
Olefin epoxidation using electricity as renewable power in a bromide-mediated electrochemical process†
Hongyang
Tang
,
Jef R.
Vanhoof
and
Dirk
De Vos
 *
*
Centre for Membrane Separations, Adsorption, Catalysis and Spectroscopy for Sustainable Solutions (cMACS), KU Leuven, Celestijnenlaan 200F, 3001 Leuven, Belgium. E-mail: dirk.devos@kuleuven.be
First published on 22nd November 2022
Abstract
Electrochemical bromide-mediated olefin epoxidation is a viable strategy to generate freely available bromine (FAB) in situ while minimizing process waste by avoiding chemical oxidants and reducing the use of hazardous reagents. Herein, an electrochemical bromide-mediated olefin epoxidation is presented, wherein bromide plays a double role, as an electrolyte, but also as a mediator that is easily oxidized even at moderate anodic voltage. A quaternary ammonium salt is introduced as an organic electrolyte. The amount of applied charge and the concentrations of electrolyte and olefin were tuned to maximize the efficiency and selectivity of the reaction. Target compounds like 1,2-epoxycyclohexane were obtained in high yields of up to 93%, and a broad substrate scope was demonstrated. Additionally, this process can avoid the formation of dibrominated compounds (e.g., 1,2-dibromocyclohexane), which are typically undesirable dead-end products.
Introduction
Olefin epoxidation is an important process for providing highly reactive epoxy motifs to be used in pharmaceuticals, food additives, perfumes, and agrochemicals.1 Ethylene oxide, propylene oxide, styrene oxide, and cyclohexene oxide have high volume demands in the chemical feedstock rankings every year, and demand for these compounds is constantly increasing.1,2 The chlorohydrin-based method (using ClO−), and stoichiometric oxidations with e.g. m-chloroperbenzoic acid (mCPBA) are traditional methods to achieve olefin epoxidation.3,4 These methods are not environmentally friendly due to the formation of wastes of chloride salts or organic acids. Alternatively, oxidants like tert-butyl hydroperoxide (TBHP) or hydrogen peroxide (H2O2) can be used in combination with certain d0 transition metals. The use of molecular oxygen (O2) is also documented, but its use is limited to exceptional cases like ethylene or butadiene.5–7 Although these alternative oxidation reactions are more efficient and environmentally friendly, they still have drawbacks such as the significant cost of the peroxides, catalyst deactivation, and over-oxidation if O2 is used.8 Recently, a greener electrocatalyzed method was reported that utilized manganese oxide nanoparticles to promote cyclooctene epoxidation, but only linear and cyclic olefins were explored, and only 50% conversion and 30% faradaic efficiency were obtained, which is unsatisfactory for commercial application.9 In summary, the presently known olefin epoxidation methods still present some problems, including high cost, side product formation, difficult separation of the catalyst from the homogeneous system, low effectiveness, and generation of large quantities of acidic or chlorinated waste, which means that not all processes are environmentally friendly.Bromide is one of the key constituents of water matrices and is highly susceptible to oxidation processes. While HOCl acts as a more reactive electrophile for the initial attack of the olefin, ring closure after formation of the halohydrin is easier with the Br version.10 However most efforts have focused on the chlorohydrin-based olefin epoxidation.11 Notably, BASF started the synthesis of ethylene oxide using the chlorohydrin process in 1914, which was replaced by hydrogen peroxide in subsequent progress.12 The electrochemical halohydrin method was first proposed in 1966.13 Currently, there is significant interest in using electricity as a renewable energy source for chemical transformations, including the formation of epoxides. Sargent et al. proposed a chloride-mediated electrochemical synthesis of ethylene and propylene oxides at high current density. However, after the actual electrochemical reaction, the alkali produced in the cathodic chamber needs to be mixed with the chlorohydrin (e.g., 2-Cl-ethanol) formed in the acidic anode chamber. Thus, a separate step is required to obtain ethylene oxide, since the base cannot pass in a sufficient amount through the AEM to enter the anode chamber.14 In a similar, hybrid process in simulated seawater,15 the same problem was encountered. Free available chlorine (FAC) usually shows excellent activity only in acidic conditions, while free available bromine (FAB) can provide good activity even in alkaline conditions.16 However, bromide-mediated protocols have barely received attention for epoxidation.
Here we present an electrochemical bromide-mediated olefin epoxidation. Starting with cyclohexene as a model reactant, the kinetics and mechanism are investigated in detail. In optimal conditions, cyclohexene epoxide is formed with high yield, selectivity, and faradaic efficiency. The addition of an extra base is not required since sufficient alkalinity is generated in situ. The scope of the method is documented extensively; besides cyclohexene, a variety of aliphatic and alicyclic olefins with di- or trisubstituted double bonds can be epoxidized.
Experimental
We used an undivided glass reactor with working and counter electrodes (Fig. S2†). All experiments used a power supply from TENMA (72-10480) (Fig. S5†). Epoxide products were identified and quantified via gas chromatography with flame ionization detection (GC-FID) and gas chromatography-mass spectrometry (GCMS), with mCPBA as a reference reactant to confirm epoxide formation.16 Reactions were conducted galvanostatically for 1–6 h at 298 K in a carefully tuned aqueous–organic solvent mixture. Sodium bromide (NaBr) and tetraethylammonium tetrafluoroborate (Et4NBF4) were used as simple electrolytes.Results and discussion
Reaction pathway and faradaic efficiency
We investigated the bromide-mediated electrochemical olefin epoxidation process, starting by selecting optimal electrodes with cyclohexene (1a) as the model substrate. Bromide plays a dual role as both an electrolyte and as a mediator that is easily oxidized even at moderate anodic voltage. The amount of applied charge, the ratio of solvents, and the concentrations of electrolyte and olefin were tuned to maximize efficiency and selectivity. The initial experiment was conducted in acetonitrile and deionized water (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at room temperature at a constant current of 10 mA cm−2 with carbon felt (CF) as the anode, platinum (Pt) foil as the cathode, and NaBr as the sole electrolyte. Two products were identified in this reaction: the target oxidation product 1,2-epoxycyclohexane (1b) and 1,2-dibromocyclohexane.
:1) at room temperature at a constant current of 10 mA cm−2 with carbon felt (CF) as the anode, platinum (Pt) foil as the cathode, and NaBr as the sole electrolyte. Two products were identified in this reaction: the target oxidation product 1,2-epoxycyclohexane (1b) and 1,2-dibromocyclohexane.
Next, a diverse series of electrodes were tested under the initial conditions, showing a significant beneficial impact on the selectivity for 1b when a Pt foil anode and a Pt foil cathode were used. Although most of the common anode materials were tested, several of the traditional anodes, including graphite and BDD, are not beneficial for the selectivity of the reaction for 1b (Fig. 1).
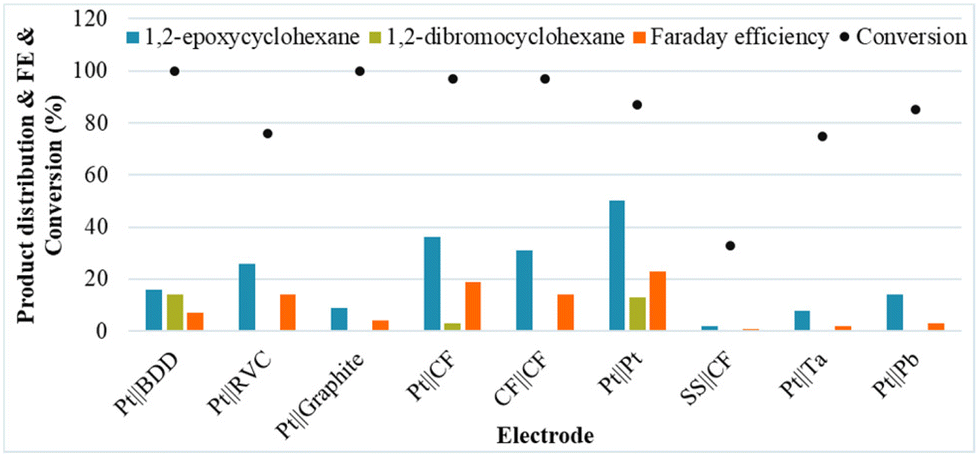 | ||
| Fig. 1 Product distribution with various electrodes. Reaction conditions: cathode||anode, constant current of 10 mA cm−2, 1a (50 mM), NaBr (1.0 eq.), MeCN/H2O (4:1, 6 ml), room temperature, 3 h. Yield was determined by GC analysis using octane as the external standard compared with the desired cyclohexene oxide. | ||
The effect of the electrolytes on the yield of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond epoxidation was investigated and plotted in Fig. 2. The concentrations of tetraethylammonium tetrafluoroborate (Et4NBF4) and sodium bromide (NaBr) were varied from 0 to 3 equivalents. In the reaction without NaBr, no product is observed, and without Et4NBF4, yields of only 47% for 1,2-epoxycyclohexane and 13% for 1,2-dibromocyclohexane were observed. As shown in Fig. 2a, the effect of additional Et4NBF4 on the yield of 1,2-epoxycyclohexane is very strong: 1,2-epoxycyclohexane yield increased from 47% to 82% (at its peak), then decreased. Additionally, the addition of tetraethylammonium tetrafluoroborate leads to the complete disappearance of the principal by-product 1,2-dibromocyclohexane. Apparently, the presence of BF4− as an alternative anion, close to the reactive bromonium intermediate, instead of Br−, makes the addition of a second bromide less likely. The effect of the concentration of NaBr was investigated and the results are shown in Fig. 2b. Product yield results show that the optimum concentration of NaBr is 1.5 equivalents, at which point the epoxide yield is maximal. When the bromide concentration increases, the electrical conductivity also increases; however, at concentrations larger than the optimum conditions, other species could be formed, such as the brown-coloured Br3−, which can be expected to be less reactive with the olefin. These results indicate that a suitable concentration of electrolyte favours the two-electron oxidation of bromide needed for obtaining epoxides in the aqueous–organic mixed solvent system. Application of the appropriate bromide amount also allows to maximize faradaic efficiency. This may be attributed to NaBr completely dissolving and transferring electrons in the aqueous solvent, and to Et4NBF4 promoting electron transfer and also reducing the charge load for electro-migration of NaBr in the reaction system, making NaBr more favourable as the mediator to form FAB. Fig. 2c shows the effect of different electrolytes on both cyclohexene conversion and selectivity toward epoxide. It is noted that with larger alkyl chain quaternary ammonium salts, such as nBu4NPF6, nBu4NBF4, etc., more dibromides will be produced. Halogenated quaternary ammonium salts were also tested, but there was no improvement. Sodium chloride can also generate some epoxide under these conditions, but the reactivity and yield are much lower than with sodium bromide.
C bond epoxidation was investigated and plotted in Fig. 2. The concentrations of tetraethylammonium tetrafluoroborate (Et4NBF4) and sodium bromide (NaBr) were varied from 0 to 3 equivalents. In the reaction without NaBr, no product is observed, and without Et4NBF4, yields of only 47% for 1,2-epoxycyclohexane and 13% for 1,2-dibromocyclohexane were observed. As shown in Fig. 2a, the effect of additional Et4NBF4 on the yield of 1,2-epoxycyclohexane is very strong: 1,2-epoxycyclohexane yield increased from 47% to 82% (at its peak), then decreased. Additionally, the addition of tetraethylammonium tetrafluoroborate leads to the complete disappearance of the principal by-product 1,2-dibromocyclohexane. Apparently, the presence of BF4− as an alternative anion, close to the reactive bromonium intermediate, instead of Br−, makes the addition of a second bromide less likely. The effect of the concentration of NaBr was investigated and the results are shown in Fig. 2b. Product yield results show that the optimum concentration of NaBr is 1.5 equivalents, at which point the epoxide yield is maximal. When the bromide concentration increases, the electrical conductivity also increases; however, at concentrations larger than the optimum conditions, other species could be formed, such as the brown-coloured Br3−, which can be expected to be less reactive with the olefin. These results indicate that a suitable concentration of electrolyte favours the two-electron oxidation of bromide needed for obtaining epoxides in the aqueous–organic mixed solvent system. Application of the appropriate bromide amount also allows to maximize faradaic efficiency. This may be attributed to NaBr completely dissolving and transferring electrons in the aqueous solvent, and to Et4NBF4 promoting electron transfer and also reducing the charge load for electro-migration of NaBr in the reaction system, making NaBr more favourable as the mediator to form FAB. Fig. 2c shows the effect of different electrolytes on both cyclohexene conversion and selectivity toward epoxide. It is noted that with larger alkyl chain quaternary ammonium salts, such as nBu4NPF6, nBu4NBF4, etc., more dibromides will be produced. Halogenated quaternary ammonium salts were also tested, but there was no improvement. Sodium chloride can also generate some epoxide under these conditions, but the reactivity and yield are much lower than with sodium bromide.
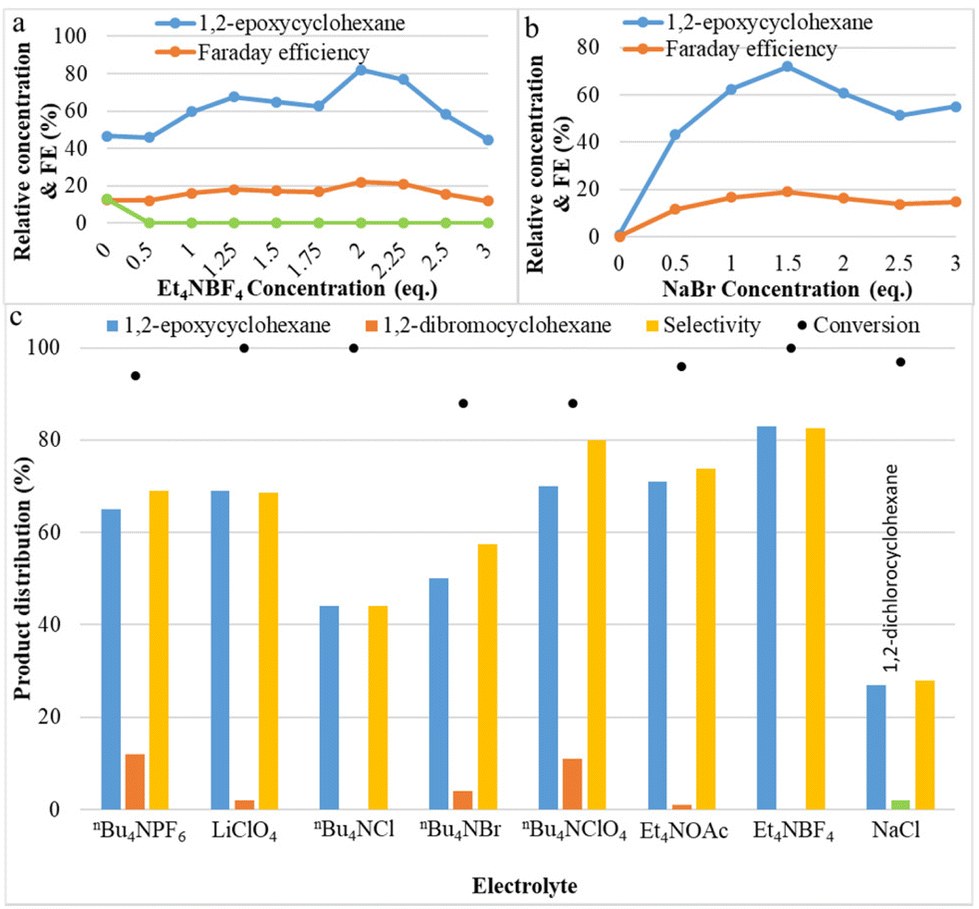 | ||
| Fig. 2 Reaction conditions: Pt as anode and Pt as cathode, room temperature, MeCN:H2O (3:2, 6 ml); (a) different Et4NBF4 concentrations: constant current of 10 mA cm−2, 1a (25 mM), NaBr (1.5 eq.), Et4NBF4 concentration from 0–3 eq., 3 h; (b) different NaBr concentrations: constant current of 10 mA cm−2, 1a (25 mM), Et4NBF4 (2 eq.), NaBr concentration from 0–3 eq., 3 h; (c) different electrolytes: constant current of 10 mA cm−2, 1a (25 mM), NaBr (1.5 eq. (or NaCl)), electrolyte (2 eq.), 3 h. | ||
Systematic experiments show that the observed reactivity is consistent with a mechanism involving H2O as the oxygen source; its concentration plays an important role in the epoxide product selectivity. We explored various factors that control selectivity in this bromide-mediated oxidation. Fig. 3a shows that the acetonitrile/deionized water ratio has a strong influence on the reaction pathway. It was observed that the yield of 1,2-epoxycyclohexane increases rapidly and then slightly decreases as the water content is increased, while the yield of by-product (1,2-dibromocyclohexane) increases initially and then decreases with a decrease in the acetonitrile/deionized water ratio. No product was observed when no water was present, and all by-products disappeared when the ratio of acetonitrile/deionized water was 3:2 or lower. A possible explanation for this phenomenon is that a high content of deionized water in the reaction system is beneficial for the oxygen transfer process to form the epoxide because it results in the formation of HOBr, which attacks the CC bond of cyclohexene.17,18 Another possibility is that water suppresses the reaction between FAB and cyclohexene which forms undesirable dibrominated products. The product selectivity results led us to conclude that an optimum acetonitrile/deionized water ratio of 3:2 maximizes epoxide yields and minimizes dibromide yields.
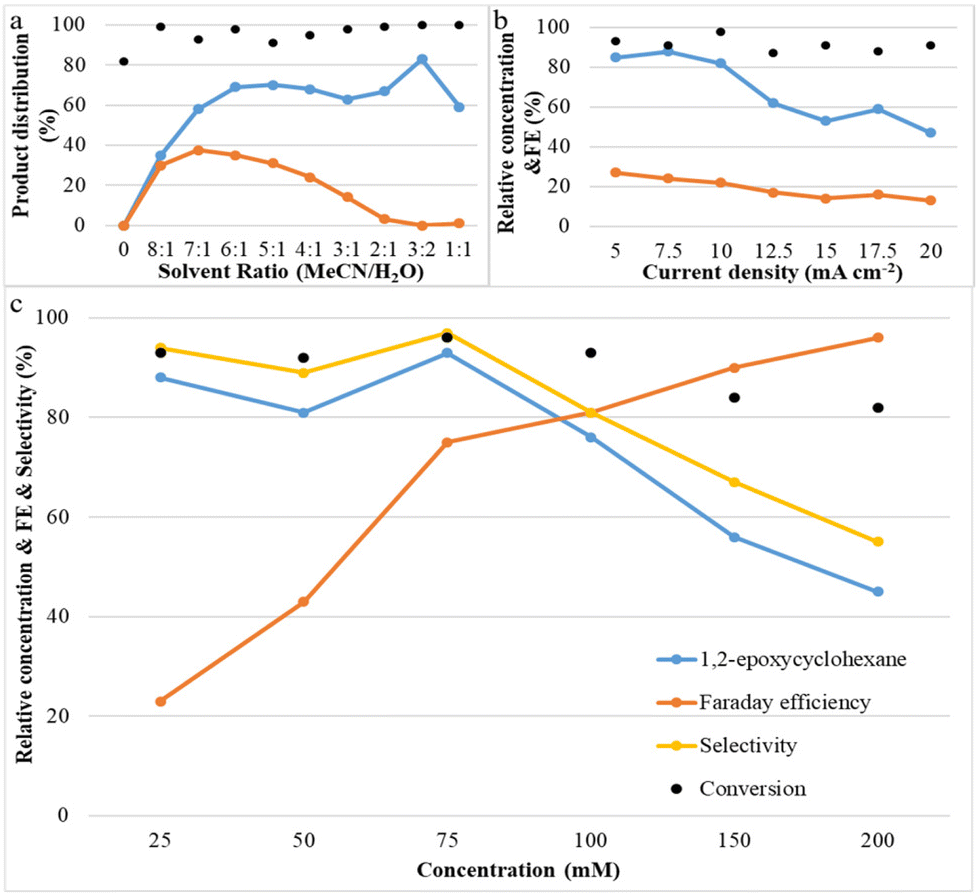 | ||
| Fig. 3 Reaction conditions: Pt as anode and Pt as cathode, room temperature; (a) different solvent ratios of MeCN and H2O: constant current of 10 mA cm−2, 1a (25 mM), Et4NBF4 (2 eq.), NaBr (1.5 eq.); the ratio of MeCN:H2O is varied from 6:0 to 1:1, 3 h; (b) different current densities: constant current of 5–20 mA cm−2, 1a (25 mM), Et4NBF4 (2 eq.), NaBr (1.5 eq.), MeCN:H2O (3:2, 6 ml), 7.46 Faraday mol−1. (c) different cyclohexene concentrations: 1a from 25–200 mM, constant current of 7.5 mA cm−2, Et4NBF4 (2 eq.), NaBr (1.5 eq.), MeCN:H2O (3:2, 6 ml), 4 h. | ||
Fig. 3b shows the effect of current density on both cyclohexene conversion and epoxide yields. With increasing current density, initially, the epoxide yield changes very little, but above 7.5 mA cm−2 the epoxide yield decreases rapidly. At the same time, it was also found that cyclohexene conversion was maintained at a similar level. A possible explanation for this phenomenon is that when the reaction proceeds at high current density, there is a lot of bromonium formed, but initially not enough OH− is available to successfully form the bromohydrin and finally the epoxide. This indicates that a low and stable current density contributes most significantly to olefin epoxidation. Fig. 3c shows that with increasing cyclohexene concentration, the conversion of cyclohexene and the epoxide selectivity and yield remain at nearly a constant level initially; they only decrease above 75 mM. However, the faradaic efficiency increases continuously with increasing concentration. This is due to the fact that a higher concentration of cyclohexene can make full use of the available current to react with free available bromine in the reaction medium to effect cyclohexene oxidation.
Kinetic study
To understand the mechanism of the electrochemical olefin epoxidation reaction, the compound concentrations and the pH were followed over time in a kinetic experiment (Fig. 4).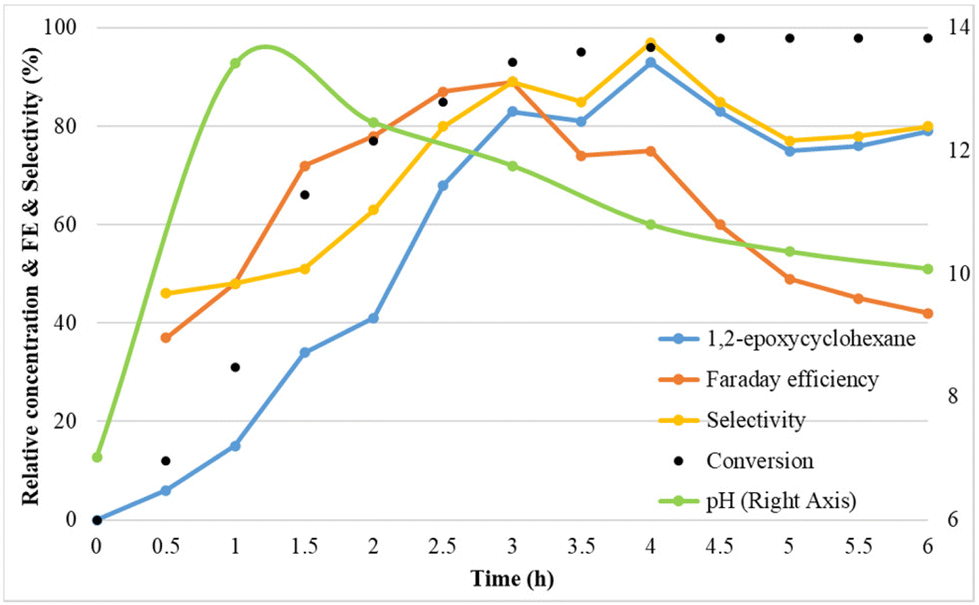 | ||
| Fig. 4 Reaction conditions: Pt as anode and Pt as cathode, constant current of 7.5 mA cm−2, 1a (75 mM), Et4NBF4 (2 eq.), NaBr (1.5 eq.), MeCN:H2O (3:2, 6 ml), room temperature. | ||
In the kinetic study, initially the cyclohexene epoxide yield, the faradaic efficiency and also the epoxide selectivity all increased. At 4 h, already >90% yield is reached, and this eventually increases up to 93%, with excellent selectivity. The faradaic efficiency reached its maximum of 89% after 3 h. Interestingly, the pH increases from its initial value of 7, to a high of 13.4 after 1 h, and then steadily decreases again towards 10 (Fig. S3†). This confirms that the cathode produces sufficient alkalinity to effect the ring closure towards the epoxide. This may be due to the initial hydrogen evolution causing a rapid pH rise, followed by a drop in pH due to bromohydrin formation and ring closure process.
Mechanistic proposal
As a possible mechanism (Scheme 1), we propose that the bromide is initially anodically oxidized to furnish an oxidized ‘Br+’ species like Br2, HOBr or BrO−, resulting in an electrochemical umpolung. Meanwhile, H2O is reduced on the cathode, and hydrogen and OH− are released. Eventually, the bromide is eliminated under the alkaline conditions, to obtain the final product epoxycyclohexane.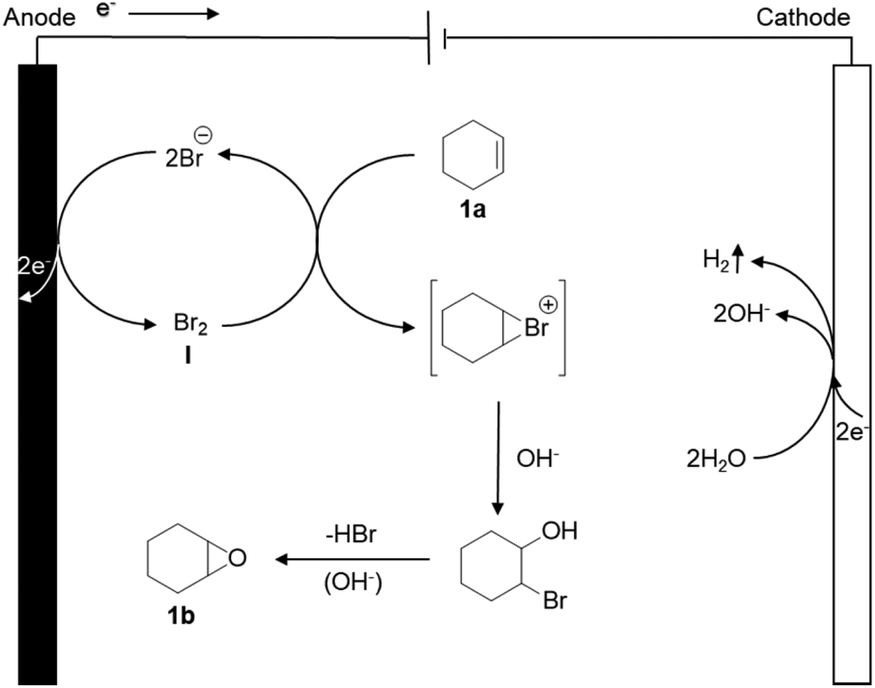 | ||
| Scheme 1 Proposed mechanism for the electrochemical bromine-mediated olefin epoxidation. | ||
Substrate scope and gram scale experiment
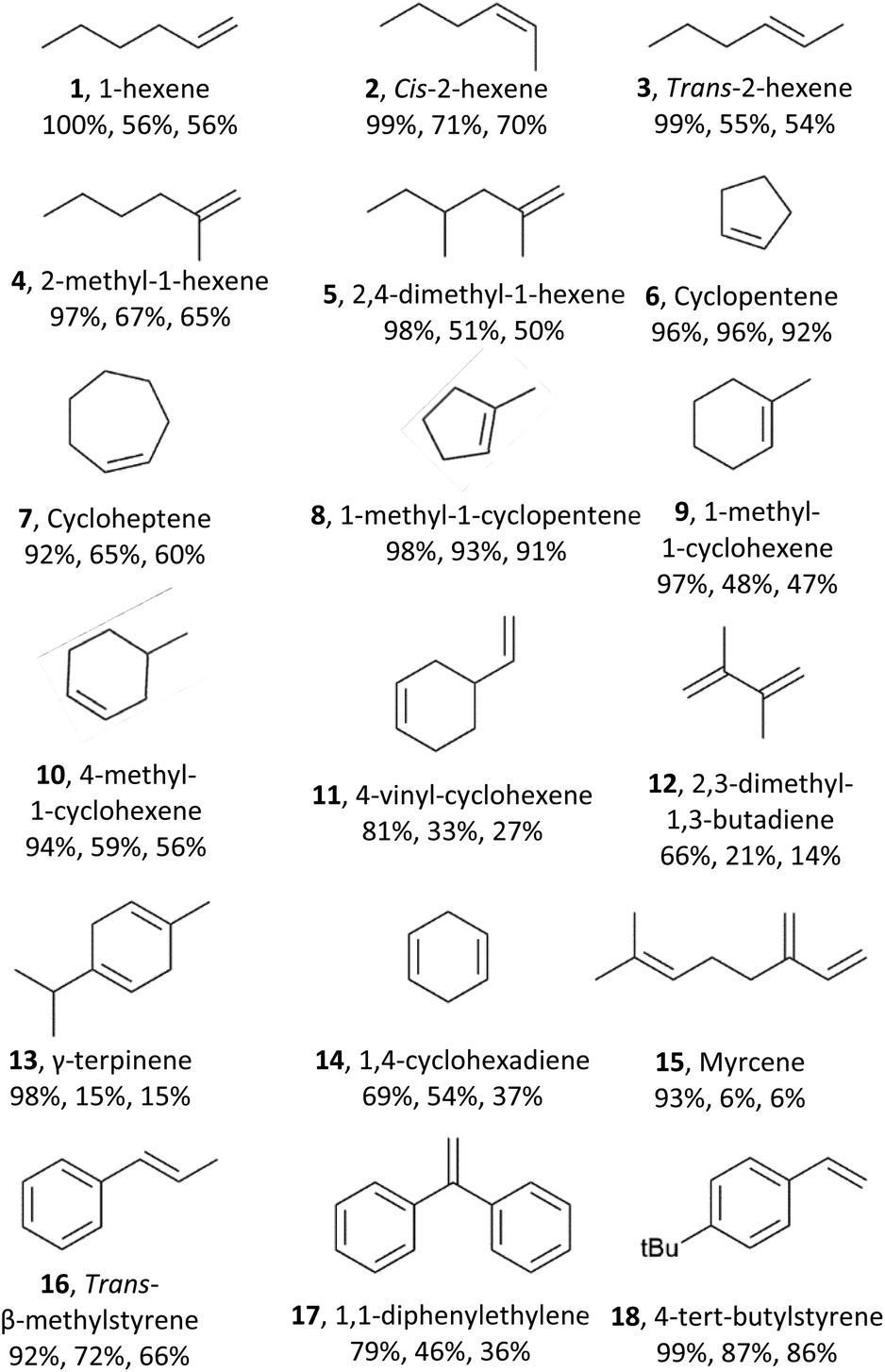
Next, we explored the substrate scope of the electrochemical olefin epoxidation using the optimized reaction conditions. Excellent product yields were achieved with a variety of different olefins. The aliphatic olefins were assessed first. A substrate such as 1-hexene (1) was tolerated, providing the corresponding product in a 56% yield. As shown in Table 1, epoxides 2 and 3 were generated in a stereospecific fashion from cis-2-hexene (70% yield) and trans-2-hexene (54% yield), respectively. This is in line with the formation of a cyclic bromonium adduct (see Scheme 1), followed by attack of the hydroxyl anion from the opposite side, and finally, stereospecific cyclization of the bromohydrin via an SN2 reaction. A series of mono- and di-substituted olefins all performed well in this transformation, as did various methyl-substituted compounds such as 2-methyl-1-hexene (4, 65% yield) and 2,4-dimethyl-1-hexene (5, 50% yield). Alicyclic olefins also performed well, including cyclopentene (6) and cycloheptene (7) with corresponding product yields of 91% and 47%, respectively. Mono-substituted cyclo-olefins were also confirmed as acceptable substrates, and the yields with 1-methyl-1-cyclopentene (8), 1-methyl-1-cyclohexene (9), 4-methyl-cyclohexene (10), and 4-vinyl-cyclohexene (11) were 91%, 47%, 56% and 27%, respectively. In dienes with almost identical electron density at their CC bonds, the observed regioselectivity mainly arises from their different steric accessibilities. This was further confirmed by our findings for 4-vinylcyclohexene (11), 2,3-dimethyl-1,3-butadiene (12), γ-terpinene (13), 1,4-cyclohexadiene (14), and myrcene (15). Furthermore, aromatic olefins were explored including trans-β-methylstyrene (16), 1,1-diphenylethylene(17), and 4-tert-butylstyrene (18), which also showed good yields.
| Reaction conditions: Pt as anode and Pt as cathode, constant current of 7.5 mA cm−2, olefin substrates (75 mM), Et4NBF4 (2 eq.), NaBr (1.5 eq.), MeCN:H2O (3:2, 6 ml), 4 h, 2.49 Faraday mol−1, room temperature. The percentages correspond to conversion, selectivity, product yield, respectively. |
|---|
|
The scope of the method has thus been documented extensively; besides cyclohexene, a variety of aliphatic, alicyclic, and aromatic olefins, with mono-, di- or trisubstituted double bonds can be epoxidized efficiently. However, the compatibility with other functional groups, e.g., alcohols, is limited. In the future, we will also report in detail on the epoxidation of a series of biobased terpenic and sesquiterpenic olefins.
Finally, we also demonstrated that the reaction could be scaled up easily (Scheme 2). At a one gram scale, the epoxidation reaction proceeded smoothly to afford product 1b in 85% yield. We also developed a simple method to work up the product to high purity with an extractive approach, effectively eliminating the organic cations.
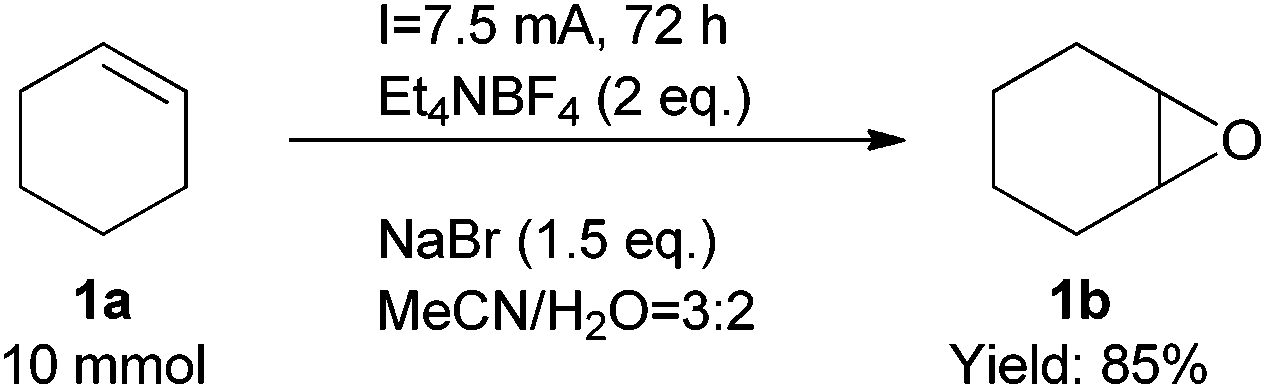 | ||
| Scheme 2 Gram scale synthesis. | ||
Conclusions
The electrochemical, bromide-mediated epoxidation of cyclohexene has the potential to combine high yields, excellent selectivity, and high faradaic efficiencies. Based on electrochemical kinetics, a reaction mechanism was unraveled, indicating that the formation of the target epoxide is directly realized under alkaline conditions. This approach, using H2O as the source of oxygen atoms, opens up a new synthetic route even for poorly activated olefins.Author contributions
Under supervision of D. D. V., H. Y. T. was responsible for the design and analysis of the experiments. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was funded by grants from the China Scholarship Council (CSC, grant number: 201806070154 for H. T). J. V. is grateful to FWO-SB funding (FWO Grant Number 1SB1522N). D. D. V. is grateful to FWO-Vlaanderen for research grants.References
- Nexant Process Evaluation Research Planning (PERP) Report-Propylene oxide, 2018.
- A. S. Sharma, V. S. Sharma, H. Kaur and R. S. Varma, Green Chem., 2020, 22, 5902–5936 RSC.
- D. Swern, Organic Peroxides, Wiley Interscience, New York, 1971, vol. 2 Search PubMed.
- C. Kim, T. G. Traylor and C. L. Perrin, J. Am. Chem. Soc., 1998, 120, 9513–9516 CrossRef CAS.
- T. Katsuki and K. B. Sharpless, J. Am. Chem. Soc., 1980, 102, 5974–5976 CrossRef CAS.
- D. E. De Vos, S. de Wildeman, B. F. Sels, P. J. Grobet and P. A. Jacobs, Angew. Chem., Int. Ed., 1999, 38, 980–983 CrossRef CAS.
- L. Wang, B. Zhang, W. Zhang, J. Zhang, X. Gao, X. Meng and F. S. Xiao, Chem. Commun., 2013, 49, 3449–3451 RSC.
- V. Polshettiwar and T. N. Asefa, Synthesis and applications, Wiley Online Library, 2013 Search PubMed.
- K. Jin, J. H. Maalouf, N. Lazouski, N. Corbin, D. Yang and K. Manthiram, J. Am. Chem. Soc., 2019, 141, 6413–6418 CrossRef CAS.
- J. Li, J. Jiang, T. Manasfi and U. von Gunten, Water Res., 2020, 187, 116424 CrossRef CAS.
- N. Nuraje, R. Asmatulu and G. Mul, Green photo-active nanomaterials: sustainable energy and environmental remediation, Royal Society of Chemistry, 2015 Search PubMed.
- S. Rebsdat and D. Mayer, Ethylene oxide, in Ullmann's Encyclopedia of Industrial Chemistry, 2000 Search PubMed.
- J. A. M. Leduc, U.S. Patent 3288692, 1966.
- W. R. Leow, Y. Lum, A. Ozden, Y. Wang, D. H. Nam, B. Chen, J. Wicks, T. T. Zhuang, F. Li, D. Sinton and E. H. Sargent, Science, 2020, 368, 1228–1233 CrossRef CAS.
- M. Chung, K. Jin, J. S. Zeng and K. Manthiram, ACS Catal., 2020, 10, 14015–14023 CrossRef CAS.
- F. Lian, K. Xu and C. Zeng, Chem. Rec., 2021, 21, 2290–2305 CrossRef CAS PubMed.
- K. Rossen, R. P. Volante and P. J. Reider, Tetrahedron Lett., 1997, 38, 777–778 CrossRef CAS.
- B. F. Sels, D. E. De Vos and P. A. Jacobs, J. Am. Chem. Soc., 2001, 123, 8350–8359 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc02883b |
| This journal is © The Royal Society of Chemistry 2022 |