
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Predictive high-throughput screening of PEGylated lipids in oligonucleotide-loaded lipid nanoparticles for neuronal gene silencing†
Apoorva
Sarode‡
 a,
Yuchen
Fan‡
a,
Amy E.
Byrnes‡
b,
Michal
Hammel
c,
Greg L.
Hura
cd,
Yige
Fu
a,
Ponien
Kou
a,
Chloe
Hu
a,
Flora I.
Hinz
b,
Jasmine
Roberts
b,
Stefan G.
Koenig
a,
Karthik
Nagapudi
a,
Casper C.
Hoogenraad
b,
Tao
Chen
a,
Dennis
Leung
a and
Chun-Wan
Yen
*a
a,
Yuchen
Fan‡
a,
Amy E.
Byrnes‡
b,
Michal
Hammel
c,
Greg L.
Hura
cd,
Yige
Fu
a,
Ponien
Kou
a,
Chloe
Hu
a,
Flora I.
Hinz
b,
Jasmine
Roberts
b,
Stefan G.
Koenig
a,
Karthik
Nagapudi
a,
Casper C.
Hoogenraad
b,
Tao
Chen
a,
Dennis
Leung
a and
Chun-Wan
Yen
*a
aSmall Molecule Pharmaceutical Sciences, Genentech Inc., 1 DNA Way, South San Francisco, CA-94080, USA. E-mail: yenc3@gene.com
bDepartment of Neuroscience, Genentech, Inc., South San Francisco, CA 94080, USA
cMolecular Biophysics and Integrated Bioimaging Division, Lawrence Berkeley National Lab, Berkeley, CA, USA
dChemistry and Biochemistry Department, University of California Santa Cruz, Santa Cruz, CA, USA
First published on 4th February 2022
Abstract
Lipid nanoparticles (LNPs) are gaining traction in the field of nucleic acid delivery following the success of two mRNA vaccines against COVID-19. As one of the constituent lipids on LNP surfaces, PEGylated lipids (PEG-lipids) play an important role in defining LNP physicochemical properties and biological interactions. Previous studies indicate that LNP performance is modulated by tuning PEG-lipid parameters including PEG size and architecture, carbon tail type and length, as well as the PEG-lipid molar ratio in LNPs. Owing to these numerous degrees of freedom, a high-throughput approach is necessary to fully understand LNP behavioral trends over a broad range of PEG-lipid variables. To this end, we report a low-volume, automated, high-throughput screening (HTS) workflow for the preparation, characterization, and in vitro assessment of LNPs loaded with a therapeutic antisense oligonucleotide (ASO). A library of 54 ASO-LNP formulations with distinct PEG-lipid compositions was prepared using a liquid handling robot and assessed for their physiochemical properties as well as gene silencing efficacy in murine cortical neurons. Our results show that the molar ratio of anionic PEG-lipid in LNPs regulates particle size and PEG-lipid carbon tail length controls ASO-LNP gene silencing activity. ASO-LNPs formulated using PEG-lipids with optimal carbon tail lengths achieved up to 5-fold lower mRNA expression in neurons as compared to naked ASO. Representative ASO-LNP formulations were further characterized using dose–response curves and small-angle X-ray scattering to understand structure–activity relationships. Identified hits were also tested for efficacy in primary murine microglia and were scaled-up using a microfluidic formulation technique, demonstrating a smooth translation of ASO-LNP properties and in vitro efficacy. The reported HTS workflow can be used to screen additional multivariate parameters of LNPs with significant time and material savings, therefore guiding the selection and scale-up of optimal formulations for nucleic acid delivery to a variety of cellular targets.
1. Introduction
The past few years have seen unprecedented growth in the development of oligonucleotide therapeutics that regulate pathological targets historically deemed undruggable by traditional small molecules and biologics.1,2 These therapeutics include aptamers that bind to and alter protein function, short-interfering RNA (siRNA) and microRNA (miRNA) that interfere with coding and non-coding RNA, and antisense oligonucleotides (ASOs) which upregulate or downregulate protein expression, or modulate mRNA splicing in a design-dependent manner.3–5 However, several formulation and intracellular delivery challenges need to be addressed to fully realize the therapeutic potential of these molecules.6–9Lipid nanoparticles (LNPs) are one of the most advanced drug delivery platforms that can overcome the challenges associated with oligonucleotide delivery. LNPs not only provide a stable matrix to protect the encapsulated nucleic acid cargo from in vivo degradation, but they also improve therapeutic efficacy by enhancing intracellular transport.10 The clinical potential of LNP therapeutics was established in 2018 upon U.S. Food and Drug Administration (FDA) approval of the first RNAi-LNP formulation, ONPATTRO®.11 More recently, LNP-mediated delivery was utilized in two mRNA vaccines in response to the SARS-CoV-2 (COVID-19) pandemic.12,13 In addition to these FDA-authorized therapeutics, there has been a dramatic increase in both clinical and preclinical LNP formulation development.14,15 Among these, LNPs are being explored for central nervous system (CNS) targeting, owing to the global burden caused by neurodegenerative diseases and glioma.16 Preliminary in vitro studies show that pathological genes can be silenced in neurons using LNP-based nucleotide formulations, and ongoing investigations are focused on developing novel LNP compositions that enhance blood–brain barrier penetration to ultimately achieve enhanced in vivo efficacy.17–20
LNP structure and nucleic acid cargo delivery are regulated by four major components: ionizable lipids, helper phospholipids, cholesterol, and polyethylene glycol-lipids (PEG-lipids).21 Cationic ionizable lipids promote the encapsulation of negatively charged nucleic acids during LNP assembly and aid in cytosolic delivery of cargo at endosomal pH (5.5–6.5).22–27 Helper phospholipids and cholesterol increase LNP structural stability, promote membrane fusion, and enhance endosomal escape.23,28–30 The impact of PEG-lipids is multi-faceted and contributes to what has been termed the ‘PEG-dilemma’.31 PEG-lipids control particle size distributions during the self-assembly of LNPs and prevent aggregation.32–35 PEG also prolongs in vivo LNP circulation time by acting as a steric barrier to the adsorption of plasma proteins.22,36–38 While extending half-life increases therapeutic exposure, the hydrophilic PEG corona may also hinder interactions between the particle surface and the lipophilic cell membrane, resulting in poor cellular internalization.37 Furthermore, PEG can block transport protein binding, which is essential for LNP cellular internalization via receptor-mediated endocytosis.39 To overcome the PEG dilemma and achieve optimal intracellular delivery, it is necessary to understand how PEG-lipid characteristics regulate LNP size, structure, and subsequent cargo delivery. Previous studies show LNP efficacy is modulated by several PEG-lipid variables including PEG size, architecture, molar ratio, carbon tail type, and length.33–36,38,40–47 However, most published studies focus on a limited set of PEG variables on account of the extensive resources required for manual formulation workflows. Due to the wide screening space for PEG-lipids, a comprehensive study encompassing multiple PEG-lipid types and variables is necessary to empirically understand how PEG-lipids impact LNP structure and function.
An automated high-throughput screening (HTS) workflow to prepare and characterize LNPs can be used to comprehensively assess how PEG-lipid parameters regulate LNP function by allowing complex LNP screening designs, while saving substantial time and materials.32 In this study, we generated a library of 54 ASO-LNPs using 18 different PEG-lipids across the phosphoglyceride, diglyceride, and ceramide families, at varying PEG-lipid molar ratios (1, 3, and 5 mol%) using a high-throughput workflow. The impact of PEG-lipid parameters – molecular weight (M.W.), carbon-tail length, and molar ratios – on ASO-LNP size and polydispersity was assessed. Additional PEG-lipid variables including PEG architecture, lipid tail saturation, PEG-lipid charge, and linker chemistry were also analyzed. Following physicochemical characterization, formulations were screened in primary mouse cortical neurons for ASO-mediated regulation of mRNA expression as a function of LNP composition. Target engagement dynamics for representative formulations were evaluated using dose–response curves and these data were correlated to the LNP structures determined by small-angle X-ray scattering (SAXS). In addition, the translatability of ASO-LNP physical properties and in vitro performance was validated in primary murine microglia and after scale-up using a microfluidic technique for manufacturing. This study presents the first systematic HTS approach that correlates the impact of PEGylating agents on LNP structure and subsequent in vitro ASO delivery efficacy in multiple primary brain cell types. These predictive results support the use of HTS workflows during early-stage development of LNP formulations. This HTS-based formulation approach can also be widely applied to additional LNP screening efforts when identifying optimal formulations for a target cell type or tissue of interest.
2. Materials and methods
2.1 High-throughput preparation and characterization of ASO-loaded LNPs
Cholesterol, 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), and all linear PEGylated lipids for LNP formulations were purchased from Avanti Polar Lipids (AL, USA). Branched PEGylated lipid N-[2′,3′-bis(methylpolyoxyethyleneoxy)propane-1′-oxycarbonyl]-1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE-2arm-PEG-2k) was commercially sourced from NOF America Corporation (NY, USA). The PEGylated lipids screened in this study are listed in Table 1. The ionizable lipid dilinoleylmethyl-4-dimethylaminobutyrate (DLin-MC3-DMA; MC3) was purchased from MedChemExpress (NJ, USA). A murine neuron-targeting 17-mer ASO (M.W. 5635 g mol−1, Na-salt form) with phosphorothioate backbone was custom synthesized by BioSpring GmbH (Frankfurt, Germany) using solid-phase synthesis. All other reagents were DNase/RNase-free and used from their commercial sources without further purification.| PEG lipid # | PEG-lipid name | Lipid family | Charge | C-tail length | C-tail saturation | PEG size (Da) | PEG architecture |
|---|---|---|---|---|---|---|---|
| 1 | DMPE (C14:0)-PEG-0.55k | Phosphoglyceride-PE | Negative | 14 | Saturated | 550 | Linear |
| 2 | DMPE (C14:0)-PEG-1k | Phosphoglyceride-PE | Negative | 14 | Saturated | 1000 | Linear |
| 3 | DMPE (C14:0)-PEG-2k | Phosphoglyceride-PE | Negative | 14 | Saturated | 2000 | Linear |
| 4 | DPPE (C16:0)-PEG-1k | Phosphoglyceride-PE | Negative | 16 | Saturated | 1000 | Linear |
| 5 | DPPE (C16:0)-PEG-2k | Phosphoglyceride-PE | Negative | 16 | Saturated | 2000 | Linear |
| 6 | DSPE (C18:0)-PEG-0.55k | Phosphoglyceride-PE | Negative | 18 | Saturated | 550 | Linear |
| 7 | DSPE (C18:0)-PEG-1k | Phosphoglyceride-PE | Negative | 18 | Saturated | 1000 | Linear |
| 8 | DSPE (C18:0)-PEG-2k | Phosphoglyceride-PE | Negative | 18 | Saturated | 2000 | Linear |
| 9 | DSPE (C18:0)-2 arm-PEG-2k | Phosphoglyceride-PE | Negative | 18 | Saturated | 2000-B | Branched |
| 10 | DOPE (C18:1)-PEG-0.55k | Phosphoglyceride-PE | Negative | 18 | Unsaturated | 550 | Linear |
| 11 | DOPE (C18:1)-PEG-1k | Phosphoglyceride-PE | Negative | 18 | Unsaturated | 1000 | Linear |
| 12 | DOPE (C18:1)-PEG-2k | Phosphoglyceride-PE | Negative | 18 | Unsaturated | 2000 | Linear |
| 13 | DMG (C14:0)-PEG-2k | Diglyceride | Neutral | 14 | Saturated | 2000 | Linear |
| 14 | DSG (C18:0)-PEG-2k | Diglyceride | Neutral | 18 | Saturated | 2000 | Linear |
| 15 | Ceramide (C8)-PEG-0.75k | Ceramide | Neutral | 8 | Saturated | 750 | Linear |
| 16 | Ceramide (C8)-PEG-2k | Ceramide | Neutral | 8 | Saturated | 2000 | Linear |
| 17 | Ceramide (C16)-PEG-0.75k | Ceramide | Neutral | 16 | Saturated | 750 | Linear |
| 18 | Ceramide (C16)-PEG-2k | Ceramide | Neutral | 16 | Saturated | 2000 | Linear |
LNPs with various PEGylated lipids were prepared using a high-throughput approach reported previously.32 Briefly, the ASO was dissolved at 93.9 μg mL−1 in citrate buffer (25 mM, pH 4.0) and dispensed at 150 μL per well in a 96-well plate (Greiner Bio One 655101, NC, USA) using a TECAN Freedom EVO robotic liquid handler (Tecan Life Sciences, NC, USA). Using the automation setup, different lipid mixtures composed of MC3, DSPC, cholesterol, and respective PEG-lipid analogs were prepared in ethanol at a molar ratio of 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:(50 − X):X, where X = 1, 3, or 5; and a total lipid concentration of 4 mM was used to maintain N:P = 2 for the resulting ASO-LNPs. N:P is defined as the molar ratio of positively-chargeable amine (N) groups in the ionizable lipid to negatively-charged phosphate (P) groups on the nucleic acid backbone. The lipid mixtures were prepared in a 12-channel reservoir plate (Axygen RES-MW12-LP or -HP, NC, USA), and 50 μL of the lipid phases were injected into the ASO solutions in the 96-well plate using the robot (speed = 0.5 mL s−1, followed by 10 cycles of mixing with 0.1 mL volume per cycle), resulting in 1 mM total lipids per well at the ethanol:aqueous phase volume ratio of 1:3. Each LNP formulation was prepared in triplicate. The ASO-loaded LNPs were diluted in phosphate-buffered saline (PBS, pH 7.4) to achieve a final concentration of 1 μM total ASO per well. The free ASO amounts were measured using a size exclusion (SEC) high-performance liquid chromatography (HPLC) system (Agilent Technologies, Santa Clara, CA) with Tosoh TSKGel UP-SW2000 (4.6 mm × 150 mm dimension, particle size of 2.0 μm, and pore size of 12.5 nm). The total ASO payload in each formulation was quantified using Quant-iT™ Oligreen™ ssDNA assay (Thermo Fisher Scientific) following LNP disruption at 37 °C using 0.2% RNase-free Triton™ X-100 (MilliporeSigma, St. Louis, MO). The determined free and total ASO concentrations were used to calculate the encapsulation efficiency (EE%) for the identified hit formulations. Small aliquots of each sample were transferred into a glass-bottom 96-well plate (Greiner Bio-One 655892, NC, USA) and further diluted 50× in PBS for characterization of their particle size distributions by dynamic light scattering (DLS) using a DynaPro plate reader III (Wyatt Technology, CA, USA).
:10:(50 − X):X, where X = 1, 3, or 5; and a total lipid concentration of 4 mM was used to maintain N:P = 2 for the resulting ASO-LNPs. N:P is defined as the molar ratio of positively-chargeable amine (N) groups in the ionizable lipid to negatively-charged phosphate (P) groups on the nucleic acid backbone. The lipid mixtures were prepared in a 12-channel reservoir plate (Axygen RES-MW12-LP or -HP, NC, USA), and 50 μL of the lipid phases were injected into the ASO solutions in the 96-well plate using the robot (speed = 0.5 mL s−1, followed by 10 cycles of mixing with 0.1 mL volume per cycle), resulting in 1 mM total lipids per well at the ethanol:aqueous phase volume ratio of 1:3. Each LNP formulation was prepared in triplicate. The ASO-loaded LNPs were diluted in phosphate-buffered saline (PBS, pH 7.4) to achieve a final concentration of 1 μM total ASO per well. The free ASO amounts were measured using a size exclusion (SEC) high-performance liquid chromatography (HPLC) system (Agilent Technologies, Santa Clara, CA) with Tosoh TSKGel UP-SW2000 (4.6 mm × 150 mm dimension, particle size of 2.0 μm, and pore size of 12.5 nm). The total ASO payload in each formulation was quantified using Quant-iT™ Oligreen™ ssDNA assay (Thermo Fisher Scientific) following LNP disruption at 37 °C using 0.2% RNase-free Triton™ X-100 (MilliporeSigma, St. Louis, MO). The determined free and total ASO concentrations were used to calculate the encapsulation efficiency (EE%) for the identified hit formulations. Small aliquots of each sample were transferred into a glass-bottom 96-well plate (Greiner Bio-One 655892, NC, USA) and further diluted 50× in PBS for characterization of their particle size distributions by dynamic light scattering (DLS) using a DynaPro plate reader III (Wyatt Technology, CA, USA).
2.2 Animals
Timed pregnant C57BL/6N mice, dams, and pups from Charles River Laboratories (MA, USA) were used to prepare dissociated neuronal and microglial cultures. Upon delivery, a controlled 14:10 hour light:dark cycle was maintained, and animals were given unrestricted access to food and water. All animal studies were conducted in accordance with local regulations and the NRC Guide for the Care and Use of Laboratory Animals followed at IBioBA-CONICET. Animal studies were authorized and approved by the local as well as Genentech Inc. Institutional Animal Care and Use Committees.
2.3 Murine cortical neuron and microglia cell cultures
Mouse embryonic cortical neurons were cultured as described previously.48 Briefly, cortices from day 15 C57BL/6N embryos (E15) were dissected and washed thrice with pre-cooled Hank's balanced salt solution (HBSS; Invitrogen, CA, USA). Cortical tissue was incubated for 10 minutes at 37 °C in HBSS supplemented with 0.25% trypsin (Invitrogen) and DNase I (Roche CustomBiotech, IN, USA). Tissue was washed thrice with cold HBSS and triturated in plating media containing DNAse I (Gibco Neurobasal medium (Thermo Fisher Scientific, MA, USA), 20% heat-inactivated horse serum (Thermo Fisher Scientific), 25 mM sucrose, and 0.25% Gibco GlutaMAX (Thermo Fisher Scientific)). Dissociated cells were centrifuged at 125g for 5 minutes at 4 °C. Cells were resuspended in a plating medium and plated in 96-well, poly-L-lysine-coated (PLL; Sigma-Aldrich, MO, USA) plates. After 24 hours, the plating medium was replaced with Neurobasal medium supplemented with 1% B-27 (Thermo Fisher Scientific) and 0.25% Gibco GlutaMAX. Cells were maintained in an incubator at 37 °C with 5% CO2 and the medium was renewed using 50% exchange every 3–4 days to maintain cell health.Mouse microglia were cultured as described previously with the following alterations.49,50 Cortices from day 3 C57BL/6N pups (P3) were dissected, stripped of meninges, and washed thrice with pre-cooled HBSS (Invitrogen). Cortical tissue was incubated for 10 minutes at 37 °C in Gibco 0.25% trypsin–EDTA (Thermo Fisher Scientific). Cortices were washed thrice in culture medium (Gibco Dulbecco's Modified Eagle Medium (DMEM), high glucose supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin (Thermo Fisher Scientific)) and triturated. The dissociated cells were 70 μm strained and the cell suspension was centrifuged at 700g for 5 minutes at room temperature. The cell pellet was resuspended in the culture medium and plated in PLL-coated 225 cm2 flasks at a density of 8 cortices per flask. Cultures were maintained at 37 °C with 5% CO2. After 4 days, the attached cells were washed thrice with PBS and a fresh culture medium was added. Confluent astrocyte cultures containing suspended microglia were agitated on a rotary shaker at 200 rpm for 2 hours at 37 °C with 5% CO2 after an additional 7 days. The resulting microglia cell suspension was centrifuged at 700g for 5 minutes at room temperature. Cells were resuspended in culture medium, counted, and plated in 96-well PLL-coated plates (Sigma-Aldrich).
2.4 Cell treatments and RT-qPCR gene knockdown analysis
Murine cortical neurons or microglia were plated at a density of 3 × 105 cells per mL in 96-well plates. PBS, ASO, or ASO-loaded LNPs were added to the cortical neuron and microglia cultures at DIV6–8 and DIV1, respectively, at the concentrations indicated below and were incubated for 24 hours at 37 °C with 5% CO2. HTS samples were diluted to 1 μM ASO in PBS, then added directly to the neuron or microglia cell culture medium to achieve a final treatment dose of 50 nM ASO per well. Alternatively, ASO or ASO-LNPs were pre-diluted in neuronal medium to twice the desired final concentration, and 50% media exchange was performed to achieve the desired dosing range for the EC50 study. Neurons were treated with free ASO at final concentrations of 1, 10, 50, 250, 1000, 5000, and 10000 nM to analyze gymnosis and ASO-LNPs were added at 0.2, 1, 5, 25, 50, and 100 nM (final) ASO concentration.
Gene expression was analyzed after treatment for 24 hours using the TaqMan™ Fast Advanced Cells-to-CT™ Kit (Invitrogen, Thermo Fisher Scientific) following the manufacturer's instructions. Briefly, neuronal or microglial cultures were washed with PBS and lysed in the presence of DNase I. The lysis reactions were stopped and reverse transcription was performed at 37 °C for 30 minutes followed by a 95 °C 5 minutes inactivation using a Veriti™ 96-well Fast Thermal Cycler (Applied Biosystems, Thermo Fisher Scientific). Forty cycles of quantitative PCR (qPCR) were conducted at 95 °C for 1 s and 60 °C for 20 s using either a QuantStudio Flex or a ViiA 7 real-time PCR system (Applied Biosystems, Thermo Fisher Scientific). A FAM-MGB mouse probe targeting a neuronal/microglial gene of interest was used to quantify ASO-mediated mRNA knockdown efficiency. Gene expression values were calculated using the comparative delta delta Ct (−ΔΔCt) method using housekeeper gene (NeuN, neurons or CSF1R, microglia) and PBS-treated Ct values for normalization. VIC-MGB mouse probes for Rbfox3/NeuN (Mm01248771_m1) and Csf1r (Mm01266652_m1) were obtained from Life Technologies, Thermo Fisher Scientific.
2.5 Small-angle X-ray scattering
SAXS data were collected in the high throughput mode (HT-SAXS) using the Advanced Light Source SIBYLS beamline 12.3.1 at the Lawrence Berkeley National Laboratory (CA, USA)51 on representative ASO-LNP formulation hits at a total lipid concentration of 1 mM. X-ray wavelength was set at λ = 1.216 Å, and the sample-to-detector distance was 2070 mm, resulting in a scattering vector, q, ranging from 0.01 Å−1 to 0.45 Å−1. The scattering vector is defined as q = 4πsinθ/λ, where 2θ is the scattering angle. Experiments were performed at 20 °C as described elsewhere.52 Briefly, the sample was exposed for 10 s with the detector framing at 0.3 s to maximize the signal while merging the SAXS signal using the SAXS FrameSlice application (https://bl1231.als.lbl.gov/ran). No radiation damage was observed during the 10 s exposure, and all collected frames were merged. The merged SAXS profile was further processed using the SCÅTTER package (https://www.bioisis.net/tutorials) and plotted using the OriginPro 2015 (OriginLab Corporation, MA, USA).
2.6 Microfluidic preparation and characterization of ASO-loaded LNPs
A microfluidic mixing method was used for the scale-up preparation of selected ASO-LNP formulations that were identified as either positive or negative hits by the HTS approach.32 Lipid mixtures that replicate the selected LNP compositions from HTS were manually prepared and dissolved in ethanol at a total lipid concentration of 4 mM. The ethanol stream was rapidly mixed with an aqueous stream containing 93.9 μg mL−1 ASO dissolved in citrate buffer (25 mM, pH 4.0) using a microfluidic laminar mixing device (NanoAssemblr™ Benchtop, Precision NanoSystems, BC, Canada) at a 1:3 volume ratio, and a total flow rate of 12 mL min−1. The formulated LNPs were purified by centrifugal ultrafiltration (MWCO 10 kDa; Amicon, MilliporeSigma, MA, USA) at 2000g for 30 min to remove free ASO and lipids, followed by buffer exchange to RNase-free PBS. Purified formulations were analyzed for their particle size distributions using DLS and ASO payload concentration as described in Section 2.1.
2.7 Statistical analysis
Data plotting and statistical analysis were performed using Prism 9.2.0 (GraphPad Software, San Diego, CA). All results are presented as mean ± SEM, n = 3 containing 3 averaged internal replicates. To determine EC50 values for ASO dose–response treatments, data were analyzed using non-linear regression, and curves were fitted using log (inhibitor concentration) vs. response, variable slope (four parameters) equations. The impact of various PEG-lipid parameters on ASO-LNP delivery and subsequent mRNA expression was analyzed using a non-parametric Kruskal–Wallis test followed by post hoc comparisons using Dunn's test, at a 95% confidence interval (p < 0.05). Linear regression analyses on the particle size datasets were performed using Microsoft® Excel 16.39. Mann–Whitney test was used at the 95% confidence interval (p < 0.05) to compare the average hydrodynamic diameters of the best and worst 10% LNP hits identified based on their in vitro efficacies.3. Results and discussion
3.1 Formulation of ASO-LNP library with diverse PEG-lipids
In our HTS lipid library design, DLin-MC3-DMA (MC3), DSPC, and cholesterol were chosen as the constituent lipids for all ASO-LNP formulations, while the PEG-lipid type and content were varied (Fig. 1A). The combination of MC3, DSPC, and cholesterol (Fig. 1B) is used in ONPATTRO®, the first FDA-approved siRNA-LNP formulation,53–55 and is also a benchmark for oligonucleotide delivery in many preclinical LNP model systems.25,56,57 To systematically understand the effect of PEG-lipid properties on LNP cargo delivery, several PEG-lipid analogs were incorporated into the ASO-LNPs (Table 1). PEG-lipids commonly used in drug delivery applications were chosen from anionic phosphoglycerides, as well as neutral diglycerides and ceramides.58,59 While all of these lipid families are biologically relevant, their chemistries differ significantly. Both phosphoglycerides and diglycerides are composed of fatty acids attached to a glycerol backbone with or without a terminal phosphatidyl ester group, respectively.60 In contrast, ceramide lipids contain fatty acids conjugated to a sphingoid base through an amide bond.61 These lipid families also have distinct linker chemistries for conjugation of the PEG moiety to the lipidic anchor. Typically, PEG is conjugated to phosphoglyceride lipids via a carbamate linkage resulting in a net negative charge on the phosphate group at physiological pH.47 Diglyceride and ceramide lipid anchors are conjugated to PEG via ether and ester linkages, respectively.47,62 In order to understand the impact of linker chemistry on ASO-LNP structure and function, multiple analogs from each of these PEG-lipid families were included while also varying C-tail anchor lengths and/or PEG chain sizes (Fig. 1C). The impacts of PEG architecture (linear or branched) and PEG-lipid C-tail saturation were also assessed. | ||
| Fig. 1 ASO-LNP formulation library. (A) Lipid nanoparticles were prepared using a liquid handling robot by rapidly mixing an aqueous phase containing ASO with an ethanol phase containing dissolved lipid mixtures with varying PEG-lipid compositions. (B and C) Each lipid mixture comprised of the ionizable lipid MC3, helper lipid DSPC, and cholesterol, in combination with a distinct PEG-lipid selected from the phosphoglyceride-PE, diglyceride, or ceramide families to generate an ASO-LNP library of 54 unique formulations. | ||
In addition to investigating different PEG-lipid species, PEG-lipid molar ratio with respect to the total lipid content in the formulation was also varied. The molar ratio of PEG-lipids was tuned to 1, 3, or 5 mol% by adjusting the molar ratio of cholesterol in the lipid mixture.63 Using these PEG-lipid parameters, a library of 54 distinct ASO-LNP formulations was prepared using a 96-well plate-based HTS workflow for physicochemical characterization and in vitro efficacy evaluation.
3.2 PEG content governs ASO-LNP particle size distribution
Previous studies suggest that PEG-lipids play an important role in particle size regulation during self-assembly.32–35 For the HTS-generated ASO-LNPs, DLS measurements generally indicated there is a negative correlation between particle sizes and molar ratios of PEGylated lipids in the LNP formulations, with mean hydrodynamic diameters ranging between 50–220 nm across the library (Fig. 2A and B). LNPs with short PEG-lipids (PEG M.W. <1000 Da) formulated at 1 mol%, e.g. #6 and #10, had the largest particle diameter of 212 nm. In contrast, LNPs containing 5 mol% of long PEG-lipids generally produced small nanoparticles. For example, PEG-lipid #9 showed the smallest mean particle diameter of 52 nm at 5 mol%. This trend is likely due to increased steric hindrance by the PEG chains when they are present at high molar ratios or M.W., which leads to smaller particle sizes by preventing particle growth and aggregation. In general, smaller nanoparticles were formed as PEGylated-lipid molar ratios were increased from 1 to 5%. Specifically, anionic PEG-lipids showed a distinct decrease in particle diameters with increasing PEG sizes and PEG molar ratios in the LNP composition (Fig. 2A). These findings suggest that both the repulsive forces between charged head groups and the steric barrier of PEG chains regulate particle size distributions of LNPs formulated with anionic PEG-lipids. In contrast, LNPs prepared with neutral diglyceride or ceramide PEG-lipids showed only a weak dependence of particle size on PEG content, with no correlation seen for formulations #13 and #16 (Fig. 2B). Additionally, a comparison of linear and branched PEG variants of DSPE-PEG-2k (#8 and #9, respectively) showed no significant effects of PEG architecture on hydrodynamic diameter (Fig. 2A).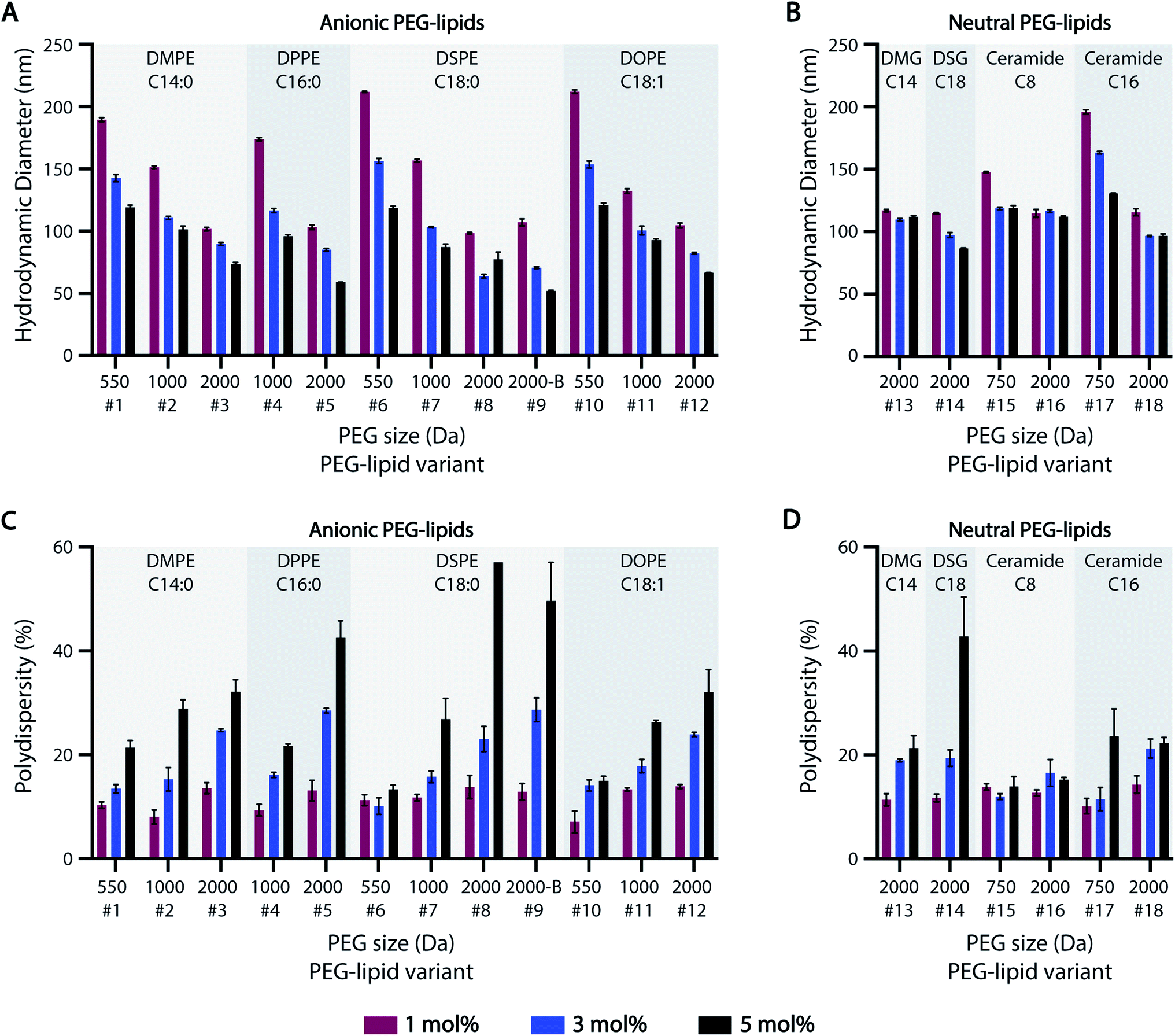 | ||
| Fig. 2 Particle size distribution of ASO-LNP formulations. (A and B) Particle sizes and (C and D) polydispersities of ASO-LNPs containing different subsets of anionic or neutral PEG-lipids, as determined by DLS measurements. | ||
While increasing PEG-lipid molar ratio showed charge-dependent effects on particle diameter, PEG-lipid content had an overall positive correlation with LNP polydispersity (% PD) (Fig. 2C and D). Notable exceptions to this trend were #6, #15, and #16. Formulations #15 and #16 contained neutral ceramide-C8 PEG-lipids, which also showed little to no correlation between PEG-lipid content and particle size (Fig. 2B). In agreement with our previous data,32 PEG-lipids formulated at 5 mol% with long PEG arms (2000 Da) had high polydispersity, possibly due to the presence of micellar PEG-lipid sub-populations.64,65
The effect of lipidic anchor groups with different C-tail features was also analyzed (Fig. 2). For ASO-LNPs formulated with the same molar ratio of PEG-lipids, lipid tail length (DMPE vs. DPPE vs. DSPE, DSG vs. DMG, and Cer-C8 vs. Cer-C16), saturation levels (DSPE vs. DOPE), and linker chemistries (DMPE vs. DMG, and DSPE vs. DSG) did not significantly affect LNP sizes or polydispersity indices. These data contrast with the distinct impact of the hydrophilic PEG blocks on LNP properties. Similar observations have been previously reported for siRNA-LNPs prepared with PEG-lipids containing 14, 16, or 18-carbon chains.36
Taken together, these data suggest that LNP size distribution is primarily dependent on surface stabilizing PEG content rather than lipid-tail attributes of PEG-lipids, and this PEG-dependence is especially dominant in anionic PEG-lipid scaffolds.
3.3 PEG-lipid carbon-tails differentially regulate ASO-LNP in vitro efficacy
The global burden from neurodegenerative diseases has contributed to a dramatic increase in brain-targeting therapeutics, including ASOs.66,67 While ASOs alter mRNA expression levels or splicing in a design-dependent manner, increasing cellular uptake and delivery efficiency is essential to improve ASO efficacy.68,69 To this end, our HTS-generated ASO-LNP library was used to examine how various PEG-lipid parameters affect downstream ASO delivery and efficacy in primary murine cortical neurons. Cortical neurons are a relevant model for many neurodegenerative diseases and enhancing ASO delivery to these historically difficult-to-transfect cells is essential for therapeutic development.The majority of ASO-LNP formulations reduced mRNA expression levels in cortical neurons relative to gymnosis (naked ASO) controls, suggesting that encapsulating ASOs in LNPs improves intracellular delivery (Fig. 3A). Among the various PEG-lipid characteristics examined, carbon-tail length played a significant role in determining the extent of mRNA downregulation, with longer carbon chains resulting in a stepwise increase in mRNA expression levels (Fig. 3B). This can be explained by the poor PEG-shedding properties exhibited by PEG-lipids with longer hydrophobic lipid anchors, as extensively demonstrated in previous literature.34,36,44,70,71 PEG-lipids with short acyl chains have faster desorption from the LNP surface owing to favorable thermodynamics for cleaving anchoring bonds. This trend was observed for both anionic phosphoglyceride PEG-lipids, as well as neutral diglyceride and ceramide PEG-lipid-based formulations (Fig. S1A†). Comparison of phosphoglyceride PEG-lipids with saturated and unsaturated carbon-tails showed no major impact on mRNA expression with PEG sizes <1000 Da (#6 vs. #10). However, mRNA reduction improved for 1000 and 2000 Da PEG-lipid variants when a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond was introduced in the lipid tail (#7 vs. #11, #8 vs. #12), possibly due to the relatively lower stability of the unsaturated chains allowing for faster PEG-lipid shedding (Fig. S1B†). Furthermore, in vitro efficacy was also affected by the PEG-linker chemistry of the PEG-lipids. For example, DMG-C14-PEG-2k (#13) showed a higher reduction in mRNA expression compared to DMPE-(14:0)-PEG-2k (#3), at all molar ratios studied. This result can be attributed to the tailored structure of DMG–PEG facilitating rapid PEG dissociation from the LNP surface, which enhances transfection.24,72 Similarly, ceramide-C16-PEG-2k (#18) performed better than the homologous phosphoglyceride PEG-lipid DPPE-(16:0)-PEG-2k (#5). These data are in congruence with previous studies reporting favorable conformational properties of ceramide-PEGs, and their better exchangeability with lipid cell membranes in comparison to their anionic phosphoglyceride counterparts.42,43,62 Furthermore, the rapidly hydrolyzing ester linkages between the PEG and ceramide backbone may lead to faster PEG shedding from the LNP surface, as compared with amide-linked PEG in phosphoglycerides.73 Thus, these data show that ASO-LNP delivery not only depends on PEG-lipid tail length, but also on the PEG-lipid linker chemistry.
C bond was introduced in the lipid tail (#7 vs. #11, #8 vs. #12), possibly due to the relatively lower stability of the unsaturated chains allowing for faster PEG-lipid shedding (Fig. S1B†). Furthermore, in vitro efficacy was also affected by the PEG-linker chemistry of the PEG-lipids. For example, DMG-C14-PEG-2k (#13) showed a higher reduction in mRNA expression compared to DMPE-(14:0)-PEG-2k (#3), at all molar ratios studied. This result can be attributed to the tailored structure of DMG–PEG facilitating rapid PEG dissociation from the LNP surface, which enhances transfection.24,72 Similarly, ceramide-C16-PEG-2k (#18) performed better than the homologous phosphoglyceride PEG-lipid DPPE-(16:0)-PEG-2k (#5). These data are in congruence with previous studies reporting favorable conformational properties of ceramide-PEGs, and their better exchangeability with lipid cell membranes in comparison to their anionic phosphoglyceride counterparts.42,43,62 Furthermore, the rapidly hydrolyzing ester linkages between the PEG and ceramide backbone may lead to faster PEG shedding from the LNP surface, as compared with amide-linked PEG in phosphoglycerides.73 Thus, these data show that ASO-LNP delivery not only depends on PEG-lipid tail length, but also on the PEG-lipid linker chemistry.
 | ||
| Fig. 3 ASO-LNP gene silencing efficacy screening in primary murine cortical neurons. (A) Gene silencing was analyzed using RT-qPCR. Data were normalized to NeuN and PBS-treated controls, and are shown as relative to gymnosis (dotted line). ASO-LNP formulations were re-grouped based on their (B) C-tail length, (C) PEG size, and (D) molar ratio of the PEG-lipids to assess the significance of each parameter on ASO-LNP in vitro efficacy (Kruskal–Wallis test and Dunn's test, *p < 0.05, ***p < 0.001, ****p < 0.0001, ns: non-significant). | ||
Unlike C-tail length, PEG M.W. and mol% did not have a significant bearing on LNP delivery and ASO-mediated mRNA downregulation (Fig. 3C and D). For example, no major impact was observed across PEG-lipid analogs containing various PEG sizes and 18-C tails formulated at 5 mol% PEG-lipid (Fig. S1C†). Similarly, mRNA expression was not affected by different PEG architectures of the PEG-lipids, e.g. linear (#8) vs. branched (#9) variants of DSPE-PEG-2k. However, additional lipids with branched PEG arms should be investigated to establish a reliable correlation between PEG architecture and LNP in vitro efficacy.
Thus, these HTS data suggest that the gene-silencing efficacy of PEGylated ASO-LNPs strongly depends on their PEG-shedding ability, as governed by the C-tail features and linker chemistry, to allow desirable membrane interactions for intracellular delivery. Future studies will focus on understanding the mechanism of ASO-LNP cellular internalization and endosomal escape that results in ASO-mediated mRNA downregulation.
3.4 HTS can be leveraged to identify ASO-LNP behavioral trends
As demonstrated, our HTS approach allows for the quick preparation and characterization of diverse ASO-LNP formulations in a 96-well plate format. This high-throughput workflow can be seamlessly extended to evaluate LNP delivery in additional target cell lines (see Section 3.6). Furthermore, owing to the small working volume and parallel sample handling, our HTS approach leads to significant material and time savings. It also generates robust datasets by directly comparing ASO-LNP formulations in identical environments, thus minimizing processing variations.In addition to the empirical benefits, our HTS approach also offers data analysis and interpretation advantages. First, physical characterization (Fig. 2) and efficacy data (Fig. 3) involving numerous formulations are generated in short timeframes. Second, comprehensive screening allows for the identification of correlations that may be masked with approaches involving narrow sample sizes. In this study, our HTS datasets showed that hydrophilic blocks of PEG-lipids govern LNP particle size distributions (Fig. 2 and S2†), whereas the hydrophobic lipid tails regulate the cellular interactions necessary for LNP uptake and ASO-mediated mRNA downregulation (Fig. 3). These behavioral trends can also be defined quantitatively using predictive correlations and regression analyses. For instance, linear regression models corroborated the significance of PEG-lipid charge, molar ratio, and PEG size on LNP particle size distributions (p < 0.05), as compared to the non-significant impact of different C-tail attributes (Sections S1A and S1B). The correlation accuracy, as represented by R2 values of 0.869 and 0.700 for anionic and neutral PEG-lipids respectively, can be further improved using iterative screening of wider sample sets, in combination with higher-order curve fittings or advanced machine learning algorithms.74
Our HTS approach is also useful for rank-ordering ASO-LNP formulations based on mRNA downregulation (Fig. 4A). On a heat map, formulations with relative mRNA expression levels lower or higher than gymnosis are depicted as positive (green) or negative (red) hits, respectively. Correlating ASO-LNP efficacy with the corresponding DLS measurements (Fig. 4B) revealed that a 100–150 nm particle size range was optimal for achieving reduced mRNA levels in cortical neurons (Fig. 4C and D). Formulations within this range can be referenced back to their constituent PEG-lipid features to identify optimal LNP compositions for neuronal delivery. Taken together, these HTS findings suggest that a combination of PEG-lipids with shorter C-tails and lower PEG-lipid mol% in LNPs can result in improved in vitro activity, which is in agreement with findings from previous LNP literature.53,75,76
 | ||
| Fig. 4 Behavioral trends across the ASO-LNP HTS library. The HTS datasets from Fig. 2 and 3 are presented using color-coded heat maps to rank-order the 54 ASO-LNP formulations based on their in vitro efficacies (A) and average particle sizes (B). Representative positive and negative hits are highlighted by dotted and solid rectangles, respectively. (C and D) The correlation between LNP diameter and relative mRNA expression in the top and bottom 10% of ASO-LNP hits suggests an optimal size range of 100–150 nm for efficacious formulations (Mann–Whitney test, *p < 0.05). | ||
3.5 ASO-LNP hits exhibit distinct delivery dynamics and structural features
Motivated by the clinical relevance of their constituent PEG-lipids, further characterization of ASO-LNP formulations #13-1% (neutral PEG) and #8-3% (anionic PEG) was performed. Diglyceride DMG(C14)-PEG-2k (PEG-lipid in #13-1%) has been used in many preclinical and clinical LNP formulations, including ONPATTRO®. Thus, there is a mounting interest in understanding the function of this PEG-lipid analog.53–55 On the other hand, DSPE(18:0)-PEG-2k (PEG-lipid in #8-3%) has been widely used for nanoparticle surface functionalization, owing to its established clinical performance in DOXIL liposomal formulations.77 Formulation #13-1% reduced mRNA expression in cortical neurons by ∼80%, whereas #8-3% showed no significant change when normalized to gymnosis (Fig. 3A). The delivery dynamics and internal structures of these ASO-LNPs were further examined to understand their observed efficacy differences in this study.Dose–response curves were generated in cortical neurons to validate the differential activities of ASO-LNPs #13-1% and #8-3%, and the results were compared to gymnosis. Treating cortical neurons with free ASO resulted in an EC50 value of 1216 nM (Fig. 5A). In contrast, ASO-LNPs showed varied efficacies based on composition. While the positive hit #13-1% had an EC50 value of 3.6 nM, the negative hit #8-3% did not reach a 50% reduction in mRNA expression over the same dosing range (Fig. 5B). Taken together, these data showed that ASO activity is concentration-dependent during gymnosis and LNP delivery. Furthermore, in this cell culture system, LNPs can dramatically and differentially improve ASO delivery as compared to ASO internalized via gymnosis, depending on the PEG-lipid used. Further in vivo studies are needed to understand the translatability of these in vitro efficacy findings.
 | ||
| Fig. 5 Characterization of representative ASO-LNP positive and negative hits. Dose-response curves were generated in cortical neurons by analyzing relative mRNA expression using RT-qPCR for (A) gymnosis, and (B) positive (green) and negative (red) ASO-LNP hits. Data were normalized to NeuN and PBS-treated control cells. (C) Structural features of the positive (green) and negative (red) ASO-LNP hits were analyzed using small-angle X-ray scattering. The repeat spacing of lipid/ASO/water layer lamellar phase is reflected as the peak maxima at q ∼ 0.13 Å−1. A lower intensity peak of #8-3% LNP compared to the #13-1% LNP corresponds to smaller periodicity along with the lamellar phase. The broadening of the peaks for #8-3% LNP suggest a disorder of the lamellar phase. | ||
The representative positive and negative ASO-LNP hits, #13-1% and #8-3%, were further characterized using SAXS to discern the underlying structure–function relationship governing their distinct cellular activities (Fig. 5C). Both LNP formulations showed the presence of one prominent peak in the q range of 0.1–0.2 Å−1, derived from the repetition of lipid membrane/ASO/water layers in the particle structure.78 This is in agreement with previously observed SAXS profiles of cationic lipid–DNA complexes (lipoplexes) coated with PEG-lipids42 or siRNA-loaded LNPs.79,80 The repeat spacing d (thickness of lipid/ASO/water layer) can be quantified from the SAXS profiles as d = 2π/q. Thus, the peak positions at q = 0.129 Å−1 for #13-1% ASO-LNPs and q = 0.131 Å−1 for #8-3% ASO-LNPs indicate a relatively thicker lamellar layer for the #13-1% formulation as compared to #8-3% formulation (48.7 Å for #13-1% vs. 47.8 Å for #8-3%). Moreover, the most significant structural differences between the two formulations can be elucidated through the width and height of their respective SAXS peaks. Lower peak height, as observed for #8-3% ASO-LNPs, correlates to a fewer number of the lamellar layers, and is consistent with the significantly smaller particle size (63.9 nm for #8-3% vs. 116.9 nm for #13-1%, Fig. 2A and B). Furthermore, we hypothesize that broadening of the peak and increased signal intensity when q < 0.11 Å−1 suggests a disordered LNP core structure for #8-3% ASO-LNPs; whereas the #13-1% ASO-LNPs adopt a more ordered lamellar structure that promotes cellular transfection. The absence of the ‘nucleic acid peak’, which is a fingerprint of the lipoplex structure generated by the nucleic acid packed in a hexagonal lattice,81,82 suggests that the ASO is not densely packed within the LNPs and may lead to a faster intracellular release of the cargo. Based on these preliminary SAXS analyses, it is hypothesized that the equilibrium between lamellar layers and a disordered core plays an essential role in determining the delivery efficacy of ASO-LNPs. The precise characterization of the structural organization of ASO-LNPs will be the subject of in-depth investigations in the future.
3.6 HTS results translate to additional cell culture systems and scaled-up formulations
Our HTS approach, like all screening assays, only becomes valuable if the results can be smoothly translated across additional cell culture systems and scaled-up formulations. Therefore, in order to validate the translatability of our HTS results, three positive hits (#13-1%, #16-3%, #1-5%) and three negative hits (#10-1%, #8-3%, #9-5%) were chosen from the HTS library based on their in vitro efficacies. To ensure robust validation of HTS predictability across a broad range of LNP features, the representative formulations included diglyceride (#13), ceramide (#16), and phosphoglyceride (#1) PEG-lipids with different lipid tail saturation levels (#10 vs. #8), PEG architectures (#9), and PEG-lipid contents (1, 3, 5 mol%).To determine if the results generated in cortical neurons translate across unique cell types and culture systems, the six hit formulations were tested for ASO-mediated mRNA downregulation in murine microglia cultures. Microglia are the tissue-resident macrophages of the brain and are key targets for treating neurodegenerative diseases.83 Furthermore, unlike neurons, microglia require serum-containing media to thrive in culture and are highly phagocytic, making these cells a unique comparison to our previous HTS data. Formulations #13-1%, #16-3%, and #1-5% reduced mRNA levels in microglia by ∼80%. In contrast, formulations #10-1%, #8-3%, and #9-5% had no activity (Fig. 6A). These data are consistent with our efficacy screening results in cortical neurons (Fig. 6A and 3) and support our methodology as a broadly applicable screening tool across cell culture systems, including in the presence or absence of serum proteins.
 | ||
| Fig. 6 ASO-LNP properties translate across cell types and formulation scales. (A) Selected positive and negative ASO-LNP hits were screened in primary microglia for their gene silencing activity as analyzed using RT-qPCR. Data are normalized to CSF1R and PBS-treated controls, and are shown relative to gymnosis (dotted line). Gene silencing results in cortical neurons were obtained from the HTS experiment shown in Fig. 3. (B) Particle sizes and (C) in vitro gene silencing efficacies of the selected ASO-LNPs were compared across the two formulation scales and techniques to validate the translatability of LNP performance. Results are presented as mean ± SEM, n = 3 containing 3 averaged internal replicates. | ||
We also studied the translatability of our HTS results to scaled up formulations using a microfluidic mixer commonly used in manufacturing.80,84,85 The ASO-LNP hits were scaled up 10× to generate ∼2 mL of each formulation using NanoAssemblr™ Benchtop with the same compositions as their HTS counterparts. DLS characterization showed that the particle sizes were comparable across both formulation techniques (Fig. 6B). ASO encapsulation efficiencies (EE%) of the six hit LNP formulations prepared by HTS and microfluidic methods were comparable with a slight decreasing trend for higher PEG molar ratios (Fig. S3†), which aligns with our previously reported results.32 Importantly, the scaled-up ASO-LNP formulations also showed comparable mRNA expression in mouse neurons, relative to their corresponding HTS formulations (Fig. 6C). Irrespective of the preparation technique, positive hit formulations (#13-1%, #16-3%, and #1-5%) showed a 4 to 5-fold reduction in relative mRNA expression, compared to negative hit formulations (#10-1%, #8-3%, and #9-5%). Taken together, these results highlight the reliable and predictive nature of our HTS approach in translating ASO-LNP physical characteristics and in vitro efficacies. This translation is especially noteworthy considering the processing differences between the HTS and microfluidics methods such as mixing patterns, downstream processing, final buffer environment, and preparation scale. Despite these differences, the HTS approach reliably predicted ASO-LNP physicochemical properties and cellular efficacy of microfluidics-based formulations. Microfluidics technology has been adapted for use in preclinical and clinical trials due to the ability to scale LNP-based drug delivery vehicles from milliliters to liters. Thus, our predictive HTS approach can be used to screen formulations prior to scale-up, saving significant resources that are typically required for optimizing formulations on the microfluidics platform.
4. Conclusions
We designed an HTS approach to understand how ASO-LNP size distribution and cellular delivery vary as a function of PEG-lipid characteristics including PEG size, PEG-lipid content, and carbon-tail structures. The identified ASO-LNP positive hit formulations decreased mRNA expression by 4 to 5-fold in primary cortical neurons and microglia compared to gymnosis. These data demonstrate the translatability of our HTS methodology across unique biologically-relevant cell types and culture systems, including in the presence and the absence of serum proteins. In addition, a smooth translation of size distributions as well as cellular activity between HTS formulations and those prepared using the microfluidic mixing technology was demonstrated, validating HTS as a predictive tool to guide formulation development. By narrowing down hits using HTS, promising formulations can be scaled-up for in vivo safety analysis and efficacy profile generation with significant time and material savings. Therefore, with the ability to screen a wide range of LNP parameters and effectively predict in vitro performance, this HTS approach represents a promising and cost-effective strategy to rapidly optimize multivariate LNP-based formulations and advance selected candidates during early-stage development.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Ben Chih and Mr Kevin Granger for their contributions towards the early development of the in vitro efficacy assay, Dr Alexandre Goyon for the SEC method development to quantify free ASO, and Dr Kelly Zhang for the review of this manuscript. Funding for the SIBYLS beamline at the Advanced Light Source was provided in part by the Offices of Science and Biological and Environmental Research, U.S. Department of Energy, under Contract DE-AC02-05BH11231 and NIGMS grant P30 GM124169-01, ALS-ENABLE. Schematics available from https://BioRender.com were used in the preparation of the graphical abstract.References
- T. C. Roberts, R. Langer and M. J. A. Wood, Advances in oligonucleotide drug delivery, Nat. Rev. Drug Discovery, 2020, 19, 673–694 CrossRef CAS PubMed.
- K. Sridharan and N. J. Gogtay, Therapeutic nucleic acids: current clinical status, Br. J. Clin. Pharmacol., 2016, 82, 659–672 CrossRef PubMed.
- D. D. Fusco, V. Dinallo, I. Marafini, M. M. Figliuzzi, B. Romano and G. Monteleone, Antisense Oligonucleotide: Basic Concepts and Therapeutic Application in Inflammatory Bowel Disease, Front. Pharmacol., 2019, 10, 305 CrossRef PubMed.
- S. Vasudevan, Posttranscriptional Upregulation by MicroRNAs: Posttranscriptional Upregulation by MicroRNAs, Wiley Interdiscip Rev Rna, 2011, 3, pp. 311–330 Search PubMed.
- K. Chen, B. S. Zhao and C. He, Nucleic Acid Modifications in Regulation of Gene Expression, Cell Chem. Biol., 2016, 23, 74–85 CrossRef CAS PubMed.
- M. Elsabahy, A. Nazarali and M. Foldvari, Non-Viral Nucleic Acid Delivery: Key Challenges and Future Directions, Curr. Drug Delivery, 2011, 8, 235–244 CrossRef CAS PubMed.
- B. Shi, M. Zheng, W. Tao, R. Chung, D. Jin, D. Ghaffari and O. C. Farokhzad, Challenges in DNA Delivery and Recent Advances in Multifunctional Polymeric DNA Delivery Systems, Biomacromolecules, 2017, 18, 2231–2246 CrossRef CAS PubMed.
- B. Nogrady, The challenge of delivering RNA-interference therapeutics to their target cells, Nature, 2019, 574, S8–S9 CrossRef CAS PubMed.
- L. Johannes and M. Lucchino, Current Challenges in Delivery and Cytosolic Translocation of Therapeutic RNAs, Nucleic Acid Ther., 2018, 28, 178–193 CrossRef CAS PubMed.
- V. Patravale, P. Dandekar and R. Jain, Nanopart. Drug Delivery, 2012, 29–85 Search PubMed.
- D. Adams, A. Gonzalez-Duarte, W. D. ORiordan, C.-C. Yang, M. Ueda, A. V. Kristen, I. Tournev, H. H. Schmidt, T. Coelho, J. L. Berk, K.-P. Lin, G. Vita, S. Attarian, V. Planté-Bordeneuve, M. M. Mezei, J. M. Campistol, J. Buades, T. H. Brannagan III, B. J. Kim, J. Oh, Y. Parman, Y. Sekijima, P. N. Hawkins, S. D. Solomon, M. Polydefkis, P. J. Dyck, P. J. Gandhi, S. Goyal, J. Chen, A. L. Strahs, S. V. Nochur, M. T. Sweetser, P. P. Garg, A. K. Vaishnaw, J. A. Gollob and O. B. Suhr, Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis, N. Engl. J. Med., 2018, 379, 11–21 CrossRef CAS PubMed.
- H. Ledford, US authorization of first COVID vaccine marks new phase in safety monitoring, Nature, 2020, 588, 377–378 CrossRef CAS PubMed.
- S. P. Teo, Review of COVID-19 mRNA Vaccines: BNT162b2 and mRNA-1273, J. Pharm. Pract., 2021, 089719002110096 CrossRef PubMed.
- T. T. H. Thi, E. J. A. Suys, J. S. Lee, D. H. Nguyen, K. D. Park and N. P. Truong, Lipid-Based Nanoparticles in the Clinic and Clinical Trials: From Cancer Nanomedicine to COVID-19 Vaccines, NATO Adv. Study Inst. Ser., 2021, 9, 359 CAS.
- T. Wang, Y. Suita, S. Miriyala, J. Dean, N. Tapinos and J. Shen, Advances in Lipid-Based Nanoparticles for Cancer Chemoimmunotherapy, Pharm, 2021, 13, 520 CAS.
- R. Shankar, M. Joshi and K. Pathak, Lipid Nanoparticles: A novel approach for brain targeting, Pharm. Nanotechnol., 2018, 6, 81–93 CrossRef CAS PubMed.
- J. A. Kulkarni, P. R. Cullis and R. van der Meel, Lipid Nanoparticles Enabling Gene Therapies: From Concepts to Clinical Utility, Nucleic Acid Ther., 2018, 28, 146–157 CrossRef CAS PubMed.
- S. D. Campbell, K. J. Regina and E. D. Kharasch, Significance of Lipid Composition in a Blood–Brain Barrier–Mimetic PAMPA Assay, J. Biomol. Screening, 2013, 19, 437–444 CrossRef PubMed.
- T. Andresen, J. Bruun, T. B. Larsen, R. I. Jølck, R. Eliasen, R. Holm and T. Gjetting, Investigation of enzyme-sensitive lipid nanoparticles for delivery of siRNA to blood–brain barrier and glioma cells, Int. J. Nanomed., 2015, 10, 5995–6008 CrossRef PubMed.
- R. M. Ray, A. H. Hansen, M. Taskova, B. Jandl, J. Hansen, C. Soemardy, K. V. Morris and K. Astakhova, Enhanced target cell specificity and uptake of lipid nanoparticles using RNA aptamers and peptides, Beilstein J. Org. Chem., 2021, 17, 891–907 CrossRef CAS PubMed.
- Let's talk about lipid nanoparticles, Nat. Rev. Mater., 2021, 6, 99 Search PubMed.
- M. A. Maier, M. Jayaraman, S. Matsuda, J. Liu, S. Barros, W. Querbes, Y. K. Tam, S. M. Ansell, V. Kumar, J. Qin, X. Zhang, Q. Wang, S. Panesar, R. Hutabarat, M. Carioto, J. Hettinger, P. Kandasamy, D. Butler, K. G. Rajeev, B. Pang, K. Charisse, K. Fitzgerald, B. L. Mui, X. Du, P. Cullis, T. D. Madden, M. J. Hope, M. Manoharan and A. Akinc, Biodegradable Lipids Enabling Rapidly Eliminated Lipid Nanoparticles for Systemic Delivery of RNAi Therapeutics, Mol. Ther., 2013, 21, 1570–1578 CrossRef CAS PubMed.
- R. Kanasty, J. R. Dorkin, A. Vegas and D. Anderson, Delivery materials for siRNA therapeutics, Nat. Mater., 2013, 12, 967–977 CrossRef CAS PubMed.
- S. C. Semple, A. Akinc, J. Chen, A. P. Sandhu, B. L. Mui, C. K. Cho, D. W. Y. Sah, D. Stebbing, E. J. Crosley, E. Yaworski, I. M. Hafez, J. R. Dorkin, J. Qin, K. Lam, K. G. Rajeev, K. F. Wong, L. B. Jeffs, L. Nechev, M. L. Eisenhardt, M. Jayaraman, M. Kazem, M. A. Maier, M. Srinivasulu, M. J. Weinstein, Q. Chen, R. Alvarez, S. A. Barros, S. De, S. K. Klimuk, T. Borland, V. Kosovrasti, W. L. Cantley, Y. K. Tam, M. Manoharan, M. A. Ciufolini, M. A. Tracy, A. de Fougerolles, I. MacLachlan, P. R. Cullis, T. D. Madden and M. J. Hope, Rational design of cationic lipids for siRNA delivery, Nat. Biotechnol., 2010, 28, 172–176 CrossRef CAS PubMed.
- M. Jayaraman, S. M. Ansell, B. L. Mui, Y. K. Tam, J. Chen, X. Du, D. Butler, L. Eltepu, S. Matsuda, J. K. Narayanannair, K. G. Rajeev, I. M. Hafez, A. Akinc, M. A. Maier, M. A. Tracy, P. R. Cullis, T. D. Madden, M. Manoharan and M. J. Hope, Maximizing the Potency of siRNA Lipid Nanoparticles for Hepatic Gene Silencing InVivo, Angew. Chem., Int. Ed., 2012, 51, 8529–8533 CrossRef CAS PubMed.
- K. T. Love, K. P. Mahon, C. G. Levins, K. A. Whitehead, W. Querbes, J. R. Dorkin, J. Qin, W. Cantley, L. L. Qin, T. Racie, M. Frank-Kamenetsky, K. N. Yip, R. Alvarez, D. W. Y. Sah, A. de Fougerolles, K. Fitzgerald, V. Koteliansky, A. Akinc, R. Langer and D. G. Anderson, Lipid-like materials for low-dose, in vivo gene silencing, Proc. Natl. Acad. Sci., 2010, 107, 1864–1869 CrossRef CAS PubMed.
- Y.-B. Hu, E. B. Dammer, R.-J. Ren and G. Wang, The endosomal-lysosomal system: from acidification and cargo sorting to neurodegeneration, Transl. Neurodegener., 2015, 4, 18 CrossRef PubMed.
- I. S. Zuhorn, U. Bakowsky, E. Polushkin, W. H. Visser, M. C. A. Stuart, J. B. F. N. Engberts and D. Hoekstra, Nonbilayer phase of lipoplex–membrane mixture determines endosomal escape of genetic cargo and transfection efficiency, Mol. Ther., 2005, 11, 801–810 CrossRef CAS PubMed.
- T. M. Allen and P. R. Cullis, Liposomal drug delivery systems: From concept to clinical applications, Adv. Drug Delivery Rev., 2013, 65, 36–48 CrossRef CAS PubMed.
- J. J. Lu, R. Langer and J. Chen, A Novel Mechanism Is Involved in Cationic Lipid-Mediated Functional siRNA Delivery, Mol. Pharmaceutics, 2009, 6, 763–771 CrossRef CAS PubMed.
- Y. Fang, J. Xue, S. Gao, A. Lu, D. Yang, H. Jiang, Y. He and K. Shi, Cleavable PEGylation: a strategy for overcoming the PEG dilemma in efficient drug delivery, Drug Delivery, 2017, 24, 22–32 CrossRef CAS PubMed.
- Y. Fan, C.-W. Yen, H.-C. Lin, W. Hou, A. Estevez, A. Sarode, A. Goyon, J. Bian, J. Lin, S. G. Koenig, D. Leung, K. Nagapudi and K. Zhang, Automated high-throughput preparation and characterization of oligonucleotide-loaded lipid nanoparticles, Int. J. Pharm., 2021, 599, 120392 CrossRef CAS PubMed.
- R. C. Ryals, S. Patel, C. Acosta, M. McKinney, M. E. Pennesi and G. Sahay, The effects of PEGylation on LNP based mRNA delivery to the eye, PLoS One, 2020, 15, e0241006 CrossRef CAS PubMed.
- V. Kumar, J. Qin, Y. Jiang, R. G. Duncan, B. Brigham, S. Fishman, J. K. Nair, A. Akinc, S. A. Barros and P. V. Kasperkovitz, Shielding of Lipid Nanoparticles for siRNA Delivery: Impact on Physicochemical Properties, Cytokine Induction, and Efficacy, Mol. Ther.--Nucleic Acids, 2014, 3, e210 CrossRef PubMed.
- D. Pozzi, V. Colapicchioni, G. Caracciolo, S. Piovesana, A. L. Capriotti, S. Palchetti, S. D. Grossi, A. Riccioli, H. Amenitsch and A. Laganà, Effect of polyethyleneglycol (PEG) chain length on the bio–nano-interactions between PEGylated lipid nanoparticles and biological fluids: from nanostructure to uptake in cancer cells, Nanoscale, 2013, 6, 2782–2792 RSC.
- B. L. Mui, Y. K. Tam, M. Jayaraman, S. M. Ansell, X. Du, Y. Y. C. Tam, P. J. Lin, S. Chen, J. K. Narayanannair, K. G. Rajeev, M. Manoharan, A. Akinc, M. A. Maier, P. Cullis, T. D. Madden and M. J. Hope, Influence of Polyethylene Glycol Lipid Desorption Rates on Pharmacokinetics and Pharmacodynamics of siRNA Lipid Nanoparticles, Mol. Ther.--Nucleic Acids, 2013, 2, e139 CrossRef CAS PubMed.
- J. S. Suk, Q. Xu, N. Kim, J. Hanes and L. M. Ensign, PEGylation as a strategy for improving nanoparticle-based drug and gene delivery, Adv. Drug Delivery Rev., 2016, 99, 28–51 CrossRef CAS PubMed.
- H. Lee and R. G. Larson, Adsorption of Plasma Proteins onto PEGylated Lipid Bilayers: The Effect of PEG Size and Grafting Density, Biomacromolecules, 2016, 17, 1757–1765 CrossRef CAS PubMed.
- H. Ishiwata, S. B. Sato, A. Vertut-Doï, Y. Hamashima and K. Miyajima, Cholesterol derivative of poly(ethylene glycol) inhibits clathrin-independent, but not clathrin-dependent endocytosis, Biochim. Biophys. Acta, Mol. Cell Res., 1997, 1359, 123–135 CrossRef CAS.
- T. Suzuki, Y. Suzuki, T. Hihara, K. Kubara, K. Kondo, K. Hyodo, K. Yamazaki, T. Ishida and H. Ishihara, PEG shedding-rate-dependent blood clearance of PEGylated lipid nanoparticles in mice: faster PEG shedding attenuates anti-PEG IgM production, Int. J. Pharm., 2020, 588, 119792 CrossRef CAS PubMed.
- S. Chen, Y. Y. C. Tam, P. J. C. Lin, M. M. H. Sung, Y. K. Tam and P. R. Cullis, Influence of particle size on the in vivo potency of lipid nanoparticle formulations of siRNA, J. Controlled Release, 2016, 235, 236–244 CrossRef CAS PubMed.
- F. Shi, L. Wasungu, A. Nomden, M. C. A. Stuart, E. Polushkin, J. B. F. N. Engberts and D. Hoekstra, Interference of poly(ethylene glycol)–lipid analogues with cationic-lipid-mediated delivery of oligonucleotides; role of lipid exchangeability and non-lamellar transitions, Biochem. J., 2002, 366, 333–341 CrossRef CAS PubMed.
- A. Lechanteur, T. Furst, B. Evrard, P. Delvenne, P. Hubert and G. Piel, PEGylation of lipoplexes: the right balance between cytotoxicity and siRNA effectiveness, Eur. J. Pharm. Sci., 2016, 93, 493–503 CrossRef CAS PubMed.
- L. Y. Song, Q. F. Ahkong, Q. Rong, Z. Wang, S. Ansell, M. J. Hope and B. Mui, Characterization of the inhibitory effect of PEG-lipid conjugates on the intracellular delivery of plasmid and antisense DNA mediated by cationic lipid liposomes, Biochim. Biophys. Acta, Biomembr., 2002, 1558, 1–13 CrossRef CAS.
- F. Mastrotto, C. Brazzale, F. Bellato, S. D. Martin, G. Grange, M. Mahmoudzadeh, A. Magarkar, A. Bunker, S. Salmaso and P. Caliceti, In Vitro and in Vivo Behavior of Liposomes Decorated with PEGs with Different Chemical Features, Mol. Pharmaceutics, 2020, 17, 472–487 CAS.
- K. Abe, K. Higashi, K. Watabe, A. Kobayashi, W. Limwikrant, K. Yamamoto and K. Moribe, Effects of the PEG molecular weight of a PEG-lipid and cholesterol on PEG chain flexibility on liposome surfaces, Colloids Surf., A, 2015, 474, 63–70 CrossRef CAS.
- M. S. Webb, D. Saxon, F. M. P. Wong, H. J. Lim, Z. Wang, M. B. Bally, L. S. L. Choi, P. R. Cullis and L. D. Mayer, Comparison of different hydrophobic anchors conjugated to poly(ethylene glycol): effects on the pharmacokinetics of liposomal vincristine, Biochim. Biophys. Acta, Biomembr., 1998, 1372, 272–282 CrossRef CAS.
- V. Swarup, F. I. Hinz, J. E. Rexach, K. Noguchi, H. Toyoshiba, A. Oda, K. Hirai, A. Sarkar, N. T. Seyfried, C. Cheng, S. J. Haggarty, R. Ferrari, J. D. Rohrer, A. Ramasamy, J. Hardy, D. G. Hernandez, M. A. Nalls, A. B. Singleton, J. B. J. Kwok, C. Dobson-Stone, W. S. Brooks, P. R. Schofield, G. M. Halliday, J. R. Hodges, O. Piguet, L. Bartley, E. Thompson, E. Haan, I. Hernández, A. Ruiz, M. Boada, B. Borroni, A. Padovani, N. J. Cairns, C. Cruchaga, G. Binetti, R. Ghidoni, L. Benussi, G. Forloni, D. Albani, D. Galimberti, C. Fenoglio, M. Serpente, E. Scarpini, J. Clarimón, A. Lleó, R. Blesa, M. L. Waldö, K. Nilsson, C. Nilsson, I. R. A. Mackenzie, G.-Y. R. Hsiung, D. M. A. Mann, J. Grafman, C. M. Morris, J. Attems, T. D. Griffiths, I. G. McKeith, A. J. Thomas, E. Jaros, P. Pietrini, E. D. Huey, E. M. Wassermann, M. C. Tierney, A. Baborie, P. Pastor, S. Ortega-Cubero, C. Razquin, E. Alonso, R. Perneczky, J. Diehl-Schmid, P. Alexopoulos, A. Kurz, I. Rainero, E. Rubino, L. Pinessi, E. Rogaeva, P. St. George-Hyslop, G. Rossi, F. Tagliavini, G. Giaccone, J. B. Rowe, J. C. M. Schlachetzki, J. Uphill, J. Collinge, S. Mead, A. Danek, V. M. V. Deerlin, M. Grossman, J. Q. Trojanowski, S. Pickering-Brown, P. Momeni, J. van der Zee, M. Cruts, C. V. Broeckhoven, S. F. Cappa, I. Leber, A. Brice, D. Hannequin, V. Golfier, M. Vercelletto, B. Nacmias, S. Sorbi, S. Bagnoli, I. Piaceri, J. E. Nielsen, L. E. Hjermind, M. Riemenschneider, M. Mayhaus, G. Gasparoni, S. Pichler, B. Ibach, M. N. Rossor, N. C. Fox, J. D. Warren, M. G. Spillantini, H. R. Morris, P. Rizzu, P. Heutink, J. S. Snowden, S. Rollinson, A. Gerhard, A. Richardson, A. C. Bruni, R. Maletta, F. Frangipane, C. Cupidi, L. Bernardi, M. Anfossi, M. Gallo, M. E. Conidi, N. Smirne, R. Rademakers, M. Baker, D. W. Dickson, N. R. Graff-Radford, R. C. Petersen, D. Knopman, K. A. Josephs, B. F. Boeve, J. E. Parisi, B. L. Miller, A. M. Karydas, H. Rosen, W. W. Seeley, J. C. van Swieten, E. G. P. Dopper, H. Seelaar, Y. A. L. Pijnenburg, P. Scheltens, G. Logroscino, R. Capozzo, V. Novelli, A. A. Puca, M. Franceschi, A. Postiglione, G. Milan, P. Sorrentino, M. Kristiansen, H.-H. Chiang, C. Graff, F. Pasquier, A. Rollin, V. Deramecourt, T. Lebouvier, L. Ferrucci, D. Kapogiannis, M. Grossman, V. M. V. Deerlin, J. Q. Trojanowski, J. J. Lah, A. I. Levey, S. Kondou and D. H. Geschwind, Identification of evolutionarily conserved gene networks mediating neurodegenerative dementia, Nat. Med., 2019, 25, 152–164 CrossRef CAS PubMed.
- K. Frei, S. Bodmer, C. Schwerdel and A. Fontana, Astrocyte-derived interleukin 3 as a growth factor for microglia cells and peritoneal macrophages, J. Immunol., 1950, 1986(137), 3521–3527 Search PubMed.
- D. Giulian and T. Baker, Characterization of ameboid microglia isolated from developing mammalian brain, J. Neurosci., 1986, 6, 2163–2178 CrossRef CAS PubMed.
- K. N. Dyer, M. Hammel, R. P. Rambo, S. E. Tsutakawa, I. Rodic, S. Classen, J. A. Tainer and G. L. Hura, High-throughput SAXS for the characterization of biomolecules in solution: a practical approach, Methods Mol. Biol., 2014, 1091, 245–258 CrossRef CAS PubMed.
- G. L. Hura, A. L. Menon, M. Hammel, R. P. Rambo, F. L. P. II, S. E. Tsutakawa, F. E. J. Jr, S. Classen, K. A. Frankel, R. C. Hopkins, S. Yang, J. W. Scott, B. D. Dillard, M. W. W. Adams and J. A. Tainer, Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS), Nat. Methods, 2009, 6, 606–612 CrossRef CAS PubMed.
- A. J. Sinegra, M. Evangelopoulos, J. Park, Z. Huang and C. A. Mirkin, Lipid Nanoparticle Spherical Nucleic Acids for Intracellular DNA and RNA Delivery, Nano Lett., 2021, 21, 6584–6591 CrossRef CAS PubMed.
- Q. Cheng, T. Wei, L. Farbiak, L. T. Johnson, S. A. Dilliard and D. J. Siegwart, Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR–Cas gene editing, Nat. Nanotechnol., 2020, 15, 313–320 CrossRef CAS PubMed.
- X. Zhang, V. Goel and G. J. Robbie, Pharmacokinetics of Patisiran, the First Approved RNA Interference Therapy in Patients With Hereditary Transthyretin-Mediated Amyloidosis, J. Clin. Pharmacol., 2019, 60, 573–585 Search PubMed.
- S. Sabnis, E. S. Kumarasinghe, T. Salerno, C. Mihai, T. Ketova, J. J. Senn, A. Lynn, A. Bulychev, I. McFadyen, J. Chan, Ö. Almarsson, M. G. Stanton and K. E. Benenato, A Novel Amino Lipid Series for mRNA Delivery: Improved Endosomal Escape and Sustained Pharmacology and Safety in Non-human Primates, Mol. Ther., 2018, 26, 1509–1519 CrossRef CAS PubMed.
- K. J. Hassett, K. E. Benenato, E. Jacquinet, A. Lee, A. Woods, O. Yuzhakov, S. Himansu, J. Deterling, B. M. Geilich, T. Ketova, C. Mihai, A. Lynn, I. McFadyen, M. J. Moore, J. J. Senn, M. G. Stanton, Ö. Almarsson, G. Ciaramella and L. A. Brito, Optimization of Lipid Nanoparticles for Intramuscular Administration of mRNA Vaccines, Mol. Ther.--Nucleic Acids, 2019, 15, 1–11 CrossRef CAS PubMed.
- H. Alrbyawi, I. Poudel, R. P. Dash, N. R. Srinivas, A. K. Tiwari, R. D. Arnold and R. J. Babu, Role of Ceramides in Drug Delivery, AAPS PharmSciTech, 2019, 20, 287 CrossRef PubMed.
- D. M. Lambert, Rationale and applications of lipids as prodrug carriers, Eur. J. Pharm. Sci., 2000, 11, S15–S27 CrossRef CAS PubMed.
- B. Y. Tao, Bioprocessing for Value-Added Products from Renewable Resources, 2007, pp. 611–627 Search PubMed.
- M. Martí, M. Lis, J. A. Navarro, R. Ramírez, L. Coderch, J. Valldeperas and J. L. Parra, Medical and Healthcare Textiles, Part Vii Smart Mater Technologies, 2010, pp. 509–516 Search PubMed.
- V. Krishnan, K. Peng, A. Sarode, S. Prakash, Z. Zhao, S. K. Filippov, K. Todorova, B. R. Sell, O. Lujano, S. Bakre, A. Pusuluri, D. Vogus, K. Y. Tsai, A. Mandinova and S. Mitragotri, Hyaluronic acid conjugates for topical treatment of skin cancer lesions, Sci. Adv., 2021, 7, eabe6627 CrossRef CAS PubMed.
- M. Kim, M. Jeong, S. Hur, Y. Cho, J. Park, H. Jung, Y. Seo, H. A. Woo, K. T. Nam, K. Lee and H. Lee, Engineered ionizable lipid nanoparticles for targeted delivery of RNA therapeutics into different types of cells in the liver, Sci. Adv., 2021, 7, eabf4398 CrossRef CAS PubMed.
- M. Johnsson and K. Edwards, Liposomes, Disks, and Spherical Micelles: Aggregate Structure in Mixtures of Gel Phase Phosphatidylcholines and Poly(Ethylene Glycol)-Phospholipids, Biophys. J., 2003, 85, 3839–3847 CrossRef CAS PubMed.
- K. K. Gill, A. Kaddoumi and S. Nazzal, PEG–lipid micelles as drug carriers: physiochemical attributes, formulation principles and biological implication, J. Drug Targeting, 2014, 23, 222–231 CrossRef PubMed.
- G. N. Collaborators, V. L. Feigin, E. Nichols, T. Alam, M. S. Bannick, E. Beghi, N. Blake, W. J. Culpepper, E. R. Dorsey, A. Elbaz, R. G. Ellenbogen, J. L. Fisher, C. Fitzmaurice, G. Giussani, L. Glennie, S. L. James, C. O. Johnson, N. J. Kassebaum, G. Logroscino, B. Marin, W. C. Mountjoy-Venning, M. Nguyen, R. Ofori-Asenso, A. P. Patel, M. Piccininni, G. A. Roth, T. J. Steiner, L. J. Stovner, C. E. I. Szoeke, A. Theadom, S. E. Vollset, M. T. Wallin, C. Wright, J. R. Zunt, N. Abbasi, F. Abd-Allah, A. Abdelalim, I. Abdollahpour, V. Aboyans, H. N. Abraha, D. Acharya, A. A. Adamu, O. M. Adebayo, A. M. Adeoye, J. C. Adsuar, M. Afarideh, S. Agrawal, A. Ahmadi, M. B. Ahmed, A. N. Aichour, I. Aichour, M. T. E. Aichour, R. O. Akinyemi, N. Akseer, A. Al-Eyadhy, R. A.-S. Salman, F. Alahdab, K. A. Alene, S. M. Aljunid, K. Altirkawi, N. Alvis-Guzman, N. H. Anber, C. A. T. Antonio, J. Arabloo, O. Aremu, J. Ärnlöv, H. Asayesh, R. J. Asghar, H. T. Atalay, A. Awasthi, B. P. A. Quintanilla, T. B. Ayuk, A. Badawi, M. Banach, J. A. M. Banoub, M. A. Barboza, S. L. Barker-Collo, T. W. Bärnighausen, B. T. Baune, N. Bedi, M. Behzadifar, M. Behzadifar, Y. Béjot, B. B. Bekele, A. B. Belachew, D. A. Bennett, I. M. Bensenor, A. Berhane, M. Beuran, K. Bhattacharyya, Z. A. Bhutta, B. Biadgo, A. Bijani, N. Bililign, M. S. B. Sayeed, C. K. Blazes, C. Brayne, Z. A. Butt, I. R. Campos-Nonato, C. Cantu-Brito, M. Car, R. Cárdenas, J. J. Carrero, F. Carvalho, C. A. Castañeda-Orjuela, F. Castro, F. Catalá-López, E. Cerin, Y. Chaiah, J.-C. Chang, I. Chatziralli, P. P.-C. Chiang, H. Christensen, D. J. Christopher, C. Cooper, P. A. Cortesi, V. M. Costa, M. H. Criqui, C. S. Crowe, A. A. M. Damasceno, A. Daryani, V. D. la Cruz-Góngora, F. P. D. la Hoz, D. D. Leo, G. T. Demoz, K. Deribe, S. D. Dharmaratne, D. Diaz, M. T. Dinberu, S. Djalalinia, D. T. Doku, M. Dubey, E. Dubljanin, E. E. Duken, D. Edvardsson, Z. El-Khatib, M. Endres, A. Y. Endries, S. Eskandarieh, A. Esteghamati, S. Esteghamati, F. Farhadi, A. Faro, F. Farzadfar, M. H. Farzaei, B. Fatima, S.-M. Fereshtehnejad, E. Fernandes, G. T. Feyissa, I. Filip, F. Fischer, T. Fukumoto, M. Ganji, F. G. Gankpe, M. A. Garcia-Gordillo, A. K. Gebre, T. G. Gebremichael, B. K. Gelaw, J. M. Geleijnse, D. Geremew, K. E. Gezae, M. Ghasemi-Kasman, M. Y. Gidey, P. S. Gill, T. K. Gill, E. T. Girma, E. V. Gnedovskaya, A. C. Goulart, A. Grada, G. Grosso, Y. Guo, R. Gupta, R. Gupta, J. A. Haagsma, T. B. Hagos, A. Haj-Mirzaian, A. Haj-Mirzaian, R. R. Hamadeh, S. Hamidi, G. J. Hankey, Y. Hao, J. M. Haro, H. Hassankhani, H. Y. Hassen, R. Havmoeller, S. I. Hay, M. I. Hegazy, B. Heidari, A. Henok, F. Heydarpour, C. L. Hoang, M. K. Hole, E. H. Rad, S. M. Hosseini, G. Hu, E. U. Igumbor, O. S. Ilesanmi, S. S. N. Irvani, S. M. S. Islam, M. Jakovljevic, M. Javanbakht, R. P. Jha, Y. B. Jobanputra, J. B. Jonas, J. J. Jozwiak, M. Jürisson, A. Kahsay, R. Kalani, Y. Kalkonde, T. A. Kamil, T. Kanchan, M. Karami, A. Karch, N. Karimi, A. Kasaeian, T. D. Kassa, Z. Y. Kassa, A. Kaul, A. T. Kefale, P. N. Keiyoro, Y. S. Khader, M. A. Khafaie, I. A. Khalil, E. A. Khan, Y.-H. Khang, H. Khazaie, A. A. Kiadaliri, D. N. Kiirithio, A. S. Kim, D. Kim, Y.-E. Kim, Y. J. Kim, A. Kisa, Y. Kokubo, A. Koyanagi, R. V. Krishnamurthi, B. K. Defo, B. K. Bicer, M. Kumar, B. Lacey, A. Lafranconi, V. C. Lansingh, A. Latifi, C. T. Leshargie, S. Li, Y. Liao, S. Linn, W. D. Lo, J. C. F. Lopez, S. Lorkowski, P. A. Lotufo, R. M. Lucas, R. Lunevicius, M. T. Mackay, N. B. Mahotra, M. Majdan, R. Majdzadeh, A. Majeed, R. Malekzadeh, D. C. Malta, N. Manafi, M. A. Mansournia, L. G. Mantovani, W. März, T. P. Mashamba-Thompson, B. B. Massenburg, K. K. V. Mate, C. McAlinden, J. J. McGrath, V. Mehta, T. Meier, H. G. Meles, A. Melese, P. T. N. Memiah, Z. A. Memish, W. Mendoza, D. T. Mengistu, G. Mengistu, A. Meretoja, T. J. Meretoja, T. Mestrovic, B. Miazgowski, T. Miazgowski, T. R. Miller, G. Mini, E. M. Mirrakhimov, B. Moazen, B. Mohajer, N. M. G. Mezerji, M. Mohammadi, M. Mohammadi-Khanaposhtani, R. Mohammadibakhsh, M. Mohammadnia-Afrouzi, S. Mohammed, F. Mohebi, A. H. Mokdad, L. Monasta, S. Mondello, Y. Moodley, M. Moosazadeh, G. Moradi, M. Moradi-Lakeh, M. Moradinazar, P. Moraga, I. M. Velásquez, S. D. Morrison, S. M. Mousavi, O. S. Muhammed, W. Muruet, K. I. Musa, G. Mustafa, M. Naderi, G. Nagel, A. Naheed, G. Naik, F. Najafi, V. Nangia, I. Negoi, R. I. Negoi, C. R. J. Newton, J. W. Ngunjiri, C. T. Nguyen, L. H. Nguyen, D. N. A. Ningrum, Y. L. Nirayo, M. R. Nixon, B. Norrving, J. J. Noubiap, M. N. Shiadeh, P. S. Nyasulu, O. S. Ogah, I.-H. Oh, A. T. Olagunju, T. O. Olagunju, P. R. Olivares, O. E. Onwujekwe, E. Oren, M. O. Owolabi, M. PA, A. H. Pakpour, W.-H. Pan, S. Panda-Jonas, J. D. Pandian, S. K. Patel, D. M. Pereira, M. Petzold, J. D. Pillay, M. A. Piradov, G. V. Polanczyk, S. Polinder, M. J. Postma, R. Poulton, H. Poustchi, S. Prakash, V. Prakash, M. Qorbani, A. Radfar, A. Rafay, A. Rafiei, F. Rahim, V. Rahimi-Movaghar, M. Rahman, M. H. U. Rahman, M. A. Rahman, F. Rajati, U. Ram, A. Ranta, D. L. Rawaf, S. Rawaf, N. Reinig, C. Reis, A. M. N. Renzaho, S. Resnikoff, S. Rezaeian, M. S. Rezai, C. M. R. González, N. L. S. Roberts, L. Roever, L. Ronfani, E. M. Roro, G. Roshandel, A. Rostami, P. Sabbagh, R. L. Sacco, P. S. Sachdev, B. Saddik, H. Safari, R. Safari-Faramani, S. Safi, S. Safiri, R. Sagar, R. Sahathevan, A. Sahebkar, M. A. Sahraian, P. Salamati, S. S. Zahabi, Y. Salimi, A. M. Samy, J. Sanabria, I. S. Santos, M. M. S. Milicevic, N. Sarrafzadegan, B. Sartorius, S. Sarvi, B. Sathian, M. Satpathy, A. R. Sawant, M. Sawhney, I. J. C. Schneider, B. Schöttker, D. C. Schwebel, S. Seedat, S. G. Sepanlou, H. Shabaninejad, A. Shafieesabet, M. A. Shaikh, R. A. Shakir, M. Shams-Beyranvand, M. Shamsizadeh, M. Sharif, M. Sharif-Alhoseini, J. She, A. Sheikh, K. N. Sheth, M. Shigematsu, R. Shiri, R. Shirkoohi, I. Shiue, S. Siabani, T. J. Siddiqi, I. D. Sigfusdottir, R. Sigurvinsdottir, D. H. Silberberg, J. P. Silva, D. G. A. Silveira, J. A. Singh, D. N. Sinha, E. Skiadaresi, M. Smith, B. H. Sobaih, S. Sobhani, M. Soofi, I. N. Soyiri, L. A. Sposato, D. J. Stein, M. B. Stein, M. A. Stokes, M. B. Sufiyan, B. L. Sykes, P. Sylaja, R. Tabarés-Seisdedos, B. J. T. Ao, A. Tehrani-Banihashemi, M.-H. Temsah, O. Temsah, J. S. Thakur, A. G. Thrift, R. Topor-Madry, M. Tortajada-Girbés, M. R. Tovani-Palone, B. X. Tran, K. B. Tran, T. C. Truelsen, A. G. Tsadik, L. T. Car, K. N. Ukwaja, I. Ullah, M. S. Usman, O. A. Uthman, P. R. Valdez, T. J. Vasankari, R. Vasanthan, Y. Veisani, N. Venketasubramanian, F. S. Violante, V. Vlassov, K. Vosoughi, G. T. Vu, I. S. Vujcic, F. S. Wagnew, Y. Waheed, Y.-P. Wang, E. Weiderpass, J. Weiss, H. A. Whiteford, T. Wijeratne, A. S. Winkler, C. S. Wiysonge, C. D. A. Wolfe, G. Xu, A. Yadollahpour, T. Yamada, Y. Yano, M. Yaseri, H. Yatsuya, E. M. Yimer, P. Yip, E. Yisma, N. Yonemoto, M. Yousefifard, C. Yu, Z. Zaidi, S. B. Zaman, M. Zamani, H. Zandian, Z. Zare, Y. Zhang, S. Zodpey, M. Naghavi, C. J. L. Murray and T. Vos, Global, regional, and national burden of neurological disorders, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016, Lancet Neurol., 2019, 18, 459–480 CrossRef.
- C. D. Wurster and A. C. Ludolph, Antisense oligonucleotides in neurological disorders, Ther. Adv. Neurol. Disord., 2018, 11, 1756286418776932 Search PubMed.
- Y. Wang, J. Pang, Q. Wang, L. Yan, L. Wang, Z. Xing, C. Wang, J. Zhang and L. Dong, Delivering Antisense Oligonucleotides across the Blood-Brain Barrier by Tumor Cell-Derived Small Apoptotic Bodies, Adv. Sci., 2021, 8, 2004929 CrossRef CAS PubMed.
- T. Nagata, C. A. Dwyer, K. Yoshida-Tanaka, K. Ihara, M. Ohyagi, H. Kaburagi, H. Miyata, S. Ebihara, K. Yoshioka, T. Ishii, K. Miyata, K. Miyata, B. Powers, T. Igari, S. Yamamoto, N. Arimura, H. Hirabayashi, T. Uchihara, R. I. Hara, T. Wada, C. F. Bennett, P. P. Seth, F. Rigo and T. Yokota, Cholesterol-functionalized DNA/RNA heteroduplexes cross the blood–brain barrier and knock down genes in the rodent CNS, Nat. Biotechnol., 2021, 1–8 CAS.
- H. Akita, R. Ishiba, R. Togashi, K. Tange, Y. Nakai, H. Hatakeyama and H. Harashima, A neutral lipid envelope-type nanoparticle composed of a pH-activated and vitamin E-scaffold lipid-like material as a platform for a gene carrier targeting renal cell carcinoma, J. Controlled Release, 2015, 200, 97–105 CrossRef CAS PubMed.
- Y. Xu, M. Ou, E. Keough, J. Roberts, K. Koeplinger, M. Lyman, S. Fauty, E. Carlini, M. Stern, R. Zhang, S. Yeh, E. Mahan, Y. Wang, D. Slaughter, M. Gindy, C. Raab, C. Thompson and J. Hochman, Quantitation of Physiological and Biochemical Barriers to siRNA Liver Delivery via Lipid Nanoparticle Platform, Mol. Pharmaceutics, 2014, 11, 1424–1434 CrossRef CAS PubMed.
- A. Akinc, W. Querbes, S. De, J. Qin, M. Frank-Kamenetsky, K. N. Jayaprakash, M. Jayaraman, K. G. Rajeev, W. L. Cantley, J. R. Dorkin, J. S. Butler, L. Qin, T. Racie, A. Sprague, E. Fava, A. Zeigerer, M. J. Hope, M. Zerial, D. W. Sah, K. Fitzgerald, M. A. Tracy, M. Manoharan, V. Koteliansky, A. de Fougerolles and M. A. Maier, Targeted Delivery of RNAi Therapeutics With Endogenous and Exogenous Ligand-Based Mechanisms, Mol. Ther., 2010, 18, 1357–1364 CrossRef CAS PubMed.
- J. J. Wheeler, L. Palmer, M. Ossanlou, I. MacLachlan, R. W. Graham, Y. P. Zhang, M. J. Hope, P. Scherrer and P. R. Cullis, Stabilized plasmid-lipid particles: construction and characterization, Gene Ther., 1999, 6, 271–281 CrossRef CAS PubMed.
- P. J. Harrison, H. Wieslander, A. Sabirsh, J. Karlsson, V. Malmsjö, A. Hellander, C. Wählby and O. Spjuth, Deep-learning models for lipid nanoparticle-based drug delivery, Nanomedicine, 2021, 16, 1097–1110 CrossRef CAS PubMed.
- C. D. Sago, M. P. Lokugamage, F. Z. Islam, B. R. Krupczak, M. Sato and J. E. Dahlman, Nanoparticles That Deliver RNA to Bone Marrow Identified by in Vivo Directed Evolution, J. Am. Chem. Soc., 2018, 140, 17095–17105 CrossRef CAS PubMed.
- M. P. Lokugamage, C. D. Sago, Z. Gan, B. R. Krupczak and J. E. Dahlman, Constrained Nanoparticles Deliver siRNA and sgRNA to T Cells In Vivo without Targeting Ligands, Adv. Mater., 2019, 31, e1902251 CrossRef PubMed.
- V. Makwana, J. Karanjia, T. Haselhorst, S. Anoopkumar-Dukie and S. Rudrawar, Liposomal doxorubicin as targeted delivery platform: Current trends in surface functionalization, Int. J. Pharm., 2020, 593, 120117 CrossRef PubMed.
- E. Quagliarini, S. Renzi, L. Digiacomo, F. Giulimondi, B. Sartori, H. Amenitsch, V. Tassinari, L. Masuelli, R. Bei, L. Cui, J. Wang, A. Amici, C. Marchini, D. Pozzi and G. Caracciolo, Microfluidic Formulation of DNA-Loaded Multicomponent Lipid Nanoparticles for Gene Delivery, Pharm, 2021, 13, 1292 CAS.
- K. Aburai, K. Hatanaka, S. Takano, S. Fujii and K. Sakurai, Characterizing an siRNA-Containing Lipid-Nanoparticle Prepared by a Microfluidic Reactor: Small-Angle X-ray Scattering and Cryotransmission Electron Microscopic Studies, Langmuir, 2020, 36, 12545–12554 CrossRef CAS PubMed.
- M. Y. Arteta, T. Kjellman, S. Bartesaghi, S. Wallin, X. Wu, A. J. Kvist, A. Dabkowska, N. Székely, A. Radulescu, J. Bergenholtz and L. Lindfors, Successful reprogramming of cellular protein production through mRNA delivered by functionalized lipid nanoparticles, Proc. Natl. Acad. Sci., 2018, 115, 201720542 Search PubMed.
- I. Koltover, T. Salditt, J. O. Rädler and C. R. Safinya, An Inverted Hexagonal Phase of Cationic Liposome-DNA Complexes Related to DNA Release and Delivery, Science, 1998, 281, 78–81 CrossRef CAS PubMed.
- L. Wang, R. Koynova, H. Parikh and R. C. MacDonald, Transfection Activity of Binary Mixtures of Cationic O-Substituted Phosphatidylcholine Derivatives: The Hydrophobic Core Strongly Modulates Physical Properties and DNA Delivery Efficacy, Biophys. J., 2006, 91, 3692–3706 CrossRef CAS PubMed.
- M. Prinz, S. Jung and J. Priller, Microglia Biology: One Century of Evolving Concepts, Cell, 2019, 179, 292–311 CrossRef CAS PubMed.
- E. Kastner, R. Kaur, D. Lowry, B. Moghaddam, A. Wilkinson and Y. Perrie, High-throughput manufacturing of size-tuned liposomes by a new microfluidics method using enhanced statistical tools for characterization, Int. J. Pharm., 2014, 477, 361–368 CrossRef CAS PubMed.
- Y. Haseda, L. Munakata, J. Meng, R. Suzuki and T. Aoshi, Microfluidic-prepared DOTAP nanoparticles induce strong T-cell responses in mice, PLoS One, 2020, 15, e0227891 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00712b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |