
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic C–H to C–M (M = Al, Mg) bond transformations with heterometallic complexes
Maria
Batuecas†
 ,
Nikolaus
Gorgas†
and
Mark R.
Crimmin
*
,
Nikolaus
Gorgas†
and
Mark R.
Crimmin
*
Department of Chemistry, Molecular Sciences Research Hub, Imperial College London, 80 Wood Lane, Shepherds Bush, London, W12 0BZ, UK. E-mail: m.crimmin@imperial.ac.uk
First published on 17th August 2020
Abstract
C–H functionalisation is one of the cornerstones of modern catalysis and remains a topic of contemporary interest due its high efficiency and atom-economy. Among these reactions, C–H borylation, that is the transformation of C–H to C–B bonds, has experienced a fast development because of the wide utility of organoboron reagents as synthetic intermediates. The mechanistic background is now well-understood and the role of transition metal boryl or σ-borane intermediates in this transformation is well documented. This mini-review focuses on efforts made by our group, and others, to establish palladium- and calcium-catalysed methods for C–H metalation employing heavier main group elements (M = Al, Mg). These are new catalytic reactions first accomplished in our group that we have termed C–H alumination and magnesiation respectively. Unusual heterometallic complexes have been identified as key on-cycle intermediates and their unique reactivity is discussed in the context of new catalytic pathways for C–H functionalisation. Hence, this mini-review summarises the recent progress in the area of C–H metalation reactions as well as the new opportunities that may arise from this concept.
Introduction
The transformation of simple hydrocarbons into more complex and valuable products via catalytic C–H bond functionalisation has revolutionised modern synthetic chemistry. This type of reactivity has quickly become one of the cornerstones of modern catalysis as it provides a straightforward and highly efficient strategy to form C–C or C–X bonds from C–H bonds (X = heteroatom). Catalytic C–H functionalisation processes have found widespread applications ranging from upgrading simple hydrocarbons to sophisticated late-stage functionalisation of complex molecules.1 Despite the widespread development of C–H functionalisation reactions, further improvements in scope, catalyst activity, and catalyst selectivity are still desirable.2 The majority of catalytic methods requires high metal catalyst loading,3 which limits their industrial applications. Furthermore the presence of directing groups is typically used to achieve high site selectivity.4 To address these limitations, a number of new base-metal catalysts5–7 and new approaches to selectivity control, e.g. through non-covalent interactions, have recently emerged.8–10Particular progress has been achieved in the field of transition metal (TM) catalysed C–H borylation, that is the transformation of C–H to C–B bonds. Due to the high utility of organoboron reagents as synthetic intermediates, C–H borylation is now widely employed in the construction of natural products, active pharmaceutical ingredients, and building blocks for organic polymers.11 A number of transition-metal12,13 and main-group14–16 catalysts have been reported for C–H borylation of a range of substrates including heteroarenes, arenes, alkenes and alkanes. The mechanistic background of transition metal catalysed C–H bond borylation has been extensively studied over the past few decades and is well-understood. Thermodynamically, the formation of C–B bonds provides a driving force in these reactions.17 Kinetically, C–H borylation reactions proceed via low energy pathways in which transition metal boryl species often play an essential role. The intrinsic properties of boryl ligands facilitate the elementary C–H activation and C–B bond formation steps. For example, the strong σ-donor ability of the boryl ligand can stabilise high oxidation state transition metal complexes necessary for redox-based catalytic cycles.18 On the other hand, the accessible p-orbital of the boron atom of these ligands is decisive for C–H/B–H σ-bond metathesis mechanisms occurring at redox-neutral transition metal centres.19
Beyond boron, organometallic reagents based on main group metals such as aluminium or magnesium have been an important aspect of synthetic chemistry since its beginnings.20 By analogy to C–H borylation, gaining access to synthetically valuable compounds via the direct metalation of simple hydrocarbons constitutes an attractive goal (Scheme 1). Recent efforts have focused on using main group species as stoichiometric reagents capable of breaking strong C–H, C–C and C–X bonds.21–23 A particular exciting aspect of this work has been the exploration of bimetallic systems in which cooperative effects between two main group metals play a role in determining reactivity.24 Further advancements can be expected if one could modify the scope and selectivity of these reactions with the aid of a TM catalyst.25 In this mini-review, we will address this new approach and show how the combined action of two metals can lead to new reaction pathways thus providing new opportunities in terms of reactivity and selectivity. Since direct access to new organometallic reagents is now available their potential synthetic utility will be also considered.
 | ||
| Scheme 1 Outline of metal-catalysed C–H metalation reactions. | ||
Catalytic C–H metalation reactions
C–H alumination
Our interest in C–H metalation reactions arose from the discovery of a palladium bis(phosphine) complex as a catalyst for the dehydrocoupling of sp2 C–H and Al–H bonds.26 Reaction of fluoroarenes and heteroarenes with aluminium dihydride Al-1 in the presence of a catalytic amount of [Pd(PCy3)2] affords the corresponding metallated product in high yield. This new transformation was discovered as part of a mechanistic study on a related catalytic reaction involving the transformation of sp2 C–F to C–Al bonds of perfluoroarenes using [Pd(PCy3)2]. During this investigation we discovered that a two-step process was in operation, likely involving an initial hydrodefluorination of the fluoroarene, followed by C–H alumination of the resulting C–H bond. In accordance with this and with the thermochemistry calculated for the C–H alumination of pentafluorobenzene with Al-1, we found that this transformation was an extremely facile reaction and one that showed high selectivity (Scheme 2a and b).27 Thus when 2,3,5,6-tetrafluorobiaryls were used as substrates metalation at the single C–H position took place. Similarly, high selectivity was also observed for the C–H alumination of heteroarenes such as benzofuran, furans, or N-methyl indole resulting in the selective functionalisation in the 2-position of these heterocycles. When the 2-position was blocked, as in the case of 1,2-dimethylimidazole, functionalisation of the remote positions of the heterocycle by C–H alumination still took place.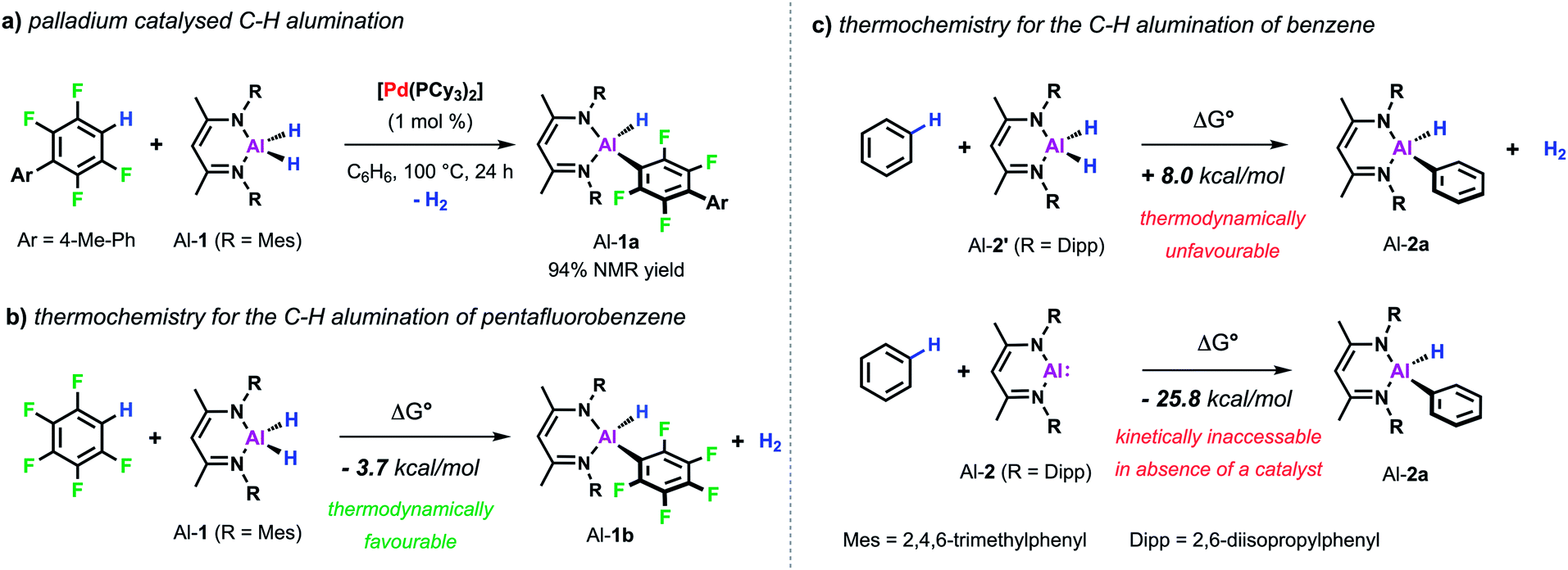 | ||
| Scheme 2 (a) Pd-catalysed C–H alumination of fluoroarenes. Calculated thermochemistry for C–H alumination of (b) pentafluorobenzene and (c) benzene, M06L-GD3 functional and hybrid (Al, SDDAll; 6-31G**, C, H, N) basis set. | ||
These first examples demonstrating the viability of C–H alumination under catalytic conditions stimulated efforts to expand this new type of reaction to less reactive unfunctionalised aromatic substrates. However, calculations on the thermochemistry of the reaction of the aluminium dihydride Al-2′ with benzene to afford the C–Al–H product Al-2a accompanied by the liberation of dihydrogen reveal an endergonic process. The recent developments in the chemistry of main-group compounds in low oxidation states has demonstrated these compounds show unusual reactivity due to the presence of accessible frontier molecular orbitals with a small HOMO–LUMO gap.28–31 Accordingly, oxidative addition to the parent low-valent aluminium(I) reagent Al-232 is calculated to be thermodynamically favourable (Scheme 2c). In the absence of a catalyst Al-2 does not react with a C–H bond of benzene.33 However, the group of Fischer reported that, when reacted with a Ni(0) precursor in C6H6, the related Al(I) reagent AlCp* readily inserts into the C–H bond of the solvent.34 Carbon–hydrogen bond functionalisation was proposed to take place via an initial oxidative addition to the transition metal centre followed by the migration of both the hydride and phenyl group to the appended aluminylene ligand. However, this reaction is not catalytic since the resulting H–Al–Ph moiety remains coordinated to the transition metal.
Similarly, the reactivity of Al-2 is significantly enhanced by the addition of [Pd(PCy3)2].35 The stoichiometric reaction of low-valent Al(I) complex Al-2 with the Pd(0) catalyst precursor in cyclohexane yields complex Pd/Al-1 in which two aluminylene ligands are bound to the transition metal centre (Scheme 3a). Formation of this complex could also be observed in benzene but it proved to be unstable as the reaction with the solvent resulted in the clean formation of the respective C–H alumination product as well as the regeneration of [Pd(PCy3)2]. The reaction could be run with [Pd(PCy3)2] as a catalyst and takes place at room temperature with loadings as low as 0.1 mol% (Scheme 3b). This is remarkable when compared with the rather harsh reaction conditions required for the C–H alumination of fluoroarenes and heteroarenes with the parent aluminium dihydride Al-1 reported before.
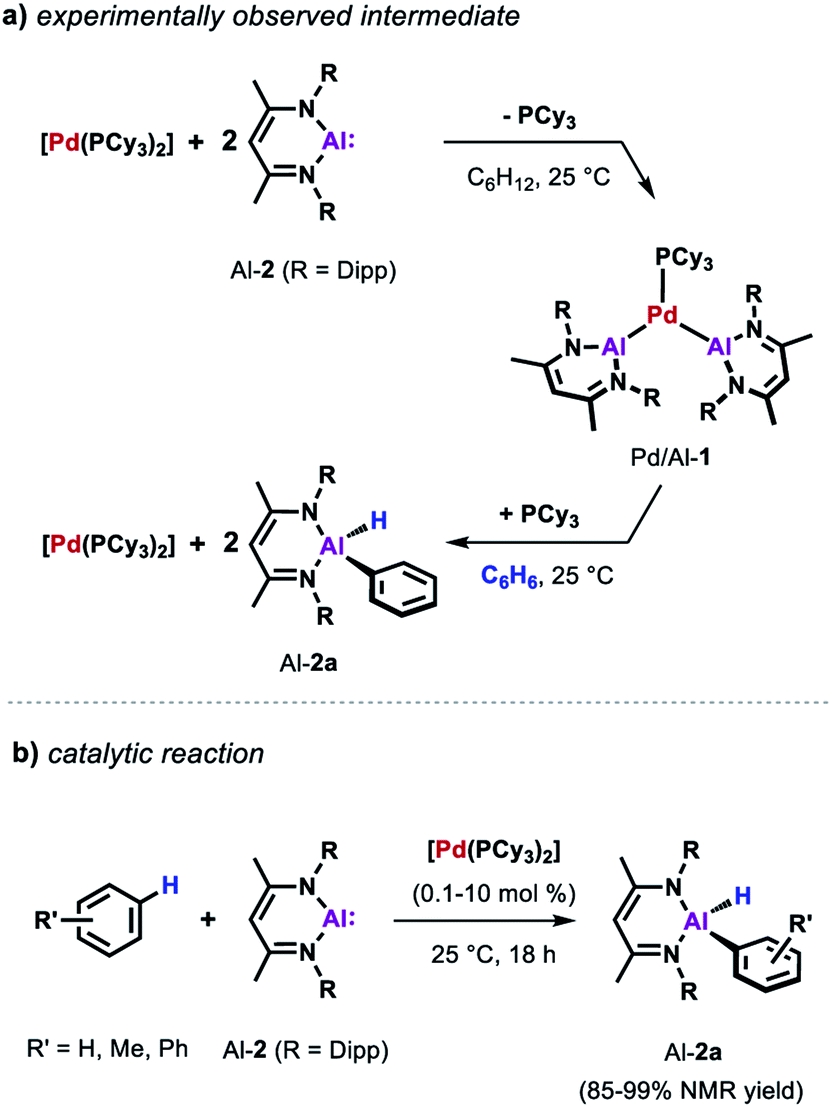 | ||
| Scheme 3 (a) Reaction of [Pd(PCy)3] with Al-2. (b) Catalytic alumination of benzene. | ||
Further insight into the mechanism of the palladium catalysed alumination of unfunctionalised arenes with Al-2 could be obtained by means of DFT calculations (Scheme 4a). The calculations were initiated from Pd/Al-2 formed from dissociation of the PCy3 ligand from Pd/Al-1. Pd/Al-2 is a two-coordinate 14-electron fragment with a bent geometry. Association of benzene with this fragment can occur through formation of a weak encounter complex with the ligand sphere. C–H bond cleavage occurs by a classical three-centred oxidative addition transition state (Pd/Al-3) affording the cis-Pd(II) intermediate Pd/Al-4. This is close in energy to its trans-isomer Pd/Al-5, from which the reaction proceeds via an unusual double migration step (Pd/Al-6) in which the Al–C and the Al–H bonds are formed in a concerted ligand transfer of both Ph and H from palladium to the aluminium centre to yield intermediate Pd/Al-7. This step proceeds with a two-electron transfer from the aluminium to the palladium centre (Pd(II)/Al(I) to Pd(0)/Al(III)). Exchange of the σ-bounded alane product by another aluminylene ligand finally closes the catalytic cycle. The calculated barriers as well as kinetic experiments suggest C–H bond cleavage at the Pd(0) centre is the turnover limiting step in the reaction.
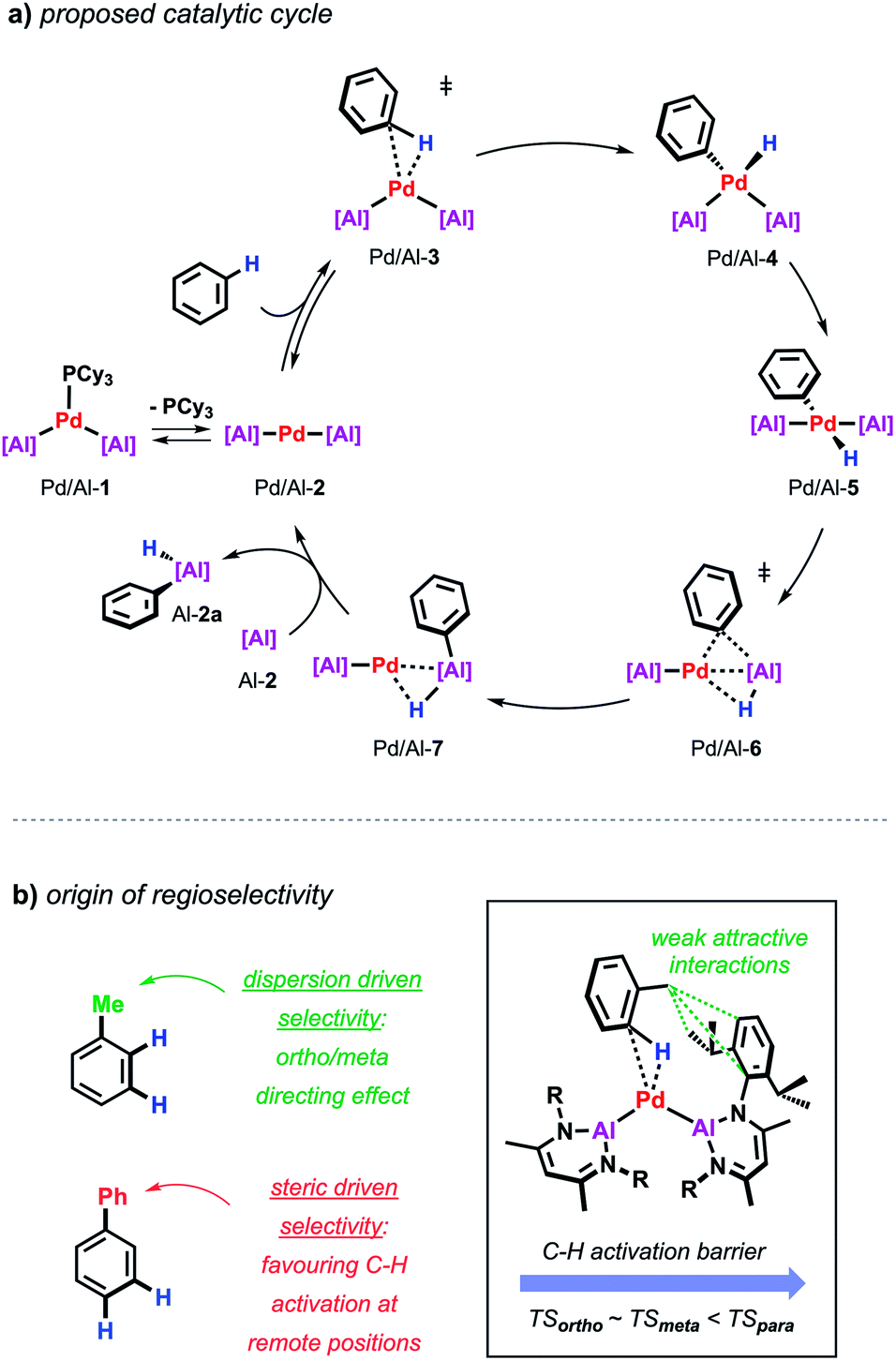 | ||
| Scheme 4 (a) Proposed catalytic cycle for the Pd-catalysed alumination of unfunctionalised arenes. (b) Origin of regioselectivity in Pd-catalysed alumination of arenes. | ||
During these studies an unusual mode of regiocontrol was observed for the Pd/Al system. The Pd-catalysed reaction of Al-2 with methyl-substituted arenes such as toluene or xylenes takes place primarily in the ortho- and meta-position which is complementary to traditional C–H borylation methodologies preferably affording the respective meta- and para-functionalised products.36–38 The origin of regioselectivity could be rationalised by the difference in energy of the transition states for the C–H bond activation step and can be traced back to non-covalent interactions between the methyl group of the arene and the aromatic substituents on the aluminylene ligands. In case of a phenyl substituent where attractive dispersive interactions are not expected, a switch of selectivity can be observed resulting in the activation of the remote meta/para-positions of the substrate probably controlled by steric effects as established for related C–H borylations (Scheme 4b).39
More recently, the group of Harder showed that also catalytic amounts of a molecular calcium hydride complex can promote the oxidative addition of the sp2 C–H bonds of unactivated arenes (benzene, toluene and xylene) to Al-2 at room temperature (Scheme 5).33 In contrast to palladium catalysed reaction, C–H bond cleavage is thought to be facilitated by the combined action of a nucleophilic Al centre and arene activation by π-coordination to a Lewis acidic Ca centre. Similar to anionic alumanyl complexes that are capable of breaking strong C–H bond of benzene,23,40,41 an unusual low positive charge on Al could be identified through calculations on the relevant Al–H–Ca species Ca/Al-1. Remarkably, the Ca-catalysed alumination of toluene occurs almost exclusively at the meta-position of the substrate, consistent with a mechanism involving nucleophilic attack of the aluminium reagent on an arene with an electron-donating substituent.42,43
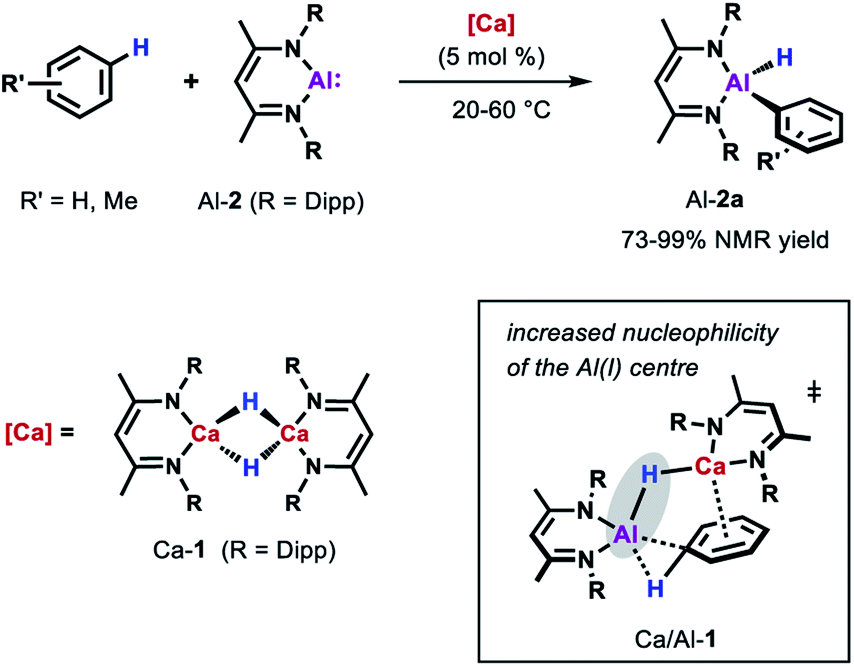 | ||
| Scheme 5 Ca-catalysed alumination of arenes. | ||
Recent studies have revealed that the C–H activation step also plays an important role in reactions that break strong C–O and C–F bonds. In the presence of palladium catalyst, C–O and C–F alumination reactions with Al-2 can also occur via a C–H activation process. This step has an impact on the scope and selectivity of the transformations as C–H bonds act as a transient directing group, being broken and remade along the reaction pathway (Scheme 6).44,45
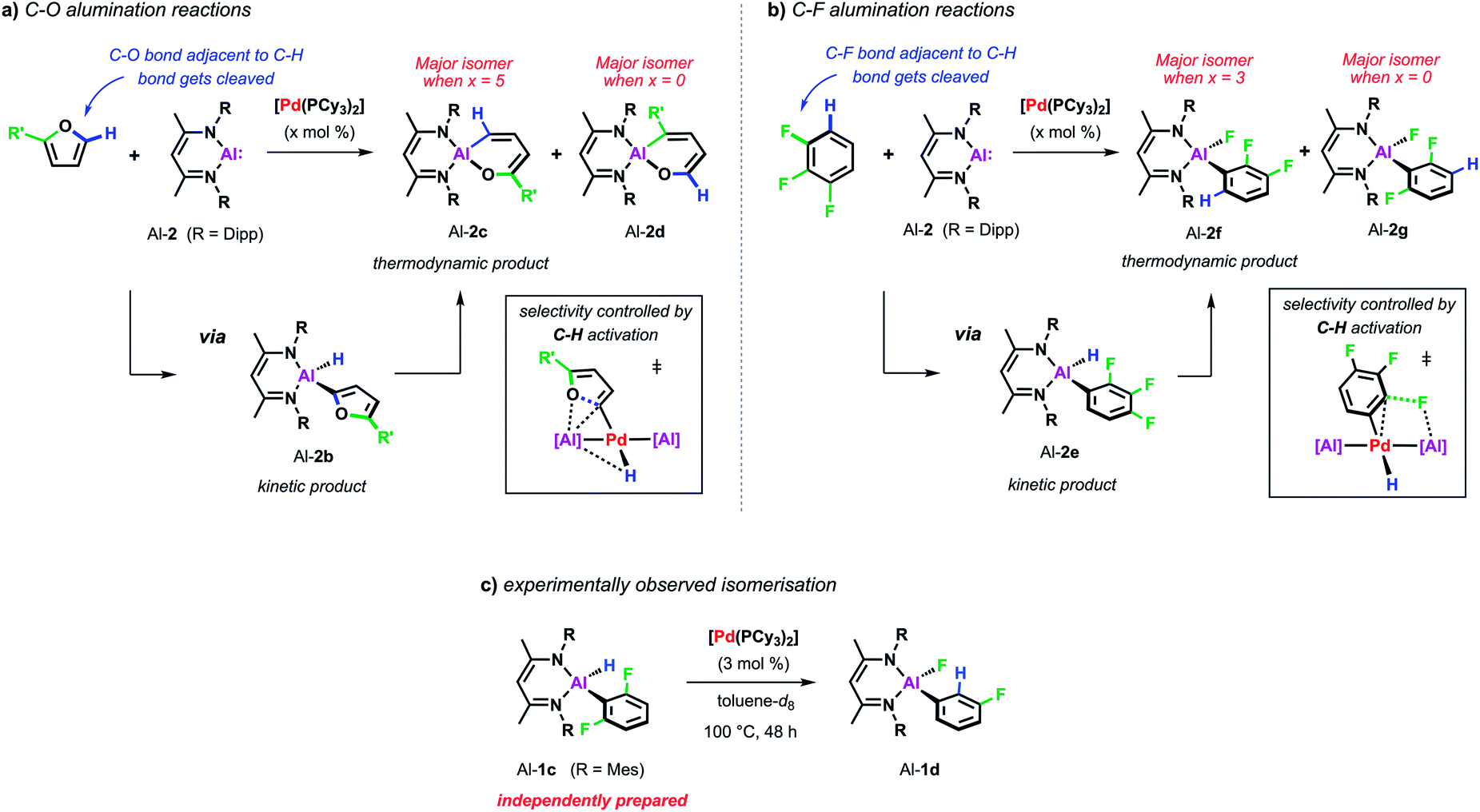 | ||
| Scheme 6 (a) Pd-catalysed C–O alumination of furans. (b) Pd-catalysed C–F alumination of fluoroarenes. (c) Pd-catalysed isomerisation from Al-1c to Al-1d. | ||
For example, the non-catalysed reaction of Al-2 with 2-methylfuran at 80 °C affords two isomers (Al-2c and Al-2d) of the ring-expanded metallacycle (Scheme 6a). The major species (Al-2d) results from insertion into the more hindered sp2 C–O bond. DFT calculations suggest a mechanism proceeding via an initial (4 + 1) cycloaddition of Al-2 and the furan followed by the rearrangement of the resulting bicyclic intermediate. Selectivity originates from the latter step and can be traced back to the electronic influence of the substituent weakening the adjacent sp2 C–O bond. Higher and complementary selectivity is obtained in the presence of catalytic [Pd(PCy3)2]. At room temperature, C–H alumination at the 2-position of the substrate affords a kinetic product (Al-2b) which, under more forcing conditions, converts into the thermodynamic ring-expanded aluminium cycle Al-2c (Scheme 6a). In contrast to the non-catalysed reaction, C–O bond alumination occurs exclusively at the reaction site adjacent to the intermediately activated C–H position. Mechanistic studies revealed that not only the C–H alumination step but also the rearrangement of the kinetic into the thermodynamic product is promoted by the palladium catalyst. DFT calculations suggested that the C–H aluminated intermediate can be re-activated at palladium following an alternative high energy pathway that leads to the thermodynamic product. As the key step in this mechanism, C–O bond cleavage involves the attack of an aluminium based metalloligand on the 2-palladated heterocycle. The C–H bond guides the catalyst to the adjacent site by being broken and remade along the reaction pathway, this mechanism explains the unusually high selectivity of the palladium catalysed reaction. The catalytic approach allows expansion of the substrate scope beyond furans to 2,3-dihydrofuran and 3,4-dihydropyran which do not react cleanly with Al-2 in absence of [Pd(PCy3)2].
A role for reversible breaking of a C–H bond could also be identified for the C–F alumination reaction of fluoroarenes with Al-2 (Scheme 6b). In absence of [Pd(PCy3)2], the aluminium(I) reagent does not show any appreciable reactivity towards low-fluorine-content substrates such as 1,2,3-trifluorobenzene at room temperature.46 At higher temperatures, the non-catalysed reaction of this substrate affords the oxidative addition product in moderate yield and as a mixture of two regioisomers (Al-2f and Al-2g). The catalytic approach instead allows the C–F alumination of mono-, di- or trifluorobenzenes to take place under exceptionally mild conditions and with high selectivity. For example, while the major product of the non-catalysed reaction of 1,2,3-trifluorobenzene results mainly from reaction of the central C–F bond (Al-2g), the complementary regioisomer (Al-2f), in which the aluminium fragment is installed next to an existing C–H bond, is preferably formed under catalytic conditions. Mechanistic studies in combination with DFT calculations suggest a stepwise C–H to C–F functionalisation process which leads the catalyst to a C–F bond adjacent to a reactive C–H site and thus accounts for the observed regioselectivity. Experimental support for C–H functionalisation playing a role in catalysis was obtained by the identification of a 100% atom efficient palladium catalysed isomerisation of the kinetic C–H alumination product Al-1c to the thermodynamic C–F alumination product Al-1d (Scheme 6c).
Despite the promising progress in the catalytic C–H metalation using low-valent aluminium reagent Al-2 the implementation of the respective hydride species (Al-1) is still desirable due to their higher stability and more convenient and efficient preparation. Although being endergonic based on DFT calculations (Scheme 2c) catalytic C–H alumination of benzene with Al-1 could be realised experimentally by conducting the reaction under static vacuum as removal of the liberated H2 permits catalytic turnover (Scheme 7a).35 The same approach has been successfully applied on the C–O and C–F alumination of furans and fluoroarenes respectively.44,45
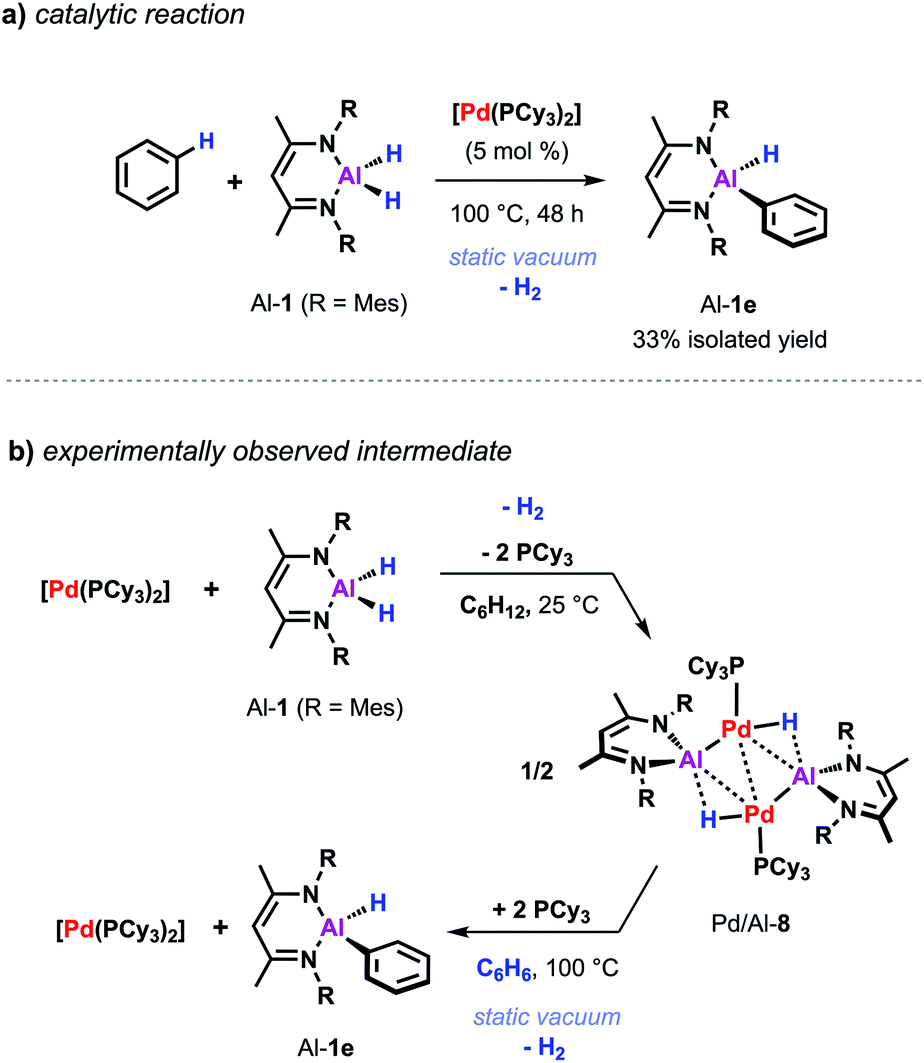 | ||
| Scheme 7 (a) Pd-catalysed C–H alumination of benzene with Al-1. (b) Stoichiometric reactivity of [Pd(PCy3)2] with Al-1. | ||
While a consistent mechanistic picture could be established for the alumination of sp2 C–H bonds with Al-2, it remains unclear if and how palladium aluminylene intermediates are formed when using Al-1 as a stoichiometric reagent.47 For example, one might imagine an elementary step that takes place in a similar but reverse fashion to the aforementioned double migration step from Pd/Al-5 (Scheme 8a). Experimental evidence for such a concerted double H–M–H bond activation at a transition metal centre has recently been provided by Aldridge and co-workers demonstrating that the analogous gallium dihydride is prone to react with a Rh(I) precursor to form the respective Rh(III) dihydride Rh/Ga-3 featuring a neutral Ga(I) gallylene ligand (Scheme 8b).48 An alternative metal-mediated dehydrogenation of gallium dihydride was reported by the same group and involves the oxidative addition of Ga–H to metal carbonyl complexes followed by subsequent loss of H2 under photochemical conditions.49
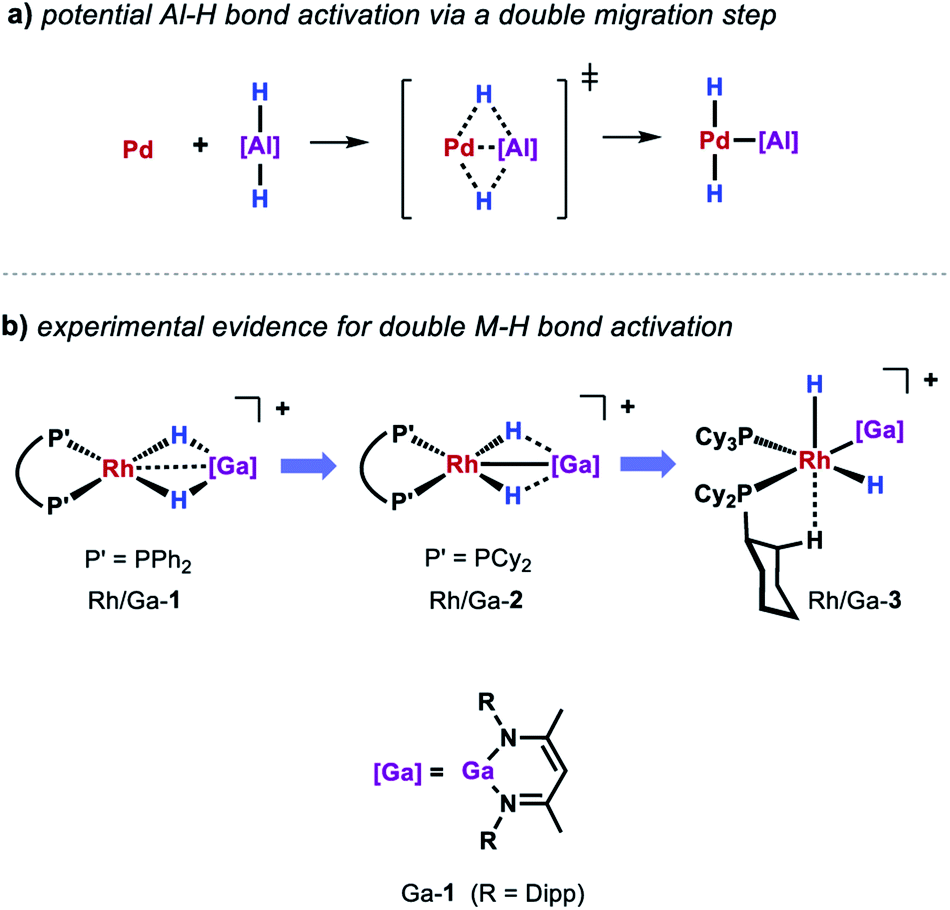 | ||
| Scheme 8 (a) Potential pathway for the activation of Al-1 at Pd(0). (b) Experimental evidence for a double migration step through structural snapshots of the progressing Ga–H bond activation at Rh(I). | ||
The equivalent process in the Pd/Al system is yet to be observed. Nevertheless, stoichiometric experiments unveiled Al–H bond activation and partial dehydrogenation of the aluminium dihydride when reacted with diverse palladium precursor complexes.47 For example, room temperature reaction of the aluminium dihydride Al-1 with [Pd(PCy3)2] led to the isolation of the tetrametallic complex Pd/Al-8 (Scheme 7b).35 The position and number of the hydride ligands in Pd/Al-8 were confirmed by single crystal neutron diffraction studies of an analogue complex featuring 2,6-xylyl substituents on the metalloligand.47 Formation of this complex involves partial dehydrogenation of Al-1 and proceeds with liberation of H2. Heating Pd/Al-8 in benzene under static vacuum results in the regeneration of [Pd(PCy3)2] accompanied by C–H alumination of the solvent. Nevertheless, it remains unclear at the moment if Pd/Al-8 constitutes an on-cycle intermediate in dehydrogenative alumination reactions.
C–H magnesiation
The same palladium diphosphine complex also proved effective in the catalytic reaction of a low-valent magnesium complex with C–H bonds.50 The reaction of Mg-1 with benzene in presence of catalytic [Pd(PCy3)2] results in the formation of the kinetically stabilised organomagnesium hydride Mg-1a (Scheme 9a). In the solid state, this species exists as a dimer in which two equivalent magnesium sites are bridged by 3-centre, 2-electron Mg–H–Mg and Mg–C–Mg bonds. The reaction takes place at room temperature whereas, even at high temperatures, no reaction is observed in the absence of a catalyst. While Mg-1 and related species are emerging as versatile reagents in synthesis,22,51 this is the first example of their use in the activation of inert carbon–hydrogen bonds. Preliminary kinetics experiments suggest that breaking of the C–H bond is not likely to be the turnover limiting step. The value of KIE obtained of 1.1 contrasts that reported for the C–H alumination of benzene with low-valent aluminium complexes described above which showed a large primary KIE of 5.8 ± 0.1.35 These findings indicate that magnesiation of benzene may be mechanistically distinct from a related reaction involving the low-valent aluminium reagent Al-2.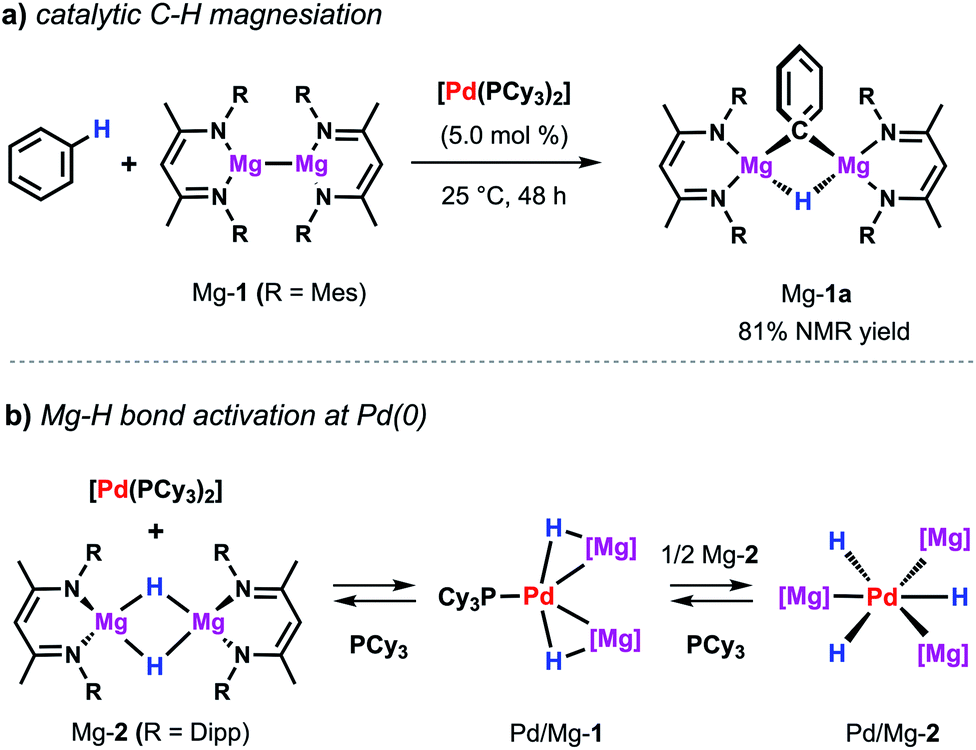 | ||
| Scheme 9 (a) Pd-catalysed magnesiation of benzene. (b) Stoichiometric reactivity of [Pd(PCy3)2] with Mg-2. | ||
Dehydrogenative magnesiation of benzene using the parent dimeric magnesium hydride complex (Mg-2) did not lead to the clean formation of Mg-1a. Nevertheless, stoichiometric reactions of Mg-2 with [Pd(PCy3)2] revealed the reversible cleavage of the Mg–H bond at the transition metal centre (Scheme 9b).52 Stepwise substitution of the phosphine ligands could be observed in solution leading to an equilibrium between the Pd(0) precursor as well as the two new bimetallic hydride complexes Pd/Mg-1 and Pd/Mg-2 mapping the progressing Mg–H bond activation process. Near complete cleavage of the magnesium hydride bonds occurs in Pd/Mg-2 leading to the formation of pairs of σ-donor (H−) and σ-acceptor (Mg+) ligands at a Pd(0) centre adopting an unusual hexagonal planar coordination geometry. The heterometallic hydride complexes undergo H/D-exchange in benzene-d6 demonstrating that these species are essentially competent for C–H activation reactions.
Perspective
In this mini-review, we summarised recent efforts to extend the concept of established C–H borylation methodologies to C–H metalation reactions that allow the direct functionalisation of aromatic sp2 C–H bonds into C–Al and C–Mg bonds in a straightforward and atom efficient way. These new reactions currently rely on the implementation of a palladium catalyst. Mechanistic studies disclosed new reaction pathways in which heterometallic intermediates play a central role. We believe that these first examples are just the beginning of a new chapter in C–H activation chemistry.Future efforts will undeniably focus on the diversification of the new catalytic transformations. These may comprise an expansion of the substrate scope from aromatic and heteroaromatic compounds to other hydrocarbons including the functionalisation of sp3 C–H bonds. Moreover, mechanistic understanding as well as the knowledge about the factors that determine reactivity and selectivity can be exploited on a general basis and used for rational design of novel catalytic systems. As exemplified by the calcium hydride mediated C–H alumination discovered by the group of Harder, the use of precious metals such as palladium is not a prerequisite to overcome the kinetic barriers in these reactions. This approach could lead to new developments in the growing field of base metal catalysis.
In order to exploit the synthetic potential of these organometallated reagents, the development of efficient derivatisation methods has to be considered. Transition metal catalysed cross-coupling reactions are likely to play a central role in this respect. Organozinc and -magnesium compounds are widely used reagents in Negishi- and Kumada-type C–C cross-coupling protocols. As these reactions occur in presence of the same palladium pre-catalyst, one might envision two step processes that combine the C–H activation and C–C bond formation steps in a one-pot reaction. Especially in view of widely employed Negishi cross-coupling reactions, an expansion of the current alumination and magnesiation reactions to the direct formation of C–Zn bonds might largely increase the applicability of these new C–H metalation methodologies. On the other hand, the direct access to C–Al derivatives provides an entirely new perspective in this field and may potentially lead to the development of a new class of transition metal catalysed coupling reactions.
In the light of ongoing attempts tending to improve the scope, efficiency and selectivity of C–H borylation catalysts we described how some of these challenges may be addressed by translating the established concept into new methodologies. The catalytic C–H metalation processes presented herein clearly differ from conventional borylation reactions. The new reaction pathways give rise to complementary reactivity and selectivity as illustrated by the distinct site selectivity for toluene and xylenes or the ring opening of heterocycles proceeding with predictable selectivity. Finally, the more pronounced polarity of C–M bonds in the resulting organometallic reagents will inevitably provide new opportunities to organic synthesis reaching beyond the widespread application of established boronate esters.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to all current and former group members who contributed to the work highlighted in this article. We are grateful for the ERC (StG: Fluorofix 677367) and EPSRC (EP/S036628/1) for funding. NG is grateful to the Austrian Science Fund (FWF) for an Erwin Schrödinger Postdoctoral Fellowship (Project No. J-4399). Martí Garçon is thanked for DFT calculations for the thermodynamic presented in Scheme 2.References
- J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369–375 CrossRef CAS PubMed.
- F. Roudesly, J. Oble and G. Poli, J. Mol. Catal. A: Chem., 2017, 426, 275–296 CrossRef CAS.
- T. Gensch, M. J. James, T. Dalton and F. Glorius, Angew. Chem., Int. Ed., 2018, 57, 2296–2306 CrossRef CAS.
- C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603–6743 RSC.
- B. Su, Z. C. Cao and Z. J. Shi, Acc. Chem. Res., 2015, 48, 886–896 CrossRef CAS PubMed.
- S. St John-Campbell and J. A. Bull, Adv. Synth. Catal., 2019, 361, 3662–3682 CrossRef CAS.
- P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS PubMed.
- M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. Van Leeuwen, Chem. Soc. Rev., 2014, 43, 1660–1733 RSC.
- N. R. Mote and S. H. Chikkali, Chem.–Asian J., 2018, 13, 3623–3646 CrossRef CAS PubMed.
- K. T. Mahmudov, A. V. Gurbanov, F. I. Guseinov and M. F. C. Guedes da Silva, Coord. Chem. Rev., 2019, 387, 32–46 CrossRef CAS.
- D. G. Hall, Boronic acids: preparation and applications in organic synthesis, medicine and materials, Wiley-VCH, 2011 Search PubMed.
- I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed.
- L. Xu, G. Wang, S. Zhang, H. Wang, L. Wang, L. Liu, J. Jiao and P. Li, Tetrahedron, 2017, 73, 7123–7157 CrossRef CAS.
- M.-A. Legare, M.-A. Courtemanche, E. Rochette and F.-G. Fontaine, Science, 2015, 349, 513–516 CrossRef CAS PubMed.
- Y.-L. Liu, G. Kehr, C. G. Daniliuc and G. Erker, Chem.–Eur. J., 2017, 23, 12141–12144 CrossRef CAS PubMed.
- J. S. McGough, J. Cid and M. J. Ingleson, Chem.–Eur. J., 2017, 23, 8180–8184 CrossRef CAS PubMed.
- P. R. Rablen and J. F. Hartwig, J. Am. Chem. Soc., 1996, 118, 4648–4653 CrossRef CAS.
- H. Tamura, H. Yamazaki, H. Sato and S. Sakaki, J. Am. Chem. Soc., 2003, 125, 16114–16126 CrossRef CAS PubMed.
- C. E. Webster, Y. Fan, M. B. Hall, D. Kunz and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 858–859 CrossRef CAS PubMed.
- Main Group Metals in Organic Synthesis, ed. H. Yamamoto and K. Oshima, Wiley, 2004 Search PubMed.
- T. Chu, I. Korobkov and G. I. Nikonov, J. Am. Chem. Soc., 2014, 136, 9195–9202 CrossRef CAS PubMed.
- C. Jones, Nat. Rev. Chem., 2017, 1, 59 CrossRef CAS.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Nature, 2018, 557, 92–95 CrossRef CAS PubMed.
- S. D. Robertson, M. Uzelac and R. E. Mulvey, Chem. Rev., 2019, 119, 8332–8405 CrossRef CAS PubMed.
- M. J. Butler and M. R. Crimmin, Chem. Commun., 2017, 53, 1348–1365 RSC.
- W. Chen, T. N. Hooper, J. Ng, A. J. P. White and M. R. Crimmin, Angew. Chem., Int. Ed., 2017, 56, 12687–12691 CrossRef CAS PubMed.
- S. D. Pike, M. R. Crimmin and A. B. Chaplin, Chem. Commun., 2017, 53, 3615–3633 RSC.
- T. Chu and G. I. Nikonov, Chem. Rev., 2018, 118, 3608–3680 CrossRef CAS PubMed.
- K. Hobson, C. J. Carmalt and C. Bakewell, Chem. Sci., 2020, 11, 6942–6956 RSC.
- M. Zhong, S. Sinhababu and H. W. Roesky, Dalton Trans., 2020, 49, 1351–1364 RSC.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202007530.
- C. Cui, H. W. Roesky, H.-G. Schmidt, M. Noltemeyer, H. Hao and F. Cimpoesu, Angew. Chem., Int. Ed., 2000, 39, 4274–4276 CrossRef CAS PubMed.
- S. Brand, H. Elsen, J. Langer, S. Grams and S. Harder, Angew. Chem., Int. Ed., 2019, 58, 15496–15503 CrossRef CAS PubMed.
- T. Steinke, C. Gemel, M. Cokoja, M. Winter and R. A. Fischer, Angew. Chem., Int. Ed., 2004, 43, 2299–2302 CrossRef CAS PubMed.
- T. N. Hooper, M. Garçon, A. J. P. White and M. R. Crimmin, Chem. Sci., 2018, 9, 5435–5440 RSC.
- J.-Y. Cho, C. N. Iverson and M. R. Smith, J. Am. Chem. Soc., 2000, 122, 12868–12869 CrossRef CAS.
- J.-Y. Cho, M. K. Tse, D. Holmes, R. E. Maleczka Jr and M. R. Smith, Science, 2002, 295, 305–308 CrossRef CAS PubMed.
- M. K. Tse, J.-Y. Cho and M. R. Smith, Org. Lett., 2001, 3, 2831–2833 CrossRef CAS PubMed.
- J. F. Hartwig, Chem. Soc. Rev., 2011, 40, 1992–2002 RSC.
- S. Kurumada, S. Takamori and M. Yamashita, Nat. Chem., 2020, 12, 36–39 CrossRef CAS PubMed.
- S. Grams, J. Eyselein, J. Langer, C. Färber and S. Harder, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202006693.
- J. Hicks, P. Vasko, A. Heilmann, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202008557.
- S. Kurumada, K. Sugita, R. Nakano and M. Yamashita, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202009138.
- F. Rekhroukh, W. Chen, R. Brown, A. J. P. White and M. Crimmin, Chem. Sci., 2020, 11, 7842–7849 RSC.
- T. N. hooper, R. Brown, F. Rekhroukh, M. Garçon, A. J. P. White, P. J. Costa and M. Crimmin, Chem. Sci., 2020, 11, 7850–7857 RSC.
- O. Kysliak, H. Görls and R. Kretschmer, Chem. Commun., 2020, 56, 7865–7868 RSC.
- T. N. Hooper, S. Lau, W. Chen, R. K. Brown, M. Garçon, K. Luong, N. S. Barrow, A. S. Tatton, G. A. Sackman, C. Richardson, A. J. P. White, R. I. Cooper, A. J. Edwards, I. J. Casely and M. R. Crimmin, Chem. Sci., 2019, 10, 8083–8093 RSC.
- J. A. B. Abdalla, A. Caise, C. P. Sindlinger, R. Tirfoin, A. L. Thompson, A. J. Edwards and S. Aldridge, Nat. Chem., 2017, 9, 1256–1262 CrossRef CAS PubMed.
- J. Turner, J. A. B. Abdalla, J. I. Bates, R. Tirfoin, M. J. Kelly, N. Phillips and S. Aldridge, Chem. Sci., 2013, 4, 4245–4250 RSC.
- M. Garçon, A. J. P. White and M. R. Crimmin, Chem. Commun., 2018, 54, 12326–12328 RSC.
- A. Stasch and C. Jones, Dalton Trans., 2011, 40, 5659–5672 RSC.
- M. Garçon, C. Bakewell, G. A. Sackman, A. J. P. White, R. I. Cooper, A. J. Edwards and M. R. Crimmin, Nature, 2019, 574, 390–393 CrossRef PubMed.
Footnote |
| † These authors contributed equally and are listed in alphabetical order. |
| This journal is © The Royal Society of Chemistry 2021 |