
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric β-arylation of cyclopropanols enabled by photoredox and nickel dual catalysis†
Jianhua
Wang
 and
Xiaoxun
Li
*
and
Xiaoxun
Li
*
Department of Medicinal Chemistry, Key Laboratory of Chemical Biology (Ministry of Education), NMPA Key Laboratory for Technology Research and Evaluation of Drug Products, School of Pharmaceutical Sciences, Cheeloo College of Medicine, Shandong University, Jinan, Shandong 250012, China. E-mail: xli@sdu.edu.cn
First published on 17th February 2022
Abstract
The enantioselective functionalization and transformation of readily available cyclopropyl compounds are synthetically appealing yet challenging topics in organic synthesis. Here we report an asymmetric β-arylation of cyclopropanols with aryl bromides enabled by photoredox and nickel dual catalysis. This dual catalytic transformation features a broad substrate scope and good functional group tolerance at room temperature, providing facile access to a wide array of enantioenriched β-aryl ketones bearing a primary alcohol moiety in good yields with satisfactory enantioselectivities (39 examples, up to 83% yield and 90% ee). The synthetic value of this protocol was illustrated by the concise asymmetric construction of natural product calyxolane B analogues.
Introduction
Cyclopropanes are versatile building blocks and participate in a diverse range of fundamental transformations in organic synthesis by taking advantage of their intrinsic high ring strain.1 Among these cyclopropane derivatives, cyclopropanols, which are usually considered as precursors of ketone homoenolates, have gained emerging interest as three-carbon synthons for new bond formation and natural product synthesis in recent years.2 In the presence of transition metal (TM) catalysis or single electron transfer (SET) catalysis, they have been widely applied in the synthesis of β-functionalized ketones by involving intermediates of homoenolates or β-keto radicals generated via ring-opening pathways, albeit mostly in racemic forms (Scheme 1A).3,4 Despite recent advances,5 the development of catalytic asymmetric strategies to access various enantioenriched β-functionalized ketones through ring-opening of cyclopropanols has proven to be quite challenging yet highly desirable.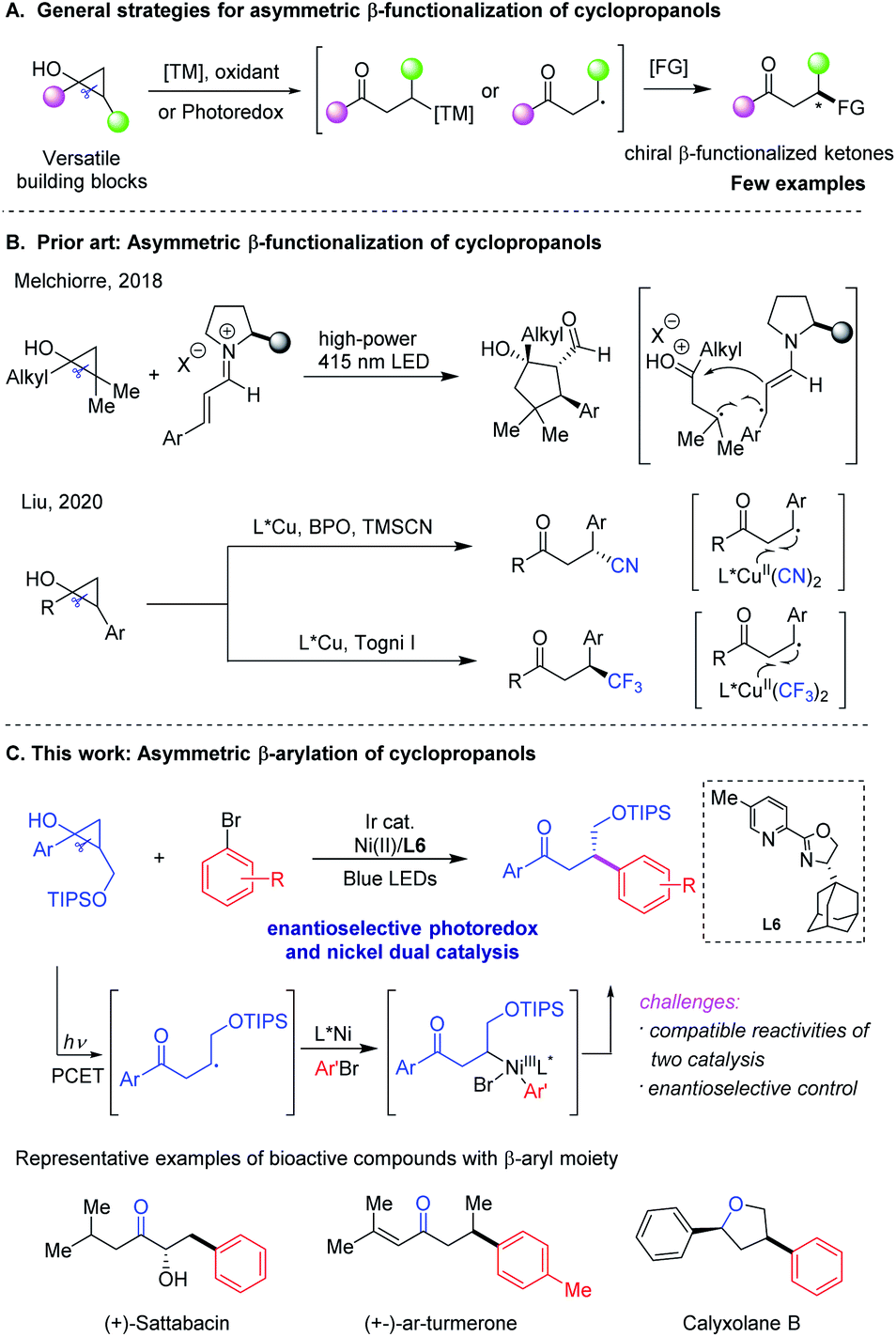 | ||
| Scheme 1 Asymmetric β-functionalization of cyclopropanols. | ||
Over the past decade, photoredox catalysis has become a powerful and reliable tool for developing innovative synthetic methodologies, designing and constructing interesting bioactive molecules and valuable functional materials.6 In this context, there has been tremendous progress in the area of photoredox dual catalysis with transition metals,7 Lewis acids,8 organocatalysts,9 and eletrocatalysts,10 providing attractive strategies for the stereoselective and divergent preparation of structurally diverse chiral molecules and pharmacophores. Recently, seminal studies have been disclosed toward the asymmetric β-functionalization of cyclopropanols by exploiting these novel catalytic protocols. In 2018, the Melchiorre group reported their pioneering work on the enantioselective photochemical organocascade process that converted cyclopropanols and α, β-unsaturated aldehydes into densely functionalized cyclopentanols with excellent stereoselectivities (Scheme 1B).11 Later, Liu and coworkers achieved the asymmetric benzylic trifluoromethylation and cyanation of racemic aryl-substituted cyclopropanols via copper-mediated radical ring-opening pathways, which provided robust synthetic methods to access chiral β-CF3 and β-CN ketones in good yields with high enantioselectivities, respectively (Scheme 1B).12 In 2020, Huang and Rueping disclosed a general strategy for the remote arylation of ketones from cyclic alcohols via photoredox and nickel catalysis, although cyclopropanol was reported as only a single non-enantioselective example.13 In spite of these exciting advances, asymmetric β-arylation of cyclopropanols remain unexplored, highlighting the need to develop catalytic enantioselective arylation methods while considering the wide prevalence of β-aryl ketone derivatives in bioactive compounds and pharmaceuticals.14
In recent years, photoredox/nickel dual catalysis has enabled the non-asymmetric construction of different types of C–C or C-heteroatom bonds under mild reaction conditions, providing attractive alternatives to challenging synthetic metal-catalyzed cross-coupling reactions.15 Encouraged by recent progress in such enantioselective dual catalysis16 and our continuous interest in nickel-catalyzed asymmetric radical transformation,17 we envisioned that engaging a chiral nickel catalyst photocatalytically might serve as a general platform for enantioselective β-functionalization of cyclopropanols to access a series of value-added β-functionalized ketones in a stereoselective fashion (Scheme 1C). To realize this hypothesis, two major challenging issues need to be addressed. First, the reactivity of highly reactive alkyl radicals generated via ring-opening of cyclopropanols should be compatible with nickel involved oxidative addition of aryl electrophiles in order to avoid the competing homo-coupling reactions, undesired dimerization of alkyl radicals and other byprocesses. Second, the enantioselective photoredox and nickel dual catalysis remains largely undeveloped.16 Here we presented our study on asymmetric β-arylation of cyclopropanols enabled by this dual mode catalysis (Scheme 1C).
Results and discussion
We initiated our investigation by using cyclopropanol 1a bearing a primary alcohol moiety and 4-bromoacetophenone 2a as model substrates (Table 1). After an extensive survey of different reaction parameters, including photoredox catalysts, nickel complexes, chiral ligands and variation of alcohol protection groups,18 the best conditions were established when reacting cyclopropanol 1a (3 equiv.) and 4-bromoacetophenone 2a (1 equiv.) with photocatalyst [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 [E1/2(*P/P−) = 1.21 V vs. SCE in MeCN]19 in the presence of a simple chiral Ni(OAc)2/Py-(oxazoline) catalyst, collidine as the base and DMF as the solvent under blue-light irradiation, furnishing the desired chiral β-aryl ketone 3a in 75% yield with a 93![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 enantiomeric ratio at room temperature (entry 1). Notably, common bis(oxazoline) catalysts L1 and L2 were not effective for this reaction (entries 2–3) and other Py-(oxazoline) catalysts L3 to L5 gave similar yields (70–80%), albeit in diminished enantioselectivities (entries 4–6). The use of other nickel catalysts, such as NiBr2, afforded 3a in lower yield and ee (entry 7). Encouraged by recent progress in the area of proton-coupled electron transfer (PCET),20 we examined various bases which have been frequently applied for the ring-opening transformations of cyclic alcohols through the PCET process.18 Commercially available base PnBu3Et+(EtO)2POO− generated 3a in 67% yield with 74% ee (entry 8). Next, the screening of solvents showed that DMF was the optimal solvent for this asymmetric dual reaction (entry 9). Surprisingly, the yield was decreased to 31% when the reaction was conducted at 10 °C due to the very low conversion, albeit with a 90:10 enantiomeric ratio (entry 10). Finally, control experiments indicated that the photocatalyst, nickel catalyst, base, and blue-light irradiation were essential for the success of this reaction (entry 11).
:7 enantiomeric ratio at room temperature (entry 1). Notably, common bis(oxazoline) catalysts L1 and L2 were not effective for this reaction (entries 2–3) and other Py-(oxazoline) catalysts L3 to L5 gave similar yields (70–80%), albeit in diminished enantioselectivities (entries 4–6). The use of other nickel catalysts, such as NiBr2, afforded 3a in lower yield and ee (entry 7). Encouraged by recent progress in the area of proton-coupled electron transfer (PCET),20 we examined various bases which have been frequently applied for the ring-opening transformations of cyclic alcohols through the PCET process.18 Commercially available base PnBu3Et+(EtO)2POO− generated 3a in 67% yield with 74% ee (entry 8). Next, the screening of solvents showed that DMF was the optimal solvent for this asymmetric dual reaction (entry 9). Surprisingly, the yield was decreased to 31% when the reaction was conducted at 10 °C due to the very low conversion, albeit with a 90:10 enantiomeric ratio (entry 10). Finally, control experiments indicated that the photocatalyst, nickel catalyst, base, and blue-light irradiation were essential for the success of this reaction (entry 11).
|
|
|||
|---|---|---|---|
| Entry | Variation from standard conditions | Yieldsb | erc |
| a Unless otherwise indicated, all reactions were performed with 1a (0.30 mmol), 2a (0.10 mmol), [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (1.5 mmol%), Ni(OAc)2 (20 mmol%), ligand (22 mmol%), collidine (300 mmol%) in DMF(5 mL), blue LEDs (465 nm), 25 °C, 48 h under a nitrogen atmosphere. b Isolated yields. c Determined by HPLC analysis. Ad: adamantanyl group. | |||
| 1 | None | 75% | 93:7 |
| 2 | L1 instead of L6 | 0 | — |
| 3 | L2 instead of L6 | 20% | 60:40 |
| 4 | L3 instead of L6 | 70% | 55:45 |
| 5 | L4 instead of L6 | 80% | 88:12 |
| 6 | L5 instead of L6 | 77% | 90:10 |
| 7 | NiBr2.glyme instead of Ni(OAc)2 | 47% | 90:10 |
| 8 | PnBu3Et+(EtO)2POO− instead of collidine | 67% | 87:13 |
| 9 | DMA instead of DMF | 71% | 88:12 |
| 10 | 10 °C instead of 25 °C | 31% | 90:10 |
| 11 | No PC, or no Ni, or no light, or no base | 0 | — |
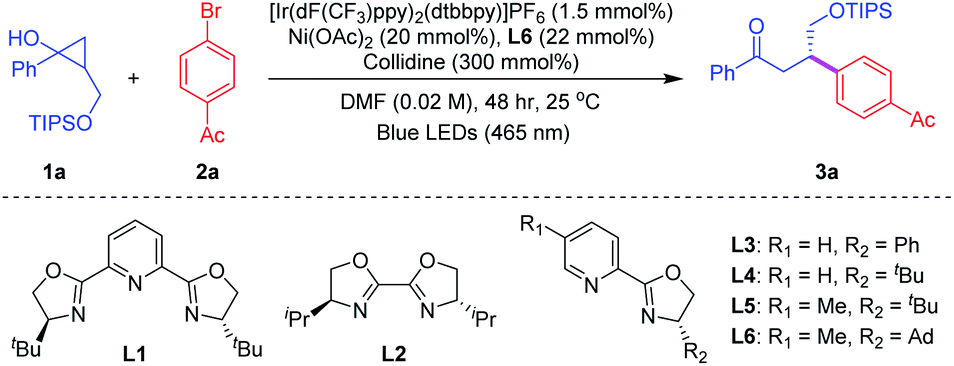
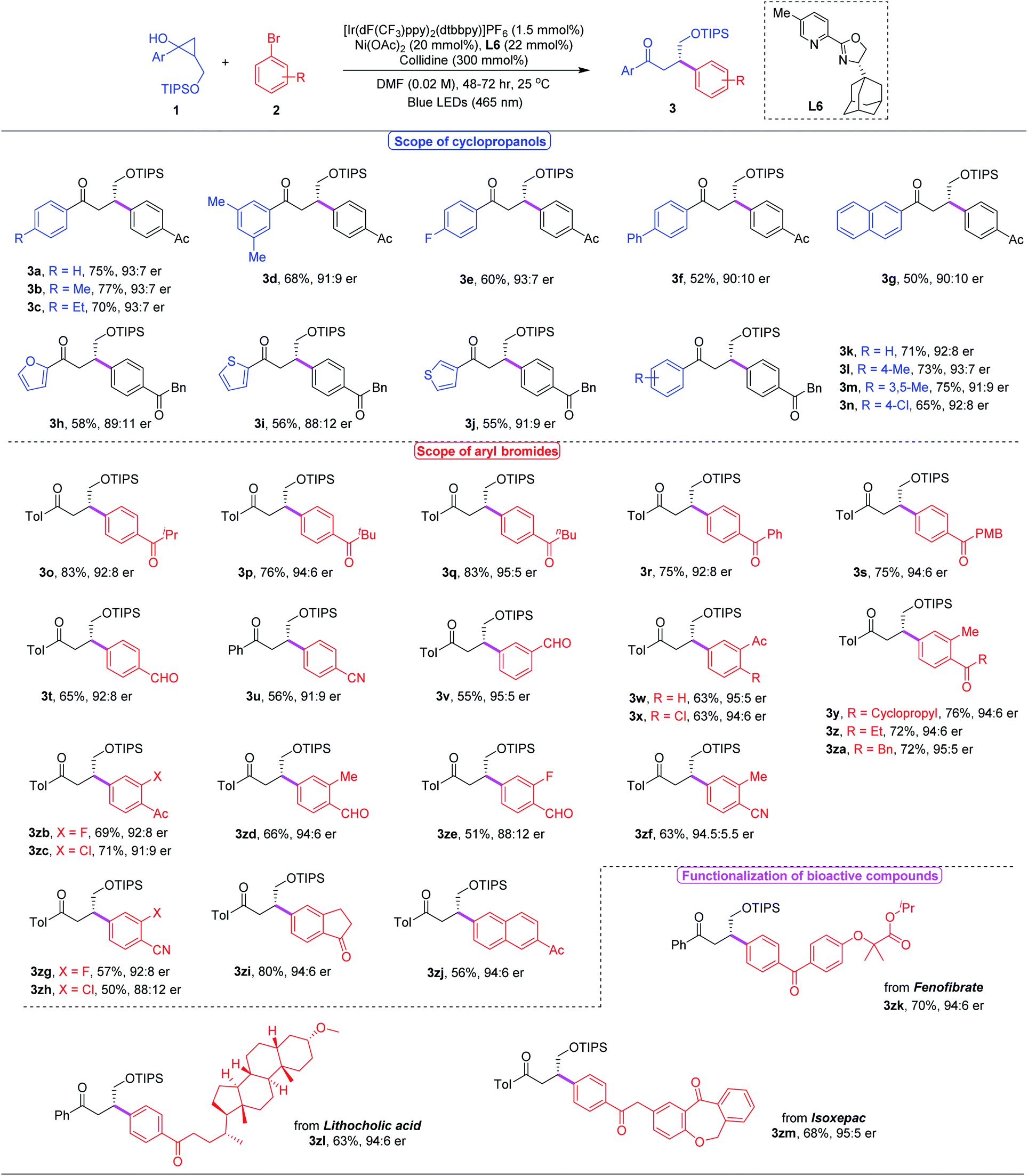
With the optimal reaction conditions in hand, we investigated the substrate scope of this asymmetric β-arylation of cyclopropanols (Table 2). In general, the enantioselectivity was sensitive to the 2-substitution groups of cyclopropanols. When the 2-methyl or 2-benzyl substituted cyclopropane-1-ols were used as the substrates, the corresponding products were obtained in 80% yield with 40% ee, and 67% yield with 36% ee, respectively. First, the generality of cyclopropanols bearing the primary alcohol moiety was evaluated. A broad range of substituted cyclopropanols bearing an electron-withdrawing (e.g. F or Cl) or electron-donating (e.g. alkyl substituents) group on the aryl moiety were well tolerated, and delivered the corresponding chiral ketones in 52–77% yields with 80–86% ee (3a–3f, 3k–3n). Cyclopropanol 1g with the 2-naphthyl group also proceeded to afford the desired product 3g in 50% yield with a 90:10 er. In addition, reactions with different heteroaryl moieties, such as 2-furyl, 2-thiofuryl and 3-thiofuryl groups, participated well to give the desired ketones in comparable yields and enantioselectivities (3h–3j). Next, the scope of commercially available aryl bromides was examined with cyclopropanol 2a under the optimized reaction conditions. Typically, electron-deficient aryl bromides were beneficial for this asymmetric photoredox/nickel dual catalytic system. A wide variety of functional groups including ketones, aldehydes, cyanides and halides were well tolerated to provide the desired β-aryl ketones with satisfying results (3o–3zi). Moreover, 2-naphthyl substituted aryl bromide 2zj also successfully yielded the corresponding ketone 3zj in 56% yield with a 94:6 er. However, heteroaryl bromides, including 4-bromo-2-(trifluoromethyl)pyridine, 5-bromo-2-furaldehyde and 5-bromooxindole, failed to generate the desired products under current reaction conditions, presumably because of their low reactivities. Encouraged by the mild reaction conditions and broad functional group tolerance, we applied this asymmetric protocol to the late-stage functionalization of complex bioactive molecules. To our delight, a number of complicated compounds, such as fenofibrate, lithocholic acid and isoxepac derived bromides, all proceeded well with cyclopropanol 1a, delivering the highly functionalized chiral ketones in 63–70% yields with up to 90% ee (3zk–3zm).
| a Unless otherwise indicated, all reactions were performed under the following conditions: cyclopropanols 1 (0.3 mmol), aryl bromides 2 (0.1 mmol), [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (1.5 mmol%), Ni(OAc)2 (20 mmol%), L6 (22 mmol%), collidine (300 mmol%), DMF(5 mL), blue LEDs (465 nm), 48–72 h, 25 °C. Isolated yields. The er values were determined by HPLC analysis. |
|---|
|
To demonstrate the synthetic value of this asymmetric β-arylation protocol, a 1.0 mmol scale reaction was carried out, providing the chiral ketone 3a in 70% yield with 93:7 er (Scheme 2A). Then, further derivatization of the products was performed. Direct deprotection of the TIPS group of ketone 3m afforded the chiral primary alcohol 4 in 88% yield (Scheme 2B). Interestingly, an enantioselective reduction of 3zd in the presence of the (R)-CBS catalyst and borane, followed by deprotection of TIPS would generate chiral tri-hydroxy product 5 in 82% yield with 96% ee (Scheme 2C). The absolute configuration of 5 was determined to be 1S, 3S by X-ray diffraction.21 Subsequently, Brønsted acid catalyzed stereospecific benzylic substitution occurred, furnishing the concise synthesis of a marine natural product ent-calyxolane B analog 6 in 80% yield (Scheme 2C).22 Moreover, subjecting 3a to the Wittig reaction, followed by deprotection of TIPS could generate γ-hydroxy ene 8 in 75% overall yield with 88% ee (Scheme 2D). With product 8 in hand, the indomethacin ester 9 was easily obtained in 95% yield.
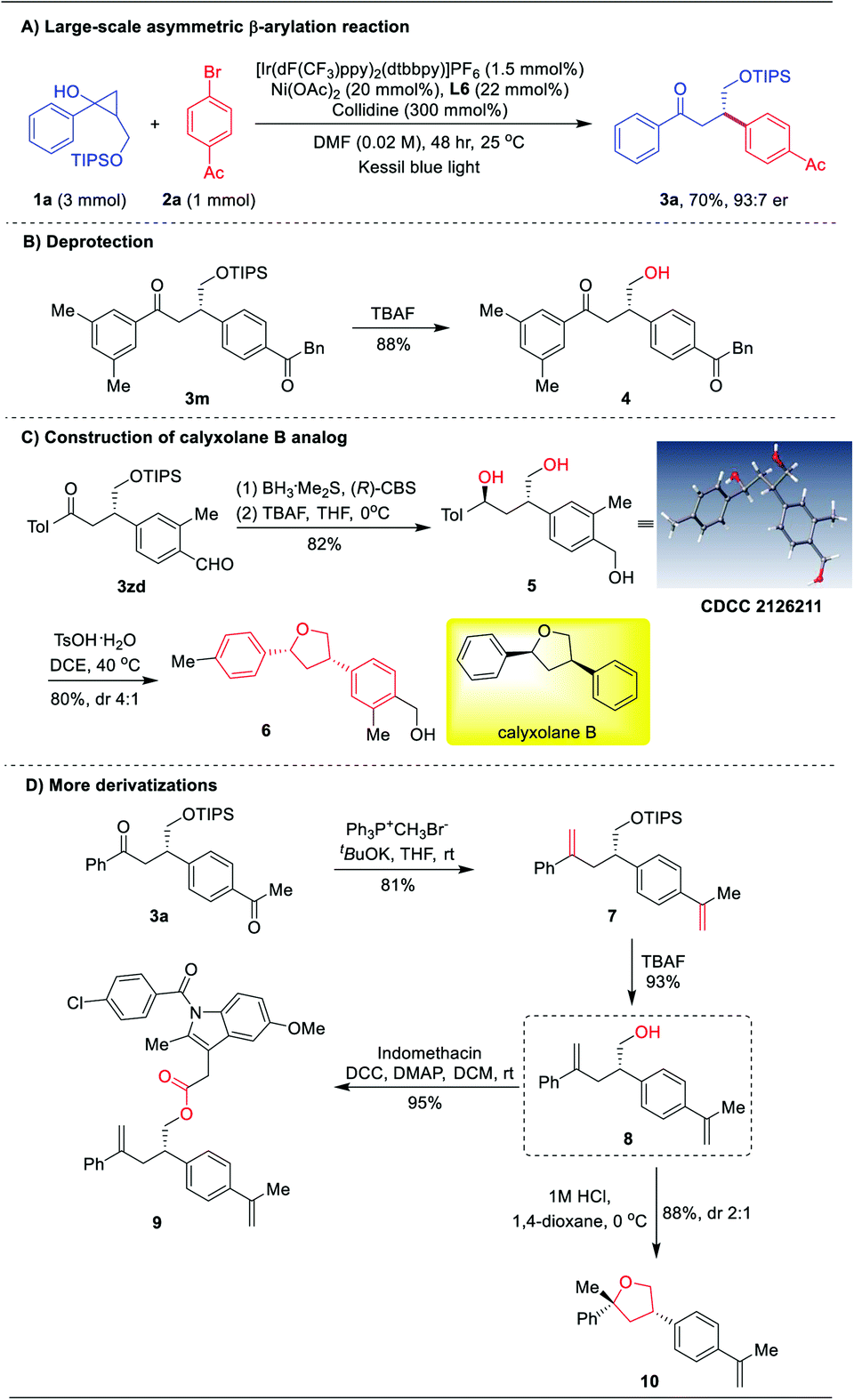 | ||
| Scheme 2 Large-scale experiment and derivatization of chiral β-aryl ketones. | ||
Alternatively, treatment of 8 with hydrogen chloride solution (1.0 M in 1,4-dioxane) led to tetrahydrofuran derivative 10 (Scheme 2D), which is a privileged structural unit in natural products and bioactive compounds.23
To gain more insight into the reaction pathway, a number of control experiments were performed. First, both trans-cyclopropanol 1f′ and cis-cyclopropanol 1f′′ were subjected to the standard conditions, producing the corresponding product 3f in a similar yield with the same enantioselectivity (Scheme 3A). TBS protected cyclopropanol 11 failed to generate product 3b, indicating that the ring-opening process of cyclopropanols underwent a PECT pathway (Scheme 3B). Furthermore, we conducted the reaction under standard conditions in the presence of TEMPO (2,2,6,6-tetra-methyl-1-piperidinyloxy) as a radical scavenger. As expected, the reaction was completely shut down, and the TEMPO adduct 12 was detected by HRMS (Scheme 3C).18 In addition, when acrylonitrile 13 was used instead of aryl bromide, the Giese-type product 14 was obtained in 52% yield under the standard conditions, albeit in the racemic form (Scheme 3D). All these results strongly supported that the asymmetric photoredox/nickel dual catalysis process involved a radical pathway.
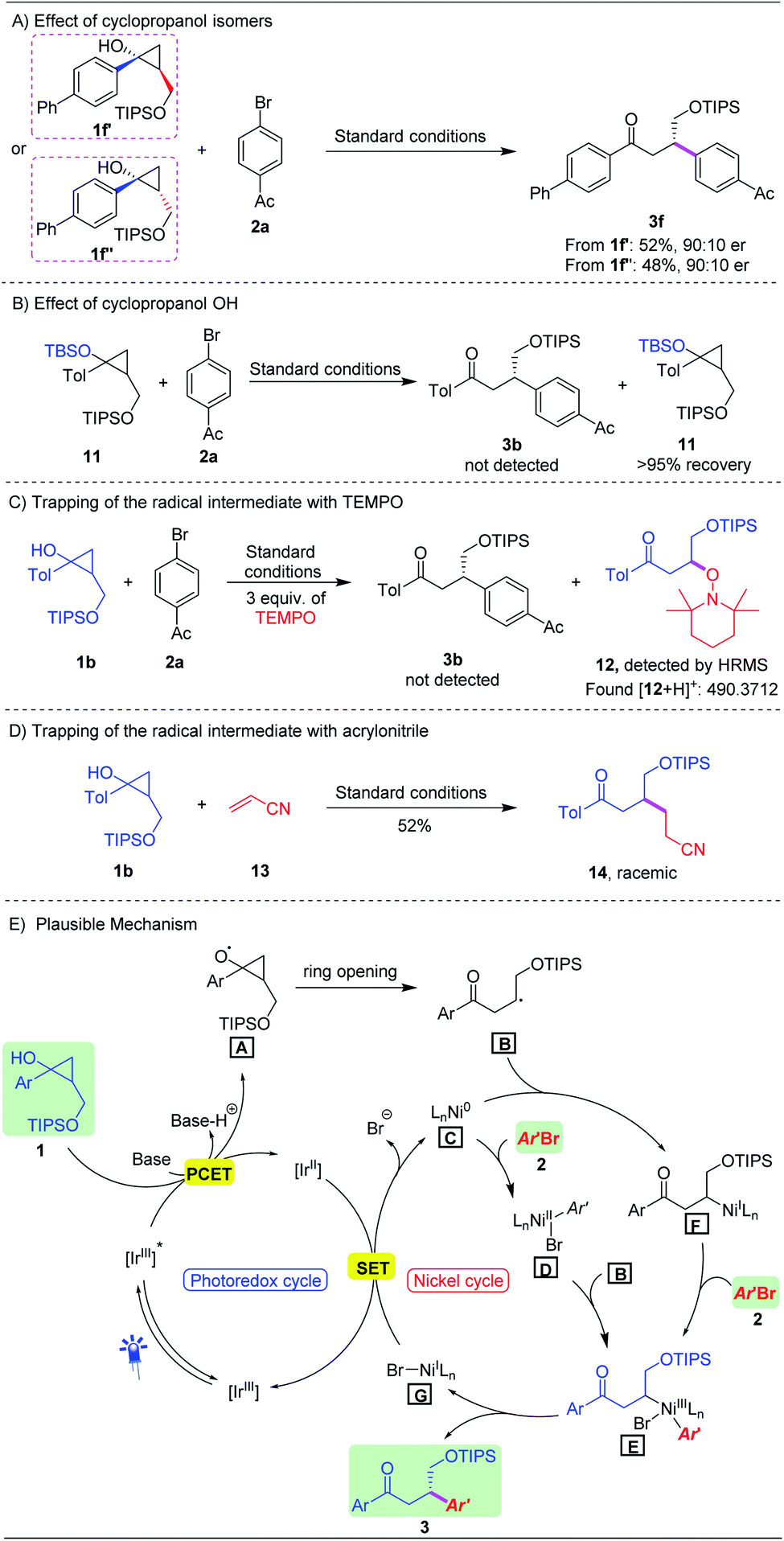 | ||
| Scheme 3 Preliminary mechanistic studies. | ||
On the basis of the preliminary mechanistic studies and literature reports,20 we proposed a plausible mechanism for this dual catalytic asymmetric β-arylation reaction as outlined in Scheme 3E. Upon blue-light irradiation, photocatalyst [Ir(dF(CF3)ppy)2(dtbbpy)]+ was promoted to its excited state *[Ir(dF(CF3)ppy)2(dtbbpy)]+, which would oxidize the cyclopropanols 1via a PCET process to generate the key alkoxy radical intermediate A. Subsequent ring opening through β-scission of A led to secondary alkyl radical B. For the nickel catalysis cycle, two possible pathways are involved. Chiral Ni(0) complex C underwent oxidative addition with aryl bromides 2 to give intermediate D, which then reacted with alkyl radical B to form Ni(III) complex E. Alternatively, alkyl radical B could couple with Ni(0) C to yield an alkyl Ni(I) intermediate F which was expected to undergo oxidative addition with aryl bromides, generating chiral Ni(III) intermediate E. At this stage, we could not be able to rule out any pathway. Next, reductive elimination from chiral Ni(III) complex E would forge the enantioenriched Csp3–Csp2 bonds of β-aryl ketones 3 along with expelling the corresponding Ni(I) complex G, and the latter could be reduced by the reduced form of the photocatalyst, thus completing the dual catalytic cycle.
Conclusions
In conclusion, we have developed an enantioselective β-arylation of cyclopropanols enabled by asymmetric photoredox/nickel dual catalysis. This method provides a convenient strategy to assemble a diverse array of highly valuable chiral β-aryl ketones from readily available cyclopropanols and aryl bromides at room temperature. Moreover, the value of this protocol was demonstrated by the synthesis of calyxolane and tetrahydrofuran analogues. Studies towards expanding the generality of this asymmetric dual catalysis are ongoing.Data availability
Data for this work, including optimization tables, general experimental procedures, characterization data (NMR and HPLC spectra) for all new compounds and X-ray data are provided in the ESI.†Author contributions
X. Li designed the project. J. Wang performed the experiments and analyzed the data. X. Li and J. Wang wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support for this work from the National Natural Science Foundation of China (21901142), the Natural Science Foundation of Shandong Province (ZR2019QB001) and the Fundamental Research Funds of Shandong University (21310088963023, 2020QNQT007, and 2020QNQT009).Notes and references
- For selected reviews, see: (a) M. Rubin, M. Rubina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117 CrossRef CAS PubMed; (b) G. Fumagalli, S. Stanton and J. F. Bower, Chem. Rev., 2017, 117, 9404 CrossRef CAS PubMed; (c) J. Wang, S. A. Blaszczyk, X. Li and W. Tang, Chem. Rev., 2021, 121, 110 CrossRef CAS PubMed; (d) V. Pirenne, B. Muriel and J. Waser, Chem. Rev., 2021, 121, 227–263 CrossRef CAS PubMed; (e) B. Biletskyi, P. Colonna, K. Masson, J. L. Parrain, L. Commeiras and G. Chouraqui, Chem. Soc. Rev., 2021, 50, 7513 RSC.
- For selected reviews, see: (a) O. G. Kulinkovich, Chem. Rev., 2003, 103, 2597 CrossRef CAS PubMed; (b) X. Cai, W. Liang and M. J. Dai, Tetrahedron, 2019, 75, 193 CrossRef CAS; (c) T. R. McDonald, L. R. Mills, M. S. West and S. A. L. Rousseaux, Chem. Rev., 2021, 121, 3 CrossRef CAS PubMed.
- (a) T. Seiser and N. Cramer, J. Am. Chem. Soc., 2010, 132, 5340 CrossRef CAS PubMed; (b) P. P. Das, K. Belmore and J. K. Cha, Angew. Chem., Int. Ed., 2012, 51, 9517 CrossRef CAS PubMed; (c) N. Nithiy and A. Orellana, Org. Lett., 2014, 16, 5854 CrossRef CAS PubMed; (d) D. G. Kananovich, Y. A. Konik, D. M. Zubrytski, I. Järving and M. Lopp, Chem. Commun., 2015, 51, 8349 RSC; (e) R. Ren, Z. Wu, Y. Xu and C. Zhu, Angew. Chem., Int. Ed., 2016, 55, 2866 CrossRef CAS PubMed; (f) L. R. Mills, L. M. B. Arbelaez and S. A. L. Rousseaux, J. Am. Chem. Soc., 2017, 139, 11357 CrossRef CAS PubMed; (g) H. Zhang, G. Wu, H. Yi, T. Sun, B. Wang, Y. Zhang, G. Dong and J. Wang, Angew. Chem., Int. Ed., 2017, 56, 3945 CrossRef CAS; (h) Z. Ye, X. Cai, J. Li and M. J. Dai, ACS Catal., 2018, 8, 5907 CrossRef CAS PubMed; (i) J. Yang, Y. Sekiguchi and N. Yoshikai, ACS Catal., 2019, 9, 5638 CrossRef CAS; (j) L. R. Mills, C. Zhou, E. Fung and S. A. L. Rousseaux, Org. Lett., 2019, 21, 8805 CrossRef CAS PubMed; (k) B.-Q. Cheng, S.-X. Zhang, Y.-Y. Cui, X.-Q. Chu, W. Rao, H. Xu, G.-Z. Han and Z.-L. Shen, Org. Lett., 2020, 22, 5456 CrossRef CAS PubMed.
- (a) J. Jiao, L. X. Nguyen, D. R. Patterson and R. A. Flowers, Org. Lett., 2007, 9, 1323 CrossRef CAS PubMed; (b) Y.-F. Wang and S. Chiba, J. Am. Chem. Soc., 2009, 131, 12570 CrossRef CAS PubMed; (c) Y.-F. Wang, K. K. Toh, E. P. J. Ng and S. Chiba, J. Am. Chem. Soc., 2011, 133, 6411 CrossRef CAS PubMed; (d) A. Ilangovan, S. Saravanakumar and S. Malayappasamy, Org. Lett., 2013, 15, 4968 CrossRef CAS PubMed; (e) H. Zhao, X. Fan, J. Yu and C. Zhu, J. Am. Chem. Soc., 2015, 137, 3490 CrossRef CAS PubMed; (f) Y. Li, Z. Ye, T. M. Bellman, T. Chi and M. J. Dai, Org. Lett., 2015, 17, 2186 CrossRef CAS PubMed; (g) Z. Ye and M. J. Dai, Org. Lett., 2015, 17, 2190 CrossRef CAS; (h) F.-Q. Huang, J. Xie, J.-G. Sun, Y.-W. Wang, X. Dong, L.-W. Qi and B. Zhang, Org. Lett., 2016, 18, 684 CrossRef CAS PubMed; (i) X. Hu, G.-X. Li, G. He and G. Chen, Org. Chem. Front., 2019, 6, 3205 RSC; (j) Y.-H. Zhang, W.-W. Zhang, Z.-Y. Zhang, K. Zhao and T.-P. Loh, Org. Lett., 2019, 21, 5101 CrossRef CAS PubMed; (k) J.-L. Zhan, M.-W. Wu, D. Wei, B.-Y. Wei, Y. Jiang, W. Yu and B. Han, ACS Catal., 2019, 9, 4179 CrossRef CAS; (l) T. Kikuchi, K. Yamada, T. Yasui and Y. Yamamoto, Org. Lett., 2021, 23, 4710 CrossRef CAS PubMed; (m) M. Vellakkaran, T. Kim and S. Hong, Angew. Chem., Int. Ed., 2022, 61, e202113658 CrossRef CAS PubMed.
- (a) M. V. Barysevich, V. V. Kazlova, A. G. Kukel, A. I. Liubina, A. L. Hurski, V. N. Zhabinskii and V. A. Khripach, Chem. Commun., 2018, 54, 2800 RSC; (b) Q. Tan, Z. Yang, D. Jiang, Y. Cheng, J. Yang, S. Xi and M. Zhang, Angew. Chem., Int. Ed., 2019, 58, 6420 CrossRef CAS PubMed; (c) M. V. Laktsevich-Iskryk, N. A. Varabyeva, V. V. Kazlova, V. N. Zhabinskii, V. A. Khripach and A. L. Hurski, Eur. J. Org. Chem., 2020, 2431 CrossRef CAS; (d) C. Lou, X. Wang, L. Lv and Z. Li, Org. Lett., 2021, 23, 7608 CrossRef CAS PubMed; (e) Y. C. Lu and J. G. West, ACS Catal., 2021, 11, 12721 CrossRef CAS; (f) Y. Sekiguchi and N. Yoshikai, J. Am. Chem. Soc., 2021, 143, 18400 CrossRef CAS PubMed; (g) W. Zhou, T. Zhou, M. Tian, Y. Jiang, J. Yang, S. Lei, Q. Wang, C. Zhang, H. Qiu, L. He, Z. Wang, J. Deng and M. Zhang, J. Am. Chem. Soc., 2021, 143, 19975 CrossRef CAS PubMed; (h) Q. Zhang, S.-W. Zhou, C.-Y. Shi and L. Yin, Angew. Chem., Int. Ed., 2021, 60, 26351 CrossRef CAS PubMed.
- For selected reviews, see: (a) M. Fagnoni, D. Dondi, D. Ravelli and A. Albini, Chem. Rev., 2007, 107, 2725 CrossRef CAS PubMed; (b) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC; (c) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828 CrossRef CAS PubMed; (d) D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed; (e) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898 CrossRef CAS PubMed; (f) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS PubMed; (g) M. D. Kärkäs, J. A. Jr Porco and C. R. J. Stephenson, Chem. Rev., 2016, 116, 9683 CrossRef PubMed; (h) F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190 RSC; (i) C. Prentice, J. Morrisson, A. D. Smith and E. Zysman-Colman, Beilstein J. Org. Chem., 2020, 16, 2363 CrossRef PubMed; (j) L. Buglioni, F. Raymenants, A. Slattery, S. D. A. Zondag and T. Noël, Chem. Rev., 2022, 122, 2752–2906 CrossRef CAS PubMed.
- For selected reviews, see: (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (b) J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS; (c) M. D. Abreu, P. Belmont and E. Brachet, Eur. J. Org. Chem., 2020, 1327 CrossRef; (d) A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS PubMed.
- (a) J. Du, K. L. Skubi, D. M. Schultz and T. P. Yoon, Science, 2014, 344, 392 CrossRef CAS PubMed; (b) T. R. Blum, Z. D. Miller, D. M. Bates, I. A. Guzei and T. P. Yoon, Science, 2016, 354, 1391 CrossRef CAS PubMed; (c) X. Huang and E. Meggers, Acc. Chem. Res., 2019, 52, 833 CrossRef CAS PubMed; (d) K. Zhang, L.-Q. Lu, Y. Jia, Y. Wang, F.-D. Lu, F. Pan and W.-J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 13375 CrossRef CAS PubMed; (e) Y. Li, M. Lei and L. Gong, Nat. Catal., 2019, 2, 1016 CrossRef CAS.
- (a) C. M. Holden and P. Melchiorre, Photochemistry and Excited-State Reactivity of Organocatalytic Intermediates in Photochemistry, Royal Society of Chemistry, Cambridge. UK, 2019, vol. 47, p. 344 Search PubMed; (b) S.-H. Xiang and B. Tan, Nat. Commun., 2020, 11, 3786 CrossRef CAS PubMed.
- J. Liu, L. Lu, D. Wood and S. Lin, ACS Cent. Sci., 2020, 6, 1317 CrossRef CAS PubMed.
- Ł. Woźniak, G. Magagnano and P. Melchiorre, Angew. Chem., Int. Ed., 2018, 57, 1068 CrossRef PubMed.
- (a) C. Jiang, L. Wang, H. Zhang, P. Chen, Y.-L. Guo and G. Liu, Chem, 2020, 6, 2407 CrossRef CAS; (b) L. Wu, L. Wang, P. Chen, Y.-L. Guo and G. Liu, Adv. Synth. Catal., 2020, 362, 2189 CrossRef.
- L. Huang, T. Ji and M. Rueping, J. Am. Chem. Soc., 2020, 142, 3532 CrossRef CAS PubMed.
- (a) G. N. Roth, A. Chandra and M. G. Nair, J. Nat. Prod., 1998, 61, 542 CrossRef CAS PubMed; (b) W.-Z. Weng and B. Zhang, Chem.–Eur. J., 2018, 24, 10934 CrossRef CAS PubMed.
- For selected reviews, see: (a) J. K. Matsui, S. B. Lang, D. R. Heitz and G. A. Molander, ACS Catal., 2017, 7, 2563 CrossRef CAS PubMed; (b) J. A. Milligan, J. P. Phelan, S. O. Badir and G. A. Molander, Angew. Chem., Int. Ed., 2019, 58, 6152 CrossRef CAS PubMed; (c) C. Zhu, H. Yue, L. Chu and M. Rueping, Chem. Sci., 2020, 11, 4051 RSC; (d) S. O. Badir and G. A. Molander, Chem, 2020, 6, 1327 CrossRef CAS PubMed.
- For selected reviews, see: (a) A. Lipp, S. O. Badir and G. A. Molander, Angew. Chem., Int. Ed., 2020, 60, 1714 CrossRef PubMed; (b) H.-H. Zhang, H. Chen, C. Zhu and S. Yu, Sci. China: Chem., 2020, 63, 637 CAS . For recent reports, see:; (c) X. Cheng, H. Lu and Z. Lu, Nat. Commun., 2019, 10, 3549 CrossRef PubMed; (d) L. Huan, X. Shu, W. Zu, D. Zhong and H. Huo, Nat. Commun., 2021, 12, 3536 CrossRef CAS PubMed.
- M. Lv and X. Li, ACS Catal., 2021, 11, 14829 CrossRef CAS.
- See ESI† for details..
- M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl, R. A. Pascal, G. G. Malliaras and S. Bernhard, Chem. Mater., 2005, 17, 5712 CrossRef CAS.
- For selected reviews, see: (a) E. C. Gentry and R. R. Knowles, Acc. Chem. Res., 2016, 49, 1546 CrossRef CAS PubMed; (b) N. Hoffmann, Eur. J. Org. Chem., 2017, 1982 CrossRef CAS; (c) E. Tsui, H. Wang and R. R. Knowles, Chem. Sci., 2020, 11, 11124 RSC; (d) P. R. D. Murray, J. H. Cox, N. D. Chiappini, C. B. Roos, E. A. McLoughlin, B. G. Hejna, S. T. Nguyen, H. H. Ripberger, J. M. Ganley, E. Tsui, N. Y. Shin, B. Koronkiewicz, G. Qiu and R. R. Knowles, Chem. Rev., 2022, 122, 2017–2291 CrossRef CAS PubMed . For recent reports, see:; (e) C. B. Roos, J. Demaerel, D. E. Graff and R. R. Knowles, J. Am. Chem. Soc., 2020, 142, 5974 CrossRef CAS PubMed.
- Crystallographic data of compounds 6 (CCDC 2126211).†.
- (a) A. D. Rodríguez, O. M. Cóbar and O. L. Padilla, J. Nat. Prod., 1997, 60, 915 CrossRef PubMed; (b) C. Kim, B. Roh and H. G. Lee, Chem. Sci., 2021, 12, 3668 RSC; (c) A. Bunrit, P. Srifa, T. Rukkijakan, C. Dahlstrand, G. Huang, S. Biswas, R. A. Watile and J. S. M. Samec, ACS Catal., 2020, 10, 1344 CrossRef CAS.
- (a) A. Lorente, J. Lamariano-Merketegi, F. Albericio and M. Alvarez, Chem. Rev., 2013, 113, 4567 CrossRef CAS PubMed; (b) L. N. Chavan, P. S. Mainkar and S. Chandrasekhar, Eur. J. Org. Chem., 2019, 6890 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2126211. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc07237d |
| This journal is © The Royal Society of Chemistry 2022 |