
Tuning the electronic communication of the Ru–O bond in ultrafine Ru nanoparticles to boost the alkaline electrocatalytic hydrogen production activity at large current density†
Weihang
Feng
,
Yongqiang
Feng
 *,
Yingrui
He
,
Junsheng
Chen
,
Hai
Wang
,
Tianmi
Luo
,
Yuzhu
Hu
,
Chengke
Yuan
,
Liyun
Cao
,
Liangliang
Feng
and
Jianfeng
Huang
*,
Yingrui
He
,
Junsheng
Chen
,
Hai
Wang
,
Tianmi
Luo
,
Yuzhu
Hu
,
Chengke
Yuan
,
Liyun
Cao
,
Liangliang
Feng
and
Jianfeng
Huang
School of Materials Science and Engineering, Shaanxi Key Laboratory of Green Preparation and Functionalization for Inorganic Materials, Shaanxi University of Science and Technology, Xi'an, 710021, People's Republic of China. E-mail: fengyq@sust.edu.cn
First published on 4th July 2022
Abstract
The rational design and synthesis of efficient electrocatalysts for the hydrogen evolution reaction (HER) are of great importance for alkaline hydrogen production. This study describes a nanocomposite material design, in which ultrafine and small (2.0–3.5 nm) Ru nanoparticles coordinated with O atom sites are supported on a carbon matrix formed by C60(OH)n (Ru–O/C-600). The Ru–O bonds create a channel of electron communication to facilitate charge transfer and improve the conductivity of electrocatalysts. The ultrafine and evenly-distributed Ru nanoparticles provide a high density of active sites to extend electrochemical surface areas. Ru–O/C-600 achieved a low overpotential of 32 mV at a current density of 10 mA cm−2 with a small Tafel slope of 51.8 mV dec−1 and long-term stability of 50 h. Moreover, Ru–O/C-600 also gave an output of 500 and 1000 mA cm−2 with an overpotential of 242 and 383 mV for practical use. These findings open up new avenues for developing Ru-based hybridization materials with enhanced electron transfer and abundant active sites for HER performance.
Introduction
Hydrogen (H2) is a form of clean energy, and hydrogen production by water splitting1 is an important green hydrogen production method.2 Platinum (Pt) is currently the best choice for electrode catalysts for hydrogen production by water splitting,3 but it is scarce and expensive.4 Researchers are pursuing the development of inexpensive and convenient catalysts for hydrogen evolution (HER) to replace Pt.5 Non-noble metal catalysts, such as carbides,6 nitrides,7 oxides,8 phosphates,9 sulfides10 and transition metal borides,11 have been extensively explored as alternatives to Pt. In recent years, cost-effective precious metals (Ir,12 Ru,13 Pd,14etc.), with electronic configurations similar to Pt, have been investigated as reliable substitutes with efficient HER ability.Ruthenium (Ru) in the form of nanoparticles,15 alloys16,17 and single atoms18,19 showed HER activity in alkaline solution comparable to that of commercial platinum catalysts. Ru nanoparticles have promoted catalytic performance via suitable hetero-atom doping, adjusting the morphology and reconstituting the nanostructure.20 According to a previous study, Youn and Choi successfully synthesized Ru/C nanoparticles and probed HER activity during the phase-transition process (RuΔccp/C⋯RuΔc→h/C⋯RuΔhcp/C),21 suggesting that atomic interactions on nanoparticle surfaces were activated by phase-transitions, which could optimize the HER performance in alkaline media. Moreover, the introduction of nonmetallic elements (N,22,23 P,24 S,25 O,26etc.) and anions can change the metal coordination environment at the interface by adjusting the charge distribution, thus affecting its catalytic performance. Zhou and his group prepared Ru/Co3O4 by cation replacement and controlled reduction, which further proved that the introduction of O improved the intrinsic HER activity.27 Although several viable improvements have been made, there is a scarcity of research on the synergistic effects between oxygen coordination and the distribution of metal nanoparticles.28
Herein, we first synthesized Ru–O/C-600 electrocatalysts by liquid-phase precipitation and pyrolysis methods. Ru–O/C-600 possesses enhanced HER catalytic activity with an overpotential of 32 mV and a current density of 10 mA cm−2, which is more favorable than that of 20 wt% Pt/C (46 mV). For large current density, Ru–O/C-600 delivered 500 and 1000 mA cm−2 with the overpotential of 242 and 383 mV, respectively. Researched via structural characterization and chemical state analysis, Ru nanoparticles were anchored on carbon substrates with Ru–O bonds through the strong electron absorption of the surface hydroxyl group on C60(OH)n, and the ultrafine and small particle size of Ru nanoparticles has a significant positive effect on enhancing the HER activity of the material. Our study provides a possible way to explore the catalytic activity of Ru by regulating surrounding bonds between nanoparticles and substrates and producing efficient Ru-based HER catalysts.
Experimental
Chemicals and reagents
All reagents were used as received without any further purification. Ruthenium trichloride (RuCl3), Pt/C (20 wt%), Nafion solution (5%) and potassium hydroxide (KOH, ≧95%) were purchased from Sigma-Aldrich. Fullerene (C60) was purchased from Xiamen Funano New Material Technology Co. Ltd. Toluene (C7H8, ≧99.5%), isopropyl alcohol (IPA, (CH3)2CHOH, ≧99.7%), ethanol (EtOH, CH3CH2OH, 99.7%), methanol (MeOH, CH3OH, 99%), tetrabutylammonium hydroxide (TBAH, 50% in water) and hydrogen peroxide (H2O2, 40%) were received from Sinopharm Chemical Reagent Co. Ltd. Deionized water (DW, 18.25 MΩ cm−1) was obtained from the ultra-pure purification system (ULUPURE, UPDR-I-10T).The preparation of C60(OH)n
In the preparation process of C60(OH)n,29 first, 200 mg of C60 was dissolved in a 250 mL flask containing 150 mL toluene, then 5 mL of 50% TBAH and 10 mL of H2O2 were added to the toluene solution. After stirring at 80 °C for 5 days, the upper toluene layer turned into a colourless transparent solution and the lower water layer turned into a bright yellow solution. The yellow and turbid aqueous solution at the bottom of the flask was separated using a separation funnel and condensed by evaporation. The obtained yellow-brown solid was freeze-dried and dehydrated under vacuum to give a bright yellow sample of C60(OH)n powder.30The preparation of Ru–O/C-600, Ru–O/C-500 and Ru–O/C-700
Ru–O/C-600 HER electrocatalyst was prepared by condensation reflux and solid-phase sintering methods. Firstly, 400 mg of C60(OH)n and 40 mg RuCl3 were completely dissolved in 200 mL of deionized water, and then the mixture was placed in a round-bottom flask at 120 °C for condensation and reflux for 6 h. After cooling to room temperature, the precursor of Ru–O/C-600 was obtained by centrifugal extraction and filtration. The grey-black powder obtained was placed in a corundum porcelain boat under the protection of Ar/H2 (5%) mixture in a tubular furnace, heated from room temperature to 600 °C at a heating rate of 5 °C min−1, and kept for 3 h. After cooling to room temperature, the black powder was collected and ground to obtain the target product Ru–O/C-600 electrocatalyst.In contrast, Ru–O/C-500 and Ru–O/C-700 electrocatalysts were synthesized by changing the temperature of the tube furnace to 500 °C and 700 °C under the same experimental conditions; Ru/C-600 without the Ru–O bond was synthesized by using C60 instead of C60(OH)n to clarify the effect of oxygen on the Ru species under the same experimental conditions.
Structural characterization
X-ray diffraction (XRD) patterns of the electrocatalysts were obtained on a Rigaku D/max-2200PC diffractometer (Japan) using Cu Kα radiation (λ = 1.5406 Å). High-resolution Transmission Electron Microscope (HRTEM) images and EDS mapping images were recorded using a JEOL JEM-2010 field-emission transmission electron microscope with an accelerating voltage of 200 kV. The chemical bonding states and compositions of the samples were processed by Fourier transform infrared spectroscopy (FT-IR) in the range of 4000 to 400 cm−1 on a Bruker vector-80 installation. Raman spectra were collected on a Renishaw-inVia Microscopic confocal laser Raman spectrometer with 532 nm as the excitation laser. The pyrolysis process of the precursors was characterized by thermogravimetry and differential thermal analysis (TG/DTA) using Universal V4.5A TA Instruments from room temperature to 800 °C in a N2 atmosphere with a heating rate of 5 °C min−1. Deionized water (DW, 18.25 MΩ cm−1) was obtained from the ultra-pure purification system (ULUPURE, UPDR-I-10T).Electrochemical measurements
The HER tests were performed on the electrochemical workstation (CHI660E, Chenhua, Shanghai) using a three-electrode system in N2-saturated 1 M KOH. A glassy carbon electrode (GCE, 0.0706 cm2) was used as the working electrode. The mass loading of each sample on the electrode was calculated to be about 0.7 mg cm−2. For the large-scale current measurement, the catalyst ink was drop-casted on the surface of carbon fiber paper (0.3 × 0.3 mm, 0.09 mm2) with a loading amount of 0.56 mg cm−2. The graphite rod was used as a counter electrode and a saturated Hg/HgO electrode as a reference electrode. All potentials collected in this work were calibrated against the reversible hydrogen electrode (RHE), with Pt foil as the working electrode and Pt wire as the counter electrode. The potentials were obtained via the equation E(RHE) = E(Hg/HgO) + 0.932 (in 1 M KOH).
The electrochemical impedance spectroscopy (EIS) measurement was performed within the frequency range from 100 kHz to 0.1 Hz at a potential corresponding to the current density of 10 mA cm−2. The cyclic voltammogram (CV) curves were obtained in 1 M KOH for the HER (0.2 to 0.3 V vs. RHE) and OER (0.95 to 1.05 V vs. RHE) in the non-faradaic region with scanning rates of 2, 4, 6, 8, 10 and 12 mV s−1. Double layer capacity (Cdl) was obtained by plotting the current difference of the CV curves. The electrochemical active surface area (ECSA) was determined using the following equation:
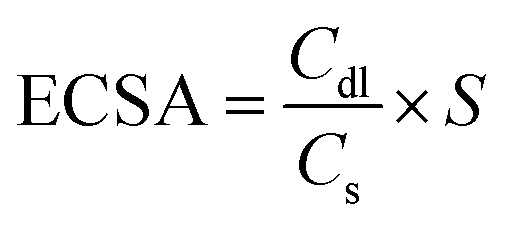 | (1) |
Results and discussion
Structure characterizations and chemical properties of Ru–O/C-600
The Ru–O/C-600 catalyst was well prepared via a simple reflux condensation treatment followed by pyrolysis under an Ar/H2 atmosphere as schematically illustrated in Fig. 1. During the reflux condensation step, the Ru3+ tended to coordinate with C60(OH)n. After that, C60(OH)n was then decomposed into bowl-like fragments according to the thermogravimetric analysis (TGA) (Fig. S1†) under pyrolysis, and the coordinated Ru3+ were simultaneously reduced to Ru nanoparticles. Synchronously, the decomposed bowl-like fragments from C60(OH)n were transferred to the O-doped carbon matrix.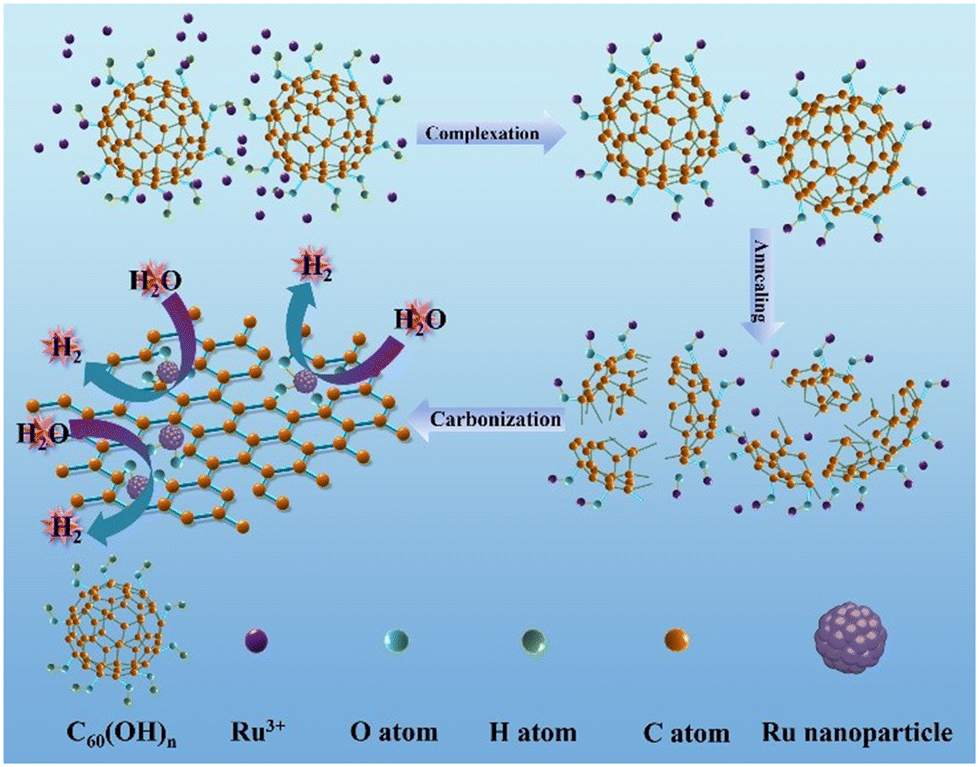 | ||
| Fig. 1 Schematic of the synthetic procedure of the Ru–O/C-600 sample. | ||
Fig. 2a shows the XRD patterns of Ru–O/C-600, Ru–O/C-500 and Ru–O/C-700 electrocatalysts. The XRD patterns of the three samples were very similar, with three strong peaks at 38.4°, 42.2° and 44.0°, corresponding to (100), (002) and (101) lattices of Ru (JCPDS No: 06-0663), and two weak peaks located at 58.3° and 69.4° belonging to the (102) and (110) facets of Ru. It is worth noting that at low diffraction angles (15°–30°), the XRD curves of Ru–O/C-600 and Ru–O/C-700 samples exhibited broadening peaks with weak intensity, which could be identified as the graphitized carbon substrate transformed from C60(OH)n after pyrolysis. However, the XRD curve of Ru–O/C-500 samples did not have such a wide peak, which can be attributed to incomplete graphitization at low temperature, 500 °C. In Fig. S2,† the XRD pattern of the Ru/C-600 sample displayed characteristic peaks of C60 and Ru species, proving that C60 existed in Ru/C-600 rather than decomposition at 600 °C. In the Fourier transform infrared spectroscopy (FT-IR) of the three samples (Fig. 2b), the –C–O/–C–C, –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, –CO and –C–H signals, located at around 1200–1350, 1579, 1695–1843 and 3400 cm−1, proved that the carbon substrate, which was transformed by C60(OH)n, existed in all three samples. Additionally, in Fig. 2c, for Ru–O/C-600, Ru–O/C-500 and Ru–O/C-700 electrocatalysts, it was obvious that the D and G peaks of carbon were located at 1350 and 1597 cm−1, respectively. The ID/IG intensity ratios of Ru–O/C-600, Ru–O/C-500 and Ru–O/C-700 samples were 0.82, 0.98 and 0.78 (Table S1†), respectively. According to previous studies, the lower ID/IG value revealed a higher graphitic degree and outstanding conductivity,31 and the higher ID/IG value indicated more defects in the carbon matrix,32 which demonstrated that plentiful O atoms or metal atoms (Ru) were doped into the carbon framework. Therefore, the Ru–O/C-600 electrocatalyst, with a suitable ID/IG value, provided more active sites and accelerated electron transportation to promote its electrocatalytic activity.
C, –CO and –C–H signals, located at around 1200–1350, 1579, 1695–1843 and 3400 cm−1, proved that the carbon substrate, which was transformed by C60(OH)n, existed in all three samples. Additionally, in Fig. 2c, for Ru–O/C-600, Ru–O/C-500 and Ru–O/C-700 electrocatalysts, it was obvious that the D and G peaks of carbon were located at 1350 and 1597 cm−1, respectively. The ID/IG intensity ratios of Ru–O/C-600, Ru–O/C-500 and Ru–O/C-700 samples were 0.82, 0.98 and 0.78 (Table S1†), respectively. According to previous studies, the lower ID/IG value revealed a higher graphitic degree and outstanding conductivity,31 and the higher ID/IG value indicated more defects in the carbon matrix,32 which demonstrated that plentiful O atoms or metal atoms (Ru) were doped into the carbon framework. Therefore, the Ru–O/C-600 electrocatalyst, with a suitable ID/IG value, provided more active sites and accelerated electron transportation to promote its electrocatalytic activity.
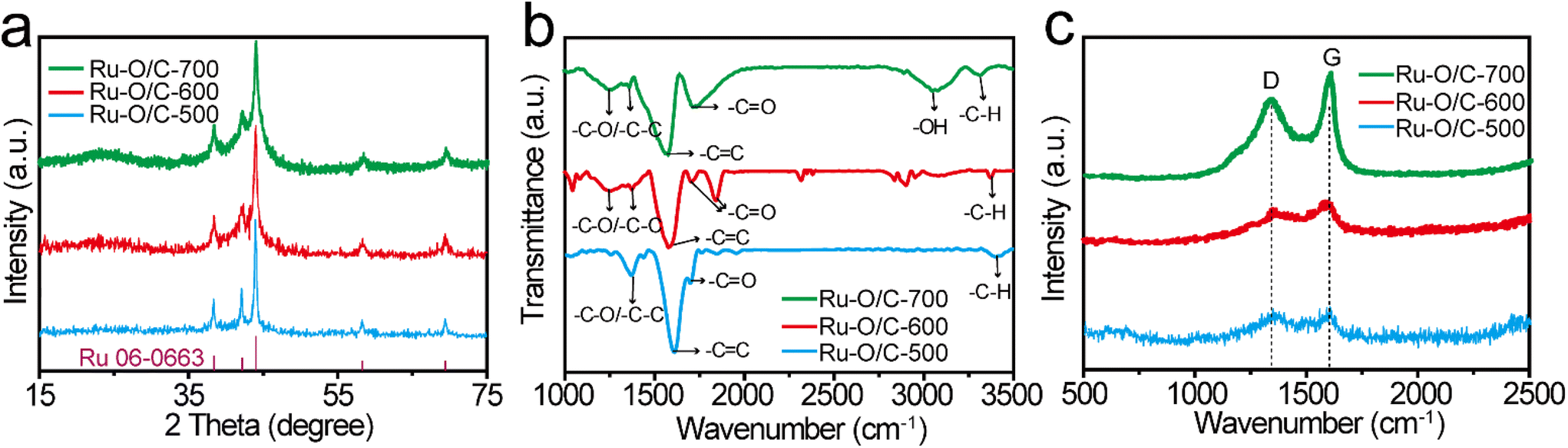 | ||
| Fig. 2 Structure characterization of Ru–O/C-600, Ru–O/C-700 and Ru–O/C-500. (a) XRD patterns, (b) FT-IR spectra and (c) Raman spectra of Ru–O/C-600 (red), Ru–O/C-700 (green) and Ru–O/C-500 (blue). | ||
Fig. 3a shows the HRTEM images of Ru–O/C-600, in which bright yellow areas are marked as Ru nanoparticles with uniform particle sizes. By measuring the lattice fringe, it was confirmed that the characteristic lattice fringe is the (101) crystal plane of the hexagonal crystal phase Ru, and the corresponding crystal plane spacing was 0.214 nm. It was believed that Ru nanoparticles were evenly distributed on the carbon matrix. In addition, the corresponding selected electron diffraction pattern (Fig. 3b) depicted Ru (002) crystal plane diffraction spots, which further confirmed the crystal structure of Ru nanoparticles. Moreover, scanning electron microscopy (SEM) images of Ru–O/C-600 (Fig. 3c) illustrated that Ru nanoparticles were modified on the carbon matrix. HAADF-TEM images and element mapping (Fig. 3d) clearly showed that Ru in the Ru–O/C-600 particles was uniformly distributed on the O-doped carbon substrate, consistent with the previous XRD, FT-IR and Raman results. Fig. 3e, f and g revealed the particle size of Ru nanoparticles in three different samples. When the pyrolysis temperature was 500 °C, the particle size of Ru nanoparticles in Ru–O/C-500 samples was small and densely distributed on the carbon substrate in Fig. 3e, and most Ru nanoparticles ranged in size from 2.0 nm to 3.5 nm. Fig. 3e and S3a† also show that the crystallinity of Ru nanoparticles in the Ru–O/C-500 sample was very poor, and the lattice fringe could not be observed. When the pyrolysis temperature rose to 600 °C, the sizes of most Ru nanocrystalline particles were still between 2.0 nm and 3.5 nm in Fig. 3f. It was confirmed that Ru nanoparticles in the Ru–O/C-600 sample maintained the characteristics of small particle size and dense distribution on the carbon substrate while improving the crystallinity and exposing the crystal planes. Lattice stripes were visible. When the pyrolysis temperature rose to 700 °C, Ru nanoparticles in Ru–O/C-700 samples showed an aggravated agglomeration phenomenon, and the particle size growth became large and uneven in Fig. 3g. Fig. S3b† depicted that Ru nanoparticles in the Ru–O/C-700 sample had good crystallinity, and Ru (101) crystal planes were exposed with a spacing of 0.205 nm. Therefore, it could be inferred that the optimal temperature for the growth of Ru nanoparticles in the three samples was 600 °C. At this temperature, the crystallinity of Ru nanoparticles was improved, while the particle size was small and dense on the carbon layer. In Fig. S4,† the TEM image showed the lattices of Ru (100) and C60 (420) in the Ru/C-600 sample. The Ru and C60 nanoparticles were in different sizes. The ICP-AES analysis of Ru–O/C-600, Ru–O/C-500 and Ru–O/C-700 showed that the content of Ru in Ru–O/C-600 (30.81%) was higher than that in Ru–O/C-500 and Ru–O/C-700 (25.33% and 28.79%), and the high metal content was conducive to the enhancement of the HER activity.
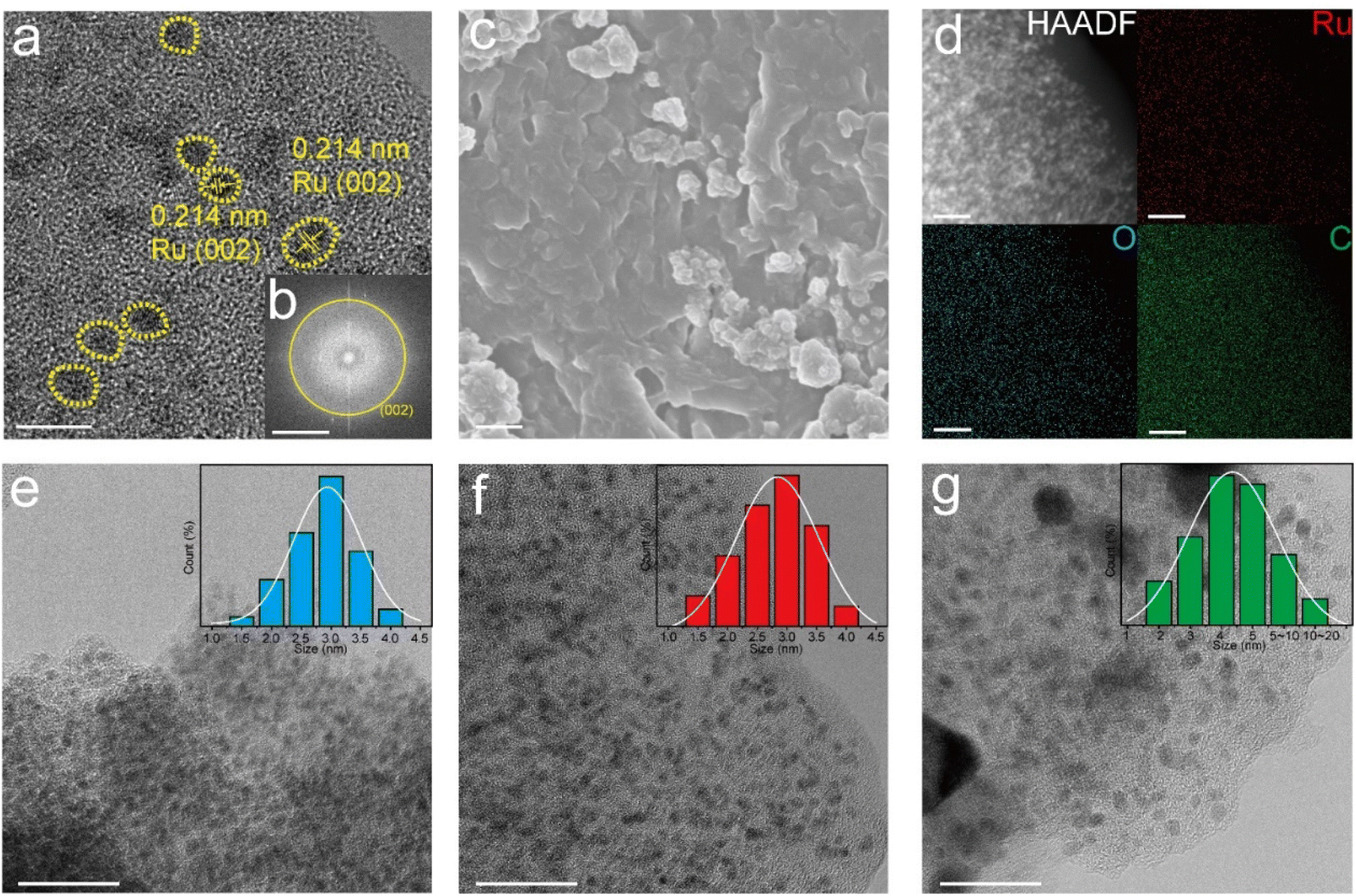 | ||
| Fig. 3 (a) HRTEM, (b) SAED, (c) SEM and (d) HAADF-TEM images and the corresponding elemental mapping of Ru, C and O for Ru–O/C-600. HRTEM images of (e) Ru–O/C-500, (f) Ru–O/C-600 and (g) Ru–O/C-700. Scale bar: (a–g) 5 nm, 21/nm, 1 μm, 20 nm, 20 nm, 20 nm, and 20 nm, respectively. | ||
The XPS results further evaluated the elemental valence states and electronic structures of Ru–O/C-500, Ru–O/C-600 and Ru–O/C-700 samples. As shown in Fig. S5,† Ru–O/C-500, Ru–O/C-600 and Ru–O/C-700 samples contained Ru, C and O elements, which were consistent with TEM mapping results. According to Fig. S6,† the peaks, appearing in the C 1s spectrum of Ru–O/C-600 samples, could be attributed to C–C/CC (284.6 eV), C–O (285.7 eV) and CO (287.5 eV) species,33,34 respectively, which demonstrated that O atoms were anchored to the carbon layer. The three C species could also be found in Ru–O/C-500 and Ru–O/C-700 samples in corresponding locations. Moreover, in the Ru 3d spectrum, the peaks of Ru0 were located at 280.4 eV and 284.5 eV,35 and the peaks of Ru4 were located at 281.1 eV and 285.5 eV. In the Ru 3p spectrum, due to the different orientations of electrons in the electron spin–orbital interactions, the Ru 3p orbital was split into two orbitals Ru 3p3/2 and Ru 3p1/2. The peaks at 462.7 eV and 484.9 eV of Ru–O/C-500 could be attributed to Ru0 species (Fig. 4a), and peaks at 466.7 eV and 487.1 eV could be attributed to Ru4+ species with surface oxidation.36
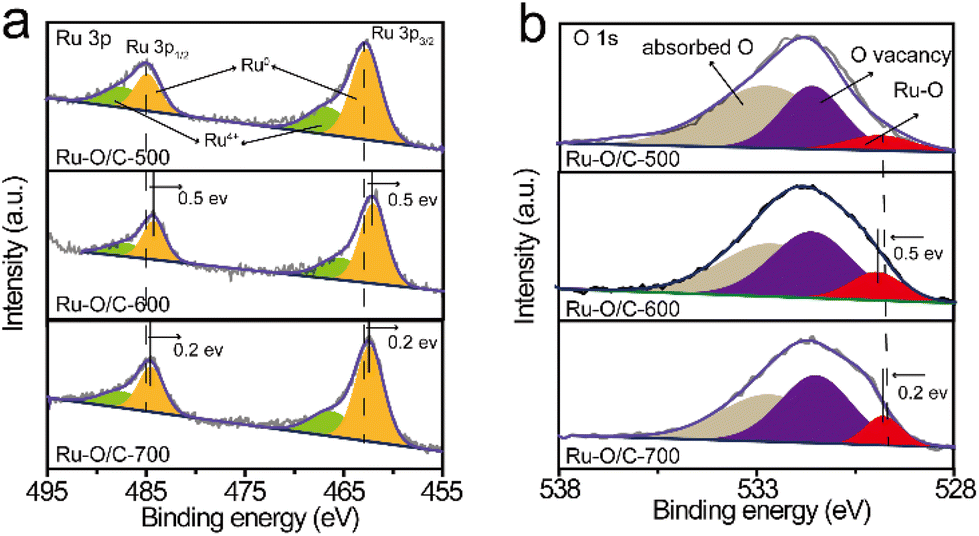 | ||
| Fig. 4 High-resolution XPS spectra for (a) Ru 3p and (b) O 1s of Ru–O/C-500, Ru–O/C-600 and Ru–O/C-700. | ||
Compared with the Ru–O/C-500 sample, the peak of the Ru0 species in Ru–O/C-600 and Ru–O/C-700 samples appeared at 462.2 eV and 484.4 eV, and shifted by 0.5 eV and 0.2 eV to the direction of low binding energy, respectively. It demonstrated that Ru0 in Ru–O/C-600 and Ru–O/C-700 samples were in the state of gaining electrons, becoming electron acceptors. Similarly, surface oxidized Ru4+ species also appeared in the Ru–O/C-600 and Ru–O/C-700 samples. Fig. 4b shows the valence analysis of element O in the three samples. In the O 1s spectrum of Ru–O/C-500, peaks at 529.9 eV, 531.7 eV and 532.6 eV could be attributed to the metal–oxygen bond (Ru–O), O vacancy and absorbed hydroxyl or H2O, respectively.4,37 It was supposed that the entry of O into the Ru lattice during the pyrolysis of C60(OH)n caused the formation of the oxygen coordination vacancy. In contrast, these three O elements were also presented in Ru–O/C-600 and Ru–O/C-700 samples. Among them, the Ru–O bond migrated with 0.5 eV and 0.2 eV in the direction of high binding energy, which gave solid evidence that O in Ru–O/C-600 and Ru–O/C-700, as the electron donor, lost part of the charges. Notably, the larger shift of the Ru–O binding energy in Ru–O/C-600 as compared with Ru–O/C-700 could be attributed to the facile charge communication between Ru and O in the former, while for the latter the destruction of the Ru–O bond at 700 °C led to the decrease in such electronic communication. In this case, Ru–O bonds could create the electron pathway to accelerate electron transfer between Ru and O, which facilitated the electrocatalytic HER performance. Thus, the strong electron communication between Ru and O in the Ru–O/C-600 sample probably represented more outstanding HER performance.
HER performance of Ru–O/C-600
To prove the electrocatalytic performance of the synthesized Ru–O/C-600 catalyst, a standard three-electrode system was used to test the HER in a 1 M KOH aqueous solution. A Hg/HgO electrode was used as the reference electrode, a carbon rod was used as the counter electrode, and a glassy carbon electrode was used as the working electrode. For accuracy, calibration with the RHE reference to the Hg/HgO reference electrode in 1 M KOH media was done prior to all tests38 (Fig. S7†).For comparison, Ru–O/C-500, Ru–O/C-700 and commercial 20% Pt/C were tested as references. All test data were collected after the electrode cycle until a stable performance was obtained. Fig. 5a was the linear sweep voltammetry curve (LSV) at the scanning rate of 1 mV s−1. It could be seen that the overpotential (η10) of Ru–O/C-600 was only 32 mV at the current density of 10 mA cm−2 (the corresponding efficiency of solar energy to hydrogen was 12.3%), which was far lower than the other comparison samples Ru–O/C-500 (190 mV), Ru–O/C-700 (75 mV) and commercial 20% Pt/C (46 mV). Besides, the LSV curves normalized by Ru content (Fig. S8†) demonstrated that Ru–O/C-600 outperformed Ru–O/C-500 and Ru–O/C-700. Furthermore, compared with Ru–O/C-600, the η10 for the sample of Ru/C-600 without a Ru–O bond increased to 124 mV (Fig. S9†), much higher than that of Ru–O/C-600 (32 mV), demonstrating the positive regulatory effect of the O atom on Ru species for improving the HER performance. As shown in Fig. 5b, the Tafel slopes of Ru–O/C-600, Ru–O/C-500, Ru–O/C-700 and commercial 20% Pt/C were 51.8, 236.9, 99.8 and 61.2 mV dec−1, respectively. Among them, the Tafel slope of Ru–O/C-600 was the smallest, according to the Volmer–Heyrovsky process, which indicated the strongest inherent activity and the fastest reaction kinetics of the electrocatalytic HER.39 The bar chart in Fig. 5c shows that the Ru–O/C-600 sample had the lowest overpotential Tafel slope compared with other samples, which advocated the remarkable electrocatalytic hydrogen evolution activity. To further examine the electrochemical performance of the Ru–O/C-600 sample, the specific surface area of its electrochemical activity was measured by the double-layer capacitance method (Cdl) in Fig. 5d and Fig. S10.† CV testing is an effective method for the determination of the Cdl of the catalyst.40 The Cdl value of Ru–O/C-600 was 117.5 mF cm−2. Compared with Ru–O/C-500 (1.5 mF cm−2), Ru–O/C-700 (10.8 mF cm−2) and commercial 20% Pt/C (26.1 mF cm−2), Ru–O/C-600 had the largest double capacitance value, which probably benefited from the ultrafine size, homogeneous distribution and advantageous crystallinity of Ru nanoparticles. The HER dynamics and charge transfer between the electrode and electrolyte interface were analysed by the EIS method.41,42The charge transfer resistance (Rct) is related to the interface charge transfer process of the electrode. In general, the lower the Rct, the faster the hydrogen production. The Nyquist diagram of the Ru–O/C-600 sample (Fig. 5e) showed a smaller semicircle diameter. Compared with Ru–O/C-500 (81.5 Ω), Ru–O/C-700 (40.5 Ω) and 20% Pt/C (29.8 Ω), the sample had an excellent Rct of 12.8 Ω (Table S2†). A dominant index of the electrocatalyst was its catalytic stability, which meant long catalytic times. The Ru–O/C-600 electrocatalyst showed satisfactory durability as measured in long-term chronograph amperes (Fig. 5f).43,44 It could be seen that the activity of the Ru–O/C-600 electrocatalyst remained stable for at least 50 h with almost no decay. On the other hand, after 2000 cycles of CV, the HER polarization curve only showed slight attenuation. Finally, the overpotential of Ru–O/C-600 at 10 mA cm−2 current density was compared with other recently reported Ru-based HER electrocatalysts in alkaline media (Fig. 5g). The results illustrated that the Ru–O/C-600 electrocatalyst was superior to most Ru-based HER electrocatalysts recently reported (Table S3†). In conclusion, compared with the single-phase Ru–O/C-500, Ru–O/C-700 and Ru/C-600, Ru–O/C-600 with small particle size, uniform distribution, obvious crystallinity and electron communication between Ru and O of Ru nanoparticles had distinguished HER performance.
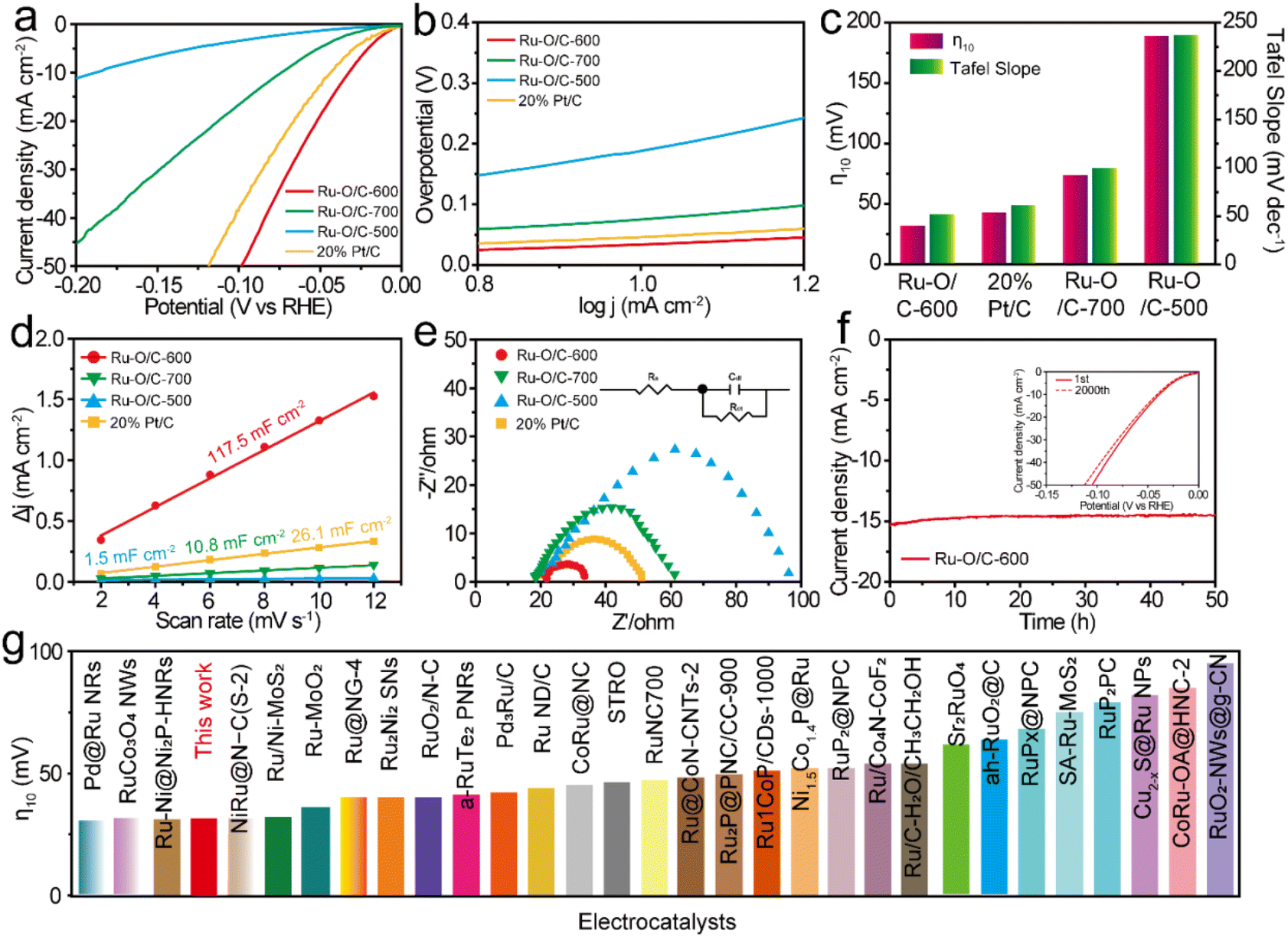 | ||
| Fig. 5 HER performance tests in 1 M KOH: (a) LSV curves; (b) Tafel slopes; (c) comparison of η10 (red column) and Tafel slope (green column); (d) the corresponding plots of current density difference against scan rates; (e) Nyquist plots during the HER test for (a) 20% Pt/C, (b) Ru–O/C-500, (c) Ru–O/C-700 and (d) Ru–O/C-600. (f) Stability test of Ru–O/C-600 (inset: LSV curves before and after 2000 CV cycles). (g) Comparison of the overpotential at 10 mA cm−2 (η10) in 1 M KOH for Ru–O/C-600 with recently-reported Ru-based HER electrocatalysts. | ||
The XPS results showed the chemical states in the Ru–O/C-600 catalyst after long-time cyclic (2000 cycles CV) stability testing (Fig. S11†), illustrating that the electron transfer environment between Ru and O was not damaged, which further confirmed the excellent stability of Ru–O/C-600 (Fig. 6a and b). The TEM images and elemental mapping of the samples confirmed that the morphology of the Ru–O/C-600 electrocatalyst remained intact and the particle size was uniform after 50 h, almost the same as before (Fig. 6c and Fig. S12†).
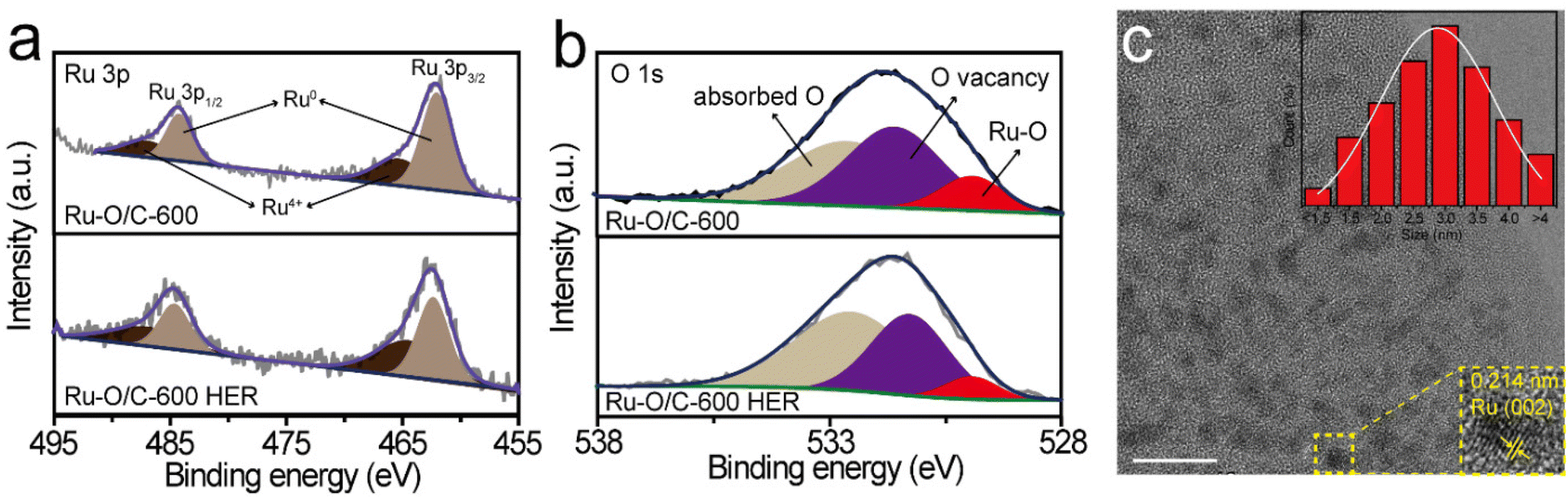 | ||
| Fig. 6 The Ru–O/C-600 results after long-time stability testing. High-resolution XPS spectra of (a) Ru 3p and (b) O 1s. (c) TEM image. Scale bar: 10 nm. | ||
For practical purposes, the HER performance under industrial-level current output at 1000 mA cm−2 was investigated by coating the catalyst powder onto the surface of carbon fiber paper (CFP). As shown in Fig. 7a, Ru–O/C-600 displayed excellent HER activity in the range of 1000 mA cm−2 in alkaline conditions. Specifically, it could reach current densities of 500 and 1000 mA cm−2 with overpotentials of 242 and 383 mV, respectively, much lower than those (358 and 599 mV) of 20% Pt/C (Fig. 7b). Furthermore, compared with Ru–O/C-600, the samples of Ru–O/C-500, Ru–O/C-700 and Ru/C-600 reached 891, 902 and 443 mA cm−2 with an overpotential of 600 mV, which showed much worse HER performance under a relatively large-scale current output. Moreover, after continuous i–t operating for 40 h, the LSV curves of Ru–O/C-600 in alkaline conditions showed 257 and 395 mV, respectively, at the current densities of 500 and 1000 mA cm−2 with negligible decay (dashed lines in Fig. 7a). Chronoamperometric measurement proved that the electrocatalytic activity of Ru–O/C-600 could be maintained for at least for 40 h under the current density of 1000 mA cm−2 in 1 M KOH (Fig. 7c). Overall, considering the high activity and favorable durability, Ru–O/C-600 would be a convincing alternative HER catalyst to Pt/C for practical hydrogen production.
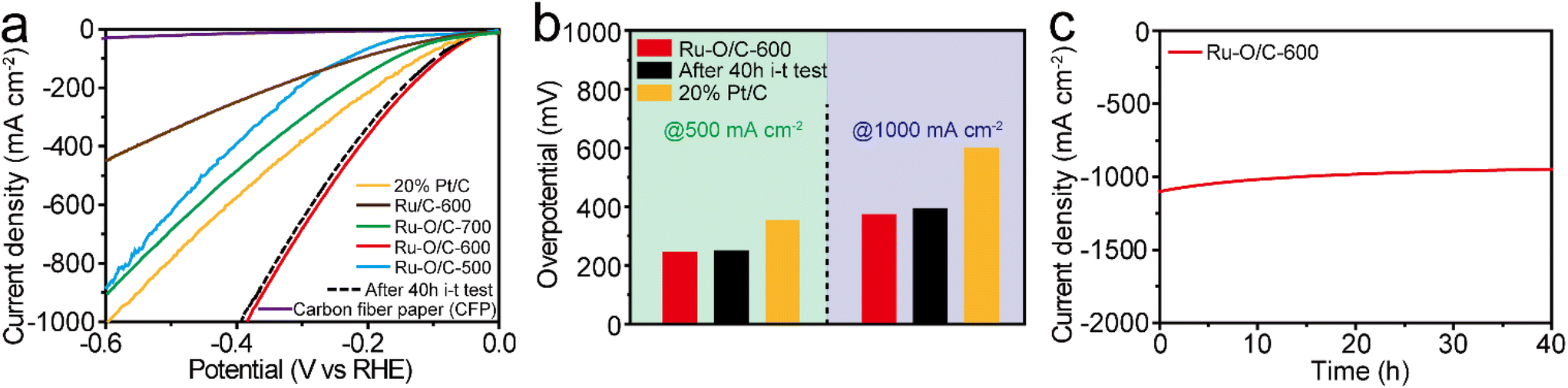 | ||
| Fig. 7 HER performance under large-current conditions. (a) LSV curves of Ru–O/C-600, 20% Pt/C and carbon fiber paper (CFP) in 1 M KOH; the dashed lines in (a) refer to the LSV curves of Ru–O/C-600 CFP after the 40 h i–t test. (b) Comparison of the overpotentials at the current densities of 500 and 1000 mA cm−2 for Ru–O/C-600 and 20% Pt/C, both in 1 M KOH. (c) Long-term chronoamperometric testing of Ru–O/C-600 in 1 M KOH at a current density of 1000 mA cm−2. | ||
Conclusions
In summary, we first synthesized C60(OH)n yellow powder by a liquid–liquid interface precipitation method, and prepared the Ru3+ and C60(OH)n precursor by a condensation reflux method. After high-temperature pyrolysis, the Ru–O/C-600 electrocatalyst was obtained. Ru–O/C-600 exhibited outstanding HER activity in alkaline medium. Ru–O/C-600 required only 32 mV overpotential in 1 M KOH to achieve a current density of 10 mA cm−2, which exceeded the widely used commercial 20% Pt/C. Furthermore, the Ru–O/C-600 catalyst can provide 500 and 1000 mA cm−2 with an overpotential of 242 and 383 mV. After characterization of the internal structure and chemical composition, the Ru–O/C-600 electrocatalyst had a high HER activity due to the following reasons: (1) in Ru–O/C-600, evenly distributed Ru nanoparticles have a small particle size (2.0–3.5 nm) with good crystallinity, exposing the active catalytic crystal surface to increase in the specific surface area of the electrochemical activity. The reaction kinetics between the electrocatalyst and electrolyte were accelerated. (2) The charge transfer between Ru and O regulated the electronic structure of the electrocatalyst, promoted the electron transfer rate in Ru–O/C-600, reduced the charge transfer resistance, and enhanced the intrinsic catalytic activity of the material. Therefore, the feasible work of Ru–O/C-600 provided a reliable and novel direction for designing and controlling the electronic structure of nanoparticle electrocatalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (52072226, 52073166), Scientific Research Program Funded by Shaanxi Provincial Education Department (No. 20JY001), the Xi'an Key Laboratory of Green Manufacture of Ceramic Materials Foundation (No. 2019220214SYS017CG039), the Key Program for International S&T Cooperation Projects of Shaanxi Province (2020KW-0382020GHJD-04), Science and Technology Program of Xi'an, China (2020KJRC0009) and Science and Technology Resource Sharing Platform of Shaanxi Province (2020PT-022). Dr Y. Q. Feng was grateful for the support from the Science and Technology Youth Stars Project of Shaanxi Province (2021KJXX-35).Notes and references
- Y. Feng, R. Wang, P. Dong, X. Wang, W. Feng, J. Chen, L. Cao, L. Feng, C. He and J. Huang, Enhanced electrocatalytic activity of nickel cobalt phosphide nanoparticles anchored on porous N-doped fullerene nanorod for efficient overall water splitting, ACS Appl. Mater. Interfaces, 2021, 13, 48949–48961 CrossRef CAS PubMed.
- J. Mahmood, F. Li, S. M. Jung, M. S. Okyay, I. Ahmad, S. J. Kim, N. Park, H. Y. Jeong and J. B. Baek, An efficient and pH-universal ruthenium-based catalyst for the hydrogen evolution reaction, Nat. Nanotechnol., 2017, 12, 441–446 CrossRef CAS PubMed.
- S. Anantharaj, P. E. Karthik, B. Subramanian and S. Kundu, Pt Nanoparticle anchored molecular self-assemblies of DNA: An extremely stable and efficient HER electrocatalyst with ultralow Pt content, ACS Catal., 2016, 6, 4660–4672 CrossRef CAS.
- P. Zhai, M. Xia, Y. Wu, G. Zhang, J. Gao, B. Zhang, S. Cao, Y. Zhang, Z. Li, Z. Fan, C. Wang, X. Zhang, J. T. Miller, L. Sun and J. Hou, Engineering single-atomic ruthenium catalytic sites on defective nickel-iron layered double hydroxide for overall water splitting, Nat. Commun., 2021, 12, 4587 CrossRef CAS PubMed.
- Y. Feng, P. Dong, L. Cao, X. Wang, J. Wang, H. Wang, W. Feng, J. Chen, L. Feng, C. He and J. Huang, Defect-rich bimetallic yolk–shell metal-cyanide frameworks as efficient electrocatalysts for oxygen evolution reactions, J. Mater. Chem. A, 2021, 9, 2135–2144 RSC.
- X. Wu, Z. Wang, D. Zhang, Y. Qin, M. Wang, Y. Han, T. Zhan, B. Yang, S. Li, J. Lai and L. Wang, Solvent-free microwave synthesis of ultra-small Ru-Mo2C@CNT with strong metal-support interaction for industrial hydrogen evolution, Nat. Commun., 2021, 12, 4018 CrossRef CAS PubMed.
- Y. Chen, J. Yu, J. Jia, F. Liu, Y. Zhang, G. Xiong, R. Zhang, R. Yang, D. Sun, H. Liu and W. Zhou, Metallic Ni3Mo3N porous microrods with abundant catalytic sites as efficient electrocatalyst for large current density and superstability of hydrogen evolution reaction and water splitting, Appl. Catal., B, 2020, 272, 118956 CrossRef CAS.
- J. Zhang, X. Shang, H. Ren, J. Chi, H. Fu, B. Dong, C. Liu and Y. Chai, Modulation of inverse spinel Fe3O4 by phosphorus doping as an industrially promising electrocatalyst for hydrogen evolution, Adv. Mater., 2019, 31, 1905107 CrossRef CAS PubMed.
- D. Zhao, K. Sun, W. C. Cheong, L. Zheng, C. Zhang, S. Liu, X. Cao, K. Wu, Y. Pan, Z. Zhuang, B. Hu, D. Wang, Q. Peng, C. Chen and Y. Li, Synergistically interactive pyridinic-N-MoP sites: identified active centers for enhanced hydrogen evolution in alkaline solution, Angew. Chem., Int. Ed., 2020, 59, 8982–8990 CrossRef CAS PubMed.
- S. H. Yu, P. K. Gogoi, A. Rath, H. Dai, Z. Q. Cavin Ng, K. Suenaga, S. J. Pennycook and D. H. C. Chua, In situ derived highly active NiS2 and MoS2 nanosheets on NiMoO4 microcuboids via controlled surface sulfidation for high-current-density hydrogen evolution reaction, Electrochim. Acta, 2021, 389, 138733 CrossRef CAS.
- H. Sun, X. Xu, Z. Yan, X. Chen, L. Jiao, F. Cheng and J. Chen, Superhydrophilic amorphous Co–B–P nanosheet electrocatalysts with Pt-like activity and durability for the hydrogen evolution reaction, J. Mater. Chem. A, 2018, 6, 22062–22069 RSC.
- F. Shen, Y. Wang, G. Qian, W. Chen, W. Jiang, L. Luo and S. Yin, Bimetallic iron-iridium alloy nanoparticles supported on nickel foam as highly efficient and stable catalyst for overall water splitting at large current density, Appl. Catal., B, 2020, 278, 119327 CrossRef CAS.
- B. Lu, L. Guo, F. Wu, Y. Peng, J. E. Lu, T. J. Smart, N. Wang, Y. Z. Finfrock, D. Morris, P. Zhang, N. Li, P. Gao, Y. Ping and S. Chen, Ruthenium atomically dispersed in carbon outperforms platinum toward hydrogen evolution in alkaline media, Nat. Commun., 2019, 10, 631 CrossRef CAS PubMed.
- Y. Jia, T. H. Huang, S. Lin, L. Guo, Y. M. Yu, J. H. Wang, K. W. Wang and S. Dai, Stable Pd-Cu Hydride catalyst for efficient hydrogen evolution, Nano Lett., 2022, 22, 1391–1397 CrossRef CAS PubMed.
- L. Deng, F. Hu, M. Ma, S. C. Huang, Y. Xiong, H. Y. Chen, L. Li and S. Peng, Electronic modulation caused by interfacial Ni-O-M (M=Ru, Ir, Pd) bonding for accelerating hydrogen evolution kinetics, Angew. Chem., Int. Ed., 2021, 60, 22276–22282 CrossRef CAS PubMed.
- Q. Wu, M. Luo, J. Han, W. Peng, Y. Zhao, D. Chen, M. Peng, J. Liu, F. M. F. de Groot and Y. Tan, Identifying electrocatalytic sites of the nanoporous copper–ruthenium alloy for hydrogen evolution reaction in alkaline electrolyte, ACS Energy Lett., 2019, 5, 192–199 CrossRef.
- W. Feng, Y. Feng, J. Chen, H. Wang, Y. Hu, T. Luo, C. Yuan, L. Cao, L. Feng and J. Huang, Interfacial electronic engineering of Ru/FeRu nanoparticles as efficient trifunctional electrocatalyst for overall water splitting and Zn-air battery, Chem. Eng. J., 2022, 437, 135456 CrossRef CAS.
- X. Chen, J. Wan, J. Wang, Q. Zhang, L. Gu, L. Zheng, N. Wang and R. Yu, Atomically Dispersed Ruthenium on Nickel Hydroxide Ultrathin Nanoribbons for Highly Efficient Hydrogen Evolution Reaction in Alkaline Media, Adv. Mater., 2021, 33, e2104764 CrossRef PubMed.
- Y. Feng, W. Feng, J. Wan, J. Chen, H. Wang, S. Li, T. Luo, Y. Hu, C. Yuan, L. Cao, L. Feng, J. Li, R. Wen and J. Huang, Spherical vs. planar: Steering the electronic communication between Ru nanoparticle and single atom to boost the electrocatalytic hydrogen evolution activity both in acid and alkaline, Appl. Catal., B, 2022, 307, 121193 CrossRef CAS.
- J. A. Trindell, Z. Duan, G. Henkelman and R. M. Crooks, Well-Defined Nanoparticle Electrocatalysts for the Refinement of Theory, Chem. Rev., 2020, 120, 814–850 CrossRef CAS PubMed.
- J. Kim, H. J. Kim, B. Ruqia, M. J. Kim, Y. J. Jang, T. H. Jo, H. Baik, H. S. Oh, H. S. Chung, K. Baek, S. Noh, M. Jung, K. J. Kim, H. K. Lim, Y. S. Youn and S. I. Choi, Crystal phase transition creates a highly active and stable RuCx nanosurface for hydrogen evolution reaction in alkaline media, Adv. Mater., 2021, 33, 2105248 CrossRef CAS PubMed.
- S. W. Sun, G. F. Wang, Y. Zhou, F. B. Wang and X. H. Xia, High-performance Ru@C4N electrocatalyst for hydrogen evolution reaction in both acidic and alkaline solutions, ACS Appl. Mater. Interfaces, 2019, 11, 19176–19182 CrossRef CAS PubMed.
- J. Chen, J. Huang, R. Wang, W. Feng, H. Wang, T. Luo, Y. Hu, C. Yuan, L. Feng, L. Cao, K. Kajiyoshi, C. He, Y. Liu, Z. Li and Y. Feng, Atomic ruthenium coordinated with chlorine and nitrogen as efficient and multifunctional electrocatalyst for overall water splitting and rechargeable zinc-air battery, Chem. Eng. J., 2022, 441, 136078 CrossRef CAS.
- F. Yu, H. Zhou, Y. Huang, J. Sun, F. Qin, J. Bao, W. A. Goddard 3rd, S. Chen and Z. Ren, High-performance bifunctional porous non-noble metal phosphide catalyst for overall water splitting, Nat. Commun., 2018, 9, 2551 CrossRef PubMed.
- C.-F. Li, J.-W. Zhao, L.-J. Xie, Y. Wang, H.-B. Tang, L.-R. Zheng and G.-R. Li, N coupling with S-coordinated Ru nanoclusters for highly efficient hydrogen evolution in alkaline media, J. Mater. Chem. A, 2021, 9, 12659–12669 RSC.
- D. Cao, J. Wang, H. Xu and D. Cheng, Construction of dual-site atomically dispersed electrocatalysts with Ru-C5 single atoms and Ru-O4 nanoclusters for accelerated alkali hydrogen evolution, Small, 2021, 17, 2101163 CrossRef CAS PubMed.
- Z. Liu, L. Zeng, J. Yu, L. Yang, J. Zhang, X. Zhang, F. Han, L. Zhao, X. Li, H. Liu and W. Zhou, Charge redistribution of Ru nanoclusters on Co3O4 porous nanowire via the oxygen regulation for enhanced hydrogen evolution reaction, Nano Energy, 2021, 85, 105940 CrossRef CAS.
- L. Zhang, R. Si, H. Liu, N. Chen, Q. Wang, K. Adair, Z. Wang, J. Chen, Z. Song, J. Li, M. N. Banis, R. Li, T. K. Sham, M. Gu, L. M. Liu, G. A. Botton and X. Sun, Atomic layer deposited Pt-Ru dual-metal dimers and identifying their active sites for hydrogen evolution reaction, Nat. Commun., 2019, 10, 4936 CrossRef PubMed.
- K. Kokubo, S. Shirakawa, N. Kobayashi, H. Aoshima and T. Oshima, Facile and scalable synthesis of a highly hydroxylated water-soluble fullerenol as a single nanoparticle, Nano Res., 2010, 4, 204–215 CrossRef.
- A. Dawid, K. Górny and Z. Gburski, Water solvent effect on infrared and raman spectra of C60(OH)24 fullerenol isomers: DFT Study, J. Phys. Chem. C, 2017, 121, 2303–2315 CrossRef CAS.
- Q. Cheng, C. Hu, G. Wang, Z. Zou, H. Yang and L. Dai, Carbon-defect-driven electroless deposition of Pt atomic clusters for highly efficient hydrogen evolution, J. Am. Chem. Soc., 2020, 142, 5594–5601 CrossRef CAS PubMed.
- Y. Xie, C. Feng, Y. Guo, A. Hassan, S. Li, Y. Zhang and J. Wang, Dimethylimidazole and dicyandiamide assisted synthesized rich-defect and highly dispersed CuCo-Nx anchored hollow graphite carbon nanocages as efficient trifunctional electrocatalyst in the same electrolyte, J. Power Sources, 2022, 517, 230721 CrossRef CAS.
- G. Chen, P. Liu, Z. Liao, F. Sun, Y. He, H. Zhong, T. Zhang, E. Zschech, M. Chen, G. Wu, J. Zhang and X. Feng, Zinc-mediated template synthesis of Fe-N-C electrocatalysts with densely accessible Fe-Nx active sites for efficient oxygen reduction, Adv. Mater., 2020, 32, 1907399 CrossRef CAS PubMed.
- S. Zhang, M. Jin, T. Shi, M. Han, Q. Sun, Y. Lin, Z. Ding, L. R. Zheng, G. Wang, Y. Zhang, H. Zhang and H. Zhao, Electrocatalytically active Fe-(O-C2)4 single-atom sites for efficient reduction of nitrogen to ammonia, Angew. Chem., Int. Ed., 2020, 59, 13423–13429 CrossRef CAS PubMed.
- L. Zhang, H. Jang, Y. Wang, Z. Li, W. Zhang, M. G. Kim, D. Yang, S. Liu, X. Liu and J. Cho, Exploring the dominant role of atomic- and nano-ruthenium as active sites for hydrogen evolution reaction in both acidic and alkaline media, Adv. Sci., 2021, 8, 2004516 CrossRef CAS PubMed.
- H. Song, M. Wu, Z. Tang, J. S. Tse, B. Yang and S. Lu, Single atom ruthenium-doped CoP/CDs nanosheets via splicing of carbon-dots for robust hydrogen production, Angew. Chem., Int. Ed., 2021, 60, 7234–7244 CrossRef CAS PubMed.
- B. H. R. Suryanto, Y. Wang, R. K. Hocking, W. Adamson and C. Zhao, Overall electrochemical splitting of water at the heterogeneous interface of nickel and iron oxide, Nat. Commun., 2019, 10, 5599 CrossRef CAS PubMed.
- C. Wei, R. R. Rao, J. Peng, B. Huang, I. E. L. Stephens, M. Risch, Z. J. Xu and Y. Shao-Horn, Recommended practices and benchmark activity for hydrogen and oxygen electrocatalysis in water splitting and fuel cells, Adv. Mater., 2019, 31, 1806296 CrossRef PubMed.
- J. Wang, F. Xu, H. Jin, Y. Chen and Y. Wang, Non-noble metal-based carbon composites in hydrogen evolution reaction: fundamentals to applications, Adv. Mater., 2017, 29, 1605838 CrossRef PubMed.
- D. Voiry, M. Chhowalla, Y. Gogotsi, N. A. Kotov, Y. Li, R. M. Penner, R. E. Schaak and P. S. Weiss, Best practices for reporting electrocatalytic performance of nanomaterials, ACS Nano, 2018, 12, 9635–9638 CrossRef CAS PubMed.
- J. Ge, J. Zheng, J. Zhang, S. Jiang, L. Zhang, H. Wan, L. Wang, W. Ma, Z. Zhou and R. Ma, Controllable atomic defect engineering in layered NixFe1−x(OH)2 nanosheets for electrochemical overall water splitting, J. Mater. Chem. A, 2021, 9, 14432–14443 RSC.
- L. Wang, L. Zhang, W. Ma, H. Wan, X. Zhang, X. Zhang, S. Jiang, J. Zheng and Z. Zhou, In situ anchoring massive isolated Pt atoms at cationic vacancies of α-NixFe1−x(OH)2 to regulate the electronic structure for overall water splitting, Adv. Funct. Mater., 2022, 2203342 CrossRef.
- H. Li, X. Han, S. Jiang, L. Zhang, W. Ma, R. Ma and Z. Zhou, Controllable fabrication and structure evolution of hierarchical 1T-MoS2 nanospheres for efficient hydrogen evolution, Green Energy Environ., 2022, 7, 314–323 CrossRef.
- W. Ma, H. Wan, Li Zhang, J. Zheng and Z. Zhou, Single-atom catalysts for electrochemical energy storage and conversion, J. Energy Chem., 2021, 63, 170–194 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2qi00847e |
| This journal is © the Partner Organisations 2022 |