
Construction of nano receptors for ubiquitin and ubiquitinated proteins based on the region-specific interactions between ubiquitin and polydopamine†
Zezhou
Li‡
a,
Xinyi
Li‡
a,
Wei
Xian‡
b,
Huaisyuan
Xie
a,
Ying
Sun
a,
Yuxuan
Zhang
a,
Jiayu
Wang
a,
Hongwei
Li
a,
Changwen
Jin
a,
Xiaoyun
Liu
*b,
Zhiwei
Zhu
 *a and
Meiping
Zhao
*a
*a and
Meiping
Zhao
*a
aBeijing National Laboratory for Molecular Sciences, MOE Key Laboratory of Bioorganic Chemistry and Molecular Engineering, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China. E-mail: mpzhao@pku.edu.cn; zwzhu@pku.edu.cn
bDepartment of Microbiology and Infectious Disease Center, School of Basic Medical Sciences, Peking University Health Science Center, Beijing 100191, China. E-mail: xiaoyun.liu@bjmu.edu.cn
First published on 25th April 2022
Abstract
Ubiquitination is a prevalent post-translational modification that controls a multitude of important biological processes. Due to the low abundance of ubiquitinated proteins, highly efficient separation and enrichment approaches are required for ubiquitinome analysis. In this work, we disclose the region-specific interactions between the hydrophobic patch of ubiquitin and polydopamine. Taking advantage of this inherent binding property, we have constructed surface-imprinted magnetic nanoparticles (NPs) for ubiquitin by sequential dopamine polymerization and surface PEGylation. The obtained molecularly imprinted polymer (MIP) NPs showed a binding constant of 2.6 × 106 L mol−1 for the template ubiquitin. The bound ubiquitin could be quantitatively released by heating to 70 °C at pH 2.0 or 90 °C at neutral (pH 7.0) conditions. The MIP NPs exhibited nano receptor-like property which not only effectively blocked the formation of branched ubiquitin chains but also selectively separated ubiquitin from the bacterial cell lysates. By incubating the MIP NPs with the lysates of 293T cells, totally 529 ubiquitinated proteins were captured, among which 287 proteins were not identified by the anti-ubiquitin monoclonal antibodies (mAbs). With the distinct merits of low cost and high stability, the as-prepared MIP NPs may be utilized either separately or as an important complement to the mAbs for the purification and enrichment of ubiquitin and ubiquitinated proteins from complex biological samples. Furthermore, due to the flexibility in modification of the binding sites during or after the imprinting reactions, the results of this work also paved the way for generation of artificial receptors for branched ubiquitin chains and polyubiquitinated proteins with higher avidity and specificity.
Introduction
Molecular recognition of proteins plays a critical role in bio-systems and life science research.1 Over the years, biomolecules, including antibodies and aptamers, and small synthetic molecules, have been utilized for specific recognition of proteins.2,3 However, they show drawbacks of high cost, poor stability, or limited binding affinity or specificity.3–5 Molecularly imprinted polymers (MIPs), which selectively capture the molecules with which act as the templates during the production, are regarded as synthetic analogues to the natural, protein-based antibodies or receptors.6,7 Compared with antibodies, MIPs show the advantages of physical robustness, low cost, and pre-determined selectivity, thus have found wide applications in separation, chem-/biosensing, enzyme mimicking, etc.8–12Ubiquitin (Ub) is a small regulatory protein (76-amino acid, 8.6 kDa) present in all eukaryotic organisms.13–15 It can be further polymerized to form a variety of ubiquitin chains by combination of different connecting modules, just like a string of special “codes” that can be recognized by specific interacting proteins.16,17 Ubiquitination is an essential and prevalent post-translational modification that affects many important cellular processes.18 In addition to being a signal for degradation by the proteasome,19 ubiquitination also leads to a variety of other responses, such as gene expression, DNA repair, protein trafficking, and immune responses.20 Due to the low abundance of ubiquitin-modified proteins in cells, highly efficient and facile methods for the isolation and enrichment of ubiquitin and ubiquitinated proteins from cell lysates are critical for ubiquitinome analysis. Existing approaches for ubiquitin enrichment are mainly based on antibodies, ubiquitin binding domains (UBDs) and Ub epitope-tag expressing systems.21 However, the antibodies and UBDs are expensive and fragile while tagging ubiquitin in mammalian cells may interfere with normal cellular functions. Moreover, antibodies and UBDs can only identify a part of the ubiquitinated substrates, leading to the loss of a significant amounts of ubiquitin-associated species. Therefore, it is of great importance to develop novel high-affinity, stable and easily accessible purification tools for the enrichment of ubiquitin and ubiquitinated proteins under different conditions.
Dopamine (DA) is a biocompatible monomer which can self-polymerize in alkaline medium at ambient temperature to generate adhesive polydopamine (PDA) films on various solid materials.22–25 In our preliminary study, we observed that ubiquitin molecules could be strongly adsorbed on the surface of PDA (Fig. 1(a)). Further investigations with nuclear magnetic resonance (NMR) titration revealed that the hydrophobic patch of ubiquitin and peripheral residues were mainly involved in the binding to PDA. Taking advantage of this region-specific interactions, we immobilized ubiquitin templates in uniform orientations on the surface of PDA preformed over silica coated magnetic nanoparticles (SiMNPs). Then we further performed surface imprinting reactions with dopamine as the functional monomer to construct specific binding cavities for ubiquitin. After the polymerization, we modified the non-imprinted sites with methoxypoly(ethylene glycol) amine (mPEG-NH2) before removal of the templates. The resultant MIP NPs showed high specificity and affinity to ubiquitin, which efficiently blocked the ubiquitin polymerization reactions and selectively separated ubiquitin from the bacterial cell lysates. Moreover, the MIP NPs successfully captured ubiquitinated proteins that were not identified by the anti-ubiquitin monoclonal antibodies (mAbs) from 293T cell lysates, indicating high potential applicability to comprehensive ubiquitinome analysis.
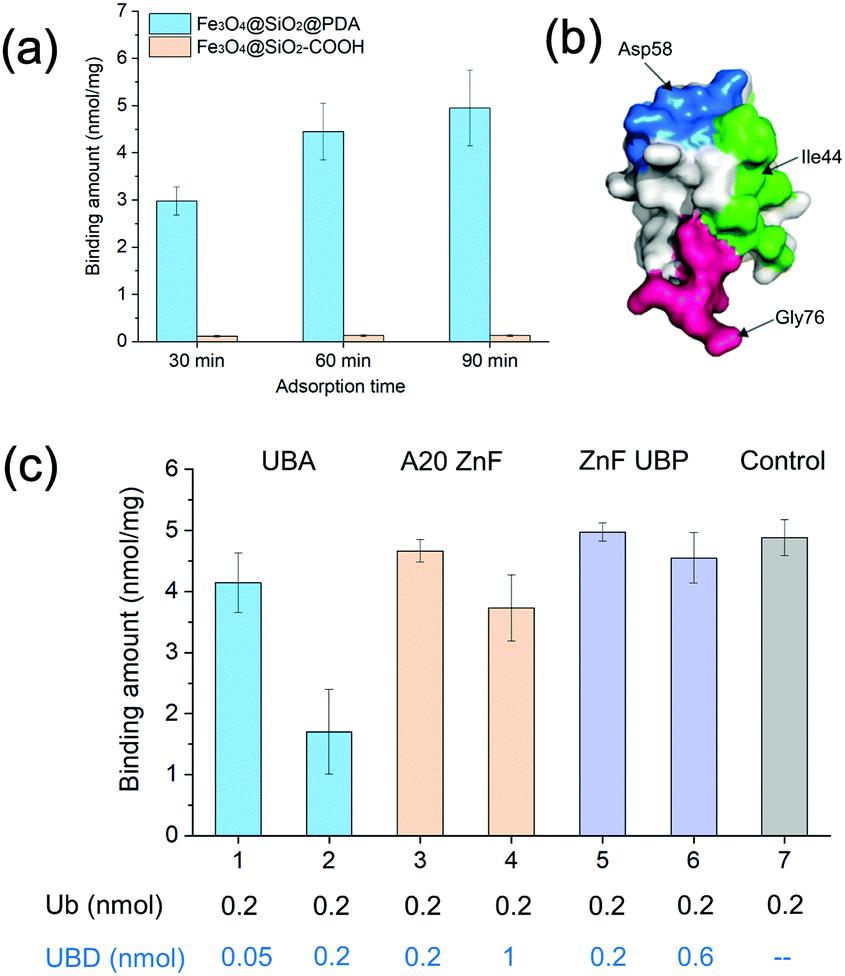 | ||
| Fig. 1 (a) Binding amounts of ubiquitin on Fe3O4@SiO2@PDA and Fe3O4@SiO2@–COOH at different time intervals. (b) Schematic diagram of the ubiquitin epitopes. (c) Binding amounts of ubiquitin on Fe3O4@SiO2@PDA in the presence of different UBDs. | ||
Materials and methods
Materials
Dopamine (DA) and methoxypoly(ethylene glycol) amine (mPEG-NH2) were purchased from Sigma Chemical Co. (St. Louis. MO). Fe3O4@SiO2–COOH magnetic nanoparticles (SiMNPs) were purchased from PuriMag Biotech (Xiamen, China). Trypsin was purchased from Promega Biotech Co. (Beijing). N,N-Diisopropylethylamine (DIPEA) was purchased from Aladdin Biochemical Technology Co. (Shanghai, China). The ubiquitin sequence was subcloned into pGEX-6p-1 vector. Then ubiquitin was expressed in E. coli and purified as described previously.26Nuclear magnetic resonance (NMR) titration of 15N-labeled ubiquitin with unlabeled polydopamine nanoparticles
15N-labeled ubiquitin (15N-Ub) and polydopamine (PDA) nanoparticles (NPs) were synthesized according to previous studies.26,27 Before titration, the PDA NPs were concentrated to about 2.0 mM in phosphate-buffered saline (PBS). The NMR titration was carried out on a Bruker AVANCE III 600 MHz NMR spectrometer at 25 °C. The concentration of 15N-Ub was 0.5 mM with 10% v/v D2O pre-added. The 1H-NMR and 1H–15N heteronuclear single-quantum correlation (HSQC) spectra were collected by incubation of the mixed samples for 1 h.Synthesis of surface-imprinted magnetic nanoparticles (NPs) for ubiquitin
Fe3O4@SiO2–COOH NPs (SiMNPs, 0.2 mg mL−1) were dispersed in 20 mM Tris–HCl buffer (pH 8.0). After ultrasonication, dopamine (0.1 mg mL−1) was added and the self-polymerization was carried out at room temperature for 1 h. The resultant NPs were isolated under an external magnetic field and washed by 20 mM Tris–HCl buffer (pH 8.0) for three times. The obtained NPs were incubated with 1.0 μM ubiquitin at room temperature for 1 h and then washed three times with 20 mM Tris–HCl buffer (pH 8.0) to remove the non-adsorbed ubiquitin. The resultant NPs were resuspended in 20 mM Tris–HCl buffer (pH 8.0) at a concentration of 0.2 mg mL−1, to which dopamine was added at a concentration of 0.1 mg mL−1. The polymerization was carried out at 37 °C for 2 h. The resultant NPs were washed three times with 20 mM Tris–HCl buffer (pH 8.0) and resuspended in 50 mM Tris–HCl buffer (pH 9.0). mPEG-NH2 was added at a concentration of 25 mg mL−1 to react with the non-imprinted sites on the surface of PDA overnight at 37 °C. MIP NPs were obtained after removal of ubiquitin template by trypsin digestion. The non-imprinted (NIP) NPs were prepared following the same procedures as above without the addition of ubiquitin templates.Release of ubiquitin bound on the MIP NPs
To investigate the capability of different solutions in dissociation of ubiquitin from the MIP NPs, 1.0 μM ubiquitin was incubated with 0.2 mg mL−1 MIP NPs in 20 mM Tris–HCl buffer (pH 8.0) for 30 min. After removal of the supernatant, the MIP NPs were incubated with different solutions for 30 min to desorb the bound ubiquitin. The recovered ubiquitin was quantified by LC-MS/MS after trypsin digestion.Isothermal titration calorimetry assay
Isothermal titration calorimetry (ITC) experiments were performed using a microcalorimeter (MicroCal ITC200). Prior to measurement, each solution was degassed to remove air bubbles. The NPs (2 mg mL−1) dispersed in PBS buffer were loaded in a 0.3 mL ITC cell at 25 °C. Ubiquitin (38 μL, 50 μM) in the same buffer was titrated into the cell (2 μL each time, except for the first injection of 0.4 μL). The enthalpy (ΔH) and the binding constant (Ka) were obtained through fitting the titration curves to a one-site binding model. The Kd and ΔG values were calculated from 1/Ka and ΔG = −RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(Ka), respectively, where R is the gas constant. ΔS was calculated from ΔG = ΔH − TΔS.
ln(Ka), respectively, where R is the gas constant. ΔS was calculated from ΔG = ΔH − TΔS.
Enrichment of ubiquitin by the MIP NPs and proteomic analyses of ubiquitinated proteins
10 mg of MIP NPs were incubated with cell lysates at 4 °C for 6 h. Then the NPs were isolated under an external magnetic field and resuspended in SDS-PAGE loading buffer. The adsorbed proteins were fully denatured and dissolved after heating at 95 °C for 5 min. After SDS-PAGE separation, the proteins were processed into 8 gel bands, and subjected to in-gel trypsin digestion as previously described.28 Peptides were extracted, vacuum dried, and resuspended in solvent A containing 97% H2O, 3% acetonitrile (ACN), and 0.1% formic acid (FA) for proteomic analyses. Liquid chromatography-tandem mass spectrometry (LC-MS/MS) experiments were performed on a hybrid ion trap-Orbitrap mass spectrometer (LTQ-Orbitrap Velos; Thermo Scientific) coupled with nanoflow reversed-phase liquid chromatography (EASY-nLC 1000, Thermo Scientific).Results and discussion
Investigation on the interactions between ubiquitin and polydopamine nanoparticles
Dopamine can undergo oxidative polymerization at room temperature and deposit on the surface of substrates without the need of other cross-linkers or initiators.29–31 The reaction mechanism involves the formation of heteroaromatic 5,6-dihydroxyindole and its oxidized product 5,6-indolequinone, which undergo branching reactions at positions 2, 3, 4 and 7. The resultant isomeric dimers or higher order oligomers then self-assemble to form thin film coating of substrates. We first performed dopamine self-polymerization in the presence of Fe3O4@SiO2–COOH magnetic nanoparticles (SiMNPs). Then the resultant SiMNPs@PDA (0.2 mg mL−1) was incubated with 1.0 μM ubiquitin and the Ub remained in the supernatant was quantified by trypsin digestion and LC–MS/MS. Surprisingly, after about 60 min, more than 90% of the added ubiquitin was absorbed on the SiMNPs@PDA, while the SiMNPs hardly adsorbed any ubiquitin (Fig. 1(a)), implying strong interactions between ubiquitin and the surface of PDA.To determine the epitope of ubiquitin that dominates its binding to the surface of PDA, we expressed three different UBDs (Fig. S1, ESI†), including UBA domain (from Ubiquilin protein), A20-ZnF domain (from Rabex-5 protein) and ZnF-UBP domain (from USP5), which specifically bind to the Ile44 patch, Asp 58 patch and diglycine patch of ubiquitin with similar affinity (Kd = 1–20 μM), respectively (Fig. 1(b)). The binding capacity of ubiquitin on SiMNPs@PDA in the presence of different amounts of the three UBDs were compared in Fig. 1(c). Clearly, addition of the UBA domain at a molar ratio of 1:1 exhibited the highest inhibitory effect on the binding of ubiquitin by PDA, suggesting that the Ile44 patch of ubiquitin was substantially involved in the interactions with PDA.
To further identify the specific amino acid residues on ubiquitin that have been involved in the interactions with PDA, we expressed and purified 15N-labeled ubiquitin (15N-Ub) and recorded the 1H–15N heteronuclear single-quantum correlation (HSQC) spectra which could reflect the chemical shift signals of 15N-Ub amido bonds or NH2 groups in the protein side chain. With the addition of unlabeled PDA NPs into the 15N-labeled ubiquitin solution, the NMR signal intensity notably attenuated at a factor greater than the dilution factor, indicating the formation of Ub-PDA complexes with no observable NMR signals (Fig. 2(a)). Some of the 1H–15N HSQC spectra peaks of 15N-labeled ubiquitin were significantly shifted when the ratio of Ub:PDA increased to 1:0.3 (Fig. 2(b)).
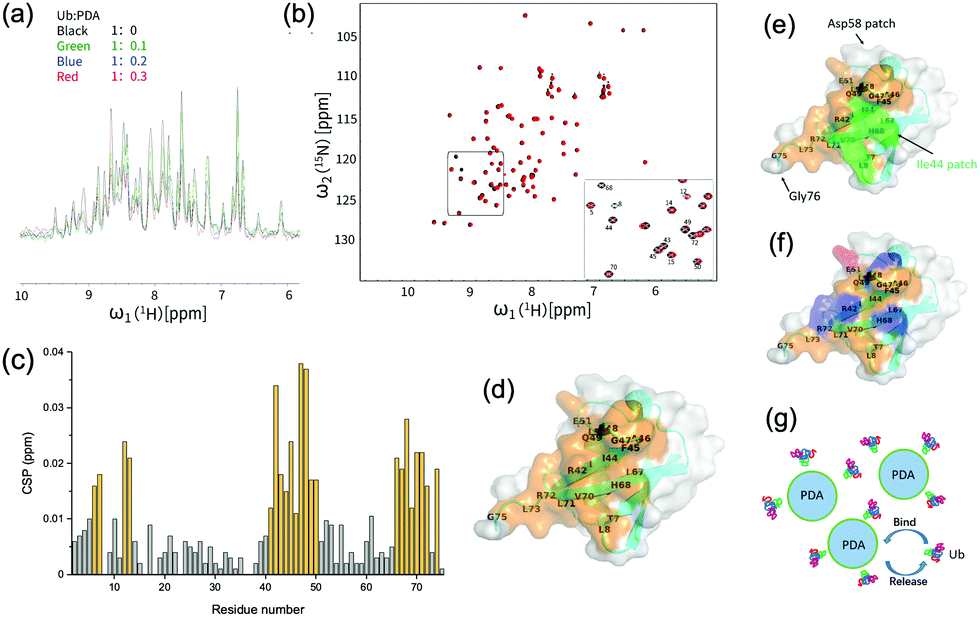 | ||
| Fig. 2 (a) 1H-NMR spectra of ubiquitin during the titration with polydopamine (PDA). (b) 1H–15N HSQC spectra of ubiquitin with or without the addition of PDA at a molar ratio of Ub:PDA = 1:0.3. (c) Chemical shift perturbation (CSP) of 1H–15N HSQC spectra of ubiquitin with the addition of PDA at a molar ratio of Ub:PDA = 1:0.3. (d) A schematic diagram of the ubiquitin binding surface. The residues with CSP ≥ 0.012 ppm were shown in orange. (e) The ubiquitin residues with CSP ≥ 0.012 ppm shown in orange and Ile44 patch shown in green. The Asp58 patch and Gly76 patch were also indicated. (f) The acidic (shown in red) and basic (shown in blue) residues among the ubiquitin residues with CSP ≥ 0.012 ppm. (g) Schematic diagram of the interactions between PDA and Ub. | ||
The changes in the local environment of the amino acid residues in ubiquitin could be seen from the chemical shift perturbation (CSP) data (Fig. 2(c)). The residues with a CSP of ≥0.012 ppm included T7, L8, I13, T14, R42, I43, I44, F45, A46, G47, K48, Q49, L50, E51, L69, L67, H68, V70, L71, R72, L73, G75. A schematic diagram of the ubiquitin binding surface was obtained by marking these residues on the three-dimensional model (Fig. 2(d)). More than half of the residues on the binding surface were hydrophobic residues, including I43, L69, I13, A46, L71, L67, L8, I44, L50, V70, L73, and F45, among which Ile44 patch (L8, I44, H68, V70) was involved (Fig. 2(e)). These results strongly proved the important roles of the Ile44 patch and other hydrophobic residues in the interactions with PDA. Moreover, as shown in Fig. 2(f), the positively charged residues (R42, K48, R72, H68) may have electrostatic interactions with the negatively charged PDA surface, while the residues T7, T14, R42, F45, K48, Q49, H68, and R72 could also interact with the surface of PDA via hydrogen or van der Waals bonds. These interactions made additional contribution to the binding of ubiquitin to PDA (Fig. 2(g)). For comparison, we performed similar titration experiments with the 15N-Ub and unlabeled dopamine monomer, which showed negligible effects on the 1H–15N HSQC spectrum of 15N-labeled ubiquitin, demonstrating that the binding sites were the typical structures on polydopamine rather than dopamine itself (Fig. S2, ESI†).
Synthesis and characterization of the ubiquitin imprinted magnetic nanoparticles
The strong binding of Ub to PDA not only provided a region-specific binding interface, but also fixed the Ub templates on the SiMNPs in a uniform orientation without the need of other specific ligands (Fig. 3(a)). Next, we attempted to construct tailor-made nanocavities for Ub via in situ molecular imprinting. For simplicity, we only used dopamine as sole monomer in this study. We optimized the polymerization time by monitoring the removal efficiency of Ub templates by trypsin digestion after the imprinting reaction. As shown in Fig. S3 (ESI†), when the polymerization time increased to longer than 2 h, the removal rate significantly declined, indicating over polymerization which had blocked the contact of trypsin with the Ub templates. On the other hand, to differentiate the imprinted sites from the non-imprinted surface of PDA for Ub binding, we tested to modify the surface of PDA with methoxypoly(ethylene glycol) amine (mPEG-NH2) to increase its hydrophilicity. We measured the Ub binding capacity of the SiMNPs@PDA after treated with different concentrations of mPEG-NH2. From Fig. S4 (ESI†), after an overnight treatment of the SiMNPs@PDA with mPEG-NH2 at a concentration higher than 25 mg mL−1, the binding of Ub on the resultant NPs reduced to less than 10% of the original level. By modification of the MIP with mPEG-NH2 at 25 mg mL−1 before removal of the templates, the imprinting factor (IF = QMIP/QNIP) achieved as high as 4.9 at a polymerization time of 2 h (Fig. 3(b)). The results were further confirmed by Fig. S5 (ESI†).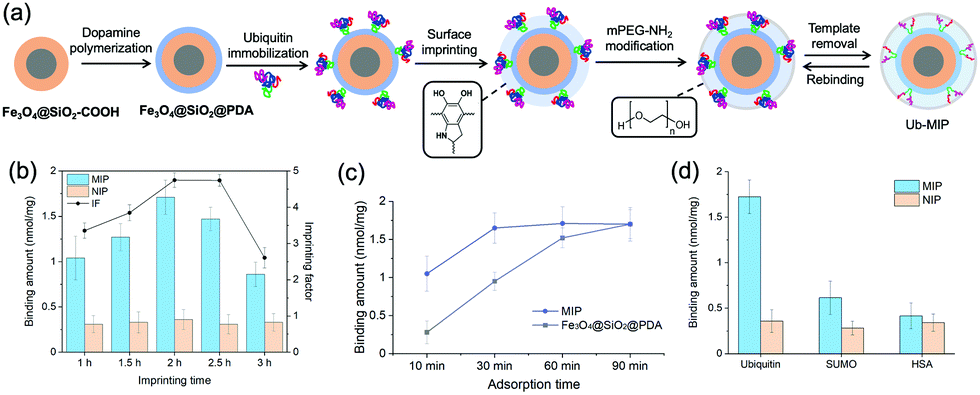 | ||
| Fig. 3 (a) Schematic illustration of construction of nano receptors for ubiquitin by surface imprinting over uniformly oriented templates on PDA preformed over silica coated magnetic nanoparticles (SiMNPs). (b) Binding amounts of ubiquitin on MIP and NIP NPs synthesized at different imprinting time and modified with mPEG-NH2 at 25 mg mL−1. (c) Comparison of the binding kinetics of ubiquitin on MIP NPs and Fe3O4@SiO2@PDA. (d) Selectivity test results of the MIP towards ubiquitin. The concentrations of MIP NPs and the tested proteins are 0.2 mg mL−1 and 0.4 μM, respectively. | ||
Fig. 3(c) compared the binding kinetics of ubiquitin on the MIP NPs and SiMNPs@PDA. Within 10 min, a large difference in the binding amounts of ubiquitin were observed between MIP and the SiMNPs@PDA. Moreover, the binding amount of Ub on MIP reached the maximum value after only 30 min, while it took about 90 min for the SiMNPs@PDA to achieve the same level. These results indicated significant enhancement of the kinetic binding performance of MIP after the imprinting process, which might be ascribed to more rapid capture of Ub by MIP followed by confinement of the molecules within the imprinted cavities. Then we evaluated the specificity of the MIP NPs by using SUMO, a small ubiquitin-like modifier and human serum albumin (HSA), a high-abundance protein in the serum as the reference proteins (Fig. 3(d)). The binding amount of ubiquitin (pI 6.9, Mw 8.6 kD) on the MIP NPs was much larger than those of SUMO (pI 5.4, Mw 11 kD) and HSA (pI 4.8, Mw 67 kD), indicating a good selectivity of the MIP towards ubiquitin.
We used isothermal titration calorimetry (ITC) to measure the binding affinity of the MIP NPs to ubiquitin. As shown in Fig. 4(a), binding of ubiquitin by the MIP NPs released significantly more heat than by the NIP NPs (Fig. 4(b)). The enthalpy, entropy, and binding constant (Ka), determined from the integration of heat, were displayed in Table S1 (ESI†). The binding constant (Ka) of ubiquitin by the MIP NPs was observed to be 2.6 × 106 L mol−1, which was more than 100 times larger than that of NIP NPs (2.2 × 104 L mol−1). From the thermodynamic parameters in Table S1 (ESI†), the binding of ubiquitin to MIP involves hydrogen and van der Waals bonds as indicated by the negative binding enthalpy (ΔH) with some conformational changes as indicated by the unfavorable entropy. By contrast, the binding of ubiquitin by NIP involves less polar interactions and more entropically favored hydrophobic interactions. The different mechanisms of binding between MIP and NIP proved the key role of the imprinting process in the formation of specific and strong binding sites. After removal of the bound Ub by trypsin digestion, we inspected the reusability of the MIP NPs. From Fig. S6 (ESI†), the MIP NPs showed stable rebinding capacity in at least three regeneration cycles.
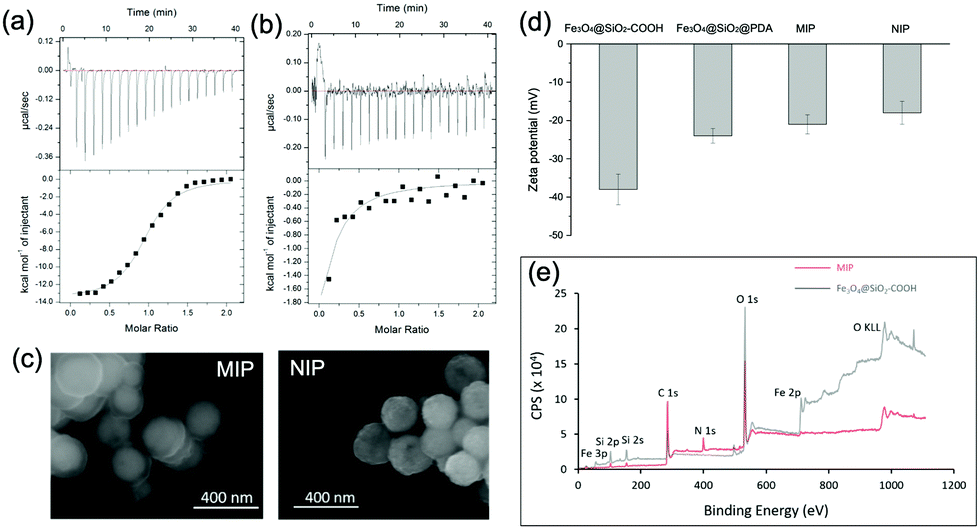 | ||
| Fig. 4 (a) and (b) ITC data of the binding of ubiquitin to (a) MIP and (b) NIP NPs. (c) SEM images of MIP and NIP NPs. (d) Zeta potential values of different NPs. (e) XPS spectra of MIP and Fe3O4@SiO2–COOH NPs. | ||
Morphology study and dissociation of Ub from the MIP NPs
We characterized the morphology of the obtained MIP NPs by scanning electron microscopy-energy dispersive spectrometer (SEM-EDS), X-ray photoelectron spectroscopy (XPS), and zeta potential analysis. Both SEM (Fig. 4(c)) and zeta potential (Fig. 4(d)) results proved the formation of a PDA layer over the SiMNPs. The thickness of the PDA shell was ca. 20 nm. From the XPS data (Fig. 4(e) and Table S2, ESI†), the nitrogen-to-carbon signal ratio (N/C) increased from 0.021 for the SiMNPs to 0.107 for the MIP NPs, which also confirmed the formation of PDA on the surface of the NPs.Though trypsin could efficiently remove the Ub bound on MIP NPs, the digestion process was relatively time-consuming and destructive. To facilitate the dissociation of Ub from the MIP NPs as intact proteins, we investigated the desorption efficiencies of different combinations of solvents and temperature conditions. From Fig. 5(a), the desorption efficiencies of 20 mM Tris–HCl (pH = 7.6), 5% HAc, and 1% N,N-diisopropylethylamine (DIPEA) were only <5%, ∼5% and ∼10%, respectively. With the addition of 3 M NaCl, the binding of Ub on the MIP NPs was even more stable. These results indicated that electrostatic interaction only made very small contribution to the adsorption of Ub on the MIP. By contrast, when we added different surfactants to these solutions, the desorption efficiencies dramatically increased to >80%, which might be attributed to either the destruction of the H-bonding, van der Waals force and hydrophobic interactions between Ub and MIP or the collapse of the conformation of Ub. However, using surfactants was sometimes unfavourable for regeneration of the MIP NPs. Moreover, they might also affect the subsequent applications of the collected Ub or ubiquitinated proteins.
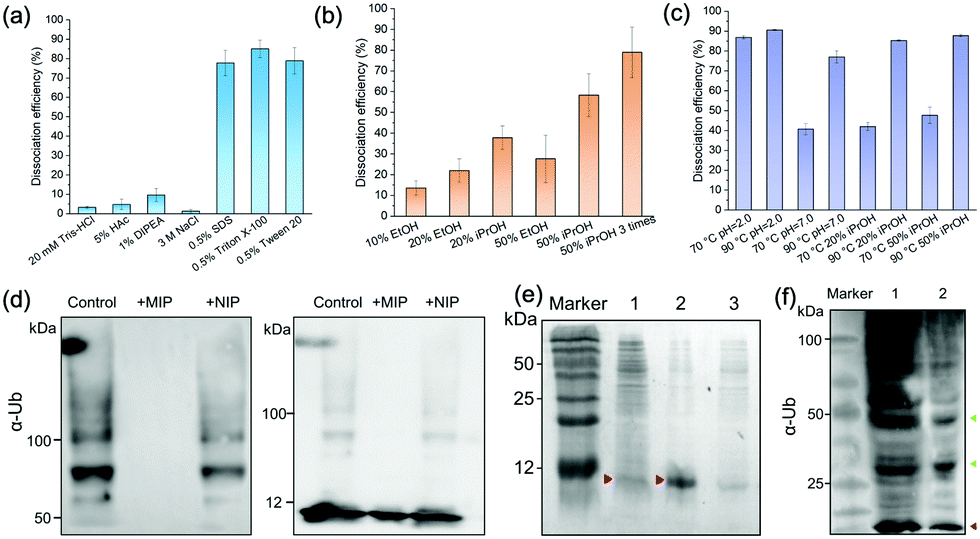 | ||
| Fig. 5 (a) Influences of pH, salt, and surfactants on the dissociation efficiency of Ub from MIP NPs. (b) Dissociation efficiency of Ub from MIP NPs by EtOH and iPrOH at different concentrations. (c) Dissociation efficiency of Ub from MIP NPs in neutral or acid solution and iPrOH solution at elevated temperature conditions. (d) SDS-PAGE analysis of ubiquitin polymerization products in the presence of MIP and NIP NPs. Left: Products above 50 kDa; Right: Products above 5 kDa. (e) SDS-PAGE analysis of bacterial cell lysates containing ubiquitin before (Lane 1), and after purification by MIP (Lane 2), or NIP (Lane 3) NPs. (f) SDS-PAGE analysis of ubiquitinated proteome from 293T cell lysates enriched by ubiquitin monoclonal antibodies (mAb, Lane 1) and MIP NPs (Lane 2). | ||
Fig. 5(b) compared the desorption efficiencies of ethanol (EtOH) and isopropanol (iPrOH) at different concentrations. Ub was observed to be soluble in 50% EtOH or 50% iPrOH, most likely because of its small molecular weight and high stability. With the increase of the concentrations of EtOH or iPrOH, the desorption efficiencies notably elevated. By using 50% iPrOH, the dissociation percentage increased to 60%, which was higher than that of 50% EtOH. Then we tried to elute the Ub bound on the MIP by 50% iPrOH for three times. A desorption efficiency close to 80% was achieved, which was as high as those of the surfactants. These data further proved that multiple interactions (H-bonding, van der Waals force and hydrophobic effects) were involved in the interactions between Ub and the PDA-based MIPs.
Theoretically, high temperature is favourable for the hydrophobic interactions and detrimental to the secondary and tertiary structures of proteins. However, ubiquitin has great thermal stability and the denatured tertiary structure caused by high temperature can be easily restored.32 So we further compared the dissociation efficiencies of Tris–HCl buffer and iPrOH solution at different pH and temperature conditions (Fig. 5(c)). At 70 °C, the desorption efficiency in acidic solution (pH 2.0) reached to >85%, much higher than that at neutral conditions. This might be attributed to the increase of the repulsion between the positively charged residues in ubiquitin in acidic conditions, which destabilized the protein and lowered the melting temperature relative to neutral pH.32,33 When the temperature further rose from 70 °C to 90 °C, the desorption efficiencies in Tris–HCl buffer (pH = 7.0), 20% iPrOH or 50% iPrOH all increased to ≥80%. Taken above results together, apart from trypsin digestion, the target ubiquitin could be reversibly recovered as an intact protein at 70 °C, pH = 2.0 or 90 °C under neutral conditions.
Application of the MIP NPs to inhibit the ubiquitin polymerization and isolate ubiquitin and ubiquitinated proteins from cell lysates
Ubiquitin tends to form poly-Ub chains with high molecular weights under the catalysis of E1, E2 and E3 ligases. We added the MIP NPs to the ubiquitin solution to test their effects on the polyubiquitylation reaction process. After magnetic separation, the polymerization products were measured by immunoblotting analysis (Fig. 5(d)). In comparison with the control solution without the addition of any NPs (Lane 1), the MIP NPs substantially inhibited the ubiquitin polymerization (Lane 2), while the NIP NPs only showed slight influences on the reaction (Lane 3). These results indicated that binding of Ub to the MIP NPs significantly blocked the polymerization activity of Ub, which provided a novel nano inhibitor against the formation of polyubiquitination.We then employed the MIP NPs to purify the bacterial cell lysates of BL21 strain that stably expressed ubiquitin. The eluted fractions were collected and measured by SDS-PAGE (Fig. 5(e)). After purification with the MIP NPs, the recovered ubiquitin gave a single band in Lane 2. In contrast, the cleanup efficiency of the NIP NPs was much lower (Lane 3).
Finally, we challenged to apply the MIP NPs to enrich ubiquitinated proteins from the lysates of 293T cells. To inspect the quantitative enrichment capability of the MIP NPs, we spiked the 293T cell lysates with known amount of ubiquitin after removal of the nuclei and cell debris. After incubation with the MIP NPs, the bound ubiquitin was eluted by 0.5% Triton X-100 and quantitatively measured by stable isotope (15N) labelled LC-MS/MS. The recovery of ubiquitin from the spiked cell lysates was found to be 91 ± 4% (n = 3).
For comparison, we used both the MIP NPs and the magnetic nanoparticles functionalized with anti-Ub monoclonal antibodies (mAb-MNPs) to isolate Ub-associated proteins from the 293T cell lysates. The captured proteins were detected by western blot as shown in Fig. 5(f). Like the mAb-MNPs (Lane 1), the MIP NPs (Lane 2) effectively isolated a large number of ubiquitinated proteins from the 293T cells, including the ubiquitin and branched ubiquitin chains. These results implied that the lysine residues that were essential for the formation of these ubiquitin chains were not involved in the interactions between ubiquitin and PDA, thus the branched ubiquitin chains were also identified. Among the isolated proteins, the largest fraction was the ubiquitin monomer (marked in red). This was consistent with the previous reports that free ubiquitin accounted for the largest fraction of total ubiquitin pool in 293T cells.21 In comparison with the bands in Lane 1 obtained by using the mAb-MNPs, the bands near 50 kDa and 25 kDa in Lane 2 (marked in green) were apparently more intense than other bands, indicating different selectivity between the MIP and mAb in binding the ubiquitinated proteins.
After SDS-PAGE separation and in-gel trypsin digestion, the peptides were extracted and subjected to proteomic analysis by LC-MS/MS (Fig. 6(a)). A total of 677 proteins were enriched and identified from the cell lysates by the MIP NPs. Among them 529 proteins were further confirmed to contain at least one ubiquitination site (Fig. 6(b)). In contrast, the mAb-MNPs have extracted 1039 proteins and 657 of them were confirmed to contain at least one ubiquitination site. Among the 446 proteins that have been extracted by both the MIP and the mAb, 242 proteins were found to be ubiquitinated proteins, most of which were involved in very important biological processes (Fig. 6(c)). In comparison with the mAb, MIP extracted 287 different ubiquitinated proteins, most of which were involved in the protein processing in endoplasmic reticulum and ribosome ubiquitin mediated proteolysis. The detailed mechanisms are under further investigation.
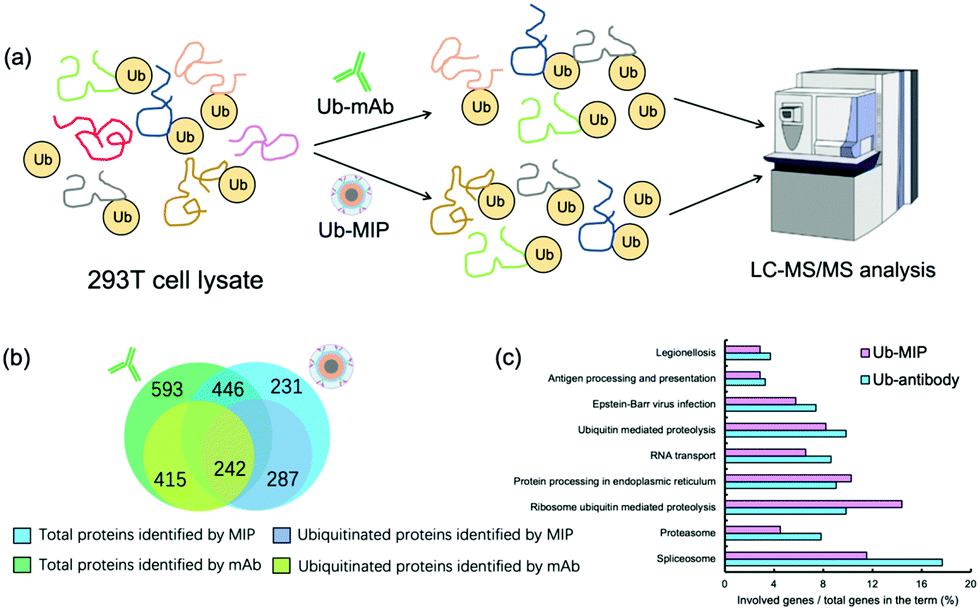 | ||
| Fig. 6 (a) Schematic illustration of the identification and enrichment of ubiquitinated proteins in 293T cell lysates by Ub-mAb or Ub-MIP followed by LC-MS/MS analysis. (b) Total and overlayed proteins and ubiquitinated proteins identified by Ub-mAb and Ub-MIP. (c) Cluster analysis of the ubiquitinated proteins identified by Ub-mAb and Ub-MIP. | ||
So far, many cellular ubiquitinated species remain undetected. In addition to anti-Ub antibodies and UBDs, antibodies that recognize the Lys-ε-Gly-Gly (diGLY) remnant generated following trypsin digestion of ubiquitylated proteins have also been employed to enrich and identify endogenously ubiquitylated proteins.34 This peptide antibody-based affinity approach is valuable and has enabled identification of a great number of ubiquitylation sites in human cells. However, some ubiquitin-like proteins will also leave identical diGLY-modified peptides after trypsinolysis of proteins modified by them, which may lead to overestimation of the ubiquitylation level. With the distinct merits of low cost and high stability, the as-prepared MIP NPs may be utilized either separately or as an important complement to the anti-Ub or anti-diGLY antibodies. In the future study, more diversified functional monomers may be further incorporated into the MIP system to construct more precise binding pockets for ubiquitin or branched ubiquitin chains.
Conclusions
In this work, we have disclosed a strong inherent region-specific interaction between polydopamine and the hydrophobic patch of ubiquitin. Employing dopamine as sole functional monomer, we have further constructed specific binding pockets for ubiquitin over silica coated magnetic nanoparticles via sequential dopamine polymerization and surface PEGylation. The resultant MIP NPs showed a receptor-like binding property to ubiquitin, with a binding constant of 2.6 × 106 L mol−1 and a reversible dissociation of the bound ubiquitin by heating the solution to 70 °C at pH 2.0. These MIP NPs effectively blocked the formation of branched ubiquitin chains and selectively separated ubiquitin from the bacterial cell lysates. Moreover, the MIP-based nano receptors captured 287 ubiquitinated proteins from the lysates of 293T cells which were not identified by the antibodies. With the distinct merits of low cost and high stability, the as-prepared MIP NPs not only offered a useful tool for the purification and enrichment of ubiquitin and ubiquitinated proteins from complex biological samples, but also paved the way for generation more precise nano receptors for branched ubiquitin chains and polyubiquitinated proteins.Author contributions
Z. Li: conceptualization, investigation, formal analysis, writing – original draft. X. Li: investigation, formal analysis, writing – original draft. W. Xian: investigation, formal analysis. H. Xie: investigation. Y. Sun: investigation. Y. Zhang: investigation. J. Wang: methodology. H. Li: investigation, validation, writing – original draft. C. Jin: resources, methodology, supervision. X. Liu: conceptualization, methodology, supervision. Z. Zhu: validation, funding acquisition. M. Zhao: conceptualization, writing – original draft, writing – review & editing, funding acquisition, supervision, project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21974005, 21775007, 21775009).References
- M. Baker, Nature, 2015, 521, 274–276 CrossRef CAS PubMed.
- H. Koide, K. Yoshimatsu, Y. Hoshino, S.-H. Lee, A. Okajima, S. Ariizumi, Y. Narita, Y. Yonamine, A. C. Weisman, Y. Nishimura, N. Oku, Y. Miura and K. J. Shea, Nat. Chem., 2017, 9, 715–722 CrossRef CAS PubMed.
- Y. Hoshino, H. Koide, K. Furuya, W. W. Haberaecker III, S.-H. Lee, T. Kodama, H. Kanazawa, N. Oku and K. J. Shea, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 33–38 CrossRef CAS PubMed.
- S. van Dun, C. Ottmann, L.-G. Milroy and L. Brunsveld, J. Am. Chem. Soc., 2017, 139, 13960–13968 CrossRef CAS PubMed.
- S. Nestora, F. Merlier, S. Beyazit, E. Prost, L. Duma, B. Baril, A. Greaves, K. Haupt and B. T. S. Bui, Angew. Chem., Int. Ed., 2016, 55, 6252–6256 CrossRef CAS PubMed.
- J. J. BelBruno, Chem. Rev., 2019, 119, 94–119 CrossRef CAS PubMed.
- H. R. Culver and N. A. Peppas, Chem. Mater., 2017, 29, 5753–5761 CrossRef CAS PubMed.
- Z. Zhang, Y. Liu, X. Zhang and J. Liu, Nano Lett., 2017, 17, 7926–7931 CrossRef CAS PubMed.
- S. Shinde, Z. El-Schich, A. Malakpour, W. Wan, N. Dizeyi, R. Mohammadi, K. Rurack, A. Gjörloff Wingren and B. Sellergren, J. Am. Chem. Soc., 2015, 137, 13908–13912 CrossRef CAS PubMed.
- T. Takeuchi, Y. Kitayama, R. Sasao, T. Yamada, K. Toh, Y. Matsumoto and K. Kataoka, Angew. Chem., Int. Ed., 2017, 56, 7088–7092 CrossRef CAS PubMed.
- G. Erturk, L. Uzun, M. A. Tumer, R. Say and A. Denizli, Biosens. Bioelectron., 2011, 28, 97–104 CrossRef PubMed.
- K. J. Jetzschmann, A. Yarman, L. Rustam, P. Kielb, V. B. Urlacher, A. Fischer, I. M. Weidinger, U. Wollenberger and F. W. Scheller, Colloids Surf., B, 2018, 164, 240–246 CrossRef CAS PubMed.
- C. M. Pickart, Annu. Rev. Biochem., 2001, 70, 503–533 CrossRef CAS PubMed.
- K. N. Swatek and D. Komander, Cell Res., 2016, 26, 399–422 CrossRef CAS PubMed.
- A. M. Mabb and M. D. Ehlers, in Annual Review of Cell and Developmental Biology, ed. R. Schekman, L. Goldstein and R. Lehmann, 2010, vol. 26, pp. 179–210 Search PubMed.
- D. Komander and M. Rape, in Annual Review of Biochemistry, ed. R. D. Kornberg, 2012, vol. 81, pp. 203–229 Search PubMed.
- M. E. French, C. F. Koehler and T. Hunter, Cell Discovery, 2021, 7, 6 CrossRef CAS PubMed.
- P. Xu, D. M. Duong, N. T. Seyfried, D. Cheng, Y. Xie, J. Robert, J. Rush, M. Hochstrasser, D. Finley and J. Peng, Cell, 2009, 137, 133–145 CrossRef CAS PubMed.
- A. Hershko and A. Ciechanover, Annu. Rev. Biochem., 1982, 51, 335–364 CrossRef CAS PubMed.
- M. D. Petroski, BMC Biochem., 2008, 9(Suppl 1), S7 CrossRef PubMed.
- Y. Shi, P. Xu and J. Qin, Mol. Cell. Proteomics, 2011, 10 Search PubMed.
- Q. Ye, F. Zhou and W. Liu, Chem. Soc. Rev., 2011, 40, 4244–4258 RSC.
- Y. Liu, K. Ai and L. Lu, Chem. Rev., 2014, 114, 5057–5115 CrossRef CAS PubMed.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- M. Zhang, X. Zhang, X. He, L. Chen and Y. Zhang, Nanoscale, 2012, 4, 3141–3147 RSC.
- C. M. Pickart and S. Raasi, Methods Enzymol., 2005, 399, 21–36 CAS.
- D. R. Amin, C. Sugnaux, K. H. A. Lau and P. B. Messersmith, Biomimetics, 2017, 2, 17 CrossRef PubMed.
- M. Hu, Y. Liu, K. Yu and X. Liu, J. Proteomics, 2014, 109, 16–25 CrossRef CAS PubMed.
- H. Lee, J. Rho and P. B. Messersmith, Adv. Mater., 2009, 21, 431–434 CrossRef CAS PubMed.
- J. Jiang, L. Zhu, L. Zhu, B. Zhu and Y. Xu, Langmuir, 2011, 27, 14180–14187 CrossRef CAS PubMed.
- Y. Liu, K. Ai and L. Lu, Chem. Rev., 2014, 114, 5057–5115 CrossRef CAS PubMed.
- O. Kerscher, R. Felberbaum and M. Hochstrasser, Annu. Rev. Cell Dev. Biol., 2006, 22, 159–180 CrossRef CAS PubMed.
- H. S. Chung, A. Shandiz, T. R. Sosnick and A. Tokmakoff, Biochemistry, 2008, 47, 13870–13877 CrossRef CAS PubMed.
- A. Fulzele and E. J. Bennett, Methods Mol. Biol., 2018, 1844, 363–384 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional materials and methods on SEM, XPS, and zeta potential measurement; recovery test. Supplemental results: SDS-PAGE analysis of the GST fusion UBDs; 1H–15N HSQC spectra of ubiquitin by titration with dopamine; optimization of the imprinting time with dopamine and the concentration of mPEG-NH2 for modification of the non-imprinted sites on PDA; reusability test on the MIP NPs. Additional tables of ITC and XPS data. See DOI: https://doi.org/10.1039/d2tb00255h |
| ‡ These authors contributed equally to this work as first authors. |
| This journal is © The Royal Society of Chemistry 2022 |