
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective covalent targeting of SARS-CoV-2 main protease by enantiopure chlorofluoroacetamide†
Daiki
Yamane
a,
Satsuki
Onitsuka
a,
Suyong
Re
b,
Hikaru
Isogai
a,
Rui
Hamada
a,
Tadanari
Hiramoto
a,
Eiji
Kawanishi
 a,
Kenji
Mizuguchi
bc,
Naoya
Shindo
*a and
Akio
Ojida
*a
a,
Kenji
Mizuguchi
bc,
Naoya
Shindo
*a and
Akio
Ojida
*a
aGraduate School of Pharmaceutical Sciences, Kyushu University, 3-1-1 Maidashi, Higashi-ku, Fukuoka 812-8582, Japan. E-mail: shindo708@phar.kyushu-u.ac.jp; ojida@phar.kyushu-u.ac.jp
bArtificial Intelligence Center for Health and Biomedical Research, National Institute of Biomedical Innovation, Health and Nutrition, 7-6-8 Saito-Asagi, Ibaraki, Osaka 567-0085, Japan
cInstitute for Protein Research, Osaka University, 3-2 Yamadaoka, Suita, Osaka 565-0871, Japan
First published on 15th February 2022
Abstract
The coronavirus disease 2019 (COVID-19) pandemic has necessitated the development of antiviral agents against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). The main protease (Mpro) is a promising target for COVID-19 treatment. Here, we report an irreversible SARS-CoV-2 Mpro inhibitor possessing chlorofluoroacetamide (CFA) as a warhead for the covalent modification of Mpro. Ugi multicomponent reaction using chlorofluoroacetic acid enabled the rapid synthesis of dipeptidic CFA derivatives that identified 18 as a potent inhibitor of SARS-CoV-2 Mpro. Among the four stereoisomers, (R,R)-18 exhibited a markedly higher inhibitory activity against Mpro than the other isomers. Reaction kinetics and computational docking studies suggest that the R configuration of the CFA warhead is crucial for the rapid covalent inhibition of Mpro. Our findings highlight the prominent influence of the CFA chirality on the covalent modification of proteinous cysteines and provide the basis for improving the potency and selectivity of CFA-based covalent inhibitors.
Introduction
The outbreak of the novel coronavirus disease 2019 (COVID-19), caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2),1,2 poses a severe threat to global public health and economy. Although efficacious vaccines are now being administered worldwide, novel SARS-CoV-2 variants of interest and concern continue to emerge.3 Thus, the development of antiviral agents against SARS-CoV-2 is urgently needed to tackle and end the current pandemic.4,5 SARS-CoV-2 has a single-stranded genomic RNA that encodes polyproteins pp1a and pp1ab. Two cysteine proteases, the main protease (Mpro, also known as 3C-like protease, 3CLpro) and the papain-like protease (PLpro), are excised from these polyproteins and further digest the polyproteins into nonstructural proteins, including crucial components of the viral replication–translation machinery. Although both proteases are essential for viral replication, the predominant role of Mpro in polyprotein processing and the lack of a human homolog has led to extensive studies to establish it as an attractive drug target against SARS-CoV-2.6,7Mpro recognizes glutamine and a hydrophobic amino acid residue at the S1 and S2 pockets in the active site, respectively, and cleaves amide bonds primarily within the Leu-Gln↓(Ser, Ala, Gly) sequence (↓: cleavage site).8 This substrate specificity is conserved across other coronaviruses, including SARS-CoV-1 and middle east respiratory syndrome coronavirus (MERS-CoV).9,10 Therefore, current efforts to develop SARS-CoV-2 Mpro inhibitors are extensively guided by the previous molecular designs, especially those targeting SARS-CoV-1 Mpro.5,6 Most of the reported SARS-CoV-2 Mpro inhibitors are peptidomimetics based on the Leu-Gln sequence, combined with an electrophilic warhead, such as α-ketoamide,11 aldehyde,12–15 ketone,16–19 vinyl sulfone,8 and nitrile,20,21 for the covalent capture of the sulfhydryl group of the catalytic Cys145. Nirmatrelvir (PF-07321332), developed by Pfizer, is one of the most advanced compounds in this category and has been recently approved by regulatory ageincies as a combination with ritonavir (Fig. 1A).21 Other than peptidomimetics, several Ugi multicomponent reaction (MCR)-generated dipeptidic compounds have been reported as inhibitors against SARS-CoV-1 and SARS-CoV-2 Mpro (Fig. 1B).22–24
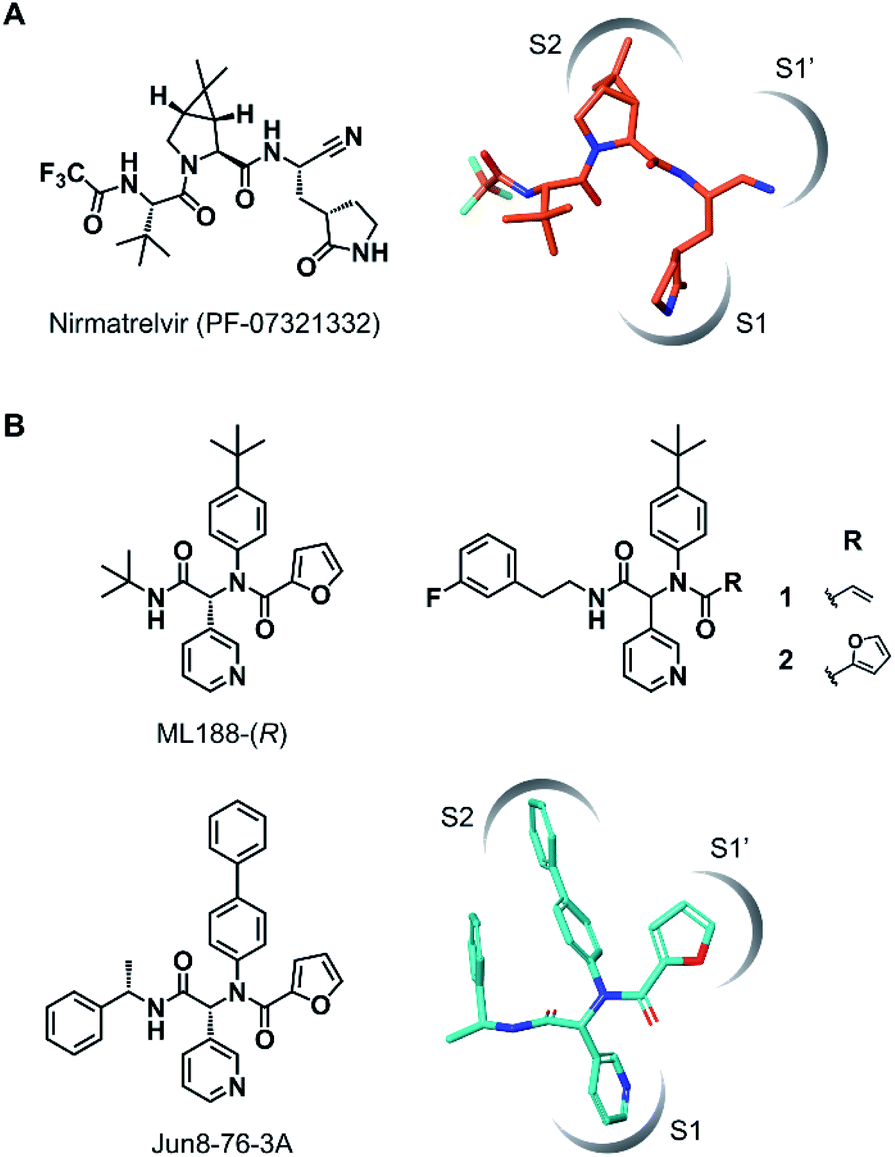 | ||
| Fig. 1 Chemical structures of SARS-CoV-2 Mpro inhibitors. (A) Structure of nirmatrelvir (PF-07321332) and its binding mode in the Mpro active site (the conformation was retrieved from the cocrystal structure, PDB code 7RFW). (B) The structures of dipeptidic Mpro inhibitors generated by Ugi multicomponent reaction and the binding mode of Jun8-76-3A in the Mpro active site (the conformation was retrieved from the cocrystal structure, PDB code 7KX5). | ||
Covalent inhibition of proteins has been a powerful strategy for achieving potent and sustained pharmacological efficacy.25 To date, a number of acrylamide-based covalent inhibitors targeting a noncatalytic cysteine of proteins have been clinically approved or under clinical trials for cancer treatment.26,27 In development of covalent inhibitors, high target selectivity should be ensured to minimize the risk of toxicity due to off-target labeling.28 This point is also crucial for designing covalent inhibitors targeting SARS-CoV-2 Mpro, as medications for infectious diseases generally require a broader safety margin than cancer treatment. We have recently introduced α-chlorofluoroacetamide (CFA) as a weakly reactive, cysteine-directed warhead for covalent inhibition of proteins including tyrosine kinases.29–31 We found that the CFA derivatives exhibited higher selectivity in covalent modification toward the targeted tyrosine kinases than the structurally related acrylamide-based inhibitors. We also demonstrated that the CFA–thiol adduct is hydrolyzed under neutral aqueous conditions to reversibly generate an unmodified cysteine, which could contribute to high target selectivity by eliminating the solvent-exposed off-target labeling. These desirable features led us to envision CFA as a valuable tool for designing selective covalent SARS-CoV-2 Mpro inhibitors.
Results and discussion
Wang et al. recently reported Jun8-76-3A as a reversible inhibitor of SARS-CoV-2 Mpro.23 The molecular architecture of Jun8-76-3A is based on ML188-(R) that was previously developed as an inhibitor of SARS-CoV-1 Mpro.22 The crystal X-ray structure analysis revealed that the 3-pyridyl group of Jun8-76-3A resides in the S1 glutamine binding pocket of SARS-CoV-2 Mpro by forming a hydrogen bond with His163. The 4-biphenyl group occupies the S2 pocket that prefers hydrophobic amino acid residues like leucine and phenylalanine. The furan-2-carboxamide moiety exerts a bifurcated interaction with the backbone NH of Gly143 in the S1′ pocket, wherein the catalytic Cys145 resides. Based on the same Ugi MCR-generated dipeptide scaffold, London et al. reported an acrylamide-based covalent SARS-CoV-2 Mpro inhibitor 1.24 We also employed this dipeptide scaffold as the starting point for the development of a CFA-based covalent inhibitor against Mpro. Ugi MCR using racemic chlorofluoroacetic acid, along with amine, aldehyde, and isocyanide, proceeded smoothly to yield CFA compound 3 as a mixture of diastereomers in a single step. The two diastereomers, 3A (less polar isomer) and 3B (more polar isomer), were separable by silica gel chromatography (Fig. 2A). The inhibitory activity of these compounds was assessed by an enzymatic assay using the recombinant SARS-CoV-2 Mpro and the fluorogenic substrate Ac-Abu-Tle-Leu-Gln-(4-methylcoumaryl-7-amide).8 This substrate allowed robust fluorescent reading compared to the FRET-based substrate possessing an Edans/Dabcyl donor–acceptor pair.32 We found that 3A and 3B displayed different inhibitory activities against Mpro (Fig. 2B); 3B exhibited moderate inhibitory activity (IC50 = 1.05 μM), whereas 3A was virtually inactive (IC50 > 20 μM). In the case of ML188 and Jun8-76-3A, it has been reported that the R configuration of the pyridyl α-carbon is crucial for Mpro inhibition.22,23 Considering that both 3A and 3B contain the R configuration at this stereocenter, our results suggest that the chirality at the CFA stereocenter also substantially impacts their inhibitory activities. Non-reactive analogs of 3 such as acetamide 4 and difluoroacetamide 5 were found to be inactive (IC50 > 20 μM), indicating that the covalent attachment at CFA contributes to the Mpro inhibitory activity of 3B in a stereospecific manner. In the same assay, we also found that acrylamide 1 showed a weaker activity (IC50 = 2.62 μM) than 3B.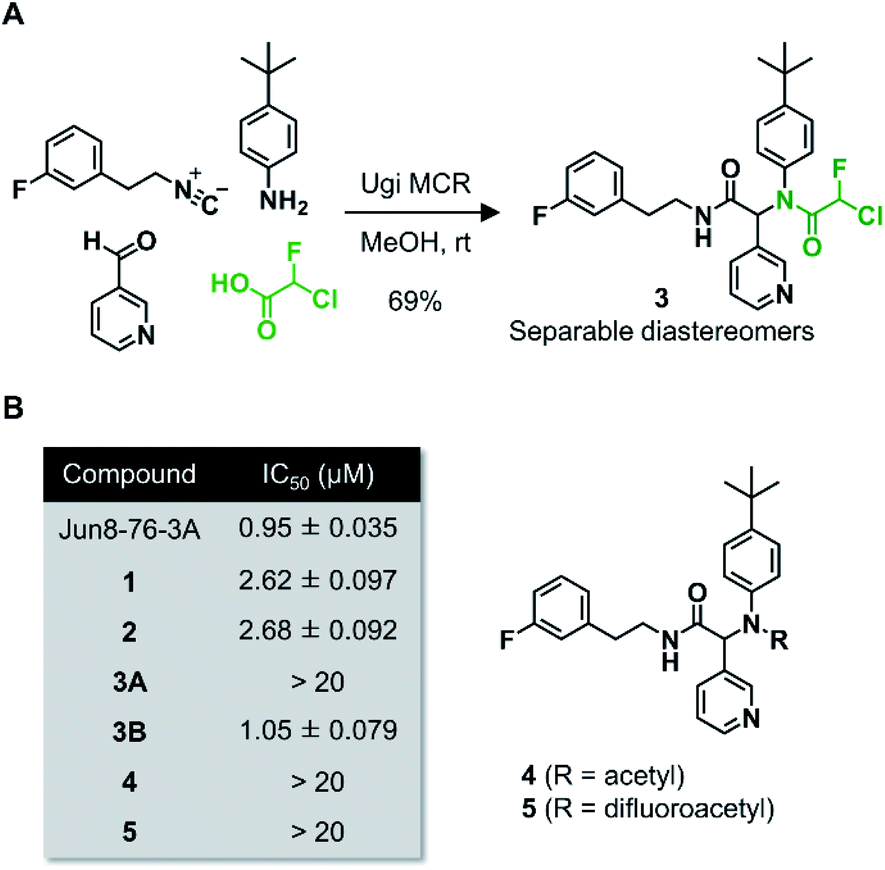 | ||
| Fig. 2 (A) Preparation of the dipeptidic inhibitor 3via Ugi multicomponent reaction (MCR). (B) Enzyme inhibitory activities of the dipeptidic inhibitors against SARS-CoV-2 Mpro. Compounds 3A and 3B are the less and more polar diastereomers, respectively. The reported IC50 values for Jun8-76-3A, 1, and 2 are 0.31, 2.95, and 2.72 μM, respectively. | ||
This observation encouraged us to subsequently conduct an SAR study to improve the potency of CFA dipeptides (Fig. 3A). We first evaluated the effect of the para-substituent of the benzene ring (R1) on inhibitory activity. As this moiety occupies the S2 pocket that primarily accommodates a leucine residue, we screened compounds 6–9 with a hydrophobic para-substituent. In the assay, less polar diastereomer A was inactive (IC50 > 20 μM), whereas more polar diastereomer B showed moderate activity in all the cases. Among them, 9B possessing a pentafluorosulfanyl (SF5) group, a thermally and chemically stable bioisostere for a tert-butyl group,33 exhibited a slightly higher Mpro inhibitory activity (IC50 = 0.90 μM) than 3B. Next, we investigated the effect of the heteroaromatic ring (R2) on the activity. The replacement of the pyridine ring of 3B with a diazine increased the potency of 11–12, whereas the 3-pyridazinyl derivative 10 was almost inactive (IC50 > 20 μM as a diastereomixture). 5-Pyrimidine 12 was the only compound with separable diastereomers, and 12B exhibited the highest activity in the series (IC50 = 0.25 μM). The effect of the substituent R3 was also investigated. The introduction of a tert-butyl group, as observed in the structure of ML188, reduced the potency (14B, IC50 = 2.70 μM). Replacement of the 3-fluorophenyl group of 3 to a 3-pyridyl group slightly increased the activity (16, IC50 = 0.73 μM as a diastereomixture). The introduction of a 7-isoquinoline ring resulted in a slight loss of activity (17B, IC50 = 1.43 μM). Combining the SAR results, we synthesized compound 18 and 19 as structurally optimized CFA inhibitors (Fig. 3B). The diastereomers were separable in both cases and the active isomers 18B and 19B exhibited potent Mpro inhibitory activities (IC50 = 0.21 and 0.14 μM, respectively). Although 19B was marginally more active than 18B, we chose 18 for further investigations because the chromatograhic separation of the diastereomixture of 18 was much easier than that of 19. Fortunately, we were able to determine the relative configuration of 18B by X-ray crystallography (Fig. S1†). The analyzed structure revealed that 18B has a syn-configuration, where the 5-pyrimidinyl group and the CFA fluorine atom are oriented toward the same direction in the drawn structure (Fig. 3C). Consequently, 18A was determined to be an anti-isomer.
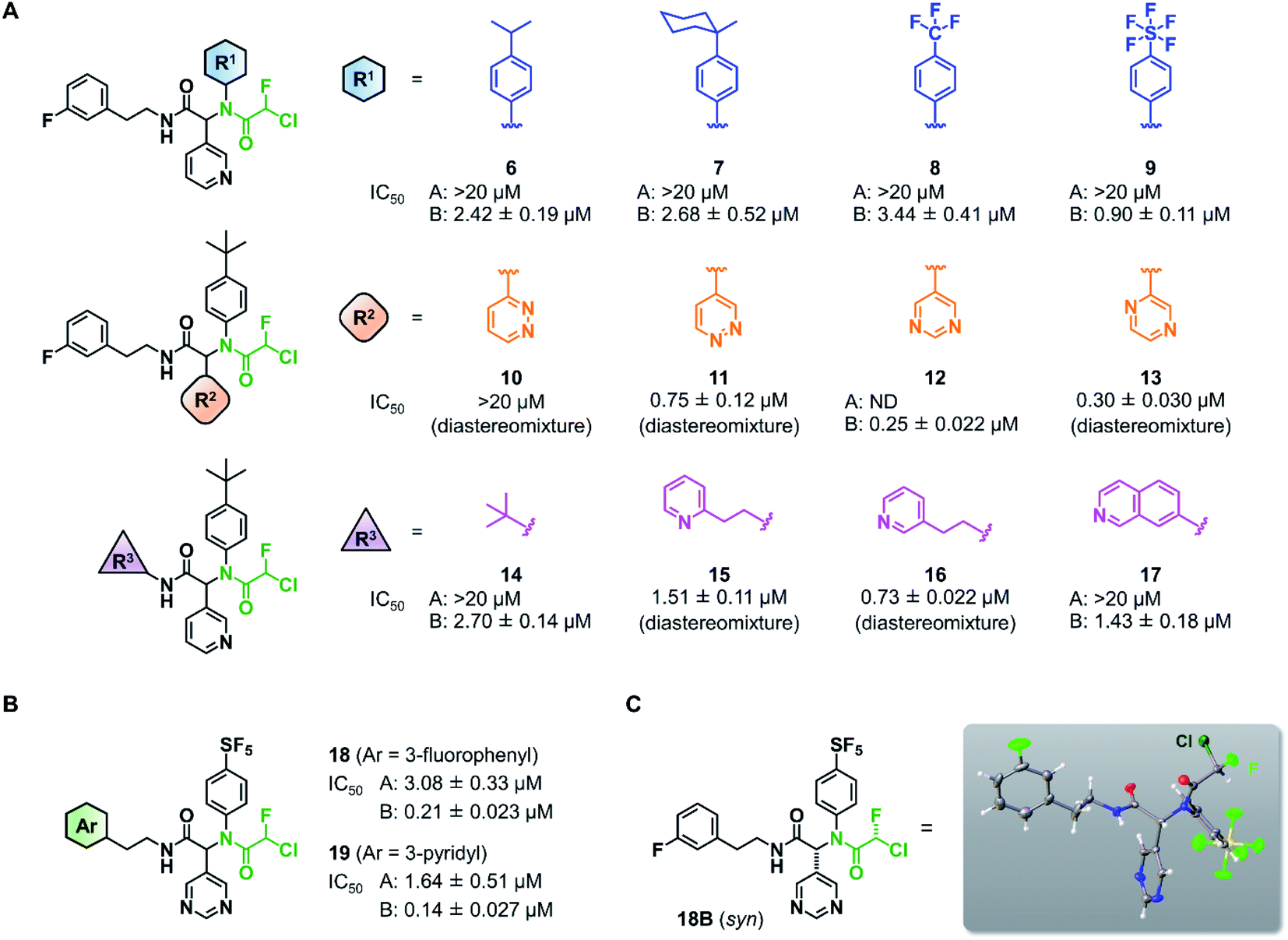 | ||
| Fig. 3 Structure–activity relationship of chlorofluoroacetamide (CFA) derivatives. (A) Screening of the CFA derivatives with the different R1, R2, and R3 substituents. ND, not determined. A and B indicate the less and more polar diastereomers separated by silica gel column chromatography. (B) The structures and inhibitory activities of the CFA dipeptides 18 and 19. (C) The relative configuration of 18B determined by X-ray crystallography (see Fig. S1†). | ||
To elucidate the relationship between the CFA chirality and the inhibitory activity, we next attempted to obtain the four stereoisomers of 18 in enantiomerically pure form using optically active chlorofluoroacetic acid.34 Wakselman previously reported the preparation of optically active chlorofluoroacetyl chloride based on the chromatographic separation of the diastereomers of CFA 20 derived from (S)-(−)-1-phenethylamine.35 However, the isolation of diastereomers of 20 proved to be laborious in our attempts due to poor separation by silica gel chromatography. Thus, we established a new method employing (R)-(−)-2-phenylglycinol as a chiral resolving agent (Scheme 1). We found that the diastereomers of CFA 21, readily accessible from commercially available (R)-(−)-2-phenylglycinol and ethyl chlorofluoroacetate, were easily separated by silica gel chromatography and afforded pure 21A and 21B on a multigram scale. X-ray crystallography determined the absolute configuration at the CFA chiral center to be R in 21B (Fig. S2†). Isolated 21A and 21B were subjected to hydrolysis in refluxing 3 M H2SO4/1,4-dioxane to afford (S)- and (R)-chlorofluoroacetic acid, respectively. Finally, Ugi MCR using enantiopure chlorofluoroacetic acids and subsequent chromatographic separation of the diastereomers yielded a set of four stereoisomers of 18.
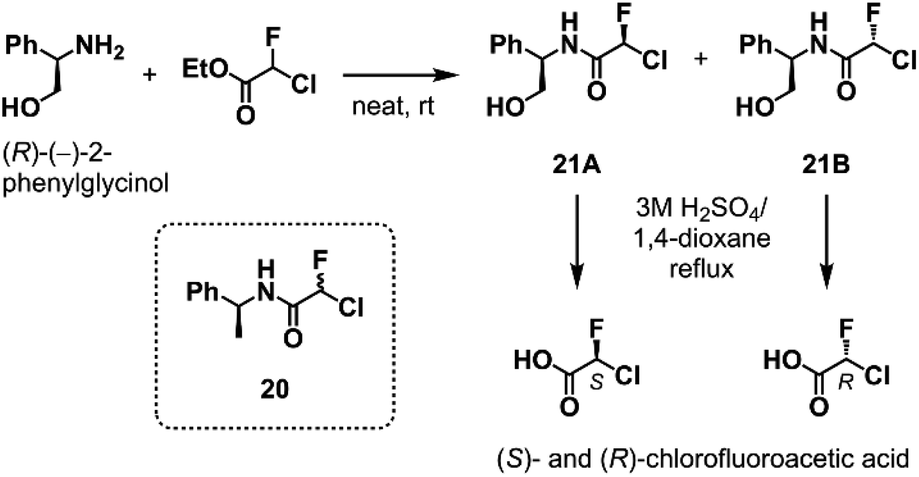 | ||
| Scheme 1 The optical resolution of chlorofluoroacetic acid using (R)-(−)-2-phenylglycinol as a chiral resolving agent. See ESI† for the detail. | ||
Having the four stereoisomers of 18 in hand, we evaluated their inhibitory activities against SARS-CoV-2 Mpro (Fig. 4). We found that (R,R)-18 (the former R indicates the absolute configuration at the Ugi MCR-generated stereocenter, and the latter at CFA) exclusively exhibited strong activity (IC50 = 0.056 μM) among the four stereoisomers (Fig. 4B and D). Notably, this activity is much higher than that of (R,S)-18 possessing an (S)-CFA warhead (IC50 = 10.62 μM), indicating that the chirality at CFA greatly influences the inhibitory activity. The IC50 values of the other isomers (S,R)-18 and (S,S)-18 were 2.24 and 14.04 μM, respectively. To further investigate these results, we performed a kinetic analysis of Mpro inhibition by (R,R)-18 and its diastereomers (S,R)- and (R,S)-18.36 All three isomers inhibited the hydrolysis of the fluorogenic substrate by Mpro in a time-dependent manner, indicating an irreversible mode of action (Fig. 4C and S3–S5†). The kinetic parameters for irreversible inactivation of Mpro are summarized in Fig. 4D. As expected, the most potent isomer (R,R)-18 inactivated Mpro with much higher efficiency (kinact/KI = 4167 M−1 s−1) than the diastereomers (S,R)- and (R,S)-18 (kinact/KI = 66.0 and 20.0 M−1 s−1, respectively). (R,R)-18 exhibited both a higher kinact value and a lower KI value than those of (R,S)-18, suggesting that the CFA chirality largely affected the reaction rate of covalent modification as well as the binding affinity with Mpro. In comparison between (R,R)-18 and (S,R)-18, the binding constant KI was significantly different (1.34 and 64.7 μM, respectively), whereas the inactivation rates were similar (kinact = 0.0056 and 0.0043 s−1, respectively). This marked difference in KI value was consistent with previous studies of ML188 and Jun8-76-3A, where the R configuration at the α-pyridine chiral center was crucial for the inhibitory activities.22,23 We also determined the kinetic parameters of acrylamide 1 (Fig. S6†). Despite higher thiol reactivity of acrylamide than CFA,29 the kinact value of 1 was significantly smaller than (R)-CFA. This result implies the mismatched disposition of the acrylamide warhead in the 1–Mpro binding complex for the Michael addition of the thiol group of Cys145.
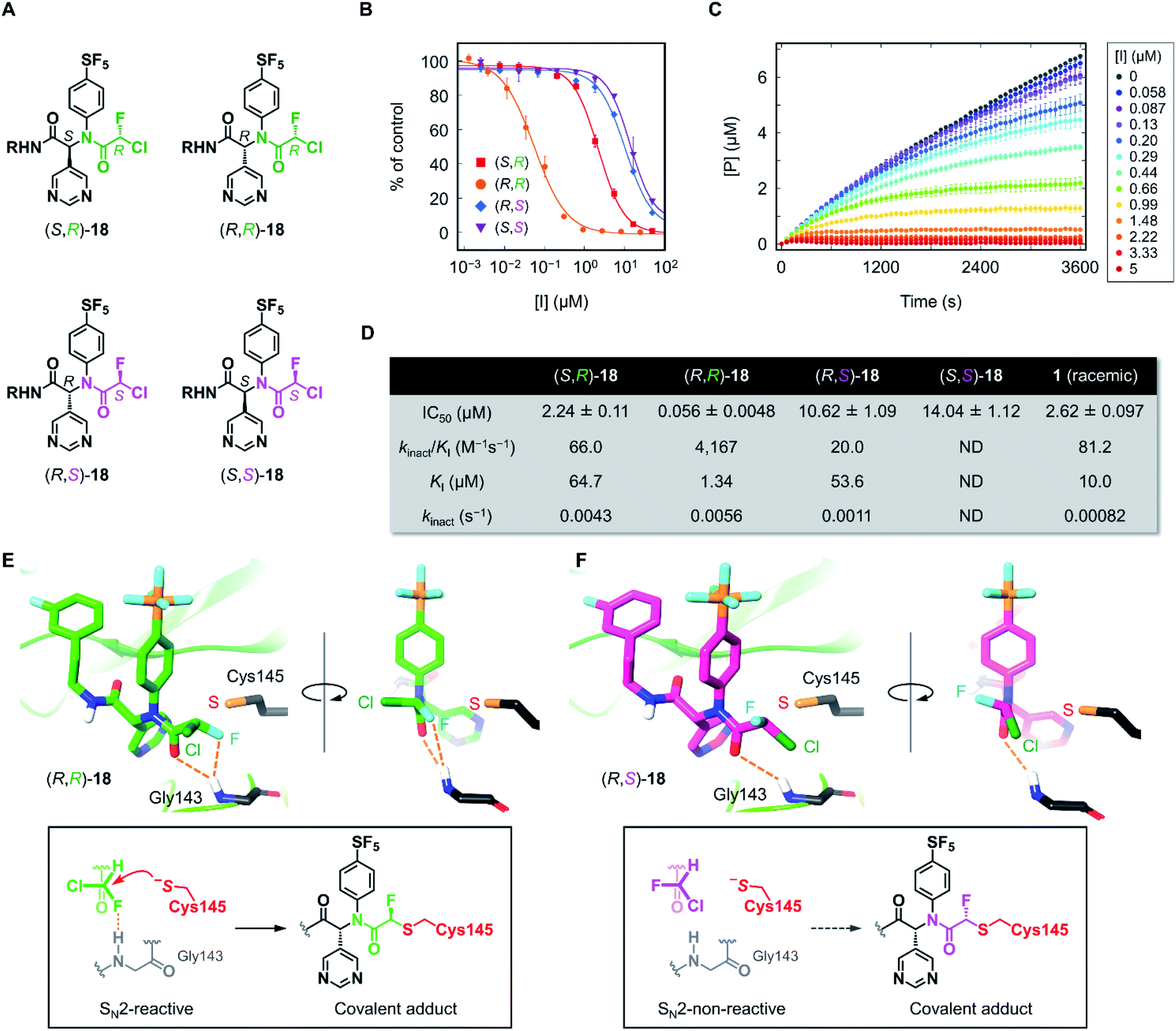 | ||
| Fig. 4 Inhibition properties of the four stereoisomers of 18 against SARS-CoV-2 Mpro. (A) The absolute configurations of the stereoisomers of 18 (R = (3-fluorophenyl)ethyl). The R and S configurations at the CFA unit are highlighted in green and magenta, respectively. (B) The inhibitory activity of the stereoisomers against SARS-CoV-2 Mpro. Each plot represents the mean of triplicate experiments ± standard deviation. (C) Time-course plot of the Mpro-catalyzed hydrolysis of the fluorogenic substrate in the presence of various concentrations of (R,R)-18. [P] represents the concentration of the hydrolyzed product of the fluorogenic peptide substrate determined by the fluorescence intensity. Each plot represents the mean of triplicate experiments ± standard error of the mean. (D) Summary of IC50 values and kinetic parameters of the chiral stereoisomers of 18. ND, not determined. (E) and (F) The computationally predicted, most stable conformations of (R,R)- and (R,S)-18 in the Mpro active site. See ESI† for a detailed computational method. | ||
To gain further insight into the effect of CFA chirality, we performed docking simulations of the stereoisomers of 18 against Mpro. Based on the X-ray cocrystal structure of (R)-1 with SARS-CoV-2 Mpro (PDB code 7NW2),24 we examined the possible binding poses and affinities of each of the four isomers, (R,R)-, (R,S)-, (S,R)-, and (S,S)-18 in the same pocket of Mpro. The predicted binding affinities for the stereoisomers fell into the range of 2 kcal mol−1 difference, but there found no clear correlation with the experimental result. To find out the cause of the higher reactivity of (R,R)-18, we assessed the occurrences of “reactive conformations” in the predicted poses for the four isomers, where we defined the pose as reactive for the SN2 reaction (backside attack of Cys145 to the CFA carbon) if the C⋯S distance <4.5 Å and Cl–C⋯S angle >150°. We found a large number of reactive conformations (46 out of 183 poses) exclusively in the low binding energy region for (R,R)-18 among the four stereoisomers (Fig. S7†). The most stable pose of (R,R)-18 was SN2-reactive, where the Cl–C⋯S angle is nearly 180° (Fig. 4E). In the complex, the CFA fluorine and the carbonyl oxygen were close proximity to the backbone NH of Gly143, which is thought to constitute the oxyanion hole in the S1′ pocket.37 This result suggests the potential contribution of NH⋯F hydrogen bond to facilitate the reaction. The bound structure of (R,R)-18 was well aligned to the X-ray crystal structure of a dichloroacetamide inhibitor Jun9-62-2R covalently bound to Mpro, which was recently reported by Wang et al. (Fig. S8†).38 On the other hand, the most stable pose of (R,S)-18 was SN2-non-reactive due to the unpreferable orientation of the C–Cl bond for the nucleophilic attack by Cys145. The rotation of the CFA unit of (R,S)-18 could give an SN2-reactive pose, but the resulting rotamer lacks any stabilizing interaction with the Mpro active site. Taken together, we infer that (R,R)-18 is more likely to become SN2-reactive as the NH⋯F hydrogen bond forces the C–Cl bond to a favorable orientation in the binding complex. The NH⋯F hydrogen bond in the (R,R)-18–Mpro complex could also contribute to increasing the electrophilic reactivity of the CFA warhead.
The inhibitory activity of (R,R)-18 against other cysteine proteases was evaluated (Fig. 5A). (R,R)-18 potently inhibited SARS-CoV-1 Mpro with an IC50 value of 0.094 μM. This result was not surprising because the structures of Mpro are highly conserved between SARS-CoV-1 and SARS-CoV-2 (96% identity of the amino acid sequence). (R,R)-18 did not inhibit SARS-CoV-2 papain-like protease (PLpro) (<1% inhibition at 20 μM). Human cathepsin B and L were not inhibited by 20 μM of (R,R)-18, whereas the noncovalent inhibitor Jun8-76-3A weakly inhibited cathepsin B (55% inhibition at 20 μM). To assess the covalent binding properties of the CFA inhibitor, we designed a clickable activity-based probe (R,R)-22 (Fig. 5B). Introduction of the ethynyl group was well tolerated, and (R,R)-22 exhibited potent inhibitory activity against SARS-CoV-2 Mpro (IC50 = 0.11 μM) comparable to that of (R,R)-18 (IC50 = 0.056 μM). In the gel-based in vitro assay, the recombinant SARS-CoV-2 Mpro was treated with (R,R)-22 (37 °C, 1 h) and conjugated to rhodamine-azide by copper-catalyzed azide–alkyne cycloaddition (CuAAC). The in-gel fluorescence analysis showed that Mpro could be detected at concentrations as low as 10 nM by 2.5 μM of (R,R)-22, and 50 nM of Mpro was labeled by (R,R)-22 in a concentration-dependent manner (0.1–10 μM). Incubation of (R,R)-22 with A431 cell lysate spiked with Mpro (50 nM) showed the main band at ∼34 kDa. This band disappeared after preincubation with the covalent inhibitors PF-00835231 (ref. 16) and (R,R)-18 (5 μM, 30 min), but not with the reversible inhibitor Jun8-76-3A (Fig. 5C and S9†). These results demonstrate the ability of the CFA-type inhibitor to covalently modify Mpro in the complex human proteome. We also synthesized acrylamide probe 23 as an alkynylated analog of 1 and evaluated its proteome-wide activity under live cell conditions (Fig. 5D and S10†). Treatment of A431 cells with (R,R)-22 or 23 (1–10 μM, 37 °C, 1 h) and the subsequent in-gel fluorescence analysis revealed that acrylamide 23 exhibited a higher level of off-target labeling compared to 22, suggesting a low indiscriminate reactivity of the dipeptidic CFA inhibitor in live cells.
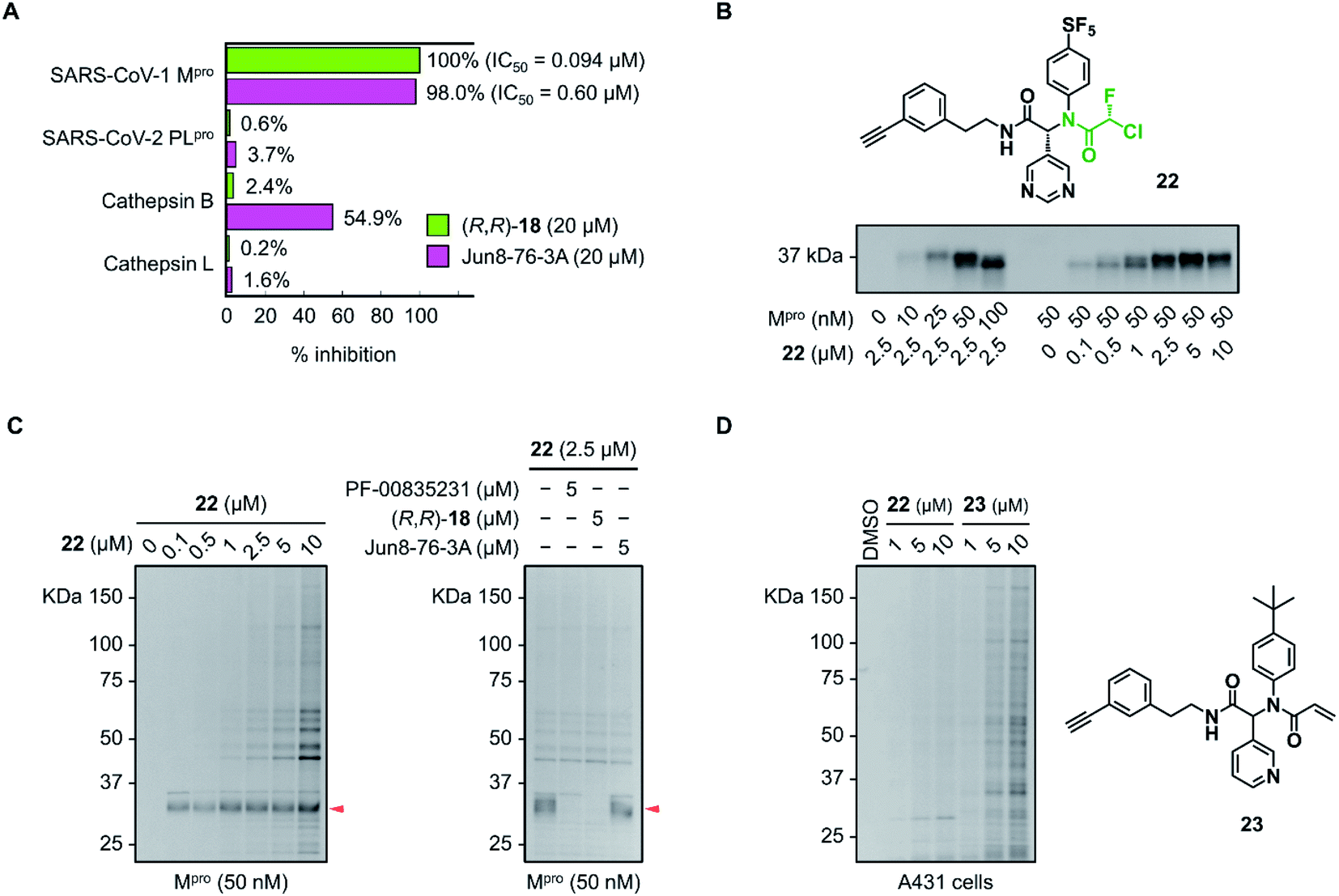 | ||
| Fig. 5 Selectivity profiles of the CFA inhibitors toward SARS-CoV-2 Mpro. (A) Inhibitory activity of (R,R)-18 and Jun8-76-3A toward the different cysteine proteases. The data are represented as the average of two independent experiments. (B) In-gel fluorescence analysis of the labeling of the recombinant Mpro with CFA probe 22 (37 °C, 1 h). (C) Reactivity profiles of (R,R)-22 toward the Mpro spiked into A431 cell lysate (37 °C, 1 h). Left: concentration-dependent labeling with (R,R)-22. Right: competition labeling by PF-00835231, (R,R)-18, and Jun8-76-3A. The red arrow indicates the fluorescence band of SARS-CoV-2 Mpro. (D) Proteome-wide reactivity profile of 22 and acrylamide probe 23 in live A431 cells (37 °C, 1 h). | ||
Conclusions
In summary, we have developed a novel irreversible inhibitor of SARS-CoV-2 Mpro using CFA as a reactive warhead. The convenient synthesis by Ugi MCR facilitated the rapid SAR study to identify the dipeptidic CFA compound 18 and 19 as potent Mpro inhibitors. Careful analysis of the diastereoselective inhibition of 18 and establishment of the synthetic protocol for enantiopure CFA derivatives revealed that (R,R)-18 exclusively shows strong inhibitory activity among the stereoisomers. The detailed reaction kinetics and computational studies suggest that the fluorine atom of the CFA warhead could participate in the reaction by forming an NH⋯F hydrogen bond in the Mpro active site that induces the stereoselective activation of (R,R)-18 for covalent modification. This reaction activation represents another possible utility of the CFA warhead, achievable by the unique characteristics of fluorine atoms, such as the solid electron-withdrawing ability.39,40 We believe that the prominent role of the CFA chirality in cysteine conjugation would provide an additional opportunity for the improvement of potency and selectivity of covalent inhibitors. Further application of the CFA chirality to the selective inhibition of other disease-associated proteins is ongoing and will be reported in due course.Data availability
Additional figures as described in the text, experimental procedures, synthetic procedures, and spectral data of the target compounds are available in the ESI.† Crystallographic data for 21B and 18B have been deposited in the Cambridge Crystallographic Data Centre (CCDC) under accession numbers CCDC 2124399 and 2124400, respectively.Author contributions
N. S. and A. O. conceived the project and designed the experiments. N. S., D. Y., S. O., H. I., R. H., T. H., and E. K. synthesized the compounds. N. S., D. Y., and S. O. performed enzymatic experiments. S. R. and K. M. carried out computational experiments. N. S. and A. O. wrote the manuscript with inputs from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Grant-in-Aid for Scientific Research on Innovative Areas “Chemistry for Multimolecular Crowding Biosystems” (JSPS KAKENHI Grant No. JP17H06349) and Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED under Grant Number JP18am0101091. N. S. acknowledges Grant-in-Aid for Scientific Research B (JSPS KAKENHI Grant No. 19H02854), AMED under Grant Number JP21ak0101121, and Grant for Basic Science Research Projects from the Sumitomo Foundation, for their financial supports. A. O. acknowledges AMED under Grant Number JP20fk0108519. We thank Dr Taisuke Matsumoto (Institute for Materials Chemistry and Engineering, Kyushu University) for X-ray diffraction experiments.Notes and references
- F. Wu, S. Zhao, B. Yu, Y.-M. Chen, W. Wang, Z.-G. Song, Y. Hu, Z.-W. Tao, J.-H. Tian, Y.-Y. Pei, M.-L. Yuan, Y.-L. Zhang, F.-H. Dai, Y. Liu, Q.-M. Wang, J.-J. Zheng, L. Xu, E. C. Holmes and Y.-Z. Zhang, Nature, 2020, 579, 265–269 CrossRef CAS PubMed.
- P. Zhou, X.-L. Yang, X.-G. Wang, B. Hu, L. Zhang, W. Zhang, H.-R. Si, Y. Zhu, B. Li, C.-L. Huang, H.-D. Chen, J. Chen, Y. Luo, H. Guo, R.-D. Jiang, M.-Q. Liu, Y. Chen, X.-R. Shen, X. Wang, X.-S. Zheng, K. Zhao, Q.-J. Chen, F. Deng, L.-L. Liu, B. Yan, F.-X. Zhan, Y.-Y. Wang, G.-F. Xiao and Z.-L. Shi, Nature, 2020, 579, 270–273 CrossRef CAS PubMed.
- W. T. Harvey, A. M. Carabelli, B. Jackson, R. K. Gupta, E. C. Thomson, E. M. Harrison, C. Ludden, R. Reeve, A. Rambaut, C. G. U. Consortium, S. J. Peacock and D. L. Robertson, Nat. Rev. Microbiol., 2021, 19, 409–424 CrossRef CAS PubMed.
- C. Gil, T. Ginex, I. Maestro, V. Nozal, L. Barrado-Gil, M. Á. Cuesta-Geijo, J. Urquiza, D. Ramírez, C. Alonso, N. E. Campillo and A. Martinez, J. Med. Chem., 2020, 63, 12359–12386 CrossRef CAS PubMed.
- T. Pillaiyar, S. Meenakshisundaram and M. Manickam, Drug Discovery Today, 2020, 25, 668–688 CrossRef CAS PubMed.
- R. Cannalire, C. Cerchia, A. R. Beccari, F. S. Di Leva and V. Summa, J. Med. Chem., 2020 DOI:10.1021/acs.jmedchem.0c01140.
- Z. Jin, X. Du, Y. Xu, Y. Deng, M. Liu, Y. Zhao, B. Zhang, X. Li, L. Zhang, C. Peng, Y. Duan, J. Yu, L. Wang, K. Yang, F. Liu, R. Jiang, X. Yang, T. You, X. Liu, X. Yang, F. Bai, H. Liu, X. Liu, L. W. Guddat, W. Xu, G. Xiao, C. Qin, Z. Shi, H. Jiang, Z. Rao and H. Yang, Nature, 2020, 582, 289–293 CrossRef CAS PubMed.
- W. Rut, K. Groborz, L. Zhang, X. Sun, M. Zmudzinski, B. Pawlik, X. Wang, D. Jochmans, J. Neyts, W. Młynarski, R. Hilgenfeld and M. Drag, Nat. Chem. Biol., 2021, 17, 222–228 CrossRef CAS PubMed.
- K. Anand, J. Ziebuhr, P. Wadhwani, J. R. Mesters and R. Hilgenfeld, Science, 2003, 300, 1763–1767 CrossRef CAS PubMed.
- A. Wu, Y. Wang, C. Zeng, X. Huang, S. Xu, C. Su, M. Wang, Y. Chen and D. Guo, Virus Res., 2015, 208, 56–65 CrossRef CAS PubMed.
- L. Zhang, D. Lin, X. Sun, U. Curth, C. Drosten, L. Sauerhering, S. Becker, K. Rox and R. Hilgenfeld, Science, 2020, 368, 409–412 CrossRef CAS PubMed.
- W. Dai, B. Zhang, X.-M. Jiang, H. Su, J. Li, Y. Zhao, X. Xie, Z. Jin, J. Peng, F. Liu, C. Li, Y. Li, F. Bai, H. Wang, X. Cheng, X. Cen, S. Hu, X. Yang, J. Wang, X. Liu, G. Xiao, H. Jiang, Z. Rao, L.-K. Zhang, Y. Xu, H. Yang and H. Liu, Science, 2020, 368, 1331–1335 CrossRef CAS PubMed.
- J. Qiao, Y.-S. Li, R. Zeng, F.-L. Liu, R.-H. Luo, C. Huang, Y.-F. Wang, J. Zhang, B. Quan, C. Shen, X. Mao, X. Liu, W. Sun, W. Yang, X. Ni, K. Wang, L. Xu, Z.-L. Duan, Q.-C. Zou, H.-L. Zhang, W. Qu, Y.-H.-P. Long, M.-H. Li, R.-C. Yang, X. Liu, J. You, Y. Zhou, R. Yao, W.-P. Li, J.-M. Liu, P. Chen, Y. Liu, G.-F. Lin, X. Yang, J. Zou, L. Li, Y. Hu, G.-W. Lu, W.-M. Li, Y.-Q. Wei, Y.-T. Zheng, J. Lei and S. Yang, Science, 2021, 371, 1374–1378 CrossRef CAS PubMed.
- K. S. Yang, X. R. Ma, Y. Ma, Y. R. Alugubelli, D. A. Scott, E. C. Vatansever, A. K. Drelich, B. Sankaran, Z. Z. Geng, L. R. Blankenship, H. E. Ward, Y. J. Sheng, J. C. Hsu, K. C. K. B. Zhao, H. S. Hayatshahi, J. Liu, P. Li, C. A. Fierke, C.-T. K. Tseng, S. Xu and W. R. Liu, ChemMedChem, 2021, 16, 942–948 CrossRef CAS PubMed.
- W. Dai, D. Jochmans, H. Xie, H. Yang, J. Li, H. Su, D. Chang, J. Wang, J. Peng, L. Zhu, Y. Nian, R. Hilgenfeld, H. Jiang, K. Chen, L. Zhang, Y. Xu, J. Neyts and H. Liu, J. Med. Chem., 2021 DOI:10.1021/acs.jmedchem.0c02258.
- R. L. Hoffman, R. S. Kania, M. A. Brothers, J. F. Davies, R. A. Ferre, K. S. Gajiwala, M. He, R. J. Hogan, K. Kozminski, L. Y. Li, J. W. Lockner, J. Lou, M. T. Marra, L. J. Mitchell, B. W. Murray, J. A. Nieman, S. Noell, S. P. Planken, T. Rowe, K. Ryan, G. J. Smith, J. E. Solowiej, C. M. Steppan and B. Taggart, J. Med. Chem., 2020, 63, 12725–12747 CrossRef CAS PubMed.
- S. Hattori, N. Higashi-Kuwata, H. Hayashi, S. R. Allu, J. Raghavaiah, H. Bulut, D. Das, B. J. Anson, E. K. Lendy, Y. Takamatsu, N. Takamune, N. Kishimoto, K. Murayama, K. Hasegawa, M. Li, D. A. Davis, E. N. Kodama, R. Yarchoan, A. Wlodawer, S. Misumi, A. D. Mesecar, A. K. Ghosh and H. Mitsuya, Nat. Commun., 2021, 12, 668 CrossRef CAS PubMed.
- B. Bai, A. Belovodskiy, M. Hena, A. S. Kandadai, M. A. Joyce, H. A. Saffran, J. A. Shields, M. B. Khan, E. Arutyunova, J. Lu, S. K. Bajwa, D. Hockman, C. Fischer, T. Lamer, W. Vuong, M. J. van Belkum, Z. Gu, F. Lin, Y. Du, J. Xu, M. Rahim, H. S. Young, J. C. Vederas, D. L. Tyrrell, M. J. Lemieux and J. A. Nieman, J. Med. Chem., 2021 DOI:10.1021/acs.jmedchem.1c00616.
- S. Konno, K. Kobayashi, M. Senda, Y. Funai, Y. Seki, I. Tamai, L. Schäkel, K. Sakata, T. Pillaiyar, A. Taguchi, A. Taniguchi, M. Gütschow, C. E. Müller, K. Takeuchi, M. Hirohama, A. Kawaguchi, M. Kojima, T. Senda, Y. Shirasaka, W. Kamitani and Y. Hayashi, J. Med. Chem., 2021 DOI:10.1021/acs.jmedchem.1c00665.
- J. Breidenbach, C. Lemke, T. Pillaiyar, L. Schäkel, G. Al Hamwi, M. Diett, R. Gedschold, N. Geiger, V. Lopez, S. Mirza, V. Namasivayam, A. C. Schiedel, K. Sylvester, D. Thimm, C. Vielmuth, L. P. Vu, M. Zyulina, J. Bodem, M. Gütschow and C. E. Müller, Angew. Chem., Int. Ed., 2021, 60, 10423–10429 CrossRef CAS PubMed.
- D. R. Owen, C. M. N. Allerton, A. S. Anderson, L. Aschenbrenner, M. Avery, S. Berritt, B. Boras, R. D. Cardin, A. Carlo, K. J. Coffman, A. Dantonio, L. Di, H. Eng, R. Ferre, K. S. Gajiwala, S. A. Gibson, S. E. Greasley, B. L. Hurst, E. P. Kadar, A. S. Kalgutkar, J. C. Lee, J. Lee, W. Liu, S. W. Mason, S. Noell, J. J. Novak, R. S. Obach, K. Ogilvie, N. C. Patel, M. Pettersson, D. K. Rai, M. R. Reese, M. F. Sammons, J. G. Sathish, R. S. P. Singh, C. M. Steppan, A. E. Stewart, J. B. Tuttle, L. Updyke, P. R. Verhoest, L. Wei, Q. Yang and Y. Zhu, Science, 2021, 374, 1586–1593 CrossRef CAS PubMed.
- J. Jacobs, V. Grum-Tokars, Y. Zhou, M. Turlington, S. A. Saldanha, P. Chase, A. Eggler, E. S. Dawson, Y. M. Baez-Santos, S. Tomar, A. M. Mielech, S. C. Baker, C. W. Lindsley, P. Hodder, A. Mesecar and S. R. Stauffer, J. Med. Chem., 2013, 56, 534–546 CrossRef CAS PubMed.
- N. Kitamura, M. D. Sacco, C. Ma, Y. Hu, J. A. Townsend, X. Meng, F. Zhang, X. Zhang, M. Ba, T. Szeto, A. Kukuljac, M. T. Marty, D. Schultz, S. Cherry, Y. Xiang, Y. Chen and J. Wang, J. Med. Chem., 2021 DOI:10.1021/acs.jmedchem.1c00509.
- D. Zaidman, P. Gehrtz, M. Filep, D. Fearon, R. Gabizon, A. Douangamath, J. Prilusky, S. Duberstein, G. Cohen, C. D. Owen, E. Resnick, C. Strain-Damerell, P. Lukacik, C.-M. Consortium, H. Barr, M. A. Walsh, F. von Delft and N. London, Cell Chem. Biol., 2021, 28, 1795–1806 CrossRef CAS PubMed.
- J. Singh, R. C. Petter, T. A. Baillie and A. Whitty, Nat. Rev. Drug Discov, 2011, 10, 307–317 CrossRef CAS PubMed.
- F. M. Ferguson and N. S. Gray, Nat. Rev. Drug Discovery, 2018, 17, 353–377 CrossRef CAS PubMed.
- D. Kim, J. Y. Xue and P. Lito, Cell, 2020, 183, 850–859 CrossRef CAS PubMed.
- U. P. Dahal, R. S. Obach and A. M. Gilbert, Chem. Res. Toxicol., 2013, 26, 1739–1745 Search PubMed.
- N. Shindo, H. Fuchida, M. Sato, K. Watari, T. Shibata, K. Kuwata, C. Miura, K. Okamoto, Y. Hatsuyama, K. Tokunaga, S. Sakamoto, S. Morimoto, Y. Abe, M. Shiroishi, J. M. M. Caaveiro, T. Ueda, T. Tamura, N. Matsunaga, T. Nakao, S. Koyanagi, S. Ohdo, Y. Yamaguchi, I. Hamachi, M. Ono and A. Ojida, Nat. Chem. Biol., 2019, 15, 250–258 CrossRef CAS PubMed.
- M. Sato, H. Fuchida, N. Shindo, K. Kuwata, K. Tokunaga, X.-L. Guo, R. Inamori, K. Hosokawa, K. Watari, T. Shibata, N. Matsunaga, S. Koyanagi, S. Ohdo, M. Ono and A. Ojida, ACS Med. Chem. Lett., 2020, 11, 1137–1144 CrossRef CAS PubMed.
- C. Miura, N. Shindo, K. Okamoto, K. Kuwata and A. Ojida, Chem. Pharm. Bull., 2020, 68, 1074–1081 CrossRef CAS PubMed.
- C.-J. Kuo, Y.-H. Chi, J. T.-A. Hsu and P.-H. Liang, Biochem. Biophys. Res. Commun., 2004, 318, 862–867 CrossRef CAS PubMed.
- M. F. Sowaileh, R. A. Hazlitt and D. A. Colby, ChemMedChem, 2017, 12, 1481–1490 CrossRef CAS PubMed.
- G. Bellucci, G. Berti, A. Borraccini and F. Macchia, Tetrahedron, 1969, 25, 2979–2985 CrossRef CAS.
- H. Molines and C. Wakselman, Synthesis, 1984, 1984, 838–839 CrossRef.
- P. J. Tonge, ACS Infect. Dis., 2019, 5, 796–808 CrossRef PubMed.
- J. Lee, L. J. Worrall, M. Vuckovic, F. I. Rosell, F. Gentile, A.-T. Ton, N. A. Caveney, F. Ban, A. Cherkasov, M. Paetzel and N. C. Strynadka, Nat. Commun., 2020, 11, 5877 CrossRef CAS PubMed.
- A Ugi MCR-generated dipeptide inhibitor of SARS-CoV-2 Mpro with dichloroacetamide warhead (Jun9-62-2R) has been reported while this manuscript was under review. However, none of their active compounds possessed a warhead with chirality at the reaction center, C. Ma, Z. Xia, M. D. Sacco, Y. Hu, J. A. Townsend, X. Meng, J. Choza, H. Tan, J. Jang, M. V. Gongora, X. Zhang, F. Zhang, Y. Xiang, M. T. Marty, Y. Chen and J. Wang, J. Am. Chem. Soc., 2021, 143, 20697–20709 CrossRef CAS PubMed.
- D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC.
- P. Zhou, J. Zhou, F. Tian and Z. Shang, J. Chem. Inf. Model., 2009, 49, 2344–2355 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplemental figures, experimental methods, and synthetic procedures. CCDC 2124399 and 2124400. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc06596c |
| This journal is © The Royal Society of Chemistry 2022 |