
Redox-copolymers for the recovery of rare earth elements by electrochemically regenerated ion-exchange†
Haley
Vapnik‡
 ,
Johannes
Elbert‡
and
Xiao
Su
*
,
Johannes
Elbert‡
and
Xiao
Su
*
Department of Chemical and Biomolecular Engineering, University of Illinois at Urbana-Champaign, Urbana, Illinois 61801, USA. E-mail: x2su@illinois.edu
First published on 23rd August 2021
Abstract
Rare earth elements (REEs) play an essential role in our modern society, being critical resources for electronic devices and renewable energy technologies. Efficient platforms for REE recovery and purification are essential to resource security and environmental management. Imparting electrochemical control over an adsorbent system can lead to higher modularity and sustainability, by enabling adsorbent regeneration without the use of additional chemicals. For the reversible capture and release of REEs, we design and synthesize a redox-copolymer, poly(ferrocenylpropyl methacrylamide-co-methacrylic acid) (P(FPMAm-co-MAA)), that combines an ion-exchange carboxyl group for REE adsorption, and a redox-active ferrocene moiety for regeneration based on electrochemical control. By molecularly tuning the copolymer composition, efficient adsorption uptake could be achieved alongside electrochemically regenerated adsorbent reuse. The copolymer electrodes exhibited an Y(III) adsorption capacity of 69.4 mg Y per g polymer, and electrochemically mediated regeneration efficiency close to 100% through oxidation of the ferrocenium units. The copolymer sorbent showed stoichiometric binding for cerium (Ce), neodymium (Nd), europium (Eu), gadolinium (Gd), and dysprosium (Dy) based on carboxyl active sites. Our work provides a proof of concept for electrochemically regenerable ion-exchange copolymers for REE recovery, and we envision generalized applications of this concept in electrifying ion-exchange systems and cation-selective separations.
1. Introduction
Rare earth elements (REEs) are a group of 17 chemically similar elements consisting of 15 lanthanides plus scandium (Sc) and yttrium (Y).1–3 The unique magnetic, phosphorescent, and catalytic properties of REEs make them irreplaceable components in a growing technology market. They play a key role in many products, from computers and smartphones to rechargeable batteries, lasers, and electric automobiles.1,2 Over the past decades, the consumption of REEs has steadily increased due to their uses in new materials and renewable technologies.4–12 The worldwide production of REEs has been geographically limited,3,13 with many countries mostly relying on imported REEs from limited supply sources.14–16 Thus, both the European Commission17 and the U. S. Department of Energy (DOE) consider REEs to have critical supply risk, especially for neodymium (Nd), europium (Eu), terbium (Tb), dysprosium (Dy), and yttrium (Y).18,19 With increasing demand, restricted supply, and low recycling rate, there is an urgent need for efficient REE recovery technologies.Commonly used methods for REE separation include chemical precipitation, liquid–liquid extraction, ion-exchange, and adsorption.20–22 However, many of these conventional approaches produce significant hazardous waste, require excess chemical input, and often have challenges in molecular selectivity.20 Functional surfaces can be a pathway for molecular selectivity, due to the library of specific adsorption groups which have proven affinity with REEs,20,21 including carboxyl and hydroxyl groups.20,21,23–28 However, adsorption often requires significant chemical consumption during regeneration of adsorbent materials, including addition of acids and bases.
Electrochemical separations offer a green alternative for REE recovery, allowing for regeneration of adsorbents without the use of additional chemicals, fully promoted by electron-transfer.29–32 Redox-active materials in particular have been shown to promote selective molecular binding.33,34 Redox-active polymers containing ferrocenyl groups have shown high selectivity towards anionic contaminants, including metal oxyanions of arsenic and chromium, among others.29,30,33,35–37 However, these redox-polymer platforms have mostly been restricted to applications in anion separations, with limited attention devoted to cation-selective separations. These limitations are partly due to a lack of redox-active polymers that are able to sustain a negative charge in aqueous solutions, especially at neutral or acidic pH values. Thus, there is significant demand for functional polymer design concepts for cation-selective systems, especially for recovery of REEs and other critical elements which are often present as species with a positive charge in solution.
Recently, redox-copolymers have been used to modulate hydrophobicity and electrostatic interactions orthogonally for electrosorption of perfluoroalkyl substances (PFAS), by leveraging binding groups for PFAS with redox-active TEMPO groups for hydrophobicity and electrostatic control.31 Unlike homopolymers, copolymers can combine different functionalities from distinct monomer units to leverage synergistic effects. Here, we propose the combination of carboxyl groups with ferrocene groups within a single redox-copolymer, to combine traditional ion-exchange effects with redox-mediated release. Poly(methacrylic acid) (PMAA) for example has been shown to efficiently adsorb metal cations by an ion exchange mechanism.20,21,26 At the same time, polymers with ferrocene moieties in the side chain, such as poly(vinyl ferrocene) and ferrocene based (meth)acrylates, show remarkable redox properties, due to the high stability of the ferrocene (Fc)/ferrocenium (Fc+) redox couple.38–40
We propose a system that combines the uptake of traditional ion exchange resins analogous to PMAA for REE capture, and regeneration by electrodesorption and charge-repulsion based on ferrocenium groups (Fig. 1). Next, we describe the synthesis and characterization of a redox copolymer, poly(ferrocenylpropyl methacrylamide-co-methacrylic acid) (P(FPMAm-co-MAA)), and its application in the recovery of important rare-earth elements, with a focus on yttrium (Y), dysprosium (Dy), neodymium (Nd), europium (Eu), cerium (Ce), and gadolinium (Gd). We discuss the effect of different copolymer compositions on electrochemical regeneration and adsorption uptake of the REEs. This work serves as a proof of concept for electrochemically regenerated ion-exchange copolymers to enable more sustainable cation-selective separations.
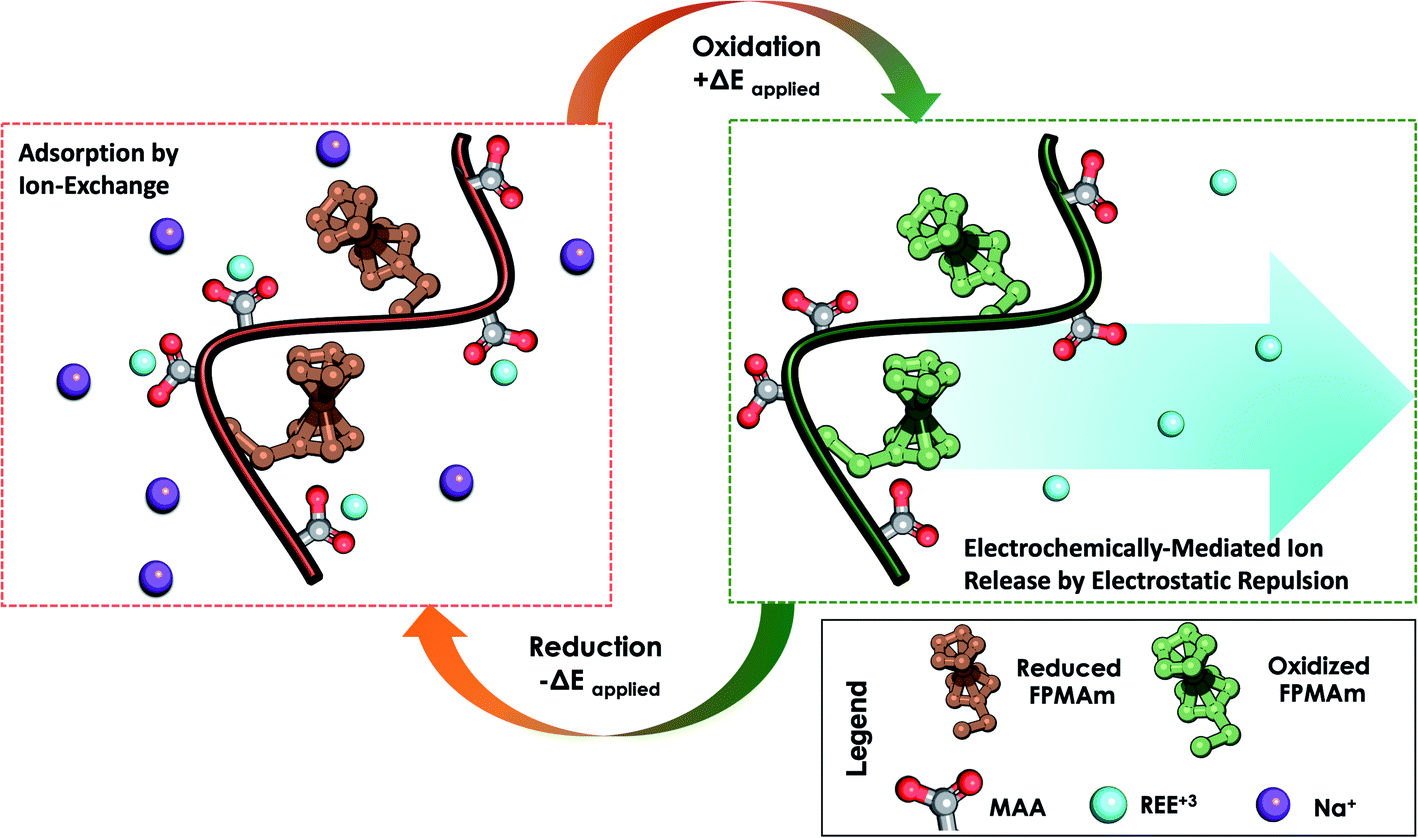 | ||
| Fig. 1 Overview of the reversible capture and release of REEs by P(FPMAm-co-MAA) through electrochemically regenerated ion exchange. During adsorption, REE ions are captured by chemical ion exchange. During desorption, ferrocene (Fc) is oxidized to ferrocenium (Fc+) electrochemically, allowing for desorption of the REE ions through electrostatic repulsion. Reduction of ferrocenium (Fc+) back to ferrocene (Fc) is required for electrode cycling. | ||
2. Experimental
2.1. Synthesis and characterization of P(FPMAm-co-MAA)
2.2. Preparation of P(FPMAm-co-MAA)-CNT electrodes
To fabricate immobilized ferrocene electrodes, two stocks were prepared: stock A consisting of 12 mg of P(FPMAm-co-MAA) (P1) in 3 mL of deionized (DI) water and 60 μL of 1 M NaOH, and stock B consisting of 12 mg of CNT (multiwalled carbon nanotubes), 1.2 mg of 1,3-BDSA in 1.5 mL of dimethylformamide (DMF). Stock A was stirred and heated until the polymer was fully dissolved while stock B was sonicated for 30 min in ice water. P1/CNT (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio) was prepared by mixing stocks A and B and sonicating for 4 h in an ice-bath. Once prepared, a titanium-grade 1 mesh (titanium screen, Fuel Cell Store) cut into rectangles (1 cm × 2 cm, 53 μm thick) was drop-coated with the solution containing the active material, with about 40–50 μL for each drop, and left to dry at 95 °C in an oven between drops, with a final total coated area of active material of 1 cm2. After coating, the polymer-coated electrodes were left overnight to ensure that all solvent was evaporated. The electrodes were left in an oven at 140 °C for 3 h to activate the crosslinker. Total coating mass was determined by the weight difference of the substrate before and after coating in a high-accuracy analytical balance. The coated electrodes were then connected to a copper wire with copper tape. These functionalized electrodes with the P1 polymer will be referred to as P1-CNT. Electrodes coated with other materials used in this paper were assembled in the same fashion as P1-CNT electrodes with some having variations to the coating solutions. Further details on materials and instruments for characterization can be found in Section SI-1,† and detailed electrode preparation is presented in Section SI-2.†
:1 ratio) was prepared by mixing stocks A and B and sonicating for 4 h in an ice-bath. Once prepared, a titanium-grade 1 mesh (titanium screen, Fuel Cell Store) cut into rectangles (1 cm × 2 cm, 53 μm thick) was drop-coated with the solution containing the active material, with about 40–50 μL for each drop, and left to dry at 95 °C in an oven between drops, with a final total coated area of active material of 1 cm2. After coating, the polymer-coated electrodes were left overnight to ensure that all solvent was evaporated. The electrodes were left in an oven at 140 °C for 3 h to activate the crosslinker. Total coating mass was determined by the weight difference of the substrate before and after coating in a high-accuracy analytical balance. The coated electrodes were then connected to a copper wire with copper tape. These functionalized electrodes with the P1 polymer will be referred to as P1-CNT. Electrodes coated with other materials used in this paper were assembled in the same fashion as P1-CNT electrodes with some having variations to the coating solutions. Further details on materials and instruments for characterization can be found in Section SI-1,† and detailed electrode preparation is presented in Section SI-2.†
2.3. Adsorption experiments
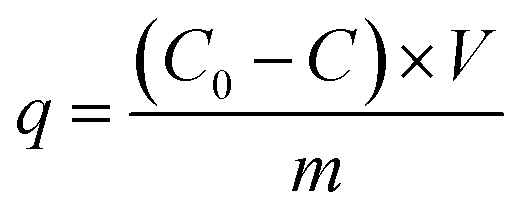 | (1) |
2.4. Electrochemically mediated regeneration
For desorption tests, 10 mL of solution with a background electrolyte concentration of 20 mM NaCl was used. Chronoamperometry was used for all desorption tests in this work. ICP-OES was used to determine the amount of REE ions desorbed into solution. The electrode regeneration experiments were carried out similarly with Y(III) as the model REE ion. After electrochemical desorption, the P1-CNT was reduced to neutralize the positive charge and reuse the electrode for cation adsorption. Chronopotentiometry was used for electrode reduction steps, with an applied current of −0.025 mA under a nitrogen purge (procedure optimized after trial and error). The electrodes could then be subsequently used for further adsorption/desorption cycles.3. Results and discussion
3.1. Synthesis and characterization of P(FPMAm-co-MAA)
To overcome the challenge of regeneration of traditional carboxylic acid-based adsorbents for REEs, we aimed to combine the carboxylic acid moiety with a redox-active ferrocene moiety in a copolymer. The electrochemical oxidation of the neutral ferrocene to the positively charged ferrocenium ion should introduce enough positive charges on the polymer to compensate for the negative charges of the carboxylate anions, and consequently displace the REE cations once bound. To achieve this goal, methacrylic acid (MAA) was chosen as the carboxylic acid monomer, due to its large-scale commercial availability and low cost. Ferrocenylpropyl methacrylamide (FPMAm) was chosen as the ferrocene-containing monomer, because it has been proven to have a remarkable cycling stability in aqueous redox flow batteries and does not have the limitations of vinyl ferrocene in a free radical polymerization process.40–42The synthesis of FPMAm starting from ferrocene (Scheme 1) was improved to increase the overall yield from 51%, reported previously,40 to 65%. The limitation of the previous synthesis was the reduction of the nitrile to the amine with LiAlH with a yield of only 60%. By combining this step and the hydrogenation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond into a single hydrogenation step using RANEY®-nickel as the catalyst, the yield was improved to 86% and the synthesis shortened by a whole step. Further improvements were achieved by replacing the time and resource intensive chromatographic purification of the 2-cyanovinyl ferrocene with a simple recrystallization protocol and replacing the expensive methacryloyl chloride with much cheaper methacrylic anhydride for the last step of the synthesis. FPMAm and MAA were copolymerized by free radical copolymerization (Scheme 2), yielding a copolymer with different ratios of FPMAm and MAA and thus different ratios of sites for REE binding and redox-active groups.
C double bond into a single hydrogenation step using RANEY®-nickel as the catalyst, the yield was improved to 86% and the synthesis shortened by a whole step. Further improvements were achieved by replacing the time and resource intensive chromatographic purification of the 2-cyanovinyl ferrocene with a simple recrystallization protocol and replacing the expensive methacryloyl chloride with much cheaper methacrylic anhydride for the last step of the synthesis. FPMAm and MAA were copolymerized by free radical copolymerization (Scheme 2), yielding a copolymer with different ratios of FPMAm and MAA and thus different ratios of sites for REE binding and redox-active groups.
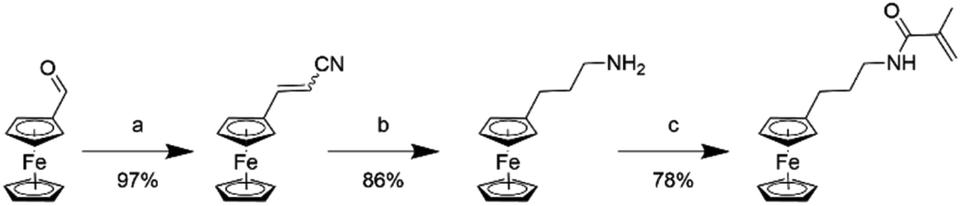 | ||
| Scheme 1 Synthesis of the FPMAm monomer: (a) acetonitrile/water, KOH, reflux, 2 h; (b) RANEY®-nickel, H2 at 8 bar, NH4OH, EtOH, 40 °C, 4 days; (c) methacrylic anhydride, NEt3, CH2Cl2, 0 °C, 16 h. | ||
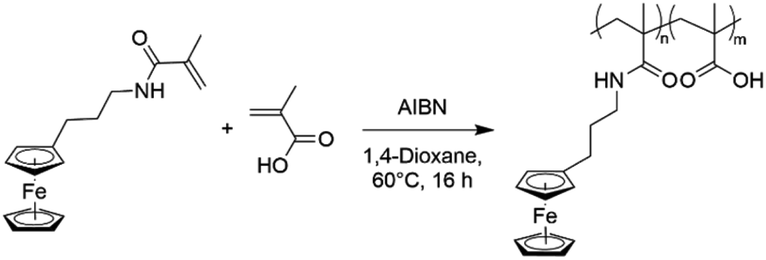 | ||
| Scheme 2 Free-radical copolymerization of FPMAm with MAA to yield P(FPMAm-co-MAA). | ||
The ratio of monomers determined by 1H NMR spectroscopy and the P(FPMAmx-co-MAA100−x) used in this study are summarized in Table 1, with x indicating the content of FPMAm in mol%. Furthermore, a copolymer of MAA and hydrophobic benzyl methacrylate (P(BzMA52-MAA48)) was synthesized as a non-redox active control. Measurement of the molecular weight was attempted by gel permeation chromatography, and only the polymer with low MAA content (P2) yielded consistent results (Fig. S28†). The polymer with a higher MAA content (P1) formed micelles in DMF, as shown by dynamic light scattering (DLS) measurements. This micellization was due to the high content of carboxylate groups, which are not well solubilized in DMF, and the organosoluble FPMAm repeating units stabilizing the micelles. When P1 was dissolved in water it also formed micelles with the hydrophobic ferrocene moieties forming the core of the micelles.
| Monomer ratio [mol% FPMAm] | ||
|---|---|---|
| Targeta | Measuredb | |
| a Targeted monomer ratio at the start of the reaction. b The FPMAm content was determined by 1H NMR spectroscopy. c Mol% BzMA. | ||
| P(FPMAm44-co-MAA56) (P1) | 50 | 44 |
| P(FPMAm69-co-MAA31) (P2) | 70 | 69 |
| P(FPMAm26-co-MAA74) (P3) | 30 | 26 |
| PFPMAm (P4) | 100 | 100 |
| P(BzMA52-MAA48) (P5) | 50c | 52c |
3.2. Fabrication and characterization of P(FPMAm-co-MAA)-CNT electrodes
The main challenge in the fabrication of stable electrodes for use in water is the water solubility of the polymers – especially in the oxidized state. To prevent dissolution of the polymer, crosslinking of the polymer film was performed. 1,3-Benzenedisulfonyl azide (1,3-BDSA) was used as a crosslinker. By heating to temperatures > 120 °C, the sulfonyl azide groups of this crosslinker formed reactive nitrene species that inserted into the C–H bonds of the polymer, thus crosslinking the polymer.43 Without the crosslinker, P1 was seen to quickly dissolve out of the P1-CNT electrodes due to the water solubility of PMAA (Fig. S1a†).The stability and redox activity of the P1-CNT electrode was investigated using cyclic voltammetry (CV) (Section SI-3†). Redox activity in the redox-copolymer was confirmed through CVs, with clear reversible oxidation and reduction of the ferrocenyl groups observed (Fig. 2). Crosslinking with 1,3-BDSA was used to prevent P1 (and the other MAA containing polymers) from dissolving into the aqueous solution and therefore stabilize the P1-CNT electrodes. The ratios of cross-linking reagent/P1 tested were 0, 0.05, 0.1, and 0.2 wt%. Even with crosslinking, the P1-CNT electrodes display some initial loss of active material due to incomplete crosslinking. However, the charge of the electrodes stabilized quickly and led to a stable operational charge of ∼80% of the initial charge. On the other hand, the polymer in the P1-CNT electrode without crosslinking quickly dissolved completely (Fig. S1 and S2†), showing the importance of optimizing crosslinking compositions. The optimal ratio of cross-linking reagent/P1 (and all polymers containing MAA) of 0.1 wt% was used throughout the rest of the work. The elemental mapping images of the P1-CNT electrode were recorded, and its surface morphology was examined (Fig. S7 and Table S3a†) to confirm the stability of the polymer in the coating and also show the microporous morphology of the electrode.
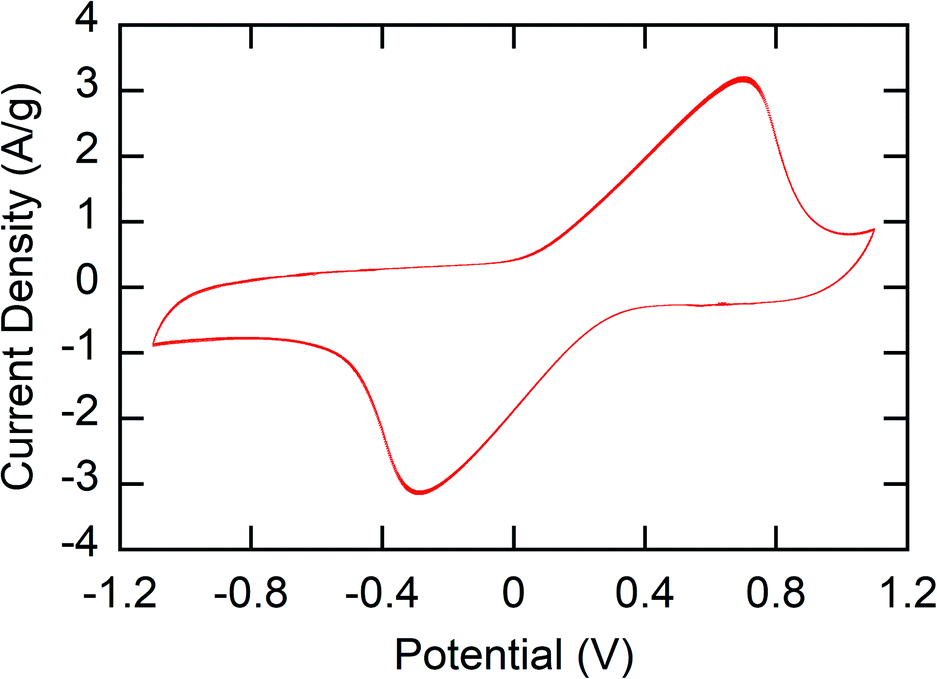 | ||
| Fig. 2 Cyclic voltammetry of the P1-CNT/Ti electrode's final 10 cycles (from over 100 cycles) in the presence of 100 mM NaClO4. The potential range chosen was from −1.1 to 1.1 V with a scan rate of 50 mV s−1. | ||
3.3. REE adsorption
Based on experimental results (Fig. 3a), the Y ion uptake increased with increasing methacrylic acid content in carboxylic acid moieties. Negligible Y uptake was observed on electrodes coated with the active material without carboxylic acid groups (P(FPMAm)-CNT and CNT controls), providing strong evidence that carboxylic acid moieties were solely responsible for Y interaction, and the ferrocenyl groups and CNT provided little to no uptake by themselves, and there was no secondary adsorption onto the experimental setup itself. The concentration drop of Y (in ppm) for all the controls is summarized in Section SI-17.† The ratio of Y ion uptake to carboxyl adsorption sites (Fig. 3b) was close to the theoretical value of 1/3 for the formation of a RE(RCOO)3 complex, showing the high accessibility and utilization of the redox-copolymer adsorption sites. A detailed description of the adsorption site calculation is given in Section SI-6.† P1 and P(FPMAm70-co-MAA30) (P2) have utilized most REE binding sites within 1 h. P(FPMAm26-co-MAA74) (P3) showed a slightly lower binding site utilization. This could either indicate a slower adsorption kinetics or be attributed to increased steric or coulombic interactions. Adsorption experiments of REEs of interest (Y, Nd, Eu, Gd, Dy, and Ce) with Na ions were performed on P1-CNT electrodes. Nd, Eu, Gd, Dy, and Ce were tested under the same conditions as yttrium (10 mL of aqueous solution containing 1 mM RE(Cl)3 and 20 mM NaCl, 1 h run time, open circuit, pH of 6, room temperature). All REEs tested showed significant uptake using P1-CNT (Fig. 3c), with the ion to adsorption site ratio close to 1/3 as well (Fig. 3d), indicating similar mechanisms for these trivalent cations. For Dy and Ce, there was a higher ratio of adsorbed REE ions per adsorption site, indicating that fewer carboxylic acid groups were binding each cation, which may hint at other possible coordination geometries at play with these two elements. Further investigation of the ligand binding mechanisms will be carried out in future work for these elements.
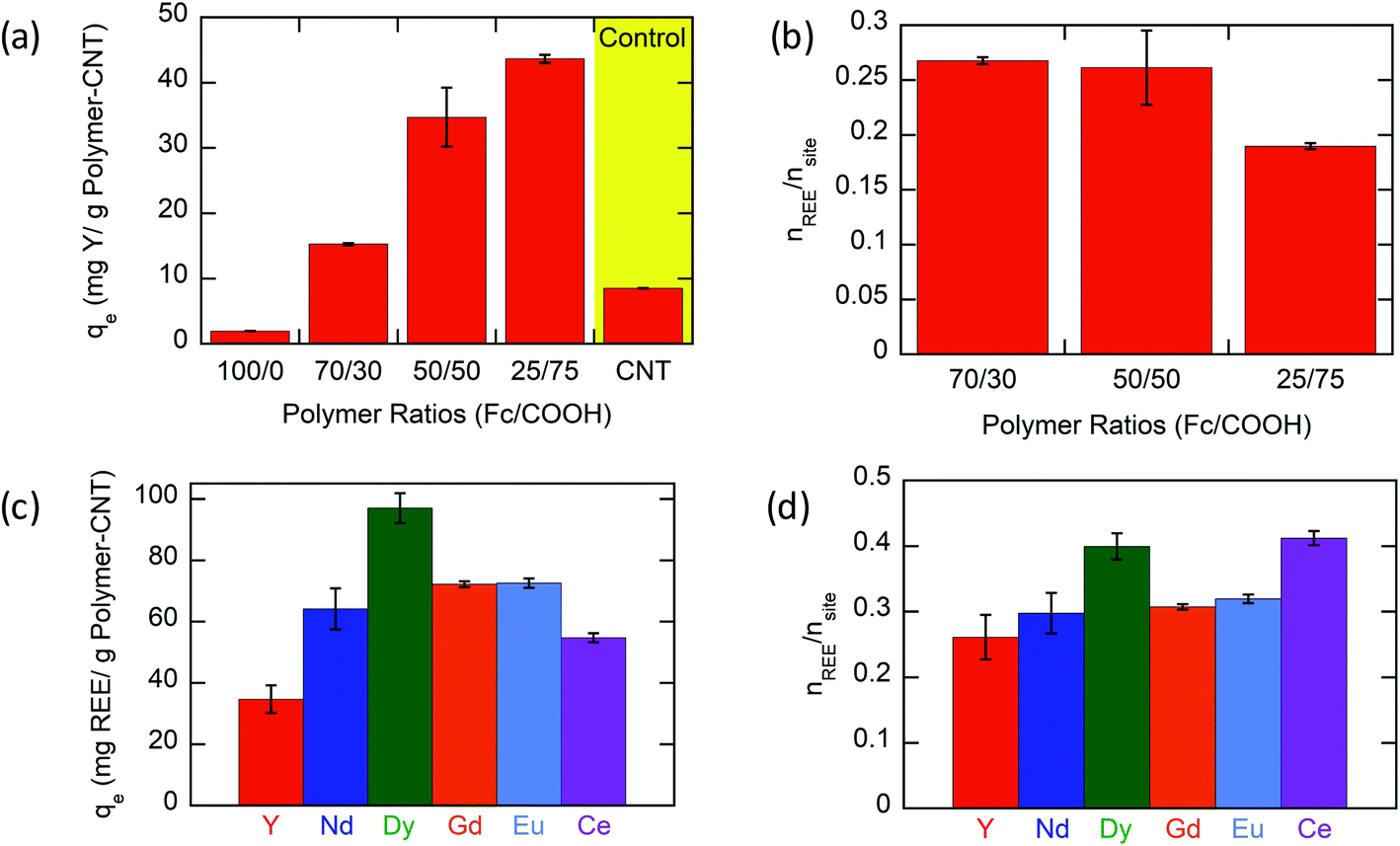 | ||
| Fig. 3 The removal of REE ions. Running conditions are as follows: 1 h, open circuit, 10 mL aqueous solution with 1 mM RE(Cl)3 and 20 mM NaCl. (a) Uptake of Y(III) onto polymer-CNT/Ti electrodes, with copolymers of various ratios of ferrocenyl groups to carboxylic acid groups (with the control being CNT/Ti electrodes without any polymer). (b) Number of Y(III) moieties adsorbed to the number of available adsorption sites (carboxylic acid sites) on polymer-CNT (P1-CNT) electrodes with polymers having various ratios of ferrocenyl groups to carboxylic acid groups. (c) Uptake of various REEs using P1-CNT. (d) Ratio of RE(III) adsorbed to the available adsorption sites based on carboxylic acid units in the polymer. | ||
The influence of ionic strength was investigated to evaluate various aqueous conditions in practical applications. The effect of ionic strength on the adsorption of Y(III) was tested at various initial concentrations of NaCl from 5 to 100 mM. As shown in Fig. S31,† the adsorption capacity decreases at higher concentrations of Na+, but the degree of reduction is extremely small. These results show that Y(III) has a stronger binding force than Na+ for the adsorption sites and that P1-CNT electrodes can be used to remove REEs from aqueous waste streams containing various amounts of salt.
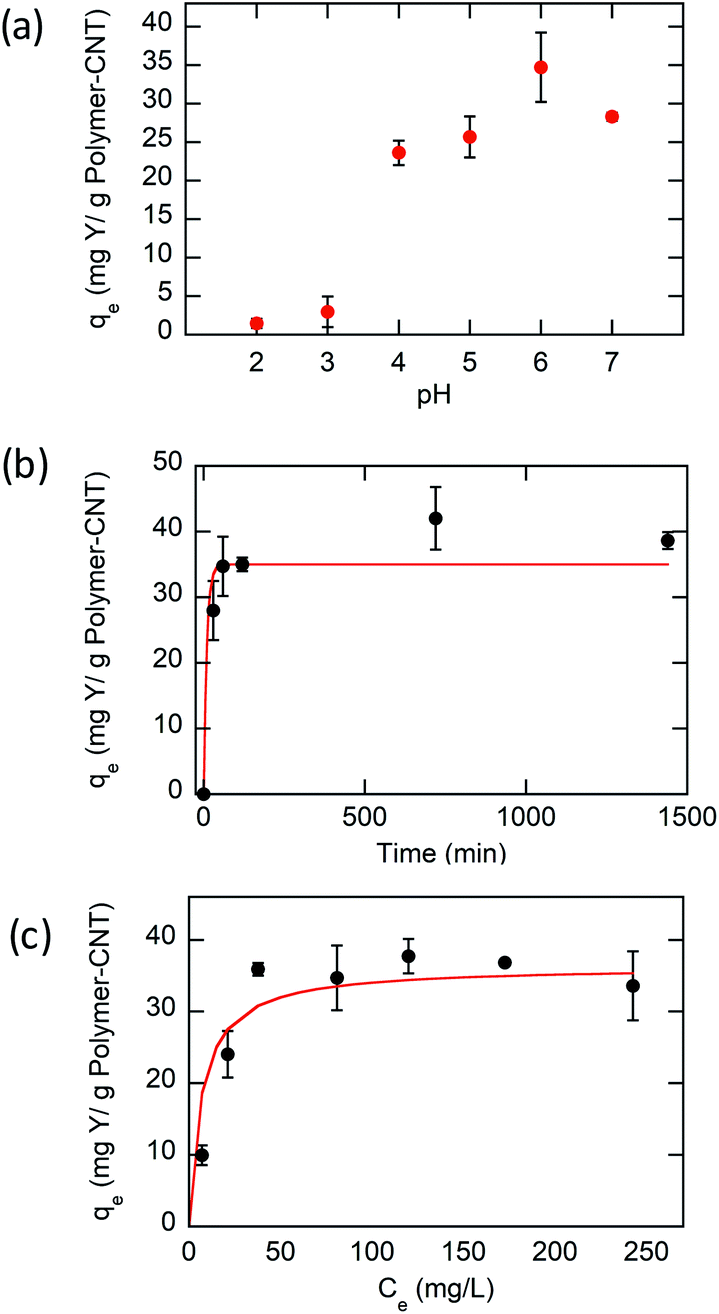 | ||
| Fig. 4 (a) Effect of pH on Y(III) adsorption uptake on P1-CNT. (b) Kinetic data and pseudo-first-order model for the adsorption. (c) Adsorption isotherm and Langmuir fit line of Y(III) adsorption on P1-CNT. | ||
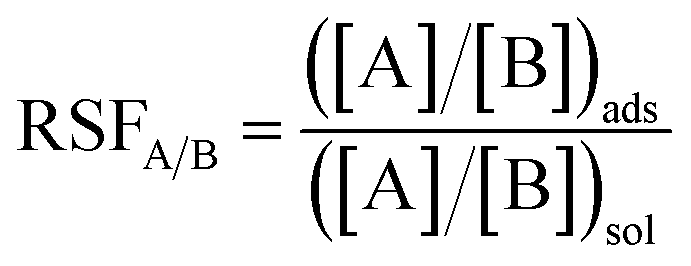 | (2) |
| Ion A | Ion B | RSFA/B |
|---|---|---|
| Y3+ | Na+ | 610.3 ± 6.33 |
| Dy3+ | Y3+ | 1.85 ± 0.087 |
| Y3+ | Ce3+ | 1.35 ± 0.046 |
3.4. Redox-mediated regeneration and cycling
Most importantly, we evaluate the electrochemical regeneration of our redox-copolymer systems for the bound REEs. In practical column adsorption applications, stripping of adsorbed ions is required for reuse of the adsorbent in the subsequent adsorption. While traditional ion exchange resins require stripping with a strong acid or base, thus generating an additional hazardous and corrosive waste stream, our proposed combination of carboxylic acid and redox active ferrocene moieties within a single copolymer allows for solely electrochemically controlled regeneration of the adsorbent. Compared to traditional adsorbents, our electrochemical approach only requires electricity for desorption, thus eliminating the need for highly acidic or alkaline stripping reagents. Bound REE ions can be released into solution through electrostatic repulsion of the positively charged ferrocenium ions, through electrochemically driven oxidation. The electrosorption of REE loaded P1-CNT electrodes was investigated under various applied positive potentials (Fig. 5a) for 1 h for releasing adsorbed Y(III) into 20 mM NaCl electrolyte. As depicted in Fig. 5a, P1-CNT exhibited almost full regeneration when applying +0.8 V during the first cycle desorption without using any acid or other stripping chemical additives. The successful adsorption and release of Y(III) on P1-CNT was also confirmed through X-ray photoelectron spectroscopy (XPS) (Fig. 5d) as well as scanning electron microscopy (SEM) and energy-dispersive spectroscopy (EDS) analyses (Section SI-7†), by monitoring the increase in yttrium content after adsorption, and its close to complete removal after electrochemical regeneration. Regeneration of P1-CNT was also tested with other REEs (Fig. 5c), with loaded P1-CNT releasing adsorbed RE(III) into 20 mM NaCl upon applying +0.8 V for 1 h. Regeneration parameters for P1-CNT were set using Y(III) as the model for REE ions.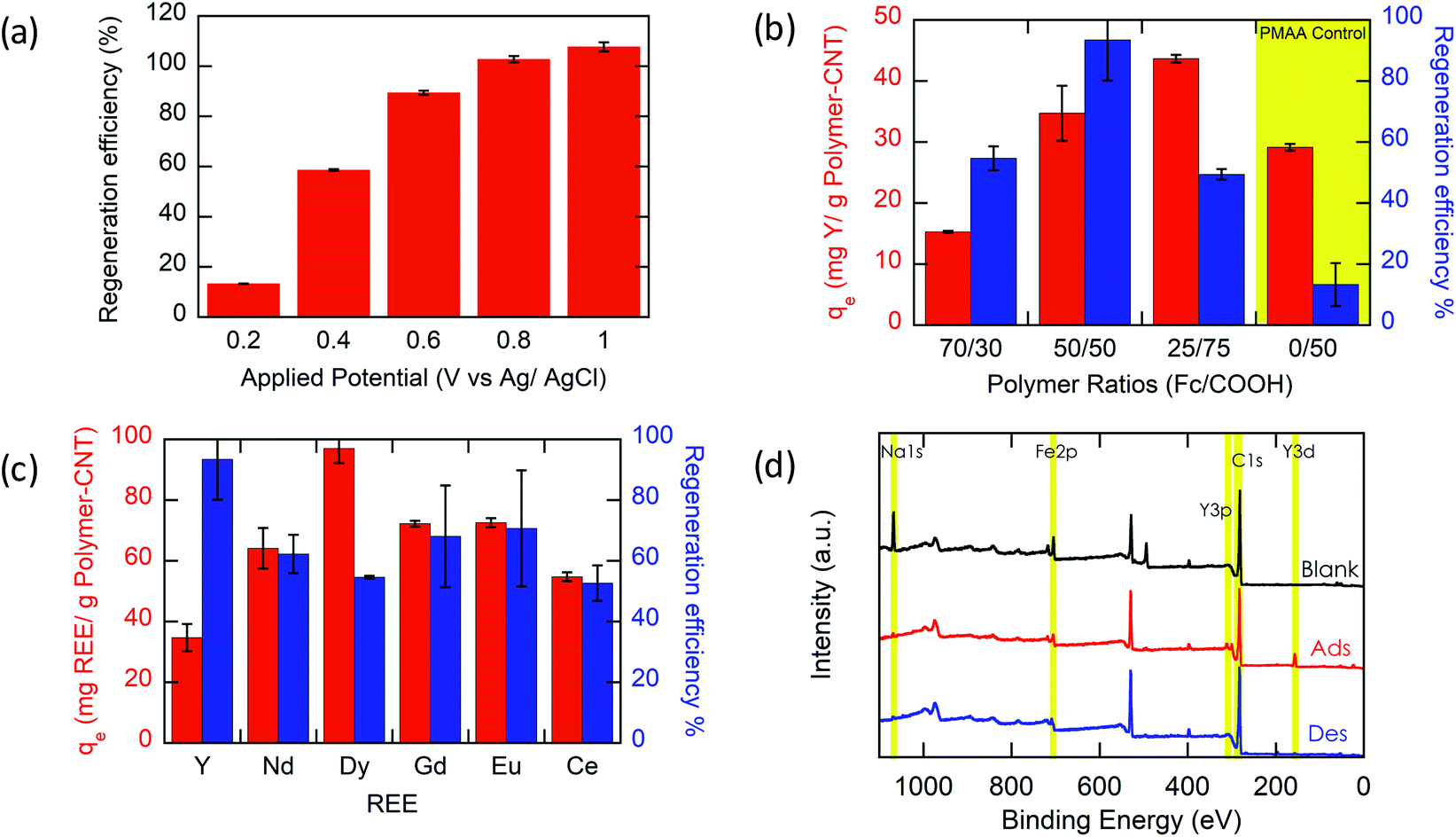 | ||
| Fig. 5 The regeneration efficiency of REE ions. All adsorption prior to regeneration was conducted using standard adsorption parameters listed earlier (1 h, open circuit, 10 mL aqueous solution with 1 mM RE(Cl)3 and 20 mM NaCl). All desorption conditions are as follows: 1 h, 10 mL aqueous solution with 20 mM NaCl. (a) Desorption of Y(III) from loaded P1-CNT electrodes at various applied potentials. (b) Adsorption/desorption of Y(III) with polymer-CNT electrodes with polymers having various ratios of ferrocenyl groups to carboxyl groups. Desorption with an applied potential of +0.8 V vs. Ag/AgCl. (c) REE uptake/regeneration efficiency using P1-CNT. (d) XPS surface analysis of P1-CNT before REE adsorption (blank), after Y(III) adsorption (Ads), and after Y(III) adsorption/desorption (Des). | ||
To prove the importance of the redox-active groups for release of REE ions, a copolymer with 52 mol% BzMA instead of FPMAm (P(BzMA52-MAA48), P5) was used, with BzMA being a non-redox control. P5-CNT showed a similar uptake to P1, but poor regeneration at 0.8 V (Fig. 5b), confirming that the redox-active ferrocene moieties are critical to promoting electrochemically regenerated release. Experiments varying the ratio of carboxylic acid to ferrocenyl groups showed that with increasing carboxylic acid group (binding site) content compared to ferrocene, desorption became less efficient (Fig. 5b), reaffirming the idea that the oxidation of ferrocene groups to ferrocenium cations is the driving force of the ion desorption process. XPS was used to estimate the ratio between reduced and oxidized ferrocene sites from the high-resolution Fe 2p spectra of the surface of P1-CNT (Fig. 6a–c). The oxidation degrees of Fe on the surface of pristine (or blank) P1-CNT and P1-CNT after adsorption of Y(III) in 1 mM YCl3 and 20 mM NaCl for 1 h were similar to each other, with both showing that most of the Fc sites were in a reduced state. After regeneration (desorption) of the P1-CNT electrode via chronoamperometry at +0.8 V (vs. Ag/AgCl) in 20 mM NaCl for 1 h, over 70% of the Fc sites were oxidized. This result further suggests that the mechanism for desorption can be attributed to the oxidation of ferrocene.
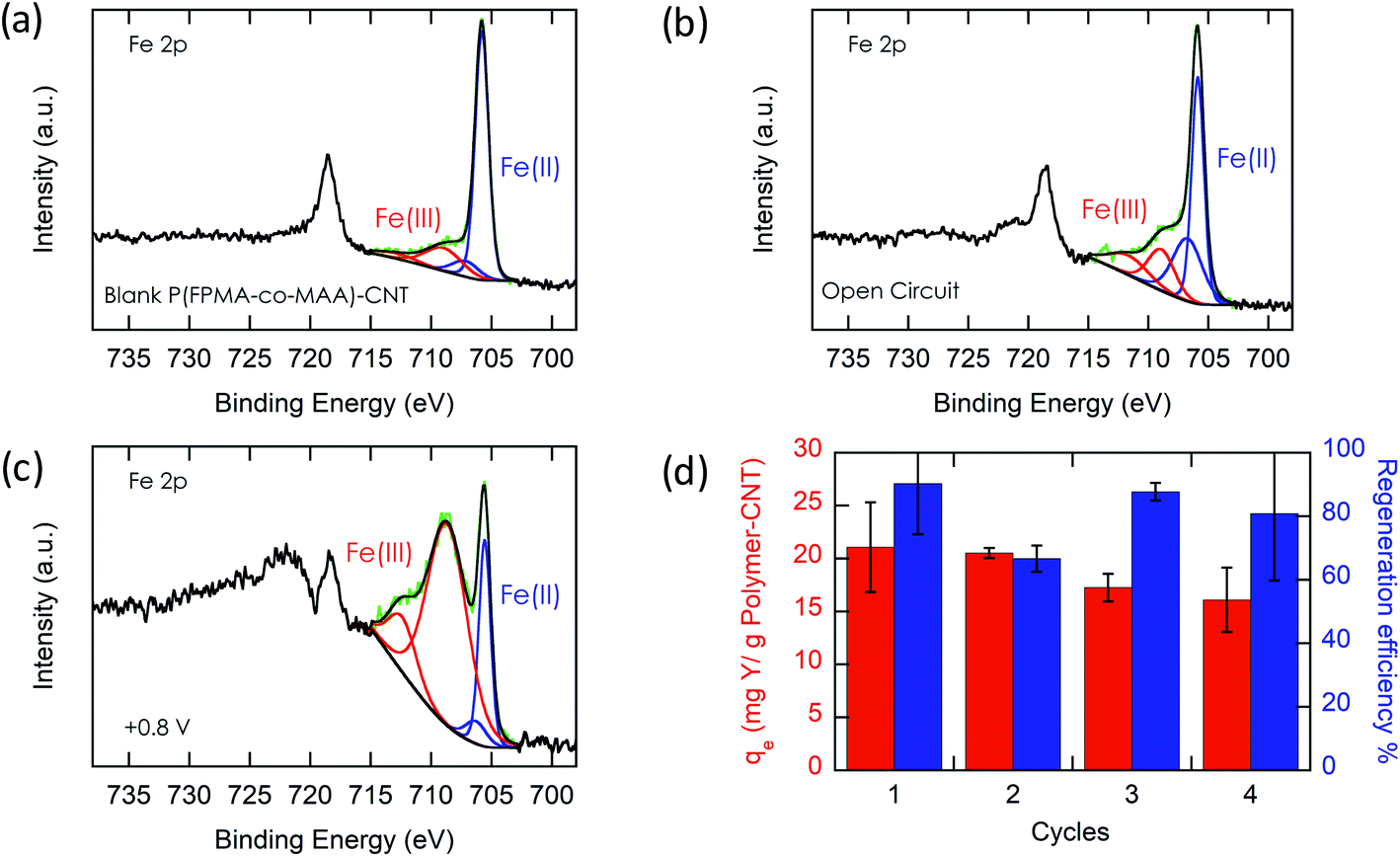 | ||
| Fig. 6 High resolution Fe 2p spectra of the surface of P1-CNT at different stages of the adsorption/desorption process. (a) Pristine (blank) P1-CNT. (b) P1-CNT used for adsorption of Y(III) under the following conditions: 1 h, open circuit, 10 mL aqueous solution with 1 mM YCl3 and 20 mM NaCl. (c) P1-CNT after adsorption of Y(III) regenerated (desorbed) for 1 h in 10 mL aqueous solution containing 20 mM NaCl with an applied potential of +0.8 V vs. Ag/AgCl. (d) Adsorption–desorption cycling runs using P1-CNT with adsorption of Y(III) under the following conditions: 1 h, open circuit, 10 mL aqueous solution with 1 mM YCl3 and 20 mM NaCl and desorption for 1 h in 10 mL aqueous solution containing 20 mM NaCl with an applied potential of +0.8 V vs. Ag/AgCl. | ||
Along with regeneration, reusability of the P1-CNT electrode is needed. As XPS analysis indicated in Fig. 6a–c, the number oxidized Fc+ species increased after the application of positive potential during regeneration. To allow for sufficient uptake in the following cycles, the oxidized Fc+ needs to be reduced back to Fc to start a new cycle of REE adsorption. Reducing the P1-CNT electrode completely is crucial to prevent excessive positive charges from repelling the REE cations, limiting the REE uptake.
The results of four adsorption–desorption cycles are shown in Fig. 6d. P1-CNT showed a minor decrease in uptake over 4 cycles but retained its regeneration efficiency. Desorption capabilities remained relatively constant over the course of the recycling runs, averaging around 80% recovery of the REE by electrochemical regeneration. Finally, an evaluation of energy integration and consumption for desorption was carried out (see Section SI-12 for calculation details†). We estimated that an energy consumption of about 79.3 kcal mol−1 Y was incurred for desorption at 0.8 V for 1 h. The use of lower potentials for oxidation showed lower energy consumption per mol of desorbed Y; however, these had significantly lower REE recovery, demonstrating that further process optimization can help decrease overall electricity consumption in the future, by improved selection of operational parameters. In parallel, molecular design of both ligands and redox-active groups can be expected to progressively increase the regeneration efficiency and reversibility.
4. Conclusions
In summary, we propose a new redox-copolymer as a promising proof-of-concept platform for electrochemically regenerated ion-exchange for REE recovery. Redox-copolymers containing carboxylic acid and ferrocene moieties were synthesized and studied for their effectiveness in recovery of REEs (Y, Nd, Eu, Gd, Dy, and Ce) from aqueous solutions. The copolymer showed increasing REE adsorption capacities with increasing content of MAA (the REE binding group), with a 50/50 ratio of ferrocenyl groups to carboxylic acid groups providing an optimal balance between uptake and electrochemical regeneration. Adsorption of Y(III) on P1-CNT showed an equilibrium capacity of 69.4 mg Y(III) per g polymer at an optimal pH of 6. Electrochemical desorption of the adsorbed REE from the electrodes was achieved using a positive potential vs. Ag/AgCl, to release the bound cation by electrostatic repulsion without the need for additional stripping reagents, achieving close to full regeneration under electrochemical conditions. The adsorption capacity of the recycled P1-CNT electrodes remained relatively constant during four consecutive cycles, confirming the structural stability of the redox-active copolymer. In conclusion, our work demonstrates that redox-copolymers can be a tunable functional material platform for electrochemically regenerable REE adsorption, providing modularity and sustainability to chemical adsorption systems, and pointing to future applicability in REE valorization from waste streams, chemical manufacturing separations, and even secondary recovery from various hydrometallurgical streams. We expect this fundamental concept of combining redox-groups with chemical ligands to be expanded to broader ion-exchange processes in the future.Author contributions
H. V.: conceptualization, investigation, methodology, visualization, writing – original draft, writing – review and editing. J. E.: conceptualization, investigation, methodology, resources, visualization, writing – original draft, writing – review and editing. X. S.: conceptualization, project administration, supervision, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences Separation Science program, under Award Number DE-SC0021409. The Q-Tof Ultima mass spectrometer was purchased in part with a grant from the National Science Foundation, Division of Biological Infrastructure (DBI-0100085). SEM and XPS characterizations were carried out in the Frederick Seitz Materials Research Laboratory Central Research Facilities, University of Illinois. The authors thank Angelique Klimek and Jing Lian Ng for their assistance with the experiments, Jemin Jeon for NMR measurements, and Dr Kwiyong Kim for assistance in energy calculations. The authors also thank Dr Roddel Remy for help with polymer characterization.References
- K. Binnemans, P. T. Jones, B. Blanpain, T. Van Gerven, Y. Yang, A. Walton and M. Buchert, J. Clean. Prod., 2013, 51, 1–22 CrossRef CAS.
- A. Chakhmouradian and F. Wall, Elements, 2012, 8, 333–340 CrossRef CAS.
- B. S. Van Gosen, P. L. Verplanck, R. R. Seal Ii, K. R. Long and J. Gambogi, Critical Mineral Resources of the United States—Economic and Environmental Geology and Prospects for Future Supply: U.S. Geological Survey Professional Paper Report 1802, Reston, VA, 2017 Search PubMed.
- J. Gu, Z.-Q. Zhao, Y. Ding, H.-L. Chen, Y.-W. Zhang and C.-H. Yan, J. Am. Chem. Soc., 2013, 135, 8363–8371 CrossRef CAS PubMed.
- N. Davydov, R. Zairov, A. Mustafina, V. Syakayev, D. Tatarinov, V. Mironov, S. Eremin, A. Konovalov and M. Mustafin, Anal. Chim. Acta, 2013, 784, 65–71 CrossRef CAS PubMed.
- N. A. Shamsutdinova, A. T. Gubaidullin, B. M. Odintsov, R. J. Larsen, V. D. Schepkin, I. R. Nizameev, R. R. Amirov, R. R. Zairov, S. N. Sudakova, S. N. Podyachev, A. R. Mustafina and A. S. Stepanov, ChemistrySelect, 2016, 1, 1377–1383 CrossRef CAS.
- R. R. Zairov, N. A. Shamsutdinova, A. N. Fattakhova, A. V. Pyataev, A. F. Abdullin, A. V. Gerasimov, A. T. Gubaidullin and A. R. Mustafina, Russ. Chem. Bull., 2016, 65, 1325–1331 CrossRef CAS.
- N. A. Alarfaj and M. F. El-Tohamy, J. AOAC Int., 2019, 99, 380–385 CrossRef PubMed.
- Q. Ju, D. Tu, Y. Liu, R. Li, H. Zhu, J. Chen, Z. Chen, M. Huang and X. Chen, J. Am. Chem. Soc., 2012, 134, 1323–1330 CrossRef CAS PubMed.
- N. Venkatachalam, T. Yamano, E. Hemmer, H. Hyodo, H. Kishimoto and K. Soga, J. Am. Ceram. Soc., 2013, 96, 2759–2765 CrossRef CAS.
- E. G. Moore, J. Xu, C. J. Jocher, E. J. Werner and K. N. Raymond, J. Am. Chem. Soc., 2006, 128, 10648–10649 CrossRef CAS PubMed.
- H.-X. Mai, Y.-W. Zhang, R. Si, Z.-G. Yan, L.-d. Sun, L.-P. You and C.-H. Yan, J. Am. Chem. Soc., 2006, 128, 6426–6436 CrossRef CAS PubMed.
- B. S. Van Gosen, P. L. Verplanck, K. R. Long, J. Gambogi and R. R. Seal Ii, The Rare-Earth Elements: Vital to Modern Technologies and Lifestyles, Report 2014-3078, Reston, VA, 2014 Search PubMed.
- U. S. G. Survey, Mineral Commodity Summaries 2015, U.S. Geological Survey, Reston, VA, 2015 Search PubMed.
- USGS, Mineral Commodity Summaries 2020, U.S. Geological Survey, 2020 Search PubMed.
- U. S. G. Survey, Mineral Commodity Summaries 2021, U.S. Geological Survey, Reston, VA, 2021 Search PubMed.
- E. Commission, Report on Critical Raw Materials for the EU, Report of the Ad hoc Working Group on Defining Critical Raw Materials E. Commission, European Commission, 2014 Search PubMed.
- U. S. D. O. Energy, Critical Materials Strategy Summary, U.S. Department of Energy, 2010 Search PubMed.
- U. S. D. O. Energy, Critical Materials Strategy, U.S. Department of Energy, 2011 Search PubMed.
- Y. Zhu, Y. Zheng and A. Wang, Int. J. Biol. Macromol., 2015, 72, 410–420 CrossRef CAS PubMed.
- I. Anastopoulos, A. Bhatnagar and E. C. Lima, J. Mol. Liq., 2016, 221, 954–962 CrossRef CAS.
- J. Song, T. Huang, H. Qiu, X. Niu, X.-M. Li, Y. Xie and T. He, Desalination, 2018, 440, 18–38 CrossRef CAS.
- A. A. Galhoum, M. G. Mafhouz, S. T. Abdel-Rehem, N. A. Gomaa, A. A. Atia, T. Vincent and E. Guibal, Nanomaterials, 2015, 5, 154–179 CrossRef PubMed.
- F. Zhao, E. Repo, Y. Meng, X. Wang, D. Yin and M. Sillanpää, J. Colloid Interface Sci., 2016, 465, 215–224 CrossRef CAS PubMed.
- F. Wang, J. Zhao, X. Wei, F. Huo, W. Li, Q. Hu and H. Liu, J. Chem. Technol. Biotechnol., 2014, 89, 969–977 CrossRef CAS.
- M. Torab-Mostaedi, M. Asadollahzadeh, A. Hemmati and A. Khosravi, Res. Chem. Intermed., 2015, 41, 559–573 CrossRef CAS.
- J. Roosen, J. Spooren and K. Binnemans, J. Mater. Chem. A, 2014, 2, 19415–19426 RSC.
- J. Li, A. Gong, F. Li, L. Qiu, W. Zhang, G. Gao, Y. Liu and J. Li, RSC Adv., 2018, 8, 39149–39161 RSC.
- X. Su, A. Kushima, C. Halliday, J. Zhou, J. Li and T. A. Hatton, Nat. Commun., 2018, 9, 4701 CrossRef PubMed.
- K. Kim, S. Cotty, J. Elbert, R. Chen, C.-H. Hou and X. Su, Adv. Mater., 2020, 32, 1906877 CrossRef CAS PubMed.
- K. Kim, P. Baldaguez Medina, J. Elbert, E. Kayiwa, R. D. Cusick, Y. Men and X. Su, Adv. Funct. Mater., 2020, 30, 2004635 CrossRef CAS.
- R. Candeago, K. Kim, H. Vapnik, S. Cotty, M. Aubin, S. Berensmeier, A. Kushima and X. Su, ACS Appl. Mater. Interfaces, 2020, 12, 49713–49722 CrossRef CAS PubMed.
- X. Su and T. A. Hatton, Phys. Chem. Chem. Phys., 2017, 19, 23570–23584 RSC.
- P. D. Beer and P. A. Gale, Angew. Chem., Int. Ed., 2001, 40, 486–516 CrossRef CAS PubMed.
- X. Su, H. J. Kulik, T. F. Jamison and T. A. Hatton, Adv. Funct. Mater., 2016, 26, 3394–3404 CrossRef CAS.
- R. Chen, T. Sheehan, J. L. Ng, M. Brucks and X. Su, Environ. Sci.: Water Res. Technol., 2020, 6, 258–282 RSC.
- R. Chen, J. Feng, J. Jeon, T. Sheehan, C. Rüttiger, M. Gallei, D. Shukla and X. Su, Adv. Funct. Mater., 2021, 2009307 CrossRef CAS.
- M. Gallei and C. Rüttiger, Chem.–Eur. J., 2018, 24, 10006–10021 CrossRef CAS PubMed.
- R. Pietschnig, Chem. Soc. Rev., 2016, 45, 5216–5231 RSC.
- P. S. Borchers, M. Strumpf, C. Friebe, I. Nischang, M. D. Hager, J. Elbert and U. S. Schubert, Adv. Energy Mater., 2020, 10, 2001825 CrossRef CAS.
- M. H. George and G. F. Hayes, Polymer, 1974, 15, 397–400 CrossRef CAS.
- M. H. George and G. F. Hayes, J. Polym. Sci., Polym. Chem. Ed., 1976, 14, 475–488 CrossRef CAS.
- D. A. Baker, G. C. East and S. K. Mukhopadhyay, J. Appl. Polym. Sci., 2001, 79, 1092–1100 CrossRef CAS.
- H. Ni, H. Kawaguchi and T. Endo, Colloid Polym. Sci., 2007, 285, 819–826 CrossRef CAS.
- S. Azizian, J. Colloid Interface Sci., 2004, 276, 47–52 CrossRef CAS PubMed.
- I. Langmuir, J. Am. Chem. Soc., 1918, 40, 1361–1403 CrossRef CAS.
- S. J. Allen, G. McKay and J. F. Porter, J. Colloid Interface Sci., 2004, 280, 322–333 CrossRef CAS PubMed.
- H. Moon and W. Kook Lee, J. Colloid Interface Sci., 1983, 96, 162–171 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta03334d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |