
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of ion mobility on the redox and catalytic properties of Cu ions in zeolites
Matteo
Signorile
 ,
Elisa
Borfecchia
,
Silvia
Bordiga
and
Gloria
Berlier
*
,
Elisa
Borfecchia
,
Silvia
Bordiga
and
Gloria
Berlier
*
Department of Chemistry and NIS Centre, Università di Torino, Via P. Giuria 7, Torino, 10125, Italy. E-mail: gloria.berlier@unito.it
First published on 19th August 2022
Abstract
This contribution aims at analysing the current understanding about the influence of Al distribution, zeolite topology, ligands/reagents and oxidation state on ions mobility in Cu-zeolites, and its relevance toward reactivity of the metal sites. The concept of Cu mobilization has been originally observed in the presence of ammonia, favouring the activation of oxygen by formation of NH3 oxo-bridged complexes in zeolites and opening a new perspective about the chemistry in single-site zeolite-based catalysts, in particular in the context of the NH3-mediated Selective Catalytic Reduction of NOx (NH3-SCR) processes. A different mobility of bare Cu+/Cu2+ ions has been documented too, showing for Cu+ a better mobilization than for Cu2+ also in absence of ligands. These concepts can have important consequences for the formation of Cu-oxo species, active and selective in other relevant reactions, such as the direct conversion of methane to methanol. Here, assessing the structure, the formation pathways and reactivity of Cu-oxo mono- or multimeric moieties still represents a challenging playground for chemical scientists. Translating the knowledge about Cu ions mobility and redox properties acquired in the context of NH3-SCR reaction into the field of direct conversion of methane to methanol can have important implications for a better understanding of transition metal ions redox properties in zeolites and for an improved design of catalysts and catalytic processes.
 Matteo Signorile | Matteo Signorile obtained the PhD in Chemistry and Materials Sciences at University of Torino in 2017. His research is devoted to the in situ/operando characterization and simulation of microporous materials and their properties as adsorbents and catalysts. He developed his research activity in collaboration with both academic and industrial partners. Since 2020, he was enrolled as assistant professor in Physical Chemistry at University of Torino. He is (co)author of more than 40 papers on peer-reviewed journals and of 2 patents. |
 Elisa Borfecchia | Elisa Borfecchia obtained her PhD degree in 2013 at the University of Turin. During her PhD and afterward, her research centred on characterizing functional materials by X-ray spectroscopy. From 2016, she worked at Haldor Topsøe A/S and she was subsequently awarded a Marie Curie Fellowship to join the Catalysis group at Oslo University. From 2019, she has been assistant professor in Physical Chemistry at the University of Turin, where she is currently Associate Professor. She authored >80 peer-reviewed papers and performed >50 experiments at synchrotrons, aimed at disclosing local structure and reactivity of metal centres in heterogeneous catalysts. |
 Silvia Bordiga | Silvia Bordiga is Full Professor in Physical Chemistry at the University of Turin. From 2012 to 2020 she was Prof. II at the University of Oslo. She received many awards: 2017-French Chemical Society prize; 2019-Francois Gault Lectureship from EFCATS; 2018–2019-Chemistry European Fellow; 2019-Wilhelm Manchot Research Professorship at the Chemistry Department at the TUM University, Germany; 2021-Premio Antonio Feltrinelli from Accademia dei Lincei. Her scientific activity is mainly devoted to the characterization of the physical–chemical properties of high surface area nanostructured materials used as heterogeneous catalysts, materials for adsorption, separation and storage, through in situ spectroscopic studies. |
 Gloria Berlier | Gloria Berlier is full Professor in Physical Chemistry at the University of Turin. She has specialized in the use of advanced characterization techniques related to surface and solid state science. Focus is on the spectroscopic investigation (often coupled to modeling) of the electronic and geometrical structure of surface sites also in reaction conditions, and of the mechanisms ruling adsorption processes as fundamental step for applications in catalysis and nanomedicine. She is co-author of 130 papers on peer-reviewed journals. |
1. Introduction
Ions mobility in solid state is crucial for several technological applications for energetic purposes, ranging from solid electrolytes to catalysis.1–3 In the latter field, the ion mobility of cations in zeolite catalysts (including Cu-exchanged ones) is still a largely unexplored field, with potentially dramatic implications in optimization and innovation of thousands of industrial processes. As a matter of fact, zeolites play crucial roles in various fields including chemical industry, petroleum refining and selective adsorption and separation. Apart from their practical applications, they have attracted the attention and curiosity of researchers along the decades because of their void intracrystalline volume consisting of channels and cages, allowing diffusion, adsorption and reaction of molecules, which are strongly affected by the microporous confinement effect.4–7 Another fascinating aspect of the inner structure of zeolites is that it can stabilize cations in geometries and even oxidation states which are not usually observed in solids, solution or homogeneous complexes.8,9 Protons represent a particular and important class of cations stabilized in zeolites, conferring them a specific Brønsted acidity,10–13 which is crucial for many applications.14–16 The acid strength of protons in zeolites depends on the zeolite topology, composition and proton location in specific crystallographic positions.12,17,18 However, the relation of proton acid strength and concentration on the catalytic activity is not straightforward,10 also in relation to proton mobility,19 which can be mediated by interaction with reactants or adsorbed molecules, such as water17,20,21 or ammonia.22–24Metal cations are stabilized in specific crystallographic positions in the channels and cages of zeolites, depending on the nature of the metal cation combined with the topology and chemical composition of the material. In principle, X-ray diffraction is the main technique to describe cations location in ordered materials, but X-ray absorption spectroscopy (XAS) has played a major role in recent years in describing the local coordination of metal ions in zeolites.25,26 Infrared spectroscopy coupled to the use of probe molecules has also been extensively applied in the field, providing a huge amount of (indirect) detailed information on cations distribution, Lewis acidity and coordinative unsaturation in different zeolites.27,28 On the other hand, analysis of the perturbation of T–O–T skeletal vibrations (T for Si or Al tetrahedral framework atom) has allowed some authors to discriminate among the different location and accessibility to molecules of extra framework cations.29–33
Most of the above mentioned reports are based on a static description of the cations, which can be described as framework coordinated, meaning that they are stabilized in specific positions by the electrostatic interactions related to the presence of the negative charge delocalized on the framework oxygen atoms connected to Al3+ heteroatoms (labelled as Z in the following). However, in analogy with what briefly mentioned for protons, the mobility of cations in zeolites is not a new concept: ions redistribution has been observed for instance as a consequence of dehydration treatments34 or oxidation reactions.35 Another fascinating example is the complexation of Cu+ ions in Cu-ZSM-5 by CO, forming homogeneous-like structures where the metal ion is partially detached from the framework.8,36,37
Keeping in mind this knowledge about metal ions in zeolite, this perspective deals with the recent findings about mobilization of Cu ions in zeolites under reaction conditions, with particular attention to the role of reactants and ligands on the ions redox properties and reactivity in the two most studied reactions over Cu-zeolites in recent years: the Selective Catalytic Reduction of NOx mediated by NH3 (NH3-SCR) and the direct conversion of methane to methanol (DMTM).
2. Redox reaction by Cu zeolites
The redox behaviour of Cu ions in zeolites, reversibly changing their oxidation state from Cu2+ to Cu+ as a function of the treatment atmosphere has been studied for decades.38–40 Their potentiality as catalysts was first investigated for the direct decomposition of nitric oxide to nitrogen and oxygen,41 but practical applications were limited by the low hydrothermal stability of the studied Cu-zeolites (mainly Cu-ZSM-5) under reaction conditions. The turning point was the discovery of the excellent stability, coupled with high activity and selectivity at low temperature (200 °C) in the NH3-SCR reaction of Cu-CHA (also referred to as Cu-SSZ-13 in case of low aluminium content materials),42 a small-pore zeolite characterized by the chabazite topology.43,44The NH3-SCR reaction is the basis of an important technology for the removal of NOx from diesel vehicles and stationary plant exhaust gases.45 It is a redox reaction following eqn (1) and (2) for standard and fast SCR, respectively:
| 4NO + 4NH3 + O2 → 4N2 + 6H2O | (1) |
| 4NH3 + 2NO + 2NO2 → 4N2 + 6H2O | (2) |
The reaction mechanism is still a fascinating (and debated) subject of investigation.46,47 Some of the key aspects of the mechanism most debated in the literature have been: (i) whether the catalytically relevant Cu are monomeric or dimeric (even, perhaps very small oligomeric clusters); (ii) whether redox cycling of the catalytic centres is involved and (iii) whether both Cu ion sites and Brønsted acid sites synergically provide the catalytic functionality.48 A step forward came with the work by Janssens et al. showing the consistency of a mechanism based on a redox cycle between monomeric framework coordinated Cu2+ and Cu+ ions (Fig. 1, left panel).49 The mechanism is based on the following hypotheses: (i) in each reaction step, the mass balance is maintained, and the complete catalytic cycle is consistent with the total stoichiometry of the SCR reaction (eqn (1)); (ii) adsorption from and desorption to the gas phase can take place only with stable molecules (O2, NO, NO2, NH3, N2, and H2O) implying that adsorption or desorption of fragments is not allowed; (iii) the oxidation state for the active Cu ion changes from 2+ to 1+ in the reduction part, and from 1+ to 2+ in the oxidation part; (iv) in each step, the charge balance is maintained and the total charge of the Cu complexes in the zeolite is always 1+, allowing for coordination to a single negatively charge Z site in the zeolite. The reversible change of oxidation state was measured by dividing the catalytic cycle into a reduction (NO/NH3 atmosphere) and an oxidation (NO/O2) half cycle. This redox concept has been widely accepted in the scientific field, even though the reaction mechanism has been later enriched by the breakthrough hypothesis that at low temperatures mobile NH3-solvated Cu+ complexes, linear Cu+(NH3)2,50,51 can activate O2 with formation of [(Cu2+)2(NH3)4O2]2+ species, implying a quasi-homogenous catalytic process (Fig. 1, right panel, part b).3,52
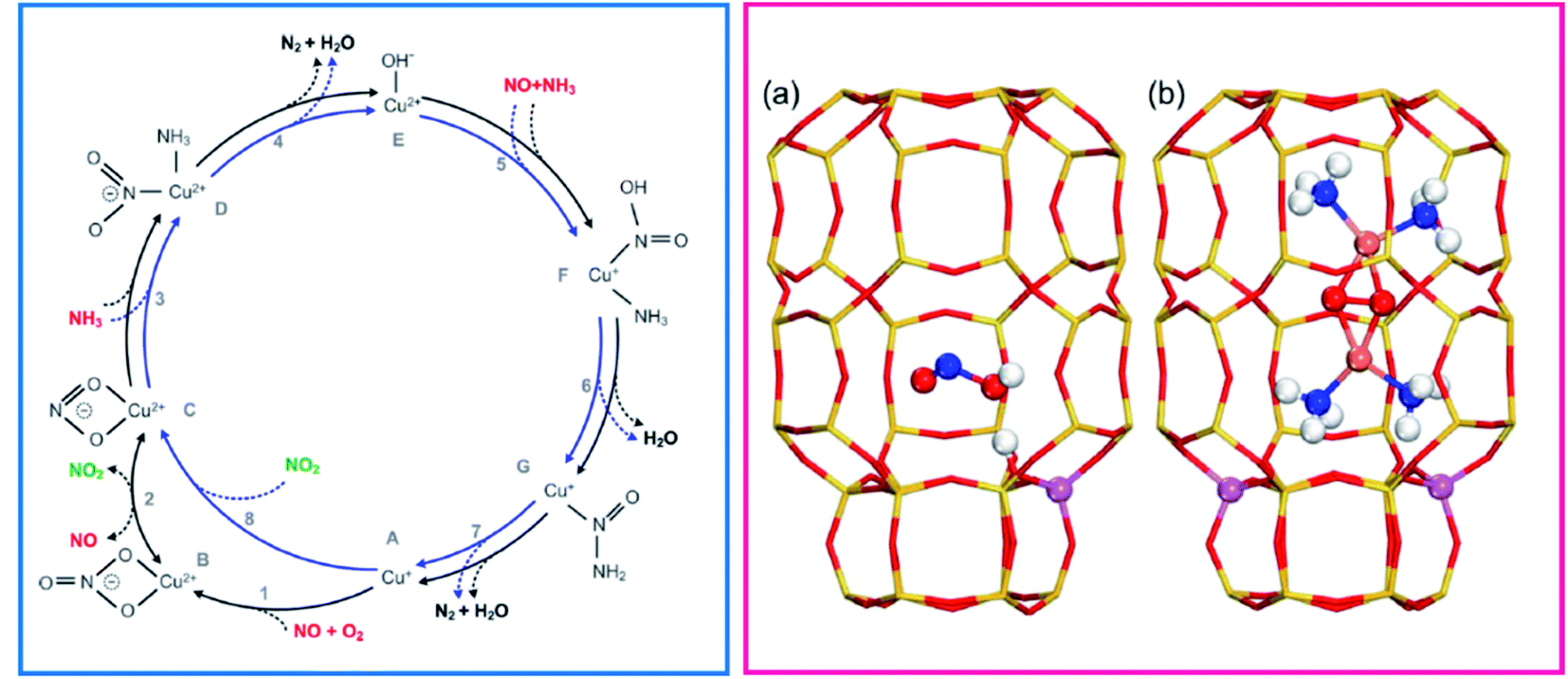 | ||
| Fig. 1 (Left) Proposed reaction scheme for the NH3-SCR reaction in a Cu-zeolite involving Cu2+ and Cu+ framework coordinated single cations. Adapted with permission from ref. 49. Copyright 2015 American Chemical Society. The inner blue circle represents the fast SCR cycle. Reactants are indicated in red, reaction products in black, and the proposed NO2 intermediate in green. (Right) Side view of the chabazite cages showing the Al position in the case with (a) one and (b) two atoms. (a) Is shown with a HONO molecule adsorbed on the Brønsted acid site. (b) Shows a [(Cu2+)2(NH3)4O2]2+ complex with two Al atoms situated in one six-membered ring. Atom colour codes: Cu (pink), Si (yellow), Al (purple), O (red), N (blue), and H (white). Adapted with permission from ref. 53. Copyright 2020 American Chemical Society. | ||
The O2 activation by Cu+ in the oxidation half-cycle is an important step, since Cu+ can deliver only one electron to close the cycle and form Cu2+, while O2 activation and dissociation requires four electrons. This implies the participation of other electron sources, such as NO or other intermediates.49,54,55 This conundrum can be solved with the formation of a side-on μ-η2,η2-peroxo diamino dicopper(II) complex, with general formula [(Cu2+)2(NH3)4O2]2+, which can react with NO giving the N2 and H2O products closing the cycle.56,57 More recently, mobile [Cu2+(OH)(NH3)x]+ complexes formed by NH3 solvation of ZCu2+OH ions (stabilized by one framework negative charge), have been proposed to be relevant in the reduction half-cycle, as discussed in mode detail in Section 3.58,59 Finally, the formation and separation of Cu pairs assisted by NH3 solvation is the core of one of the recently proposed mechanisms for NH3-SCR, which includes the role of Brønsted sites to facilitate the decomposition to N2 and H2O of HONO and H2NNO intermediates formed on Cu sites (Fig. 1, right panel, part a).53
The formation of multinuclear CuxOy centres has demonstrated a particular relevance also for reactions other than NH3-SCR, e.g. in DMTM. The first report concerning the use of Cu-zeolites in this process appeared in 2005 on Cu-ZSM-5.60 Since then, researchers have focused their efforts on the optimization of the process and related reaction yield, due its huge environmental and economic potential impact.61,62 Apart from Cu-ZSM-5 (MFI), the most studied (and best performing) zeolites for this reaction are Cu-MOR (mordenite),61,63–65 Cu-CHA,66,67,67 and Cu-MAZ (mazzite).68 With respect to NH3-SCR, naturally implemented as a continuous process, most of the studies presently available in the literature have focused on stepwise DMTM process, where reagents (O2, CH4, H2O) are fed sequentially rather than simultaneously, also adopting optimized temperature-pressure conditions at each step. Despite this conceptual difference, both the processes takes advantage of the redox cyclability of the Cu+/Cu2+ couple, also featuring similar intermediate species as stabilized by the host zeolite framework.
There is a general agreement in the literature about the role of dimeric60,61,63,64,69,70 (or multimeric)65,71,72 Cu-oxo complexes as active sites in the DMTM conversion. These should be able to selectively react with CH4 forming adsorbed methoxy groups that can be hydrolysed by water steam to release methanol. The Cu-oxo species are formed during a thermal treatment (activation) of Cu2+-zeolites in O2, air, N2O or even H2O (ref. 61) or can be formed by reaction of two vicinal framework interacting Cu+ sites (formed during an activation in inert conditions) with the oxidizing agent.66,71,73 The two Cu+ sites are then reformed by reaction of Cu-oxo active sites with methane and subsequent methanol release (Fig. 2).61
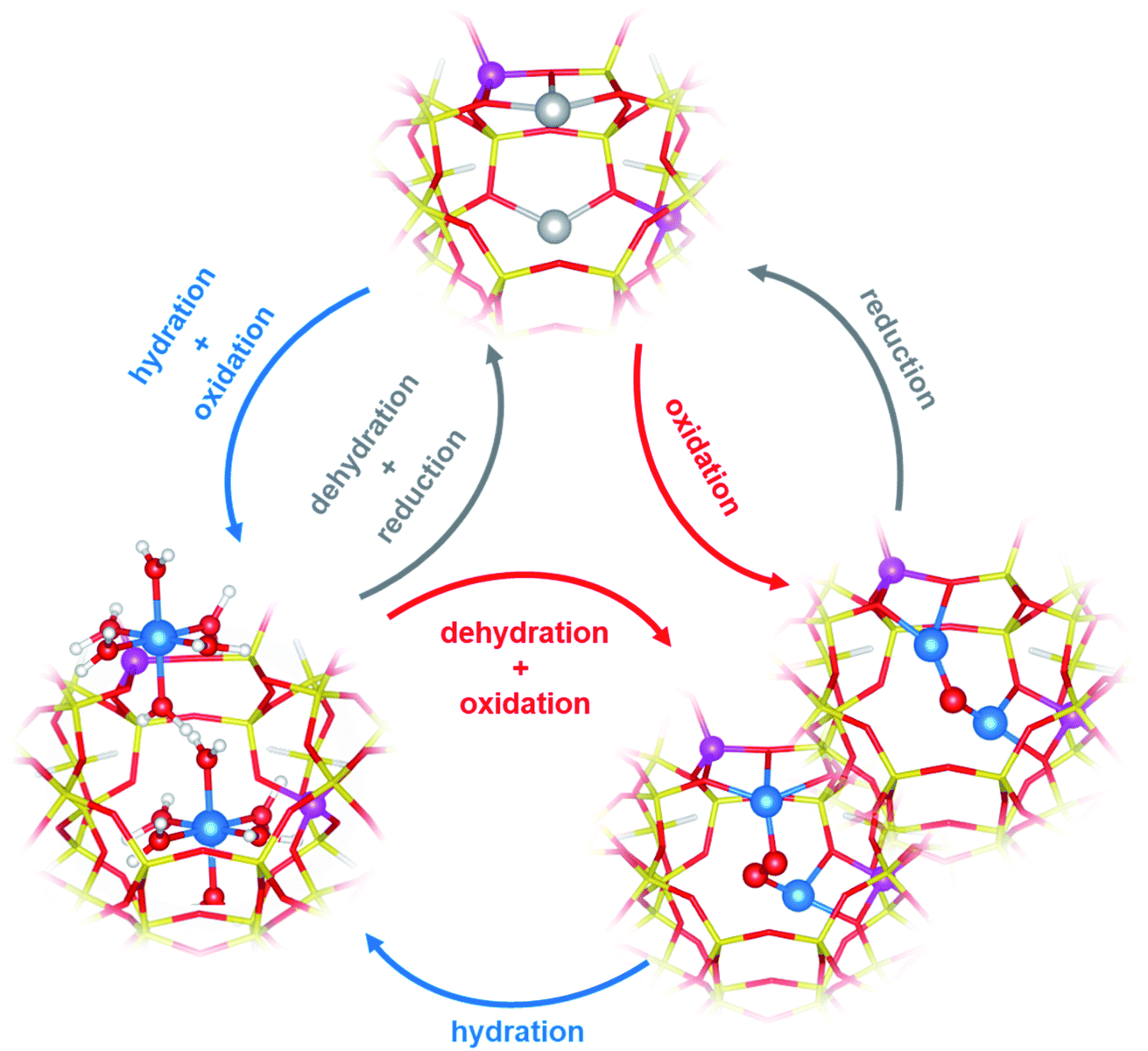 | ||
| Fig. 2 Examples of redox cycles involving Cu2+/Cu+ ions in DMTM. Colour code: blue, Cu2+; gray, Cu+; yellow, Si; magenta, Al; red, O; white, H. Unpublished figure, structural models from ref. 81. | ||
The identification of the active sites for DMTM is an intriguing and challenging research topic, due to the difficulty in the precise identification of Cu-oxo species in heterogenous systems such as Cu-zeolites.66,74,75 An overview and critical discussion of the main proposed structures and reaction mechanism (with the relevant characterization techniques) can be found in ref. 62. Even if many authors have initially proposed that this is a single-site reaction, most of the recent relevant literature points to the fact that a multitude of copper sites (CuxOy) are always formed in activated Cu-exchanged zeolites, only a fraction of which can convert methane to methanol.76 The zeolite topology, Si/Al ratio and Al distribution strongly affect the Cu sites distribution and related activity. Among the multitude of proposed structures (μ-oxo, μ-hydroxo, peroxo, superoxo, etc.)62,65,66,77 dimeric Cu2Oy sites featuring vicinal Cu2+ could be proposed for mechanisms involving Cu2+/Cu+ redox cycles, as obtained by dehydration of the pristine Cu-zeolite under appropriate oxidative/reductive environmental conditions (Fig. 2).62,71,76,78–80
Interestingly, formation of CuxOy moieties by activation in O2 or other oxidants, and subsequent reaction with methane with breaking of the multimeric cores and formation of framework interacting Cu+ ions, imply that Cu ions are reversibly moving in the zeolite cages during the redox cycle.71
3. Cu at the equilibrium: influence of framework Al content and distribution
A main parameter defining the positioning of Cu within the microporous framework of a zeolite is certainly the Al distribution as a substituent of Si. In fact, Al can occupy positions within the framework that are very different in topological terms, e.g., belonging to different ring systems/cages/channels. Furthermore, the distribution of Al sites is strictly related to the speciation of hosted cations, since it controls the degree of neighbouring of the cationic sites, thus their possibility to establish mutual interactions. The peculiarities in the local environment of Al sites are obvious in the case the investigated zeolite topology provides multiple independent crystallographic tetrahedral sites. This is the case for common topologies such as Mordenite (MOR) or ZSM-5 (MFI). The Al siting in H-MOR has been deeply investigated by correlating the different perturbation of the O–H stretching vibration of its Brønsted acid sites to the position of the Al bearing the proton. In brief, the asymmetry of the ν(O–H) band has been described as due to the overlap of two distinct components, representative for Brønsted acid sites located in the 8- and 12-membered rings of the MOR framework (giving rise to the low and high frequency shoulders of the band, respectively).80,82,83 The case of ZSM-5 is even more complex, due to the higher number of symmetry independent tetrahedral sites (12 vs. 4 in MOR). Nonetheless, the distribution of monovalent cations (including Cu+, Zn+, Cd+, Cs+) has been investigated by several techniques in order to infer the existence of preferential substitutional sites for Al. High resolution X-ray diffraction studies conducted on Cs+-ZSM-5 allowed identifying a subset of highly probable substitutional positions for Al, namely crystallographic positions T2, T7 (most favoured), T8, T10.83 More recently, Morra et al. combined EPR/HYSCORE outcomes with DFT simulations to further screen over the most probable Al site on Zn+-ZSM-5 and Cd+-ZMS-5.84 By exchanging the samples with 17O, an extremely detailed characterization of the local environment surrounding the paramagnetic cation was possible through the reconstruction of the hyperfine coupling tensor. On the basis of DFT simulations, it was finally possible to recognize as the local structure of the cations is significantly affected by both the nature of the cation and the local topology of the zeolite framework around it. As an example, Zn+ preferably coordinates to the framework by interacting with two O atoms adjacent to Al; Cd+, instead, is able to interact with more framework O, up to 5 in the case the Al site sits in a crystallographic position within a “pocket-like” region of the framework. A similar behaviour is observed for Cu+, as shown in Fig. 3.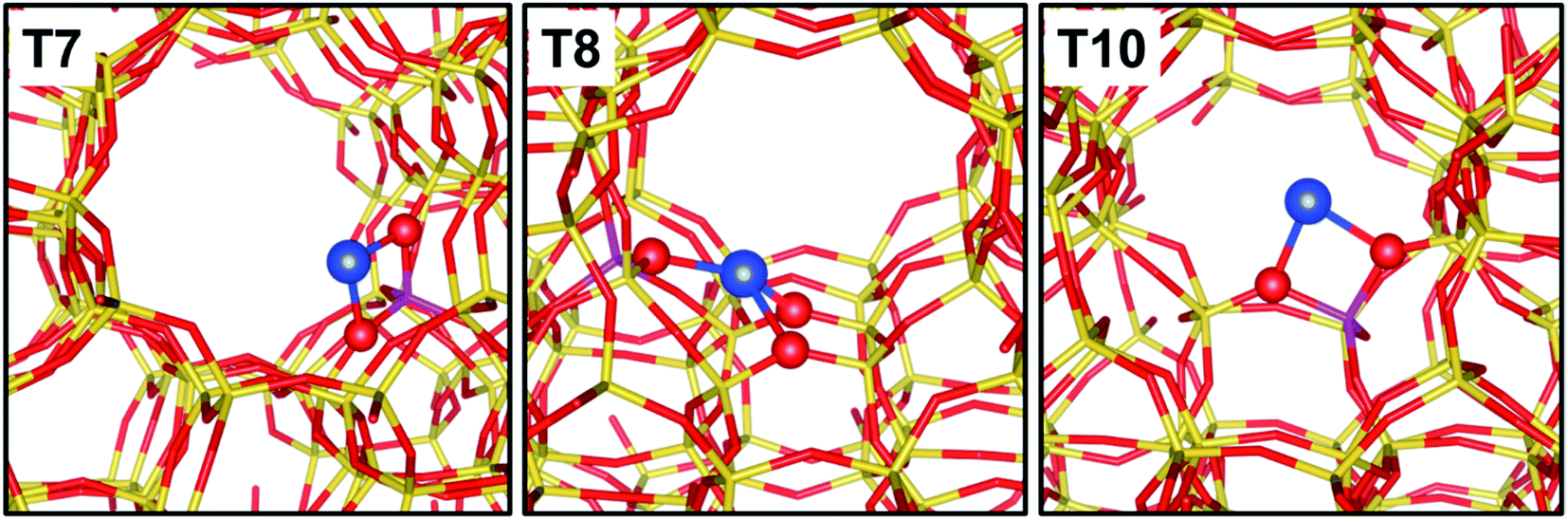 | ||
| Fig. 3 Graphical representation of Cu+ sites balancing Al sitting in the T7, T8 and T10 crystallographic positions of ZSM-5. Colour code: blue, Cu; yellow, Si; magenta, Al; red, O. Unpublished figure, structural models obtained from DFT calculations as reported in ref. 37. | ||
In MFI framework, site T7 is exposed at the crossing of the sinusoidal and the straight channels, and hosts the charge balancing Cu+ cation in the intersection cavity with a bridged configuration over two framework O atoms. Site T8 is placed on the wall of the straight channel, with a relatively flat local environment: accordingly, the Cu+ hosted by this site coordinates with more framework O atoms compared to other positions. Finally, site T10 is positioned on a step of the sinusoidal channel, with a Cu+ coordination similar to that described for site T7.
Though the Al siting could seem related to complex/low symmetry zeolite topologies only, this is also relevant for simpler/highly symmetric structures, e.g., chabazite (CHA). CHA features a single symmetry independent tetrahedral site, so that the local environment for an isolated cation is expected to be closely identical across the whole material. Nevertheless, CHA turns into a complicated system because of the possible neighbouring of Al sites (and thus the related cations), as a consequence of its relatively low Si/Al ratios and the small unit cell. Simultaneously considering multiple Al sites scales-up the siting problem to a combinatorial one, where ideally all symmetry-independent combinations for Al positioning are contributing to its overall distribution. In this regard, the composition of the sample is a fundamental variable, together with the crystallographic properties of the framework, toward the effective understanding of the Al/cations distribution in a zeolite. Paolucci et al.85 provided a milestone study in this context, where the effect of Si/Al and Cu/Al ratios on the equilibrium speciation of the Cu cations in Cu-CHA was assessed through an extensive computational screening. By computing the formation ΔG of the most relevant Cu+/Cu2+ species hosted in zeolites (also including Cu2+ oxygenated, hydroxylated and hydrated sites), a phase diagram describing the equilibrium fraction of redox active ZCu2+OH vs. total Cu as a function of Si/Al and Cu/Al ratios was obtained. Taking advantage of this results, a precise design of the Cu sites speciation is achievable by simply tuning the compositional synthetic parameters. Such model was independently validated by Martini et al.,86 who analysed by means of XAS a pool of Cu-CHA characterized by a systematic variation of Si/Al and Cu/Al. Through a Multivariate Curve Resolution – Alternating Least Squares (MCR-ALS) analysis, the variations in the speciation of Cu sites along a thermal treatment under an inert atmosphere was achieved and described also with the support of a computational-assisted identification of the singular pure components extracted by the analysis algorithm. In this way, the preference of Al-rich samples toward Z2Cu2+ (stabilized by two vicinal Al atoms) was experimentally demonstrated in a quantitative way. In agreement with previsions by Paolucci et al., as shown in Fig. 4, a low content of Cu further pushes the equilibrium towards Z2Cu2+, often described as “redox resistant” Cu.
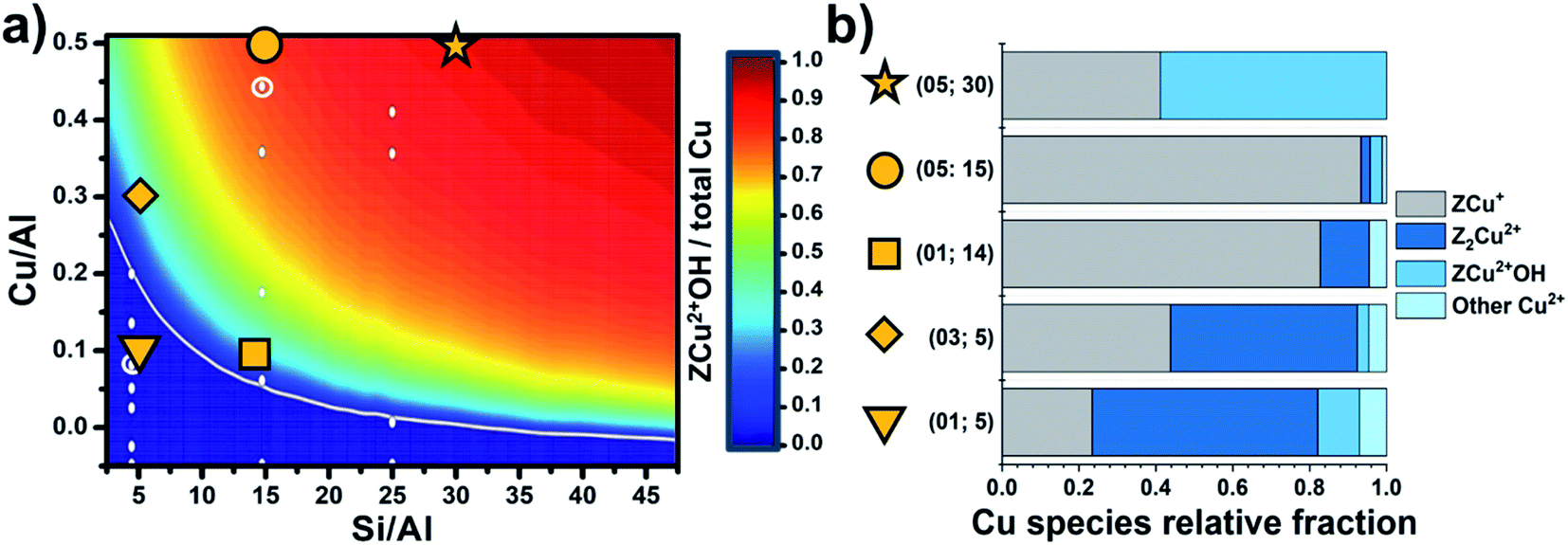 | ||
| Fig. 4 (a) Predicted Cu site compositional phase diagram versus Si/Al and Cu/Al ratios. Color scale indicates predicted fraction of ZCu2+OH. Adapted with permission from ref. 85. Copyright 2016 American Chemical Society. (b) Relative fractions of Cu species; the label “other Cu2+” refers to very minor contributions from residual, partially hydrated Cu2+ species. Unpublished figure reporting data from ref. 86. | ||
ZCu2+OH sites turn predominant in high silica samples. Finally, samples with intermediate Si/Al demonstrated an interesting reducibility, that has been interpreted as the result of the following process: (i) low temperature (<200 °C) dehydration of Cu-aquo cations to form ZCu2+OH sites; and (ii) at higher temperature (>200 °C) further dehydration of ZCu2+OH via water/oxygen elimination and consequent reduction of Cu2+ to Cu+ (i.e., forming a ZCu+ site). Since the experiment was conducted in inert environment (He flow), the reduction of Cu is most probably occurring via condensation reactions involving neighbouring ZCu2+OH centres, so their spatial relations within the CHA framework are of outmost importance. In fact, if ZCu2+OH are too far from each other, as a consequence of the long distances among Al sites (as expected in Al-poor samples), thus their mutual reactivity turns unfavoured. Conversely, if Si/Al is tuned in a way the density of Al per cell is approximately 1 (so still limiting the formation of neighbouring Al sites and the formation of redox inactive ZCu2+), most of ZCu2+OH could be sufficiently close to promote the reactions required for their conversion into reduced ZCu+ sites.40,66 Concerning the auto-reduction process, it is worth noticing that the true phenomena underlying the reduction of Cu in zeolites under an inert environment are still subject of debate; in particular, a key role in the effectiveness of reduction is played by traces of hydrocarbons commonly pre-adsorbed within the zeolite pores, that could offer much more effective reductive reaction paths than bare condensation of ZCu2+OH.40 Nonetheless, in an optimal composition sample (Si/Al = 15, Cu/Al = 0.5), almost 90% of Cu can be reduced, with a significant impact on subsequent reactivity thanks to its elevated redox cyclability.66
4. Cu mobility: relation with oxidation state and ligands coordination sphere
As mentioned above, the role of NH3 in solvating and mobilizing Cu+ and Cu2+ ions in the low temperature NH3-SCR reaction has been proposed to be pivotal, in both the oxidation and reduction half-cycles.3,52,58,59 The ability of NH3 to solvate the cations, replacing framework oxygen atoms (Ofw) from their first-shell coordination sphere has been proven by X-ray absorption and emission spectroscopies (XAS and XES).50,51 More in detail, Extended X-ray Absorption Fine structure (EXAFS) showed the disappearance of the fingerprint for framework coordinated Cu-sites (second-shell peak at ∼2.3 Å in the phase-uncorrected modulus of FT-EXAFS spectrum) when a Cu-CHA catalysts was contacted with the NH3-SCR reactants below 250 °C. This was accompanied by a raise in the pre-edge peak at ∼8982.5 eV, deriving from 1s → 4p transition in the linear Cu+(NH3)2 complex in the corresponding X-ray absorption near edge structure (XANES) spectrum, and by modifications in the XES spectra compatible with the substitution of Ofw ligand with N atoms from the NH3 molecule (Fig. 5).51 Above 250 °C the spectral features indicated the formation of framework coordinated Cu ions, as a consequence of NH3 ligands desorption.87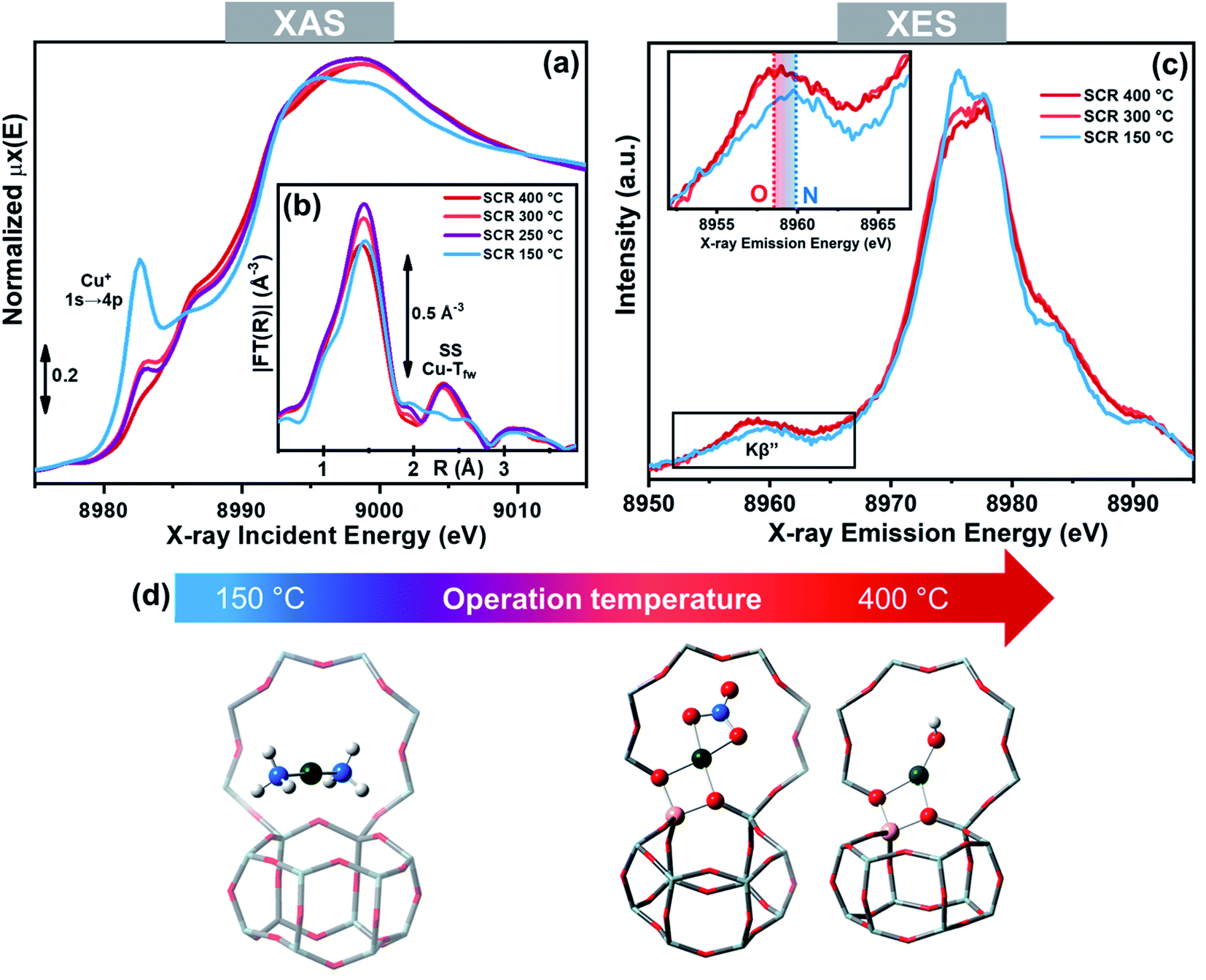 | ||
| Fig. 5 Operando XAS and XES spectra of a Cu-CHA catalyst (Cu/Al = 0.48; Si/Al = 15) during NH3-SCR in the 150–400 °C range. (a) Cu K-edge XANES. (b) Magnitudes of FT-EXAFS spectra; (c) valence-to-core Cu Kβ XES; the inset reports a magnification of the Kβ′′ satellite peak, highly sensitive to the type of ligands. (d) Structural models of the main Cu-species identified by X-ray spectroscopy in the low- and high-temperature range under NH3-SCR conditions, including Cu+(NH3)2 and ZCu2+OH/ZCu2+NO3, respectively; colour code: green, Cu; grey, Si; yellow, Al; red, O; white, H. Unpublished figure reporting data from ref. 51. | ||
The potential impact of Cu ions mobilization in the NH3-SCR reaction was pointed out by Gao et al., who proposed the formation of transient [(Cu2+)2(NH3)4O2]2+ complexes by DFT calculations.52 Paolucci et al. calculated the probability of formation of these dimeric complexes as a function of Cu volumetric density (and corresponding Cu–Cu average distance within the zeolite). They assumed that one Cu+(NH3)2 complex can migrate between adjacent chabazite cages to meet another Cu+(NH3)2 and activate together one O2 molecule. Their calculations indicate that electrostatic interactions dominate the complexes mobility, since the positively charged Cu+(NH3)2 species are stabilized by the framework negative charge, and can only travel up to 9 Å apart from the framework Al heteroatom inducing the framework negative charge, on kinetically relevant timescales.3 On the other hand, the long-range mobility of NH3 solvated Cu+ ions has been demonstrated by preparing active Cu-exchanged zeolites by a very effective solid-state ion-exchange process.88,89 It has been shown that Cu+ ions can be mobilized and travel within the zeolite by heating at low temperature (250 °C) a physical mixture of the zeolite and Cu2O or CuO in an NH3 (or NO/NH3) gas atmosphere.88 The charge neutrality for this (slow) pathway is proposed to be maintained via exchange of H+, in the form of NH4+, from the zeolite to the Cu2O surface, where water can be formed.90
The limitations to the mobility on kinetically relevant timescales of NH3-solvated Cu complexes related to the electrostatic interactions with the negatively charged framework have been considered by Villamaina et al. in their recent study.58 The authors have used dry CO oxidation to probe Cu2+ dimers in Cu-CHA catalysts in the presence of NH3. CO oxidation to CO2 is a two-electron oxidation, while the Cu reduction from Cu2+ to Cu+ is a one-electron reduction. This implies that CO2 formation in dry CO oxidation requires either two monomeric Cu2+ sites or a dimeric Cu-oxo species with one removable oxygen atom.58 By exploiting the same concept, Li et al. have made a breakthrough in the interpretation of UV-Vis spectra of Cu2+ sites in CHA, by ab initio molecular dynamics and time-dependent density functional theory calculations. Dry CO oxidation in this case was used to reduce Cu2+ dimers or oligomers to Cu+, leaving behind isolated ZCu2+OH sites with a characteristic spectroscopic identity.91
On the other hand, dry CO oxidation in the presence of NH3 allowed Villamaina et al. to propose the formation of oxygen bridged Cu2+ diamino complexes, with general formula [(NH3)x–Cu2+–O–Cu2+(NH3)x]2+, by reaction of two [Cu2+(OH)(NH3)x]+ mobile species, formed by NH3 solvation of ZCu2+OH sites. This hypothesis was based on the correlation between the amount of CO2 evolved in the dry CO oxidation of the [(NH3)x–Cu2+–O–Cu2+(NH3)x]2+ complexes and the amount of ZCu2+OH sites expected on the basis of the catalysts composition.58 This very elegant work thus suggests the importance of mobile NH3-solvated complexes also in the NH3-SCR reduction half cycle. What is relevant in this perspective is the hypothesis that ZCu2+OH sites plays a different role with respect to Z2Cu2+ sites (which are also solvated by NH3), based on the idea that the corresponding Cu2+(NH3)4 species are mobile within just one CHA cage, while no inter-cage diffusion could be possible due to the electrostatic tethering of the divalent complexes.3 This hypothesis should however be supported by theoretical calculations, to compute the energetic barriers for intercage diffusion of the [Cu2+(OH)(NH3)x]+ and Cu2+(NH3)4 complexes, which could be affected by the flexibility of the complex itself, thus influencing its mobility.
An important aspect which is often neglected when studying the behaviour of Cu-zeolites in the NH3-SCR reaction is the presence (and role)53,92 of protons, which are always present since the Cu/Al ratio of exchanged Cu-CHA is always below 1. In the presence of NH3, protons are transferred from Brønsted sites to the adsorbed NH3 molecules, with formation of NH4+,92,93 which are thus always present under NH3-SCR conditions. In situ electrical impedance spectroscopy (IS) has been used to study proton transfer under NH3-SCR reaction conditions, showing that adsorbed NH3 molecules serve as vehicle-like carriers for the long-range ion conduction across the zeolite lattice, leading to increased proton conductivities.24 This observation is at the basis of the use of Cu-CHA as sensor for the direct NH3-SCR conversion, which was evaluated according to the change of ionic conductivity with the variation of gas conditions.94 Moreover, it was reported that the high short-range mobility of NH3-solvated Cu+ species contributing predominantly to the ionic conductivity at 200 °C, hindered the sensing of NH3-SCR.94
Besides NH3, Cu ions in zeolites could be mobilized by the other components of the NH3-SCR reaction, such as NO, O2 and NO2, which are involved in the oxidation half cycle.49 Millan et al. have studied the interaction of Cu2+ and Cu+ ions in Cu-CHA and Cu-SAPO-34 with these molecules and some reaction intermediates using ab initio molecular dynamics (AIMD) simulations, with the aim to find a unique fingerprint for the state of copper under reaction conditions.32 The calculations indicated that with the exception of NH3 most reactants form stable complexes with framework coordinated Cu ions, without mobilizing them. These include nitrites and nitrates intermediates, in agreement with the proposed structures based on XAS, UV-Vis and IR spectroscopies (Fig. 5d, right).95,96
Interestingly, the formation of mobile mixed ligand [Cu2+(NH3)3(NO3)]+ complexes upon low temperature reaction of a NO/NH3 mixture with pre-formed framework coordinated Cu nitrates has been recently showed by some of us, confirming the strong mobilizing effect of NH3 also in the presence of other ligands.97 IR spectroscopy showed the transformation of a nitrate ligand from the chelating bidentate (1610 and 1575 cm−1) to a monodentate structure (1430 and 1325 cm−1) during the reaction, while the modifications in the local geometry and ligand nature around the Cu2+ ions were testified by changes in the d–d and charge-transfer regions, respectively, of the corresponding Diffuse Reflectance (DR) UV spectra (Fig. 6a and b, from blue to red). The presence of Cu-coordinated NH3 ligands in the final state is testified by the IR band at 1624 cm−1. The fingerprint peak at ca. 3 Å in the phase-uncorrected FT-EXAFS spectrum indicated the presence of Cu-nitrates in chelating bidentate configuration (multiple-scattering contribution)49,96 and the second-shell peak at ∼2.3 Å characteristic of framework coordinated ions disappeared with formation of the mixed-ligand solvated complex (Fig. 6c). Interestingly, DFT calculations confirmed the stability of nitrate ligands in the monodentate conformation in [Cu2+(NH3)3(NO3)]+ complexes (Fig. 6d) when solvated by ammonia.97
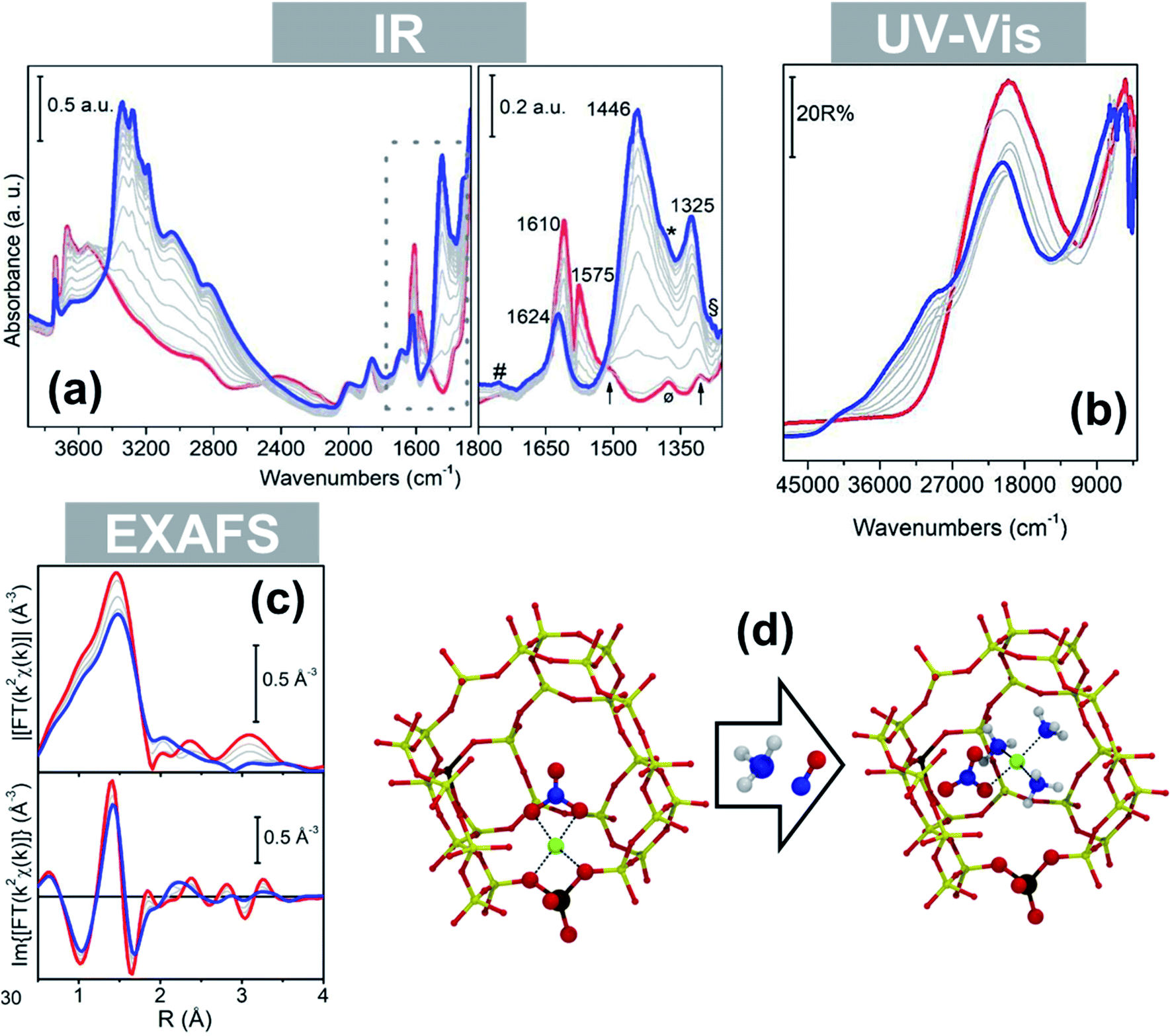 | ||
| Fig. 6 In situ spectroscopy of a Cu-CHA catalyst (Si/Al = 15; Cu/Al = 0.5) during NO/NH3 interaction at 50 °C (grey thin and blue thick lines: intermediates and final state, respectively) after equilibration in NO/O2 (red thick curves): (a) infrared; (b) DR UV-Vis; (c) magnitudes of FT-EXAFS spectra. (d) Structural model of a framework coordinated chelating bidentate Cu-nitrate and DFT optimized structure of the mobile mixed-ligand [Cu2+(NH3)3(NO3)]+ complex in the chabazite cage. Colour code: green, Cu; yellow, Si; brown, Al; red, O; blue, N; white, H. Adapted from ref. 97 with permission from John Wiley and Sons, copyright 2019. | ||
Millan et al. supported their dynamic AIMD calculations with the analysis of the infrared region between 800–1000 cm−1 region (the so-called window region) in the presence of different reactants. This allowed them to identify the modifications of the bands associated with the framework asymmetric T–O–T vibrations perturbed by the presence of coordinated cations as a fingerprint for ion mobilization.32 The same spectroscopic region has been investigated to study the interaction with SO2, an important poisoning agent for Cu-CHA catalysts, with Cu ions under different conditions.33
Besides the mobility induced by ligand molecules, bare Cu ions also show a certain degree of mobility, strongly dependent on their oxidation state as well as on temperature. Andersen et al. studied the combined effect of oxidation state of the Cu ions and their distribution in the CHA framework through a quasi-simultaneous powder XRD and XANES approach.98 They concluded that the reduction of Cu2+ to Cu+ is directly correlated to the migration of said species from an accessible 8-membered ring to the smaller double-6-rings typical of the CHA topology. In a recent study by Deplano et al., the tendency of Cu ions to form ion pairs as a function of their oxidation state was discussed.81 By performing a detailed wavelet transform analysis, a method enabling the discrimination of contributions overlapping in the R space if visualized by conventional FT-EXAFS, of the XAS data obtained on the same Cu-MOR sample, subjected to different redox pretreatments, the preference toward the formation of Cu neighbouring sites (Cu–Cu distance <3.5 Å) in the presence of Cu2+ was proved. Cu+ ions, instead, do not exhibit a clear mutual interaction fingerprint, testifying their tendency to segregate from each other within the zeolitic framework. Though indirectly, the possibility to form Cu pairs upon oxidation confirms a redox-related mobility, with relevant implication in the reactivity of the Cu sites (e.g., favouring the formation of oxidizing CuxOy species). The case studies presented herein demonstrated the redox-state dependent mobility of Cu ions at the atomic scale.
However, no information on the possible spatial redistribution of ions at larger scales is achievable through such bulk-averaging techniques. A pioneering study by Weckhuysen and coworkers reported a nanotomography study of Cu-CHA and Cu-ZSM-5 used as NH3-SCR catalysts.99 Through a 3D reconstruction of the Cu and Al distribution within fresh samples and spent catalysts, the authors could determine as the deactivation of these catalysts under operational conditions takes place as a consequence of dealumination and aggregation of Cu and Al. This process is more impactful on Cu-ZSM-5, where Cu and Al segregate from the zeolitic framework by forming a new copper aluminate phase (CuAl2O4 spinel). Nonetheless, no information is provided on the effect of redox behaviour of Cu ions on such mobilization. Spatial displacement of Cu ions can be inferred via techniques sampling a very specific portion of the sample, e.g., electron-based methods featuring high surface sensitivity. As an example, in Fig. 7, the comparison of the Total Electron Yield (TEY) Cu L3-edge NEXAFS with K-edge XANES spectra is provided for a Cu-CHA sample (Si/Al = 5, Cu/Al = 0.3) heated under inert flow up to 350 °C.
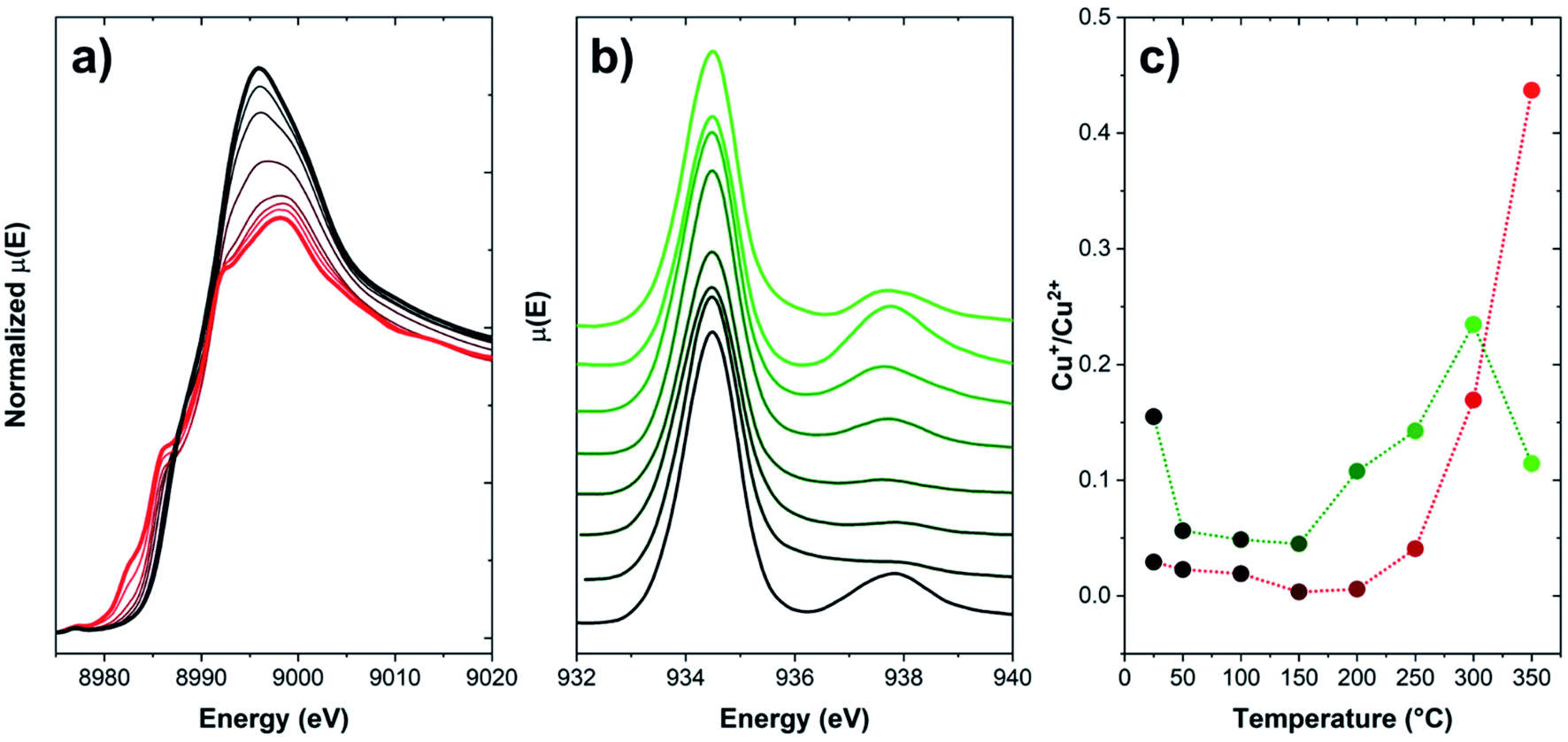 | ||
| Fig. 7 (a) Normalized Cu K-edge XANES of a Cu-CHA sample (Si/Al = 5, Cu/Al = 0.3), collected during heating from RT to 350 °C (temperature evolution from black to red) under a He flow; (b) Cu L3-edge TEY NEXAFS spectra of the same sample, collected during heating from RT to 350 °C (temperature evolution from black to green) under a He flow; and (c) Cu+/Cu2+ fraction from both techniques reported as a function of temperature. Fractions for XANES have been computed on the basis of data from ref. 86. Fractions for NEXAFS have been estimated as the intensity ratio of the features associated to Cu+ (ca. 938 eV) and Cu2+ (ca. 934.5 eV). Unpublished figure reporting data from ref. 86 and unpublished data. | ||
Upon heating in inert environment, auto-reduction phenomena take place as already discussed in Section 2, as clearly highlighted by the development of XANES features in the 8981–8985 eV range, typical of Cu+ sites in non-linear, low-coordination-number environments. At 350 °C, the Cu+/Cu2+ ratio is 0.44. This trend is apparently disconfirmed by the spectral evolution observed in Cu L3-edge NEXAFS spectra: similarly to K-edge XANES data, the amount of Cu+ initially decreases, thus increases again above 150 °C. Conversely, as the temperature increases above 300 °C, the Cu+/Cu2+ ratio drops again, suggesting an unlikely (since occurring in inert atmosphere) re-oxidation of Cu+ to Cu2+. This trend can be clarified by considering the peculiarities of detection methods adopted in each measurement. Cu K-edge XANES, collected in transmission mode as described in ref. 86, are intrinsically bulk-sensitive data, since the entire specimen (here, a self-supporting pellet) is sampled by the X-ray beam. In the case of the L3-edges NEXAFS data, since the low energy of incoming photons makes transmission measurement unfeasible under ambient conditions, a specific measurement cell allowing the collection of the NEXAFS signal in TEY has been specifically developed by the staff of the APE-HE beamline (Elettra Sincrotrone Trieste), where the measurements here reported have been performed.100 Since the signal is generated by photoelectrons, the technique is intrinsically surface sensitive (typical sampling depth of 5 nm), e.g., as occurring for XPS and related photoelectron spectroscopies. Accordingly, the decrease of the Cu+ concentration at high temperature, not justifiable on a barely chemical basis, can be effectively explained by a migration of the reduced Cu+ ions toward the core of the zeolite particles, whereas their surface remains populated by scarcely reducible Cu2+ sites. This observation, also confirmed by previous XPS studies,101 further highlights the superior mobility of Cu+ as compared to Cu2+, pointing out as long-range migration phenomena of Cu ions are possible at sufficiently high temperatures, even in the absence of solvating ligands, when they are found in the reduced state.
5. Exploiting Cu mobility in catalysis
Cu ions mobility within zeolite frameworks is well known from the fundamental point of view, as demonstrated by the different examples discussed in the previous chapter. Nevertheless, the role of Cu mobility toward the catalytic activity of Cu zeolites is not yet fully understood, in particular in the case of innovative processes as DMTM. In the field of NH3-SCR, the recent advances in characterization and simulation techniques allowed for the recognition of the active role of mobile Cu species within the catalytic cycle. In this framework, the presence of specific ligands (i.e., NH3) plays a key role in both mobilizing Cu ions (in the form of amino complexes) and promoting their redox reactivity (i.e., reduction under opportune conditions). Recent developments in the understanding of species involved in NH3-SCR demonstrated as mobilized Cu-amino complexes are capable of interacting with other molecules/ligands, including the formation of Cu-oxo bridged species acting as oxidizing intermediates during NOx abatement.Interestingly, the mobility of NH3-solvated Cu ions is described as short-range, as compared to the mobility of (water or ammonia solvated) protons, and is affected by the oxidation state of the cation. The same effect of the oxidation state on the long-range mobility of Cu2+/Cu+ ions under high temperature conditions98 highlights the influence on the phenomenon of the electrostatic potential in the zeolite internal surface, which is strongly affected by Al distribution. An important aspect that still deserves some consideration is the timescale of the cations intercage diffusion, which could be compared with the turnover frequency (TOF) of the related catalytic reactions. The timescale of the DMTM reaction (particularly in the cyclic variant) is extremely long.64 TOF values for this process are hardly reported; their calculations is not straightforward since one should normalize the turnover number to the cycle length, which include several discrete steps (oxidation, CH4 exposure, methanol extraction through water steam). Sushkevich et al. calculated kinetic constants for the consumption of namely “fast” (dimeric) and “slow” Cu sites during methane exposure at 220 °C in Cu-MOR, resulting in values of 1.4 × 10−3 s−1 and 3.1 × 10−4 s−1, respectively.76 As an example of TOF values in the NH3-SCR reaction we mention the recent work by Martini et al., reporting values between 1.0 × 10−3 and 1.5 × 10−2 mol molCu−1 s−1 for Cu-CHA catalysts with different chemical composition at 200 °C.102 While data about the diffusion timescale of protons and molecules such as water and ammonia are available in the literature,19,21–23,103 similar information related to Cu ions mobility are less straightforward. Rizzotto et al. measured by in situ electrical IS the relaxation times for local ion motion in zeolite. According to their studies, long-range ion transport is characterized by frequencies in the range 1–100 s−1, with values around 100 s−1 for short-range (local) ion transport.104 The relaxation time of ion motion in H and Cu-CHA zeolites in presence of NH3 at 200 °C were found around 7 × 10−5 s and 5 × 10−6 s, respectively, corresponding to frequencies of ca. 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and 200000 s−1. This could be an indirect measurement of the mobility of NH3 solvated Cu ions. On the other hand, the experimental self-diffusion coefficient (Ds) of NH3 at 100 °C was shown to decrease slightly passing from H- to Cu-CHA zeolites (4.90 × 10−9vs. 1.35 × 10−9 m2 s−1), with a major effect to NH3 diffusion related to confinement (2.36 × 10−6 m2 s−1 for unconfined NH3).
000 and 200000 s−1. This could be an indirect measurement of the mobility of NH3 solvated Cu ions. On the other hand, the experimental self-diffusion coefficient (Ds) of NH3 at 100 °C was shown to decrease slightly passing from H- to Cu-CHA zeolites (4.90 × 10−9vs. 1.35 × 10−9 m2 s−1), with a major effect to NH3 diffusion related to confinement (2.36 × 10−6 m2 s−1 for unconfined NH3).
Interestingly, though much less investigated, the presence of Cu-amino complexes does not seem to promote significantly the reactivity in DMTM. Dihn et al. showed as an additional cofeed of NH3 to the reaction mixture used in continuous DMTM (18% CH4/0.1% O2/3% H2O in He) at 270 °C does not enhances reactivity, rather it suppresses the conversion.105 Methanol production is not restored even in case NH3 is fully desorbed from Cu sites and those are fully reoxidized to Cu2+. Eventually, reactivity is recovered only after raising the temperature up to 400 °C in inert atmosphere: the high reactivation temperature was related to the requirement of restoring also free Brønsted acid sites, otherwise blocked by NH3 through the formation of NH4+ cations. The importance of H+ was also investigated computationally by Palagin et al., showing as the proton-transfer occurring from a Brønsted acid site to a bridged Cu2+ hydride could facilitate the release of molecular hydrogen in continuous anaerobic oxidation of methane over Cu-MOR.106
These results point out the synergistic cooperation of Cu ions and acid H+ sites as the main requirement for an effective conversion of methane to methanol in a continuous DMTM process, similarly to the most recent understanding about NH3-SCR mechanism. In our perspective, achieving a more detailed understanding of the effect of Cu mobilization in DMTM is advisable to reach a better control over this challenging reaction. Unavoidably, an explicit consideration of the role of water, often referred to as a simple extraction medium, has to be taken in account in relation to its role as solvating ligand for cations and proton carrier, affecting the ionic conductivity of zeolites in a broad temperature range.
Data availability
Data for this paper, including the spectra reported in Fig. 7 are available at Zenodo at https://doi.org/10.5281/zenodo.7015254.Author contributions
Matteo Signorile: conceptualization, investigation, data curation, writing – original draft, writing – review & editing. Elisa Borfecchia: investigation, data curation. Silvia Bordiga: conceptualization, resources. Gloria Berlier: conceptualization, data curation, writing – original draft, writing-review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has received funding from the European Union's Horizon 2020 Excellence Science ERC-Synergy program 2019-CUBE: “Unravelling the secrets of Cu-based catalysts for C–H activation” (grant agreement No. 856446), from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 955839 (CHASS) and from the Italian ministry of University and Research PRIN2017-MOSCATo: “Cutting-edge X-ray methods and models for the understanding of surface site reactivity in heterogeneous catalysts and sensors” (project ID 2017KKP5ZR). This work reflects only the authors' view, and the European Agency is not responsible for any use that can be made of the information it contains. The authors gratefully acknowledge Gabriele Deplano, Andrea Martini, Chiara Negri and Mario Leopoldo Rivera Salazar for their help in data collection, analysis and conceptualization. We are thankful to Michele Cutini for DFT calculations, conceptualization and structures in Fig. 6.References
- A. Manthiram, X. Yu and S. Wang, Nat. Rev. Mater., 2017, 2, 16103 CrossRef CAS.
- E. D. Wachsman and K. T. Lee, Science, 2011, 334, 935–939 CrossRef CAS PubMed.
- C. Paolucci, I. Khurana, A. A. Parekh, S. Li, A. J. Shih, H. Li, J. R. Di Iorio, J. D. Albarracin-Caballero, A. Yezerets, J. T. Miller, W. N. Delgass, F. H. Ribeiro, W. F. Schneider and R. Gounder, Science, 2017, 357, 898–903 CrossRef CAS PubMed.
- E. G. Derouane, J. Mol. Catal. A: Chem., 1998, 134, 29–45 CrossRef CAS.
- G. Sastre and A. Corma, J. Mol. Catal. A: Chem., 2009, 305, 3–7 CrossRef CAS.
- Y. Chai, W. Dai, G. Wu, N. Guan and L. Li, Acc. Chem. Res., 2021, 54, 2894–2904 CrossRef CAS PubMed.
- I. Derycke, J. P. Vigneron, P. Lambin, A. A. Lucas and E. G. Derouane, J. Chem. Phys., 1991, 94, 4620–4627 CrossRef CAS.
- C. Lamberti, G. Turnes Palomino, S. Bordiga, G. Berlier, F. D'Acapito and A. Zecchina, Angew. Chem., Int. Ed., 2000, 39, 2138–2141 CrossRef CAS.
- G. Qi, J. Xu, J. Su, J. Chen, X. Wang and F. Deng, J. Am. Chem. Soc., 2013, 135, 6762–6765 CrossRef CAS PubMed.
- M. Boronat and A. Corma, Catal. Lett., 2015, 145, 162–172 CrossRef CAS.
- R. A. van Santen, in Studies in Surface Science and Catalysis, Elsevier Science Publ B V, Amsterdam, Netherlands, 1994, vol. 85, pp. 273–294 Search PubMed.
- M. Trachta, R. Bulánek, O. Bludský and M. Rubeš, Sci. Rep., 2022, 12, 7301 CrossRef CAS PubMed.
- S. Bordiga, C. Lamberti, F. Bonino, A. Travert and F. Thibault-Starzyk, Chem. Soc. Rev., 2015, 44, 7262–7341 RSC.
- E. T. C. Vogt and B. M. Weckhuysen, Chem. Soc. Rev., 2015, 44, 7342–7370 RSC.
- A. Corma, Chem. Rev., 1995, 95, 559–614 CrossRef CAS.
- G. Busca, Chem. Rev., 2007, 107, 5366–5410 CrossRef CAS PubMed.
- S. Bordiga, L. Regli, C. Lamberti, A. Zecchina, M. Bjørgen and K. F. Lillerud, J. Phys. Chem. B, 2005, 109, 7724–7732 CrossRef CAS PubMed.
- G. A. V. Martins, G. Berlier, S. Coluccia, H. O. Pastore, G. B. Superti, G. Gatti and L. Marchese, J. Phys. Chem. C, 2007, 111, 330–339 CrossRef CAS.
- M. E. Franke, M. Sierka, U. Simon and J. Sauer, Phys. Chem. Chem. Phys., 2002, 4, 5207–5216 RSC.
- H. Jobic, A. Tuel, M. Krossner and J. Sauer, J. Phys. Chem., 1996, 100, 19545–19550 CrossRef CAS.
- M. Wang, N. R. Jaegers, M. S. Lee, C. Wan, J. Z. Hu, H. Shi, D. Mei, S. D. Burton, D. M. Camaioni, O. Y. Gutiérrez, V. A. Glezakou, R. Rousseau, Y. Wang and J. A. Lercher, J. Am. Chem. Soc., 2019, 141, 3444–3455 CrossRef CAS PubMed.
- N. K. Moroz, Y. V. Seryotkin, I. S. Afanassyev and V. V. Bakakin, J. Struct. Chem., 2002, 43, 595–600 CrossRef CAS.
- J. Kanellopoulos, C. Gottert, D. Schneider, B. Knorr, D. Prager, H. Ernst and D. Freude, J. Catal., 2008, 255, 68–78 CrossRef CAS.
- P. Chen and U. Simon, Catalysts, 2016, 6, 204 CrossRef.
- E. Borfecchia, P. Beato, S. Svelle, U. Olsbye, C. Lamberti and S. Bordiga, Chem. Soc. Rev., 2018, 47, 8097–8133 RSC.
- E. Borfecchia, K. A. Lomachenko, F. Giordanino, H. Falsig, P. Beato, A. V. Soldatov, S. Bordiga and C. Lamberti, Chem. Sci., 2015, 6, 548–563 RSC.
- A. Zecchina and C. Otero Areán, Chem. Soc. Rev., 1996, 25, 187–197 RSC.
- C. Lamberti, A. Zecchina, E. Groppo and S. Bordiga, Chem. Soc. Rev., 2010, 39, 4951–5001 RSC.
- Z. Sobalík, Z. Tvarůžková and B. Wichterlová, J. Phys. Chem. B, 1998, 102, 1077–1085 CrossRef.
- M. Lemishka, J. Dedecek, K. Mlekodaj, Z. Sobalik, S. Sklenak and E. Tabor, Pure Appl. Chem., 2019, 91, 1721–1732 CrossRef CAS.
- E. Broclawik, J. Datka, B. Gil and P. Kozyra, Phys. Chem. Chem. Phys., 2000, 2, 401–405 RSC.
- R. Millan, P. Cnudde, A. E. J. Hoffman, C. W. Lopes, P. Concepción, V. Van Speybroeck and M. Boronat, J. Phys. Chem. Lett., 2020, 11, 10060–10066 CrossRef CAS PubMed.
- Y. Jangjou, Q. Do, Y. Gu, L. G. Lim, H. Sun, D. Wang, A. Kumar, J. Li, L. C. Grabow and W. S. Epling, ACS Catal., 2018, 8, 1325–1337 CrossRef CAS.
- P. Norby, F. I. Poshni, A. F. Gualtieri, J. C. Hanson and C. P. Grey, J. Phys. Chem. B, 1998, 102, 839–856 CrossRef CAS.
- Y. Itho, S. Nishiyama, S. Tsuruya and M. Masai, J. Phys. Chem., 1994, 98, 960–967 CrossRef CAS.
- A. Zecchina, S. Bordiga, G. T. Palomino, D. Scarano, C. Lamberti and M. Salvalaggio, J. Phys. Chem. B, 1999, 103, 3833–3844 CrossRef CAS.
- G. Deplano, M. Signorile, V. Crocellà, N. G. Porcaro, C. Atzori, B. G. Solemsli, S. Svelle and S. Bordiga, ACS Appl. Mater. Interfaces, 2022, 14, 21059–21068 CrossRef CAS PubMed.
- C. C. Chao and J. H. Lunsford, J. Chem. Phys., 1972, 57, 2890–2898 CrossRef CAS.
- G. T. Palomino, A. Zecchina, E. Giamello, P. Fisicaro, G. Berlier, C. Lamberti and S. Bordiga, Stud. Surf. Sci. Catal., 2000, 130, 2915–2920 CrossRef.
- V. L. Sushkevich and J. A. Van Bokhoven, Chem. Commun., 2018, 54, 7447–7450 RSC.
- M. Iwamoto, H. Furukawa, Y. Mine, F. Uemura, S. I. Mikuriya and S. Kagawa, J. Chem. Soc., Chem. Commun., 1986, 1272–1273 RSC.
- S. I. Zones, US Pat., 4544538A, 1985 Search PubMed.
- J. H. Kwak, R. G. Tonkyn, D. H. Kim, J. Szanyi and C. H. F. Peden, J. Catal., 2010, 275, 187–190 CrossRef CAS.
- F. Gao and J. Szanyi, Appl. Catal., A, 2018, 560, 185–194 CrossRef CAS.
- F. Gramigni, T. Selleri, I. Nova and E. Tronconi, React. Chem. Eng., 2019, 4, 1165–1178 RSC.
- M. Bendrich, A. Scheuer, R. E. Hayes and M. Votsmeier, Appl. Catal., B, 2018, 222, 76–87 CrossRef CAS.
- Y. Feng, X. Wang, T. V. W. Janssens, P. N. R. Vennestrøm, J. Jansson, M. Skoglundh and H. Grönbeck, ACS Catal., 2021, 11, 14395–14407 CrossRef CAS.
- A. M. Beale, F. Gao, I. Lezcano-Gonzalez, C. H. F. Peden and J. Szanyi, Chem. Soc. Rev., 2015, 44, 7371–7405 RSC.
- T. V. W. Janssens, H. Falsig, L. F. Lundegaard, P. N. R. Vennestrøm, S. B. Rasmussen, P. G. Moses, F. Giordanino, E. Borfecchia, K. A. Lomachenko, C. Lamberti, S. Bordiga, A. Godiksen, S. Mossin and P. Beato, ACS Catal., 2015, 5, 2832–2845 CrossRef CAS.
- I. Lezcano-Gonzalez, D. S. Wragg, W. A. Slawinski, K. Hemelsoet, A. Van Yperen-De Deyne, M. Waroquier, V. Van Speybroeck and A. M. Beale, J. Phys. Chem. C, 2015, 119, 24394–24403 CrossRef.
- K. A. Lomachenko, E. Borfecchia, C. Negri, G. Berlier, C. Lamberti, P. Beato, H. Falsig and S. Bordiga, J. Am. Chem. Soc., 2016, 138, 12025–12028 CrossRef CAS PubMed.
- F. Gao, D. Mei, Y. Wang, J. Szanyi and C. H. F. Peden, J. Am. Chem. Soc., 2017, 139, 4935–4942 CrossRef CAS PubMed.
- L. Chen, T. V. W. Janssens, P. N. R. Vennestrøm, J. Jansson, M. Skoglundh and H. Grönbeck, ACS Catal., 2020, 10, 5646–5656 CrossRef CAS.
- L. Chen, H. Falsig, T. V. W. Janssens and H. Grönbeck, J. Catal., 2018, 358, 179–186 CrossRef CAS.
- T. Selleri, M. P. Ruggeri, I. Nova and E. Tronconi, Top. Catal., 2016, 59, 678–685 CrossRef CAS.
- C. Negri, T. Selleri, E. Borfecchia, A. Martini, K. A. Lomachenko, T. V. W. Janssens, M. Cutini, S. Bordiga and G. Berlier, J. Am. Chem. Soc., 2020, 142, 15884–15896 CrossRef CAS PubMed.
- A. Oda, H. Shionoya, Y. Hotta, T. Takewaki, K. Sawabe and A. Satsuma, ACS Catal., 2020, 10, 12333–12339 CrossRef CAS.
- R. Villamaina, U. Iacobone, I. Nova, M. P. Ruggeri, J. Collier, D. Thompsett and E. Tronconi, ChemCatChem, 2020, 12, 3843–3848 CrossRef CAS.
- W. Hu, F. Gramigni, N. D. Nasello, N. Usberti, U. Iacobone, S. Liu, I. Nova, X. Gao and E. Tronconi, ACS Catal., 2022, 12, 5263–5274 CrossRef CAS.
- M. H. Groothaert, P. J. Smeets, B. F. Sels, P. A. Jacobs and R. A. Schoonheydt, J. Am. Chem. Soc., 2005, 127, 1394–1395 CrossRef CAS PubMed.
- V. L. Sushkevich, D. Palagin, M. Ranocchiari and J. A. Van Bokhoven, Science, 2017, 356, 523–527 CrossRef CAS PubMed.
- M. A. Newton, A. J. Knorpp, V. L. Sushkevich, D. Palagin and J. A. Van Bokhoven, Chem. Soc. Rev., 2020, 49, 1449–1486 RSC.
- E. Mae Alayon, M. Nachtegaal, M. Ranocchiari and J. A. Van Bokhoven, Chem. Commun., 2012, 48, 404–406 RSC.
- D. K. Pappas, A. Martini, M. Dyballa, K. Kvande, S. Teketel, K. A. Lomachenko, R. Baran, P. Glatzel, B. Arstad, G. Berlier, C. Lamberti, S. Bordiga, U. Olsbye, S. Svelle, P. Beato and E. Borfecchia, J. Am. Chem. Soc., 2018, 140, 15270–15278 CrossRef CAS PubMed.
- S. Grundner, M. A. C. Markovits, G. Li, M. Tromp, E. A. Pidko, E. J. M. Hensen, A. Jentys, M. Sanchez-Sanchez and J. A. Lercher, Nat. Commun., 2015, 6, 7546 CrossRef PubMed.
- D. K. Pappas, E. Borfecchia, M. Dyballa, I. A. Pankin, K. A. Lomachenko, A. Martini, M. Signorile, S. Teketel, B. Arstad, G. Berlier, C. Lamberti, S. Bordiga, U. Olsbye, K. P. Lillerud, S. Svelle and P. Beato, J. Am. Chem. Soc., 2017, 139, 14961–14975 CrossRef CAS PubMed.
- M. J. Wulfers, S. Teketel, B. Ipek and R. F. Lobo, Chem. Commun., 2015, 51, 4447–4450 RSC.
- A. J. Knorpp, A. B. Pinar, M. A. Newton, V. L. Sushkevich and J. A. van Bokhoven, ChemCatChem, 2018, 10, 5593–5596 CrossRef CAS.
- P. Vanelderen, B. E. R. Snyder, M. L. Tsai, R. G. Hadt, J. Vancauwenbergh, O. Coussens, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, J. Am. Chem. Soc., 2015, 137, 6383–6392 CrossRef CAS PubMed.
- P. J. Smeets, R. G. Hadt, J. S. Woertink, P. Vanelderen, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, J. Am. Chem. Soc., 2010, 132, 14736–14738 CrossRef CAS PubMed.
- A. Martini, M. Signorile, C. Negri, K. Kvande, K. A. Lomachenko, S. Svelle, P. Beato, G. Berlier, E. Borfecchia and S. Bordiga, Phys. Chem. Chem. Phys., 2020, 22, 18950–18963 RSC.
- D. Palagin, A. J. Knorpp, A. B. Pinar, M. Ranocchiari and J. A. Van Bokhoven, Nanoscale, 2017, 9, 1144–1153 RSC.
- M. L. Tsai, R. G. Hadt, P. Vanelderen, B. F. Sels, R. A. Schoonheydt and E. I. Solomon, J. Am. Chem. Soc., 2014, 136, 3522–3529 CrossRef CAS PubMed.
- G. Berlier, V. Crocellà, M. Signorile, E. Borfecchia, F. Bonino and S. Bordiga, in Structure and Reactivity of Metals in Zeolite Materials, ed. J. P. Pariente and M. Sanchez-Sanchez, 2018, vol. 178, pp. 91–154 Search PubMed.
- C. Negri, M. Signorile, N. G. Porcaro, E. Borfecchia, G. Berlier, T. V. W. Janssens and S. Bordiga, Appl. Catal., A, 2019, 578, 1–9 CrossRef CAS.
- V. L. Sushkevich, M. Artsiusheuski, D. Klose, G. Jeschke and J. A. van Bokhoven, Angew. Chem., Int. Ed., 2021, 60, 15944–15953 CrossRef CAS PubMed.
- K. A. Lomachenko, A. Martini, D. K. Pappas, C. Negri, M. Dyballa, G. Berlier, S. Bordiga, C. Lamberti, U. Olsbye, S. Svelle, P. Beato and E. Borfecchia, Catal. Today, 2019, 336, 99–108 CrossRef CAS.
- E. Borfecchia, D. K. Pappas, M. Dyballa, K. A. Lomachenko, C. Negri, M. Signorile and G. Berlier, Catal. Today, 2019, 333, 17–27 CrossRef CAS.
- E. M. C. Alayon, M. Nachtegaal, A. Bodi, M. Ranocchiari and J. A. Van Bokhoven, Phys. Chem. Chem. Phys., 2015, 17, 7681–7693 RSC.
- S. Prodinger, K. Kvande, B. Arstad, E. Borfecchia, P. Beato and S. Svelle, ACS Catal., 2022, 12, 2166–2177 CrossRef CAS.
- G. Deplano, A. Martini, M. Signorile, E. Borfecchia, V. Crocellà, S. Svelle and S. Bordiga, Angew. Chem., Int. Ed., 2021, 60, 25891–25896 CrossRef CAS PubMed.
- A. Bhan, A. D. Allian, G. J. Sunley, D. J. Law and E. Iglesia, J. Am. Chem. Soc., 2007, 129, 4919–4924 CrossRef CAS PubMed.
- C. W. Kim, N. H. Heo and K. Seff, J. Phys. Chem. C, 2011, 115, 24823–24838 CrossRef CAS.
- E. Morra, M. Signorile, E. Salvadori, S. Bordiga, E. Giamello and M. Chiesa, Angew. Chem., Int. Ed., 2019, 58, 12398–12403 CrossRef CAS PubMed.
- C. Paolucci, A. A. Parekh, I. Khurana, J. R. Di Iorio, H. Li, J. D. Albarracin Caballero, A. J. Shih, T. Anggara, W. N. Delgass, J. T. Miller, F. H. Ribeiro, R. Gounder and W. F. Schneider, J. Am. Chem. Soc., 2016, 138, 6028–6048 CrossRef CAS PubMed.
- A. Martini, E. Borfecchia, K. A. Lomachenko, I. A. Pankin, C. Negri, G. Berlier, P. Beato, H. Falsig, S. Bordiga and C. Lamberti, Chem. Sci., 2017, 8, 6836–6851 RSC.
- E. Borfecchia, C. Negri, K. A. Lomachenko, C. Lamberti, T. V. W. Janssens and G. Berlier, React. Chem. Eng., 2019, 4, 1067–1080 RSC.
- S. Shwan, M. Skoglundh, L. F. Lundegaard, R. R. Tiruvalam, T. V. W. Janssens, A. Carlsson and P. N. R. Vennestrøm, ACS Catal., 2015, 5, 16–19 CrossRef CAS.
- A. K. S. Clemens, A. Shishkin, P. A. Carlsson, M. Skoglundh, F. J. Martínez-Casado, Z. Matej, O. Balmes and H. Härelind, ACS Catal., 2015, 5, 6209–6218 CrossRef CAS.
- L. Chen, J. Jansson, M. Skoglundh and H. Grönbeck, J. Phys. Chem. C, 2016, 120, 29182–29189 CrossRef CAS.
- H. Li, C. Paolucci, I. Khurana, L. N. Wilcox, F. Göltl, J. D. Albarracin-Caballero, A. J. Shih, F. H. Ribeiro, R. Gounder and W. F. Schneider, Chem. Sci., 2019, 10, 2373–2384 RSC.
- I. Lezcano-Gonzalez, U. Deka, B. Arstad, A. Van Yperen-De Deyne, K. Hemelsoet, M. Waroquier, V. Van Speybroeck, B. M. Weckhuysen and A. M. Beale, Phys. Chem. Chem. Phys., 2014, 16, 1639–1650 RSC.
- F. Giordanino, E. Borfecchia, K. A. Lomachenko, A. Lazzarini, G. Agostini, E. Gallo, A. V. Soldatov, P. Beato, S. Bordiga and C. Lamberti, J. Phys. Chem. Lett., 2014, 5, 1552–1559 CrossRef CAS PubMed.
- P. Chen, V. Rizzotto, A. Khetan, K. Xie, R. Moos, H. Pitsch, D. Ye and U. Simon, ACS Appl. Mater. Interfaces, 2019, 11, 8097–8105 CrossRef CAS PubMed.
- C. Negri, P. S. Hammershøi, T. V. W. Janssens, P. Beato, G. Berlier and S. Bordiga, Chem.–Eur. J., 2018, 24, 12044–12053 CrossRef CAS PubMed.
- C. Tyrsted, E. Borfecchia, G. Berlier, K. A. Lomachenko, C. Lamberti, S. Bordiga, P. N. R. Vennestrøm, T. V. W. Janssens, H. Falsig, P. Beato and A. Puig-Molina, Catal. Sci. Technol., 2016, 6, 8314–8324 RSC.
- C. Negri, E. Borfecchia, M. Cutini, K. A. Lomachenko, T. V. W. Janssens, G. Berlier and S. Bordiga, ChemCatChem, 2019, 11, 3828–3838 CrossRef CAS.
- C. W. Andersen, E. Borfecchia, M. Bremholm, M. R. V. Jørgensen, P. N. R. Vennestrøm, C. Lamberti, L. F. Lundegaard and B. B. Iversen, Angew. Chem., Int. Ed., 2017, 56, 10367–10372 CrossRef CAS PubMed.
- J. E. Schmidt, R. Oord, W. Guo, J. D. Poplawsky and B. M. Weckhuysen, Nat. Commun., 2017, 8, 1666 CrossRef PubMed.
- C. Castán-Guerrero, D. Krizmancic, V. Bonanni, R. Edla, A. Deluisa, F. Salvador, G. Rossi, G. Panaccione and P. Torelli, Rev. Sci. Instrum., 2018, 89, 054101 CrossRef PubMed.
- E. S. Shpiro, W. Grünert, R. W. Joyner and G. N. Baeva, Catal. Lett., 1994, 24, 159–169 CrossRef CAS.
- A. Martini, C. Negri, L. Bugarin, G. Deplano, R. K. Abasabadi, K. A. Lomachenko, T. V. W. Janssens, S. Bordiga, G. Berlier and E. Borfecchia, J. Phys. Chem. Lett., 2022, 13, 6164–6170 CrossRef CAS PubMed.
- A. J. O'Malley, I. Hitchcock, M. Sarwar, I. P. Silverwood, S. Hindocha, C. R. A. Catlow, A. P. E. York and P. J. Collier, Phys. Chem. Chem. Phys., 2016, 18, 17159–17168 RSC.
- V. Rizzotto, P. Chen and U. Simon, Catalysts, 2018, 8, 162–184 CrossRef.
- K. T. Dinh, M. M. Sullivan, K. Narsimhan, P. Serna, R. J. Meyer, M. Dincă and Y. Román-Leshkov, J. Am. Chem. Soc., 2019, 141, 11641–11650 CrossRef CAS PubMed.
- D. Palagin, V. L. Sushkevich and J. A. Van Bokhoven, ACS Catal., 2019, 9, 10365–10374 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |