
A simple simulation-derived descriptor for the deposition of polymer-wrapped carbon nanotubes on functionalized substrates†
Zhizhang
Shen
a,
Jonathan H.
Dwyer
a,
Jian
Sun
b,
Katherine R.
Jinkins
b,
Michael S.
Arnold
b,
Padma
Gopalan
ab and
Reid C.
Van Lehn
 *a
*a
aDepartment of Chemical and Biological Engineering, University of Wisconsin-Madison, 1415 Engineering Drive, Madison, WI 53706, USA. E-mail: vanlehn@wisc.edu
bDepartment of Materials Science and Engineering, University of Wisconsin-Madison, 1509 University Avenue, Madison, WI 53706, USA
First published on 15th June 2022
Abstract
Controlling the deposition of polymer-wrapped single-walled carbon nanotubes (s-CNTs) onto functionalized substrates can enable the fabrication of s-CNT arrays for semiconductor devices. In this work, we utilize classical atomistic molecular dynamics (MD) simulations to show that a simple descriptor of solvent structure near silica substrates functionalized by a wide variety of self-assembled monolayers (SAMs) can predict trends in the deposition of s-CNTs from toluene. Free energy calculations and experiments indicate that those SAMs that lead to maximum disruption of solvent structure promote deposition to the greatest extent. These findings are consistent with deposition being driven by solvent-mediated interactions that arise from SAM-solvent interactions, rather than direct s-CNT-SAM interactions, and will permit the rapid computational exploration of potential substrate designs for controlling s-CNT deposition and alignment.
Because of their excellent electrical, mechanical, thermal, and optical properties, 1-D single-walled carbon nanotubes (CNTs) are promising materials for a variety of applications.1 In particular, field-effect transistors (FETs) utilizing semiconducting single-walled carbon nanotubes (s-CNTs) as the channel material2,3 exhibit exceptional electronic characteristics, including high charge mobility4,5 and high current carrying capacity.6 Major obstacles to the creation of ideal s-CNT arrays for large-scale integrated circuits and systems include (1) the need to separate mixtures of metallic and semiconducting s-CNTs and (2) imperfect alignment and positioning of s-CNTs.7 It has been discovered that the wrapping of conjugated polymers, especially poly[(9,9-dioctylfluorenyl-2,7-diyl)-alt-co-(6,6′-[2,2′-bipyridine])] (PFO-BPy), allows for the selective dispersion of s-CNTs with specific diameters and chirality.8 Because of their flexiblity with respect to substrate options, solution deposition methods coupled with various other methods have been used to achieve s-CNT alignment on substrates.9–18 Functionalizing the s-CNT or the substrates enables control of the spatial localization of s-CNT deposition, largely via covalent bonding.19–21 Large biomolecules such as DNA have also been used to select different species of CNTs22 and place CNTs at specific positions and specific orientations.23,24 Recent experimental studies have also demonstrated well-aligned s-CNT arrays with tunable densities.25,26 Nonetheless, a facile and scalable technique that simultaneously accomplishes selective sorting, perfect alignment, and local positioning of s-CNT is still lacking, in part due to incompatibility in the solvents and surfactants used for sorting, alignment, and placement methods.20 More fundamentally, the driving forces for polymer-wrapped s-CNT deposition are still not well understood.
We recently showed experimental evidence that PFO-BPy wrapped s-CNTs deposit to different degrees on bare silica and on silica substrates functionalized with four different self-assembled monolayers (SAMs).27 Deposition was influenced by SAM composition and choice of organic solvent (toluene or chloroform), with similar trends in deposition as a function of SAM composition observed in both solvents. Experimental characterization of these surfaces revealed no apparent correlation between the water contact angle – a traditional experimental descriptor of surface hydrophobicity – and deposition trends. Atomistic molecular dynamics (MD) simulations instead revealed that solvent structure plays an important role in determining the affinity of a SAM surface for the PFO-BPy wrapped s-CNTs. However, these trends were observed for only four SAMs. In this work, we extend our prior simulation approach to a wider variety of SAMs to investigate the impact of solvent-mediated structural forces on s-CNT deposition. We find that variations in the solvent density near each SAM due to interactions with the substrate lead to oscillations that predict s-CNT deposition in both simulations and experiments. We define a simple descriptor of solvent density oscillations that qualitatively predicts CNT deposition across a range of SAM compositions, permitting simulation-guided expansion of the design space for control of s-CNT deposition.
To determine if a descriptor quantifying solvent structure can predict the deposition of PFO-BPy-wrapped s-CNTs, we focus on the deposition of s-CNTs initially dispersed in toluene. While the number density of s-CNTs deposited on SAMs in chloroform is higher than in toluene,27 chloroform may dope s-CNTs with chlorine28 which could lead to discrepancies with the simulated systems. Consequently, we simulated s-CNTs in toluene and expect that any identified trends would extend to chloroform. We similarly modeled a large set of SAMs containing organic ligands that vary in physical and chemical properties. Modifications include varying the alkyl chain length, terminating a hexyl backbone with both polar and nonpolar end groups, grafting the substrate with a mixture of two ligands, and different backbone structures. Specific ligands considered, and their naming conventions, are shown in Fig. 1.
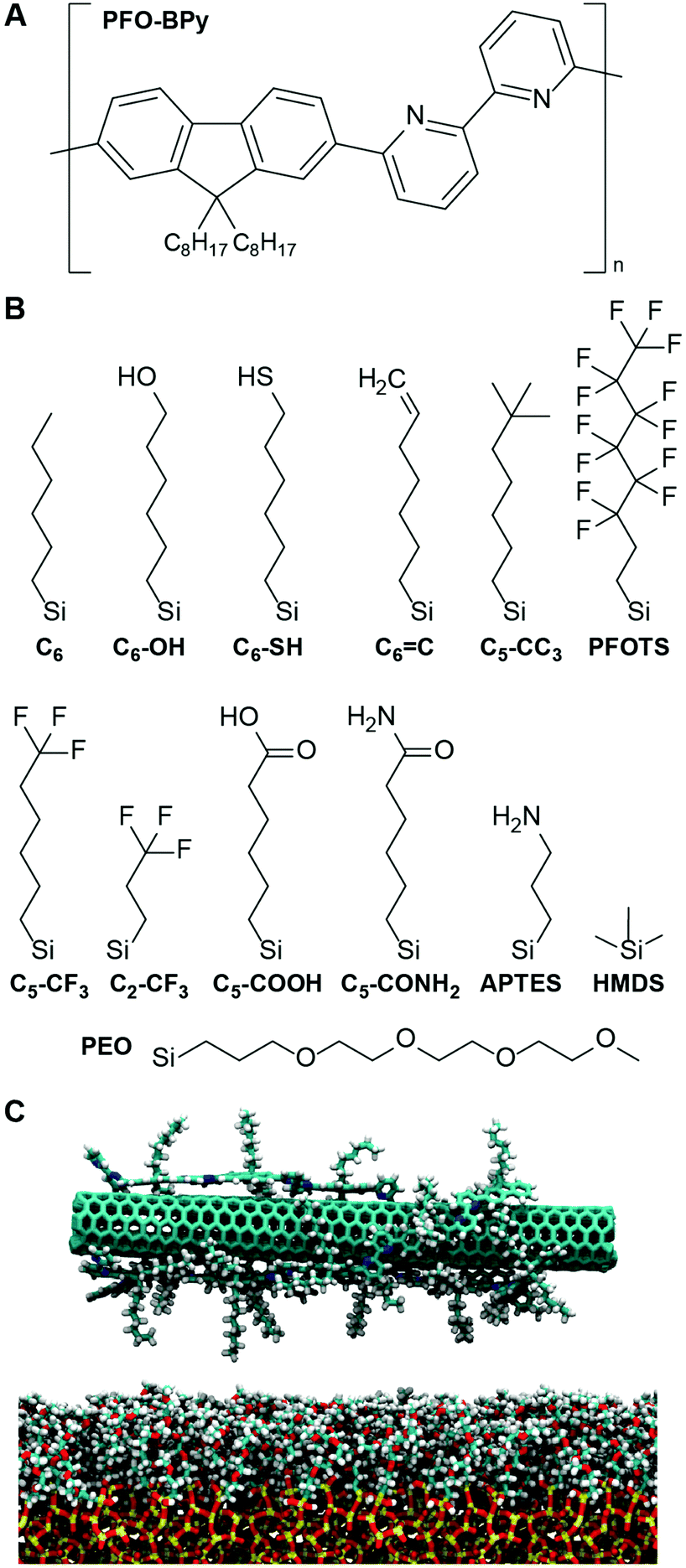 | ||
| Fig. 1 A Chemical structure of PFO-BPy. B Chemical structures of ligands considered in this work, excluding alkylsilane ligands of which only –(CH2)5CH3, abbreviated as C6, is shown. The same nomenclature is used for other ligands. For example, C6-OH stands for –(CH2)6–OH. Ligands were grafted to form SAMs on silica substrates. C Snapshot from a MD simulation of a PFO-BPy-wrapped s-CNT near a PEO SAM in toluene. Silica is shown as yellow/red, PEO as cyan/red/white, PFO-BPy as cyan/blue/white, and the CNT in cyan. Toluene is not shown. | ||
To analyze solvent structural properties, we quantified SAM-induced changes to solvent structure in the absence of the s-CNT using unbiased MD simulations. The solvent density was computed as a function of the z-component of the distance from the SAM surface, since the SAM normal is parallel to the z-axis of the simulation box (Fig. 1C). Fig. 2 shows solvent density profiles near representative SAMs containing alkylsilane ligands with either 2 or 22 carbon atoms (referred to as C2 and C22 SAMs, respectively). Both density profiles exhibit similar features: the solvent density decreases to nearly zero near the SAM, plateaus at its bulk value far from the SAM, and exhibits oscillations in the intervening region. Oscillations in solvent density are commonly observed near surfaces and are a common feature to all SAMs studied; they reflect the interplay of surface-solvent interactions, entropic effects, and the finite size of solvent molecules, as also observed in liquid-phase radial distribution functions.29 The solvent density near the C22 SAM exhibits minimal oscillations with a slight peak in the solvent density at a position near the surface. This behavior is similar to what is observed near liquid-vapor interfaces; the weak affinity of the solvent for the surface emerges as a broad density profile. Conversely, the solvent density near the C2 SAM exhibits large oscillations with a large peak near the surface. This peak indicates an affinity of the solvent molecules for the SAM, leading to their accumulation at the surface and subsequent depletion at slightly farther distances from the surface due to finite size effects. These substantial differences in solvent structure are observed despite the identical chemical functionality of the C2 and C22 SAMs.
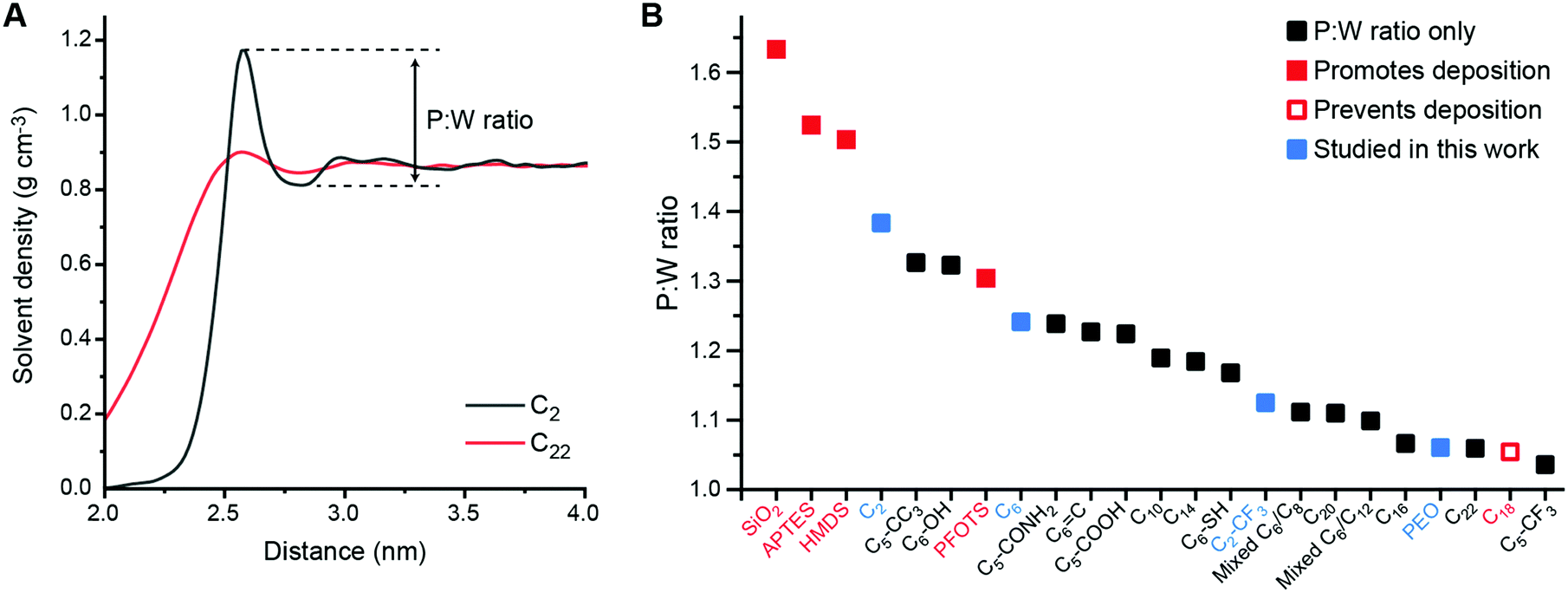 | ||
| Fig. 2 A Toluene density profile near the C2-grafted surface (black) and the C22-grafted surface (red). The first peak of the red curve was shifted to overlap with that of the black curve to help compare differences in peak:well ratios. B Peak:well (P:W) ratios for all SAM-grafted surfaces. Surfaces shown to promote (filled red) or prevent (hollow red) s-CNT deposition in past work are indicated.25 Blue squares indicate SAMs selected for additional study in this work. | ||
To compare solvent structure across SAMs, we calculated the ratio between the magnitude of the first peak to the magnitude of the first well (i.e., the local maximum and minimum at the smallest distances from the SAM) in the solvent density profile, which we refer to as the P:W ratio (Fig. 2A). This ratio provides a simple descriptor of the solvent affinity for the SAM. Fig. 2B shows computed P:W ratios for bare silica, for SAMs experimentally studied in previous work (APTES, HMDS, PFOTS, and C18)27 and for the extended set of SAMs shown in Fig. 1. We find that this ratio varies substantially across the range of simulated SAMs, exhibiting large values for the HMDS, APTES, PFOTS and bare silica surfaces that were previously shown to promote s-CNT deposition and small values for the C18 surface previously shown to prevent deposition. This observation suggests a potential relationship between this descriptor of solvent structure and deposition.
Considering all surfaces, we find that the P:W ratio decreases as the alkyl chain length increases. Surface modifications based on C6 structure (e.g., hexyl backbones terminated with different end groups) do not alter the solvent structure significantly compared to C6 except for the C5-CF3 SAM. Branching groups (C5-CC3) and the hydroxyl group (C6-OH) increase the first P:W ratio from ∼1.24 (the value for C6) to ∼1.32 while the ratio for the thiol group (C6-SH) decreases to ∼1.17. The ratios for the amide group (C5-CONH2), double bond (C6![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), and carboxyl group (C5-COOH) are similar to that for C6. However, the ratio for the trifluoromethyl group (C5-CF3) decreases significantly from ∼1.24 to ∼1.04, the smallest value for all surfaces studied in this work. A shorter alkyl chain (C2) terminated with -CF3 (C2-CF3) does increase the ratio to 1.12 compared to 1.04 for C5-CF3, but this value is still in the lower range of all surfaces. Finally, we also modeled mixtures of C6–C8 and C6–C12 ligands in 1
C), and carboxyl group (C5-COOH) are similar to that for C6. However, the ratio for the trifluoromethyl group (C5-CF3) decreases significantly from ∼1.24 to ∼1.04, the smallest value for all surfaces studied in this work. A shorter alkyl chain (C2) terminated with -CF3 (C2-CF3) does increase the ratio to 1.12 compared to 1.04 for C5-CF3, but this value is still in the lower range of all surfaces. Finally, we also modeled mixtures of C6–C8 and C6–C12 ligands in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratios. These SAMs have P:W ratios lower than their respective pure components with values similar to P:W ratios for C16 or C20. These trends highlight the interplay of physical (branching, length, component mixtures) and chemical (end group) properties on solvent structure captured through the P:W ratio. We further calculated descriptors of SAM structure (ligand tilt angles and P2 order parameters) to determine if these quantities alone predict solvent density profiles. However, no clear trend relating these order parameters to either s-CNT deposition or the P:W ratio is observed (Fig. S1 and S2, ESI†). These comparisons highlight that the analysis of solvent structure captures information not encoded within structural features of the SAM alone.
:1 molar ratios. These SAMs have P:W ratios lower than their respective pure components with values similar to P:W ratios for C16 or C20. These trends highlight the interplay of physical (branching, length, component mixtures) and chemical (end group) properties on solvent structure captured through the P:W ratio. We further calculated descriptors of SAM structure (ligand tilt angles and P2 order parameters) to determine if these quantities alone predict solvent density profiles. However, no clear trend relating these order parameters to either s-CNT deposition or the P:W ratio is observed (Fig. S1 and S2, ESI†). These comparisons highlight that the analysis of solvent structure captures information not encoded within structural features of the SAM alone.
We expect that favorable surfaces for s-CNT deposition are concentrated on the left side of the Fig. 2B (larger P:W ratios) and unfavorable surfaces on the right side (smaller ratios), motivating additional simulations and experiments to confirm the relevance of this parameter. We first used atomistic MD simulations coupled with umbrella sampling to simulate the adsorption of a single polymer-wrapped s-CNT to C2-, C6-, C2-CF3-, and PEO-grafted surfaces in toluene. The C2- and PEO-grafted surfaces were chosen because they represent surfaces expected to have high and low affinities for s-CNT adsorption based on their solvent structures (Fig. 2B). The C6- and C2-CF3-grafted surfaces were selected as examples with intermediate P:W ratios to fill gaps in our dataset. For each system, we calculated the potential of mean force (PMF) as a function of the distance along the z-axis between the center-of-mass of the s-CNT and that of the SAM-grafted surfaces. PMFs were constructed from the umbrella histograms using the weighted histogram analysis method.30 To reduce hysteresis,27 PMF curves were obtained from simulations in which initial configurations were generated by both pulling the s-CNT toward and away from the substrate (detailed in the ESI†). A negative free energy minimum in the PMF curve indicates favorable s-CNT adsorption. Due to system size limitations, these values are computed for a s-CNT that represents only a small segment (∼6 nm in length) of a full-length s-CNT (∼100 nm in length). However, for a 1D nanomaterial like a s-CNT, we expect free energies to scale linearly with the s-CNT length. Consequently, the discussion below focuses on the sign of free energy minima because numerical values are likely underestimated by an order of magnitude, suggesting that even shallow free energy minima are substantially larger than thermal energy when considering full-length s-CNTs. We thus expect that negative free energy minima for the adsorption of individual s-CNTs would be calculated for SAMs that promote favorable deposition experimentally.
Fig. 3 shows PMF curves for the adsorption of a s-CNT on C2-, C6-, PEO-, and C2-CF3-grafted surfaces in toluene. The C2 PMF exhibits oscillations with a global minimum (−0.36 ± 0.27 kcal mol−1 with standard deviations computed from bootstrapping) located at ∼3.35 nm, consistent with favorable adsorption. At closer distances, the PMF increases due to steric repulsion from the SAM. Conversely, the PMF curve for the adsorption of a s-CNT on a C6-grafted surface has two comparable shallow minima at ∼3.85 nm (−0.08 ± 0.25 kcal mol−1) and ∼4.41 nm (−0.10 ± 0.30 kcal mol−1), respectively, indicating weak adsorption. The PMF for the adsorption of a s-CNT on a PEO-grafted surface is positive and increases as the distance to the surface decreases, indicating that s-CNT interactions with the surface are uniformly repulsive and adsorption is not thermodynamically favorable. Finally, the PMF curve for the adsorption of a s-CNT on a C2-CF3-grafted surface has a global free energy minimum at ∼3.32 nm that is again negative (−0.55 ± 0.40 kcal mol−1). The locations of minima coincide with locations expected based on the solvent structure near both the SAM and s-CNT (ESI†), which is consistent with structural driving forces that emerge from perturbations to solvent structure near the surface.
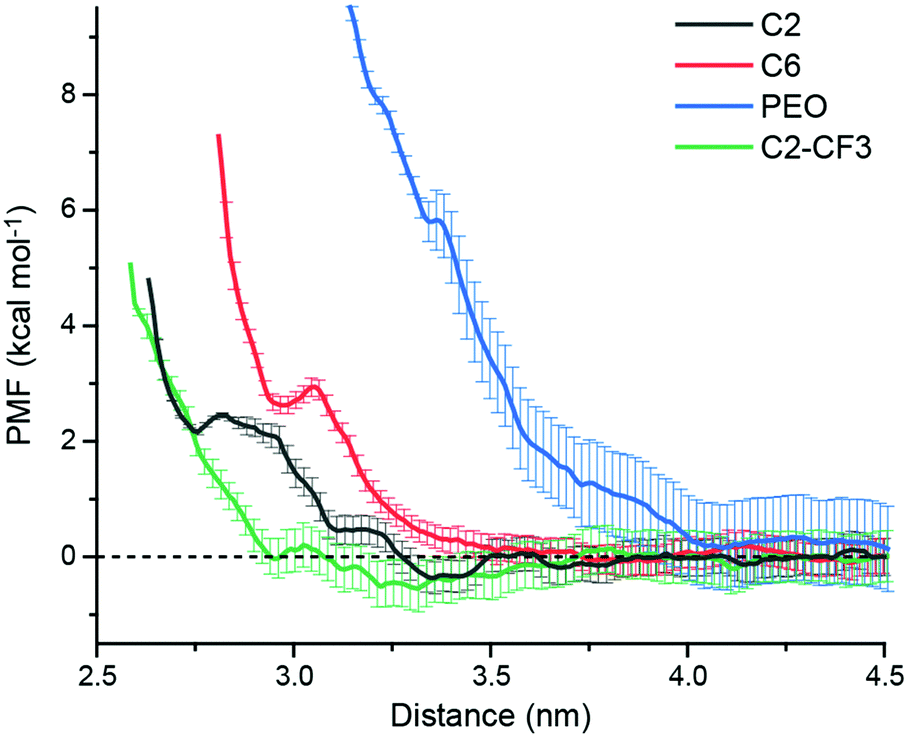 | ||
| Fig. 3 Potential of mean force (PMF) curves for s-CNT adsorption on different SAM-grafted surface in toluene. Error bars report standard deviations from bootstrapping. | ||
Together, the PMF calculations for C2-, C6-, and PEO-grafted surfaces are in good agreement with the P:W ratio predictions. The C2-grafted surface is the most favorable surface for s-CNT adsorption with the largest P:W ratio and corresponding most negative global free energy minimum; conversely, the PEO-grafted surface has the smallest P:W ratio and a monotonically increasing PMF profile. The C6-grafted surface has a moderate P:W ratio and corresponding slight affinity for s-CNT deposition as demonstrated in the small free energy minimum (∼0.1 kcal mol−1). However, the C2-CF3-grafted surface shows a strong affinity for s-CNT adsorption despite a smaller P:W ratio than that of C6. Another surface grafted with a fluorinated SAM (PFOTS, indicated in red in Fig. 2) has a smaller P:W ratio than bare silica and APTES but has been shown experimentally to have a higher affinity for s-CNT adsorption;27 we conclude that fluorinated SAMs promote stronger adsorption than would be expected from solvent structure alone, suggesting a role for additional intermolecular forces. Houston et al.'s force measurements31 revealed stronger adhesive interactions for a CF3-terminated alkanethiol monolayer than its CH3-terminated counterpart, which was ascribed to a stronger dipole moment normal to the surface caused by the CF3 end group. This stronger dipole component of the van der Waals interactions could drive stronger adsorption on C2-CF3-terminated or PFOTS-terminated surfaces in our experiments than surfaces with similar or even greater P:W ratios. In our simulations, this effect is reflected in the more negative charge of fluorine (−0.18) in the -CF3 end group than that of hydrogen (0.06) in -CH3 end groups.
We further sought to validate simulation predictions through the experimental preparation of SAM-grafted surfaces and corresponding evaluation of PFO-BPy-wrapped s-CNT deposition in toluene. Based on the PMFs and P:W ratio measurements, we selected C6 and C2-CF3 SAMs for experimental investigation to compare against s-CNT deposition on bare SiO2. These SAMs are expected to promote less deposition of s-CNTs than on bare SiO2 based on the P:W ratio but more deposition than on the C18 (OTS) SAM, which was shown to eliminate deposition completely.27 Quantifying deposition on these SAMs thus permits comparison to simulation predictions for SAMs expected to exhibit deposition behavior between previously examined extremes. They are also expected to have similar contact angles, again permitting comparison of trends for surfaces with similar macroscopic surface properties. SAMs were prepared following a previously reported procedure (described in the ESI,† Section S6).27 SAM formation was characterized by determining static water contact angles, root-mean-square (RMS) surface roughness, and thickness from ellipsometry measurements (Table 1). The RMS surface roughness and thickness values are consistent with uniform, single-monolayer SAMs. Contact angles for both SAMs are also similar and comparable to the contact angle on C18-grafted surfaces (110.8°).
| Surface | Water contact angle (°) | Thickness (nm) | RMS surface roughness (nm) | Deposition in toluene (CNTs per 10 μm2) | Deposition in chloroform (CNTs per 10 μm2) |
|---|---|---|---|---|---|
| SiO2 | <10 | N/A | 0.114 | 10.3 ± 2.4 | 51.9 ± 4.8 |
| C2-CF3 | 93.4 ± 1.2 | 0.76 ± 0.06 | 0.28 ± 0.05 | 4.8 ± 1.6 | 32.4 ± 1.1 |
| C6 | 108.1 ± 0.5 | 1.06 ± 0.04 | 0.13 ± 0.01 | 2.4 ± 0.6 | 28.6 ± 3.1 |
PFO-BPy-wrapped s-CNTs in both toluene and chloroform at a concentration of 2 μg mL−1 (preparation of s-CNT inks described in the ESI,† Section S7) were deposited on the SAMs following our past experimental procedures. The extent of s-CNT deposition was quantified via analysis of atomic force microscopy images in both chloroform and toluene to show consistency across two solvents. Example images are shown in Fig. 4 and indicate that deposition is highest on bare silica and substantially reduced on both C6- and C2-CF3-grafted surfaces (Table 1). Unlike C18-grafted surfaces, however, a non-negligible number of s-CNTs deposited on the surface. These trends are in good qualitative agreement with the simulation predictions, which also predicted weaker adsorption of single s-CNTs on these SAMs than on bare SiO2 but stronger adsorption than on C18. As noted above, the C2-CF3-grafted surface promotes more CNT deposition than the C6-grafted surface, which agrees with the PMF trend but not the P:W ratio. From this, we conclude that the P:W ratio, as a metric for solvent structure, can successfully capture qualitative deposition trends by predicting whether deposition should be near-zero, moderate, or high. PMF calculations then provide higher-fidelity predictions that capture additional non-structural forces (as observed for the fluorinated SAMs). All simulations and experiments in this work utilized planar substrates, whereas some approaches for s-CNT alignment involve deposition onto patterned substrates with variations in surface topography (e.g., substrates with lithographically defined trenches).32 Because the perturbations to solvent structure and resulting free energy minima for s-CNT adsorption occur on length scales of a few nanometers, we expect that these results will generalize to such substrates. Further exploration of the effect of surface topography on deposition trends will be a subject of future study.
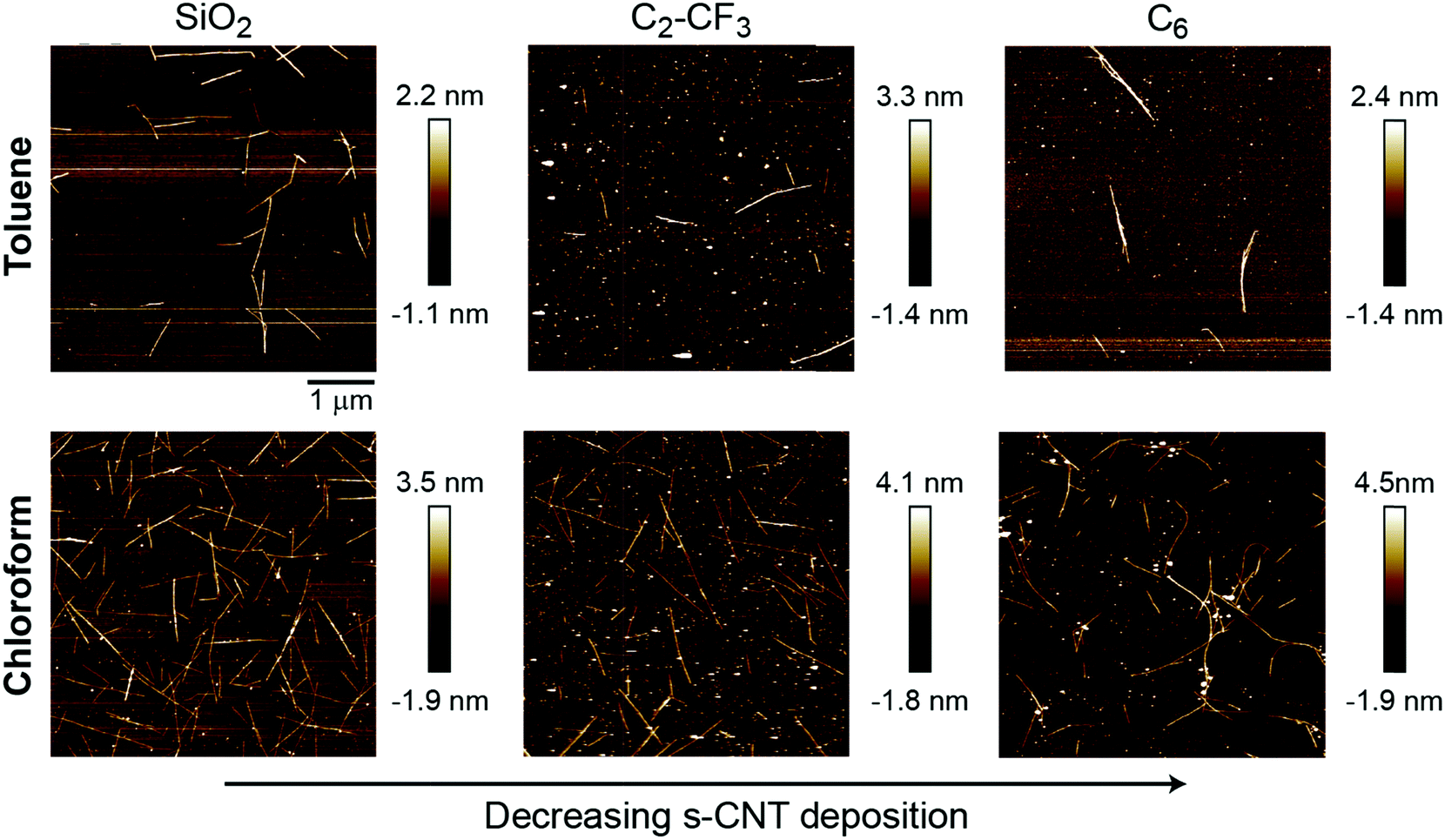 | ||
| Fig. 4 Representative atomic force microscopy images of PFO-BPy-wrapped s-CNTs deposited on bare SiO2 or surfaces grafted with C2-CF3 or C6 SAMs. Deposition was quantified for 2 μg mL−1 s-CNTs in either toluene (top row) and chloroform (bottom row) ink. s-CNTs are visible as thin white lines in the AFM images. | ||
In conclusion, we used MD simulations to examine solvent (toluene) structures near a variety of SAM-grafted silica surfaces with different chemistries. We propose a simple simulation-derived descriptor – the P:W ratio – to capture oscillatory solvent structure near these SAMs, which is associated with solvent-mediated structural forces driving deposition. Based on P:W ratios for 22 SAMs, we calculated free energies for s-CNT adsorption on 4 SAMs with strong (C2), minimal (PEO), and intermediate (C6 and C2-CF3) solvent density oscillations. C2- and C2-CF3-grafted surfaces were found to have strong affinity for s-CNT deposition as indicated by negative free energy minima. Free energy trends were found to be in good agreement with experimentally determined number densities of deposited s-CNTs. Together, these findings highlight that the P:W ratio can be used as a descriptor of s-CNT deposition that can be rapidly calculated from MD simulations prior to more accurate PMF calculations or experiments. These findings will help guide the computational design of substrates for localizing s-CNT deposition for next-generation FET fabrication.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Science Foundation (NSF) through SNM-IS award no 1727523 (K. R. J., P. G., M. S. A.) and DMR-2044997 (R. C. V.). R. C. V. also acknowledges support from the Vilas Trust through the Vilas Associates Award, provided by the Office of the Vice Chancellor for Research and Graduate Education at the University of Wisconsin-Madison. The authors gratefully acknowledge the use of facilities and instrumentation at the University of Wisconsin-Madison, Wisconsin Centers for Nanoscale Technology partially supported by the NSF through the University of Wisconsin, Materials Research Science and Engineering Center (DMR-1720415). This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number ACI-1549562.References
- A. S. R. Bati, L. P. Yu, M. Batmunkh and J. G. Shapter, Synthesis, purification, properties and characterization of sorted single-walled carbon nanotubes, Nanoscale, 2018, 10(47), 22087–22139 RSC.
- A. Bachtold, P. Hadley, T. Nakanishi and C. Dekker, Logic circuits with carbon nanotube transistors, Science, 2001, 294(5545), 1317–1320 CrossRef CAS PubMed.
- S. J. Tans, A. R. M. Verschueren and C. Dekker, Room-temperature transistor based on a single carbon nanotube, Nature, 1998, 393(6680), 49–52 CrossRef CAS.
- T. Durkop, S. A. Getty, E. Cobas and M. S. Fuhrer, Extraordinary mobility in semiconducting carbon nanotubes, Nano Lett., 2004, 4(1), 35–39 CrossRef.
- M. S. Fuhrer, B. M. Kim, T. Durkop and T. Brintlinger, High-mobility nanotube transistor memory, Nano Lett., 2002, 2(7), 755–759 CrossRef CAS.
- Z. Yao, C. L. Kane and C. Dekker, High-field electrical transport in single-wall carbon nanotubes, Phys. Rev. Lett., 2000, 84(13), 2941–2944 CrossRef CAS PubMed.
- J. Zhang, A. Lin, N. Patil, H. Wei, L. Wei, H. S. P. Wong and S. Mitra, Robust Digital VLSI using Carbon Nanotubes, IEEE T Comput Aid D, 2012, 31(4), 453–471 Search PubMed.
- S. K. Samanta, M. Fritsch, U. Scherf, W. Gomulya, S. Z. Bisri and M. A. Loi, Conjugated Polymer-Assisted Dispersion of Single-Wall Carbon Nanotubes: The Power of Polymer Wrapping, Acc. Chem. Res., 2014, 47(8), 2446–2456 CrossRef CAS PubMed.
- J. M. D'Arcy, H. D. Tran, A. Z. Stieg, J. K. Gimzewski and R. B. Kaner, Aligned carbon nanotube, graphene and graphite oxide thin films via substrate-directed rapid interfacial deposition, Nanoscale, 2012, 4(10), 3075–3082 RSC.
- J. H. Dwyer, A. Suresh, K. R. Jinkins, X. Q. Zheng, M. S. Arnold, A. Berson and P. Gopalan, Chemical and topographical patterns combined with solution shear for selective-area deposition of highly-aligned semiconducting carbon nanotubes, Nanoscale Adv., 2021, 3(6), 1767–1775 RSC.
- X. L. Li, L. Zhang, X. R. Wang, I. Shimoyama, X. M. Sun, W. S. Seo and H. J. Dai, Langmuir-Blodgett assembly of densely aligned single-walled carbon nanotubes from bulk materials, J. Am. Chem. Soc., 2007, 129(16), 4890–4891 CrossRef CAS PubMed.
- V. Sgobba, G. Giancane, D. Cannoletta, A. Operamolla, O. Hassan Omar, G. M. Farinola, D. M. Guldi and L. Valli, Langmuir-Schaefer Films for Aligned Carbon Nanotubes Functionalized with a Conjugate Polymer and Photoelectrochemical Response Enhancement, ACS Appl. Mater. Interfaces, 2014, 6(1), 153–158 CrossRef CAS PubMed.
- S. Park, G. Pitner, G. Giri, J. H. Koo, J. Park, K. Kim, H. L. Wang, R. Sinclair, H. S. P. Wong and Z. N. Bao, Large-Area Assembly of Densely Aligned Single-Walled Carbon Nanotubes Using Solution Shearing and Their Application to Field-Effect Transistors, Adv. Mater., 2015, 27(16), 2656–2662 CrossRef CAS PubMed.
- K. R. Jinkins, J. Chan, G. J. Brady, K. K. Gronski, P. Gopalan, H. T. Evensen, A. Berson and M. S. Arnold, Nanotube Alignment Mechanism in Floating Evaporative Self-Assembly, Langmuir, 2017, 33(46), 13407–13414 CrossRef CAS PubMed.
- Y. Joo, G. J. Brady, C. Kanimozhi, J. Ko, M. J. Shea, M. T. Strand, M. S. Arnold and P. Gopalan, Polymer-Free Electronic-Grade Aligned Semiconducting Carbon Nanotube Array, ACS Appl. Mater. Interfaces, 2017, 9(34), 28859–28867 CrossRef CAS PubMed.
- S. Kaida, J. Matsui, T. Sagae, Y. Hoshikawa, T. Kyotani and T. Miyashita, The production of large scale ultrathin aligned CNT films by combining AC electric field with liquid flow, Carbon, 2013, 59, 503–511 CrossRef CAS.
- J. B. Joo, R. Dillon, I. Lee, Y. D. Yin, C. J. Bardeen and F. Zaera, Promotion of atomic hydrogen recombination as an alternative to electron trapping for the role of metals in the photocatalytic production of H-2, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(22), 7942–7947 CrossRef CAS PubMed.
- K. R. Jinkins, J. Chan, R. M. Jacobberger, A. Berson and M. S. Arnold, Substrate-Wide Confined Shear Alignment of Carbon Nanotubes for Thin Film Transistors, Adv. Electron. Mater., 2019, 5(2), 1800593 CrossRef.
- C. Klinke, J. B. Hannon, A. Afzali and P. Avouris, Field-effect transistors assembled from functionalized carbon nanotubes, Nano Lett., 2006, 6(5), 906–910 CrossRef CAS PubMed.
- B. Kumar, A. L. Falk, A. Afzali, G. S. Tulevski, S. Oida, S. J. Han and J. B. Hannon, Spatially Selective, High-Density Placement of Polyfluorene-Sorted Semiconducting Carbon Nanotubes in Organic Solvents, ACS Nano, 2017, 11(8), 7697–7701 CrossRef CAS PubMed.
- J. M. Lobez and A. Afzali, Surface-Selective Directed Assembly of Carbon Nanotubes Using Side-Chain Functionalized Poly(thiophene)s, Chem. Mater., 2013, 25(18), 3662–3666 CrossRef CAS.
- G. Y. Ao, J. K. Streit, J. A. Fagan and M. Zheng, Differentiating Left- and Right-Handed Carbon Nanotubes by DNA, J. Am. Chem. Soc., 2016, 138(51), 16677–16685 CrossRef CAS PubMed.
- M. Y. Zhao, Y. H. Chen, K. X. Wang, Z. X. Zhang, J. K. Streit, J. A. Fagan, J. S. Tang, M. Zheng, C. Y. Yang, Z. Zhu and W. Sun, DNA-directed nanofabrication of high-performance carbon nanotube field-effect transistors, Science, 2020, 368(6493), 878–881 CrossRef CAS PubMed.
- H. Pei, R. J. Sha, X. W. Wang, M. Zheng, C. H. Fan, J. W. Canary and N. C. Seeman, Organizing End-Site-Specific SWCNTs in Specific Loci Using DNA, J. Am. Chem. Soc., 2019, 141(30), 11923–11928 CrossRef CAS PubMed.
- L. J. Liu, J. Han, L. Xu, J. S. Zhou, C. Y. Zhao, S. J. Ding, H. W. Shi, M. M. Xiao, L. Ding, Z. Ma, C. H. Jin, Z. Y. Zhang and L. M. Peng, Aligned, high-density semiconducting carbon nanotube arrays for high-performance electronics, Science, 2020, 368(6493), 850–856 CrossRef CAS PubMed.
- K. R. Jinkins, S. M. Foradori, V. Saraswat, R. M. Jacobberger, J. H. Dwyer, P. Gopalan, A. Berson and M. S. Arnold, Aligned 2D carbon nanotube liquid crystals for wafer-scale electronics, Sci. Adv., 2021, 7(37), eabh064 Search PubMed.
- J. H. Dwyer, Z. Z. Shen, K. R. Jinkins, W. Wei, M. S. Arnold, R. C. Van Lehn and P. Gopalan, Solvent-Mediated Affinity of Polymer-Wrapped Single-Walled Carbon Nanotubes for Chemically Modified Surfaces, Langmuir, 2019, 35(38), 12492–12500 CrossRef CAS PubMed.
- G. J. Brady, A. J. Way, N. S. Safron, H. T. Evensen, P. Gopalan and M. S. Arnold, Quasi-ballistic carbon nanotube array transistors with current density exceeding Si and GaAs, Sci. Adv., 2016, 2(9), e160124 Search PubMed.
- J. Israelachvili, Solvation Forces and Liquid Structure, as Probed by Direct Force Measurements, Acc. Chem. Res., 1987, 20(11), 415–421 CrossRef CAS.
- S. Kumar, D. Bouzida, R. H. Swendsen, P. A. Kollman and J. M. Rosenberg, The Weighted Histogram Analysis Method for Free-Energy Calculations on Biomolecules. 1. The Method, J. Comput. Chem., 1992, 13(8), 1011–1021 CrossRef CAS.
- J. E. Houston, C. M. Doelling, T. K. Vanderlick, Y. Hu, G. Scoles, I. Wenzl and T. R. Lee, Comparative study of the adhesion, friction, and mechanical properties of CF3- and CH3-terminated alkanethiol monolayers, Langmuir, 2005, 21(9), 3926–3932 CrossRef CAS PubMed.
- A. Corletto and J. G. Shapter, Nanoscale Patterning of Carbon Nanotubes: Techniques, Applications, and Future, Adv. Sci., 2021, 8(1), 2001778 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional details on simulation and experimental methods and SAM physical properties. See DOI: https://doi.org/10.1039/d2sm00572g |
| This journal is © The Royal Society of Chemistry 2022 |