
The states that hide in the shadows: the potential role of conical intersections in the ground state unimolecular decay of a Criegee intermediate†
Barbara
Marchetti
*a,
Vincent J.
Esposito
 b,
Rachel E.
Bush
a and
Tolga N. V.
Karsili
*a
b,
Rachel E.
Bush
a and
Tolga N. V.
Karsili
*a
aUniversity of Louisiana at Lafayette, Louisiana, LA 70504, USA. E-mail: barbara.marchetti1@louisiana.edu; tolga.karsili@louisiana.edu
bUniversity of Pennsylvania, Philadelphia, PA 19104, USA
First published on 6th December 2021
Abstract
Criegee intermediates are of great significance to Earth's troposphere – implicated in altering the tropospheric oxidation cycle and in forming low volatility products that typically condense to form secondary organic aerosols (SOAs). As such, their chemistry has attracted vast attention in recent years. In particular, the unimolecular decay of thermal and vibrationally-excited Criegee intermediates has been the focus of several experimental and computational studies, and it is now recognized that Criegee intermediates undergo unimolecular decay to form OH radicals. In this contribution we reveal insight into the chemistry of Criegee intermediates by highlighting the hitherto neglected multi-state contribution to the ground state unimolecular decay dynamics of the Criegee intermediate products. The two key intermediates of present focus are dioxirane and vinylhydroperoxide – known to be active intermediates that mediate the unimolecular decay of CH2OO and CH3CHOO, respectively. In both cases the unimolecular decay path encounters conical intersections, which may play a pivotal role in the ensuing dynamics. This hitherto unrecognized phenomenon may be vital in the way in which the reactivity of Criegee intermediates are modelled and is likely to affect the ensuing dynamics associated with the unimolecular decay of a given Criegee intermediate.
Introduction
Criegee intermediates are a class of highly reactive organic compounds, known most prominently as reactive atmospheric oxidants. They are distinguishable by their carbonyl oxide functional group that contains Zwitterionic (or biradical) character. Criegee intermediates are an important source of non-photolytic OH radicals,1–5 and are well-known for their role in promoting the oxidizing capacity of the troposphere and for forming low volatility products that typically condense to form secondary organic aerosols (SOAs). In the atmosphere, Criegee intermediates are formed via the oxidation of volatile alkenes with ozone.6–9 The nascent Criegee intermediates are formed highly internally excited and may undergo prompt unimolecular decay or vibrational energy relaxation via collisions with bath molecules in the troposphere (e.g., N2).10–13 If the latter prevails, the resulting state is known as a stabilized Criegee intermediate, which may then undergo unimolecular decay or bimolecular reaction with trace tropospheric gas molecules (notably H2O, SO2 and organic acids).14–17 Both processes are implicated in enhancing the oxidizing capacity of the troposphere and in forming SOAs.The chemistry of Criegee intermediates of varying molecular complexities have been at the centre of several experimental and computational studies.11,18–23 It is now recognized that the unimolecular decay of Criegee intermediates is an important source of OH radicals – which is the main oxidizer in the troposphere. The simplest Criegee intermediates, formaldehyde oxide (henceforth CH2OO) undergoes unimolecular decay to form OH radicals – as well as CO, CO2 and formic acid.24,25 This unimolecular decay is summarized in Scheme 1 and is driven by isomerization of CH2OO to form a dioxirane intermediate, which then undergoes ring-opening to form formic acid via a bisoxy biradical intermediate.24 Recent work has detected formic acid, CO, CO2 and OH products from CH2OO. The Criegee intermediates in this case is formed by initial photolysis of CH2I2 and subsequent reaction of the nascent CH2I radical with seed O2 gas molecules – forming CH2OO + I products.25
 | ||
| Scheme 1 Reaction mechanism associated with the unimolecular decay of CH2OO to form the bisoxy biradical. | ||
The simplest alkyl substituted Criegee intermediates, acetaldehyde oxide, CH3CHOO, may also undergo unimolecular rearrangement to form acetic acid, via rearrangement of an methyldioxirane intermediate. This however constitutes a minor channel in CH3CHOO and the dominant unimolecular decay pathway is intramolecular H-atom transfer to form vinylhydroperoxide (VHP), followed by unimolecular decay to form CH2CHO + OH radicals.4,26–31 This latter process is highlighted in Scheme 2 and is now understood to be a key contributor to the OH radical yield in the troposphere. Criegee intermediates with >C2 are also known to undergo unimolecular decay to form OH radicals.29,32 In such Criegee intermediates the mechanism is the same as that outlined in Scheme 2 for CH3CHOO.
 | ||
| Scheme 2 Reaction mechanism associated with the unimolecular decay of CH3CHOO to form the CH2CHO + OH radicals via a VHP intermediate. | ||
In the present study, we use state-of-the-art multi-reference quantum chemical methods to access the extent to which coupling between multiple electronic states contributes to the ground state (thermal or vibrationally excited) unimolecular decay dynamics of Criegee intermediates. In so doing, we assess the ground and excited state energy profiles of two key intermediates, dioxirane and VHP, which are known to mediate unimolecular decay in Criegee intermediates. The recognition that the ensuing dynamics occurs on multiple energy profiles may potentially influence the dynamics – including altering the product branching ratios and yields.
Computational methodology
All optimizations (ground state minima and conical intersections) were performed in the Gaussian 16 computational package.33 The (X)MS-CASPT2 computations were undertaken in the Molpro computational package.34,35The ground state minimum energy geometry of dioxirane was optimized with Density Functional Theory (DFT), using Grimme's B2PLYP-D336 functional coupled to the 6-311+G(d,p) basis set.37,38 This functional has been shown to perform well in optimizing geometries and obtaining normal modes in Criegee intermediates.29,31
Relaxed potential energy profiles along the O–O stretch coordinate (ROO) were constructed by fixing ROO at various values and optimizing the remainder of the nuclear with the Møller–Plesset second-order perturbation theory39,40 or the complete active space second-order perturbation theory (CASPT2), method coupled to the aug-cc-pVTZ basis set.41 At each relaxed geometry, multi-state complete active space second-order perturbation theory (MS-CASPT2),42 coupled to the aug-cc-pVTZ basis set, was used to obtain the ground and lowest three electronically excited state energies. The lowest four states of singlet spin-multiplicity were mixed in the MS-CASPT2 calculations and were based on a state-average complete active space self-consistent field (SA4-CASSCF) reference wavefunction. An active space consisting of twelve electrons in ten orbitals was used in the CASSCF and MS-CASPT2 calculations. The active orbitals are displayed in Fig. S1 of the ESI.†
A conical intersection between the ground and first electronically excited state of dioxirane was located at the CASSCF/6-31G(d) level of theory. In this case a smaller active space of six electrons in five orbitals was used. This smaller active space (cf. PE profiles) is adequate for describing the two-state interaction between the S0 and S1 states at a localized region, while in the PE profiles, the larger active space is required to describe the long-range PE profiles of the four coupled electronic states (vide infra).
The ground state minimum energy geometry of VHP was optimized using Grimme's B2PLYP-D3 functional coupled to the 6-311+G(d,p) basis set. Unrelaxed (rigid body) potential energy profiles along the ROO coordinate were constructed by elongating the ROO bond length and performing single-point calculations of the four lowest singlet electronic states at each geometry. All other coordinates were fixed at their equilibrium geometry. The single-point calculations were performed using Extended Multi-State complete active space second-order perturbation theory (XMS-CASPT2)43 in conjunction with the aug-cc-pVTZ basis set. All 4 states were mixed in the XMS-CASPT2 wavefunction and were based on a 4 state-averaged CASSCF (SA4-CASSCF) reference wavefunction using the same basis set. An active space of twelve electrons in nine orbitals was used, the latter of which are visualized in Fig. S2 of the ESI.† CASPT2 relaxed scans were also conducted along ROO, by fixing ROO at various values and allowing the remainder of the nuclear framework to relax to their ground state minimum energy configurations.
The active spaces for both dioxirane and VHP were selected in order describe all significant electronic effects while maintaining an appropriate computational expense. The reactant and product vertical excitation energies were benchmarked with various active spaces. These results are displayed in Table S1 and S2 of the ESI† and show that variations in the active space show only modest changes to the excitation energies.
Results and discussion
Ground state structures and vertical excitation energies
Fig. 1 presents the ground state minimum energy geometry of dioxirane and VHP – calculated at the B2PLYP-D3/6-311+G(d,p) level of theory. As previously noted,26,44 the ground state minimum energy geometry of both dioxirane and VHP are with all carbon and oxygen atoms in a common plane. In dioxirane, the carbon atom is at the centre of a pseudo-tetrahedral geometry, in which the carbon-hydrogen and carbon–oxygen bonds form a symmetric arrangement around the central carbon-atom. In VHP the hydrogen atom on the terminal OH moiety is out of the molecular plane – forming a staggered conformation. This is consistent with the molecular geometry of hydrogen peroxide which also shows a staggered conformation.45,46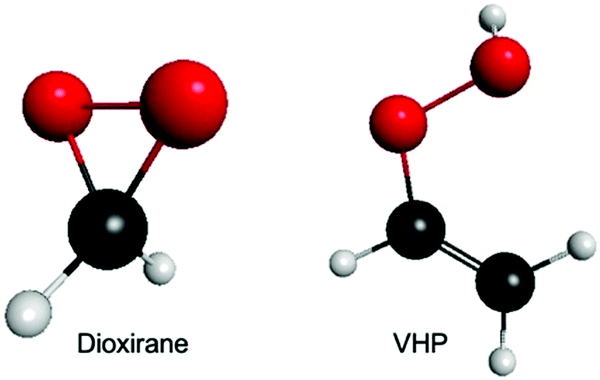 | ||
| Fig. 1 Ground state minimum energy geometry of dioxirane and VHP – calculated at the B2PLYP-D3/6-311+G(d,p) level of theory. | ||
Fig. 2 presents the dominant electronic configurations associated with the ground and excitation to the lowest three electronically excited singlet states of dioxirane and VHP.
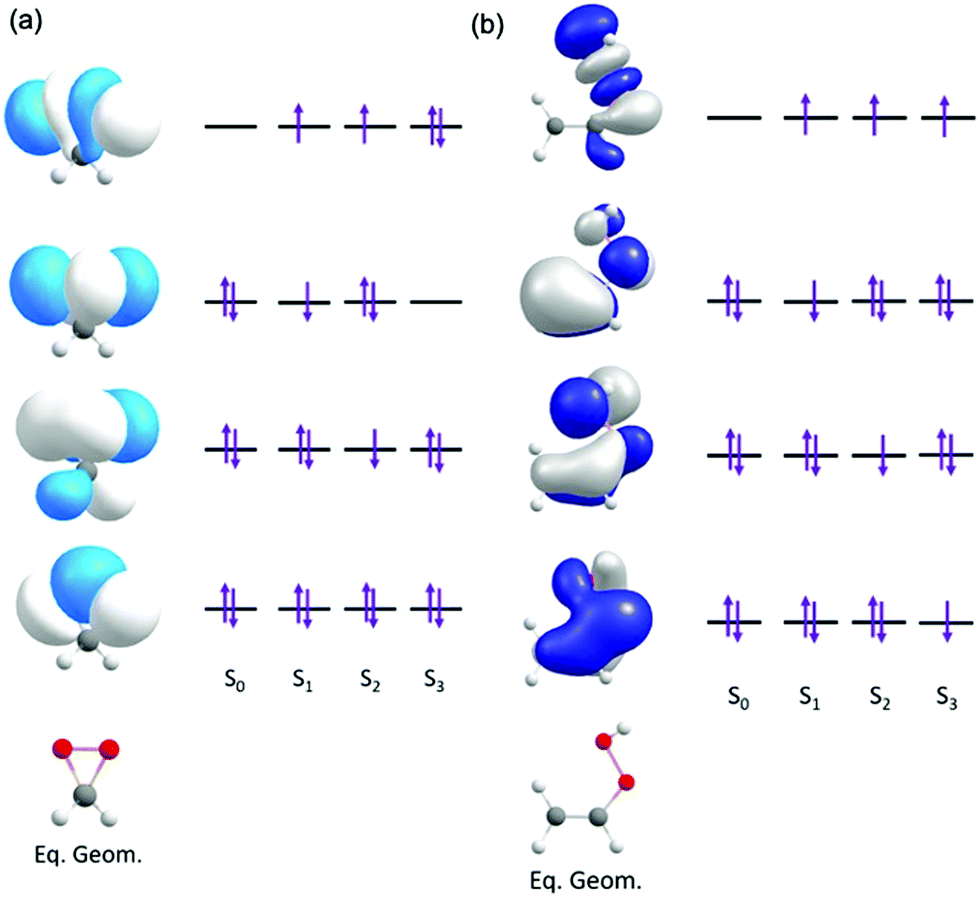 | ||
| Fig. 2 Orbitals and orbital promotions associated with the vertically excited electronic states of (a) dioxirane and (b) VHP. | ||
In dioxirane the ground state involves an expected electronic configuration in which all bonding and non-bonding valence orbitals are doubly occupied. The first and second vertically excited states involve single-excited electronic configurations, involving promotion from an out-of-plane valence orbitals to a common in-plane σ* virtual orbital. This σ* orbital is localized along the O–O bond of the dioxirane. The third excited state also arises by electron transition to the same σ* orbital but in this case the configuration is doubly-excited – with the σ* orbital doubly occupied.
In VHP the ground state also involves an expected electronic configuration in which all orbitals are doubly occupied. The excited electronic states all involve promotion of an electron to a common σ* orbital localized along the O-O bond. The first, second and third electronically excited state configurations are distinguishable by the different π orbital from which the electron is promoted.
Unimolecular decay paths
Dioxirane
Fig. 3 presents the adiabatic MS-CASPT2 potential energy (PE) profiles along the O–O stretch coordinate (henceforth ROO) of dioxirane. Panels (a) and (b) are distinguishable by the relaxation of the nuclear framework with MP2 or CASPT2, respectively. These will henceforth be referred to as MS-CASPT2/AVTZ//MP2/AVTZ and MS-CASPT2/AVTZ//CASPT2/AVTZ, respectively. Additional details are given in the methodology section. The geometry at ROO = 1.56 Å corresponds to the dioxirane reactant whilst that at ROO ∼ 2.4 Å corresponds to the bisoxy biradical – which is the intermediate precursor for forming formic acid. As shown, the PE profiles of the adiabatic ground state (blue curve in Fig. 3) follows that of a typical ground state reaction, wherein the reactant dioxirane and the product bisoxy biradical are separated by a PE barrier at ca. ROO ∼ 2.2 Å. The molecular structures of the reactant, product and saddle point (of dioxirane and the bisoxy diradical) accord well with previous single point computations by Guo and co-workers.44Fig. 3 also displays the adiabatic S1, S2 and S3 states along the ROO coordinate. In both the MS-CASPT2/AVTZ//MP2/AVTZ and MS-CASPT2/AVTZ//CASPT2/AVTZ PE profiles, all three excited states show a net decrease in PE upon increasing ROO from that of the ground state equilibrium value. This can be understood by recognizing that all vertically excited electronic states correspond to excitations to a common σ* orbital, localized on the O–O bond (as outlined in the previously section). This will inevitably lead to a long-range repulsive interaction upon decreasing ROO when viewed from the bisoxy biradical geometry. The relative gradients along ROO, of the excited electronic states at the Franck–Condon region, qualitatively increases from S1 to S3. This steepest gradient of the S3 state (cf. S1 and S2 states) can be understood by recognizing that the electronic state arises via promotion of an electron-pair to the σ* orbital (see Fig. 3) – leading to a doubly excited state configuration. This manifests in a lower bond order for S3 (cf. S1 and S2 states). Instead, the S1 and S2 states arise via a single excitation to the σ* orbital – leading to a singly occupied configuration and thus a greater O–O bond order when compared to S3.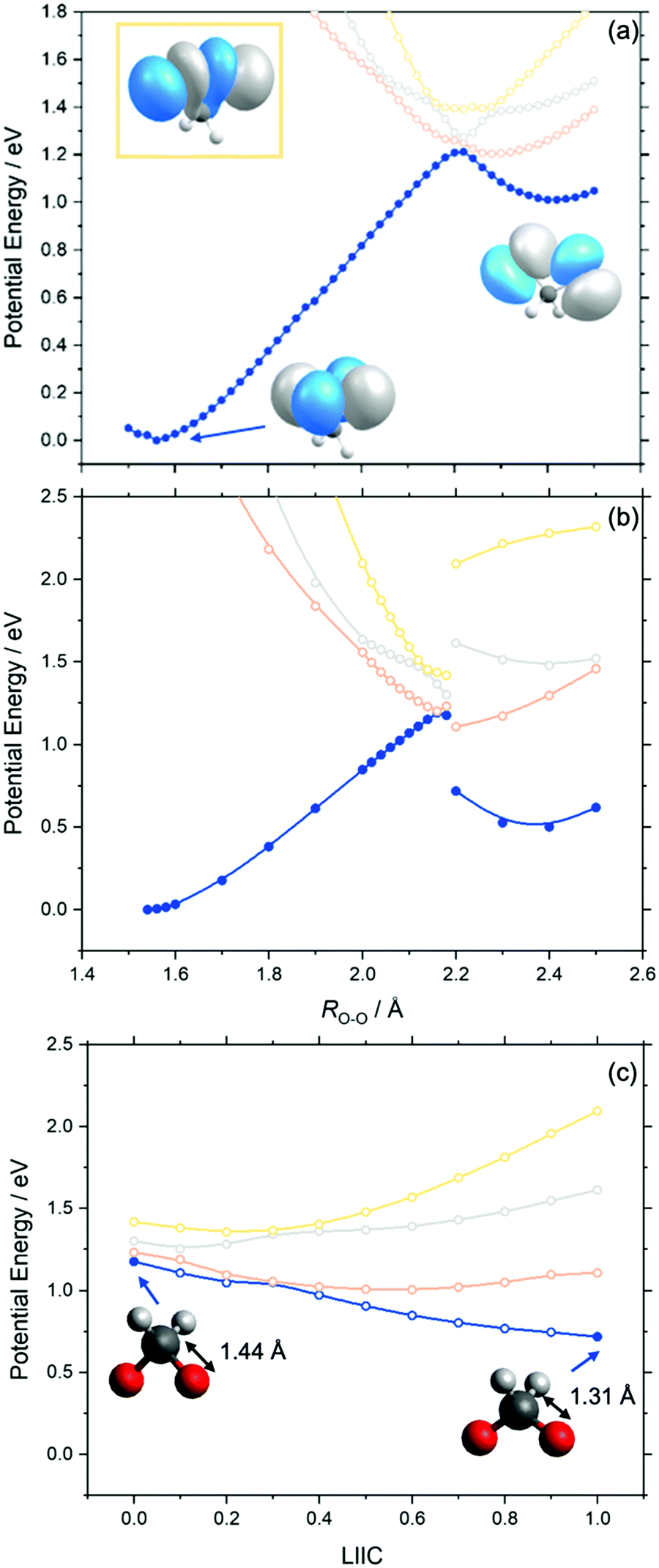 | ||
| Fig. 3 Relaxed PE profiles along the O–O stretch coordinate of dioxirane of the lowest four singlet states, calculated at the (a) MS-CASPT2/AVTZ//MP2/AVTZ and (b) MS-CASPT2/AVTZ//CASPT2/AVTZ levels of theory. The orbitals represent the highest doubly occupied molecular orbital and various points, two of which are indicated by arrow. The orbital displayed in the yellow box is the highest doubly occupied molecular orbital of the S3 state (yellow profile) at ROO = 1.56 Å. The step change between ROO = 2.18 Å and ROO = 2.2 Å represents a change in the C–O bond distance from 1.44 Å to 1.31 Å. Panel (c) shows a LIIC between this step change in energy between the relaxed ROO = 2.18 Å and ROO = 2.2 Å geometries displayed in (b). | ||
The S1 state becomes degenerate with the S0 state at ca. ROO = 2.2 Å, at which point the S2 and S3 states are also close in energy to the S0 state. This is particularly noteworthy as ROO ∼ 2.2 Å also corresponds to the transition state geometry associated with the ground state unimolecular decay of dioxirane to the bisoxy biradical. From the dioxirane parent molecule, progressive elongation of ROO leads to an adiabatic change in the ground state electronic configuration from the dioxirane to the bisoxy biradical. This change in the electronic wavefunction is illustrated by the change in the highest occupied molecular orbital upon forming the bisoxy biradical from dioxirane. The highest occupied molecular orbital of the bisoxy biradical is of anti-bonding (σ*) character and is therefore unoccupied in the ground state dioxirane. The ground state electronic configuration of the bisoxy biradical product therefore correlates with an excited electronic state configuration of the reactant dioxirane structure that is distinct from that of ground state dioxirane.
Although the PE profiles calculated at the MS-CASPT2/AVTZ//MP2/AVTZ and MS-CASPT2/AVTZ//CASPT2/AVTZ level are similar in the region between ROO = 1.56 Å and ROO = 2.18 Å, we note an important distinction at long ROO. As shown in Fig. 3(b), the MS-CASPT2/AVTZ//CASPT2/AVTZ PE profiles show a step discontinuity in the energy profiles between ROO = 2.18 Å and ROO = 2.2 Å, which arises via the reduction of the C–O bond distance from ca. RCO = 1.44 Å (at ROO ≤ 2.18 Å) to RCO = 1.31 Å (at ROO ≥ 2.2 Å). This behaviour is not captured in the MP2 relaxation due to its inherent single-reference nature. To investigate this step change in energy further, additional MS-CASPT2 PE profiles connecting the CASPT2 relaxed geometries at ROO = 2.18 Å to ROO = 2.2 Å were constructed using a Linear interpolation in internal coordinates (LIIC). Fig. 3(c) presents the resulting LIIC between the CASPT2 relaxed geometries at ROO = 2.18 Å and ROO = 2.2 Å, which shows the smooth barrierless variation of the ground state from ROO = 2.18 Å to ROO = 2.2 Å. When comparing the bisoxy biradical relative energy (cf. to the dioxirane parent molecule), calculated at the MS-CASPT2/AVTZ//MP2/AVTZ and MS-CASPT2/AVTZ//CASPT2/AVTZ levels, the latter is in excellent agreement with previous high-level CCSD(T)-F12 calculations of the energy difference between dioxirane and bisoxy.44 The variation is noteworthy since it shows that MP2 relaxation does not sufficiently describe the multi-reference nature of the bisoxy biradical (beyond ROO = 2.18 Å).
With these topographic details in mind, the ground state reaction from dioxirane to the bisoxy biradical may involve substantial non-adiabaticity as the change in the electronic wavefunction is associated with a transition state that is close in energy to the several excited states. This is reinforced by recognizing that the transition state and degeneracy at ca. ROO = 2.2 Å is associated with a conical intersection. Fig. 4 displays the optimized S0/S1 conical intersection geometry, which although performed with a different level of theory (i.e., smaller active space, basis set and CASSCF method) than the PE profiles in Fig. 3 (see Methodology section), is consistent with the same point at ROO = 2.2 Å. Any changes in the geometry are minor – as revealed by ROO = 2.157 Å in the optimized structure vs. ROO = 2.2 Å (MP2 relaxed) and ROO = 2.16 Å (CASPT2 relaxed) in Fig. 3(a) and (b), respectively. Fig. 4 also displays the g and h vectors associated with the optimized conical intersection. It is reassuring that the g-vector – i.e., the gradient-difference vector (or tuning coordinate) – is associated with the reactive coordinate. The h-vector – i.e., the derivative-coupling vector (or coupling coordinate) requires anti-symmetric wagging of the oxygen and hydrogen atoms in order to lift the degeneracy associated with the conical intersection.
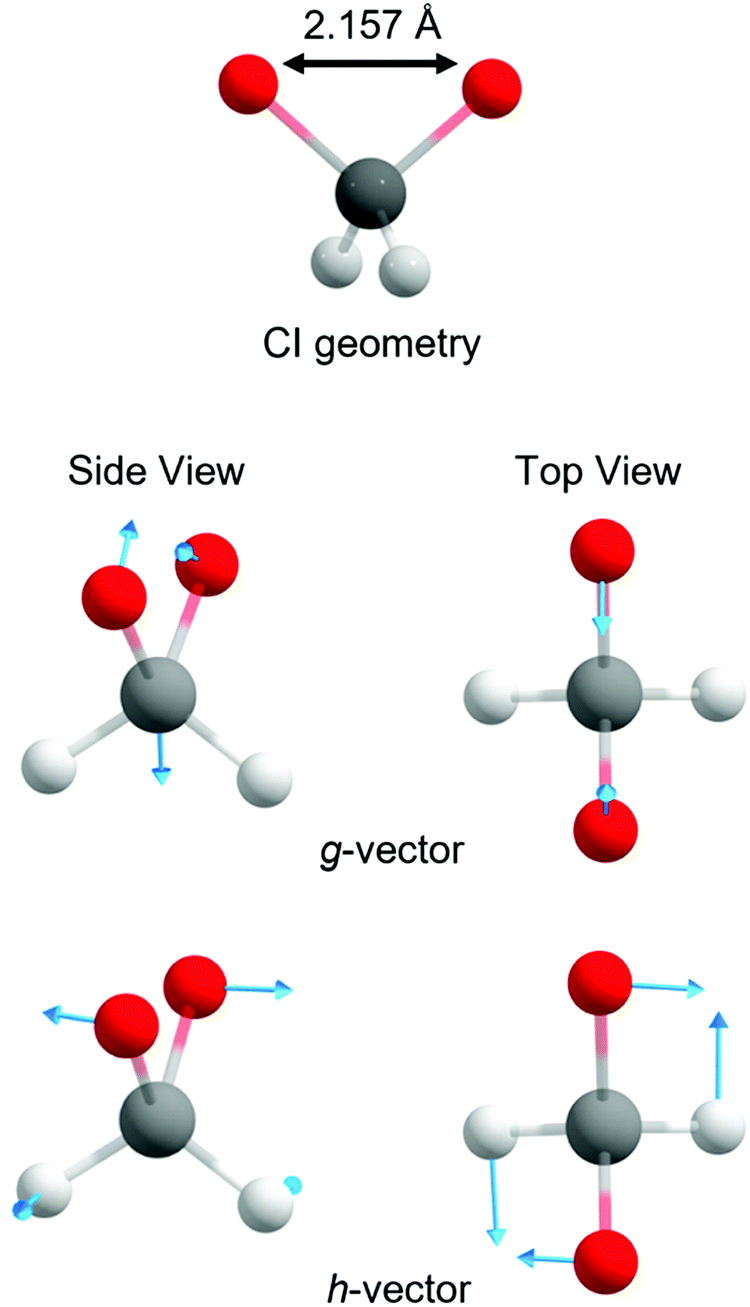 | ||
| Fig. 4 g and h vectors associated with the optimized S0/S1 conical intersection of dioxirane. | ||
The transition state at ROO = 2.2 Å in Fig. 3 is the lower part of a conical intersection and is therefore expected to undergo non-adiabatic coupling with the excited electronic states. Motion around the lower cone of the conical intersection may give rise to the geometric phase (GP) effect as the electronic wavefunction bifurcates around the point of degeneracy. The GP effect is a characteristic signature of conical intersections and arises when the adiabatic electronic wavefunction changes sign when transported around the conical intersection. The inherent GP effect may then control the exit channel energy dissipation and thus the ensuing chemistry for formic acid formation. Such effects are common in molecular reactions that are analogous to the current system of interest and it is known that such effects persist even in cases in which the conical intersection is much higher in energy than the initially excited vibrational resonance of the ground state.47 Ample studies have demonstrated that the GP needs to be considered in treating the adiabatic dynamics around a conical intersection.47–53
We make one caveat that since dioxirane is likely prepared highly vibrationally excited, the subsequent O–O bond fission to form the bisoxy biradical may not necessarily follow the minimum energy path as given by the relaxed PE profiles. Although these side routes are possible at high internal energies, the most energetically probable route is the minimum energy path.
Vinyl hydroperoxide (VHP)
Similar behaviour is observed in our next case study vinyl hydroperoxide (VHP). VHP is formed when thermalized or vibrationally excited CH3CHOO undergoes intramolecular 1,4-H-atom transfer. The nascent VHP is formed with high internal excitation – which is excess of the energetic limit for forming CH2CHO + OH products. This process is the dominant unimolecular decay path for alkyl-substituted Criegee intermediates.Fig. 5 presents the adiabatic PE profiles along the O–O stretch coordinate (ROO – identified in the molecule inset in Fig. 5(a)) of VHP. Panels (a) and (b) indicate the PE profiles along ROO at geometries at which the CCOO torsional angle (τCCOO – identified in the yellow highlight on the VHP in Fig. 5(a)) is at τCCOO = 0° and 90°, respectively. τCCOO = 0° is a planar configuration and corresponds to the minimum energy geometry displayed in Fig. 1 while τCCOO = 90° corresponds to an out-of-plane configuration.
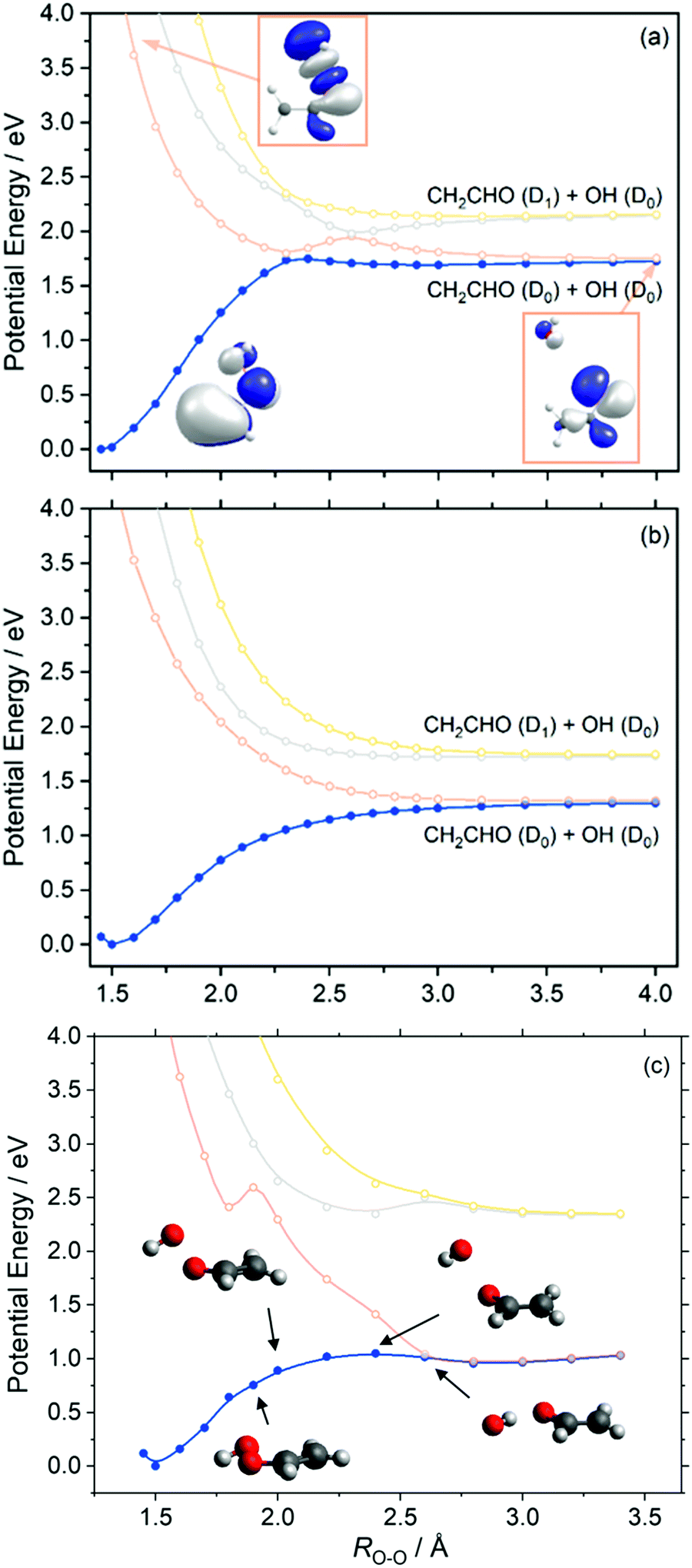 | ||
| Fig. 5 Unrelaxed adiabatic PE profile of VHP along the O–O stretch coordinate at (a) τCCOO = 0° and (b) τCCOO = 90°, calculated at the XMS-CASPT2/aug-cc-pVTZ level of theory. (c) XMS-CASPT2/AVTZ//CASPT2/AVTZ relaxed PE profiles along the ROO coordinate. | ||
Focussing on Fig. 5(a), the energy at ROO = 1.45 Å corresponds to the VHP parent molecule, while the asymptotic region beyond ROO > 2.5 Å corresponds to the OH + CH2CHO radical products. The ground state PE profile shows a characteristic topology associated with a bond dissociation process – wherein the energy rises upon increasing ROO and then plateaus as the PE profile becomes asymptotic. In the asymptotic region and under gas phase conditions, the OH and CH2CHO fragment partners are at a terminal separation. As with dioxirane, the ground state PE profile along ROO shows an avoided crossing with the S1 state at ca. ROO = 2.3 Å. As Fig. 3 implies, the degeneracy observed at ROO = 2.3 Å and τCCOO = 0° is lifted by motion along τCCOO which is consistent with coupling mode behaviour at a conical intersection. The lowest three excited states of VHP are dissociative with respect to ROO, which can be understood by considering that all three electronic states correspond to excitation into a common σ* orbital localized along the O–O bond.
To investigate the minimum energy path associated with VHP further, fully relaxed PE profiles along the ROO coordinate were undertaken. These are presented in Fig. 5(c). The CASPT2 relaxed PE profile shows that the parent VHP molecule is planar at short range ROO and undergoes progressive motion out of plane at 1.9 Å ≥ ROO ≥ 2.4 Å. This aligns with the unrelaxed PE profiles which show that the most stable configuration of VHP is planar, while motion around the crossing ROO = 2.3 Å along τCCOO shows a lower energy route to OH elimination. Although motion along τCCOO manifests in the ground state minimum energy path at 1.9 Å ≥ ROO ≥ 2.4 Å, the S1 state is unstable with respect to increasing τCCOO in this ROO range. This is highlighted by the step increase in energy from ROO = 1.8 Å to ROO = 1.9 Å in Fig. 5(c) and supported by the unrelaxed PE profiles in Fig. 5(a) and (b). At ROO = 2.6 Å, the ground state minimum energy path shows a planar geometry and a rotation of the OH moiety so as to undergo hydrogen bonding of the H-atom with the nascent vinoxy centred O-atom. Such a geometry along ROO is consistent with earlier study by Kidwell et al.54 and is attributed to a frustrated submerged transition state.
As with dioxirane, we note however that since VHP is prepared highly vibrationally excited and undergoes intramolecular vibrationally energy redistribution, the subsequent OH elimination may not necessarily follow the minimum energy path as given by the relaxed PE profiles.
As shown in the PE profiles in Fig. 5 the lowest energy asymptote corresponds to OH (D0) + CH2CHO (D0) products and correlates with an excited state electronic configuration – as indicated by the change in the highest occupied molecular orbital of the ground state VHP at ROO = 4.0 Å relative to the ground state. Again, unimolecular decay of ground state VHP clearly involves coupling of energy surfaces en route to forming OH + CH2CHO. Although motion along τCCOO lifts the degeneracy at ROO = 2.3 Å and provides a lower energy route around this crossing point, the ensuing dynamics is likely perturbed by the intersection at ROO = 2.3 Å as the evolving wavepacket bifurcates as it traverses the region around the crossing point. Unimolecular decay is therefore likely to involve substantial non-adiabaticity upon dissociation, wherein the GP effect may play a role in the ensuing dynamics as described above for dioxirane. This feature is analogous to the ground state unimolecular decay of vibrationally excited hydroxymethyl radical which is known to occur non-adiabatically.47
Conclusions
In this study, we have demonstrated the multi-state character associated with two well-known unimolecular decay paths available to Criegee intermediates. This hitherto unrecognized multi-state character, in the context of the products of Criegee intermediates, is an important recognition as it may control the nascent product yields formed upon unimolecular decay of Criegee intermediates. In 2018, Guo and co-workers discovered the role of conical intersections in the unimolecular decay of the hydroxymethyl radical and demonstrated that the geometric phase effect plays a role in controlling the rovibrational energy dissipation of the nascent formaldehyde product.47 In the context of the systems explored here, similar behaviour may effect on the product state branching in the nascent intermediates and fragments formed during the unimolecular decay of dioxirane or VHP. Providing that the nascent products react on timescales that are shorter than collisional energy relaxation, the preselected rovibrational energy distribution may have an implication on the resulting reactions of these atmospherically relevant products, but we note that these most likely undergo vibrational energy relaxation and thermalization prior to bimolecular chemistry. Notwithstanding, we demonstrate that such multi-state behaviour should be considered in future computational studies of molecular reactions that are relevant in the atmosphere or in combustion – especially in the context of Criegee intermediates.Given the details of the PE profiles above, our future plan is to generate full-dimensional energy profiles and to undertake time-dependent quantum wavepacket dynamics simulations on the subsequent energy surfaces. Such simulations will reveal the extent to which the unimolecular dynamics involves coupling to the excited state energy surfaces and thus if the reaction occurs non-adiabatically. One of the key questions that such simulations will answer is if the GP effect plays a role in the product state energy distributions of the nascent products.
The most exciting prospect is to explore how such electronic state couplings associated with the unimolecular decay of a dioxirane or a hydroperoxide change upon increasing molecular complexity of the starting Criegee intermediate. Future studies on the geometric and electronic effects of increasing molecules complexity are therefore also anticipated.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the National Science Foundation, under grant no. 2003422 (TNVK) and the National Science Foundation Graduate Research Fellowship Program under Grant No. DGE-1845298 (VJE). The authors would also like to thank the reviewers for their invaluable suggestions during the review stage.References
- L. Lu, J. M. Beames and M. I. Lester, Early time detection of OH radical products from energized Criegee intermediates CH2OO and CH3CHOO, Chem. Phys. Lett., 2014, 598, 23–27 CrossRef CAS.
- A. M. Green, V. P. Barber, Y. Fang, S. J. Klippenstein and M. I. Lester, Selective deuteration illuminates the importance of tunneling in the unimolecular decay of Criegee intermediates to hydroxyl radical products, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 12372 LP–12377 LP CrossRef PubMed.
- Y. Fang, V. P. Barber, S. J. Klippenstein, A. B. McCoy and M. I. Lester, Tunneling effects in the unimolecular decay of (CH3)2COO Criegee intermediates to OH radical products, J. Chem. Phys., 2017, 146, 134307 CrossRef PubMed.
- M. I. Lester and S. J. Klippenstein, Unimolecular Decay of Criegee Intermediates to OH Radical Products: Prompt and Thermal Decay Processes, Acc. Chem. Res., 2018, 51, 978–985 CrossRef CAS PubMed.
- R. L. Caravan, M. F. Vansco and M. I. Lester, Open questions on the reactivity of Criegee intermediates, Commun. Chem., 2021, 4, 2012–2015 Search PubMed.
- J. G. Calvert, R. Atkinson, J. A. Kerr, S. Madronich, G. K. Moortgat, T. J. Wallington and G. Yarwood, The Mechanisms of Atmospheric Oxidation of the Alkenes, Oxford University Press, 2000 Search PubMed.
- B. J. Finlayson and J. N. Pitts, Chemistry of the Upper and Lower Atmosphere, Academic Press, San Diego, 2000 Search PubMed.
- R. P. Wayne, Chemistry of atmospheres: An introduction to the chemistry of the atmospheres of earth, the planets, and their satellites, Oxford University Press, Oxford, 3rd edn, 2000 Search PubMed.
- S. E. Paulson and J. J. Orlando, The reactions of ozone with alkenes: An important source of HOx in the boundary layer, Geophys. Res. Lett., 1996, 23, 3727–3730 CrossRef CAS.
- D. Johnson and G. Marston, The gas-phase ozonolysis of unsaturated volatile organic compounds in the troposphere, Chem. Soc. Rev., 2008, 37, 699–716 RSC.
- M. S. Alam, M. Camredon, A. R. Rickard, T. Carr, K. P. Wyche, K. E. Hornsby, P. S. Monks and W. J. Bloss, Total radical yields from tropospheric ethene ozonolysis, Phys. Chem. Chem. Phys., 2011, 13, 11002–11015 RSC.
- K. M. Emmerson and N. Carslaw, Night-time radical chemistry during the TORCH campaign, Atmos. Environ., 2009, 43, 3220–3226 CrossRef CAS.
- K. M. Emmerson, N. Carslaw, D. C. Carslaw, J. D. Lee, G. McFiggans, W. J. Bloss, T. Gravestock, D. E. Heard, J. Hopkins, T. Ingham, M. J. Pilling, S. C. Smith, M. Jacob and P. S. Monks, Free radical modelling studies during the UK TORCH Campaign in Summer 2003, Atmos. Chem. Phys., 2007, 7, 167–181 CrossRef CAS.
- Criegee mechanism of ozonolysis BT - Name Reactions: A Collection of Detailed Reaction Mechanisms, ed. J. J. Li, Springer Berlin Heidelberg, Berlin, Heidelberg, 2014 Search PubMed.
- R. Criegee, Mechanism of Ozonolysis, Angew. Chem., Int. Ed. Engl., 1975, 14, 745–752 CrossRef.
- C. Geletneky and S. Berger, The Mechanism of Ozonolysis Revisited by 17O-NMR Spectroscopy, Eur. J. Org. Chem., 1998, 1625–1627 CrossRef CAS.
- O. Horie and G. K. Moortgat, Gas-Phase Ozonolysis of Alkenes. Recent Advances in Mechanistic Investigations, Acc. Chem. Res., 1998, 31, 387–396 CrossRef CAS.
- N. M. Donahue, G. T. Drozd, S. A. Epstein, A. A. Presto and J. H. Kroll, Adventures in ozoneland: down the rabbit-hole, Phys. Chem. Chem. Phys., 2011, 13, 10848–10857 RSC.
- A. Novelli, L. Vereecken, J. Lelieveld and H. Harder, Direct observation of OH formation from stabilised Criegee intermediates, Phys. Chem. Chem. Phys., 2014, 16, 19941–19951 RSC.
- J. H. Kroll, J. S. Clarke, N. M. Donahue and J. G. Anderson, Mechanism of HOx Formation in the Gas-Phase Ozone−Alkene Reaction. 1. Direct, Pressure-Dependent Measurements of Prompt OH Yields, J. Phys. Chem. A, 2001, 105, 1554–1560 CrossRef CAS PubMed.
- H. L. Huang, W. Chao and J. J. M. Lin, Kinetics of a Criegee intermediate that would survive high humidity and may oxidize atmospheric SO2, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 10857–10862 CrossRef CAS PubMed.
- D. L. Osborn and C. A. Taatjes, The physical chemistry of Criegee intermediates in the gas phase, Int. Rev. Phys. Chem., 2015, 34, 309–360 Search PubMed.
- R. Chhantyal-Pun, M. A. H. Khan, R. Martin, N. Zachhuber, Z. J. Buras, C. J. Percival, D. E. Shallcross and A. J. Orr-Ewing, Direct Kinetic and Atmospheric Modeling Studies of Criegee Intermediate Reactions with Acetone, ACS Earth Space Chem., 2019, 3(10), 2363–2371 Search PubMed.
- C. C. Womack, M. A. Martin-Drumel, G. G. Brown, R. W. Field and M. C. McCarthy, Observation of the simplest Criegee intermediate CH2OO in the gas-phase ozonolysis of ethylene, Sci. Adv., 2015, 1, 1–7 Search PubMed.
- T. Y. Chen and Y. P. Lee, Dynamics of the reaction CH2I + O2 probed: via infrared emission of CO, CO2, OH and H2CO, Phys. Chem. Chem. Phys., 2020, 22, 17540–17553 RSC.
- Y. Fang, F. Liu, V. P. Barber, S. J. Klippenstein, A. B. McCoy and M. I. Lester, Deep tunneling in the unimolecular decay of CH3CHOO Criegee intermediates to OH radical products, J. Chem. Phys., 2016, 145, 234308 CrossRef PubMed.
- Y. Fang, F. Liu, S. J. Klippenstein and M. I. Lester, Direct observation of unimolecular decay of CH3CH2CHOO Criegee intermediates to OH radical products, J. Chem. Phys., 2016, 145, 44312 CrossRef PubMed.
- Y. Fang, F. Liu, V. P. Barber, S. J. Klippenstein, A. B. McCoy and M. I. Lester, Communication: Real time observation of unimolecular decay of Criegee intermediates to OH radical products, J. Chem. Phys., 2016, 144, 61102 CrossRef PubMed.
- V. P. Barber, S. Pandit, A. M. Green, N. Trongsiriwat, P. J. Walsh, S. J. Klippenstein and M. I. Lester, Four-Carbon Criegee Intermediate from Isoprene Ozonolysis: Methyl Vinyl Ketone Oxide Synthesis, Infrared Spectrum, and OH Production, J. Am. Chem. Soc., 2018, 140, 10866–10880 CrossRef CAS PubMed.
- F. Liu, J. M. Beames, A. S. Petit, A. B. McCoy and M. I. Lester, Infrared-driven unimolecular reaction of CH3CHOO Criegee intermediates to OH radical products, Science, 2014, 345, 1596–1598 CrossRef CAS PubMed.
- V. P. Barber, S. Pandit, V. J. Esposito, A. B. McCoy and M. I. Lester, CH Stretch Activation of CH3CHOO: Deep Tunneling to Hydroxyl Radical Products, J. Phys. Chem. A, 2019, 123, 2559–2569 CrossRef CAS PubMed.
- F. Liu, J. M. Beames, A. M. Green and M. I. Lester, UV Spectroscopic Characterization of Dimethyl- and Ethyl-Substituted Carbonyl Oxides, J. Phys. Chem. A, 2014, 118, 2298–2306 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., 2016 Search PubMed.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schütz, Molpro: a general-purpose quantum chemistry program package, WIREs Comput. Mol. Sci., 2012, 2, 242–253 CrossRef CAS.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schütz, P. Celani, W. Györffy, D. Kats, T. Korona, R. Lindh, A. Mitrushenkov, G. Rauhut, K. R. Shamasundar, T. B. Adler, R. D. Amos, S. Bennie, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, E. Goll, C. Hampel, A. Hesselmann, G. Hetzer, T. Hrenar, G. Jansen, C. Köppl, S. J. R. Lee, Y. Liu, A. W. Lloyd, Q. Ma, R. A. Mata, A. J. May, S. J. McNicholas, W. Meyer, T. F. Miller, III, M. E. Mura, A. Nicklass, D. P. O’Neill, P. Palmieri, D. Peng, K. Pflüger, R. Pitzer, M. Reiher, T. Shiozaki, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson, M. Wang and M. Welborn, MOLPRO, version 1, a package of ab initio programs, 2018 Search PubMed.
- S. Grimme, Semiempirical hybrid density functional with perturbative second-order correlation, J. Chem. Phys., 2006, 124, 34108 CrossRef PubMed.
- W. J. Hehre, R. F. Stewart and J. A. Pople, Self-Consistent Molecular-Orbital Methods. I. Use of Gaussian Expansions of Slater-Type Atomic Orbitals, J. Chem. Phys., 1969, 51, 2657–2664 CrossRef CAS.
- W. J. Hehre, R. Ditchfield, R. F. Stewart and J. A. Pople, Self-Consistent Molecular Orbital Methods. IV. Use of Gaussian Expansions of Slater-Type Orbitals. Extension to Second-Row Molecules, J. Chem. Phys., 1970, 52, 2769–2773 CrossRef CAS.
- M. J. Frisch, M. Head-Gordon and J. A. Pople, Semi-direct algorithms for the MP2 energy and gradient, Chem. Phys. Lett., 1990, 166, 281–289 CrossRef CAS.
- M. J. Frisch, M. Head-Gordon and J. A. Pople, A direct MP2 gradient method, Chem. Phys. Lett., 1990, 166, 275–280 CrossRef CAS.
- T. H. Dunning, Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- J. Finley, P.-Å. Malmqvist, B. O. Roos and L. Serrano-Andrés, The multi-state CASPT2 method, Chem. Phys. Lett., 1998, 288, 299–306 CrossRef.
- A. A. Granovsky, Extended multi-configuration quasi-degenerate perturbation theory: The new approach to multi-state multi-reference perturbation theory, J. Chem. Phys., 2011, 134, 214113 CrossRef PubMed.
- J. Li and H. Guo, Full-Dimensional Potential Energy Surface and Ro-vibrational Levels of Dioxirane, J. Phys. Chem. A, 2016, 120, 2991–2998 CrossRef CAS PubMed.
- P. A. Giguère, The Infra-Red Spectrum of Hydrogen Peroxide, J. Chem. Phys., 1950, 18, 88–92 CrossRef.
- P. A. Giguère, Molecular association and structure of hydrogen peroxide, J. Chem. Educ., 1983, 60, 399 CrossRef.
- C. Xie, C. L. Malbon, D. R. Yarkony, D. Xie and H. Guo, Signatures of a Conical Intersection in Adiabatic Dissociation on the Ground Electronic State, J. Am. Chem. Soc., 2018, 140, 1986–1989 CrossRef CAS PubMed.
- C. Xie, J. Ma, X. Zhu, D. R. Yarkony, D. Xie and H. Guo, Nonadiabatic Tunneling in Photodissociation of Phenol, J. Am. Chem. Soc., 2016, 138, 7828–7831 CrossRef CAS PubMed.
- B. K. Kendrick, J. Hazra and N. Balakrishnan, Geometric Phase Appears in the Ultracold Hydrogen Exchange Reaction, Phys. Rev. Lett., 2015, 115, 153201 CrossRef CAS PubMed.
- B. K. Kendrick, J. Hazra and N. Balakrishnan, The geometric phase controls ultracold chemistry, Nat. Commun., 2015, 6, 7918 CrossRef CAS PubMed.
- J. Jankunas, M. Sneha, R. N. Zare, F. Bouakline and S. C. Althorpe, Hunt for geometric phase effects in H + HD → HD(v′, j′) + H, J. Chem. Phys., 2013, 139, 144316 CrossRef PubMed.
- J. F. E. Croft, J. Hazra, N. Balakrishnan and B. K. Kendrick, Symmetry and the geometric phase in ultracold hydrogen-exchange reactions, J. Chem. Phys., 2017, 147, 74302 CrossRef CAS PubMed.
- J. C. Juanes-Marcos, S. C. Althorpe and E. Wrede, Theoretical Study of Geometric Phase Effects in the Hydrogen-Exchange Reaction, Science, 2005, 309, 1227–1230 CrossRef CAS PubMed.
- N. M. Kidwell, H. Li, X. Wang, J. M. Bowman and M. I. Lester, Unimolecular dissociation dynamics of vibrationally activated CH3CHOO Criegee intermediates to OH radical products, Nat. Chem., 2016, 8, 509 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp02601a |
| This journal is © the Owner Societies 2022 |