
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePure silica-supported transition metal catalysts for the non-oxidative dehydrogenation of ethane: confinement effects on the stability†
Sudipta De a,
Antonio Aguilar-Tapiab,
Samy Ould-Chikha,
Andrea Zitoloc,
Jean-Louis Hazemannb,
Genrikh Shterka,
Adrian Ramireza and
Jorge Gascon*a
a,
Antonio Aguilar-Tapiab,
Samy Ould-Chikha,
Andrea Zitoloc,
Jean-Louis Hazemannb,
Genrikh Shterka,
Adrian Ramireza and
Jorge Gascon*a
aKAUST Catalysis Center (KCC), King Abdullah University of Science and Technology, Thuwal 23955, Saudi Arabia. E-mail: jorge.gascon@kaust.edu.sa
bInstitut Neel, UPR 2940 CNRS – Université Grenoble Alpes, F-38000 Grenoble, France
cSynchrotron SOLEIL, L'Orme des Merisiers, 91192 Gif-sur-Yvette, France
First published on 25th March 2022
Abstract
Designing robust catalysts for high-temperature applications has always been a critical task for chemical industries. As an example, the non-oxidative dehydrogenation of alkanes is an important chemical process that requires thermally stable metal catalysts with high resistance to metal sintering. The main obstacle is to maintain the high dispersion of the active metal centres under reaction and regeneration conditions. In an attempt to overcome this issue, here we use all-silica zeolite as a support to make nanometric and single-site metal catalysts with enhanced stability for the non-oxidative dehydrogenation of ethane. Preliminary screening of different metal catalysts suggests that Co has the highest intrinsic activity while Cr and V are highly stable against sintering and display a moderate activity. The high stability of Cr and V could be attributed to their high Gibbs energy of reduction under reaction conditions. Operando X-ray absorption spectroscopy revealed that Cr based catalysts remain as single-site monomeric species during the reaction, making it possible to increase the loading and therefore productivity. In the case of Co, we established the optimum parameters to achieve the highest activity by evaluating the effects of support, metal loading, promoter, and synthesis process.
1 Introduction
Light olefins, such as ethylene, propylene, and butenes, are important building blocks for the chemical industry.1 Among them, ethylene is the most widely used building block material, which is used for the production of polymers and other chemicals.1–3 Based on the current consumption of ethylene, its global demand is expected to reach ∼260 million tons in 2023 and ∼290 million tons by 2030.4 The conventional route for industrial ethylene production is the steam cracking of ethane and naphtha, which is energy-intensive due to its high operating temperature and causes severe CO2 emissions.5,6 Therefore, it is worth finding alternative economical and environmentally friendly routes. Increased production of shale gas associated with the reduction of light alkane prices recently encouraged the production of ethylene via the direct dehydrogenation of ethane.7–9 The catalytic dehydrogenation process is a viable alternative to the steam cracking process, which can be carried out at lower temperatures with better ethylene selectivity.Oxidative dehydrogenation is a fascinating route to produce ethylene because the process gives higher ethane conversion and consumes less energy due to the presence of oxidants such as O2, CO2, or N2O.10–12 However, the main obstacle of this process is the over-oxidation of ethane. The non-oxidative process, on the other hand, is more energy demanding, but can achieve better ethylene selectivity. However, high temperature cycling operation under oxidative (during regeneration) and reductive (during reaction) environments, often causes sintering of the active metals to form metal ensembles that induce deep dehydrogenation, including hydrogenolysis and coking.1 For example, small Pt clusters and even single Pt atoms are highly active in ethane dehydrogenation, but large Pt clusters result in coke deposition due to the strong adsorption of ethylene on the surface of the metal.13 A well-known strategy to address this issue is the use of promoters. For example, Sn is the best-known promoter for Pt-based catalysts, where Sn forms an alloy with Pt to increase the dispersion and minimize the hydrogenolysis and coking by suppressing the further dissociation of ethylene due to its fast desorption from the Pt–Sn surface.14–16 Other metal promoters, such as Zn,17,18 Ga,19 In,20,21 Mn,22 and Cu,23 as well as nonmetal promoters, such as B,24 P,25 and S,26 were also reported by many investigators to avoid metal sintering and coke formation.
Another strategy to stabilize the nanosized metal species is trapping them inside a confined space of the support. In this regard, zeolites are an excellent choice of support due to their high surface area, well-defined pore structure, and high thermal stability. Several studies have shown that zeolites can encapsulate metal nanoparticles (NPs) inside their micropores to stabilize them against thermal sintering.27–34 However, the Brønsted acidity of Al centres sometimes shows a negative effect in dehydrogenation reactions, resulting in low alkene selectivity due to cracking reactions.35 Alkali metal promoters (such as Na, K, Cs, etc.) are often used to neutralize the extra acidity of zeolites, but sometimes the added metals can poison the catalysts by altering the Lewis acidity and coordination properties of the active metals.1 As a solution, pure or high silica zeolites and other fully siliceous nano-structured solids have been found suitable for this application due to their low acidity. For example, Sayari et al. introduced the use of pure silica-based MCM-41 supported CrOx catalysts in ethane dehydrogenation.36,37 The catalyst prepared by the one-pot sol–gel method effectively encapsulated the metal species, which resulted in better activity and stability as compared to the catalyst prepared by wet impregnation method. However, the catalyst still suffered from stability in the long-term activity measurement. The main reason could be the presence of bigger pore size and one-dimensional channel of MCM-41 that were not suitable to hold the small CrOx species against sintering at high temperatures. Recent pioneering work by Corma et al. revealed that the purely siliceous MFI type zeolite has a highly suitable pore size and pore structure to stabilize nanosized bimetallic Pt–Sn species against thermal sintering.38 However, the addition of Sn and K was essential to retain the activity and dispersion of Pt for long-run propane dehydrogenation even at 600 °C.
In the present work, we make a thorough study on the activity and stability of different transition metals supported on all-silica MFI zeolite for the non-oxidative ethane dehydrogenation (EDHR). Our initial approach is to screen different monometallic catalysts to find the best candidate. In our further studies, we continue to investigate the effects of support, synthesis method, metal loading, promoter, and reaction conditions on the catalytic performances. We also perform operando X-ray absorption spectroscopic studies to find out the active species and unveil the deactivation mechanism.
2 Experimental
2.1 Catalyst characterization
Nitrogen adsorption and desorption isotherms were measured using a Micromeritics ASAP 2040 instrument at 77 K. Before measurement, the samples were degassed under vacuum at 350 °C for 3 h. The BET method was used to calculate the surface area.Powder X-ray diffraction analysis was performed using Bruker D8 Avance equipment with an operating voltage of 40 kV and Cu Kα radiation. The spectra were recorded in the 2θ range of 5–50° with a step size of 0.018° and 1.5 seconds per step.
Metal contents in the catalysts were measured by using ICP-OES on an Agilent 5100 system. For each analysis, 40 mg of sample was digested using a mixture of 6 mL aqua regia and 1 mL hydrofluoric acid at 373 K for 2 h.
Carbon contents (due to coke formation) in the deactivated catalysts were determined by CHNS elemental analysis on a Thermo Scientific FlashEA 1112 elemental analyser.
The temperature-programmed reduction (TPR) experiments were performed using a Micromeritics Autochem 2920 instrument. For each measurement, 100–120 mg of sample was first heated at 400 °C for 3 h under argon flow to remove the trace amount of water and then cooled down to 60 °C. TPR spectra were recorded by increasing the temperature of the sample to 950 °C at a heating rate of 10 °C min−1 under the gas flow of 10% H2/Ar (flow rate 50 mL min−1).
Transmission electron microscopy (TEM) of the samples was performed on an FEI Titan ST and Themis-Z microscopes by operating it at the accelerating voltage of 300 kV and with a beam current of 0.5 nA. Samples were prepared by sonicating in ethanol and dropping the suspension of the solid samples directly onto carbon-coated copper grids. Dark field imaging was performed by scanning TEM (STEM) coupled to a high-angle annular dark-field (HAADF) detector. The STEM-HAADF data were acquired with a convergence angle of 29.9 mrad and a HAADF inner angle of 30 mrad.
2.2 Method for X-ray absorption spectroscopy (XAS) analysis
Characterization of catalysts under real high-temperature reaction conditions was performed using a high-temperature reaction cell developed at the Néel Institute in Grenoble.40 Co K-edge XAS spectra were recorded under He at room temperature in order to characterize the initial state of the catalyst. Then, the cell was heated up to 650 °C at 5 °C min−1 under H2 as a pretreatment gas. During the heat treatment, XANES spectra were recorded continuously to observe the change of the metal oxidation state, and EXAFS spectra were recorded at the end. After the treatment under 30% H2 in He, the reaction mixture of 20% C2H6 in He (10 mL min−1) was flown while XANES spectra were continuously recorded. The reaction was stopped after ∼10 h and the last 6 EXAFS spectra were merged to establish the final state of the catalysts.
2.3 Syntheses of catalysts
2.4 Catalytic tests
All the catalytic tests at the atmospheric pressure were conducted in a quartz fixed-bed tubular reactor with an inner diameter of 5 mm and a length of 35 cm. 100 mg of the sieved catalyst with a size distribution of 150–250 µm was loaded in the middle of the reactor by using quartz wool. The catalyst was first activated at 650 °C for 30 min under the gas flow of 10% H2/He (flow rate 25 mL min−1), and then the gas was switched to the reactant gas, 20% C2H6/N2, at a total flow of 10 mL min−1. The gas hourly space velocity (GHSV) of ethane was 1200 mL g−1 h−1 in this case. The hydrocarbon mixture and N2 from the outlet were analysed with an online gas chromatograph equipped with a flame ionization detector (FID) and a thermal conductivity detector (TCD). The deactivated catalyst was regenerated by calcination in air at 600 °C for 1 h at a heating rate of 3 °C min−1.The catalytic tests at higher pressure were conducted in a 16 channel Flowrence reactor from Avantium. The mixed feed gas flow was distributed over 16 channels with a relative standard deviation of 2%. The quartz-made reactor tubes have an outside diameter of 3 mm, an inside diameter of 2 mm, and a length of 300 mm. The reactors were first filled with silicon carbide (SiC) particles to make sure that the catalyst bed is in the isothermal zone of the furnace. 50 mg of the sieved catalyst (150–250 µm in size) was then loaded on top of the SiC bed. Prior to feeding the reactant gas mixture of 20% C2H6/N2 (5 mL min−1), catalysts were reduced by 10% H2/He (5 mL min−1) for 30 min at 650 °C. The reactor tubes were pressurized to 7.5 bar using a membrane-based pressure controller working with N2 pressure.
Ethane conversion, ethylene selectivity, carbon balance, ethylene yield, and specific activity of the catalyst (per metal atom basis) are defined by the following equations. For the calculation, N2 was used as an internal standard to measure the change in volume of inlet/outlet gases. We provide the carbon balance value for each reaction, which reflects the amount of coke deposited on the catalyst. We include propane in the equations because we observed a small amount of propane as a product in the high-pressure reaction.
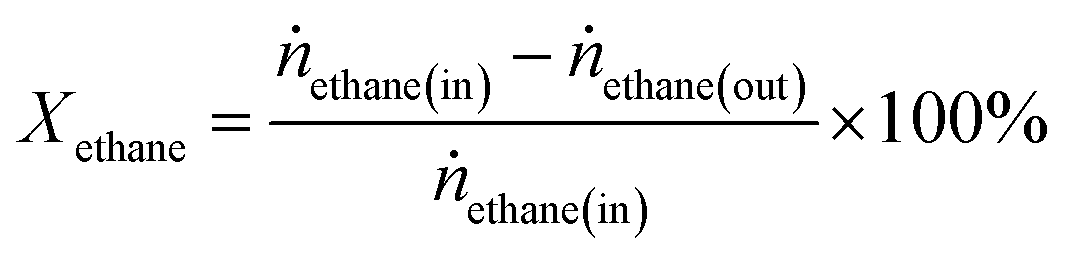 | (1) |
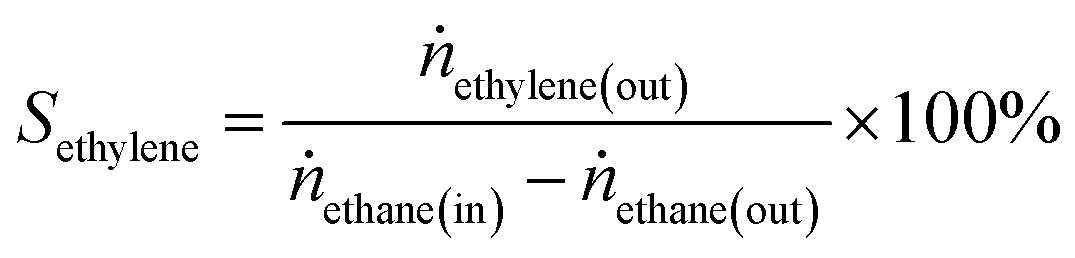 | (2) |
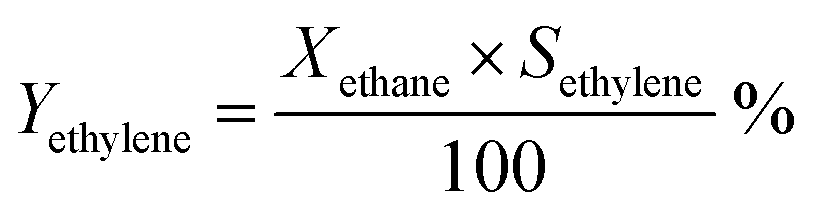 | (3) |
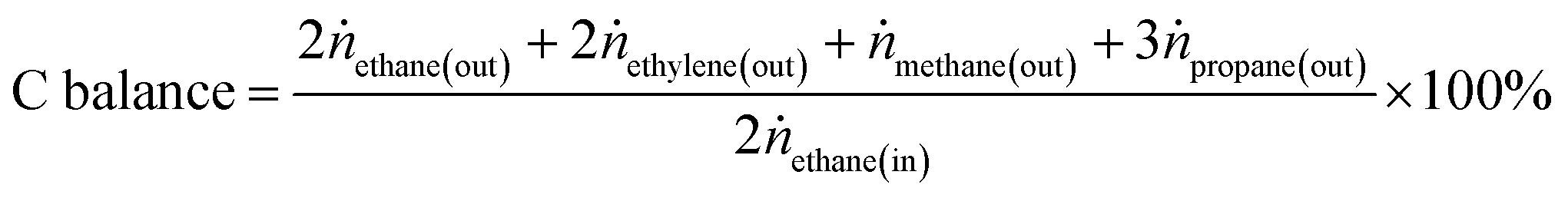 | (4) |
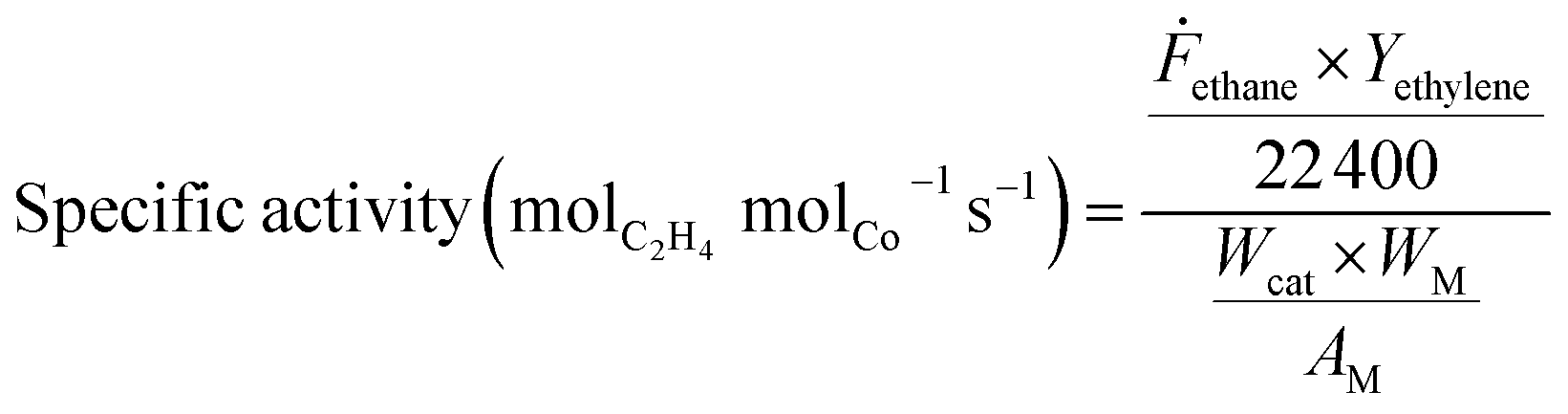 | (5) |
Here ṅx represents the molar flow of component ‘x’, Ḟethane represents the ethane flow (mL) per second in the reaction, Wcat represents the weight of the catalyst (g), WM represents the actual metal content (g) in 1 g of catalyst, AM represents the atomic weight of metal ‘M’ (g mol−1).
3 Results and discussion
3.1 Characterization and catalytic performance
As observed from the XRD results (Fig. S2†), all catalysts display an MFI structure. We started our study with a preliminary screening of different active metals to identify trends in catalytic performance. For this purpose, we chose those transition metals that are reported to be active for alkane dehydrogenation in earlier literature.1,9 Using the reported synthesis method,38 we tried to encapsulate V, Cr, Fe, Co, Ni, Ga, Mo, and Pt inside the MFI zeolite. However, we observed that the bottom-up approach for the one-step synthesis of M@MFI was not effective for all metals. We could successfully encapsulate Cr, Co, and Pt with desired metal loading, while the other metals (in complex form) remained mostly in the mother liquor during the crystallization of the zeolite. The bottom-up approach was very successful for Co and Pt, with nearly quantitative encapsulation up to 2.5 wt% loadings. However the Cr content was smaller than the theoretical loading. In the case of other metals, we applied a simple wet impregnation technique to prepare the M/MFI catalysts. To make a fair comparison of catalytic performance and stability, we loaded all the metals at a fixed M/Si atomic ratio of 0.5 (Table S1†).Catalytic EDHR was studied in a PID reactor at 650 °C and atmospheric pressure with a GHSV of 1200 mL gcat−1 h−1. Catalytic activities are summarized in Fig. 1 and Table 1. The Co@MFI catalyst exhibited the highest initial activity with >25% C2H6 conversion and >99% C2H4 selectivity and remained active for over 60 h. We observed the formation of some small NPs (∼5 nm) of Co at the end of the reaction (Fig. 2d). Cr@MFI and V/MFI also showed good C2H6 conversion and higher stability, with V based catalysts showing a lower C2H4 selectivity. The high stability of these catalysts could be attributed to the low coke formation and resistance to metal sintering. We could not find large NPs through intensive TEM analysis, which indicates that the spent catalysts maintained high dispersion of V and Cr (Fig. 2a and b). Among the other catalysts, Mo/MFI showed an acceptable initial activity but it dropped sharply. Although we did not observe any particle growth of Mo (Fig. 2g), coke formation was significant in this case, which could be the reason for deactivation. Fe, Ni, and Pt exhibited and the formation of large metal particles under reaction conditions could be observed.
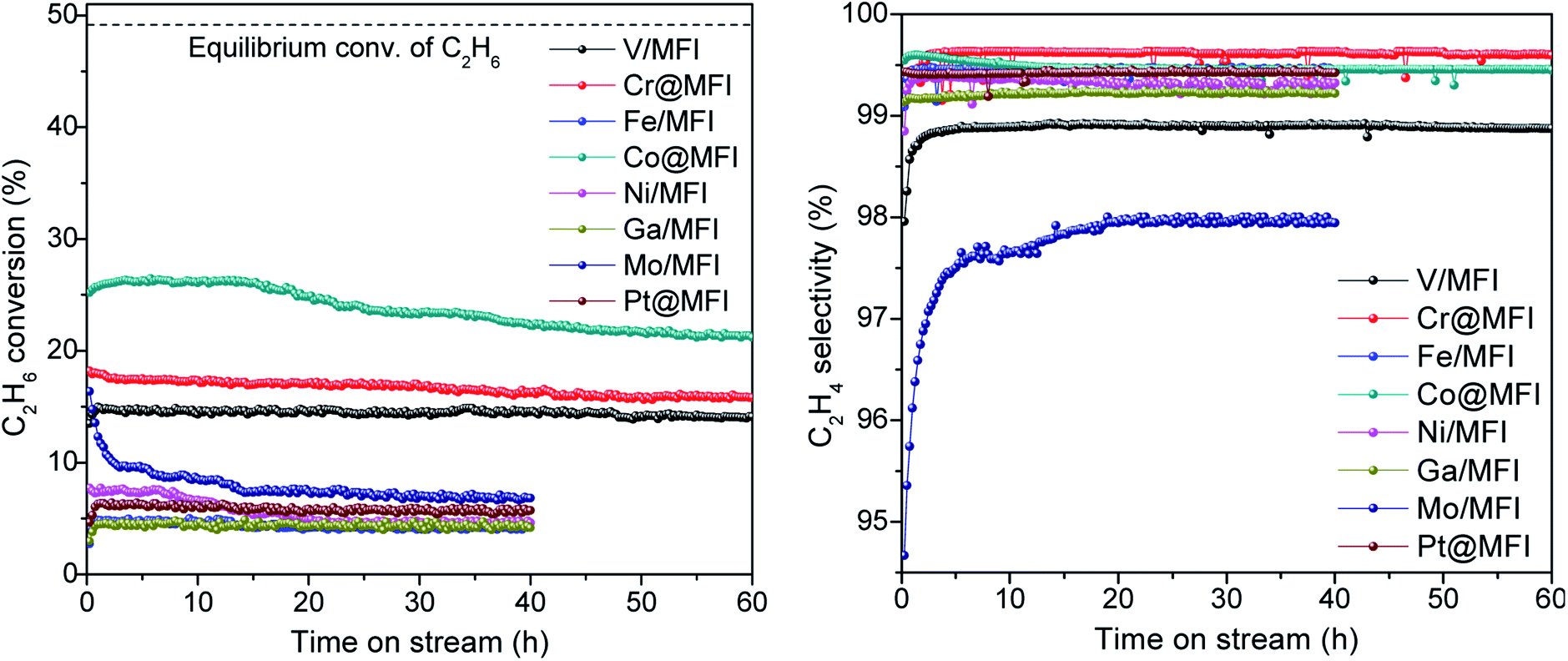 | ||
| Fig. 1 EDH activities of different metal catalysts. Reaction conditions: 100 mg catalyst, pretreatment by 10% H2/N2 for 30 min, 20% C2H6/N2, 10 mL min−1, 1 bar, 650 °C. | ||
| Catalystb | C2H6 conversion & C2H4 selectivity (%) | Specific activityc (molC2H4 molCo−1 s−1) | C balanced (%) | Deactivatione (%) | Coke amount (g gcat−1) | Size of metal NPS after EDH (nm) | |||
|---|---|---|---|---|---|---|---|---|---|
| At 1 h | At 40 h | ||||||||
| a Reaction conditions: 100 mg catalyst, pretreatment by 10% H2/N2 for 30 min, 20% C2H6/N2 as the feed gas, 10 mL min−1, 1 bar, 650 °C.b M/Si atomic ratio in all catalysts was ∼0.5.c Calculated at 1 h.d Calculated at 40 h.e Calculated at 40 h with respect to the highest C2H6 conversion. | |||||||||
| V/MFI | 15.0 | 98.7 | 14.5 | 98.9 | 2.88 | 96.3 | 3.3 | 0.009 | — |
| Cr@MFI | 17.9 | 99.4 | 16.2 | 99.6 | 3.28 | 97.7 | 11.0 | 0.003 | — |
| Fe/MFI | 4.9 | 99.4 | 4.2 | 99.4 | 0.89 | 98.1 | 16.0 | 0.023 | 17 |
| Co@MFI | 25.7 | 99.6 | 22.3 | 99.4 | 4.67 | 96.4 | 15.5 | 0.039 | 7 |
| Ni/MFI | 7.3 | 99.4 | 4.6 | 98.5 | 1.23 | 98.1 | 40.3 | 0.120 | 25 |
| Ga/MFI | 4.5 | 99.2 | 4.2 | 99.2 | 0.83 | 99.4 | 12.5 | 0.006 | — |
| Mo/MFI | 12.3 | 96.1 | 6.8 | 97.9 | 2.20 | 98.6 | 58.5 | 0.068 | — |
| Pt@MFI | 6.3 | 99.4 | 5.7 | 99.4 | 1.22 | 99.3 | 10.9 | 0.007 | 20 |
 | ||
| Fig. 2 TEM images of catalysts after EDH at 1 bar: (a) V/MFI, (b) Cr@MFI, (c) Fe/MFI, (d) Co@MFI, (e) Ni/MFI, (f) Ga/MFI, (g) Mo/MFI, (h) Pt@MFI. | ||
The differences in stability among the different catalysts can be explained when taking into consideration the Gibbs energy (ΔGr) values for the reduction of the metal oxides to their corresponding metals at the reaction temperature (Table S2†). For the calculation of Gibbs energy, we considered the reduction of stable metal oxides with the lowest oxidation state. A negative ΔGr indicates that the reduction of a metal oxide to its corresponding metal is spontaneous, which was observed in the cases of Co, Ni, and Pt, resulting in the formation of NPs (Fig. 2). We also observed Fe NPs due to the low positive value of ΔGr for FeO → Fe. In the case of other metals (V, Cr, Ga, and Mo), ΔGr is positive to highly positive, and thus we did not observe any particle formation.
Based on the comparative studies of different metals, Co showed the highest intrinsic activity and therefore we systematically studied the effects of Co loading, support, synthesis method, base promoter, and pressure on the activity and stability of the catalysts. We prepared Co@MFI catalysts at three different Co loadings. N2 physisorption analysis indicates a characteristic type I adsorption isotherms for all samples with a steep increase at relative pressures between 10−6 and 0.01, which is due to the filling of micropores (Fig. 3a).46 The parent MFI sample showed high microporosity, which was gradually reduced upon increasing the loading of Co up to 1.3 wt% (Table 2), suggesting that the CoOx species possibly occupied the micropores. Further increase of Co loading to 2.5 wt% did not change the micropore area or micropore volume proportionally, which also suggests that the MFI zeolite has a certain limit to encapsulate metal species inside the pores. The result was supported by the TEM analysis (Fig. S3†), where we observe some tiny NPs (<3 nm) in the case of 2.5Co@MFI. In the H2-TPR analysis, we observed two main reduction peaks, a broad peak at a temperature range of 300–500 °C and another at a temperature range of 600–700 °C with a shoulder at 730 °C (Fig. 4). The first peak (300–500 °C) corresponds to the sequential reduction of surface Co3O4 species to metallic Co via CoO.47 The second peak between 600 and 700 °C corresponds to the reduction of Co2+ ions in the pores of MFI zeolite and the shoulder peak could be due to the reduction of Co–SiO2 mixed oxide.48,49 On the other hand, the reduction behaviour of CoOx species in the samples 0.5Co@MFI and 1.3Co@MFI were quite similar. No peak below 600 °C was observed, which indicates the absence of surface Co3O4, meaning that the CoOx species were successfully encapsulated by the MFI zeolite at lower loading. As can be seen from temperature-programmed desorption (TPD) analysis with NH3 (Fig. S4†), the MFI support has no such acidity, which is a desired property to minimize the coke formation.
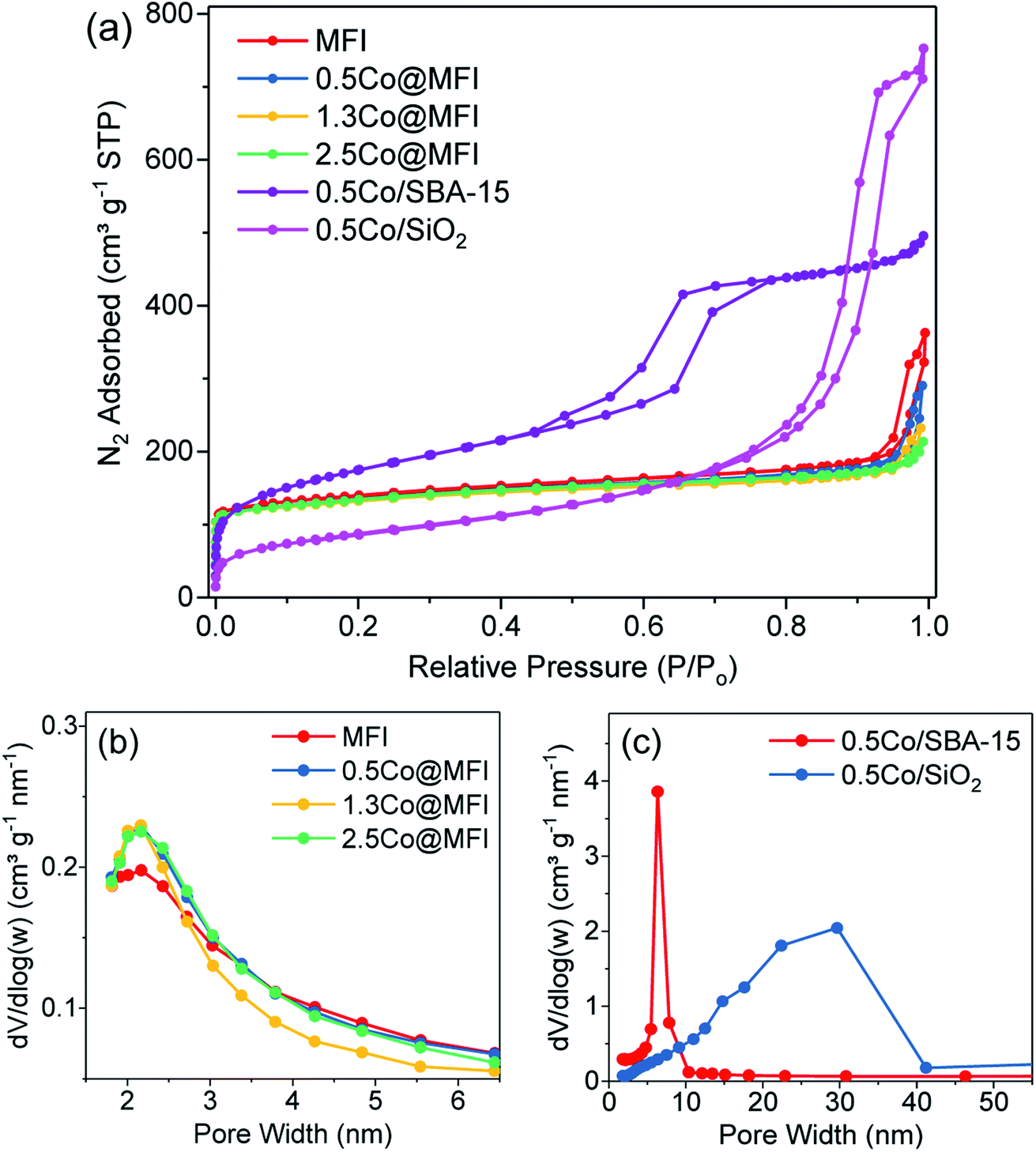 | ||
| Fig. 3 (a) Nitrogen adsorption–desorption isotherms and (b and c) pore size distribution of different silica samples. | ||
| Sample | BET surface areaa (m2 g−1) | Micropore areab (m2 g−1) | External surface area (m2 g−1) | Total pore volumec (cm3 g−1) | Micropore volume (cm3 g−1) | Pore sized (nm) |
|---|---|---|---|---|---|---|
| a The BET surface areas were calculated from the data in the relative pressure range of P/P0 = 0.05–0.25.b Micropore area and micropore volume were calculated by t-plot method.c Pore volume was estimated from the amounts absorbed at a relative pressure of P/P0 = 0.98.d Pore size distributions were calculated from adsorption branch using the BJH method. | ||||||
| MFI | 517 | 326 | 191 | 0.31 | 0.13 | 2.4 |
| 0.5Co@MFI | 498 | 313 | 185 | 0.29 | 0.13 | 2.3 |
| 1.3Co@MFI | 493 | 307 | 186 | 0.27 | 0.12 | 2.2 |
| 2.5Co@MFI | 492 | 306 | 186 | 0.27 | 0.12 | 2.2 |
| 0.5Co/SBA-15 | 596 | 60 | 536 | 0.71 | 0.04 | 6.4 |
| 0.5Co/SiO2 | 301 | 0 | 301 | 0.98 | 0 | 29.7 |
| 0.5Co@MFI_used | 478 | 316 | 162 | 0.28 | 0.13 | 2.3 |
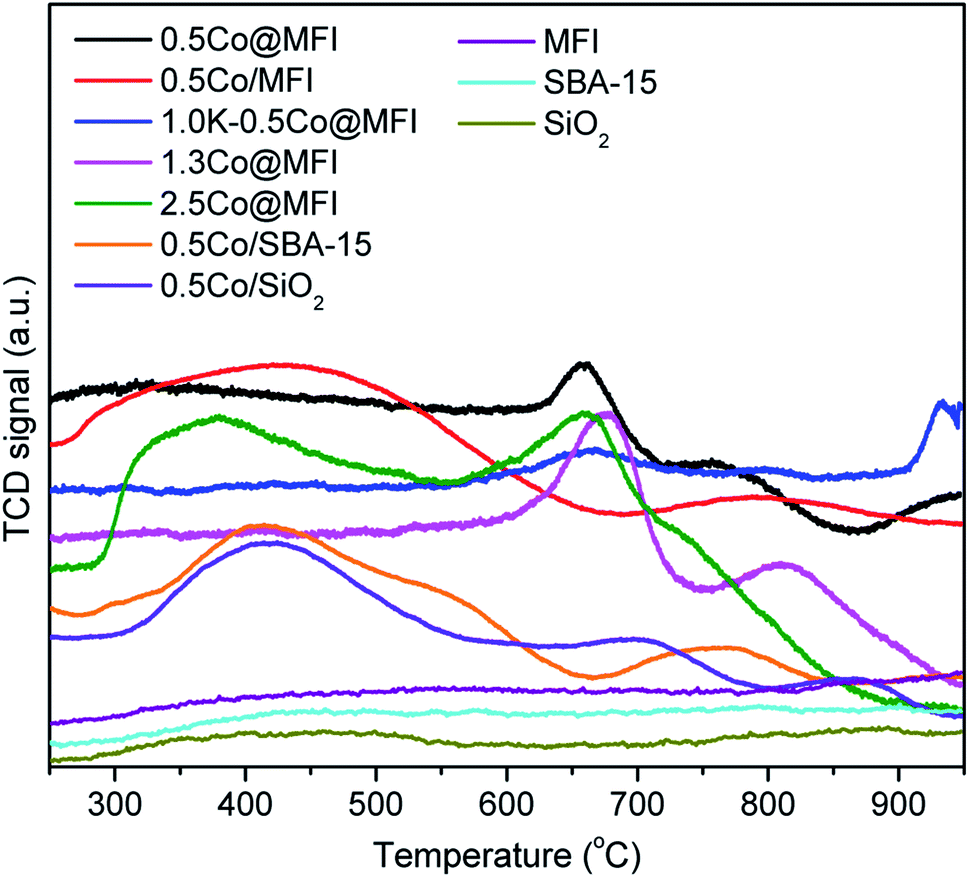 | ||
| Fig. 4 H2-TPR profiles of different Co-based catalysts and bare supports. | ||
Catalytic activity and stability of the Co@MFI catalysts with three different Co loading are summarized in Fig. 5. The initial activities of 1.3Co@MFI and 2.5Co@MFI were higher as compared to 0.5Co@MFI, however, they deactivated faster, and the C2H6 conversion level reached below 20% in 10 h. The main reason was the formation of NPs (Fig. S6†) and a decrease in the active surface of Co. On the other hand, 0.5Co@MFI maintained a steady C2H6 conversion over time with a slower deactivation rate. STEM analysis and elemental mapping were performed to compare the Co distribution between fresh and used 0.5Co@MFI catalyst (Fig. S3 and S7†), and we observed that Co was dispersed quite well along with some small NPs after the reaction. We did the N2 physisorption analysis of the spent catalyst (Fig. S8†) and found only a little drop in the total surface area because of coke formation. However, since the coke was mostly accumulated out of the MFI crystals (Fig. S7c†), the pore structures (micropore area, micropore volume, and pore size) were unchanged (Table 2), which could be another reason for its better stability. The above studies indicate that an optimal loading of Co is necessary to achieve high activity and stability.
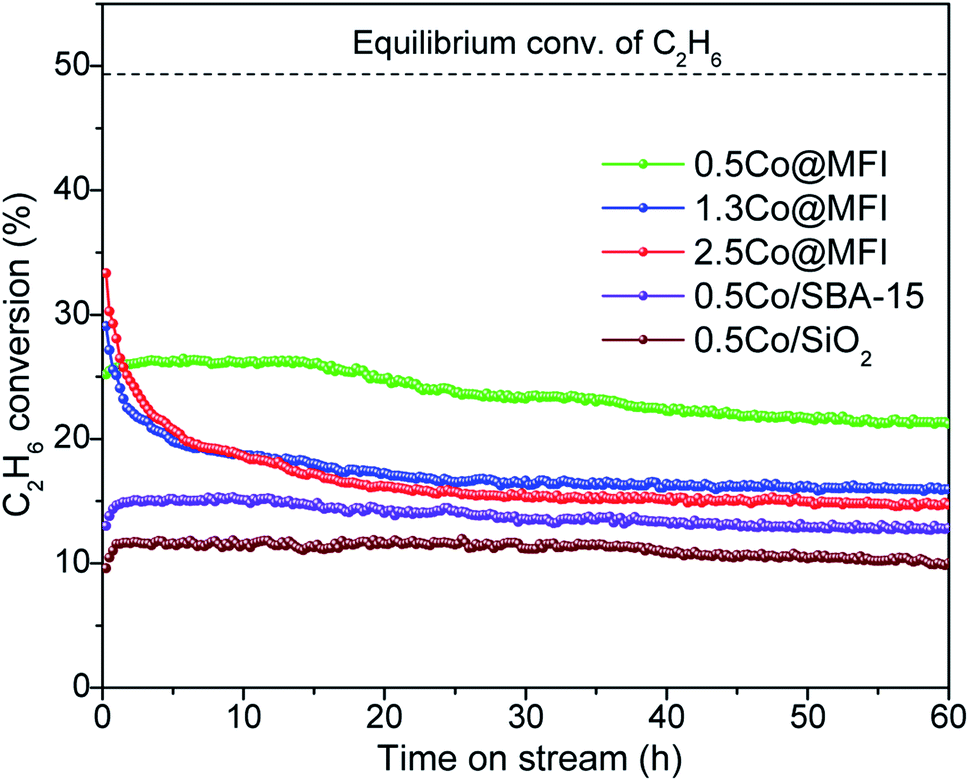 | ||
| Fig. 5 Effect of Co loading and catalyst support on the EDH activities of different Co catalysts. Reaction conditions: 100 mg catalyst, pretreatment by 10% H2/N2 for 30 min, 20% C2H6/N2, 10 mL min−1, 1 bar, 650 °C. | ||
As we have mentioned that the confinement effect has a great advantage in providing thermal stability against metal sintering, our next objective was to find out the stability of Co catalysts on other silica-based supports with different pore structures. For this purpose, we chose SBA-15 and commercial SiO2 as supports because they have a high overall surface area and both are neutral in nature. Since the 0.5Co/SBA-15 catalyst has a much lower micropore area and micropore volume compared to the 0.5Co@MFI catalyst (Table 2), the CoOx species were mostly dispersed on the surface. This was supported by the H2-TPR results, where the major reduction was seen to occur at a lower temperature. The pore structure of SBA-15 also affected the catalytic stability. The initial C2H6 conversion over the 0.5Co/SBA-15 catalyst was much lower compared to the 0.5Co@MFI catalyst, although the activity was maintained well afterward (Fig. 5). The main reason for this low initial activity was the formation of Co NPs at the stage of pretreatment with H2 (Fig. S9b†). Such confinement effect is even more prominent in the case of SiO2 supported catalyst, which showed lower initial activity than that of the 0.5Co/SBA-15 catalyst. Similar to 0.5Co/SBA-15, 0.5Co/SiO2 also formed Co NPs during H2 pretreatment because the commercial SiO2 does not have any micropore to stabilize the CoOx species against sintering (Fig. S9e†). These results are consistent with the XAS data (reported later).
An earlier report has shown that the encapsulation of metal species is more effective in the case of the bottom-up synthesis approach, which is also reflected in the stability of the catalysts.38 To check the effect of the synthesis method on the stability of our Co catalyst, we prepared a Co/MFI catalyst by simple wet impregnation with similar Co loading. The Co/MFI catalyst showed good initial conversion of C2H6 but deactivated at a little faster rate compared to the Co@MFI catalyst (Fig. 6 vs. Fig. 5). However, compared to the report on a Pt-based system, where the catalyst synthesized by wet impregnation showed fast deactivation,38 our Co-based catalyst showed less impact of synthesis method on the stability. The main reason could be the lower reducibility of Co than Pt, which led to better stability for Co under the reductive conditions. The Cr-based catalysts showed similar performance independently of the synthesis method and even at higher loading we observed that the 0.8Cr/MFI catalyst maintained the single-site nature of Cr species after the reaction.50
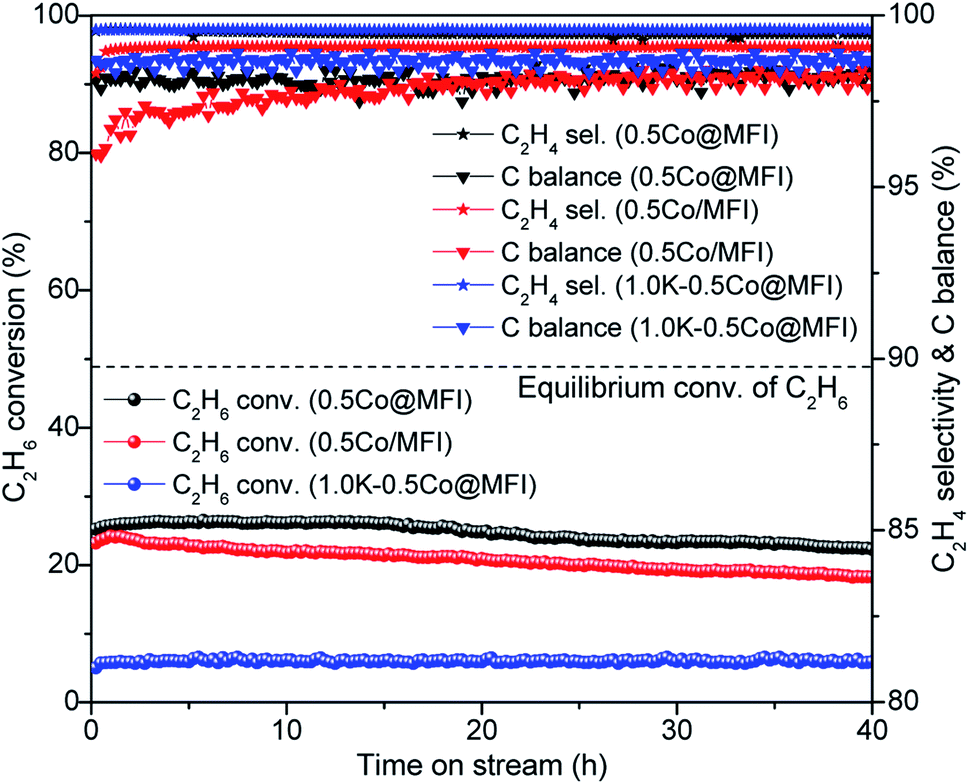 | ||
| Fig. 6 EDH performances of 0.5Co@MFI, 0.5Co/MFI, and 1.0K-0.5Co@MFI at 1 bar pressure. Reaction conditions: 100 mg catalyst, pretreatment by 10% H2/N2 for 30 min, 20% C2H6/N2, 10 mL min−1, 650 °C. | ||
Since the addition of alkali metals has been proved to be an effective way to improve catalyst stability in dehydrogenation reactions,18,38 we synthesized K-containing catalyst, 1.0K-0.5Co@MFI, with the aim of further preventing particle growth. We observed a difference in colour between the samples with and without K (Fig. S1†). In the absence of K, the light grey colour of Co@MFI catalyst turned a bit darker upon H2 treatment, which indicates the reduction of CoOx species to metallic Co. On the other hand, the calcined 1.0K-0.5Co@MFI sample was initially bluish in colour, indicating the presence of tetrahedral Co2+, which did not change the colour after H2 reduction at 650 °C. As expected, the addition of K successfully rendered the NP formation, but the activity of the catalyst was extremely low (Fig. 6). As observed from the H2-TPR results (Fig. 4), a small fraction of CoOx species could be reduced at 650 °C (which is also the pretreatment temperature of EDH) and the major fraction was reduced at above 900 °C, which means that the reduction of Co was suppressed by K. All the results indicate that metallic Co is the actual active species for the dehydrogenation and that K acts as a poison for the Co catalyst.
We examined the regeneration ability of 0.5Co@MFI catalyst for five consecutive cycles (Fig. 7). The deactivated catalyst was calcined at 600 °C in the air to remove the deposited coke and again pretreated with H2 before the next catalytic run. The catalyst lost the activity due to the gradual increase of the Co particle sizes after each run, but the C2H4 selectivity and C balance were unchanged. The initial C2H6 conversion was 27% in the 1st run, which was decreased to 18% in the 5th run with 33% loss in original activity. We observed a particle growth by TEM, but the Co particles were homogeneously distributed over the MFI support even after 5th run (Fig. S12†). XRD results also showed that the MFI structure was preserved after 5th run (Fig. S2†). Therefore, the decreased dispersion of Co could be considered as the main reason for catalyst deactivation.
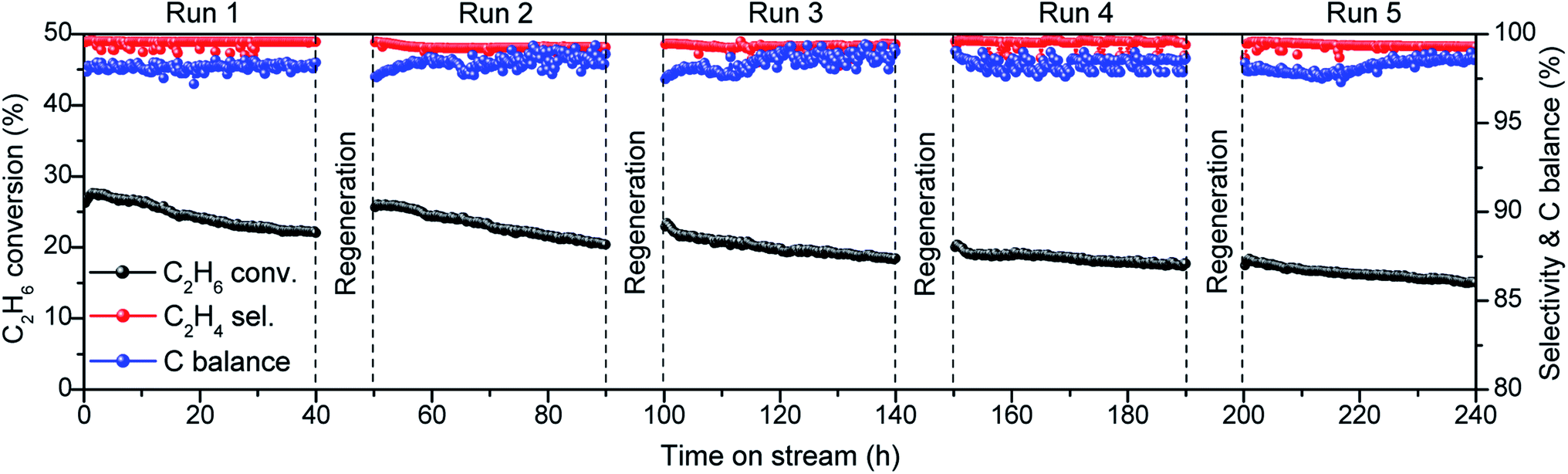 | ||
| Fig. 7 Recycling experiment with 0.5Co@MFI catalyst. Reaction conditions: 100 mg catalyst, pretreatment by 10% H2/N2 for 30 min, 20% C2H6/N2, 10 mL min−1, 1 bar, 650 °C. | ||
Although dehydrogenation at high pressure is of industrial interest, EDH at high pressure has been rarely studied.51 To extend the scope of applicability, we carried out the EDH reaction in an Avantium reactor at a total pressure of 7.5 bar and a C2H6 partial pressure of 1.5 bar. As expected, the C2H6 conversion level was lower compared to the reactions at atmospheric pressure, but we achieved a maximum C2H6 conversion of ∼21%, which is very close to the equilibrium conversion value (23%) under these conditions (Fig. 8 and Table 3). Unlike the reactions at atmospheric pressure, we observed some propylene at high pressure due to successive oligomerization-cracking processes, which also induced more coke formation. Although the 0.5Co@MFI catalyst exhibited better stability than the 1.3Co@MFI catalyst, the deactivation rate is faster in both cases compared to the reactions at 1 bar. The main reason being the formation of large Co NPs at high pressure as observed from the TEM results (Fig. S14†). We also observed more cracking products at higher Co loading (e.g., in the case of 1.3Co@MFI), which was due to the formation of bigger Co particles. A comparison can be made here with the 0.8Cr/MFI catalyst, which showed better stability and higher selectivity in the high-pressure EDHR.50
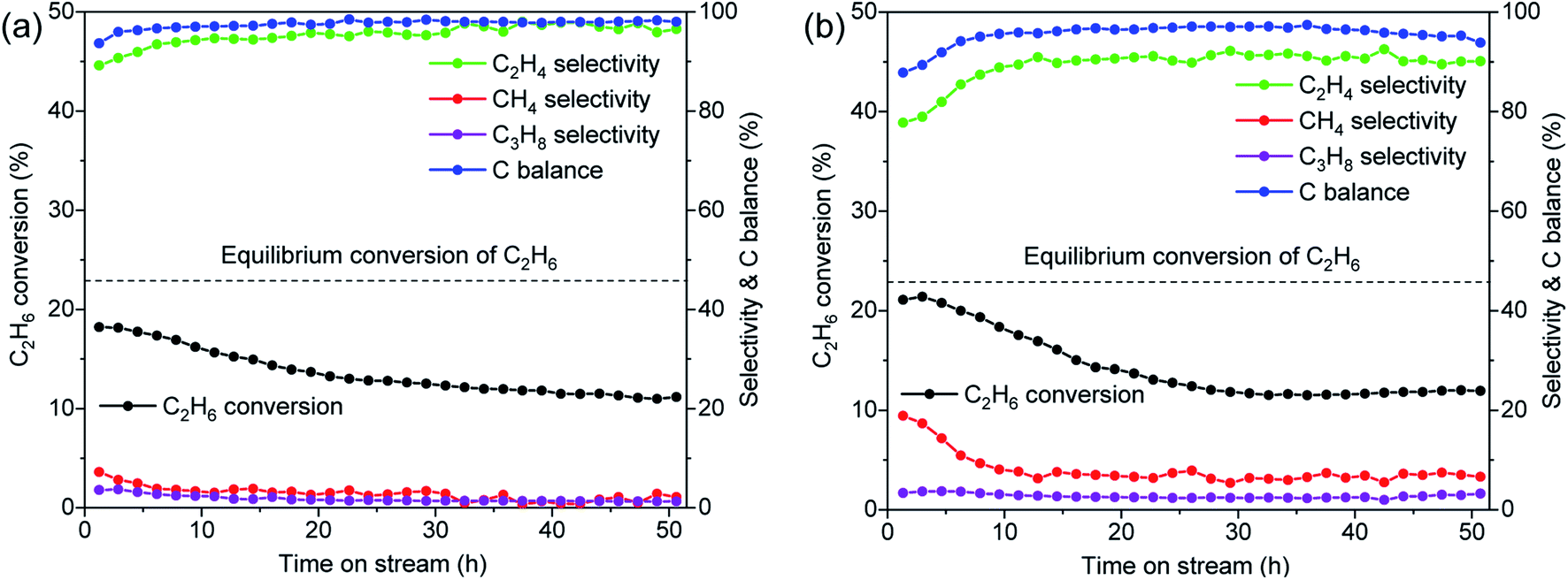 | ||
| Fig. 8 Catalytic performances of (a) 0.5Co@MFI and (b) 1.3Co@MFI catalysts at 7.5 bar pressure. Reaction conditions: 50 mg catalyst, pretreatment by 10% H2/N2 for 30 min, 20% C2H6/N2, 5 mL min−1, 650 °C. | ||
| Catalyst | C2H6 conversion & C2H4 selectivity (%) | Specific activityb (molC2H4 molCo−1 s−1) | C balancec (%) | Deactivationd (%) | Coke amount (g gcat−1) | Size of Co NPs after EDH (nm) | |||
|---|---|---|---|---|---|---|---|---|---|
| At 1 h | At 40 h | ||||||||
| a Reaction conditions: 100 mg catalyst, pretreatment by 10% H2/N2 for 30 min, 20% C2H6/N2 as the feed gas, 10 mL min−1, 1 bar, 650 °C.b Calculated at 1 h.c Calculated at 40 h.d Calculated at 40 h with respect to the highest C2H6 conversion.e 10% C2H6/N2 as the feed gas.f 40% C2H6/N2 as the feed gas.g No pretreatment with H2.h 50 mg catalyst, 5 mL min−1, 7.5 bar. | |||||||||
| 0.5Co@MFIe | 35.1 | 98.6 | 27.4 | 98.8 | 3.10 | 96.2 | 21.9 | 0.037 | — |
| 0.5Co@MFI | 25.7 | 99.6 | 22.3 | 99.4 | 4.67 | 96.4 | 15.5 | 0.039 | 7 |
| 0.5Co@MFIf | 17.3 | 99.0 | 13.2 | 99.1 | 6.13 | 98.6 | 24.6 | 0.042 | — |
| 0.5Co@MFIg | 27.9 | 99.3 | 17.1 | 99.3 | 4.96 | 95.0 | 41.6 | 0.049 | — |
| 1.3Co@MFI | 25.1 | 97.7 | 16.4 | 99.1 | 1.64 | 97.9 | 43.6 | 0.036 | 9 |
| 2.5Co@MFI | 28.1 | 97.8 | 15.1 | 99.0 | 0.97 | 97.2 | 54.7 | 0.035 | 12 |
| 0.5Co/SBA-15 | 14.6 | 99.5 | 13.3 | 99.5 | 2.50 | 97.6 | 13.1 | 0.042 | 20 |
| 0.5Co/SiO2 | 11.5 | 99.4 | 10.9 | 99.5 | 2.13 | 96.4 | 7.6 | 0.023 | 15 |
| 0.5Co/MFI | 24.2 | 99.0 | 18.3 | 99.1 | 4.40 | 98.2 | 24.4 | 0.035 | 8 |
| 1.0K-0.5Co@MFI | 5.8 | 99.6 | 5.9 | 99.5 | 1.13 | 98.6 | 9.2 | 0.002 | — |
| 0.5Co@MFIh | 18.2 | 89.2 | 11.5 | 97.8 | 2.90 | 98.0 | 36.8 | 0.224 | 7 |
| 1.3Co@MFIh | 21.1 | 77.8 | 11.6 | 90.6 | 1.10 | 96.3 | 45.8 | 0.292 | 15 |
3.2 Operando X-ray absorption studies during EDHR
The 2.5Co@MFI catalyst was first characterized at its initial state under the flow of He at room temperature (RT). Thereafter the catalyst was characterized after reduction under H2 at 650 °C for 30 min. The sample was finally characterized after 10 h of ethane dehydrogenation reaction (EDHR) conditions at 650 °C. Fig. S17† shows the evolution of Co K-edge XANES spectra as a function of the experimental conditions. The XANES spectrum of the initial state shows a weak peak in the pre-edge region due to the 1s → 3d quadrupolar transitions and the intense white line is characteristic of an oxidized state of Co sitting in octahedral environments.52,53 This is further confirmed by examining the EXAFS spectra and their corresponding Fourier transforms (FTs) (Fig. S18†), which display two scattering paths. These paths are easily attributed to oxygen and cobalt metal shell (Table S5†).After the reduction at 650 °C under H2 flow, XANES spectrum shows an increase of the pre-edge peak and the disappearance of the white line previously observed with the starting material. Comparison of XANES and EXAFS spectra recorded at 650 °C with the spectra of the references (Fig. S16†) confirms the formation of metallic Co in the sample where FT shows a unique Co–Co coordination shell corresponding to metallic Co (Fig. S18†). Fig. S18 and Table S5† show the quantitative analysis of the EXAFS spectra. The first coordination sphere of Co is fitted with 5.7 ± 1.1 Co atoms at 2.46 ± 0.01 Å. It was not possible to include metal–oxygen scattering paths in the fits, which indicates the complete reduction of all metal atoms. Finally, after 10 h under reaction conditions, the XANES spectrum looks similar to the spectrum after H2 treatment, indicating that Co remained in the metallic state after 10 h of reaction. A slight change in the amplitude of FT after 10 h can be observed (Fig. S18†), which indicates an increase in the particle size. The first coordination sphere of Co is fitted with 7.3 ± 0.8 Co atoms at 2.46 ± 0.008 Å. The higher coordination number (CN) in comparison with the one after reduction indicates the increase of the particle size during the reaction as the spectra were obtained at the same temperature, in accordance with the increase of the particle size observed by TEM.54
A similar observation was noted for the 0.5Co/SiO2 catalyst in the initial state as indicated by XANES and EXAFS spectra (Fig. S19 and S20†). However, the peak amplitude corresponding to the metal shell is much weaker than the oxide references (Fig. S16†). Thus, the material obtained after synthesis presents two key features, (i) a local structural order, and (ii) a disordered or amorphous state of a metal oxide. Reduction at 650 °C under H2 flow made a similar impact on the 0.5Co/SiO2 catalyst as observed in the case of 2.5Co@MFI. The quantitative analysis of the EXAFS spectra is shown in Table S6 and Fig. S20.† The first coordination sphere of Co is fitted with 6.9 ± 1.1 Co atoms at 2.45 ± 0.01 Å. After 10 h of EDHR, the XANES spectrum looks similar to the spectrum after reduction, indicating that Co remained in the metallic state. However, there is an increase in the amplitude of the FT after the reaction (Fig. S20†). The first coordination sphere of Co (Table 3) is fitted with 9.5 ± 1.2 Co atoms at 2.46 ± 0.01 Å. This result indicates the increase of the particle size along with the progress of the reaction.
Our next study was with the 0.5Co/MFI catalyst, which showed similar results to that of 0.5 wt% Co/SiO2 catalyst at the initial state (Fig. S21 and S22†). After treatment under H2 gas flow at 650 °C, the XANES spectrum shows an increase of the pre-edge peak and a decrease of the white line previously observed for the starting material. However, EXAFS oscillations show a modulation at low wavenumbers and its FT displays mean scattering paths that are easily attributed to a Co metal shell. The presence of a small shoulder indicates the interaction of a low-weight element with Co. The quantitative analysis of the EXAFS spectra is shown in Table S7 and Fig. S22.† The first coordination sphere of Co is fitted with 1.4 ± 1.2 O atoms at 1.9 ± 0.02 Å and 5 ± 0.8 Co atoms at 2.42 ± 0.01 Å. After 10 h of EDHR, the XANES spectrum evolved and it shows a slight increase of the pre-edge peak and the disappearance of the white line previously observed, indicating that Co was totally reduced after 10 h of reaction. The first coordination sphere of Co is fitted with and 5.9 ± 1.5 Co atoms at 2.43 ± 0.02 Å. These results confirm that Co was fully reduced and the increase of the particle size during the reaction.
The most active Co catalyst, 0.5Co@MFI, was characterized during the EDH reaction under two different pretreatment conditions and the results were compared. Prior to the reaction, one sample was treated under H2 and the other sample was treated under He. XANES spectrum at the initial state shows that Co is present in the oxidized form (Fig. 9). The H2-treated sample shows an increase of the pre-edge peak and a decrease of the white line indicating the reduction of Co. Differently, the He-treated sample shows a less intense pre-edge and lightly more intense in the white line, indicating that the Co is partially oxidized. However, in both materials, EXAFS oscillations show a modulation at low wavenumbers due to the interaction of a low weight element with Co and its FT displays mean scattering paths that are easily attributed to a Co metal shell and the presence of a small shoulder indicates the Co–O interaction. This is confirmed by the quantitative analysis of the EXAFS spectra shown in Table S8† and Fig. 9. The first coordination sphere of Co in the H2-treated sample is fitted with 1.5 ± 0.7 O atoms at 1.9 ± 0.01 Å and 5 ± 0.8 Co atoms at 2.45 ± 0.01 Å. Similarly, the first coordination sphere of Co in the He-treated sample is fitted with 1.3 ± 0.2 O atoms at 1.9 ± 0.01 Å and 1.9 ± 0.6 Co atoms at 2.46 ± 0.01 Å. After 10 h of EDHR, the H2-treated sample shows an increase of the pre-edge peak and the disappearance of the white line, indicating the total reduction of Co. The first coordination sphere of Co is fitted with and 5.6 ± 1.2 Co atoms at 2.45 ± 0.01 Å. On the other hand, the XANES spectrum of the He-treated sample features partially oxidized Co. This is further confirmed by the EXAFS oscillations that show modulation of ∼2–4 Å−1. The FT displays mean scattering paths that are easily attributed to a Co metal shell and the presence of a shoulder indicates the Co–O interaction. The first coordination sphere of Co is fitted with 0.9 ± 0.1 O atoms at 1.8 ± 0.01 Å and 5.3 ± 0.4 Co atoms at 2.45 ± 0.01 Å. In both cases, EDHR initiates the particle growth for Co, which causes deactivation.
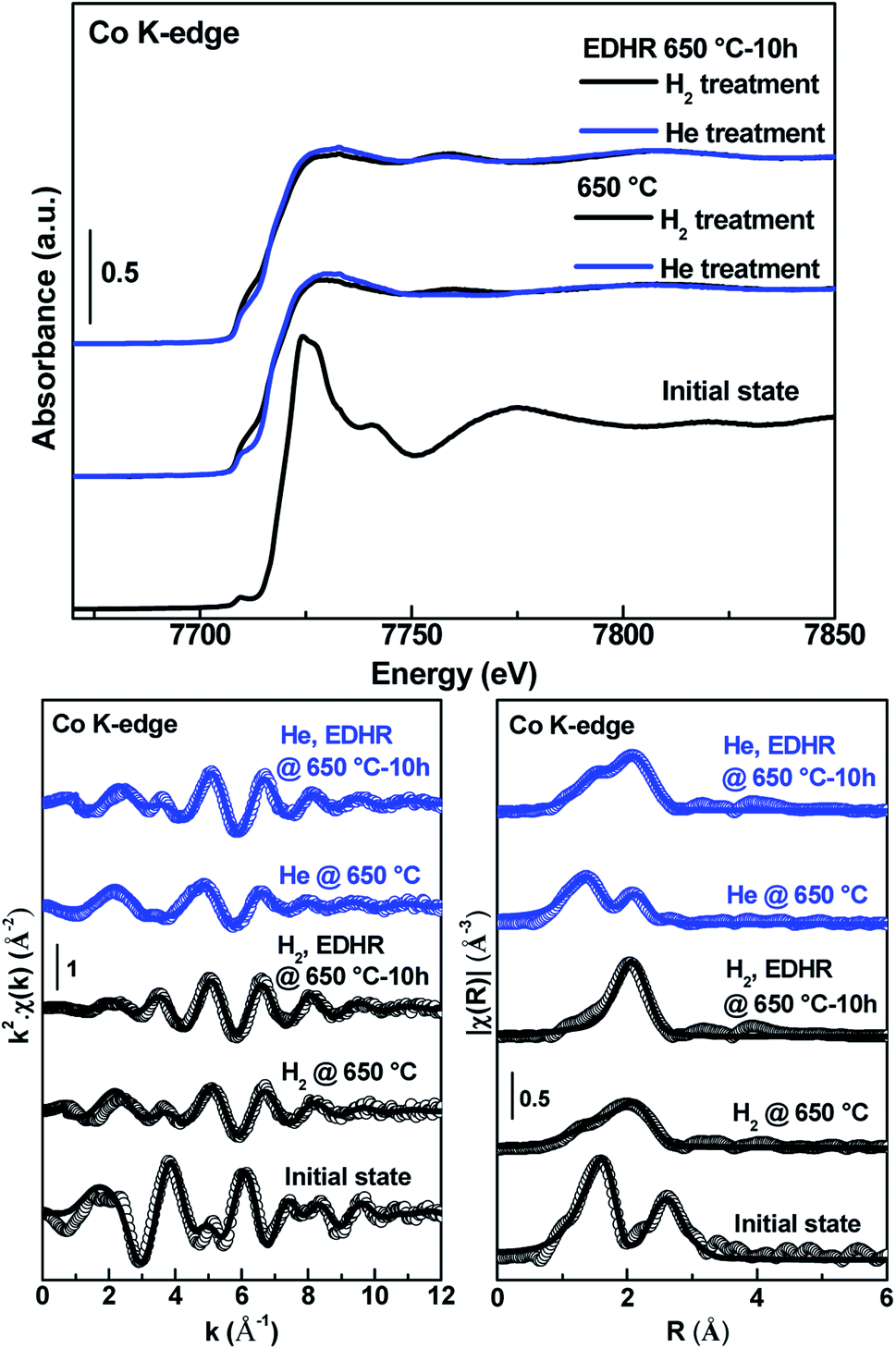 | ||
| Fig. 9 Evolution of Co K-edge XANES spectra (top) of 0.5Co@MFI at the initial state, after pretreatment, and after EDHR. k2 weighed EXAFS spectra (bottom left) and FTs (bottom right) of 0.5Co@MFI at the initial state, after H2 pretreatment, and after EDHR. | ||
We can make a correlation between the EDH activity of different Co catalysts and the change in the first shell coordination number (CN) of Co–Co (Fig. S23†). MFI-supported catalysts at the Co loading of 0.5 wt% have lower CN and they exhibit higher specific activity. The catalyst synthesized by the one-pot method (i.e., 0.5Co@MFI) shows a little lower change in CN and thus shows a little better stability. At higher Co loading (2.5 wt%), the catalyst shows higher CN at the initial stage and thus exhibits lower specific activity. The specific activity of the 2.5Co@MFI catalyst was even lower than the SiO2-supported 0.5Co/SiO2 catalyst, which is probably because 2.5Co@MFI formed a dense population of Co NPs that effectively reduced the active surface area of Co. The 0.5Co/SiO2 catalyst shows a much higher CN after H2 pretreatment as SiO2 has no confinement effect. However, the specific activity was moderate in this case, which could be due to the presence of fine Co NPs/clusters (not observed by TEM) but the actual reason is not clear.
All these results suggest that Co always gets reduced into the metallic state either during H2-pretreatment or during EDHR and it is hard to stabilize the nanometric Co species against sintering even at low loading. Therefore, although the Co-based catalysts show high intrinsic activity, their stability is always an issue under such harsh reductive conditions. In this regard, the Cr-based catalysts show little lower EDH activity, but they are extremely stable due to the high Gibbs energy value for the reduction of oxidized Cr. We made a detailed operando XAS study on the Cr/MFI system and identified a divalent monomeric Cr species as an active centre for the reaction.50 Based on the XAS results and DFT studies, we made a conclusion that the CrII(–O–Si![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) )2 species was directly involved in the first C–H activation, which is believed to be the rate-determining step in the EDH process. Importantly, this monomeric species was present in the Cr/MFI system with a Cr loading of 0.8 wt%, which is double the Co loading in the most active Co@MFI catalyst in terms of atomic loading. The most striking fact is that the 0.8Cr/MFI catalyst maintained its high activity for around 110 h without a significant deactivation and it was possible due to the high stability of the single-site Cr species. The regeneration of Cr-based catalysts is also very easy as the deactivation was caused mainly due to the coke formation. On the other hand, in addition to coke formation, the Co-based catalysts gradually form Co NPs in most cases and make the regeneration more difficult.
)2 species was directly involved in the first C–H activation, which is believed to be the rate-determining step in the EDH process. Importantly, this monomeric species was present in the Cr/MFI system with a Cr loading of 0.8 wt%, which is double the Co loading in the most active Co@MFI catalyst in terms of atomic loading. The most striking fact is that the 0.8Cr/MFI catalyst maintained its high activity for around 110 h without a significant deactivation and it was possible due to the high stability of the single-site Cr species. The regeneration of Cr-based catalysts is also very easy as the deactivation was caused mainly due to the coke formation. On the other hand, in addition to coke formation, the Co-based catalysts gradually form Co NPs in most cases and make the regeneration more difficult.
4 Conclusions
Pure silica MFI zeolite shows an excellent ability to encapsulate metal species and provide stability against thermal sintering as compared to other silica supports with similar surface areas. Preliminary studies on metal screening showed that Co has the highest intrinsic activity although its stability depends on metal loading. At a Co loading of 0.5 wt%, the Co@MFI catalyst showed the highest activity and stability, while increasing the loading resulted in faster deactivation due to the formation of bigger NPs. According to operando XAS studies, the Co-based catalysts always form a metallic Co phase either during the pretreatment or during the reaction, and therefore it is hard to maintain the initial dispersion. The synthesis method did not show a large effect on the catalytic performance, as we observed comparable activity and stability for the Co/MFI catalyst prepared by a simple wet impregnation. Cr was another metal of interest in this study due to its excellent stability and easy regenerability. The most interesting fact is that Cr can maintain its single-site nature even at higher temperatures than that used in this study, which makes it suitable for high-temperature applications under reductive conditions. The results obtained in this study indicate that the MFI silicalite can be excellent support for a diverse range of metals even where the one-pot synthesis strategy fails. We have compared the activity and stability of our Co catalyst with the earlier reported catalysts for the non-oxidative EDHR (Table S9†), which indicates that the current study can provide a direction towards designing more stable transition metal catalysts to be operated under industrially relevant conditions.Conflicts of interest
There are no conflicts to declare.Note added after first publication
This article replaces the version published on 30th March 2022, which contained errors in the display of eqn (1) and (4) in the PDF version only.References
- J. J. H. B. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Chem. Rev., 2014, 114, 10613–10653 CrossRef CAS PubMed.
- M. J. Darabi Mahboub, J.-L. Dubois, F. Cavani, M. Rostamizadeh and G. S. Patience, Chem. Soc. Rev., 2018, 47, 7703–7738 RSC.
- A. Mohsenzadeh, A. Zamani and M. J. Taherzadeh, ChemBioEng Rev., 2017, 4, 75–91 CrossRef.
- G. Dimitrakopoulos, B. Koo, B. Yildiz and A. F. Ghoniem, ACS Catal., 2021, 11, 3638–3661 CrossRef CAS.
- T. Ren, M. Patel and K. Blok, Energy, 2006, 31, 425–451 CrossRef CAS.
- D. Fairuzov, I. Gerzeliev, A. Maximov and E. Naranov, Catalysts, 2021, 11, 833 CrossRef CAS.
- E. E. Stangland, Annu. Rev. Chem. Biomol. Eng., 2018, 9, 341–364 CrossRef CAS PubMed.
- S. Kim, S. Jeong and E. Heo, Energy Sources, Part B, 2019, 14, 49–66 CrossRef CAS.
- Y. Dai, X. Gao, Q. Wang, X. Wan, C. Zhou and Y. Yang, Chem. Soc. Rev., 2021, 50, 5590–5630 RSC.
- D. Melzer, G. Mestl, K. Wanninger, Y. Zhu, N. D. Browning, M. Sanchez-Sanchez and J. A. Lercher, Nat. Commun., 2019, 10, 4012 CrossRef PubMed.
- S. Yao, B. Yan, Z. Jiang, Z. Liu, Q. Wu, J. H. Lee and J. G. Chen, ACS Catal., 2018, 8, 5374–5381 CrossRef CAS.
- Y. Zhou, J. Lin, L. Li, M. Tian, X. Li, X. Pan, Y. Chen and X. Wang, J. Catal., 2019, 377, 438–448 CrossRef CAS.
- Y. Chen and D. G. Vlachos, J. Phys. Chem. C, 2010, 114, 4973–4982 CrossRef CAS.
- A. Iglesias-Juez, A. M. Beale, K. Maaijen, T. C. Weng, P. Glatzel and B. M. Weckhuysen, J. Catal., 2010, 276, 268–279 CrossRef CAS.
- H. Xiong, S. Lin, J. Goetze, P. Pletcher, H. Guo, L. Kovarik, K. Artyushkova, B. M. Weckhuysen and A. K. Datye, Angew. Chem., Int. Ed., 2017, 56, 8986–8991 CrossRef CAS PubMed.
- J. Wu, Z. Peng and A. T. Bell, J. Catal., 2014, 311, 161–168 CrossRef CAS.
- V. J. Cybulskis, B. C. Bukowski, H.-T. Tseng, J. R. Gallagher, Z. Wu, E. Wegener, A. J. Kropf, B. Ravel, F. H. Ribeiro, J. Greeley and J. T. Miller, ACS Catal., 2017, 7, 4173–4181 CrossRef CAS.
- Q. Sun, N. Wang, Q. Fan, L. Zeng, A. Mayoral, S. Miao, R. Yang, Z. Jiang, W. Zhou, J. Zhang, T. Zhang, J. Xu, P. Zhang, J. Cheng, D.-C. Yang, R. Jia, L. Li, Q. Zhang, Y. Wang, O. Terasaki and J. Yu, Angew. Chem., Int. Ed., 2020, 59, 19450–19459 CrossRef CAS PubMed.
- G. Siddiqi, P. Sun, V. Galvita and A. T. Bell, J. Catal., 2010, 274, 200–206 CrossRef CAS.
- P. Sun, G. Siddiqi, W. C. Vining, M. Chi and A. T. Bell, J. Catal., 2011, 282, 165–174 CrossRef CAS.
- E. C. Wegener, Z. Wu, H.-T. Tseng, J. R. Gallagher, Y. Ren, R. E. Diaz, F. H. Ribeiro and J. T. Miller, Catal. Today, 2018, 299, 146–153 CrossRef CAS.
- Z. Wu, B. C. Bukowski, Z. Li, C. Milligan, L. Zhou, T. Ma, Y. Wu, Y. Ren, F. H. Ribeiro, W. N. Delgass, J. Greeley, G. Zhang and J. T. Miller, J. Am. Chem. Soc., 2018, 140, 14870–14877 CrossRef PubMed.
- G. Sun, Z.-J. Zhao, R. Mu, S. Zha, L. Li, S. Chen, K. Zang, J. Luo, Z. Li, S. C. Purdy, A. J. Kropf, J. T. Miller, L. Zeng and J. Gong, Nat. Commun., 2018, 9, 4454 CrossRef PubMed.
- M. Aly, E. L. Fornero, A. R. Leon-Garzon, V. V. Galvita and M. Saeys, ACS Catal., 2020, 10, 5208–5216 CrossRef CAS.
- Y. Gu, H. Liu, M. Yang, Z. Ma, L. Zhao, W. Xing, P. Wu, X. Liu, S. Mintova, P. Bai and Z. Yan, Appl. Catal., B, 2020, 274, 119089 CrossRef CAS.
- G. Wang, N. Sun, C. Gao, X. Zhu, Y. Sun, C. Li and H. Shan, Appl. Catal., A, 2014, 478, 71–80 CrossRef CAS.
- L. Liu, U. Díaz, R. Arenal, G. Agostini, P. Concepción and A. Corma, Nat. Mater., 2017, 16, 132–138 CrossRef CAS PubMed.
- M. Moliner, J. E. Gabay, C. E. Kliewer, R. T. Carr, J. Guzman, G. L. Casty, P. Serna and A. Corma, J. Am. Chem. Soc., 2016, 138, 15743–15750 CrossRef CAS PubMed.
- J. Zhang, L. Wang, B. Zhang, H. Zhao, U. Kolb, Y. Zhu, L. Liu, Y. Han, G. Wang, C. Wang, D. S. Su, B. C. Gates and F.-S. Xiao, Nat. Catal., 2018, 1, 540–546 CrossRef CAS.
- M. Moliner, J. Gabay, C. Kliewer, P. Serna and A. Corma, ACS Catal., 2018, 8, 9520–9528 CrossRef CAS.
- N. Wang, Q. Sun and J. Yu, Adv. Mater., 2019, 31, 1803966 CrossRef PubMed.
- S.-M. Wu, X.-Y. Yang and C. Janiak, Angew. Chem., Int. Ed., 2019, 58, 12340–12354 CrossRef CAS PubMed.
- Y. Chai, W. Shang, W. Li, G. Wu, W. Dai, N. Guan and L. Li, Adv. Sci., 2019, 6, 1900299 CrossRef PubMed.
- Y. Wang, Z.-P. Hu, X. Lv, L. Chen and Z.-Y. Yuan, J. Catal., 2020, 385, 61–69 CrossRef CAS.
- A. Janda and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 19193–19207 CrossRef CAS PubMed.
- I. Martyanov and A. Sayari, Catal. Lett., 2008, 126, 164–172 CrossRef CAS.
- T. V. M. Rao, E. M. Zahidi and A. Sayari, J. Mol. Catal. A: Chem., 2009, 301, 159–165 CrossRef CAS.
- L. Liu, M. Lopez-Haro, C. W. Lopes, C. Li, P. Concepcion, L. Simonelli, J. J. Calvino and A. Corma, Nat. Mater., 2019, 18, 866–873 CrossRef CAS PubMed.
- S. Belin, V. Briois, A. Traverse, M. Idir, T. Moreno and M. Ribbens, Phys. Scr., 2005, 2005, 980–983 CrossRef.
- A. Aguilar-Tapia, S. Ould-Chikh, E. Lahera, A. Prat, W. Delnet, O. Proux, I. Kieffer, J.-M. Basset, K. Takanabe and J.-L. Hazemann, Rev. Sci. Instrum., 2018, 89, 035109 CrossRef PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- B. W. Lee, R. Alsenz, A. Ignatiev and M. A. Van Hove, Phys. Rev. B: Condens. Matter Mater. Phys., 1978, 17, 1510–1520 CrossRef CAS.
- A. S. Andreev, J.-B. d'Espinose de Lacaillerie, O. B. Lapina and A. Gerashenko, Phys. Chem. Chem. Phys., 2015, 17, 14598–14604 RSC.
- B. Predel, Phase Equilibria, Crystallographic and Thermodynamic Data of Binary Alloys' of Landolt-Bornstein-Group IV Physical Chemistry, Springer-Verlag, Berlin, 1993 Search PubMed.
- S. De, M. V. Babak, M. J. Hülsey, W. H. Ang and N. Yan, Chem.–Asian J., 2018, 13, 1053–1059 CrossRef CAS PubMed.
- H. Ahmad Alyosef, H. Roggendorf, D. Schneider, A. Inayat, J. Welscher, W. Schwieger, T. Münster, G. Kloess, S. Ibrahim and D. Enke, J. Porous Mater., 2016, 23, 1609–1618 CrossRef CAS.
- Q. Tang, Q. Zhang, P. Wang, Y. Wang and H. Wan, Chem. Mater., 2004, 16, 1967–1976 CrossRef CAS.
- J. Li, X. Xu, Z. Hao and W. Zhao, J. Porous Mater., 2008, 15, 163–169 CrossRef CAS.
- K. Klaigaew, C. Samart, C. Chaiya, Y. Yoneyama, N. Tsubaki and P. Reubroycharoen, Chem. Eng. J., 2015, 278, 166–173 CrossRef CAS.
- S. De, S. Ould-Chikh, A. Aguilar, J.-L. Hazemann, A. Zitolo, A. Ramirez, S. Telalovic and J. Gascon, ACS Catal., 2021, 11, 3988–3995 CrossRef CAS.
- S. Dangwal, R. Liu, S. V. Kirk and S.-J. Kim, Energy Fuels, 2018, 32, 4628–4637 CrossRef CAS.
- F. de Groot, G. Vankó and P. Glatzel, J. Phys.: Condens. Matter, 2009, 21, 104207 CrossRef PubMed.
- D. Cabaret, A. Bordage, A. Juhin, M. Arfaoui and E. Gaudry, Phys. Chem. Chem. Phys., 2010, 12, 5619–5633 RSC.
- A. Jentys, Phys. Chem. Chem. Phys., 1999, 1, 4059–4063 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ta00223j |
| This journal is © The Royal Society of Chemistry 2022 |