
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High entropy metal chalcogenides: synthesis, properties, applications and future directions
Mark A.
Buckingham
 ,
Brendan
Ward-O’Brien
,
Weichen
Xiao
,
Yi
Li
,
Jie
Qu
and
David J.
Lewis
*
,
Brendan
Ward-O’Brien
,
Weichen
Xiao
,
Yi
Li
,
Jie
Qu
and
David J.
Lewis
*
Department of Materials, The University of Manchester, Oxford Raod, M13 9PL, UK. E-mail: David.lewis-4@manchester.ac.uk
First published on 23rd June 2022
Abstract
Metal oxides, sulphides, selenides and tellurides have routinely been investigated and utilised for a wide range of applications, in particular in the areas of energy (photovoltaic, thermoelectric) and catalysis (thermocatalysis, electrocatalysis and photoelectrocatalysis). A recent development in this chemical space is high entropy and entropy-stabilised inorganic materials, which took inspiration from preceding work on high entropy metal alloys (multicomponent alloys). High entropy inorganic materials typically have many (often ≥6) different cations or anions to yield a high configurational entropy, which can give unexpected structures and properties that are conducive to a broad range of applications in energy and catalysis. More recently still, high entropy metal chalcogenides have been developed and have been investigated for thermoelectric energy generation, batteries for energy storage and electrocatalysis. This review sets out to define entropic stabilisation in high entropy materials, along with a discussion of synthetic techniques to produce these compounds, focusing on high entropy metal chalcogenides including extant routes to high entropy sulfides, selenides and tellurides. The resulting advantageous properties of these materials due to their multi-element nature and the currently proposed and investigated applications of these materials are reviewed. We end the perspective with an opinion on future directions.
 Mark A. Buckingham | Mark A. Buckingham achieved his MChem (Hons) Chemistry degree from the University of Bath in 2017 and his PhD in electrochemical energy conversion at King's College London in Jan 2022. Mark is currently a post-doctoral research associate at the University of Manchester working with Dr Lewis on high entropy metal chalcogenides for electrocatalytic applications. |
 Brendan Ward-O’Brien | Brendan Ward-O’Brien is currently in the final year of his PhD at the University of Manchester working with Dr Lewis. He was awarded an MChem (Hons) degree in chemistry from The University of Liverpool in 2019. His current work is focused around high entropy lanthanide oxysulfide systems made using a molecular precursor approach. |
 Weichen Xiao | Weichen Xiao finished his undergraduate degree in Beihang University (China), obtained MEng Materials degree from the University of Manchester in 2019 and moved onto a PhD working with Dr Lewis on high entropy ceramics as functional materials. |
 Yi Li | Yi Li completed his undergraduate degree at Nanjing University of Science and Technology (China) and achieved his MSc (distinction) degree in nanomaterials from the University of Manchester in 2019. He is currently undertaking a PhD in materials science with Dr Lewis focusing on high entropy metal sulfide nanoparticles. |
 Jie Qu | Jie Qu achieved a MSc in Chemical Engineering at Sichuan University, China (2019) and is currently undertaking a PhD at The University of Manchester (UK) working with Dr Lewis on the synthesis of two-dimensional high entropy dichalcogenides and their applications as water splitting electrocatalysts. |
 David J. Lewis | David J. Lewis is Deputy Head of the Department of Materials at The University of Manchester. He obtained both his MSc (2002) and PhD (2006) degrees in chemistry from The University of Birmingham. He was appointed as Lecturer in 2016 at The University of Manchester and was promoted directly to Reader in Materials Chemistry in 2020. His research revolves around the synthesis of semiconductors and nanomaterials by chemical routes, and he has published over 100 papers in the area. |
Introduction to high entropy (HE) materials
High entropy (HE) materials were first reported in 2004 for metal alloys.1,2 The principle behind these materials is that the formation of a single material with high proportions (either equimolar or non-equimolar) of many principle elements will yield a high configurational entropy of formation as in eqn (1):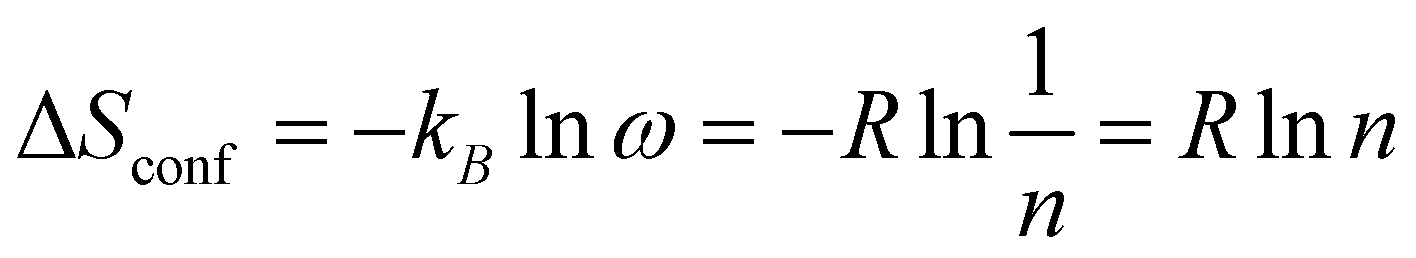 | (1) |
The high entropy stabilisation effect describes the ability to produce a single, entropy-stabilised phase over forming a multi-phase material, which can lead to materials with unexpected/rare phases formed.5–7 This effect has also led to the ability to stabilise single-atom sites for catalysis in both high entropy alloys8 and oxides.9 The sluggish diffusion effect describes the diffusion kinetics in HE alloys to be anomalously slow, which is reported to originate from fluctuations in the potential energies of lattice sites.3,10,11 However, this has been the subject of more recent debate, with other studies suggesting that diffusion is not unusually sluggish as demonstrated by observations of precipitation in many as-cast HE alloy samples, including those subjected to very rapid cooling, as well as in those quenched from high temperature heat treatments.4,12,13 For a more in-depth discussion of this debate, the reader is directed to a review by Pickering et al.12 The severe lattice distortion effect is caused by the randomly distributed nature of mismatched sized elements or ions within HE materials, which affects the thermodynamic stability, microstructure and deformation mechanisms of the synthesised HE alloys.14 The so-called cocktail effect is in essence a blanket term used to describe any emergent properties which cannot be described by any of the individual components in the HE material, and is potentially the most exciting aspect of exploring these materials and has led to a number of unexpected discoveries.4 It is important to note that these effects are not limited to HE metal alloys and have also been observed in other HE materials such as HE metal chalcogenides, borides,15–18 carbides,19–22 nitrides,22–26 silicides,27,28 phosphides/phosphates,29–31 and fluorides.32–34 Which have been extensively discussed in a recent review.35
Defining high entropy materials
In metal alloys, the state of high entropy is reported to be reached once the material is composed of at least five alloying elements in a ratio between 5–35 mol%.1 High entropy has also been reported to be achieved more quantitatively, when a material has reached a configurational entropy of > 1.5R, where R is the universal gas constant (8.314 J K−1 mol−1).3 Hence, using eqn (1) and calculating for a structure that contains a single lattice with equimolar ratios of multiple elements (such as an alloy), this condition is satisfied once the material contains five elements (Fig. 1). However, if two or more distinct sub-lattices are considered (such as in HE metal chalcogenides) eqn (1) is no longer valid and the following expression is required which accounts for the proportion of the sub-lattice contributing to the overall configurational entropy in the material:36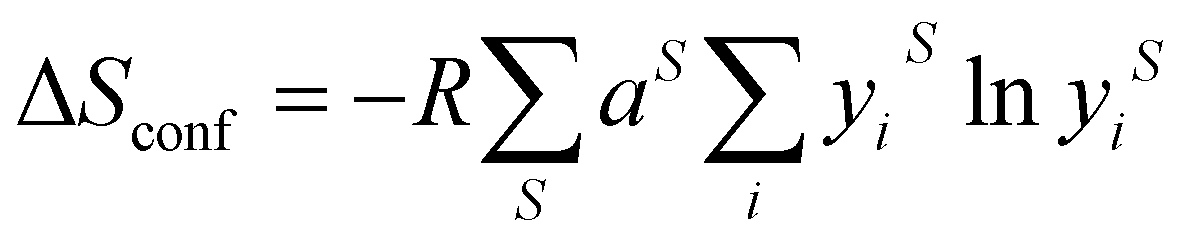 | (2) |
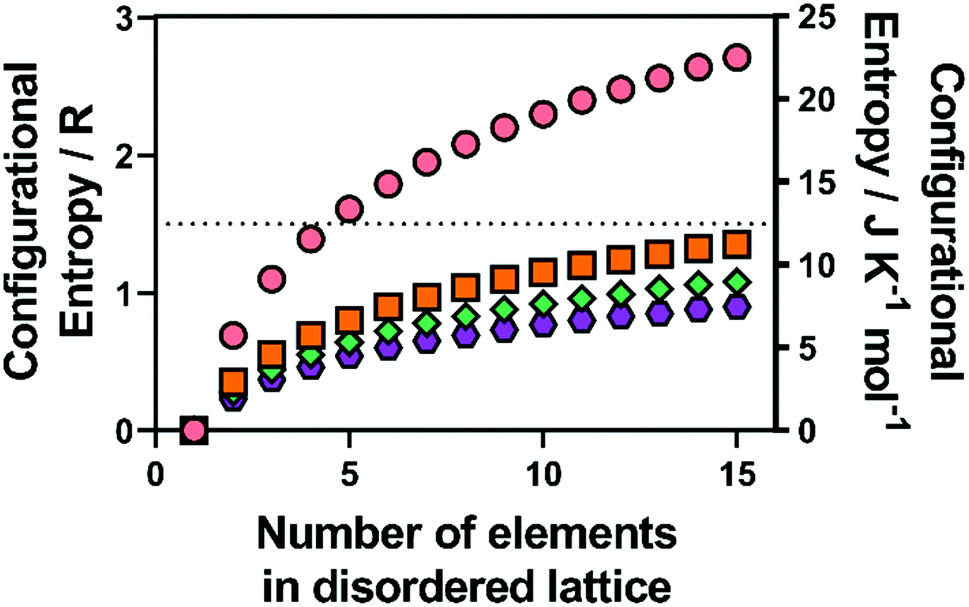 | ||
| Fig. 1 Calculated configurational entropy (in both R and J K−1 mol−1) for species with a single disordered sub-lattice (M) for (pink circles) alloys with a single lattice, and a range of common metal chalcogenide materials with a single chalcogenide sub-lattice (i.e. X is all O, S, Se or Te) for (orange squares) MX, (blue diamonds) M2X3 and (purple hexagons) MX2. | ||
A universal configurational entropy metric (EM) has been proposed by Vecchio.38 This method is proposed to account for systems with more than one sub-lattice (L) by including a lattice factor into the equation:
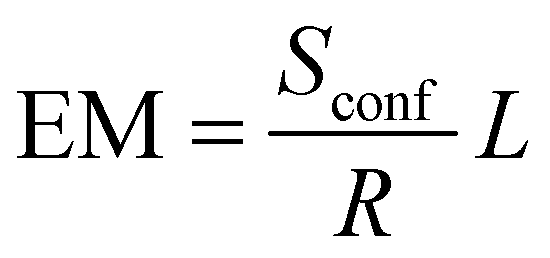 | (3) |
High entropy metal chalcogenides
The first high entropy (HE) metal chalcogenide (for the purposes of introducing HEMCs, we have elected to include oxides, however these will not be discussed throughout the rest of the review) to be developed was the hexarnary metal oxide: (MgNiCoCuZn)O.39 This material was prepared by ball milling the constituent metal oxides which were pressed into pellets and subsequently annealed in air from 700 °C up to 1100 °C.39 The authors found that at 700 °C two distinct phases (rocksalt and tenorite) were observed by powder XRD (pXRD), but once they reached 850–900 °C, a single rock salt phase was formed. With a single oxide phase and the various metals arranged randomly throughout the crystal structure on the cation site (as in Fig. 2).39 It was also found that this process (forming a single HE phase) was reversible by a low-temperature equilibration, as entropy-driven transitions are expected to be reversible.39 The authors also demonstrate that this material is truly entropically stabilized, due to the configurational entropic contribution overcoming the enthalpic penalty of phase transitions between the component metal oxides.39 As discussed above, HE metal chalcogenides represent two or more distinct sub-lattices (such as a cationic and an anionic), despite this difference between HE metal chalcogenides and HE alloys, eqn (1) was erroneously initially used to calculate the configurational entropy.39 To calculate the true configurational entropy, all sub-lattices need to be taken into account to represent the contribution of each atom to the configurational entropy of the constituent sub lattice and the sub-lattice to the overall material, as in eqn (2).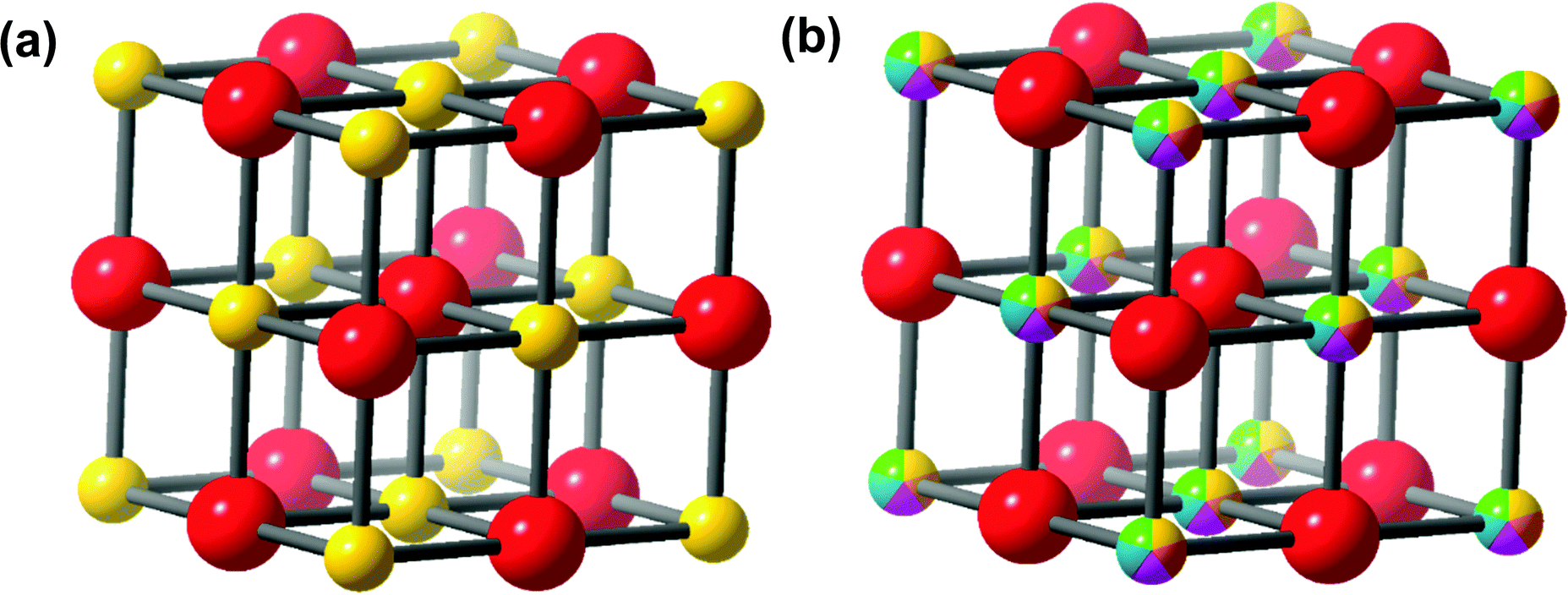 | ||
| Fig. 2 Figure showing (a) a typical binary metal chalcogenide rock salt structure with a single cation (yellow sphere) and anion (red sphere) compared to (b) a High Entropy metal chalcogenide with a rock salt structure where the various cation metals (multicolour sphere) are randomly distributed with an ordered single anionic sub-lattice (red sphere). | ||
Since the advent of inorganic HE materials, there have been numerous reports of HE metal oxides comprising of combinations of alkali, alkali-earth, transition, main group and even lanthanide metals.35,40–45 These materials have been investigated in a wide range of applications, mainly in the energy44 and catalysis40,43 fields. Due to this, far more work on HE metal oxides has been reported than for the sulphides, selenides and tellurides. This large body of work has already been extensively and comprehensively reviewed.35,40–43,45
HE metal chalcogenides with sulphide, selenide and telluride ions (S, Se, Te or a combination of the three) as the anionic sub-lattice are a more recent development.46 HE metal sulphide or disulphide materials are the most common of these more recently synthesised HE metal chalcogenides,47–52 although HE metal telluride,46 and mixed chalcogenide37,46,53–55 materials have also been reported. Like HE metal oxides, these materials have also demonstrated emergent properties for energy conversion,37,47,56 storage52 and catalytic48,49,51 applications. Recent work on these non-oxide HE metal chalcogenide systems has reported a range of synthetic routes, on both micro and nano length scales, towards a number of different crystal phases for a variety of different applications. These non-oxide HE metal chalcogenides will be the exclusive focus of this review.
Synthesis of high entropy metal chalcogenides
HE metal chalcogenides have been synthesised through a variety of routes (Fig. 3), the most common of which is elemental annealing in the presence of the relevant chalcogen (or chalcogen source) in an inert gas or vacuum (Table 1).37,53–56 This is both a high temperature (typically between 750 °C and 1000 °C)54,56 and a time consuming (anywhere between 20 h and 8 days)46,49 process. This process is also typically undertaken with a pre-annealing step consisting of either ball-milling,47 or pulverising by mortar and pestle57 to produce fine elemental powders for annealing. A drawback of these approaches is that particle size in the lower bound is around 100 nm, which precludes elemental mixing at the atomic scale prior to reaction and explains the longer times and high temperatures necessary to substantially mix the elementals by cationic diffusion through the anionic lattice. As such, the risk of phase separation may be high with these reaction types. However, this is still an effective method of producing HE metal chalcogenides, particularly those with mixed anions (i.e. a combination of S, Se, Te).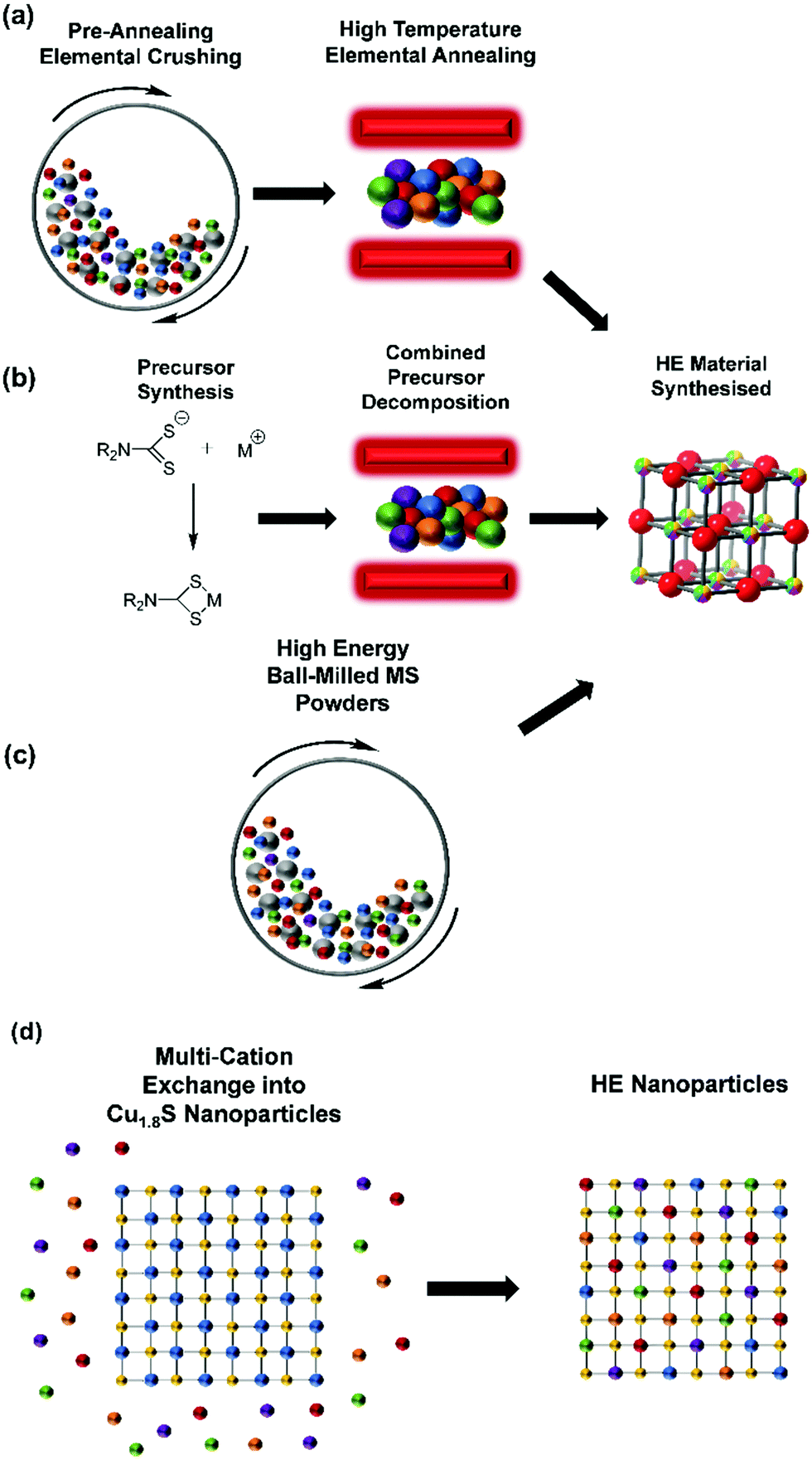 | ||
| Fig. 3 Figure showing a schematic representation of extant synthetic procedures toward high entropy metal chalcogenides. (a) elemental annealing (b) thermolysis of single source precursor cocktails (c) high energy ball-milling of individual metal sulphides (d) nanoparticle cation exchange. | ||
| HE Material | Synthetic Route | Preparation Conditions | Reported phase | Application | Ref |
|---|---|---|---|---|---|
| (Cu5SnMgGeZn)S9, (Cu3SnMgInZn)S7 | Ball-milling and Elemental Annealing | 60 h Ball-milling followed by 750 °C sintering at 50 MPa. | Tetragonal | Thermoelectric | 47 |
I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 2d 2d |
|||||
| No exact phase reported | |||||
| (FeNiCoCrMn)S2, (FeNiCoCrCu)S2, (FeNiCoCrZn)S2, (FeNiCoCrAl)S2 | Solvothermal Annealing | Solution annealing of metal salts at 150 °C for 10 h. Followed by introduction of sulphur source and a further annealing at 160 °C for 8 h. | Pyrite | Electrocatalysis | 48 |
Pa![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) |
OER | ||||
| (MoWVNbTa)S2 (Nano) | Elemental annealing | Vessel preparation: Quartz tubes etched with HF followed by 1000 °C annealing for 24 h. Sample preparation: Elemental powders introduced into the vessel and ramp annealed for 120 h. | 3R – TMDC | Electrocatalysis CO2 reduction | 49 |
| R3m | |||||
| No exact phase reported | |||||
| (ZnCoCuInGa)S (Nano) | Element Exchange | Cu1.8S nanoparticles dispersed in a solution of metal salts. 30 min annealing under vacuum at 110 °C followed by 30 min annealing under Ar at 180 °C. Further annealing at 140 °C for 30 min. | Wurtzite | Synthetic proof of concept | 50 |
| P63mc | |||||
| (CrMnFeCoNi)Sx (Nano) | Pulsed Thermal Decomposition | Solvothermal mixing of metal salts followed by pulsed thermal annealing at 1650 K for 0.055 s, then dried for 6 h in an oven. | Cubic | Electrocatalysis OER | 51 |
|
Fmm |
|||||
| No exact phase reported | |||||
| (FeMnNiCoCr)3S2, (FeMnNiTiCr)S, (FeMnNiCoCr)S, (FeMnNiCoCr)3S4, (FeMnNiCoCr)2S3, (FeMnNiCoCr)S2, (FeMnNiCoCu)S2, (FeMnNiTiCr)S2 | High-Energy Ball Milling of MS Powders | Ball-milling metal sulphide powders for either (MS2) 110 h, or (MS, M2S3, M3S4, M3S2) 60 h. | Orthorhombic | Li-ion batteries | 52 |
| Pnma | |||||
| No exact phase reported | |||||
| Pyrite | |||||
|
Pa |
|||||
| (CuAg)(SSeTe) | Elemental Annealing | Heated to 1100 °C and slowly cooled to 650 °C and held for between 5–8 days, followed by the produced ingot being crushed into a powder and plasma sintered at 600 °C under 60–65 MPa. | Rock Salt | Thermoelectric | 46 |
|
Fmm |
|||||
| Hexagonal | |||||
| P63/mmc | |||||
| No exact phase reported | |||||
| (GeSnPb)(SSeTe) | Elemental Annealing | Annealing of elements at 1000 °C for 20 h. | Rock Salt | Thermoelectric | 53 |
|
Fmm |
|||||
| (CoFeNi)(SSe) | Elemental Annealing | Elements annealed at 1273 K for 24 h. Then the temperature was cooled to 773 K and annealed for a further 72 h. | Pentlandite | Thermoelectric | 54 |
|
Fmm |
|||||
| (PbSbSn)(SSeTe) | Elemental Annealing | Elements annealed at 723 K in 7 h, then further heated to 1423 K in 7 h, then held for 6 h. The produced ingot was crushed and plasma sintered at 853 K at 50 MPa for 10 min. | Rock Salt | Thermoelectric | 37 |
|
Fmm |
|||||
| (AgPbBi)(SSeTe) | Elemental annealing | Initial annealing at 400 for 5 h, followed by 800 for 20 h. To obtain high-density samples, hot pressing was performed at 400 °C for 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) min at 70 MPa. min at 70 MPa. |
Rock salt | Thermoelectric | 66 |
|
Fmm |
|||||
| (AgInPbBi)Te0.75Se0.25, (AgInPbBi)Te0.5Se0.5 | High-pressure elemental annealing | Elemental powders were initially melted at 800 °C. Followed by high-pressure annealing at 500 °C under 3 GPa for 30 minutes. | Rock Salt | Superconductivity | 55 |
|
Fmm |
|||||
| (AgCdSnSbPb)Te5, (AgInSnSbPb)Te5, (AgCdInSnSb)Te5, (AgCdSnPbBi)Te5, (AgCdInPbBi)Te5, (AgCdInSnBi)Te5 | High-pressure elemental annealing | High-pressure annealing at 500 °C under 3 GPa for 30 minutes. | Rock Salt | Superconductivity | 67 |
|
Fmm |
|||||
| (TiVCrNbTa)S2, (Ti0.19V0.2Cr0.14Nb0.24Ta0.26)S2 | Elemental Annealing and chemical vapor transport (CVT) method | 1050 °C for 24 h. |
Pm1 |
Not tested | 68 |
| 1050 °C for 30 d. | |||||
| (TiVCrNbTa)Se2, Ti0.2V0.21Cr0.21Nb0.19Ta0.2Se2 | 1000 °C for 24 h. | Not tested | |||
| 1000 °C for 14 d. | |||||
| (Ti,V,Cr,Nb)0.8(Fe,Mn)0.2Se2, Ti0.21V0.22Cr0.23Nb0.22Fe0.08Mn0.09Se | 1000 °C for 48 h. | Not tested | |||
| (Ti,V,Cr,Nb,Ta)SSe, Ti0.16V0.15Cr0.15Nb0.24Ta0.17S1.02Se0.98 | 1000 °C for 24 h. | Not tested | |||
| No exact phase reported | |||||
| (Ti,V,Zr,Nb,Hf)Te2, Ti0.15V0.22Zr0.2Nb0.18Hf0.17Te2 | 1000 °C for 24 h. | Not tested | |||
| 1000 °C for 14 d. | |||||
| (Co,Au)0.2(Rh,Ir,Pd,Pt)0.8Te2, Co0.03Au0.06Rh0.23Ir0.24Pd0.16Pt0.28Te2 | 750 °C for 30 h. | Superconductivity | |||
| Co0.1(Rh,Ir,Pd,Pt)0.9Te2, Co0.03Rh0.28Ir0.25Pd0.19Pt0.28Te2 | 750 °C for 30 h. | Superconductivity | |||
| (Co,Rh,Ir,Pd,Pt)Te2, Co0.19Rh0.23Ir0.2Pd0.15Pt0.18Te2 | 750 °C for 30 h. | Not tested |
Other synthetic strategies have been reported for HE metal chalcogenides. Another technique is based on a solvothermal process where the solvated metal salts are heated with a sulphur source (thioacetamide) present in the solution at a significantly lower temperature (150–160 °C) than elemental annealing, but still over an extended timeframe (18 h).48 A further method of synthesising HE metal chalcogenides is through high energy ball milling of the constituent metal sulphides, often known as mechanical alloying. In this method, pre-synthesised metal sulphide materials are ball milled for between 60 h and 110 h. This method has the significant advantage of not requiring any external heating steps, and can rely on the energy generated by asperity contacts in the ball milling alone.52 However, the disadvantages of this method is the pre-synthetic step required for the respective metal sulphides, along with the extended time required for the ball-milling process.
One method that significantly reduces the time required to produce HE metal chalcogenide materials is through a single source precursor approach. This approach has been used to produce bulk lanthanide oxysulphide materials.58 This method utilises annealing of lanthanide dithiocarbamate precursors at a temperature of 900 °C, over relatively short timescales (5 hours) compared to elemental annealing. However, due to the unique chemistry of the lanthanides, oxysulphides are produced over sulphides, even in argon atmospheres unless extensive measures are taken to remove oxygen.59 Utilising single source precursors is a scalable approach which exploits a simple and clean (often used without further purification) synthetic step to produce single source precursors with pre-fabricated M–S bonds, which can be decomposed at significantly lower temperatures than the lanthanides described above (typically 200–500 °C).60,61 This advantage, coupled with the extensive library of metal single source precursors available62 makes this approach a highly appealing synthetic route towards both bulk and nanomaterial HE metal chalcogenides.
There have been other synthetic routes towards the synthesis of nanomaterial (both 2D and nanoparticles) HE metal sulphides. However, these methods represent some of the harshest and least scalable conditions reported to date. 2D HE metal sulphides based on group 5 and 6 metals have been synthesised through an elemental annealing process which requires a pre-synthetic step that involves highly toxic63 HF etching of the synthetic vessel, followed by a 24 h annealing step at 1000 °C. The synthesis of the sample (elemental annealing step) is then introduced into the prepared vessel and ramp annealed for 120 h.49
A pulsed thermal annealing synthetic route towards HE metal chalcogenides has also been developed. This method relies on utilising extremely high temperatures (1650 K) but only over short timescales (55 ms).51 However, the authors haven’t reported the number of pulses utilised in this synthetic strategy. The final step of this method is a 6 h oven dry of the produced materials,51 significantly extending the synthetic timeframe.
Another interesting synthetic route towards HE metal chalcogenide nanoparticles is through a cation exchange method.50 This method initially requires the synthesis of Cu1.8S nanoparticles, these are dispersed in a solution which contains salts of the desired metals to be exchanged with Cu to form the HE material. This solution was then heated initially under vacuum at 110 °C, then under Ar at 180 °C and again at 140 °C, with each heating step taking 30 mins.50 This method represents an interesting route towards producing HE materials through a cation-exchange reaction. Previous studies have determined the diffusion coefficients of metal ions in solids to be ca. 1 × 10−9 cm2 s−1 for Zn in Al at ca. 400 °C,64 or for Fe2+ ions in single crystal MgO at ca. 1650 °C.65 Using this value as a model for a 1D diffusion of ions in solids. These ions will travel 15 nm in 25 minutes, consistent with the observations of Schaak et al.50 However it should be noted that at this rate of diffusion, it would take ca. 115 days for these ions to travel 100 μm, which is potentially unfeasible for a scalable synthetic procedure towards bulk HE material. Therefore, to date a simple, fast, direct, and low temperature synthesis route toward bulk HE metal sulphides still evades researchers.
Properties of HE metal chalcogenides
The range of metals, chalcogenides, and synthetic procedures utilised in the preparation of both bulk and nanoscale HE metal chalcogenides has led to a wide range of materials with various crystallographic phases produced, which exhibit interesting and advantageous properties for a variety of different applications (summarised in Table 1). There are four interesting properties for high entropy alloys proposed by Yeh,3 three of these are also important for HE metal chalcogenides; the high entropy effect, the distorted lattice effect, and the cocktail effect.The high entropy effect in HE metal chalcogenides affects the ability of these materials to stabilise a single crystalline lattice phase. Therefore, the various reported crystal structures to date are discussed initially. The most commonly synthesised phase in HE metal chalcogenides is the rock salt37,53,55,56 structure (Fig. 2). Other reported phases observed in synthesised HEMCs are shandite,57 pyrite,48 wurtzite,50 and pentlandite.54
The distorted lattice effect is caused by the differing sizes of metal ions within a HEMC lattice. It is also noteworthy that if several chalcogenides are also present this distortion can also be achieved in the anionic sub-lattice as well as the cationic sub-lattice. This has been visually demonstrated by a previous HE metal chalcogenide which contained 3 metals (Pb, Sb, Sn) and 3 chalcogenides (S, Se, Te), shown here in Fig. 4. This unique property of HE materials has been demonstrated to yield significantly frustrated thermal conductivity,37 which is a significant advantage for thermoelectric materials.69 The mechanism for this thermal frustration is the scattering of phonons in the distorted lattice.37 This phonon scattering can be quantified in a 1D single atom harmonic chain by the equation:
 | (4) |
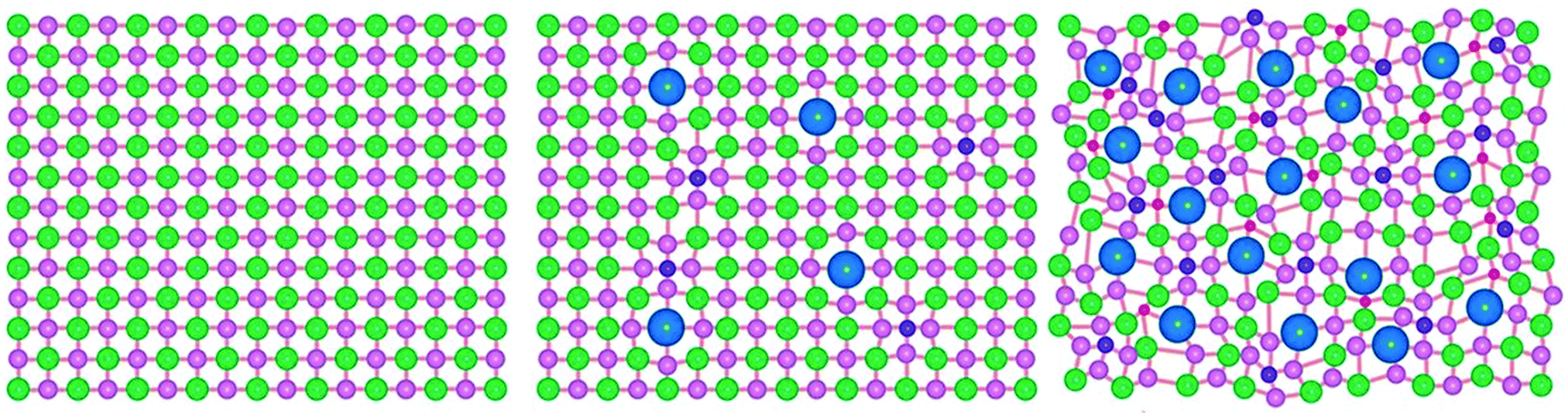 | ||
| Fig. 4 Figure visually showing the distortion in the lattice structure of a (PbSbSn)(SSeTe) HEMC where the pink, red, green, blue, and purple spheres represent Pb, Sn, Se, Te, and S atoms, respectively. Copyright permission from Science, used from ref. 37. | ||
The cocktail effect in HE materials typically describe any anomalous and unexpected properties which cannot be described by any of the individual components in the HE material. This particular property has been of significant interest in catalysis. A HE metal chalcogenide based on top-row transition metals (FeNiCoCrMnS2) has been thoroughly investigated to probe the cocktail effect in electrocatalysis.48 The presence of five transition metals initially found that stabilisation and modulation of the oxidation states of the metals was possible, with the Fe, Ni, Co and Mn all being found in both the 2+ and 3+ oxidation states, which was stabilised by the presence of other metals.48 The multi-metal, multivalent nature of this material allows a range of potential catalytic active sites. For example, with the FeNiCoCrMnS2 HEMC the Fe3+ species was found to be crucial for the oxygen evolution reaction (OER), due to the high affinity of iron to oxygen and the fast electron transfer between the Fe and the absorbed oxygen species.48
Further analysis of the HE material after OER found that the valence of the metal species was driven towards the 3+ oxidation state. Other significant alterations were also observed such as an initial reduction in crystallinity (over 30 minute period of OER activity), followed by gaining in crystallinity (by 9 hours of OER activity), this ‘self-reconstruction’ of the HE metal chalcogenide material is also accompanied by a change in morphology from a ‘yolk–shell’ structure to a significantly more porous sponge-like morphology, with a catalytic oxyhydroxide layer formed on the surface.48 Therefore, HE metal chalcogenides clearly demonstrate unique properties which are of significant benefit towards applications such as thermoelectrics and electrocatalysis.
Applications of high entropy metal chalcogenides
High Entropy metal sulphides have the widest range of applications reported to date for the non-oxide HE metal chalcogenides, having been demonstrated as thermoelectric materials,47,57 catalytic electrodes for both the oxygen evolution48,51 and CO2 reduction49 reactions, and as electrodes in Li-ion batteries.52 Thermoelectric energy generation is the most common application of these non-oxide HE metal chalcogenides and will be discussed together with the telluride and mixed chalcogenide systems in the next section.Electrodes: electrocatalysis
With respect to use of these materials as electrodes, we discussed above how the cocktail effect of having several metal ions present results in valence stabilisation of multi-redox states of several ions within these materials, leading to high catalytic performance of these materials. The significant advantage of the multi-cation approach has been previously demonstrated by comparing low entropy binary, ternary, quaternary and quinternary with high entropy hexernary metal sulphides (Fig. 5(a) and (b)).48 It is important to note here that the main catalytic site (Fe3+) was found to be stabilised by the high entropy cocktail effect of the presence of multiple cations. Increasing the number of metals in these materials was found to decrease the overpotential required to reach 100 mA cm−2. The resulting Tafel slopes of the catalysis also show the considerable advantage of the multi-cation, high entropy materials with a decrease in the Tafel slope from 57.7 mV dec−1 for FeSx to 39.1 mV dec−1 for (FeNiCoCrMn)S. This performance could not be improved with substitution of the Mn for Cu, Al or Zn (Fig. 5(c) and (d)),48 and only resulted in slightly higher overpotentials and slightly smaller Tafel slopes compared with (FeNiCoCrMn)Sx. In all cases these Tafel slopes are substantially lower than the non-HE materials investigated such as 57.7 mV dec−1 for FeSx, 52 mV dec−1 for (FeNi)Sx, 50.6 mV dec−1 for (FeNiCo)Sx and 42.5 mV dec−1 for (FeNiCoCr)Sx.48 These results (lower overpotential and smaller Tafel slopes) have also been observed in (CrMnFeCoNi)Sx, which again contains the proposed catalytic Fe sites stabilised by the cocktail effect.51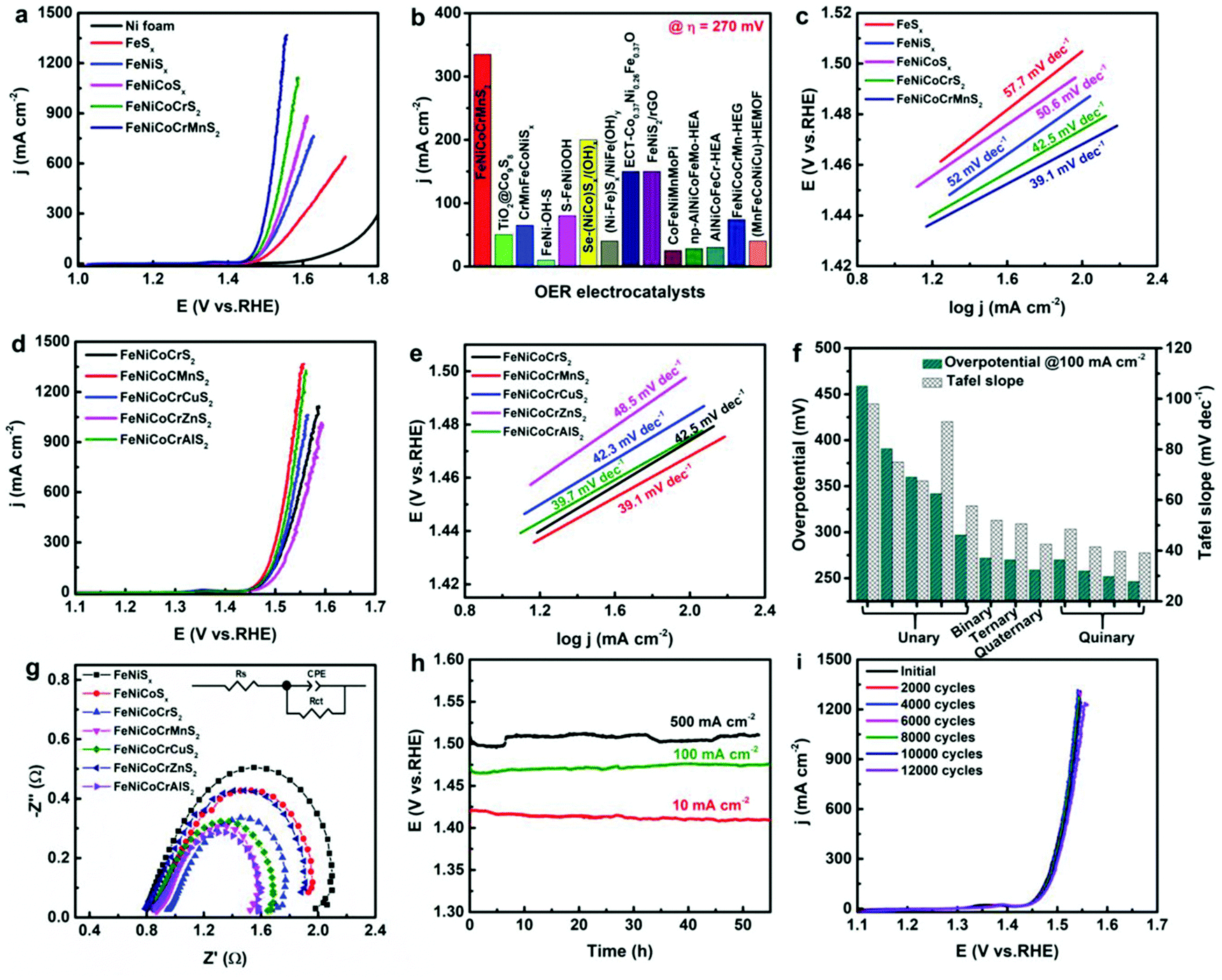 | ||
| Fig. 5 (a) Polarization curves of FeNiCoCrMnS2, FeNiCoCrS2, FeNiCoSx, FeNiSx, and FeSx. (b) Comparison of current densities at overpotential of 270 mV among FeNiCoCrMnS2 and reported transition metal sulphides, metal sulphides derivatives, and other HEMs catalysts. (c) Tafel slopes of FeNiCoCrMnS2, FeNiCoCrS2, FeNiCoSx, FeNiSx, and FeSx. (d) The polarization curves and (e) the corresponding Tafel slopes of FeNiCoCrXS2. (f) Comparison of η 100 among the obtained metal sulphide electrocatalysts. (g) EIS spectra of different transition metal sulphides. (h) Durability and (i) stability of FeNiCoCrMnS2. Copyright permission from Wiley, used from ref. 48. | ||
Electrocatalysis using a 2D HE metal disulphide has been reported for the CO2 reduction reaction (CO2RR).49 This study compared the (MoWNbTaV)S2 HE material to Ag for the CO2RR, where Ag is known to be highly efficient at the CO2 to CO conversion.71,72 The (MoWNbTaV)S2 has been reported as having a higher current density than Ag.49 However, this is compared using the geometric surface area. As (MoWNbTaV)S2 is a 2D nanomaterial it is expected to have a significantly higher electrochemically active surface area, which could account for the higher current density achieved using the 2D HE electrode.49 The (MoWNbTaV)S2 material is also possibly analogous to one of the component binary metal disulphides (MoS2, WS2etc.), a potentially more direct comparison would be the parent metal disulphide material, however the authors haven’t attempted to synthesise a mono- or dual-metal disulphide through their synthetic method as a direct comparison.49
Electrodes: energy storage
Recently, HE metal sulphides have been reported as battery anodes.52 This work compared several 5-metal disulphide materials ((FeMnNiCoCr)S2, (FeMnNiCoCu)S2 and (FeMnNiTiCr)S2) to a 5-metal monosulphide ((FeMnNiCoCr)S) and a comparable 4-metal disulphide ((FeMnNiCo)S2). Fig. 6 shows that the three HE 5-metal disulphide materials demonstrated superior discharge specific capacity to the 4-metal disulphide and HE 5-metal monosulphide, where the latter two also demonstrated poorer discharge specific capacity than the comparable CoS2.52 This significant difference has been proposed to be due to the unique ability of layered disulphide materials to facilitate the proposed two-step first discharge reaction that occurs in pyrite structured metal disulphides such as FeS2 and CoS2:73–75| MS2 + xLi+ + xe− → LixMS2 | (5) |
| LixMS2 + (4 − x)Li+ + (4 − x)e− → 2Li2S + M | (6) |
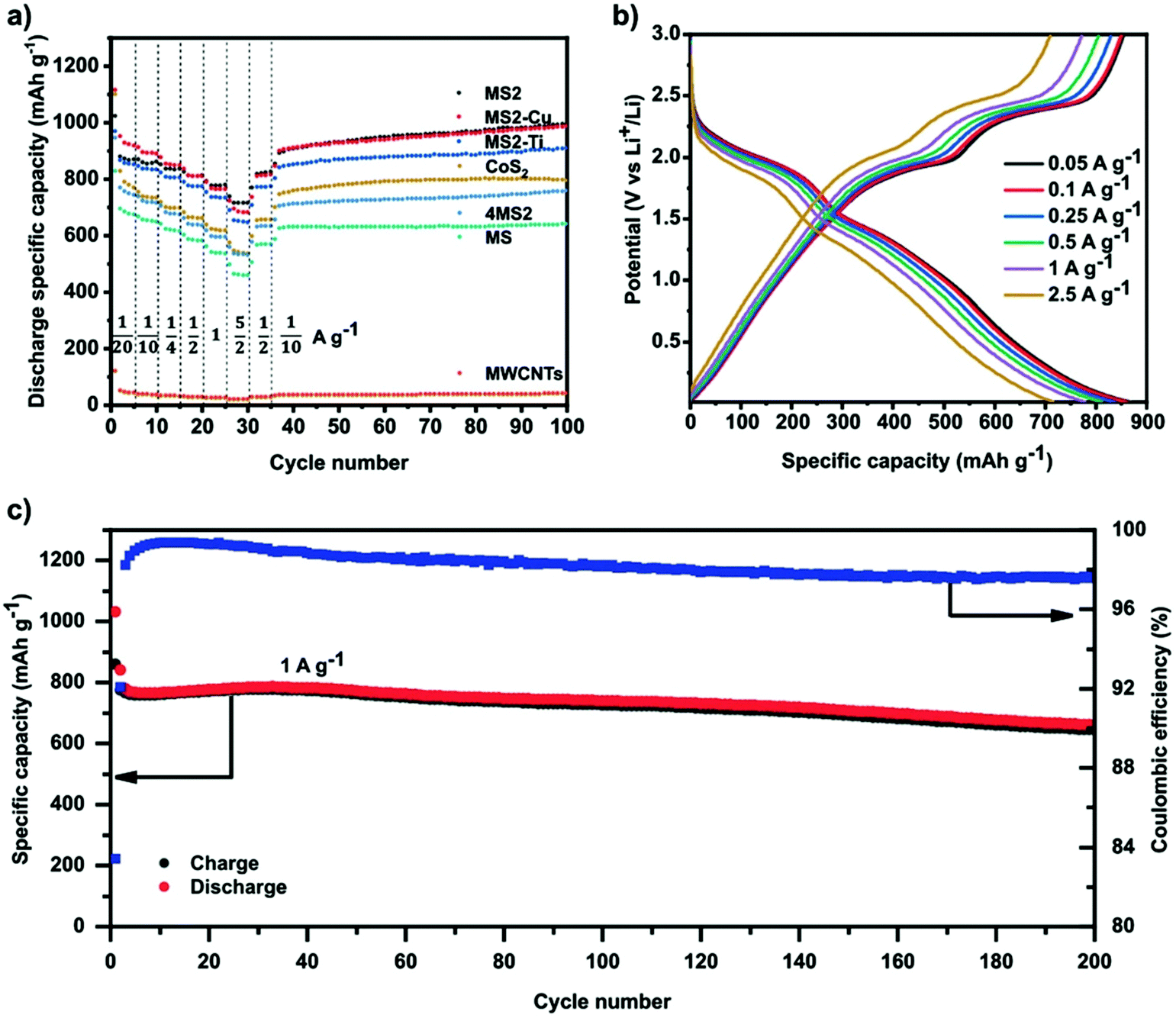 | ||
| Fig. 6 (a) Galvanostatic rate performance test of all HESs, CoS2, 4MS2, and MWCNTs half-cell at different current densities and 25 °C in the voltage range between 0.01 and 3 V versus Li+/Li. (b) Voltage profiles of MS2 half-cell at different currents. (c) Specific charge/discharge capacity of MS2 half-cell and Coulombic efficiency as a function of cycle number at 1 C and 25 °C in the range between 0.01 and 3 V. Copyright permission from Wiley, used from ref. 52. | ||
The cocktail effect and the high entropy nature of the 5-metal materials has been proposed for the increased discharge specific capacity over the 4-metal material. Although there have been no proposed key metal active sites, substituting Cr for Cu or Co for Ti didn’t have a large effect on the observed discharge specific capacity. Further work should be undertaken on these layered HE metal disulphide materials to understand the nature of this high entropy effect and which sites are active for the lithiation/delithiation processes and if these can be further optimised for improved energy storage capacities.52
Thermoelectric energy generation
Thermoelectric energy generation is by far the most common application reported for HE metal sulphides, selenides and tellurides. The ability to introduce distortion in both the cationic position (by introducing several metals of different sizes) and the anionic position (by introducing several chalcogenides of different sizes) in the lattice can lead to significant lattice distortion, which directly results in high phonon scattering ability and low thermal conductivities and resultant high unitless figure of merit values (zT),37 where zT: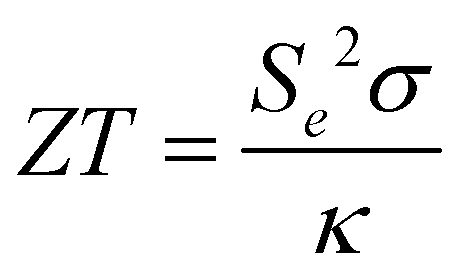 | (7) |
HE metal chalcogenides for thermoelectrics were initially investigated by Reece et al. through a two-step combination where initially a computational property analysis was used to predict a good candidate material, which was then subsequently synthesised and tested.47 For this, 47 (49 initially but two were excluded47) Cu–S containing materials were analysed, 18 with a wurtzite structure and 29 with a zinc blende structure. This comparison determined a large variation in Cu, Si, Ge and Sn, these metals were therefore incorporated into the high entropy sulphide design to reduce local strain energy. Other criteria such as the sustainability and the absence of magnetic properties of cations was also favourable. Magnetic properties contain spin which introduces additional complexity into high-entropy compounds, this was therefore determined to be unfavourable. From this analysis, two predicted HE materials were designed and synthesised, (Cu5SnMgGeZn)S9 and (Cu3SnMgInZn)S7.47 (Cu3SnMgInZn)S7 was found to be a semiconductor with very low electrical conductivity and was therefore not explored for its thermoelectric properties. The thermoelectric properties of (Cu5SnMgGeZn)S9 were assessed and the electrical conductivity showed metallic behaviour, with an electrical conductivity (σ) of ca. 1000 S cm−1 at 623 K, which is >200 S cm−1 higher than tetrahedrite ((Cu,Fe)12Sb4S13).78 However, this high electrical conductivity of (Cu5SnMgGeZn)S9 was found to reduce the Seebeck coefficient (Se), therefore additional (20%) Sn was included to reduce electrical conductivity and increase the Se forming the (Cu5Sn1.2MgGeZn)S9 species.47 Although ideally both of these parameters should be improved, the Se has a squared relationship to zT, so increases in Se are favourable over σ. The (Cu5Sn1.2MgGeZn)S9 material reached a power factor (Se2σ) of 8 μW cm−1 K−2 at 773 K. From eqn (7), (Se2σ) is the power factor and κ is the thermal conductivity. Therefore, for a high zT value, high Se and σ and low κ are favourable. The thermal conductivity of the (Cu5Sn1.2MgGeZn)S9 material was found to be low, and reduced with increasing temperature with a value of ca. 0.4 W m−1 K−1 at 773 K. A zT value of 0.58 at 723 K was obtained for n-type (Cu5Sn1.2MgGeZn)S9,47 which is significantly lower than the zT of the benchmark Bi2Te3 of 1.1 at a lower temperature of 340 K.79 Copper-based thermoelectric materials have been the subject of recent attention.80 Several of these materials have reported high zT values such as Cu1.9ZnSnS4 doped with 0.1 mol% Na which has reported a zT value of 1.6 at 800 K.81 A Cu2−xS has reported a zT of 1.9 at 970 K which are significantly higher than the Cu-based HE metal chalcogenide.82 However, the HE metal chalcogenides are comparable to other Cu-based thermoelectric materials such as the borite Cu5FeS4,83 and tetrahedrite Cu12Sb4S13,78 which have reported zT values of 0.55 and 0.56 at 543 K and 673 K, respectively.
HE metal sulphides are not the only HE metal chalcogenides investigated for thermoelectrics, HE metal tellurides have also been investigated.46,56 In these reports, the p-type AgMnGeSbTe4 has been reported with a peak power factor of ca. 12 μW cm−1 K−2 (again exact values are not reported) and a peak zT value of 1.05 at 773 K. Incorporating small quantities of (1–5 mol%) of Ag8GeTe6 into the material was found to present extra scatting centres for holes with low energy and phonons with medium wavelength, where the optimal 1 mol% Ag8GeTe6 incorporation resulted in maximum power factor and zT of ca. 14 μW cm−1 K−2 and 1.27, respectively.56 Another p-type HE metal telluride material has been reported with a maximum zT value of 1.6 for Cu0.8Ag0.2In0.5Ga0.5Te2, however this is at the high temperature of 900 K.46 Whilst these are not high entropy materials by our definition, these works still demonstrate the improved performance of thermoelectric materials by increasing the entropy of the material. Having a Te, rather than a S anionic lattice in the HE material results in increased lattice strain, which is reported to decrease thermal conductivity due to increases phonon scattering.70
Having metals of various sizes results in significant lattice distortion in the cationic position of the HE lattice. However, this is also possible in the anionic position too by using more than one chalcogenide, which has also been investigated for thermoelectrics.37,53,54 This combination of cationic and anionic lattice strain has been best demonstrated by (PbSbSn)(SSeTe) with various ratios of metal cations and chalcogenide anions to create severe lattice distortion in both cationic and anionic positions.37 In this report the n-type Pb0.89Sb0.012Sn0.1Se0.5Te0.25S0.25 HE material is reported to have a maximum power factor of ca. 16 μW cm−1 K−2 at 600 K, with an incredibly low thermal conductivity of < 0.5 W m−1 K−2 at temperatures >600 K and a maximum zT of 1.8 at 900 K.37 Another example of induced disorder in the anionic lattice producing excellent thermoelectric performance is of Pb0.935Na0.025Cd0.04Se0.5S0.25Te0.25.84 This p-type material has been reported with a high zT of 2.0 at 900 K and a high power factor of 16 μW cm−1 K−1.84 However, it should be noted that as the concentration of both Na (1.25 mol%) and Cd (2 mol%) are both below the 5 mol% threshold we have defined as required for high entropy materials, this is a dual-doped medium entropy material rather than a true high entropy metal chalcogenide.
Despite the clear advantages of utilising a combination of metals and chalcogenides, it should be noted that using any combination of metals and chalcogenides does not always result in good thermoelectric properties. A recent investigation into the n-type (Co,Fe,Ni)9(S,Se)8 only resulted in zT values < 0.03 at ca. 325 K, which decreased with further increases in temperature.54 The compiled power factor and zT values of HEMCs are shown in Fig. 7.
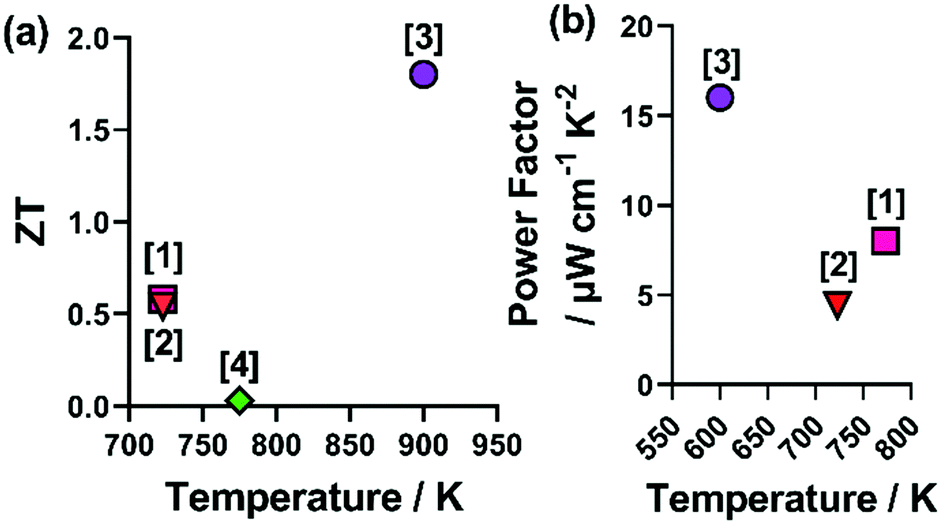 | ||
| Fig. 7 Figure showing the reported zT and power factor (PF) values for non-oxide HE metal chalcogenides. Where [1] Cu5Sn1.2MgGeZnS9, [2], (CoFeNi)(SSe), [3] (PbSbSn)(SSeTe), [4] (AgPbBi)(SSeTe) correspond to ref. 37, 47, 54 and 66. | ||
Superconductors
HEMCs have also been found to exhibit superconductivity. Mizuguchi et al. were able to show that a range of high entropy tellurides (e.g. (AgInSnSbPb)Te) exhibit superconductivity at low temperatures.67 For the HE tellurides, the temperature below which 0 resistance was measured was between 0.9–1.3 K for different metal compositions. In further work, the same research group also observed a superconducting transition in their HE selenide/telluride alloys – (Ag,In,Pb,Bi)Te1−xSex, where x = 0, 0.25 and 0,5.55 They observed an increasing critical temperature, Tc, with increasing amounts of selenium present in the material at 2.7, 2.5 and 2.0 K for the x = 0, 0.25 and 0.5 samples respectively. They observed bulk superconductivity by direct measurement of the electrical resistance over a range of temperatures as well as observing the sudden drop in the magnetisation as the material became diamagnetic below Tc. They also observed that Tc increased with increasing applied magnetic field for both the x = 0 and 0.25 samples.Summary and future outlook
The exceptional properties exhibited by the unique environment of HE metal chalcogenides clearly represent a significant advancement in the development of advanced, high-performance materials. These remarkable properties are most impactful for energy and catalysis research.Despite the significant advancement that HE metal chalcogenides represent, the scalability of these materials can only be considered if they can be synthesised as both bulk and nano (both particles and thin film) materials. A further hugely important factor towards the scalability of HE metal chalcogenides is the synthetic method, which should ideally be facile, fast, and at a low temperature. The synthetic method should also take into account the principles of green chemistry,85 to eliminate any hazardous solvents or preparatory steps.
It is also true that despite the significant promise shown for both catalysis and energy research in both conversion (as thermoelectrics) and storage (as batteries) we may have only touched upon the potential advancement of these materials, particularly the non-oxide materials. Further research needs to be undertaken to explore further catalytic reactions and operando spectroscopic techniques to determine the active metal centres and active surfaces of the materials in order to enhance this further particularly in the nano-scale for enhanced activity using less material. This area of research could also benefit significantly from further computational input, on analysis of developed materials, prediction of energy band gaps and prediction and development of materials for enhanced activity.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
B. W.-O’. B. is funded by an EPSRC DTA studentship. J. Q. Acknowledges funding from Chinese Scholarship Council (CSC) 201906240222. W. X. thanks Beihang University for the opportunity to come to Manchester.References
- J. W. Yeh, S. K. Chen, S. J. Lin, J. Y. Gan, T. S. Chin, T. T. Shun, C. H. Tsau and S. Y. Chang, Adv. Eng. Mater., 2004, 6, 299–303 CrossRef CAS.
- B. Cantor, I. T. H. Chang, P. Knight and A. J. B. Vincent, Mater. Sci. Eng., A, 2004, 375–377, 213–218 CrossRef.
- J. W. Yeh, JOM, 2013, 65, 1759–1771 CrossRef CAS.
- J. Dąbrowa, M. Zajusz, W. Kucza, G. Cieślak, K. Berent, T. Czeppe, T. Kulik and M. Danielewski, J. Alloys Compd., 2019, 783, 193–207 CrossRef.
- X. Yang and Y. Zhang, Mater. Chem. Phys., 2012, 132, 233–238 CrossRef CAS.
- R. Kozak, A. Sologubenko and W. Steurer, Z. Kristallogr., 2015, 230, 55–68 CAS.
- E. P. George, D. Raabe and R. O. Ritchie, Nat. Rev. Mater., 2019, 4, 515–534 CrossRef CAS.
- J. K. Pedersen, T. A. A. Batchelor, D. Yan, L. E. J. Skjegstad and J. Rossmeisl, Curr. Opin. Electrochem., 2021, 26, 100651 CrossRef CAS.
- H. Xu, Z. Zhang, J. Liu, C. L. Do-Thanh, H. Chen, S. Xu, Q. Lin, Y. Jiao, J. Wang, Y. Wang, Y. Chen and S. Dai, Nat. Commun., 2020, 11, 1–9 CrossRef CAS PubMed.
- M. H. Tsai and J. W. Yeh, Mater. Res. Lett., 2014, 2, 107–123 CrossRef.
- J. W. Yeh, JOM, 2015, 67, 2254–2261 CrossRef CAS.
- E. J. Pickering and N. G. Jones, Int. Mater. Rev., 2016, 61, 183–202 CrossRef CAS.
- D. B. Miracle and O. N. Senkov, Acta Mater., 2017, 122, 448–511 CrossRef CAS.
- H. Song, F. Tian, Q. M. Hu, L. Vitos, Y. Wang, J. Shen and N. Chen, Phys. Rev. Mater., 2017, 1, 1–8 CrossRef.
- M. Qin, J. Gild, H. Wang, T. Harrington, K. S. Vecchio and J. Luo, J. Eur. Ceram. Soc., 2020, 40, 4348–4353 CrossRef CAS.
- M. Qin, Q. Yan, H. Wang, C. Hu, K. S. Vecchio and J. Luo, Scr. Mater., 2020, 189, 101–105 CrossRef CAS.
- J. Gild, Y. Zhang, T. Harrington, S. Jiang, T. Hu, M. C. Quinn, W. M. Mellor, N. Zhou, K. Vecchio and J. Luo, Sci. Rep., 2016, 6, 2–11 CrossRef PubMed.
- Y. Zhang, Z. Bin Jiang, S. K. Sun, W. M. Guo, Q. S. Chen, J. X. Qiu, K. Plucknett and H. T. Lin, J. Eur. Ceram. Soc., 2019, 39, 3920–3924 CrossRef CAS.
- B. Ye, T. Wen, M. C. Nguyen, L. Hao, C. Z. Wang and Y. Chu, Acta Mater., 2019, 170, 15–23 CrossRef CAS.
- P. Sarker, T. Harrington, C. Toher, C. Oses, M. Samiee, J. P. Maria, D. W. Brenner, K. S. Vecchio and S. Curtarolo, Nat. Commun., 2018, 9, 1–10 CrossRef CAS PubMed.
- T. J. Harrington, J. Gild, P. Sarker, C. Toher, C. M. Rost, O. F. Dippo, C. McElfresh, K. Kaufmann, E. Marin, L. Borowski, P. E. Hopkins, J. Luo, S. Curtarolo, D. W. Brenner and K. S. Vecchio, Acta Mater., 2019, 166, 271–280 CrossRef CAS.
- V. Braic, M. Balaceanu, M. Braic, A. Vladescu, S. Panseri and A. Russo, J. Mech. Behav. Biomed. Mater., 2012, 10, 197–205 CrossRef CAS PubMed.
- C. W. Tsai, S. W. Lai, K. H. Cheng, M. H. Tsai, A. Davison, C. H. Tsau and J. W. Yeh, Thin Solid Films, 2012, 520, 2613–2618 CrossRef CAS.
- K. Johansson, L. Riekehr, S. Fritze and E. Lewin, Surf. Coat. Technol., 2018, 349, 529–539 CrossRef CAS.
- K. Balasubramanian, S. V. Khare and D. Gall, Acta Mater., 2018, 159, 77–88 CrossRef CAS.
- T. Jin, X. Sang, R. R. Unocic, R. T. Kinch, X. Liu, J. Hu, H. Liu and S. Dai, Adv. Mater., 2018, 30, 1–5 Search PubMed.
- J. Gild, J. Braun, K. Kaufmann, E. Marin, T. Harrington, P. Hopkins, K. Vecchio and J. Luo, J. Materiomics, 2019, 5, 337–343 CrossRef.
- Y. Liu, Y. Zhang, H. Zhang, N. Wang, X. Chen, H. Zhang and Y. Li, J. Alloys Compd., 2017, 694, 869–876 CrossRef CAS.
- X. Zhao, Z. Xue, W. Chen, Y. Wang and T. Mu, ChemSusChem, 2020, 13, 2038–2042 CrossRef CAS PubMed.
- D. Lai, Q. Kang, F. Gao and Q. Lu, J. Mater. Chem. A, 2021, 9, 17913–17922 RSC.
- H. Qiao, X. Wang, Q. Dong, H. Zheng, G. Chen, M. Hong, C. P. Yang, M. Wu, K. He and L. Hu, Nano Energy, 2021, 86, 106029 CrossRef CAS.
- T. Wang, H. Chen, Z. Yang, J. Liang and S. Dai, J. Am. Chem. Soc., 2020, 142, 4550–4554 CrossRef CAS PubMed.
- X. Chen and Y. Wu, J. Am. Ceram. Soc., 2020, 103, 750–756 CrossRef CAS.
- P. A. Sukkurji, Y. Cui, S. Lee, K. Wang, R. Azmi, A. Sarkar, S. Indris, S. S. Bhattacharya, R. Kruk, H. Hahn, Q. Wang, M. Botros and B. Breitung, J. Mater. Chem. A, 2021, 9, 8998–9009 RSC.
- S. Akrami, P. Edalati, M. Fuji and K. Edalati, Mater. Sci. Eng., R, 2021, 146, 100644 CrossRef.
- M. Hillert, Phase Equilibria, Phase Diagrams and Phase Transformations: Their Thermodynamic Basics, Cambdridge university Press, 2nd edn, 2008 Search PubMed.
- B. Jiang, Y. Yu, J. Cui, X. Liu, L. Xie, J. Liao, Q. Zhang, Y. Huang, S. Ning, B. Jia, B. Zhu, S. Bai, L. Chen, S. J. Pennycook and J. He, Science, 2021, 371, 830–834 CrossRef CAS PubMed.
- O. F. Dippo and K. S. Vecchio, Scr. Mater., 2021, 201, 113974 CrossRef CAS.
- C. M. Rost, E. Sachet, T. Borman, A. Moballegh, E. C. Dickey, D. Hou, J. L. Jones, S. Curtarolo and J. P. Maria, Nat. Commun., 2015, 6, 8485 CrossRef CAS PubMed.
- S. H. Albedwawi, A. AlJaberi, G. N. Haidemenopoulos and K. Polychronopoulou, Mater. Des., 2021, 202, 109534 CrossRef CAS.
- A. Sarkar, Q. Wang, A. Schiele, M. R. Chellali, S. S. Bhattacharya, D. Wang, T. Brezesinski, H. Hahn, L. Velasco and B. Breitung, Adv. Mater., 2019, 31, 1806236 CrossRef PubMed.
- R. Z. Zhang and M. J. Reece, J. Mater. Chem. A, 2019, 7, 22148–22162 RSC.
- Y. Gao, Y. Liu, H. Yu and D. Zou, Appl. Catal., A, 2022, 631, 118478 CrossRef CAS.
- Y. Lin, N. Luo, M. Chamas, C. Hu and S. Grasso, Int. J. Appl. Ceram. Technol., 2021, 18, 1560–1569 CrossRef CAS.
- R. Witte, A. Sarkar, R. Kruk, B. Eggert, R. A. Brand, H. Wende and H. Hahn, Phys. Rev. Mater., 2019, 3, 1–8 Search PubMed.
- R. Liu, H. Chen, K. Zhao, Y. Qin, B. Jiang, T. Zhang, G. Sha, X. Shi, C. Uher, W. Zhang and L. Chen, Adv. Mater., 2017, 29, 1702712 CrossRef PubMed.
- R. Z. Zhang, F. Gucci, H. Zhu, K. Chen and M. J. Reece, Inorg. Chem., 2018, 57, 13027–13033 CrossRef CAS PubMed.
- T. X. Nguyen, Y. Su, C. Lin and J. Ting, Adv. Funct. Mater., 2021, 2106229, 2106229 CrossRef.
- J. Cavin, A. Ahmadiparidari, L. Majidi, A. S. Thind, S. N. Misal, A. Prajapati, Z. Hemmat, S. Rastegar, A. Beukelman, M. R. Singh, K. A. Unocic, A. Salehi-Khojin and R. Mishra, Adv. Mater., 2021, 33, 1–9 CrossRef PubMed.
- C. R. McCormick and R. E. Schaak, J. Am. Chem. Soc., 2021, 143, 1017–1023 CrossRef CAS PubMed.
- M. Cui, C. Yang, B. Li, Q. Dong, M. Wu, S. Hwang, H. Xie, X. Wang, G. Wang and L. Hu, Adv. Energy Mater., 2021, 11, 1–8 Search PubMed.
- L. Lin, K. Wang, A. Sarkar, C. Njel, G. Karkera, Q. Wang, R. Azmi, M. Fichtner, H. Hahn, S. Schweidler and B. Breitung, Adv. Energy Mater., 2022, 2103090 CrossRef CAS.
- Z. Deng, A. Olvera, J. Casamento, J. S. Lopez, L. Williams, R. Lu, G. Shi, P. F. P. Poudeu and E. Kioupakis, Chem. Mater., 2020, 32, 6070–6077 CrossRef CAS.
- A. Mikuła, J. Dąbrowa, A. Kusior, K. Mars, R. Lach and M. Kubowicz, Dalton Trans., 2021, 50, 9560–9573 RSC.
- A. Yamashita, R. Jha, Y. Goto, T. D. Matsuda, Y. Aoki and Y. Mizuguchi, Dalton Trans., 2020, 49, 9118–9122 RSC.
- Z. Ma, T. Xu, W. Li, Y. Cheng, J. Li, D. Zhang, Q. Jiang, Y. Luo and J. Yang, Adv. Funct. Mater., 2021, 31, 2103197 CrossRef CAS.
- P. Mangelis, P. Vaqueiro and A. V. Powell, ACS Appl. Energy Mater., 2020, 3, 2168–2174 CrossRef CAS PubMed.
- B. Ward-O’Brien, E. J. Pickering, R. Ahumada-Lazo, C. Smith, X. L. Zhong, Y. Aboura, F. Alam, D. J. Binks, T. L. Burnett and D. J. Lewis, J. Am. Chem. Soc., 2021, 143, 21560–21566 CrossRef PubMed.
- W. L. Boncher, M. D. Regulacio and S. L. Stoll, J. Solid State Chem., 2010, 183, 52–56 CrossRef CAS.
- M. A. Buckingham, K. Norton, P. D. McNaughter, G. Whitehead, I. Vitorica-Yrezabal, F. Alam, K. Laws and D. J. Lewis, Inorg. Chem., 2022, 61, 8206 CrossRef CAS PubMed.
- M. A. Buckingham, A. L. Catherall, M. S. Hill, A. L. Johnson and J. D. Parish, Cryst. Growth Des., 2017, 17, 907 CrossRef CAS.
- J. C. Sarker and G. Hogarth, Chem. Rev., 2020, 121, 6057 CrossRef PubMed.
- G. V. Alexeeff, D. C. Lewis and N. L. Ragle, Risk Anal., 1993, 13, 63–69 CrossRef CAS PubMed.
- S. Ceresara, T. Federighi and F. Pieragostini, Phys. Status Solidi, 1966, 16, 439–447 CrossRef CAS.
- B. J. Wuensch and T. Vasilos, J. Chem. Phys., 1962, 36, 2917–2922 CrossRef CAS.
- A. Yamashita, Y. Goto, A. Miura, C. Moriyoshi, Y. Kuroiwa and Y. Mizuguchi, Mater. Res. Lett., 2021, 9, 366–372 CrossRef CAS.
- R. Kasem, K. Hoshi, R. Jha, M. Katsuno, A. Yamashita, Y. Goto, T. D. Matsuda, Y. Aoki and Y. Mizuguchi, Appl. Phys. Express, 2020, 13, 033001 CrossRef.
- T. Ying, T. Yu, Y. S. Shiah, C. Li, J. Li, Y. Qi and H. Hosono, J. Am. Chem. Soc., 2021, 143, 7042–7049 CrossRef CAS PubMed.
- M. G. Kanatzidis, Chem. Mater., 2010, 22, 648–659 CrossRef CAS.
- Y. Wu, Z. Chen, P. Nan, F. Xiong, S. Lin, X. Zhang, Y. Chen, L. Chen, B. Ge and Y. Pei, Joule, 2019, 3, 1276–1288 CrossRef CAS.
- J. Rosen, G. S. Hutchings, Q. Lu, S. Rivera, Y. Zhou, D. G. Vlachos and F. Jiao, ACS Catal., 2015, 5, 4293–4299 CrossRef CAS.
- Q. Lu, J. Rosen, Y. Zhou, G. S. Hutchings, Y. C. Kimmel, J. G. Chen and F. Jiao, Nat. Commun., 2014, 5, 1–6 Search PubMed.
- R. Fong, J. R. Dahn and C. H. W. Jones, J. Electrochem. Soc., 1989, 136, 3206–3210 CrossRef CAS.
- Q. Wang, L. Jiao, Y. Han, H. Du, W. Peng, Q. Huan, D. Song, Y. Si, Y. Wang and H. Yuan, J. Phys. Chem. C, 2011, 115, 8300–8304 CrossRef CAS.
- J. Zhao, Y. Zhang, Y. Wang, H. Li and Y. Peng, J. Energy Chem., 2018, 27, 1536–1554 CrossRef.
- M. A. Buckingham, K. Laws, J. T. Sengel and L. Aldous, Green Chem., 2020, 22, 6062 RSC.
- M. A. Buckingham, K. Laws, E. Cross, A. Surman and L. Aldous, Green Chem., 2021, 23, 8901 RSC.
- X. Lu, D. T. Morelli, Y. Xia, F. Zhou, V. Ozolins, H. Chi, X. Zhou and C. Uher, Adv. Energy Mater., 2013, 3, 342–348 CrossRef CAS.
- M. Saleemi, M. S. Toprak, S. Li, M. Johnsson and M. Muhammed, J. Mater. Chem., 2012, 22, 725–730 RSC.
- P. Qiu, X. Shi and L. Chen, Energy Storage Mater., 2016, 3, 85–97 CrossRef.
- A. Nagaoka, K. Yoshino, T. Masuda, T. D. Sparks, M. A. Scarpulla and K. Nishioka, J. Mater. Chem. A, 2021, 9, 15595–15604 RSC.
- L. Zhao, X. Wang, F. Y. Fei, J. Wang, Z. Cheng, S. Dou, J. Wang and G. J. Snyder, J. Mater. Chem. A, 2015, 3, 9432–9437 RSC.
- G. Guélou, A. V. Powell and P. Vaqueiro, J. Mater. Chem. C, 2015, 3, 10624–10629 RSC.
- B. Jiang, Y. Yu, H. Chen, J. Cui, X. Liu, L. Xie and J. He, Nat. Commun., 2021, 12, 1–8 CrossRef PubMed.
- P. Anastas and J. Warner, Green Chemistry: Theory and Practice, Oxford University Press, Oxford [England], 2000 Search PubMed.
| This journal is © The Royal Society of Chemistry 2022 |