of five acrylamide-based monomers using the Glide (Schrodinger) docking tool and studied the binding site-specific interactions to optimize imprinting of myoglobin protein.27 The simulation results established NHMAm as the most suitable monomer that formed MIPs with up to 98.9% rebinding capacity.
Centered around the electrostatic, steric and H-bond enthalpies, molecular mechanics-based programs employ scoring functions that estimate the binding free energy of the monomers within a sampled space.28 Moreover, a molecular interaction map based on the binding energy models aids in recounting the most relevant multipoint interactions of different monomers realized with the target biomolecule, which in turn governs MIP selectivity. Thus, applied scoring functions vary in their nature, i.e., using explicit force fields, H-bond placement and empirical or knowledge-based scoring. As a first step, for the purpose of selecting a suitable program for the rational design of MIPs this study has applied a ‘bottom-up’ approach to validate theoretical studies based on published experimental studies. Herein, we focus on two commonly used force fields - namely Amber and OPLS4 - applied in three software packages - Autodock 4, Vina and Glide (Schrodinger) - respectively for assessing interaction energies from monomer-template combinations employed in the study by Bedwell et al.29
Moving forward, multi-monomer simultaneous docking (MMSD) protocol was then introduced to collectively analyze the type of interactions resulting from multiple monomers in complex with a template in a pre-polymerization mixture. Thus far, molecular docking has only been applied to virtually screen libraries of individual functional monomers for guiding the synthesis of imprinted polymers. In contrast, the potential of molecular mechanics to computationally screen multiple monomer combinations is much less explored and has been used to the best knowledge of the authors for the first time in this context. Encouragingly, the developed MMSD approach can theoretically mimic the results from combinatorial synthesis/screening and thus has to potential to replace the laborious task of experimentally optimizing thousands of polymer combinations.30 Furthermore, the developed routine may also enhance the sampling, ranking and binding specificity of the monomers for minimizing the number of necessary experiments.
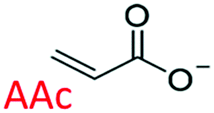
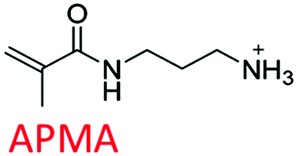
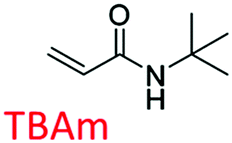
Following these fundamental considerations and by using combinatorial libraries of monomers, first the viability of the protocol in the same bottom-up approach was evaluated using a reference study that deals with the experimental optimization of polymer compositions in the initial approach.29 This study employs N-isopropylacrylamide (NIPAm) as backbone monomer in combination with acrylamide that contributes to H-bonding. Furthermore, acrylic acid (AAc), N-(3-amino-propyl) methacrylamide hydrochloride (APMA), and N-tert-butyl-acrylamide (TBAm), negative-charged, positive-charged, and hydrophobic functional monomers are employed in different molar ratios and combinations for imprinting three different types of peptides. Synchronously, the effect of each of these monomers in single docking and multi-monomer simultaneous docking experiments was evaluated, and the type of interactions in a pre-polymerization mixture that would ultimately determine the MIP performance was simulated. Consequently, the present study in addition allows insight into the binding mechanics and possible intermediates occurring during the imprinting process. The variations in docked positions and binding free energies unanimously revealed the effect of each monomer in the pre-polymerization mixture. Finally, unique interactions at multiple sites were identified that amplify the complementarity of the synthesized MIP to the entire peptide.
Materials and methods
Single monomer docking
The structure of the three target peptides (Table 1) was modelled using PEP-FOLD server,31 and a predicted local structure profile was generated that showed the probabilities at each position of the sequence assorted from helical (red), coil (blue) to extended (green) (Fig. S1, ESI†). The structural files of monomers such as acrylic acid (AAc), N-(3-amino-propyl) methacrylamide hydrochloride (APMA), N-tert-butyl-acrylamide (TBAm), and N-isopropylacrylamide (NIPAm) and N,N-methylenebis(acrylamide) (BIS) were obtained from the PubChem database. Next, Autodock tools (ADT) 1.5.7 were employed for preparation of the ligand and peptide files. Autodock, which is the most commonly employed open-source molecular docking software is based on the AMBER force field suitable for proteins, nucleic acids and other organic molecules.32 The ligand files were converted to pdbqt files after setting the torsional degrees of freedom based on the detected rotatable bonds. Polar hydrogens were added to the peptides; water molecules were removed. A grid box of suitable dimensions (Table S1, ESI†) was centered around the peptide.| Peptide | Sequence | Amino acid composition | Pl | Structural conformation | Momomers |
|---|---|---|---|---|---|
| 1 |

|
Acidic: 2, Basic: 3, Hydrophobic: 3 | 8.74 | Coiled |
|
| 2 |
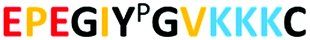
|
8.8 | Coiled |
|
|
| 3 |

|
Acidic: 2, Basic: 2, Hydrophobic: 6 | 6.66 | Helical |
|
For docking with Autodock 4 (v4.2.6), the number of energy evaluations was set to 250 million (evals) to improve the reproducibility and accuracy of the calculations.33 Furthermore, the number of docking runs was set to 100, as specified by the ga_run in the docking parameter file (.dpf). For molecular docking, the Lamarckian Genetic Algorithm (LGA) was employed for each monomer.
Similarly, for docking with Autodock Vina (v1.1.2),34 the pdbqt files of the ligand and peptide along with the grid dimensions were added in the respective configuration files. The exhaustiveness was set to 500 to improve the accuracy of the calculations.
For glide docking, the peptide structures were first prepared using Schrödinger's protein preparation wizard.35 Water molecules were deleted, bond orders were assigned and hydrogen atoms added with protonation states based on residue pKa values at normal pH (7.0) calculated using PROPKA. This was followed by optimization of hydrogen bonding patterns, and finally, minimization of the structure using the OPLS4 force field36 to remove steric hindrances and inadequate contacts. It should be noted that heavy atoms were constrained to within 0.3 Å (RMSD) of their crystallographic positions. Similarly, using the Ligprep tool the monomers shown in Table 1 were prepared for calculations with the OPLS4 force field and default settings. The possible states of ligands were generated at pH 7.0 ± 2.0 using Epik.
Docking calculations on the monomers were performed using Glide in extra-precision (XP) mode37 with default OPLS4 atomic charges and van der Waals scaling (0.8) for ligand nonpolar atoms to include modest “induced fit” effects. Docking grids were generated for each of the peptides with the grid dimensions summarized in Table S1 (ESI†).
All docking results in terms of binding energies and positions were accordingly analyzed using the BIOVA Discovery Studio Visualizer software, UCSF Chimera (v1.16), Maestro (v12.7.156) (Schrödinger).38–40
Multi-monomer simultaneous docking
For multi-monomer simultaneous docking, the scoring functions of Autodock 4 were employed. The monomer and peptide pdbqt files were prepared similarly with ADT. The docking parameter files (.dpf) of the monomers were merged in possible combinations (Table 3) with reference to the experimental combinations (Table S2, ESI†) and docking was performed with the standard LGA method. MMSD allows multiple conformations of different monomers to interact with the peptide at the same time. Each monomer is randomly initialized with its own set of variables. The docked models were visualized using UCSF chimera. The most favorable docking results were saved into.pdb files to subsequently create a 2D interaction map of each monomer-peptide interaction using LigPlot (v2.2.5).41Results and discussion
In a bottom-up approach, the present study has validated a novel in silico method for combinatorial screening and synthesis with reference to experimental data published by Bedwell et al. This study employs three different monomers (AAc, APMA and TBAm) for optimization of MIP performance for three different peptides (Table 1). Peptide 2 is phosphorylated at the TYR residue, which increases the negative charge on the peptide and is found to interact better with the positively charged monomer, APMA. In fact, in all three peptides the beneficial impact of APMA has been observed as opposed to AA (Fig. 1), although the peptides are composed of almost an equal number of positively charged amino acids (like lysine) and negatively charged amino acids (mostly glutamic acid) (Table 1). The peptides also contain hydrophobic amino acids (most in peptide 3). Hence, TBAm is supposed to play a crucial role in the polymer composition. Through molecular docking analysis, we have determined the contribution of each monomer interaction with the template, and therefore, the effect on the binding properties of the resulting MIPs.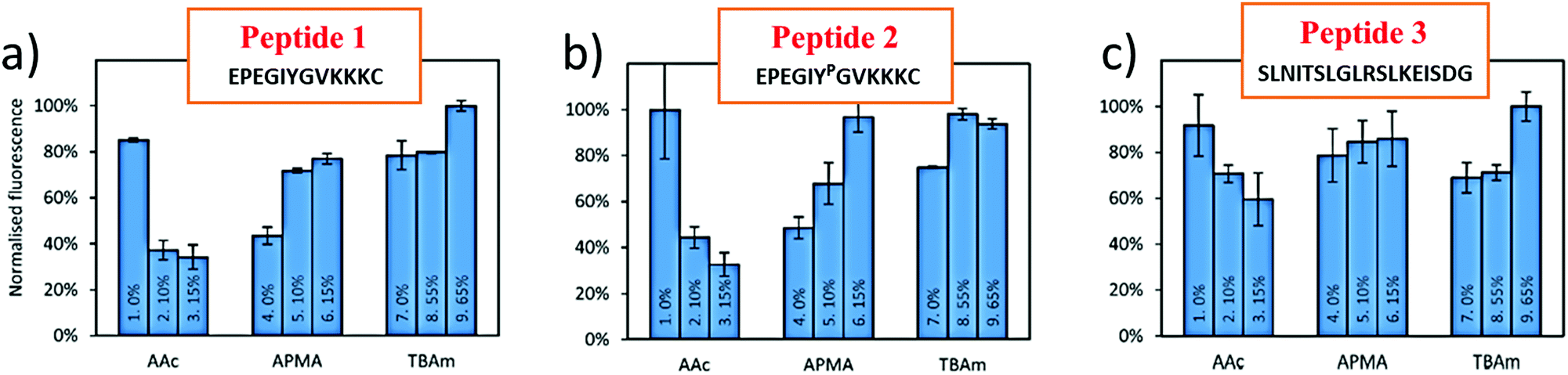 | ||
| Fig. 1 Percentage bound for each polymer composition where MIPs are formed by templating peptide 1 (a), peptide 2 (b) and peptide 3 (c). Source of experimental results: Bedwell et al. 2019.29 | ||
In the first step, three programs - namely Autodock (AD) 4, Vina and Glide - were used each based on different scoring functions to quantify the strength of the monomer–template complex. The computed binding energy values are directly proportional to the binding affinity of the monomer to the target peptide (Table 2).
| Monomer | Peptide 1 | Peptide 2 | Peptide 3 | |
|---|---|---|---|---|
| Autodock 4 | AAc | −2.84 | −2.73 | −2.91 |
| APMA | −4.66 | −4.77 | −3.39 | |
| TBAm | −4.02 | −4.02 | −2.56 | |
| Autodock Vina | AAc | −3.1 | −3.1 | −2.4 |
| APMA | −3.9 | −4.0 | −3.3 | |
| TBAm | −4.0 | −4.0 | −3.0 | |
| Glide | AAc | −5.01 | −4.21 | −2.98 |
| APMA | −3.34 | −3.51 | −2.97 | |
| TBAm | −2.83 | −2.57 | −2.36 |
In the docking results obtained with both AD 4 and Vina, the estimated free energy of binding of APMA in contrast to AAc corroborated the observed effect in the experiments (Fig. 1). However, the Glide docking results showed an opposite trend vs. Autodock and the experimental performance. AAc and TBAm showed higher binding scores than APMA, and - quite unexpectedly - value of AAc with peptide 2 was higher. The phosphorylation in peptide 2 should create an opposite effect with AAc, which corresponds to the experimental findings. The correlation to binding affinities not reflected by Glide docking (i.e., compared to Autodock 4 and Vina) can be reasoned by the different scoring functions and search algorithms of the programs. While AD4 and Glide both use a hybrid empirical/physics-based scoring function, Glide – based on OPLS force field (optimized potential for liquid simulations) – employs a local optimization search algorithm in a hierarchical search protocol. In contrast, AD 4 uses the Lamarckian genetic (search) algorithm (LGA).42 Autodock 4 uses the Amber family of force fields that sums up the inter- and intra-molecular energy resulting from H-bonding, Lennard-Jones dispersion/repulsion, electrostatics and desolvation.
In a next step, we have further investigated the two most promising Autodock programs that also reveal differences in searching capabilities/algorithms. While the obtained differences with respect to the experimental results are only minute, quite evidently AD 4 has provided better quantitative assessment of the interactions with the peptide. For example, the binding affinity of AAc is lower with peptide 2 than 1 and the correlation is only seen in AD 4 scores. Furthermore, the experiments reveal better binding of AAc with peptide 3 (Fig. 1) when compared to peptide 1 and 2, and AD Vina does not reflect a similar trend while AD 4 does. Better correlation of AD4 results as compared to Vina concerning correlation with the experimental binding affinities is also supported by other studies.33 While Vina employs a hybrid empirical/knowledge-based scoring function, similar to Glide it performs the local optimization on the most promising ligand positions. At this point, it is noteworthy that unlike in rational drug design approaches, the aim of MIP design is not to identify a strongly binding ligand in a small binding pocket, yet, to determine monomers potentially forming multi-point non-covalent (i.e., weak) interactions with the entire biomolecular template surface. Therefore, after assessing the most favorable binding position, in a next step the interactions at multiple residues were addressed.
From the above trends, it was established that the scoring functions of AD4 are most coherent with the experimental results and therefore, we have combined this program with the MMSD approach to computationally predict multi-monomer combinations that are most suitable for the MIP synthesis devised and executed in the reference article. Furthermore, comparison of the docking calculations for single monomer docking (SMD) with the MMSD aided in gaining mechanistic insight on the behavior of multiple monomers assembling around the target peptide. Similar to the polymer compositions used in the published experiments, we have also considered several such combinations (Table 3 and Tables S2 and S3, ESI†) to study the effect of more than one type of monomer in the so-called ‘pre-polymerization complex’.
| Peptide | Multi-monomer simultaneous docking | Single monomer docking | |||
|---|---|---|---|---|---|
| APMA+TBAm | AA + TBAm | AA+ APMA | AA +APMA +TBAm | ||
| Peptide 1 | −4.67 (APMA), −4.01 (TBAm) | −2.84 (AA), −4.02 (TBAm) | −2.84 (AA), −4.58 (APMA) | −2.84 (AA), −4.55 (APMA), −4.02 (TBAm) Total = −11.41 | −2.84 (AA), −4.66 (APMA), −4.02 (TBAm) |
| Peptide 2 | −4.76 (APMA), −4.02 (TBAm) | −2.79 (AA), −4.01 (TBAm) | −2.70 (AA), −4.66 (APMA) | −2.71 (AA), −4.68 (APMA), −4.02 (TBAm) Total = −11.41 | −2.73 (AA), −4.77 (APMA), −4.02 (TBAm) |
| Peptide 3 | −3.33 (APMA), − 2.57 (TBAm) | −2.87 (AA), −2.56 (TBAm) | −2.88 (AA), −3.41 (APMA) | −2.93 (AA), −3.18 (APMA), −2.56 (TBAm) Total = −8.67 | −2.91 (AA), −3.39 (APMA), −2.56 (TBAm) |
Multi-monomer simultaneous docking
To understand the effect of monomer combinations on the binding capacity of MIPs templated with peptide 1 (MIP1), peptide 2 (MIP2) and peptide 3 (MIP3), we have computed the estimated mean binding affinities for each. The binding energy values (ΔG) in the first conformational cluster of the 100 runs obtained using Autodock 4 was used for sorting and comparing outcomes (Table 3). The remaining clusters show a similar trend in terms of change in binding energy values.Among all monomers, APMA revealed the highest (negative) binding energy, which directly correlates with the performance of different polymer compositions entailing this functional monomer. The binding capacity of MIP2 is enhanced in presence of APMA in polymer composition as compared to MIP1, which is likewise observed in the computed binding energy values (Table 3, highlighted in bold). Compared to single monomer docking, MMSD reveals that the overall binding affinity of APMA with every peptide is slightly decreased when applied in a tri-monomer combination (AAc + APMA + TBAm). This reduction in binding affinity can be explained when analyzing corresponding bi-monomer combinations. When APMA or AAc are docked in combination with TBAm (APMA + TBAm or AAc + TBAm), there is little change in binding affinities hence, matching the single monomer docking outcomes. However, if AAc and APMA are docked together at the peptide surface (AAc + APMA), a slight decrease in binding energy score of APMA (Table 3, highlighted in italics) is observed indicating that the presence of AAc impacts the affinity of APMA with the peptides, yet, in a negative way. Apparently, small changes in the binding affinities of monomer-peptide combinations translate have a sizeable impact during the polymer synthesis.
The variable effect of AAc in different polymer compositions (PC) (Fig. 2) can also be interpreted from the computational results (Table 3). In the experiments, increasing concentrations of AAc have significantly affected the MIP2 performance (Fig. 2b, PC 1–3), which is also corroborated by the lowest binding affinity of AAc with peptide 2 (−2.71 kcal mol−1). Additionally, we calculated the difference in estimated binding energies of APMA and AAc in the tri-monomer combinations (AAc + APMA + TBAm). The highest difference is obtained in case of peptide 2 (BEAPMA- BEAAc (−4.68–(−2.71)) = −1.97 kcal mol−1) followed by peptide 1 (−1.71 kcal mol−1) and at the least in peptide 3 (−0.25 kcal mol−1). This also explains the largest difference in binding capacities of the MIPs in PC 1–3 templated with peptide 2 followed by peptide 1 and at last peptide 3 (Fig. 2).
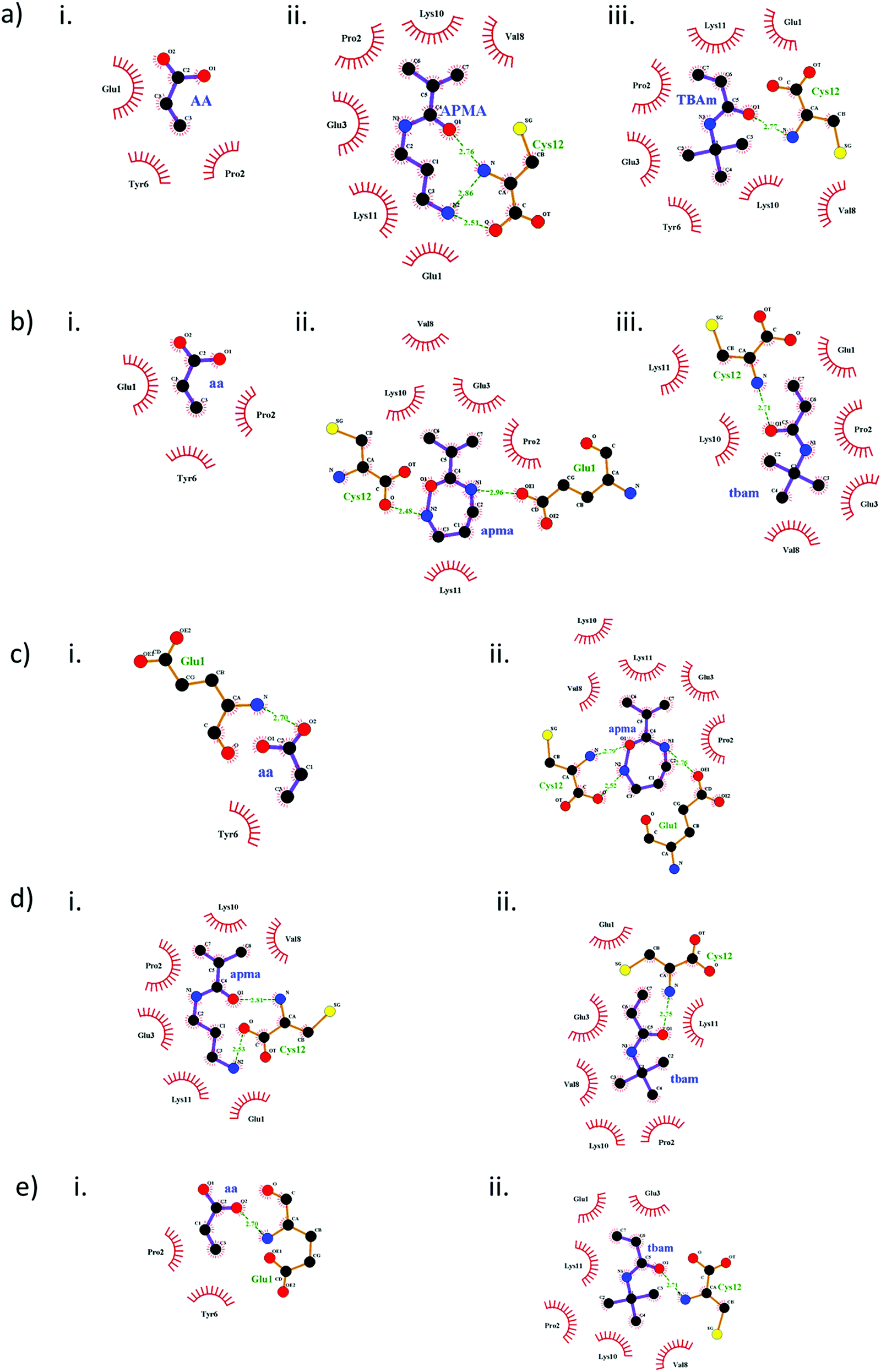 | ||
| Fig. 2 2D interaction maps representing monomer interaction with peptide 1, generated using LigPlot. Interactions resulting from single monomer docking of AAc, APMA and TBAm are shown in a–i, ii and iii, respectively. Multi-monomer simultaneous docking outcomes (b–e) are shown for tri-monomer combination (AAc + APMA + TBAm) (b), and bi-monomer combinations like AAc + APMA (c), APMA + TBAm (d) and AAc + TBAm (e). Monomers are marked in capital when docked individually and in small letters, when used in MMSD. Green dotted lines (with bond lengths) indicate H-bonds and red solid lines indicate hydrophobic interactions with the respective amino acids. The heteroatoms like N, O and S are represented by blue, red and yellow spheres, respectively. | ||
While apparently AAc has a well-established negative effect, MIPs templated with peptide 3 (MIP3) with increasing concentrations of AAc (Fig. 2c, PC 2–4) still reveal higher binding capacities compared to PC 2, 3 and 4 in MIP2 and MIP3 (Fig. 2a and b). The improved performance with MIP3 relates to theoretical results as well, as AAc binds with highest (negative) binding energy to peptide 3 (−2.93 kcal mol−1) compared to other two (−2.71 and −2.84 kcal mol−1). In case of TBAm, the high binding affinity with peptide 1 and 2 (−4.03 kcal mol−1) correlates well to PC 8 and 9 (Fig. 2a and b). However, there is a confusing trend with peptide 3, whereby [TBAm] increased from 0 to 55 mol% has little effect on the binding capacity, while an increase up to 65% shows maximum MIP performance. This is hypothesized to be resulting from the helical structure of the peptide facilitating a high number of intramolecular interactions. An increase in the concentration of TBAm may indeed affected the 3D structure of the peptide yielding improved binding with all the residues of the peptide. This may not be desirable during imprinting of epitopes, as they should be preferably imprinted in their native conformation and will therefore be the subject of more detailed future theoretical and experimental studies.
Interestingly, in another set of MMSD calculations involving NIPAM as part of the pre-polymerization mixture (Table S2, ESI†) we observed no effect on peptide 1 and 2, but peptide 3. The addition seems to slightly decrease the binding energy score of APMA similar to the effect of TBAm, and also has similar binding domains. However, no change in binding energy is evident within the combination of four monomers. As there is was control polymer (0 mol% of NIPAM) used within the experimental study, the MMSD results cannot be appropriately compared.
The diverse type of interactions resulting from monomer-peptide complexation can be mapped via the docking results. The 2D interaction maps generated from the docking models in the first cluster reveal the H-bonding and hydrophobic interactions (Fig. 2). The change in both inter- and intra–molecular interactions of monomers with peptide 1 during single monomer docking (Fig. 2a) and MMSD outcomes (Fig. 2b) correlate with the change in docking scores in Table 3. Clearly, the molecular orientation of the modelled poses changes in second case. There is a significant change in the type of interactions resulting from APMA when docked simultaneously with AAc and TBAm (Fig. 2b). The same effect is evident only when AAc and APMA are docked together (Fig. 3c). This explains the influence of AAc on the binding energy values of APMA (Table 3). There are also changes in the arrangement of interactions of TBAm with the peptide (Fig. 2a(iii) and b(iii)) and this was observed to be influenced by APMA (Fig. 2d). The type of interactions of TBAm remain unchanged when docked in combination with AAc (Fig. 2e). The overall effect on TBAm is minor compared to that on APMA. The different types and strength of H-bonding affected due to the intramolecular interactions with the latter and are the main reason for changes in the docking scores.
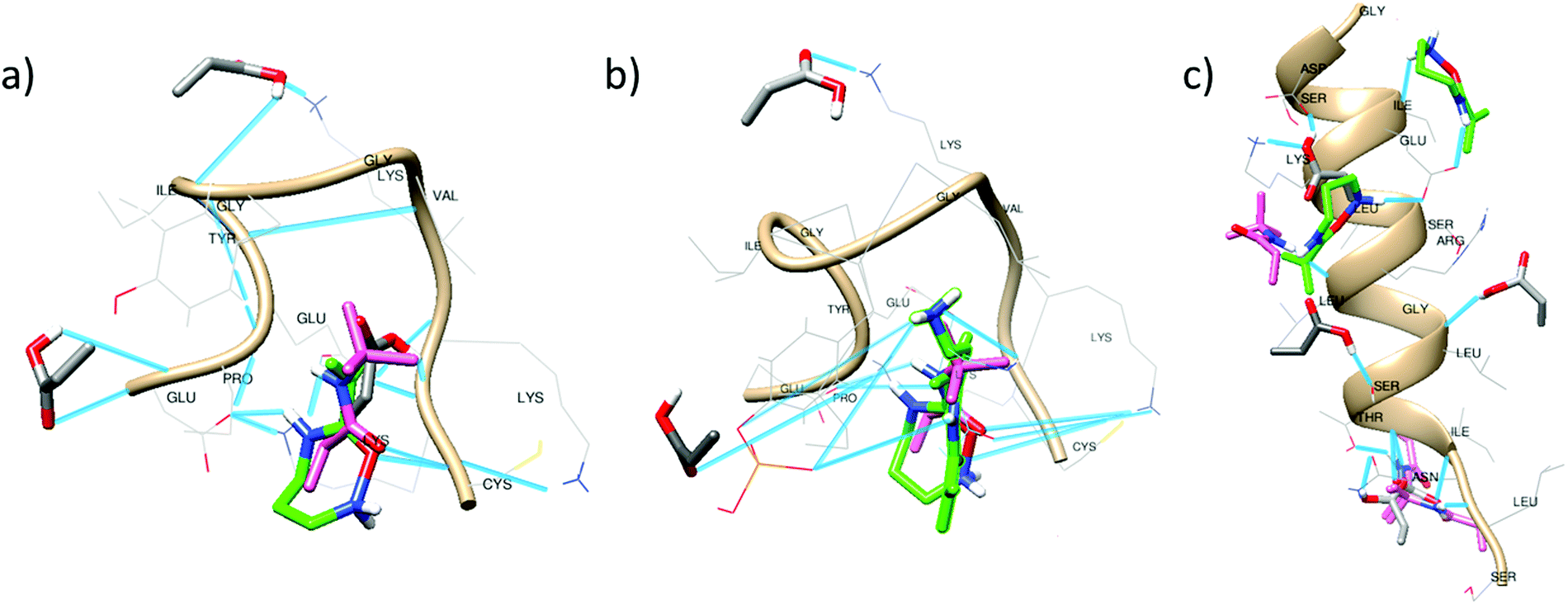 | ||
| Fig. 3 Multi–point interactions of monomers: acrylic acid (grey), APMA (green) and TBAm (pink) with peptide 1 (a), peptide 2 (b) and peptide 3 (c) as predicted by the scoring function of Autodock 4. Hydrogen bonds are represented as blue lines. | ||
A similar trend was also observed with peptide 2 and peptide 3 interacting with APMA, whereby the number and type of interactions changed in the MMSD outcomes compared to individual monomer-peptide complex predominantly influenced by AAc (Fig. S2 and S3, ESI†). Also, there is a minor effect on TBAm interacting with the peptides, both due to AAc and APMA but does not affect the docking scores in either case.
Multi–point interactions
The clusters observed under the RMSD values of 2.0 Å in the tri-monomer docking results allowed the analysis of multi-point non-covalent (i.e., weak) interactions of each monomer with the entire peptide (Fig. 3).Acrylic acid mainly interacts with the LYS and PRO residues in peptide 1 and 2 (Fig. 3a and b). H-bonding with the N-terminal of the peptide is also realizable but the intramolecular interactions in the peptides especially between LYS and GLU may affect the monomer binding capacity. APMA and TBAm occupy similar binding domains, as evident in all the docking clusters and form a strong complex that might preserve the peptide conformation during imprinting process. Intermolecular H-bonding with GLU, LYS and C-terminal (CYS) is observed in case of APMA. The number of H-bonds further increases due to the presence of phosphorylation in peptide 2 (Fig. 3b). The tert-butyl group of TBAm is oriented towards the hydrophobic residues enabling alkyl-based interactions with PRO, VAL, LYS and π–alkyl interaction with TYR residues.
In case of peptide 3, the monomers clustered over a larger area of the peptide, thereby leading to an extended number of multi–point interactions as compared to peptide 1 and 2. Also, the peptide is longer and enables a diversity of binding interactions. This may explain the comparable performance of MIP3 with MIP1 and MIP2 (Fig. 1), even though overall binding energy values are comparatively lower with peptide 3 (Table 3, highlighted in bold italics). AAc majorly interacts with LYS, ARG, SER and ASN residues. APMA forms binding domains near the C-terminal complexing with the negatively charged amino acids (ASP and GlU). TBAm interacts with the hydrophobic residues forming alkyl-based interactions with LEU, LYS and ILE residues.
Conclusions
Multi-monomer usage for advanced molecular imprinting routines requires complementary computational predictive methods that are clearly different from single monomer/ligand based virtual screening. The present small-scale study based on a bottom-up approach revealed the direct correlation of experimental MIP performance with in silico predicted multi–monomer interactions with the template. The developed MMSD approach allows screening numerous monomer combinations within a reasonable computing time, thereby minimizing the experimental efforts. It was unambiguously elucidated how each monomer affects template binding, and how individual monomers may determine the binding of other monomers to the template species – herein, peptides. Therefore, it is apparent that a suitable approximation of monomer effects using advanced MIP-oriented docking simulations significantly reduces the complexity of experimental design. Moreover, the developed strategy facilitates not only the assessment of the most favourable binding sites, but also enables analysing multi–point interactions in atomistic detail across the entire surface of the template species, which fundamentally governs the imprinting efficiency and the creation of selective binding sites. During the past decade, a variety of strategies have been applied within molecular mechanics studies to optimize molecular imprinting of proteins and peptides. To the best of our knowledge, the present study for the first time selects more suitable softwares for devising a novel in silico predictive method optimizing rational imprinted polymer design. We have recently established the utility of the AD4 scoring functions within two experimental studies, which present a direct correlation with the MIP performance.43,44 The rational screening from a library of monomers for peptide imprinting complemented with the analysis of multi–point interactions across the template surface has aided in significantly improving the specificity of the resulting MIP.Finally, using appropriate scoring functions augmented by MMSD will facilitate combinatorial screening and synthesis of highly selective MIPs at a yet unprecedent level of molecular detail and with reasonable computational efforts. The computational capacity of AD4-MMSD offers the opportunity of screening more than 100 combinations from a small library of 10 monomers, and their hierarchical sorting based on the total binding energy of the combinations for directly motivating a refined and guided synthesis.
Author contributions
Both authors have contributed equally to the preparation of this manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. Rajpal thanks the German exchange service (DAAD) for the fellowship award under the bi-nationally supervised doctoral degree program at the Institute of Analytical and Bioanalytical Chemistry, Ulm University, Germany. The authors also acknowledge support by the state of Baden-Württemberg through bwHPC and the German Research Foundation (DFG) through Grant no INST 40/575-1 FUGG (JUSTUS 2 cluster). This work was also in part supported by the Ministerium für Wissenschaft, Forschung und Kunst (MWK) Baden-Württemberg, Germany, under the Program “Special Measures against the SARS-CoV-2 Pandemic”.References
- J. J. Belbruno, Chem. Rev., 2019, 119, 94–119 CrossRef CAS PubMed.
- F. Meier and B. Mizaikoff, Artificial Receptors for Chemical Sensors, Wiley-VCH, 2010, pp. 391–437 Search PubMed.
- M. Gast, H. Sobek and B. Mizaikoff, Mater. Sci. Eng., C, 2019, 99, 1099–1104 CrossRef CAS PubMed.
- N. Karaseva, T. Ermolaeva and B. Mizaikoff, Sens. Actuators, B, 2016, 225, 199–208 CrossRef CAS.
- N. A. El Gohary, A. Madbouly, R. M. El Nashar and B. Mizaikoff, Electroanalysis, 2017, 29, 1388–1399 CrossRef.
- E. M. Saad, N. A. El Gohary, M. Abdel-Halim, H. Handoussa, R. M. El Nashar and B. Mizaikoff, Food Chem., 2021, 335, 127644 CrossRef CAS PubMed.
- S. Rajpal, S. Bhakta and P. Mishra, J. Mater. Chem. B, 2021, 9, 2436–2446 RSC.
- M. Dinc, H. Basan, T. Diemant, R. J. Behm, M. Lindén and B. Mizaikoff, J. Mater. Chem. B, 2016, 4, 4462–4469 RSC.
- S. Bhakta, M. S.-I. Seraji, S. L. Suib and J. F. Rusling, ACS Appl. Mater. Interfaces, 2015, 7, 28197–28206 CrossRef CAS PubMed.
- J. Xu, H. Miao, J. Wang and G. Pan, Small, 2020, 16, 1906644 CrossRef CAS PubMed.
- K. Yang, J. Liu, S. Li, Q. Li, Q. Wu, Y. Zhou, Q. Zhao, N. Deng, Z. Liang, L. Zhang and Y. Zhang, Chem. Commun., 2014, 50, 9521–9524 RSC.
- K. Yang, S. Li, L. Liu, Y. Chen, W. Zhou, J. Pei, Z. Liang, L. Zhang and Y. Zhang, Adv. Mater., 2019, 31, 1902048 CrossRef CAS PubMed.
- O. Hayden, P. A. Lieberzeit, D. Blaas and F. L. Dickert, Adv. Funct. Mater., 2006, 16, 1269–1278 CrossRef CAS.
- A. Rachkov and N. Minoura, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 2001, 1544, 255–266 CrossRef CAS.
- B. Fresco-Cala and B. Mizaikoff, ACS Appl. Polym. Mater., 2020, 2, 3714–3741 CrossRef CAS.
- A. Kushwaha, J. Srivastava, A. K. Singh, R. Anand, R. Raghuwanshi, T. Rai and M. Singh, Biosens. Bioelectron., 2019, 145, 111698 CrossRef CAS PubMed.
- B. Science, K. Karim, T. Cowen, A. Guerreiro, E. Piletska, M. J. Whitcombe and S. A. Piletsky, A Protocol for the Computational Design of High Affinity Molecularly Imprinted Polymer Synthetic Receptors, University of Leicester, 2017, vol. 3 Search PubMed.
- T. Cowen, K. Karim and S. Piletsky, Anal. Chim. Acta, 2016, 936, 62–74 CrossRef CAS PubMed.
- C. Dong, X. Li, Z. Guo and J. Qi, Anal. Chim. Acta, 2009, 647, 117–124 CrossRef CAS PubMed.
- R. Boroznjak, J. Reut, A. Tretjakov, A. Lomaka, A. Öpik and V. Syritski, J. Mol. Recognit., 2017, 30, e2635 CrossRef PubMed.
- S. Suryana, Mutakin, Y. Rosandi and A. N. Hasanah, Molecules, 2021, 26, 1891 CrossRef CAS PubMed.
- S. Wei, M. Jakusch and B. Mizaikoff, Anal. Bioanal. Chem., 2007, 389, 423–431 CrossRef CAS PubMed.
- S. Wei, M. Jakusch and B. Mizaikoff, Anal. Chim. Acta, 2006, 578, 50–58 CrossRef CAS PubMed.
- J. O’Mahony, B. C.-G. Karlsson, B. Mizaikoff and I. A. Nicholls, Analyst, 2007, 132, 1161–1168 RSC.
- S. Zink, F. A. Moura, P. A.-D. S. Autreto, D. S. Galvão and B. Mizaikoff, Phys. Chem. Chem. Phys., 2018, 20, 13145–13152 RSC.
- A. Molinelli, K. Nolan, M. R. Smyth, M. Jakusch and B. Mizaikoff, Int. J. Environ. Anal. Chem., 2002, 41, 5196–5204 Search PubMed.
- M.-v Sullivan, S. R. Dennison, G. Archontis, S. M. Reddy and J. M. Hayes, J. Phys. Chem. B, 2019, 123, 5432–5443 CrossRef CAS PubMed.
- S. Subrahmanyam and S. A. Piletsky, Combinatorial Methods for Chemical and Biological Sensors, Springer, New York, 2009, pp. 135–172 Search PubMed.
- T. S. Bedwell, N. Anjum, Y. Ma, J. Czulak, A. Poma, E. Piletska, M. J. Whitcombe and S. A. Piletsky, RSC Adv., 2019, 9, 27849–27855 RSC.
- H. Li and C. Li, J. Comput. Chem., 2010, 31, 2014–2020 CrossRef CAS PubMed.
- A. Lamiable, P. Thévenet, J. Rey, M. Vavrusa, P. Derreumaux and P. Tufféry, Nucleic Acids Res., 2016, 44, W449–W454 CrossRef CAS PubMed.
- G. M. Morris, H. Ruth, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- N. T. Nguyen, T. H. Nguyen, T. N.-H. Pham, N. T. Huy, M. van Bay, M. Q. Pham, P. C. Nam, V. V. Vu and S. T. Ngo, J. Chem. Inf. Model., 2020, 60, 204–211 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2009, 31, 455–461 Search PubMed.
- G. Madhavi Sastry, M. Adzhigirey, T. Day, R. Annabhimoju and W. Sherman, J. Comput.-Aided Mol. Des., 2013, 27, 221–234 CrossRef CAS PubMed.
- C. Lu, C. Wu, D. Ghoreishi, W. Chen, L. Wang, W. Damm, G. A. Ross, M. K. Dahlgren, E. Russell, C. D. von Bargen, R. Abel, R. A. Friesner and E. D. Harder, J. Chem. Theory Comput., 2021, 17, 4291–4300 CrossRef CAS PubMed.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelley, J. K. Perry, D. E. Shaw, P. Francis and P. S. Shenkin, J. Med. Chem., 2004, 47, 1739–1749 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- BIOVIA Discovery Studio Visualizer - Dassault Systèmes, https://discover.3ds.com/discovery-studio-visualizer-download, (accessed July 5, 2021).
- Schrödinger | Schrödinger is the scientific leader in developing state-of-the-art chemical simulation software for use in pharmaceutical, biotechnology, and materials research., https://www.schrodinger.com/, (accessed July 5, 2021).
- R. A. Laskowski and M. B. Swindells, J. Chem. Inf. Model., 2011, 51, 2778–2786 CrossRef CAS PubMed.
- E. D. Boittier, Y. Y. Tang, M. E. Buckley, Z. P. Schuurs, D. J. Richard and N. S. Gandhi, Int. J. Mol. Sci., 2020, 21, 5183 CrossRef CAS.
- B. Fresco-Cala, S. Rajpal, T. Rudolf, B. Keitel, R. Groß, J. Münch, A. D. Batista and B. Mizaikoff, Nanomaterials, 2021, 11, 2985 CrossRef CAS PubMed.
- A. D. Batista, S. Rajpal, B. Keitel, S. Dietl, B. Fresco-Cala, M. Dinc, R. Groß, H. Sobek, J. Münch and B. Mizaikoff, Adv. Mater. Interfaces, 2022, 9, 2101925 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tb00418f |
| This journal is © The Royal Society of Chemistry 2022 |