
Unraveling the role of solvent–precursor interaction in fabricating heteroatomic carbon cathode for high-energy-density Zn-ion storage†
Hui
Duan‡
,
Ziyang
Song‡
,
Ling
Miao
,
Liangchun
Li
 ,
Dazhang
Zhu
,
Lihua
Gan
* and
Mingxian
Liu
*
,
Dazhang
Zhu
,
Lihua
Gan
* and
Mingxian
Liu
*
Shanghai Key Lab of Chemical Assessment and Sustainability, School of Chemical Science and Engineering, Tongji University, Shanghai, 200092, P. R. China. E-mail: ganlh@tongji.edu.cn; liumx@tongji.edu.cn
First published on 23rd March 2022
Abstract
Adsorption-type carbon cathodes deliver great potential for application in aqueous Zn-based hybrid supercapacitors but systematic tailoring of carbon microstructures to optimize the supercapacitive activity of the Zn ion remains a perplexing topic. Herein, a solvent-mediated strategy has been demonstrated to design heteroatomic carbons with tailor-made features for activating Zn storage sites. The solvent–precursor interaction is optimized by a solubility parameter model and molecular growth trajectory simulations, which promote the thermodynamic solubilization (−5.93 kcal mol−1) and growth kinetics (−151.4 kcal mol−1) of the polymeric intermediates with an energy-minimized reaction roadblock. Flake-shaped carbon nanoarchitectures with substantial electrosorption spaces (1022 m2 g−1) and rich O/N motifs (6.03/6.01 wt%) are also demonstrated. Such profitable features implanted in the customized nanocarbon enable low ion migration hurdles for accommodating electroactive species and fast heteroatomic phase conversion for delivering redox response, contributing to a super-high energy density of 92.8 W h kg(cathode)−1 and an ultrastable cycle-life of 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles at 40 A g−1 in the constructed Zn-based hybrid supercapacitor. Comprehensive characterization untangles the high-kinetics chemical adsorption of N-substituted active sites and the powerful redox reactivity of the hydroxyl/carboxyl functionalities on the host Zn ions during each round-trip discharge/charge cycle. This work proposes a design concept of regulating the solvent–precursor interaction for structuring carbons toward highly efficient Zn-ion storage.
000 cycles at 40 A g−1 in the constructed Zn-based hybrid supercapacitor. Comprehensive characterization untangles the high-kinetics chemical adsorption of N-substituted active sites and the powerful redox reactivity of the hydroxyl/carboxyl functionalities on the host Zn ions during each round-trip discharge/charge cycle. This work proposes a design concept of regulating the solvent–precursor interaction for structuring carbons toward highly efficient Zn-ion storage.
Introduction
Aqueous zinc batteries have captured significant research interest in energy storage by virtue of their unique merits including superb energy delivery, excellent chemical stability of Zn in water, and high safety.1–7 However, the dissolution of electrode materials in electrolyte and parasitic reactions generally triggers irreversible capacity attenuation and degrades the work lifetime of zinc metal batteries, thus hindering their further development.8–10 Therefore, the objective being pursued is to harvest compatible features of superb capability output and long-life performance for zinc-based devices, which yet remains a considerable challenge. By introducing the capacitive charge storage mechanism in the cathode side of zinc batteries, the coupling of a capacitive cathode and a battery-type zinc anode can assemble into a hybrid Zn-based supercapacitor.11–14 As a result, the rapid plating/stripping process of Zn2+ on the Zn anode promises battery-level energy supply, while the highly reversible adsorption/desorption of Zn2+ on the carbon cathode ensures capacitor-mode power output, which creates the possibility of collaborating the advantages of two standalone technologies.15–18 The task remaining in this strategy is to develop capacitive cathode materials with the following two traits: (i) the synergy of non-faradaic/faradaic charge storage mechanisms is highly desired to match the zinc anode to achieve high specific capacity and energy density; (ii) the sluggish ion diffusion kinetics should be substantially eliminated to deliver high capacity retention and power output at large current rates.Carbon materials, benefiting from great tunability in geometry, pore structure, and surface chemistry, are extensively implemented as an important category of electrode materials for fulfilling efficient energy storage.19–23 The microstructure engineering of carbons is an effective and essential route to boost the electrode/electrolyte contact interface, lower ion diffusion length, and promote structural stability, thus enhancing the electrochemical reaction kinetics and capacitive energy storage.24–26 In addition, functionalizing porous carbon surfaces with heteroatomic motifs (e.g., O, N, and P species) is an efficient strategy to simultaneously regulate their electronic structure and chemisorption capability for boosting electrochemical ion storage.10 Solution chemistry has been frequently devoted to engineering versatile carbons with designable performances.27–29 In this process, organic molecules as starting materials undergo polymerization in solvents to prepare polymers that can be easily converted into carbons through carbonization.30–32 In comparison to chemical vapor deposition and solid phase fabrication, the solution synthesis strategy allows the design of carbon materials with a broad tailoring of their microstructures that are crucial indicators for use in energy storage.32–35 The solvent effect undoubtedly plays a crucial role during the liquid-phase carbon fabrication. Different solvent–polymer interactions or compatibility exert a notable influence on the nucleation, growth, and assembly of the polymeric intermediates, thus affecting the microstructure properties of the resultant carbons.36–39 Therefore, unraveling the role of solvent–precursor interaction to synthesize heteroatomic porous carbons with optimized microstructures would substantially promote carbon design for energy-related applications.
Herein, an efficient solvent-regulated synthesis strategy is developed to design dual heteroatom-decorated carbon cathodes with tailored microstructures for building advanced Zn-ion hybrid supercapacitors. Tuning the solvent–precursor interaction significantly shapes the dissolution behavior, polymerization energy barrier, and growth kinetics of the polymeric midbody, thereby affecting the resultant carbon features. With an increase in the solvent–polymer interactions, the carbons show gradually refined geometries from particles to sheets along with improved electrosorption spaces (from 33 to 1022 m2 g−1) and tunable O/N motifs. Using the optimized carbon cathode, a constructed Zn-ion hybrid supercapacitor delivers a superb energy supply of 92.8 W h kg(cathode)−1, an excellent power output of 28.3 kW kg−1, and an ultralong lifespan over 40000 cycles at 40 A g−1 with an ultralow capacity decay rate of 1.6% per 10000 cycles. Ex situ spectral characterization untangles the intrinsic chemical adsorption and conversion coupling mechanisms of the assembled hybrid device on the host Zn ions, in which the O/N-embedded carbon frameworks as nanoreactors demonstrate robust chemical adsorption capability for accepting electroactive species, and the hydroxyl/carboxyl groups trigger significant redox phase switching for triggering the pseudocapacitance. This work provides invaluable guidance for optimal nanostructured carbon design and sheds new insight into the mechanism of boosting Zn-ion storage.
Results and discussion
Based on the Schiff-base reaction between 1,5-diaminonaphthalene and glyoxal in different solvents (including water, tetrahydrofuran (THF), dioxane (DOA), N-methyl-2-pyrrolidone (NMP), dimethylformamide (DMF), and ethanol (EtOH)) and the subsequent carbonization procedure (Fig. S1†), various carbon materials were synthesized (termed as CX; X represents the used solvent). The identical characteristic peaks of the obtained polymers were detected in the Fourier transform infrared (FT-IR) spectra (Fig. S2a†). The absorption peak at 1615 cm−1 is assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N covalent bonds, which corroborates the successful formation of the polymers.38 After carbonization at 700 °C, the polymers exhibit varied carbonization yields of 43.0–78.8% (Fig. S2b†), in which the polymer synthesized using EtOH solvent displays optimal thermal stability. The X-ray powder diffraction (XRD) patterns depicted in Fig. S3† show two distinct broad diffraction peaks, indicating the amorphous feature of the carbons.40 Further, the morphologies of CX are well-maintained compared with the polymers (Fig. S4†), revealing good thermal stability. The scanning electron microscopy (SEM) images of CX display various geometries, including bulk for CWater (Fig. 1a), micron-scaled particles for CTHF and CNMP (Fig. 1b and d), adhesive microspheres for CDMF (Fig. 1e), thick flakes for CDOA (Fig. 1c), and thin nanosheets for CEtOH (Fig. 1f). Therefore, the reaction solvent plays a significant role in the assembly of organic precursors into versatile carbons. The corresponding element maps (Fig. S5†) show the homogeneous distribution of nitrogen and oxygen heteroatoms throughout the carbon matrix.
N covalent bonds, which corroborates the successful formation of the polymers.38 After carbonization at 700 °C, the polymers exhibit varied carbonization yields of 43.0–78.8% (Fig. S2b†), in which the polymer synthesized using EtOH solvent displays optimal thermal stability. The X-ray powder diffraction (XRD) patterns depicted in Fig. S3† show two distinct broad diffraction peaks, indicating the amorphous feature of the carbons.40 Further, the morphologies of CX are well-maintained compared with the polymers (Fig. S4†), revealing good thermal stability. The scanning electron microscopy (SEM) images of CX display various geometries, including bulk for CWater (Fig. 1a), micron-scaled particles for CTHF and CNMP (Fig. 1b and d), adhesive microspheres for CDMF (Fig. 1e), thick flakes for CDOA (Fig. 1c), and thin nanosheets for CEtOH (Fig. 1f). Therefore, the reaction solvent plays a significant role in the assembly of organic precursors into versatile carbons. The corresponding element maps (Fig. S5†) show the homogeneous distribution of nitrogen and oxygen heteroatoms throughout the carbon matrix.
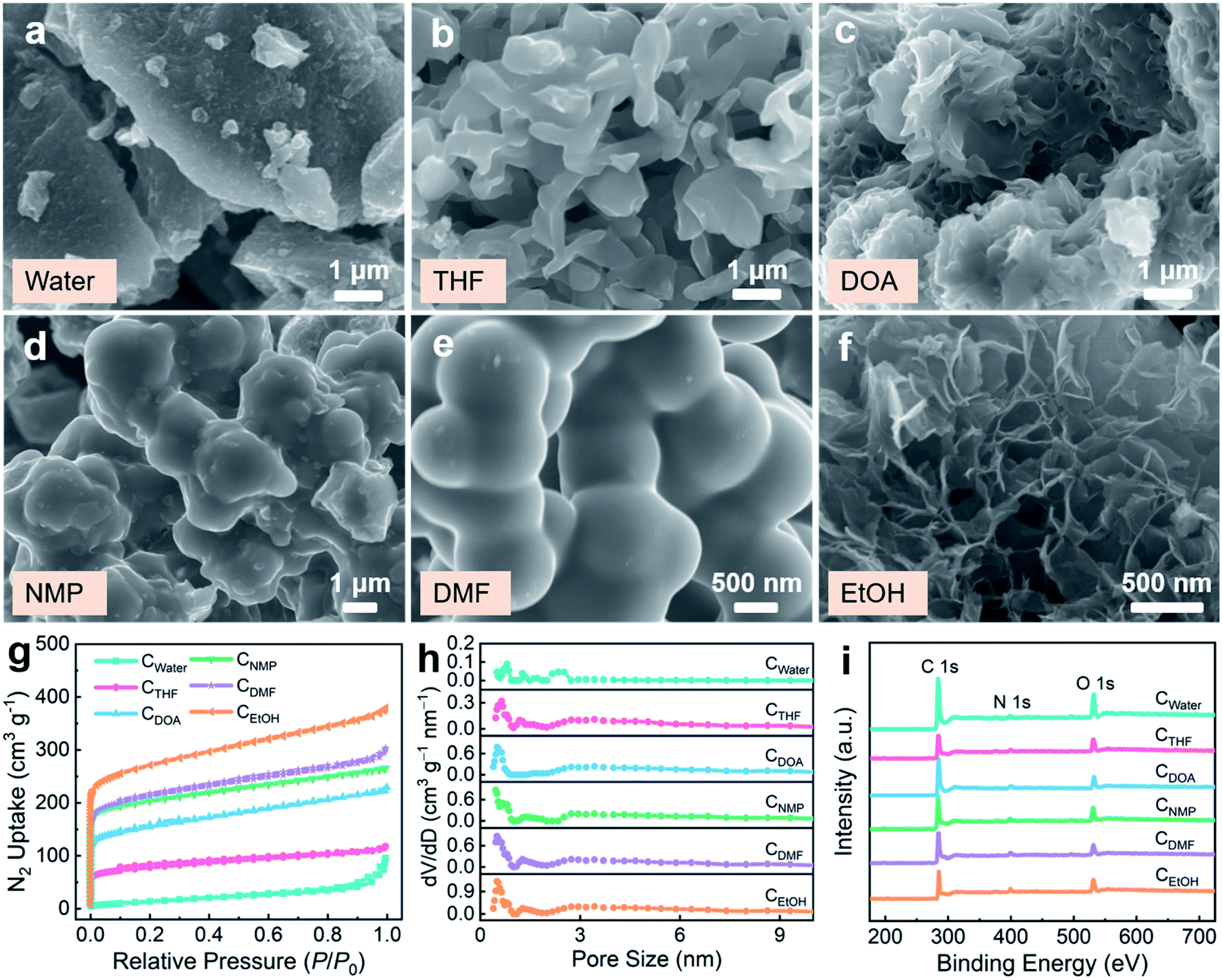 | ||
| Fig. 1 (a–f) SEM images of nanostructured carbons synthesized using (a) water, (b) THF, (c) DOA, (d) NMP, (e) DMF, and (f) EtOH as solvents. (g) Nitrogen adsorption/desorption isothermal curves, (h) pore size distributions, and (i) wide-scan XPS survey spectra of CX. | ||
The N2 adsorption/desorption measurements of CX show representative type-I isotherms with a significant increase at a relative pressure of P/P0 < 0.1 (Fig. 1g), featuring a high-density microporous structure.41,42 All the CX exhibit a similar microporous distribution of < 1 nm, together with few mesopores (Fig. 1h). Abundant micropores in these carbons are created by the decomposition of the polymers and the release of volatile matters during the heat-treatment process.43,44 The surface area of CX is consecutively but radically boosted from 33 to 1022 m2 g−1 (Table 1) by tuning the solvent polarity. CEtOH exhibits the highest surface area among these resulting carbons, which can be assigned to nanosheets arranged in the three-dimensional directions. In contrast, CDMF, CNMP, CDOA, and CTHF with less-defined architectures display relatively low surface areas (286–803 m2 g−1). In particular, a very low surface area of 33 m2 g−1 is produced in CWater due to the dense massive structure. The X-ray photoelectron spectrum (XPS) clearly depicts the strong C (284.2 eV), N (400.2 eV), and O (532.4 eV) signals (Fig. 1i). As shown in Table 1, with the increase in the polymer/solvent compatibility, the N dopant of CX is boosted from 2.78 to 6.01 wt%, while the O content declines from 10.17 to 6.03 wt%. The highest surface area of CEtOH markedly magnifies the surficial functional species exposed to electroactive ions. The high-resolution N 1s and O 1s spectra reveal the chemical natures of the heteroaromatic moieties in CEtOH (Fig. S6†). Three nitrogen signals of the N-6, N-5, and N-Q species represent pyridinic-nitrogen, pyrrolic-nitrogen, and quaternary-nitrogen, respectively.45 The N-6/N-5 species provide rich electrochemically active sites, thus effectively boosting the chemical adsorption of the electrolyte ions.46 N-Q enhances the electrical conductivity of the carbon frameworks and lowers the electron transfer barrier. The O 1s core-level spectrum can be fitted into three genres of CO (O-1), C–OH (O-2), and COOH (O-3).47,48 The O groups are capable of substantially optimizing the surface wettability, which leads to the electrolyte ions having easy access to the electrode surface.47 A surge in the heteroaromatic N/O species of CEtOH unravels the incorporation of substantial defects into the nanostructure skeleton, which is energetically compatible with the adsorbing electrolyte ions.
| Solvent | S BET | O | N | δ T | δ D | δ P | δ H | |δT| | R a | RED |
|---|---|---|---|---|---|---|---|---|---|---|
| Water | 33 | 10.17 | 2.78 | 47.9 | 15.5 | 16 | 42.4 | 22.0 | 25.60 | 5.03 |
| THF | 286 | 9.30 | 3.90 | 19.5 | 16.8 | 5.7 | 8 | 6.4 | 10.32 | 3.12 |
| DOA | 572 | 8.40 | 4.10 | 20.5 | 19 | 1.8 | 7.4 | 5.4 | 13.48 | 2.65 |
| NMP | 790 | 9.58 | 5.12 | 23.1 | 18.0 | 12.3 | 7.2 | 2.7 | 11.48 | 2.26 |
| DMF | 803 | 7.23 | 5.59 | 24.8 | 17.4 | 13.7 | 11.3 | 1.1 | 8.24 | 1.62 |
| EtOH | 1022 | 6.03 | 6.01 | 26.5 | 15.8 | 8.8 | 19.4 | 0.6 | 2.26 | 0.44 |
It was widely held that the macroscopic properties of carbons pertain to the microscopic molecular structure and the growth state of the polymer precursors.49 To deeply understand the influence mechanism of the solvent on the morphology of carbon materials, Hansen solubility parameters (HSP) are applied to estimate the solvent–polymer interaction, which depicts the interaction intensity between the associated matter as follows27
| δT2 = δD2 + δP2 + δH2 | (1) |
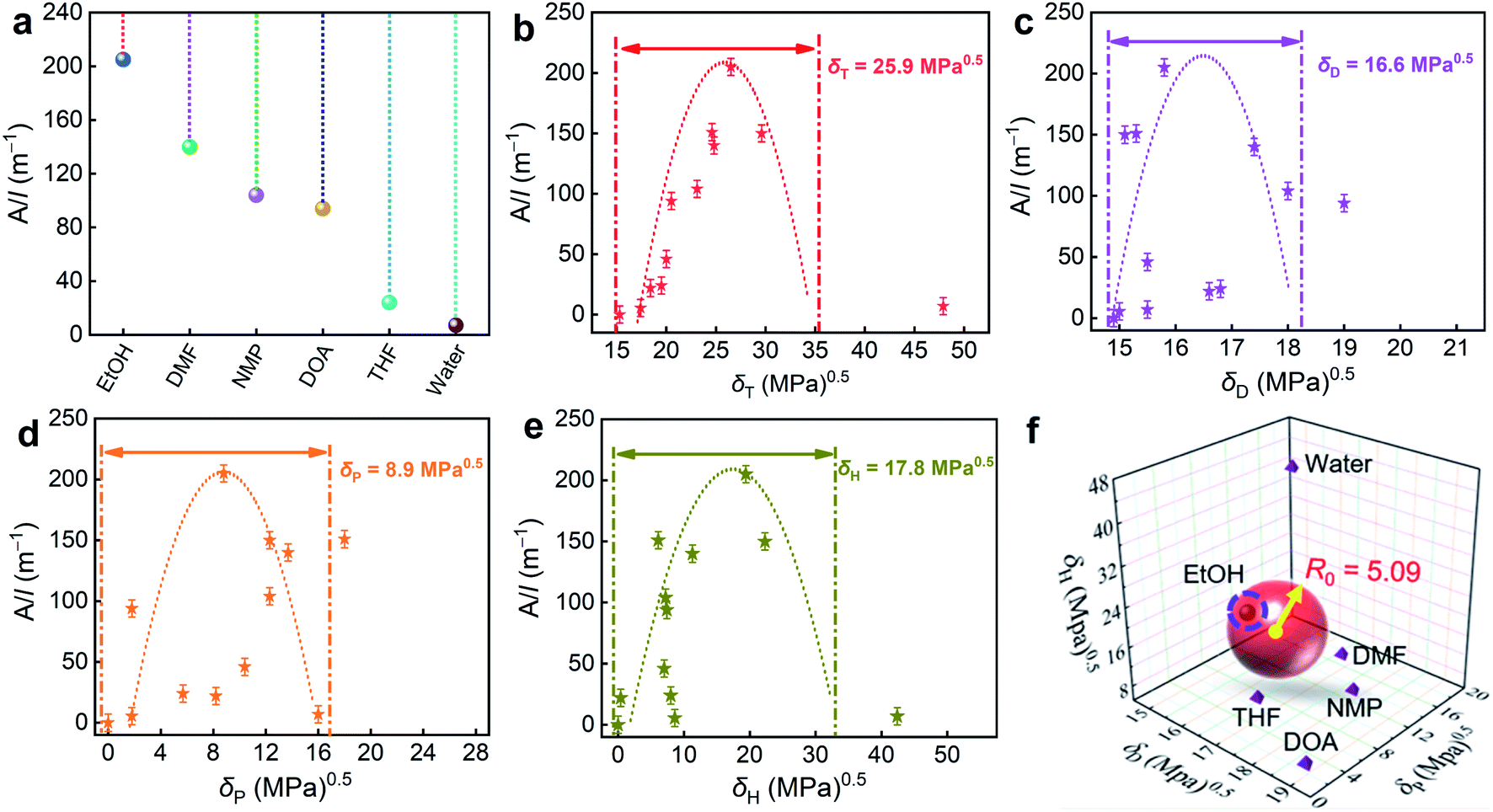 | ||
| Fig. 2 (a) UV/Vis absorbance per cell path length (A/l) of the polymer in diverse experimental solvents. (b–e) Relationships between the solvent/polymer suspension absorbance and δT, δD, δP, and δH, respectively. (f) Hansen solubility sphere of the polymer and δD, δP, and δH values of the six solvents. | ||
A closer δT value for a given material in a solvent means a stronger interaction and a better intimacy. Therefore, we further introduce the difference (|δ|) of HSP between the solvent and the polymer as theoretical guidance to go deep into the role of the solvent in the formation of the differences.49
| |δT| = |δT, polymer − δT, solvent| | (2) |
The δT values of THF, DOA, NMP, DMF, EtOH, and water are 19.5, 20.5, 23.1, 24.8, 26.5, and 47.9 (MPa)0.5, respectively (Table 1). According to the Hansen solubility parameters theory,50 if |δT| > 3, the solvent is poor and not suitable for polymer synthesis; if 1 < |δT| < 3, the solvent could be an intermediary medium; when |δT| < 1, the solvent could be an optimal candidate for polymer growth. In case of water (|δT| = 22.0), it has a higher hydrogen-bond solubility parameter than the polymer that can form strong hydrogen bonds with itself; its inability to dissolve the reaction monomer well and weak stability results in the inferior-defined blocky structure and inferior surface area (33 m2 g−1) and N doping (2.78 wt%). For THF and DOA, the |δT| values are 6.4 and 5.4 (MPa)0.5, indicating faint affinity that provides weak interactions between these two solvents and the polymer. Therefore, these two solvents are not conducive for the growing polymer, producing polymers with heterogeneous particle morphologies, finite active sites (286–572 m2 g−1), and relatively low N content (3.90–4.10 wt%). In contrast, the |δT| values for DMF and NMP are calculated to be 1.1 and 2.7, suggesting their moderate compatibility with the polymer, which contributes to improved surface areas (790–803 m2 g−1) and enriched N contents (5.12–5.59 wt%) for CDMF and CNMP. EtOH with quite a close δT to the polymer (|δT| = 0.6) shows superior matching. The formed linear polymeric intermediates prefer to associate more exclusively with EtOH during the polymerization process and subsequently tend to grow into the sheet assembly due to strong π–π interactions between the adjacent polymer chains. The nanosheet architecture has the potential to maximize the surface area for contact between the polymer and the EtOH solvent. This may be the driving force to extend the interweaving of the nanosheet into a large particle structure. Therefore, CEtOH achieves a large surface area up to 1022 m2 g−1 with optimized micro/mesoporous distribution, and high-level N doping of 6.01 wt%. These results demonstrate that the solvent nature is a striking factor governing the formation of the geometries, pore structure parameters, and functions of the fabricated carbon networks.
Furthermore, the adaptation of three individual energies (δD, δP, and δH) for the solvent/polymer was further assessed by introducing another indicator, HSP distance (Ra), which describes the distance between a solvent and a polymer in a three-dimensional Hansen space known as the Hansen solubility sphere (Fig. 2f) and depends on their separate solubility parameter (δD, δP, and δH) constituents.51
| Ra = [4(δD, solvent − δD, polymer)2 + (δP, solvent − δP, polymer)2 + (δH, solvent − δH, polymer)2].5 | (3) |
Table 1 gives the calculated Ra values of the polymer in these solvents, among all the tested solvents; the polymer has the smallest Ra value in EtOH. The closer the δ values between the polymer and the solvent, its higher stability would be due to the optimum balance of the mixing energy delivered by interactions between and within the matters.52 In a Hansen solubility sphere, the HSP of a desired solvent should be represented at the center of the sphere, and the sphere radius is called the interaction radius R0 (R0 = δT, polymer0.5). On the basis of the Hansen solubility measurement results, an ideal three-dimensional sphere model was built (Fig. 2f). The center coordinates of the sphere correspond to the solubility parameters of the polymer (δD = 16.6, δP = 8.9, and δH = 17.8). A dot in the sphere interior represents a good solvent, whereas a dot outside the sphere indicates a bad solvent. Therefore, EtOH located inside the sphere represents a good solvent for this polymer system. Another very useful parameter is the relative energy difference (RED) value defined as follows.53
| RED = Ra/R0 | (4) |
A good solvent should be inside the sphere (RED < 1), an acceptable solvent is at least near the sphere surface, while a poor solvent is outside the region of the sphere (RED > 1), indicating low affinity.50 The RED value is 0.44 for the EtOH solvent, which is much lower than that of other solvents (Table 1), confirming the similar interaction force between the polymer and the solvent, and a higher miscibility for each other.
To further unravel the solvent effects on the dissolution and growth kinetics of the polymer precursor, density functional theory-based molecular dynamics (DFT-MD) simulation was performed (Fig. 3). The dynamic simulation trajectories of the growing polymer in EtOH and water solvent were quantitatively characterized by the representatively initial and final snapshots, reflecting the stability of the polymer structures and the polymer/solvent interactions.54 On the one hand, to uncover the dissolving effect of various solvents on the polymer, the dissolving energy for each system was calculated as follows
| ΔEdissolution = Etotal − (Esolution + 2Epolymer) | (5) |
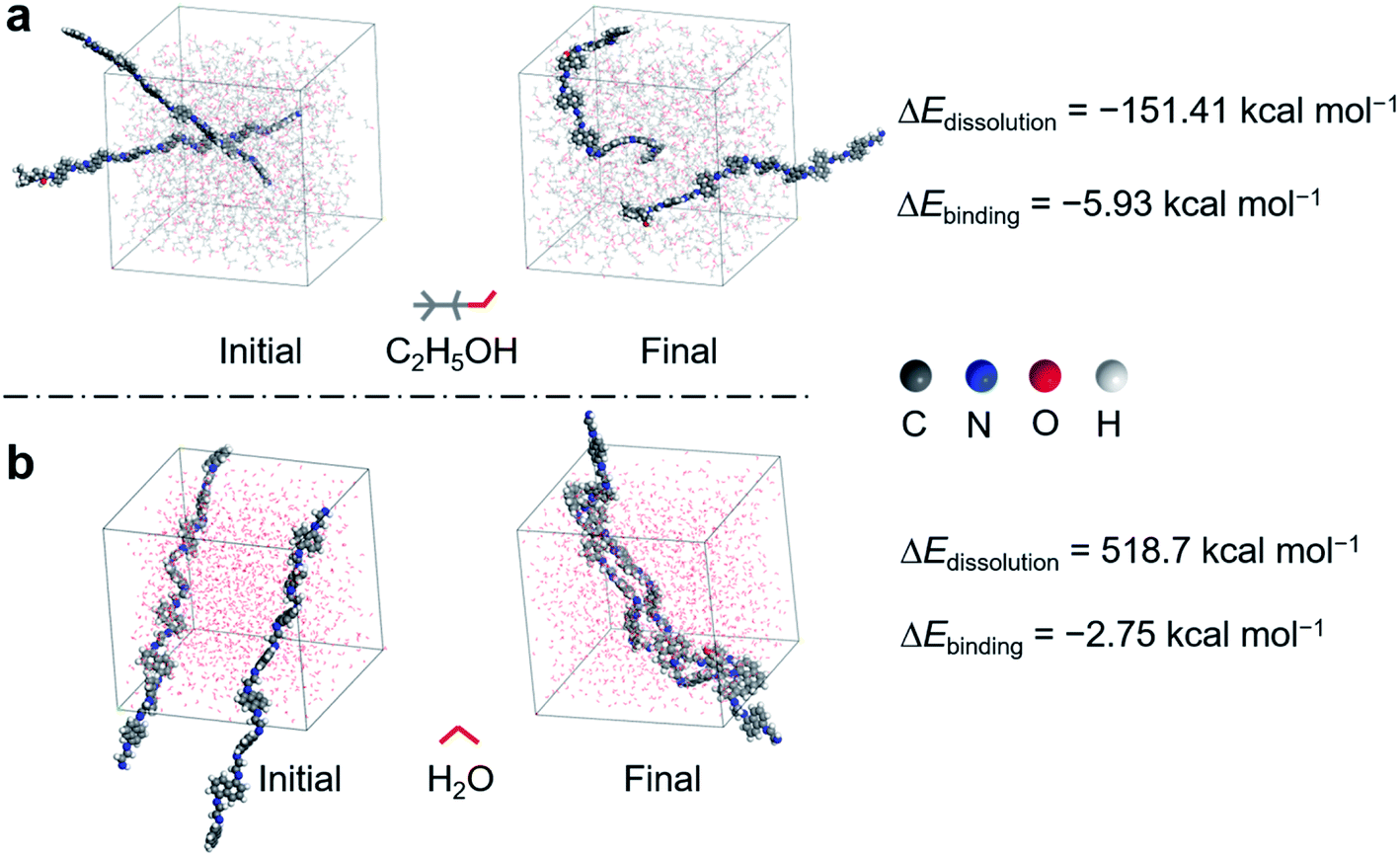 | ||
| Fig. 3 The representative initial and final snapshots for dynamic simulation trajectory of the polymer system in (a) EtOH and (b) water solvent, respectively. | ||
Besides, a COSMO solvation model was constructed to reveal the influence of both solvents on the growth kinetics of the polymer. The binding energy (ΔEbinding) in the first step of polymerization was calculated through the equation below
| ΔEbinding = Edimer − (Esolvent + Emonomer) | (6) |
Fig. 4 shows the schematic illustration of the effects of the solvent–precursor interaction on the formation of the heteroatomic carbons with varied structures and surface functional groups. Regulating the solvent–precursor interaction significantly shapes the thermodynamic solubilization and growth kinetics of the polymeric intermediates, thereby affecting the resultant features of CX. As the solvent–precursor interaction increases, CX exhibits gradually refined morphologies from particles to nanosheets along with boosted surface areas (from 33 to 1022 m2 g−1), and variable O/N functional groups (from 10.17/2.78 to 6.03/6.01 wt%).
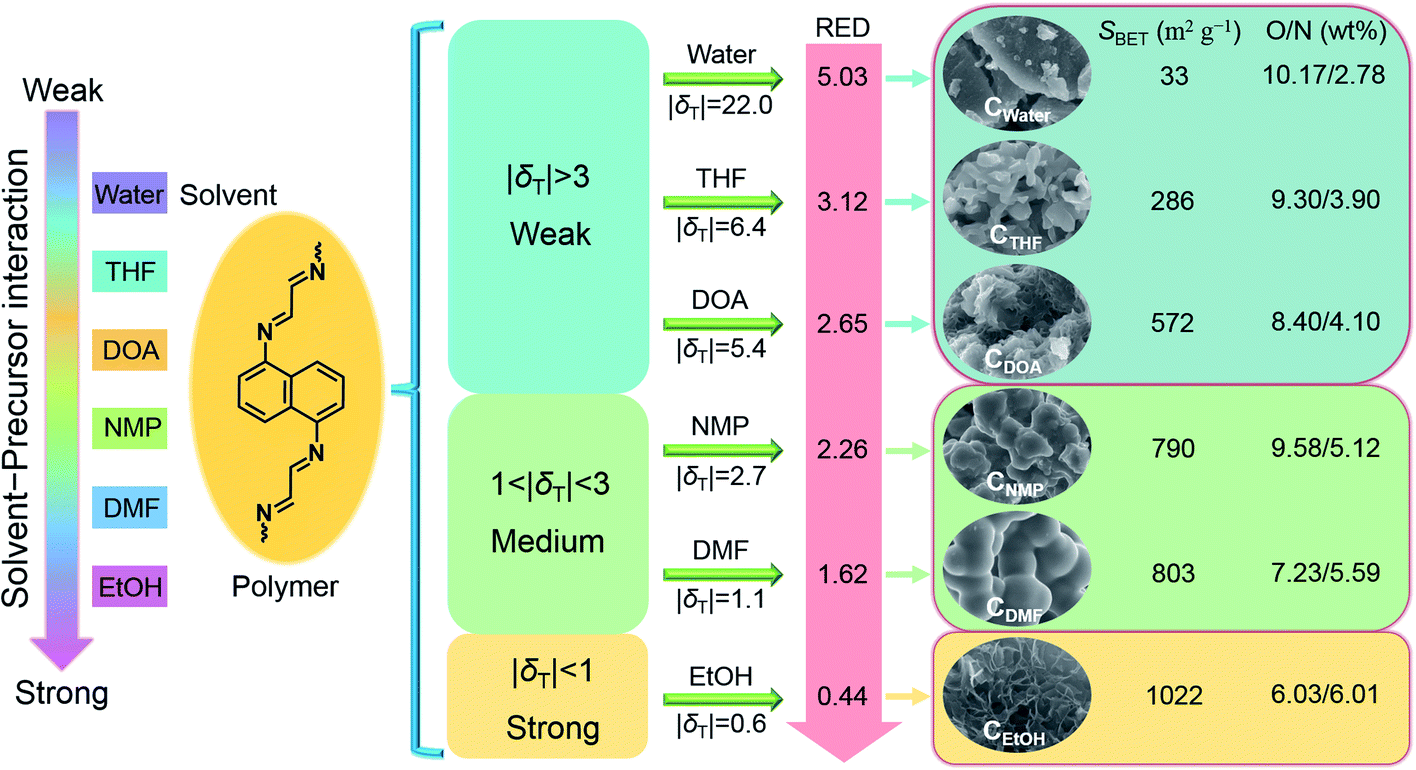 | ||
| Fig. 4 A schematic illustration of the effects of the solvent–precursor interaction on the formation of the heteroatomic carbons with varied structures and surface functional groups. | ||
The controlled tuning of morphology, pore structure, and surface chemistry of carbons is highly desirable for energy storage applications. The electrochemical performances of CX were investigated by constructing Zn-ion hybrid supercapacitors using zinc metal foil as the anode, CX as the cathode, and 3 mol L−1 Zn(SO3CF3)2 aqueous solution as the electrolyte. Rapid ion adsorption/desorption with redox responses on the CX cathode and reversible Zn/Zn2+ deposition/stripping on the Zn anode enable the hybrid devices to efficiently harvest/deliver electrical energy.55,56 The specific capacities of six aqueous Zn-ion devices were determined by galvanostatic charge–discharge (GCD) profiles (Fig. 5a). A high reversible capacity of 170 mA h g−1 at 1 A g−1 is achieved for the CEtOH-based device (30–148 mA h g−1 for other CX devices), along with a retained value of 100 mA h g−1 at 40 A g−1 (Fig. 5b), showing a superb rate capability. The coulombic efficiency of the fabricated CEtOH-based supercapacitor achieves 95.3 and 97.2% at 1 and 2 A g−1, respectively, and almost 100% at 5–40 A g−1 (Fig. S8†), indicating its superb electrochemical reversibility and capacitive behaviors. The specific capacity and cycling performance of the CX-based devices under various rates within ten cycles are shown in Fig. 5c. As the current density rises from 1 to 40 A g−1, the CEtOH-based supercapacitor displays optimal rate performance with a capacity retention of 58.7% in comparison to its counterparts (16.6–54.5%), suggesting a superb large-current tolerance. After a 40-times increase in the current density, the device capacity entirely reverts to the original level, which is indicative of high reversibility and good structural firmness. Besides, a maximum energy density of 92.8 W h kg−1 at 545 W kg−1 (based on the carbon cathodic mass) is obtained for the CEtOH-based device (Fig. 5d), which is higher than those of other five CX devices (20.3–83.6 W h kg−1) and the recently reported carbon-based Zn-ion supercapacitors (Table S2†). In addition, based on the total mass of the CEtOH cathode and Zn anode, the constructed CEtOH-based Zn-ion supercapacitor achieves an energy density of 13.6 W h kg−1. With the power output sharply rising until 28.3 kW kg−1, the device still delivers an ultrahigh energy output of 70.4 W h kg−1, implying superior charge storage capability. Significantly, the CEtOH device presents an ultralong high-rate cycle life of 93.5% capacity retention at 40 A g−1 after 40000 cycles (Fig. 5e), superior to those of the CX counterparts (37.4–88.7%). The SEM image of the CEtOH cathode after cycling (Fig. S9†) indicates the superior structural stability in the Zn-ion hybrid supercapacitor. Through overall comparison (Fig. S10a†), CEtOH achieves the best electrochemical performances due to the well-defined geometry, enhanced surface area, and surface chemistry. Besides, the constructed CEtOH-based Zn-ion hybrid supercapacitor with a voltage window of 1.8 V can light up an orange light-emitting diode (LED) lamp (Fig. S10b†), showing considerable potential for energy storage.
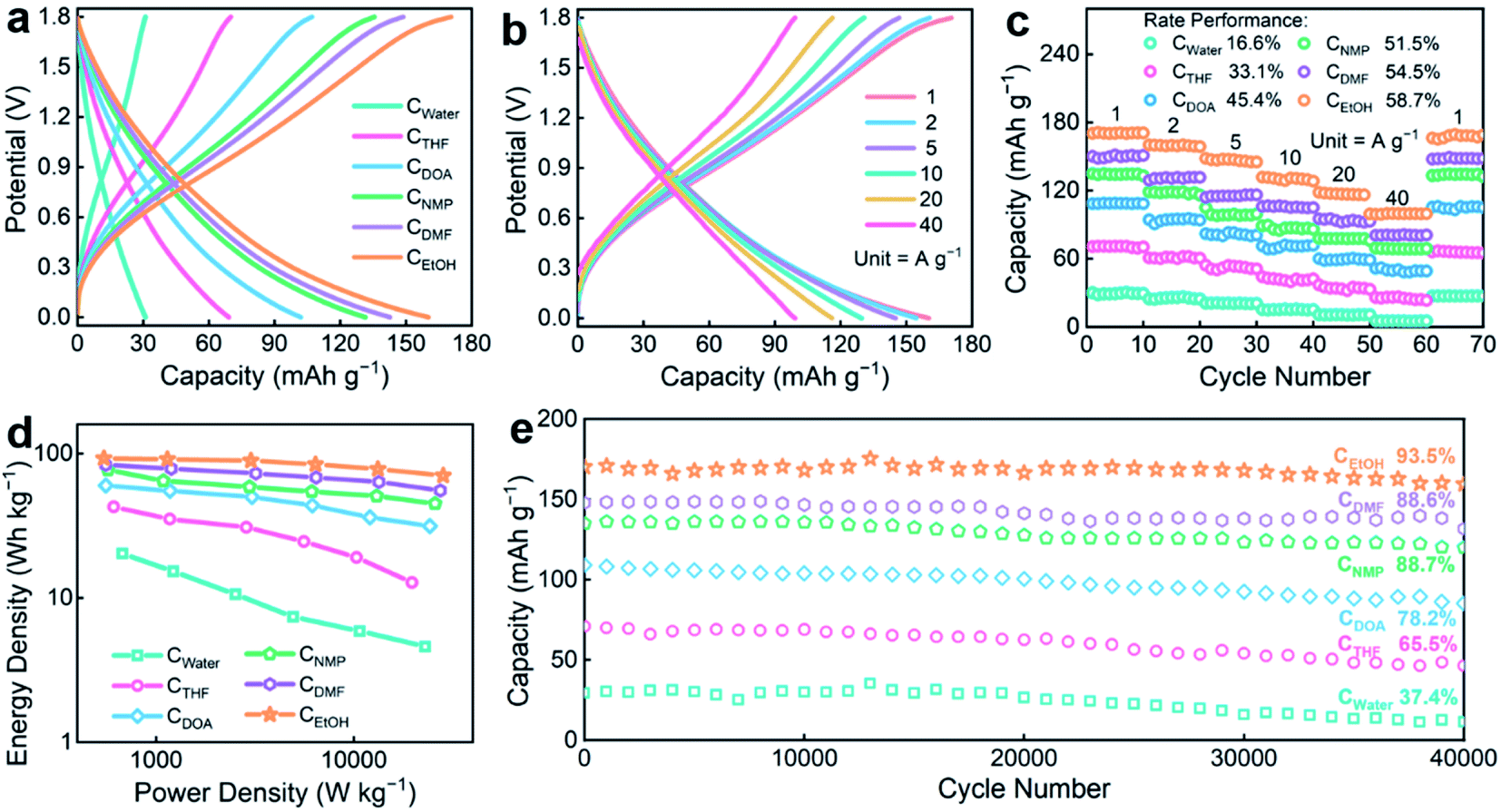 | ||
| Fig. 5 Electrochemical performances of CX-based Zn-ion hybrid supercapacitors. (a) GCD curves of CX devices at 1 A g−1. (b) GCD profiles of the CEtOH device at different current densities. (c) Rate performances. (d) Ragone plots. (e) Cycle stability after 40000 cycles at 40 A g−1. | ||
Considering the high supercapacitive activity and superior rate capability of the CEtOH device, electrochemical impedance spectroscopy (EIS) measurements were further performed to estimate the ion/electron transfer behaviors in depth (Fig. 6a). All CX devices demonstrate incomplete semicircles with linear tails, which are analogous to the features of combining typically fast-kinetics-diffusion-controlled processes as well as the pseudocapacitive process.57,58 Of the six devices, the CEtOH supercapacitor exhibits the smallest equivalent series resistance (Rs) of 1.09 Ω and charge-transfer resistances (Rct) of 15.9 Ω (Table S3†) due to the all-round improvements in the ion-accessibility, interface compatibility, and electrochemical activity of the carbon cathode. The small Rs and Rct are key attributes for CEtOH because thin carbon nanosheets furnish shortened ion diffusion and electron transport distance, and generate large active surface for ion contact. Except for the effective charge transfer dynamics, these devices also display ultrasmall diffusion resistances (σ), reflecting remarkable ion reaction kinetics. Fig. 6b indicates the linear relationship of the real part of impedance (Z′) and the reciprocal of the square root of frequency (ω−0.5) to collect the σ values from the slope of the fitted straight lines based on the following equation.59
| Z′ = σω−.5 + Rs + Rct | (7) |
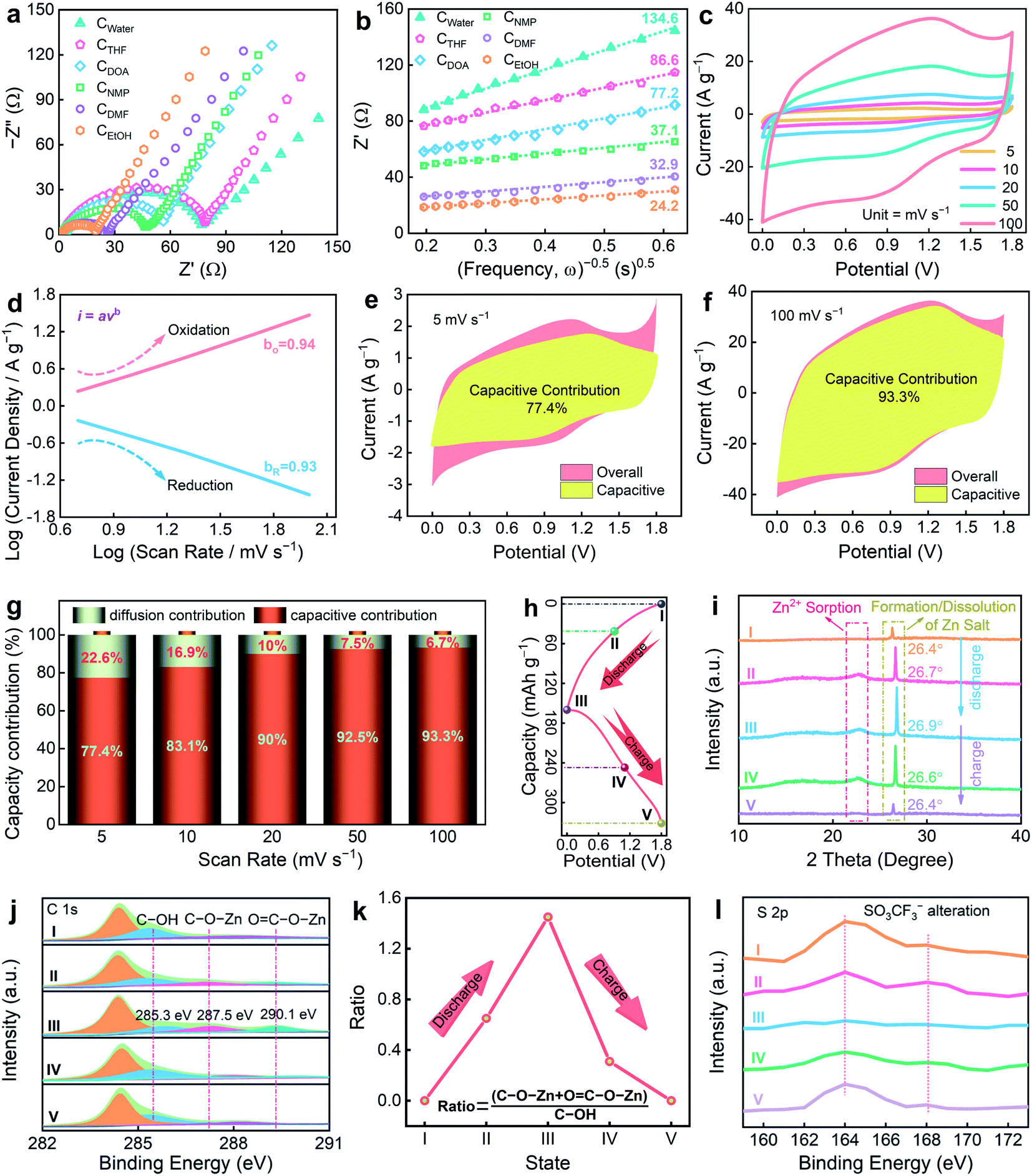 | ||
| Fig. 6 (a) Nyquist plots and (b) relationship between Z and ω−.5 in the middle frequency region of six CX-based Zn-ion supercapacitors. Charge-storage kinetics of the assembled CEtOH-based Zn-ion devices: (c) GCD profiles; (d) the relationship between current (i) and the scan rate (v); (e and f) capacitive contribution (yellow) and diffusion-controlled process at 5 and 100 mV s−1; (g) histograms of the capacity contributed by capacitive and diffusion-controlled processes; (h) a typical GCD curve; (i) XRD patterns; (j) ex situ C 1s XPS spectra; (k) the relative ratio variation of the oxygen species during the charge/discharge process; (l) ex situ S 2p XPS spectra. | ||
CEtOH yields the lowest σ value of 24.2 Ω s−0.5, followed by CDMF (32.9 Ω s−0.5), CNMP (37.1 Ω s−0.5), CDOA (77.2 Ω s−0.5), CTHF (86.6 Ω s−0.5), and CWater (134.6 Ω s−0.5). In addition, the Zn2+ diffusion coefficient (D) within the carbon cathodes can be obtained based on the following formula.60
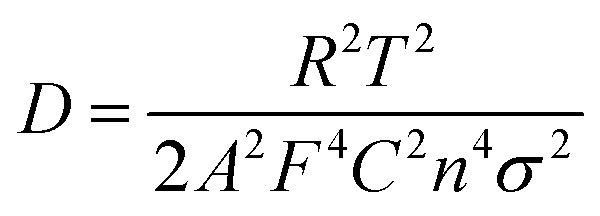 | (8) |
The smallest σ value of the CEtOH device corresponds to a DZn2+ value of 4.23 × 10−9 cm2 s−1, which is higher than that of other devices (0.76–3.11 × 10−9). This result highlights the vital role of the favorable morphology and porous structure in accelerating electrolyte infiltration and ion diffusion in CEtOH.
Quasi-rectangular CV profiles with pronounced redox signals (Fig. 6c) of the CEtOH device are indicative of ion adsorption-induced electrical double-layer capacitance and heteroatom-triggered pseudocapacitance.61,62 With ultralow ion diffusion resistivity, CEtOH is expected to possess ultrafast charge/discharge kinetics. Therefore, the ion diffusion and charge transport kinetics of the CEtOH device were further investigated. The charge-storage kinetics information can be collected by analyzing the relationship between current (i) and the scan rate (v).63
| i = kvb | (9) |
By plotting logi vs. logv, the b values were calculated to be 0.94/0.93 for the cathodic/anodic peaks in the scan-rate range of 5–100 mV s−1 (Fig. 6d), approaching that of an ideal capacitor (b = 1) and suggesting the ultrafast charge-storage behavior of the CEtOH device.64 The respective capacitance contribution from fast and slow reactions can be quantitatively differentiated based on the following equations65
| i = k1v + k2v1/2 | (10) |
| i/v1/2 = k1v1/2 + k2 | (11) |
To further clarify the charge storage mechanism of the CEtOH//Zn device, ex situ X-ray diffraction (XRD) as depicted in a GCD profile (Fig. 6h) was conducted to characterize the structural variation on the CEtOH cathode surface (Fig. 6i). Broad peaks at ∼22.4° can be observed, suggesting the significant adsorption/desorption feature of Zn2+ facilitated by N doping and the unchanged carbon skeleton.40 Sharp peaks at 26.4° correspond to the (002) plane of graphite.67 During the discharge process, the peak intensity continuously increases, along with the shift of the peak position to the low angle region. In the following charge process, the opposite trend can be observed. It is worth mentioning that the characteristic peak of the Zn(CF3SO3)2[Zn(OH)2]3·xH2O byproduct formed during the electrochemical reactions is also located at ∼26.4°. The varied peak location and intensity confirm the reversible increase and disappearance transformation of the Zn(CF3SO3)2[Zn(OH)2]3·xH2O phase on the CEtOH surface.40 Boosted Zn ion adsorption derived from strong electronegative oxygen functionalities could be a typical reason for the reversible formation/dissolution process of the zinc compound product.
To monitor the surface chemistry transformation of the CEtOH cathode during the charge/discharge course in detail, ex situ XPS measurements at different voltage states were performed (Fig. 6j). Three obvious characteristic peaks at 284.4, 285.3, and 288.1 eV at primitive condition (I) originate from C–C, C–OH, and COOH species.68–70 From 1.8 to 0 V in the discharge process, both C–OH and COOH signals undergo a continuous decrease. In the charge process from 0 to 1.8 V, they exhibit a contrary trend, indicating the highly reversible chemical reactions between the C–OH/COOH groups and Zn ions during the whole discharge/charge cycles. As a proof of concept, two new peaks at 287.5 and 290.1 eV derived from C–O–Zn and COO–Zn bonding were detected at full discharge state (III) and gradually faded away in the charge process (state IV to V), suggesting the reversible faradaic reactions of Zn ions with C–OH and COOH groups.71Fig. 6k summarizes the ratio variation trend of these two typical Zn-ion bonding groups during the charging/discharging courses, further proving the highly reversible pseudocapacitive Zn-ion storage mechanisms. The signal of S element can be used to track the trajectory of the SO3CF3− anions in the system, which proves that they contribute to capacity storage. The S signal gradually decays in the process of 1.8–0 V discharging and almost returns back to the original state in the reverse course of 0–1.8 V charging (Fig. 6l). This result indicates that the reversible desorption/adsorption process of the SO3CF3− anions takes place on the carbon surface during the full discharge/charge process and thus contributes to the electric double-layer capacitance. Besides, in view of the large spatial size of the SO3CF3− anions and a majority of <1 nm micropores in CX, it is expected to trigger severe steric hindrance for SO3CF3− diffusion/adsorption within the carbon micropores, leading to a very small amount of anion storage. The hybrid charge storage mechanisms of the CEtOH cathode involves two sources of chemical Zn2+/SO3CF3− adsorption within the porous carbon scaffold (electric double-layer capacitance) and extra redox responses (pseudocapacitance) originating from the variation in the redox states of the oxygen-containing functional groups.
Overall, the CEtOH-based Zn-ion hybrid supercapacitor achieves excellent electrochemical energy storage, which can be attributed to the structural and functional design of the carbon cathode: (i) the open carbon nanosheet assembly avoids the severe restacking of nanosheets and minimizes the loss of electroactive interfaces, enabling sufficient space for charge accumulation; (ii) the porous architecture with large surface area favors the exposure of more active sites and promotes efficient ion diffusion kinetics with a low energy obstacle, contributing to superb supercapacitive activity and rate capability; (iii) anchoring functional nitrogen groups on the carbon surfaces improves the charge distribution environment and boosts the electrode/electrolyte interface interaction, significantly facilitating the chemical adsorption capability of Zn ions for contributing to electric double-layer capacitance. Electroactive oxygen substituents drive reversibly significant redox reactions between the C–OH/COOH groups and Zn ions for harvesting pseudocapacitance. The synergistic coupling of these aspects endows the CEtOH-based Zn-ion hybrid supercapacitor with significant electrochemical performances.
Conclusion
In conclusion, an efficient solvent–precursor interaction-customized strategy is developed to design porous heteroatom-decorated carbons with tailor-made physicochemical properties. By tailoring the solvent–polymer compatibility, the polymerization kinetics and growth behaviors of the oligomers can be regulated systematically, endowing the resultant carbon materials with wide tunability of geometries (bulk, particle, sphere, and sheet), surface areas (33–1022 m2 g−1), and N/O functionalities (2.78/10.17–6.01/6.03 wt%). By constructing a solubility parameter plot and performing molecule dynamic simulations, ethanol solvent is proved to be an optimum, facilitating low binding roadblock (−5.93 kcal mol−1), and robust reaction interaction (−151.4 kcal mol−1) between the solvent and the precursor, thus achieving the accurate synthesis of carbons with optimized nanostructures. With the assistance of the optimal CEtOH cathode, the assembled Zn-ion hybrid supercapacitor achieves a battery-level energy density of 92.8 W h kg(cathode)−1 and a capacitor-level power density of 28.3 kW kg−1, and a prodigious cycle performance of 93.5% energy retention over 40000 cycles at 40 A g−1. Besides, systematic characterization confirms the intense adsorption ability of the N-embellished structural sites and robust chemical interactions of the hydroxyl/carboxyl surface groups with the Zn ions, giving ultrarapid ion reaction kinetics and superb supercapacitive charge storage. This work provides enlightening insights into strategic carbon design and electrochemical mechanism study for efficient Zn-based carbon supercapacitors.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (No. 21875165, 21905207, 22172111, and 51772216), the Science and Technology Commission of Shanghai Municipality, China (No. 22ZR1464100, 20ZR1460300, and 14DZ2261100), and the Fundamental Research Funds for the Central Universities.Notes and references
- Z. Huang, T. Wang, H. Song, X. Li, G. Liang, D. Wang, Q. Yang, Z. Chen, L. Ma, Z. Liu, B. Gao, J. Fan and C. Zhi, Angew. Chem., Int. Ed., 2021, 60, 1011–1021 CrossRef CAS PubMed.
- F. Xie, H. Li, X. Wang, X. Zhi, D. Chao, K. Davey and S. Z. Qiao, Adv. Energy Mater., 2021, 11, 2003419 CrossRef CAS.
- X. Wu, Y. Xu, C. Zhang, D. P. Leonard, A. Markir, J. Lu and X. Ji, J. Am. Chem. Soc., 2019, 141, 6338–6344 CrossRef CAS PubMed.
- W. Sun, F. Wang, S. Hou, C. Yang, X. Fan, Z. Ma, T. Gao, F. Han, R. Hu, M. Zhu and C. Wang, J. Am. Chem. Soc., 2017, 139, 9775–9778 CrossRef CAS PubMed.
- M. Tang, Q. Zhu, P. Hu, L. Jiang, R. Liu, J. Wang, L. Cheng, X. Zhang, W. Chen and H. Wang, Adv. Funct. Mater., 2021, 31, 2102011 CrossRef CAS.
- X. Li, M. Li, Q. Yang, D. Wang, L. Ma, G. Liang, Z. Huang, B. Dong, Q. Huang and C. Zhi, Adv. Energy Mater., 2020, 10, 2001394 CrossRef CAS.
- H. Li, C. Han, Y. Huang, Y. Huang, M. Zhu, Z. Pei, Q. Xue, Z. Wang, Z. Liu, Z. Tang, Y. Wang, F. Kang, B. Li and C. Zhi, Energy Environ. Sci., 2018, 11, 941–951 RSC.
- J. Yin, W. Zhang, W. Wang, N. A. Alhebshi, N. Salah and H. N. Alshareef, Adv. Energy Mater., 2020, 10, 2001705 CrossRef CAS.
- J. Zeng, L. Dong, L. Sun, W. Wang, Y. Zhou, L. Wei and X. Guo, Nano-Micro Lett., 2020, 13, 19 CrossRef PubMed.
- S. Wu, Y. Chen, T. Jiao, J. Zhou, J. Cheng, B. Liu, S. Yang, K. Zhang and W. Zhang, Adv. Energy Mater., 2019, 9, 1902915 CrossRef CAS.
- H. Zhang, Q. Liu, Y. Fang, C. Teng, X. Liu, P. Fang, Y. Tong and X. Lu, Adv. Mater., 2019, 31, 1904948 CrossRef CAS PubMed.
- X. Qiu, N. Wang, Z. Wang, F. Wang and Y. Wang, Angew. Chem., Int. Ed., 2021, 60, 9610–9617 CrossRef CAS PubMed.
- Y. Lu, Z. Li, Z. Bai, H. Mi, C. Ji, H. Pang, C. Yu and J. Qiu, Nano Energy, 2019, 66, 104132 CrossRef CAS.
- R. Yuksel, O. Buyukcakir, P. K. Panda, S. H. Lee, Y. Jiang, D. Singh, S. Hansen, R. Adelung, Y. K. Mishra, R. Ahuja and R. S. Ruoff, Adv. Funct. Mater., 2020, 30, 1909725 CrossRef CAS.
- W. Fan, J. Ding, J. Ding, Y. Zheng, W. Song, J. Lin, C. Xiao, C. Zhong, H. Wang and W. Hu, Nano-Micro Lett., 2021, 13, 59 CrossRef PubMed.
- C. C. Hou, Y. Wang, L. Zou, M. Wang, H. Liu, Z. Liu, H. F. Wang, C. Li and Q. Xu, Adv. Mater., 2021, 33, 2101698 CrossRef CAS PubMed.
- H. Tang, J. Yao and Y. Zhu, Adv. Energy Mater., 2021, 11, 2003994 CrossRef CAS.
- P. Zhang, Y. Li, G. Wang, F. Wang, S. Yang, F. Zhu, X. Zhuang, O. G. Schmidt and X. Feng, Adv. Mater., 2019, 31, 1806005 CrossRef PubMed.
- Z. Song, L. Miao, L. Ruhlmann, Y. Lv, D. Zhu, L. Li, L. Gan and M. Liu, Adv. Mater., 2021, 33, 2104148 CrossRef CAS PubMed.
- L. Peng, C. T. Hung, S. Wang, X. Zhang, X. Zhu, Z. Zhao, C. Wang, Y. Tang, W. Li and D. Zhao, J. Am. Chem. Soc., 2019, 141, 7073–7080 CrossRef CAS PubMed.
- B. Yin, S. Liang, D. Yu, B. Cheng, I. L. Egun, J. Lin, X. Xie, H. Shao, H. He and A. Pan, Adv. Mater., 2021, 33, 2100808 CrossRef CAS PubMed.
- X. Hu, G. Zhong, J. Li, Y. Liu, J. Yuan, J. Chen, H. Zhan and Z. Wen, Energy Environ. Sci., 2020, 13, 2431–2440 RSC.
- S. Herou, J. J. Bailey, M. Kok, P. Schlee, R. Jervis, D. J. L. Brett, P. R. Shearing, M. C. Ribadeneyra and M. Titirici, Adv. Sci., 2021, 8, 2100016 CrossRef CAS PubMed.
- W. Ye, L. Wang, Y. Yin, X. Fan, Y. Cheng, H. Gao, H. Zhang, Q. Zhang, G. Luo and M.-S. Wang, ACS Energy Lett., 2021, 6, 2145–2152 CrossRef CAS.
- Y. V. Kaneti, J. Zhang, Y.-B. He, Z. Wang, S. Tanaka, M. S. A. Hossain, Z.-Z. Pan, B. Xiang, Q.-H. Yang and Y. Yamauchi, J. Mater. Chem. A, 2017, 5, 15356–15366 RSC.
- J. Wang, Y. Yao, C. Zhang, Q. Sun, D. Cheng, X. Huang, J. Feng, J. Wan, J. Zou, C. Liu and C. Yu, Adv. Sci., 2021, 8, 2100120 CrossRef CAS PubMed.
- Z. Xu, X. Zhuang, C. Yang, J. Cao, Z. Yao, Y. Tang, J. Jiang, D. Wu and X. Feng, Adv. Mater., 2016, 28, 1981–1987 CrossRef CAS PubMed.
- S. Chen, D. M. Koshy, Y. Tsao, R. Pfattner, X. Yan, D. Feng and Z. Bao, J. Am. Chem. Soc., 2018, 140, 10297–10304 CrossRef PubMed.
- G. Chen, Y. Yan, J. Wang, Y. S. Ok, G. Zhong, B. Y. Guan and Y. Yamauchi, Angew. Chem., Int. Ed., 2020, 59, 19663–19668 CrossRef CAS PubMed.
- J. Yan, H. Li, K. Wang, Q. Jin, C. Lai, R. Wang, S. Cao, J. Han, Z. Zhang, J. Su and K. Jiang, Adv. Energy Mater., 2021, 11, 2003911 CrossRef CAS.
- W. Li, B. Li, M. Shen, Q. Gao and J. Hou, Chem. Eng. J., 2020, 384, 123309 CrossRef CAS.
- Z. Tang, G. Zhang, H. Zhang, L. Wang, H. Shi, D. Wei and H. Duan, Energy Storage Mater., 2018, 10, 75–84 CrossRef.
- P. Liu, Y. Gao, Y. Tan, W. Liu, Y. Huang, J. Yan and K. Liu, Nano Res., 2019, 12, 2835–2841 CrossRef CAS.
- T. Liu, Z. Zhou, Y. Guo, D. Guo and G. Liu, Nat. Commun., 2019, 10, 675 CrossRef CAS PubMed.
- C. Wang, Y. V. Kaneti, Y. Bando, J. Lin, C. Liu, J. Li and Y. Yamauchi, Mater. Horiz., 2018, 5, 394–407 RSC.
- T. N. Gao, T. Wang, W. Wu, Y. Liu, Q. Huo, Z. A. Qiao and S. Dai, Adv. Mater., 2019, 31, 1806254 CrossRef PubMed.
- Y. Li, S. Wu, L. Zhang, X. Xu, Y. Fang, J. Yi, J. Kim, B. Shen, M. Lee, L. Huang, L. Zhang, J. Bao, H. Ji and Z. Huang, Angew. Chem., Int. Ed., 2020, 59, 21525–21529 CrossRef CAS PubMed.
- J. Yu, C. Yu, W. Guo, Z. Wang, S. Li, J. Chang, X. Tan, Y. Ding, M. Zhang, L. Yang, Y. Xie, R. Fu and J. Qiu, Nano Energy, 2019, 64, 103921 CrossRef CAS.
- L. Li, F. Lu, R. Xue, B. Ma, Q. Li, N. Wu, H. Liu, W. Yao, H. Guo and W. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 26355–26363 CrossRef CAS PubMed.
- Z. Li, D. Chen, Y. An, C. Chen, L. Wu, Z. Chen, Y. Sun and X. Zhang, Energy Storage Mater., 2020, 28, 307–314 CrossRef.
- Q. Li, Z. Dai, J. Wu, W. Liu, T. Di, R. Jiang, X. Zheng, W. Wang, X. Ji, P. Li, Z. Xu, X. Qu, Z. Xu and J. Zhou, Adv. Energy Mater., 2020, 10, 1903750 CrossRef CAS.
- Y. Qian, S. Jiang, Y. Li, Z. Yi, J. Zhou, J. Tian, N. Lin and Y. Qian, Energy Storage Mater., 2020, 29, 341–349 CrossRef.
- W. Feng, N. Feng, W. Liu, Y. Cui, C. Chen, T. Dong, S. Liu, W. Deng, H. Wang and Y. Jin, Adv. Energy Mater., 2020, 10, 2003215 Search PubMed.
- D. Ni, W. Sun, Z. Wang, Y. Bai, H. Lei, X. Lai and K. Sun, Adv. Energy Mater., 2019, 9, 1900036 CrossRef.
- C. Liu, X. Huang, J. Wang, H. Song, Y. Yang, Y. Liu, J. Li, L. Wang and C. Yu, Adv. Funct. Mater., 2018, 28, 1705253 CrossRef.
- D. Qiu, J. Guan, M. Li, C. Kang, J. Wei, F. Wang and R. Yang, Adv. Sci., 2020, 7, 2001681 CrossRef CAS PubMed.
- J. Cui, J. Yin, J. Meng, Y. Liu, M. Liao, T. Wu, M. Dresselhaus, Y. Xie, J. Wu, C. Lu and X. Zhang, Nano Lett., 2021, 21, 2156–2164 CrossRef CAS PubMed.
- Y. G. Lee and G. H. An, ACS Appl. Mater. Interfaces, 2020, 12, 41342–41349 CrossRef CAS PubMed.
- J. Chen, W. Yan, E. J. Townsend, J. Feng, L. Pan, V. D. A. Hernandez and C. F. J. Faul, Angew. Chem., Int. Ed., 2019, 58, 11715–11719 CrossRef CAS PubMed.
- C. M. Hansen, Hansen Solubility Parameters: A Users Handbook, CRC, Boca Raton, 2007 Search PubMed.
- K. R. Graham, P. M. Wieruszewski, R. Stalder, M. J. Hartel, J. Mei, F. So and J. R. Reynolds, Adv. Funct. Mater., 2012, 22, 4801–4813 CrossRef CAS.
- W. Li, J. Hao, P. Zhou, Y. Liu, C. Lu and Z. Zhang, J. Appl. Polym. Sci., 2017, 134, 45405 CrossRef.
- L. Zhao, Q. Wang and K. Ma, ACS Sustainable Chem. Eng., 2019, 7, 10544–10551 CrossRef CAS.
- Z. Song, L. Miao, L. Li, D. Zhu, L. Gan and M. Liu, Carbon, 2021, 180, 135–145 CrossRef CAS.
- Q. Liu, H. Zhang, J. Xie, X. Liu and X. Lu, Carbon Energy, 2020, 2, 521–539 CrossRef CAS.
- L. Dong, X. Ma, Y. Li, L. Zhao, W. Liu, J. Cheng, C. Xu, B. Li, Q.-H. Yang and F. Kang, Energy Storage Mater., 2018, 13, 96–102 CrossRef.
- X. Zheng, L. Miao, Z. Song, W. Du, D. Zhu, Y. Lv, L. Li, L. Gan and M. Liu, J. Mater. Chem. A, 2022, 10, 611–621 RSC.
- K. Tian, J. Wang, L. Cao, W. Yang, W. Guo, S. Liu, W. Li, F. Wang, X. Li, Z. Xu, Z. Wang, H. Wang and Y. Hou, Nat. Commun., 2020, 11, 3884 CrossRef PubMed.
- Z. Song, L. Miao, L. Li, D. Zhu, Y. Lv, W. Xiong, H. Duan, Z. Wang, L. Gan and M. Liu, J. Mater. Chem. A, 2020, 8, 3717–3725 RSC.
- Y. Lu, J. Liang, S. Deng, Q. He, S. Deng, Y. Hu and D. Wang, Nano Energy, 2019, 65, 103993 CrossRef CAS.
- H. Wang, M. Wang and Y. Tang, Energy Storage Mater., 2018, 13, 1–7 CrossRef CAS.
- R. Shi, C. Han, H. Li, L. Xu, T. Zhang, J. Li, Z. Lin, C.-P. Wong, F. Kang and B. Li, J. Mater. Chem. A, 2018, 6, 17057–17066 RSC.
- Y. Liu, Y. Liu, Y. Yamauchi, Z. A. Alothman, Y. V. Kaneti and X. Wu, Batteries Supercaps, 2021, 4, 1–8 CrossRef.
- C. Chen, Y. Huang, Z. Meng, Z. Xu, P. Liu and T. Li, J. Energy Chem., 2021, 54, 482–492 CrossRef.
- C. Han, T. Zhang, J. Li, B. Li and Z. Lin, Nano Energy, 2020, 77, 105165 CrossRef CAS.
- P. Liu, W. Liu, Y. Huang, P. Li, J. Yan and K. Liu, Energy Storage Mater., 2020, 25, 858–865 CrossRef.
- L.-F. Chen, Y. Lu, L. Yu and X. W. Lou, Energy Environ. Sci., 2017, 10, 1777–1783 RSC.
- Y. Zhang, Z. Wang, D. Li, Q. Sun, K. Lai, K. Li, Q. Yuan, X. Liu and L. Ci, J. Mater. Chem. A, 2020, 8, 22874–22885 RSC.
- J. Huang, L. Wang, Z. Peng, M. Peng, L. Li, X. Tang, Y. Xu, L. Tan, K. Yuan and Y. Chen, J. Mater. Chem. A, 2021, 9, 8435–8443 RSC.
- M. Mansuera, L. Miao, Y. Qina, Z. Song, D. Zhu, H. Duan, Y. Lv, L. Li, M. Liu and L. Gan, Chin. Chem. Lett., 2022 DOI:10.1016/j.cclet.2022.03.027.
- Y. Shao, Z. Sun, Z. Tian, S. Li, G. Wu, M. Wang, X. Tong, F. Shen, Z. Xia, V. Tung, J. Sun and Y. Shao, Adv. Funct. Mater., 2020, 31, 2007843 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ta00754a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |