
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCore vs. surface labelling of mesoporous silica nanoparticles: advancing the understanding of nanoparticle fate and design of labelling strategies†
María de los Ángeles
Ramírez
ab,
Ángel Manuel
Martínez-Villacorta
ac,
Vanessa
Gómez-Vallejo
c,
Patricia
Andreozzi
a,
Galo
Soler-Illia
b,
Jordi
Llop
 *c and
S. E.
Moya
*a
*c and
S. E.
Moya
*a
aSoft Matter Nanotechnology Group, CIC biomaGUNE, Basque Research and Technology Alliance (BRTA), Paseo Miramón 194, 20014 San Sebastián, Guipúzcoa, Spain. E-mail: smoya@cicbiomagune.es
bInstituto de Nanosistemas, UNSAM, CONICET, Avenida 25 de Mayo 1021, 1650 San Martín, Buenos Aires, Argentina
cRadiochemistry and Nuclear Imaging Group, CIC biomaGUNE, Basque Research and Technology Alliance (BRTA), Paseo Miramón 194, 20014 San Sebastián, Guipúzcoa, Spain
dCentro de Investigación Biomédica en Red – Enfermedades Respiratorias (CIBERES), Av. Monforte de Lemos 3–5, 28029 Madrid, Spain
First published on 1st March 2022
Abstract
Despite great interest in the use of silica mesoporous nanoparticles (MSNs) in drug delivery little is known on their biological fate. Positron emission tomography (PET) studies of radiolabelled MSNs face a major difficulty due to the degradation of the MSNs during circulation as it is difficult to assign activity values to either the MSNs or their degradation products. Here, a PET study is conducted using two strategies of labelling. MSNs are either radiolabelled in the core by complexation with silanols from the MSNs with 89Zr, or on the MSN coating through attachment of 131I radiolabelled Lin-TT1 (AKRGARSTA), a homing peptide for targeting cancer tissue. Results from the biodistribution of MSNs with the two labels are compared, obtaining meanful information on the fate of MSNs. While MSNs accumulate in liver and spleen, MSN degradation products 89Zr or silicate bearing the radioisotope, are found in the bones and probably in lungs. A partial detachment of the peptide from the surface of the MSN is also observed. This work highlights the importance of choosing an appropriate labelling strategy for nanoparticles since core or surface labelling may result in different particle biodistribution if the labelled component degrades or the label detaches.
Introduction
Since around 2001 there has been great interest in the use of nanoparticles (NPs) for biomedical applications, drug delivery and imaging. NPs are used to transport drugs otherwise insoluble in aqueous media.1,2 They offer the advantage over other formulations that their surface can be engineered to prevent their recognition and removal by the mononuclear phagocytic system, increasing their circulation in vivo.3 Surface engineering also allows for targeting NPs to specific tissues while minimizing off-target accumulation. Multiple examples have been reported of NPs modified with targeting labels such as antibodies, peptides, and carbohydrates that provide special recognition properties and enhance their interaction with specific cells and tissues.4 In this approach, the targeting molecule usually interacts with a protein or motif present in the membrane of targeted cells.5Among a large variety of NPs, mesoporous silica nanoparticles (MSNs) have attracted attention for drug delivery as they have a large loading capacity6 and can be easily filled with drugs7 and therapeutics.2 Pores in MSNs can be functionalized with a rich chemistry that leads to the entrapment of a wide variety of molecules.8 The release of entrapped drugs occurs through diffusion from the pores to the bulk, and through the degradation of the mesoporous structure.9 MSNs have limited stability in basic and neutral pH, and progressively dissolve in physiological medium.10 Dissolution in vivo is also triggered by the relatively low concentration of silica in the whole-body volume. Although such dissolution is desired, it is expected to occur in the targeted organs or tissues. Optimally, in the application of MSNs for cancer therapy, the loaded drug must be released into the cancerous tissue and not elsewhere. To prevent the rapid degradation of MSNs, their surface can be modified with polyethylene glycol (PEG) through silane chemistry to create a physical barrier for silica dissolution, in addition to PEG acting as a stealth coating.11,12
In a recent paper we studied the degradation of silica NPs in vivo by positron emission tomography (PET) imaging using core–shell NPs, displaying a dense silica core, covered with a mesoporous shell on top, and functionalized with PEG on the outer surface.13 Core–shell NPs were either radiolabelled in the core or in the shell by attaching the positron emitter zirconium-89 (89Zr), which binds to the silanol groups present on the silica surface. Identical particles were prepared, differing only in the position of the labelling agent, which therefore are expected to have identical biodistribution. However, the biodistribution observed by PET imaging significantly differed depending on the position of the label, strongly suggesting the degradation of the shell (in the hour scale) with consequent release of the radionuclide for shell-labelled NPs.
Despite degradation, our results confirmed coincidences between the distribution patterns of shell- and core-labelled NPs, suggesting that the release of 89Zr, and consequently the degradation of the mesoporous shell, was not complete within the time window of our study. Since it is possible to obtain information on the NP location from the biodistribution pattern of the NPs labelled at the mesoporous shell, in this work we tackled the investigation of the biodistribution of MSNs without the need of a core shell structure, to evaluate if it was possible to corroborate the conclusions about the fate of the NPs from our previous paper. Additionally, moving forward towards biomedical applications, NPs were further modified with targeting moieties, which can lead to higher uptake in tumor tissue. A homing peptide, Lin-TT1 (AKRGARSTA), which targets the p32 mitochondrial protein overexpressed in the membrane of angiogenic endothelial cells and several cancer cells and can be anchored to the PEG via a free terminal cysteine, was selected as the targeting moiety.14 After the peptide is cleaved by tumoral proteases the CendR motif is exposed to the neuropilin-1 receptor that trigger internalization of NPs.15–17
To obtain complementary information about the targeting efficacy, we carried out additional biodistribution studies in a melanoma mouse model by incorporating the radiolabel in the peptide and quantifying its accumulation in different organs by ex vivo analysis. The gamma emitter iodine-131 (131I), which can be readily incorporated into tyrosine residues, was selected. With this dual labelling approach, we demonstrate in the current work that additional information on the biodistribution of MSNs can be obtained by comparing biodistribution patterns of NPs and peptide-labelled NPs, thus shedding light on the in vivo fate of MSNs. Moreover, our results show that information obtained from biodistribution and targeting studies may be easily misinterpreted, and care must be taken to design a reliable labelling strategy.
Results and discussion
Mesoporous silica nanoparticles with an average size of 140 nm were synthesized and further functionalized with PEG and a peptide linked to the end group of the PEG. The presence of PEG molecules was expected to prevent MSN interaction with blood serum and enhance circulation time.5,18 MSNs were prepared following a previously reported method based on the base catalyzed hydrolysis and condensation of tetraethyl orthosilicate over the surface of Pluronic F127 micelles.19 The modification of the surface of MSNs was carried out following well-established methods; first, the condensation of (3-aminopropyl)triethoxy silane (APTES) on the surface of NPs in ethanolic conditions providing amine groups to the surface, followed by the click reaction with the bifunctional NHS-PEG-maleimide in phosphate media (Scheme 1). The NHS ester group on one end of the PEG chains reacts with the amines grafted on the MSN surface. For peptide binding, the maleimide group on the other end of the PEG is made to react with the geminal cysteine of the peptide. | ||
| Scheme 1 Strategies for labelling MSNs core with 89Zr to perform PET imaging studies and tyrosine residue iodination with 131I in attached peptides to perform ex vivo gamma counting studies. | ||
The required equivalents of reagents to decorate the total surface of MSNs were calculated on the basis of the density of silanol groups estimated by Zhuravlev in amorphous silica (4.9 –SiOH nm−2).20 An excess of 10% was assumed for the sol–gel amination reaction with APTES. It was assumed that each amine silane could condense with three silanol groups and since the hydrodynamic diameter of PEG5000 molecule is 4.5 nm in PBS 10 mM,21 PEG do not diffuse in the pores of the structure with a smaller diameter than the PEG chains, and will be anchored on the external surface of the NPs.22 Prior to the post-grafting reactions, the surfactant template pluronic was removed in acidic methanolic media, obtaining bare MSNs.
Electron microscopy analysis showed spherical-shaped MSNs with uniform porous structure (Fig. 1). The Mobil Composition of Matter number forty-eight-type (MCM-48) mesoporous structure was characterized by nitrogen sorption measurements, revealing an average pore diameter of 2.4 nm and a specific surface area of 800 m2 g−1 (Fig. 1). The dynamic light scattering showed one homogenous population of MSNs with a hydrodynamic average size of 140 nm and average ζ-potential value of 15 mV. The thermogravimetric analysis indicates a total organic content after APTES and PEG functionalization of 5% and 8% respectively (ESI Fig. S1A and B†).
 | ||
| Fig. 1 (A) Representative TEM image of MSNs. Scale bar 100 nm and (B) isotherm of nitrogen adsorption of bare and aminated mesoporous nanoparticles. The inset corresponds to the pore size distribution curve (B). | ||
Radiochemical labelling of the core of MSNs with 89Zr
Zirconium(IV) is an oxyphilic cation with a large radius that forms stable complexes with oxygen ligands being easily incorporated into the amorphous silica oxide mesoporous structure.3Scheme 1 (upper route) shows the chelator free strategy used to label the core on the surface of the wall of MSNs. The cation acts as a hard Lewis's acid with the protonated silanol groups that act as hard donor groups.23 The incubation of 89Zr was performed with MSNs-NH2, at concentrations above the saturation of silica, preventing the hydrolyzation of the silica during the process. It was observed that raising temperature from 25 to 90 °C induced an increase in the zirconium incorporated into the silicon surface. At 70 °C, a maximum activity was reached after 60 minutes of incubation. The maximum radiochemical incorporation value observed was 95% as obtained by radio-TLC (ESI Fig. S2†) and was observed for aminated MSNs. The radiolabelling of MSNs-PEG5000 showed lower incorporation of 89Zr for the same experimental conditions. Therefore, the radiolabelling procedures were performed with aminated MSNs-NH2 before PEGylation.Radiochemical labelling the peptide at the surface of MSNs with 131I
Iodination labelling of peptides was performed as sketched in Scheme 2. 131I was coupled to the aromatic ring of the tyrosine residue in the presence of a mild oxidation agent (Iodogen). After 1 hour of incubation at room temperature and frequent shaking, the MSNs were purified by centrifugation and washed twice. The radiochemical yield of 85% was determined by radio-TLC, and the resulting MSNs showed a radiochemical purity of 99.5%. Previous results show that Lin-TT1 labelled with this methodology retain targeting properties.24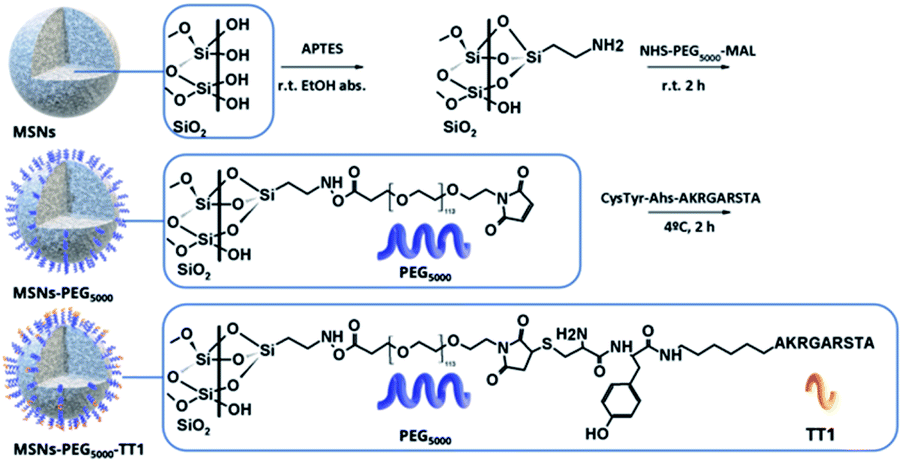 | ||
| Scheme 2 Post graph functionalization of MSNS. Initial surface functionalization of MSNs with APTES. Amine groups of APTES react with the ester group of the bifunctional PEG. The TT1 peptides are anchored through reaction of the PEG maleimide group with the germinal cysteine end modified TT1. | ||
Stability of 89Zr-MSNs under the saturation concentration of silica
The radiochemical stability of 89Zr-MSNs was evaluated in vitro by mimicking in vivo conditions. With that aim, MSNs at a concentration of 0.1 mg mL−1 were incubated in different media (Fig. 2). Results show a fast release of the radionuclide for NPs lacking PEG and for NPs functionalized with PEG5000 in all media, with >50% of the radioactivity detached from the NPs following 72 hours of incubation. This instability is probably due to the hydrolysis of the silica, which is expected at such a low concentration of NPs. At concentrations above the solubility limit of silica in water (0.145 mg mL−1 at 37 °C for silicic acid),25 free silicon species saturation is quickly reached, and the silica begins to re-precipitate, apparently stopping the dissolution process. If the MSN concentration is below the solubility limit for silica, re-precipitation is avoided, and MSNs degrade in a few hours. It has been previously shown that at a concentration of 0.1 mg mL−1, 85% of the starting MSN material is dissolved in 2 hours in simulated body fluid (SBF) before re-precipitation occurs.10 It is of note that 89Zr-MSNs functionalized with PEG5000 show high stability in SBF, with more than 90% of the radioactivity still attached to the NPs at t = 24 hours. The stability is lost when the NPs are incubated in SBF-EDTA.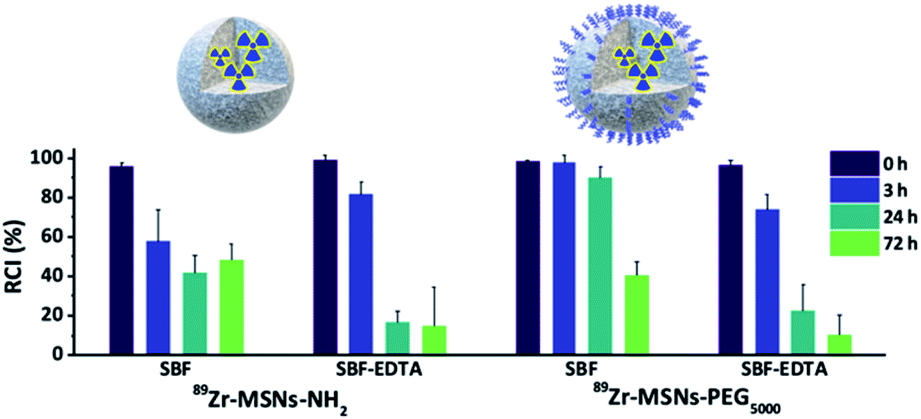 | ||
| Fig. 2 Radiochemical stability studies. Data bars show the percentage of remaining of 89Zr labelled NPs incubated at 0.1 mg mL−1 silica at 37 °C in simulated body fluid (SBF) and SBF with EDTA. | ||
Immunostaining
To evaluate the homing effect of the Lin-TT1 (ref. 26) peptide, in vitro studies were performed with B16F10 melanoma tumor cells. First, cells were incubated with the anti-p32 antibody to confirm the expression of p32 protein (green channel, ESI Fig. S3†). After confirmation of p32 protein expression, the MSNs-RhB with and without the peptide were incubated with B16F10 cells. An intense and homogeneously distributed fluorescence along the whole cell membrane fluorescence was observed for the MSNs with the homing peptide. Conversely, only a few fluorescent spots could be identified on the cell membrane for MSNs without the peptide (ESI Fig. S3†), confirming the contribution of the homing peptide to enhance attachment of MSNs to the cells.Biodistribution of core labelled MSNs with 89Zr by PET-CT
After proving the homing capacity of TT1-modified MSNs in vitro, we extended our studies in vivo using a melanoma mouse model. Experimentally, animals (n = 3 per NP type) bearing tumors with an approximate volume of 200 mm3 were intravenously injected with 3–5 MBq of 89Zr-labelled MSNs (89Zr-MSNs-PEG5000 and 89Zr-MSNs-PEG5000-TT1; 100 μL; 1 mg mL−1; dose = 5 mg kg−1) and subjected to PET scans at 30 min, 8, 24 and 48 hours after intravenous administration.To quantify the amount of radioactivity in each organ, PET-CT images were analyzed by PMOD software. Volumes of interest (VOI) were generated in the organs localized by the anatomical tomographic reference image and then, the volumes were translated to the co-registered PET images to obtain an estimation of the activity per unit of volume. Visual inspection of the images of 89Zr-MSNs-PEG5000 and 89Zr-MSNs-PEG5000-TT1 (Fig. 3A) confirms a biodistribution pattern of the MSNs with major accumulation in the liver and spleen, with presence of NPs also in the lungs at short times after administration, especially in the case of 89Zr-MSNs-PEG5000 NPs. This distribution pattern can be attributed to sequestration of the MSNs by the mononuclear phagocytic system. The accumulation of radioactivity in the bones, which is faster for the MSNs without peptide, suggests a progressive release of the radionuclide due to silica dissolution.
 | ||
| Fig. 3 (A) Maximum intensity coronal projections images of PET-CT image biodistribution of core labelled 89Zr-MSNs-PEG5000 (upper) and 89Zr-MSNs-PEG5000-TT1 (lower) after intravenous administration on xerograph B16F10 melanoma tumor bearing mice. (B) Percentage of injected dose per volume (ID cc−1) of major organs at different times. (C) Results from ex vivo measurements by γ-counter represented quantification of injected dose per gram (ID g−1) of major organ after intravenous administration of MSNs-PEG5000-131I-TT1 and labelled peptide 131I-TT1. | ||
A large accumulation of MSNs can be observed in liver (46–55% ID cc−1) and spleen (47–32% ID cc−1), some accumulation in the lungs (20–1% ID cc−1), and in femur (0.5–7.5% ID cc−1). Accumulation in liver and spleen is visibly stable throughout the duration of the study, while the concentration of radioactivity in the lung decreases with time and a progressive increase is observed in the femur, due to the accumulation of 89Zr cations in the cartilaginous joints of the femur (Fig. 3B). The increasing accumulation rate in bones indicates that the mesoporous structure of MSNs is slowly dissolved as shown in our previous work.13 Accumulation in the tumor is low, around 1% ID cc−1, and no significant differences were observed between 89Zr-MSNs-PEG5000 and 89Zr-MSNs-PEG5000-TT1. Overall, there are no major differences observed in the biodistribution of MSNs with or without the homing peptide.
A statistical comparison between pairs of organs at each time point shows significant difference in femur at 24 and 48 h (α = 0.05, 95%), while the other organs do not show differences.
Biodistribution ex vivo studies of MSNs functionalized with 131I-labelled TT1 peptide and free peptide
Since results from core labelling did not show major differences between 89Zr-MSNs-PEG5000 and 89Zr-MSNs-PEG5000-TT1, we hypothesized that this could be due to the loss of the peptide from MSNs during circulation, which would hinder targeting properties. Hence, we incorporated the radiolabel directly into the peptide. Peptides were labelled with 131I and anchored through the PEG chain to the MSN surface. Stability studies of 131I labelling (ESI Fig. S4†) show that in simulated body fluid (SBF) iodine activity remains on the peptide but is progressively lost by MSNs. In addition, the free peptide is rapidly degraded when incubated with blood serum (ESI Fig. S5†). The purified MSNs were injected at least in triplicate to the same B16F10 melanoma tumor mouse model. Parallel experiments were performed with the free peptide. For quantitative data, ex vivo experiments were conducted. The same types of NPs with the peptide labelled with 131I (MSNs-PEG5000-131I-TT1) and peptide, were administered to mice, which were subsequently sacrificed at predefined time points. At 4, 24 and 48 h after intravenous injection of MSNs, the animals were anesthetized and perfused. Organs were harvested, and the amount of radioactivity was determined by gamma (γ) counting. Results confirmed that the free peptide is eliminated through the kidneys and excreted in the urine and intestine over the first 4 hours (Fig. 3C). No presence of radioactivity could be detected in the thyroid gland, where free iodine tends to be accumulated, thus suggesting the radiochemical stability of the labelled peptide.NPs labelled through the TT1 peptide show a very different pattern from those labelled at the core. A rapid elimination of the isotope via urine was observed within the first 4 hours while there is some accumulation in the bladder and in the liver. To a large extent the biodistribution of the labelled NPs resembles that of the free labelled peptide, which is also eliminated via urine within 4 hours following injection. For the NPs, up to 15% of the injected dose was found in liver, and 30% in spleen. However, no radioactivity was observed in spleen and liver in case of the free peptide, which leads to the conclusion that the observed biodistribution for the NPs labelled with the peptide probably reflects the fate of the NPs and free peptide simultaneously, as NPs are too big to be eliminated by urine. No radioactivity was observed in the femur, which confirms that the activity observed for the experiments with the labelled core comes from released 89Zr or from degradation products from the NPs, but not from intact NPs, corroborating our hypothesis.13 No activity for the free labelled peptide could be detected in the lungs, where a high activity for the core labelled NPs was detected. This also hints that the 89Zr is present in the lung as free species or associated with the degradation products, but not as NPs. 89Zr interacts with phosphates, which is the reason it accumulates in the femur, and could also explain accumulation in the lungs, where there is a large concentration of lipids in the pulmonary surfactant.
For both core and peptide labelled NPs a limited uptake of NPs in tumors is observed. No significant differences can be appreciated from the core labelled NPs when they are functionalized with PEG or with PEG plus peptide. In part this can be understood by the hydrolysis and removal of the targeting peptide from the NPs, which reduces the potential targeting efficacy. It is also clear that NPs largely accumulate in liver/spleen and remain there, which restricts the NPs potential accumulation in the tumor to those circulating, which is a smaller fraction of the administrated NPs. Polymer nanoparticles functionalized with TT1 have shown significantly higher tumor targetability probably because of a better tumor permeability.24
Results from core labelling show that the NPs degrade liberating 89Zr or degradation products with the radioisotope that accumulate preferentially in the bones or are eliminated by urine in short times. Radiolabelling of the peptide TT1 on the surface of the NPs shows that the peptide hydrolyzes and is eliminated by urine but the biodistribution pattern shows that labelled NPs differ substantially from the free peptide in their fate, showing accumulation in liver and bladder like the core labelled NPs, which is absent in the biodistribution of the labelled peptides.
Targeting functions as well as stealth coating are normally attached in post functionalization steps following NP synthesis using click chemistry reactions and involving the formation of amides. The efficacy of the NPs to target the desired tissue will finally depend on the stability of the coating around the NP. Despite this issue being fundamental in the design of NPs for biomedical applications, studying the stability of the coating is often neglected and limited to in vitro experiments, which may not fully represent the situation in vivo. A stable coating in vitro may not be so inside the body where circulation, translocation and the interaction with multiple biomolecules and cells can result in a faster and more effective detachment of molecules from the NP surface. The lack of studies on the stability of targeting molecules and stealth is in part due to the difficulty to trace degradation of NPs in vivo and in general of tracing NPs in biological matrixes. Biodistribution studies based on either fluorescence or radiolabelling usually do not address these issues. Indeed, when the labelling is performed in the coating, such as through the targeting molecule, it is assumed that the label remains attached to the NP and that the results on the biodistribution and the efficacy of targeting correspond to the NPs when it is actually coming from the targeting molecules, which may be detached from the NP surface. Conversely, when the NP core is labelled, if the efficacy of targeting and the accumulation of NPs in the targeted tissue are low it is often assumed that the targeting molecule is not effective in directing the NPs, while it may simply be that the targeting molecule has detached.
Conclusions
We have advanced here on the understanding of the fate of mesoporous silica NPs in vivo. We have demonstrated that the biodistribution of the radiolabelled MSN core provides information of both the fate of the MSN and the products of degradation. By alternatively labelling a peptide attached for targeting MSNs to tumor tissue we have been able to identify the patterns of biodistribution that correspond to the MSN and its degradation products more clearly, showing an overwhelming accumulation of the MSNs in liver and spleen. We have also demonstrated that the targeting peptide is partially removed from the MSN and eliminated, which partly explains the practically absent targeting effect of the peptide, along with the rapid accumulation in the liver and spleen. Products of degradation of the MSNs, 89Zr or silicate bearing the radioisotope but not intact MSNs, accumulate in the bones and probably in lung. Our results here show that it is fundamentally important to choose the appropriate pathway for labelling a nanocarrier, as core labelling may provide different information from surface labelling on biodistribution. The difference in biodistribution results depends on the stability of the marker, which may indicate a false location for the nanocarrier if it detaches.Experimental
Reagents
All syntheses were carried out with the following commercially available reagents used without further purification: tetraethyl orthosilicate 98% (TEOS), (3-aminopropyl)triethoxy silane (APTES), methanol (MeOH), ethanol (EtOH), NHS-PEG-maleimide (MW 5000 Da), rhodamine-B (RhB), OH-PEG-maleimide (MW 5000 Da) and phosphate buffered saline were bought from Sigma-Aldrich. Peptide Lin-TT1 sequence: Cys-Tyr-Ahx-AKRGARSTA, was bought from Peptides International upon sequence demand order, including the incorporation of fluorescence molecule 6-carboxyfluorescein (6-FAM) for confocal microscopy studies. Simulated body fluid composition per ion (SBF, mM): 142 Na+, 5 K+, 1.5 Mg2+, 2.5 Ca2+, 103 Cl−, 4.2 HCO3−, 1 HPO42−, 0.5 SO42−, (HOCH2)3CNH2 (Tris base) at pH 7.4. Iodine-131 for peptide labelling was bought from PerkinElmer.Synthesis of mesoporous silica nanoparticles (MSNs)
MSNs were synthesized following a previously described method26 with minor modification to obtain MCM-48 (Ia![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) d porous structure) MSNs with a size of 140 nm in diameter. In brief, 0.50 g of hexadecyltrimethylammonium bromide (CTAB, 1.4 mmol, 98%) and 2.05 g Pluronic F-127 (163 μmol, 98%) were dissolved in a solution containing 96 mL ultrapure water, 43 mL of absolute ethanol (EtOH, 0.93 mol, 100%) and 11.2 mL NH4OH (180 mmol, 29%) under magnetic stirring for 30 min until complete dissolution. Under magnetic stirring of 1000 rpm, 1.95 mL of tetraethyl orthosilicate (TEOS, 8.5 mmol, 98%, 0.93 g mL−1), were poured into the vortex of the previous solution. The stirring was kept for 1 min to initiate the nucleation stage. Subsequently, the mixture was kept for 24 h at room temperature without stirring during the condensation stage.
d porous structure) MSNs with a size of 140 nm in diameter. In brief, 0.50 g of hexadecyltrimethylammonium bromide (CTAB, 1.4 mmol, 98%) and 2.05 g Pluronic F-127 (163 μmol, 98%) were dissolved in a solution containing 96 mL ultrapure water, 43 mL of absolute ethanol (EtOH, 0.93 mol, 100%) and 11.2 mL NH4OH (180 mmol, 29%) under magnetic stirring for 30 min until complete dissolution. Under magnetic stirring of 1000 rpm, 1.95 mL of tetraethyl orthosilicate (TEOS, 8.5 mmol, 98%, 0.93 g mL−1), were poured into the vortex of the previous solution. The stirring was kept for 1 min to initiate the nucleation stage. Subsequently, the mixture was kept for 24 h at room temperature without stirring during the condensation stage.
MSNs were collected as a white solid product by centrifugation at room temperature (9000 RCF, 20 min). Then, they were washed with ultrapure water three times and twice with EtOH (70%).
The extraction of CTAB was performed by acidic hydrolysis by refluxing the obtained suspension overnight in ethanolic acid mixture (EtOH, 20 mL, 100%; HCl, 4 mL, 37%), and washed three times with ultrapure water and twice with EtOH 70%. Finally, they were dried at 60 °C for storage and characterization. The removal of CTAB was observed by FTIR in the extracted MSNs due the absence of the symmetric and asymmetric stretching vibration bands of methylene chains at 2920 and 2850 cm−1 respectively (ESI Fig. S6A†).
Attachment of targeting molecules on MSNs
The post-graph step was performed according to the literature.27 Briefly, 100 mg of NPs were resuspended in 10 mL of absolute ethanol in an ultrasonic water bath for 10 min. Then, 0.6 μL of APTES (0.32 mmol, 99%, 0.946 g mL−1) was added to the suspension under magnetic stirring at room temperature. The reaction was kept for 4 hours and afterward MSNs-NH2 were collected by centrifugation and washed twice with absolute ethanol. Polyethylene glycol (PEG) graph step was performed according to the literature.12 PEG chains with molecular weights of 5000 Da were used, with double end functional groups: NHS-PEG5000-maleimide. The coupling of PEG chains was performed as follows: 10 mg of MSNs-NH2 were dispersed in 10 mL of PBS for 10 min in an ultrasonic water bath. Then, 2.5 mg of bifunctional PEG were dissolved in 100 μL of PBS and poured into the suspension. The reaction was kept for 2 hours under magnetic stirring. Then, the MSNs-PEG-maleimide were washed three times with ultrapure water and dispersed at a final concentration between 1 to 10 mg mL−1 in PBS (ESI Fig. S6B†). Subsequently, the coupling of the peptide TT1 (CY(Ahx)AKRGARSTA) to the PEG chain was performed adding 5 eq. of peptide per 1 eq. of PEG to a suspension of MSNs-PEG-maleimide 1 mg mL−1 in PBS and kept at 4 °C for 2 hours with frequent shaking. The peptide Lin-TT1 was designed to be anchored to the PEG via a free terminal cysteine, directly attached to amino hexanoic acid (Ahx) as a spacer and a tyrosine residue to enable 131I-labelling. The radiolabelled tyrosine was localized in the sequence after cysteine amino acid and spaced from the recognition fragment of the Lin-TT1 peptide. The maleimide reacts with the geminal sulfhydryl group of the peptide. MSNs-PEG-TT1 were washed three times with ultrapure water and dispersed at a final concentration of 1 mg mL−1 in PBS.Radiolabelling of the mesoporous core
The radionuclide was incorporated post synthesis both for bare (MSNs) and the PEGylated particles (MSNs-PEG). Experimentally (ESI Fig. S2A†), the suspension of NPs at 2–10 mg mL−1 in HEPES buffer were incubated with 185 MBq of 89Zr for different times (20–90 min) and temperatures (25–90 °C). At each time point an aliquot was extracted, and the NPs were centrifuged at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 RCF for 20 min and washed three times with ultrapure water. The radiochemical incorporation was determined by the ratio between the measured activity of the NPs and the activity of the collected wash. The results showed an optimal radiolabelling efficiency and stability when the bare NPs (10 mg mL−1) were incubated with the radio-cation at 70 °C for 60 min (ESI Fig. S2B†).
000 RCF for 20 min and washed three times with ultrapure water. The radiochemical incorporation was determined by the ratio between the measured activity of the NPs and the activity of the collected wash. The results showed an optimal radiolabelling efficiency and stability when the bare NPs (10 mg mL−1) were incubated with the radio-cation at 70 °C for 60 min (ESI Fig. S2B†).
To assess radiochemical integrity of the core of NPs labelled with 89Zr, MSNs were incubated in n-SBF (simulated body fluid) or n-SBF-EDTA (2 mM) at a concentration of 0.1 mg mL−1 (note that MSNs were resuspended below silica critical concentration). The radiochemical incorporation was determined by radio-iTLC as follows: a paper strip impregnated with silica was spotted with the suspension at each time point and developed with mobile phase (2:18 ACN/citric acid 20 mM, EDTA 60 mM at pH ∼ 5) and the ratio of the areas obtained after the radio-iTLC were calculated.
Radiolabelling of the peptide TT1 at the surface
The incorporation of 131I to the peptide was performed by covalent attachment to the aromatic ring of the tyrosine residue, using the mild oxidizing agent 1,3,4,6-tetrachloro-3α,6α-diphenylglycoluril (Iodogen).28 Experimentally, MSNs-PEG-TT1 were incubated at 1 mg mL−1 with 37 MBq of 131I at room temperature in a tube (Iodogen impregnated) for 60 min with frequent shaking. After incubation, the MSNs were centrifuged and washed twice with PBS and resuspended in 1 mL of PBS, to obtain radiochemical purity of 99.5% measured by radio-iTLC with a radiochemical yield of 85% (ESI Fig. S7†).Transmission electron microscopy
TEM studies were conducted in a JEOL JEM-1400 microscope operated at 120 kV. The samples were prepared by depositing a drop of nanoparticle suspension onto a copper specimen grid, coated with a holey carbon film, previously treated in a glow discharged chamber for 1 minute, and allowing it to dry.Dynamic light scattering and ζ-potential
Particle size analysis was performed using a NanoSizer (Malvern Nano-ZS, UK) with 173° scattering angle detector. Each sample was measured at least in triplicate (0.1 mg mL−1 in 10 mM NaCl). ζ-Potential measurements were carried out with the same NanoSizer equipment at 25 °C and a cell drive voltage of 60 V using a Smoluchowski model.Cell culture experiments
The B16-F10 (CRL-6475) skin melanoma cell line was bought from the American Type Culture Collection (ATCC®). Cells were cultured in RMPI 1640 (Lonza) supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin and L-glutamine (Gibco®) and kept in a humidified atmosphere of 5% CO2 at 37 °C. Cells were treated with trypsin–EDTA and seeded at 5000 cells per square centimeter.MSNs-(6-FAM)-Lin-TT1 synthesis
MSNs used for in vitro studies were labelled with the fluorescence tag molecule 6-carboxyfluorescein (6-FAM) spaced with an amino-hexanoic acid chain from the active peptide sequence. Acrylate-PEG-maleimide was incubated with the peptide in PBS and subsequently incubated in the presence of aminated NPs.Immunofluorescence cell culture experiments
105 cells per well were seeded in 8-well plates for confocal light microscopes (Ibidi®) in 0.2 mL of full RPMI medium. 24 h later the supernatant was removed, and cells were washed twice with full RPMI. A suspension of 100 μg mL−1 of NPs was prepared in RPMI (FBS 1%) and 100 μL (10 μg) were incubated for 2 hours at 37 °C (at 4 °C to evaluate membrane interaction). After the incubation time the supernatant was removed, and particles were washed twice with PBS. Cells were fixed with 200 μL paraformaldehyde (4% in saline solution, pH 7.4) for 10 min, then washed twice with PBS.Permeation of cell membranes for fluorescence microscopy
After fixation the cells were incubated for 10 minutes with 200 μL of DPBST (Triton 0.1% in DPBS) at room temperature. The supernatant was removed, and cells were washed twice with the same volume of DPBS.Blocking and immunostaining
Cells were incubated for 1 hour with 5% BSA in PBST. Then cells were incubated overnight with primary antibody (anti-p32, rabbit) 1/300 in 1% BSA in PBST. Washed twice with PBST and incubated 1 hour with the secondary antibody (goat anti-rabbit) labelled with the fluorophore Alexa-488 (490/525, green emission channel). Then, the cells were washed three times with PBST. For nuclei staining, 200 μL of DAPI 0.3 μg mL−1 in PBS were incubated for 5 minutes, then washed twice with 200 μL of DPBS.89Zr production for radiolabelling the core of MSNs
The radioisotope was produced via the 89Y (p, n) 89Zr reaction. Briefly, two enriched yttrium-89 coins (ϕ = 10 mm × 2 mm), were placed in the aluminum degrader (400 barns) case and, irradiated in a Cyclotron IBA 18/9 MeV for 2 h (20 μA). After irradiation, the disk was extracted and placed in 3 mL HCl 6 N until dissolution. The obtained liquid was passed through a chemically modified hydroxamate resin solid phase column and washed with 5 mL of HCl 6 N to remove excess of yttrium and non-reactant species.Silica MSNs radiolabelling for in vivo studies
50 μL of 89Zr (37 MBq) in oxalic acid 1 M was neutralized with 200 μL of sodium carbonate, until neutral pH. A suspension of 1 mg of silica NPs (50 μL of HEPES 10 mM) was incubated with the previous solution for 1 h at 70 °C. The crude reaction was cooled down to room temperature and separated by centrifugation and washed with 1 mL HEPES, ultrapure water and PBS buffer; then resuspended in 300 μL of PBS containing maleimide-PEG-LinTT1 or maleimide-PEG-OH. The optimization of the incubation was performed incubating at different temperatures and time points. A small aliquot was taken to perform radio-iTLC (impregnated silica thin layer chromatography), with a mobile phase of citric acid pH 5.5, EDTA 50 mM.In vivo biodistribution studies
Melanoma model tumor bearing mice C57BL/6 with an appropriate tumor volume were injected with 3.7 MBq were injected with NPs in PBS solution intravenously (100 μL, 1 mg mL−1; 5 mgNPs kgmice−1). The animals were anesthetized, and PET images were obtained after intravenous administration of labelled NPs, at 40 min and 2, 8, 24 and 48 h after administration. The total radioactivity was measured in a CRC-25R dose calibrator (Calintec, USA) to determine the dose administered in each animal.Image acquisition
For administration of labelled particles, intravenous injection by the tail vein was performed, measuring the remains in the syringe before and after injection to determine the resulting injected activity. After the PET scan, the mice were moved to the X-Cube to perform a computed tomography scan. Image acquisition was repeated at sequential times until the biodistribution profile was defined.PET image acquisition
MOLECUBES β-CUBE PET scanner incorporates thick monolithic LYSO (Lutetium Yttrium Orthosilicate) scintillators of 25 × 25 × 8 mm coupled to an array of Hamamatsu multi-pixel photon counters of 3.2 × 3.2 mm. Five rings are each provided with nine detectors, generating a cylindrical camera chamber of 45 detectors. As the light function is known to vary upon the interaction depth, the silicon photomultiplier provides 5 levels of depth-of-interaction (DOI) measurements. The 45 PET detectors generate a total trans-axial field of view (FOV) of 7.3 cm and 13 cm on the axial plane. An attenuation map of 1 μm3 voxel resolution was applied to the ordered subset expectation maximization (OSEM) reconstruction, in which a maximum likelihood clustering algorithm was iteratively applied thirty times, to reconstruct the 3D images according to each interaction with the PET detectors.CT image acquisition
MOLECUBES X-CUBE CT is a dedicated micro-CT capable of performing spiral high-resolution computerized tomography of a rodent whole-body scan with a 6.5 cm transverse and 3.5 cm axial FOV, that takes less than 5 min to scan per animal. An attenuation map of hard tissues was obtained after ten times single scatter simulation (SSS) of OSEM reconstruction.Image reconstruction and quality
CT imaging performed at 100 μm was used for performing PET attenuation and scatter corrections, to obtain a final PET reconstructed image with 1 mm resolution. After 30 iterations of iterative OSEM images were reconstructed into a 192 × 192 transverse matrix with cubic voxels of 0.4 mm. This equipment provides images with elevated sensitivity and volumetric spatial resolution of around 1 mm3 for all locations within the imaging FOV.Nuclear image analyses
Images were reconstructed applying a CT-based attenuation correction factor and radioactivity decay, and analyzed by PMOD software. Volumes of interest were manually drawn in the major organs, using CT images for anatomical reference. Volumes were transferred to PET images and the concentration of radioactivity was calculated for each organ. The values obtained were transformed to real activity (Bq cm−3) and normalized with the injected dose, to obtain the percentage of the injected dose per volume unit of organ at each time.Animal care
The animals were cared for and handled in accordance with the Guidelines for Accommodation and Care of Animals (European Convention for the Protection of Vertebrate Animals Used for Experimental and Other Scientific Purposes), internal guidelines, and experimental procedures previously approved by local authorities. Before each procedure, animals were anesthetized with a mixture of 3–5% isoflurane in O2 for induction, which was reduced to 1.5–3% by a nose cone mask before intravenous injection. During PET-CT imaging acquisitions, rodents were anesthetized and kept at 37 °C using an electric blanket, set up at the base of the animal's bed. Subsequently, CT acquisitions were performed to provide anatomical information on each animal.Ethical statement
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of CIC biomaGUNE and experiments were approved by: (i) The Animal Ethics Committee of CIC biomaGUNE; (ii) an external Ethical Committee (Órgano Habilitado IIS Biodonostia); and (iii) Diputación Foral de Guipúzcoa.Tumor inoculation
C57BL/6 mice (6–8 weeks old) were used for all in vivo experiments. B16-F10 cells were used for the tumor inoculation and were previously cultured similarly to those for the in vitro experiments. Prior to injection in vivo, cells were tested for mycoplasma using the commercially available MYCOALERT mycoplasma detection kit (Lonza). For tumor inoculation 3 × 105 cells per mouse were homogenized in Corning® Matrigel Basement Membrane Matrix High mixed with the same volume of PBS. All the material (tips, pipettes, syringes, and storage vials) and the cell suspension in Matrigel® were kept in ice until use. The cell suspension was then loaded into a syringe and 100 μL per mouse was injected into the right back side of the animals using a 26 G needle. Animals were monitored for tumor growth using an electronic digital caliper 779A series (Starrett) until the tumor size was appropriate to start the biodistribution studies (volume of 100 mm3, or 6 to 9 days since tumor inoculation).Ex vivo quantification
After 2, 24, and 48 h post injection of labelled NPs, animals were anesthetized, urine was extracted, and blood collected. For fixation of the tissues, heart puncture was performed with 20 mL of heparinized (5 UI mL−1) in saline (NaCl 154 mM) followed by a perfusion with the same volume of 4% paraformaldehyde. All the organs were weighed and measured in a 2470 WIZARD2 Automatic Gamma Counter (PerkinElmer).Author contributions
M. A. R. & A. M. M. V. did the synthesis, radiolabelling, and data analysis; V. G. V. supervised the radiolabelling; P. A. performed cell studies; G. S. I. supervised the synthesis; J. L. and S. E. M. designed the experiments, analyzed data, and wrote the manuscript. All authors provided input on the manuscript prior to submission for publication.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. E. M. and J. L. respectively thank the PID2020-114356RB-I00 and PID2020-117656RB-100 projects from the Ministry of Science and Innovation of the Government of Spain. G. S. I. acknowledges support from ANPCyT (PICT 2015-2526 and PICT 2018-4651). This work was performed under the Maria de Maeztu Units of Excellence Program from the Spanish State Research Agency – Grant no. MDM-2017-0720. We greatly acknowledge Dr Julia Cope, a PhD from CIC biomaGUNE, for kindly revising the manuscript.Notes and references
- L. F. De Oliveira, K. Bouchmella, K. D. A. Gonçalves, J. Bettini, J. Kobarg and M. B. Cardoso, Langmuir, 2016, 32, 3217–3225 CrossRef CAS PubMed.
- C. Argyo, V. Weiss, C. Bräuchle and T. Bein, Chem. Mater., 2014, 26, 435–451 CrossRef CAS.
- Q. He, J. Zhang, J. Shi, Z. Zhu, L. Zhang, W. Bu, L. Guo and Y. Chen, Biomaterials, 2010, 31, 1085–1092 CrossRef CAS PubMed.
- R. Singh and J. W. Lillard, Exp. Mol. Pathol., 2009, 86, 215–223 CrossRef CAS PubMed.
- D. E. Owens and N. A. Peppas, Int. J. Pharm., 2006, 307, 93–102 CrossRef CAS PubMed.
- J. Shen, Q. He, Y. Gao, J. Shi and Y. Li, Nanoscale, 2011, 3, 4314 RSC.
- N. K. Mal, M. Fujiwara, Y. Tanaka, T. Taguchi and M. Matsukata, Chem. Mater., 2003, 15, 3385–3394 CrossRef CAS.
- J. M. Rosenholm, C. Sahlgren and M. Linden, Nanoscale, 2010, 2, 1870–1883 RSC.
- J. G. Croissant, Y. Fatieiev and N. M. Khashab, Adv. Mater., 2017, 1604634 CrossRef PubMed.
- Q. He, J. Shi, M. Zhu, Y. Chen and F. Chen, Microporous Mesoporous Mater., 2010, 131, 314–320 CrossRef CAS.
- V. Cauda, A. Schlossbauer and T. Bein, Microporous Mesoporous Mater., 2010, 132, 60–71 CrossRef CAS.
- C. Yagüe, M. Moros, V. Grazú, M. Arruebo and J. Santamaría, Chem. Eng. J., 2008, 137, 45–53 CrossRef.
- E. Bindini, M. de los A. Ramirez, X. Rios, U. Cossío, C. Simó, V. Gomez-Vallejo, G. Soler-Illia, J. Llop and S. E. Moya, Small, 2021, 2101519, 1–10 Search PubMed.
- L. Paasonen, S. Sharma, G. B. Braun, V. R. Kotamraju, T. D. Y. Chung, Z. G. She, K. N. Sugahara, M. Yliperttula, B. Wu, M. Pellecchia, E. Ruoslahti and T. Teesalu, ChemBioChem, 2016, 17, 570–575 CrossRef CAS PubMed.
- V. Fogal, L. Zhang, S. Krajewski and E. Ruoslahti, Cancer Res., 2008, 68, 7210–7218 CrossRef CAS PubMed.
- V. Fogal, A. D. Richardson, P. P. Karmali, I. E. Scheffler, J. W. Smith and E. Ruoslahti, Mol. Cell. Biol., 2010, 30, 1303–1318 CrossRef CAS PubMed.
- K. N. Sugahara, T. Teesalu, P. P. Karmali, V. R. Kotamraju, L. Agemy, O. M. Girard, D. Hanahan, R. F. Mattrey and E. Ruoslahti, Cancer Cell, 2009, 16, 510–520 CrossRef CAS PubMed.
- G. E. Musso, E. Bottinelli, L. Celi, G. Magnacca and G. Berlier, Phys. Chem. Chem. Phys., 2015, 17, 13882–13894 RSC.
- T.-W. Kim, P.-W. Chung and V. S. Y. Lin, Chem. Mater., 2010, 22, 5093–5104 CrossRef CAS.
- L. T. T. Zhuravlev, Colloids Surf., A, 2000, 173, 1–38 CrossRef CAS.
- J. K. Armstrong, R. B. Wenby, H. J. Meiselman and T. C. Fisher, Biophys. J., 2004, 87, 4259–4270 CrossRef CAS PubMed.
- M. Bouchoucha, R. C. Gaudreault, M.-A. Fortin and F. Kleitz, Adv. Funct. Mater., 2014, 24, 5911–5923 CrossRef CAS.
- F. Chen, S. Goel, H. F. Valdovinos, H. Luo, R. Hernandez, T. E. Barnhart and W. Cai, ACS Nano, 2015, 9, 7950–7959 CrossRef CAS PubMed.
- L. Simon-Gracia, P. Scodeller, S. S. Fuentes, V. G. Vallejo, X. Rios, E. S. Sebastian, V. Sidorenko, D. Di Silvio, M. Suck, F. de Lorenzi, L. Y. Rizzo, S. Stillfried, K. Kilk, T. Lammers, S. E. Moya and T. Teesalu, Oncotarget, 2018, 9, 18682–18697 CrossRef PubMed.
- A. L.-T. Pham, D. L. Sedlak and F. M. Doyle, Appl. Catal., B, 2012, 126, 258–264 CrossRef CAS PubMed.
- P. K. Garg, K. L. Alston and M. R. Zalutsky, Bioconjugate Chem., 1995, 6, 493–501 CrossRef CAS PubMed.
- T. W. Kim, P. W. Chung and V. S. Y. Lin, Chem. Mater., 2010, 22, 5093–5104 CrossRef CAS.
- M. A. Deri, S. Ponnala, B. M. Zeglis, G. Pohl, J. J. Dannenberg, J. S. Lewis and L. C. Francesconi, J. Med. Chem., 2014, 57, 4849–4860 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00719j |
| This journal is © The Royal Society of Chemistry 2022 |