
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMain group bimetallic partnerships for cooperative catalysis†
Jose M.
Gil-Negrete
and
Eva
Hevia
 *
*
Department für Chemie und Biochemie, Universität Bern, CH3012, Bern, Switzerland. E-mail: eva.hevia@dcb.unibe.ch
First published on 16th October 2020
Abstract
Over the past decade s-block metal catalysis has undergone a transformation from being an esoteric curiosity to a well-established and consolidated field towards sustainable synthesis. Earth-abundant metals such as Ca, Mg, and Al have shown eye-opening catalytic performances in key catalytic processes such as hydrosilylation, hydroamination or alkene polymerization. In parallel to these studies, s-block mixed-metal reagents have also been attracting widespread interest from scientists. These bimetallic reagents effect many cornerstone organic transformations, often providing enhanced reactivities and better chemo- and regioselectivities than conventional monometallic reagents. Despite a significant number of synthetic advances to date, most efforts have focused primarily on stoichiometric transformations. Merging these two exciting areas of research, this Perspective Article provides an overview on the emerging concept of s/p-block cooperative catalysis. Showcasing recent contributions from several research groups across the world, the untapped potential that these systems can offer in catalytic transformations is discussed with special emphasis placed on how synergistic effects can operate and the special roles played by each metal in these transformations. Advancing the understanding of the ground rules of s-block cooperative catalysis, the application of these bimetalic systems in a critical selection of catalytic transformations encompassing hydroamination, cyclisation, hydroboration to C–C bond forming processes are presented as well as their uses in important polymerization reactions.
Introduction
s-Block organometallic reagents, epitomised by organolithium and Grignard reagents, are amongst the most widely used polar organometallic reagents in synthesis worldwide, not just in academia but also in many industrial commercial situations.1 While s-block organometallics have traditionally served stoichiometric chemistry,2 seminal work from several research groups across the world over the past two decades have uncovered the vast potential of these systems in homogeneous catalysis.3 Thus, despite their lack of redox capabilities, s-block metal complexes can efficiently catalyse a myriad of transformations, for example covering hydroamination, hydrosilylation and polymerisation to alkene hydrogenation reactions,4 that for decades have been considered the exclusive preserve of transition metal catalysis. Driven by the ever more pressing challenge of developing more sustainable chemistry as well as by academic interest, s-block metal catalysis is now a consolidated and well-established field of study which continues to grow.3 It should be noted that for many of the transformations outlined above, it has been found that heavier alkaline-earth metals such as Ca, Ba and Sr offer significantly better catalytic abilities than their Mg congener,5 which has been generally attributed to the greater polarity of the Ca–X versus Mg–X bonds (X = N, O, P, C, etc.) as well as to the greater polarizability of Ca2+versus Mg2+. Computational and structural studies have been key to understand and rationalise reactivities as well as to be able to tune catalytic ability of s-block metal complexes.6In parallel to these catalytic studies, another evolving area of s-block organometallic chemistry has been the synthesis and reactivity of mixed-metal reagents for chemical cooperativity.7 Thus, the use of heterobimetallic reagents combining a group 1 metal with a lower electropostive s/p-block metal partner such as Mg, Zn or Al has uncovered unique reactivity profiles that cannot be replicated by their single metal components. Hence, at least qualitatively, in many cases these heterobimetallic systems combine the high reactivity typically associated with group 1 organometallics with the greater regioselective control and functional group tolerance of low polarity reagents. Unsurprisingly, these bimetallic reagents have already made an impact in organic synthesis, finding widespread applications in core transformations such as deprotonative metallation, metal halogen exchange or C–C bond forming processes.8 Some key advances in this area include kinetically activated turbo Grignard reagents, iPrMgCl·LiCl, which were first reported in 2004 by Knochel9 and subsequently evolving to a wide new class of reagents in organic synthesis, being commercially available and generally offering enhanced reactivities and selectivities that are out of reach of conventional Grignard reagents.10 Through the isolation and characterisation of informative organometallic intermediates from deprotonative metallation reactions of arenes using heterobimetallic bases, Mulvey has introduced the concept of alkali-metal-mediated metallation (AMMM).7 This approach facilitates direct zincation or magnesiation of a wide range of substrates, where the cooperative effects between the metals enables, in some cases, to override directed-ortho-metallation (DoM) regioselectivities.11
Building on these stoichiometric successes, more recently, certain of these unique reactivities have been applied to catalytic regimes, providing some of the first examples of main group cooperative catalysis. This Perspective Article provides an overview on highlights of these recent advances, showcasing how cooperative effects between metals can be tailored for the design of more selective and more efficient catalysts.
Accessing s-block heterobimetallic complexes
The two main synthetic strategies most commonly used to access s-block bimetallics are salt-metathesis and interlocking co-complexation. The former reactions usually require an excess of the polar organometallic reagent which is reacted with the halide salt of the lower polarity metal in an ethereal solvent like THF or Et2O. This approach is frequently employed to prepare bimetallic systems in situ, as shown for tetraorganozincates Li2ZnR4 (1) (R = tBu, Et, Ph), which are obtained by reacting 4 equivalents of the relevant RLi reagent with ZnCl2 in THF (Scheme 1a). These mixed Li/Zn reagents have shown excellent regioselectivities and functional group tolerance for direct Zn–halogen exchange of haloarenes12 and nucleophilic alkylation/arylation of sensitive nitroolefins.13 Interestingly, in certain cases the inorganic salts which are concomitantly generated in these reactions (e.g., LiCl) can be incorporated in the structure of the mixed-metal reagent as observed in the reaction of LiTMP with MgCl2 which led to the isolation of the turbo Hauser base [(THF)4LiCl2Mg(TMP)(THF)] (2) (TMP = 2,2,6,6-tetramethylpiperidide) (Scheme 1b).14 Base 2 can be envisaged as a co-complex between the metathesis product TMPMgCl and the inorganic salt LiCl and exhibits enhanced magnesiating powers towards aromatic and heteroaromatic substrates, which are generally inert towards TMPMgCl.8a,b,15,16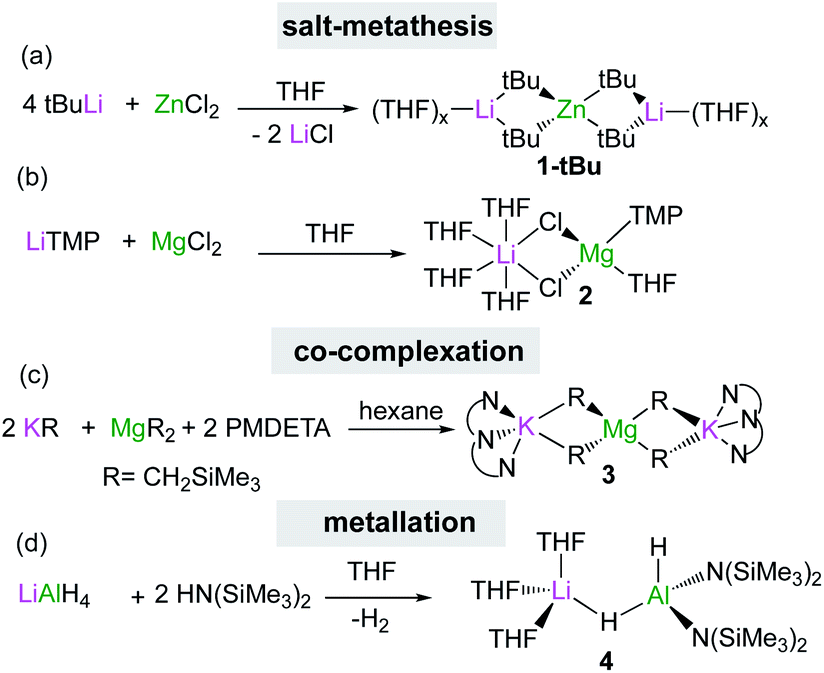 | ||
| Scheme 1 Representative examples of the main synthetic approaches to access s-block heterobimetallic complexes. | ||
An alternative strategy to salt-metathesis is the use of co-complexation by combining the monometallic components in the desired stoichiometry as exemplified in Scheme 1c for the formation of potassium magnesiate [(PMDETA)2K2MgR4] (3) (R = CH2SiMe3, PMDETA = N,N,N′,N′′,N′′-pentamethyldiethylenetriamine).17 These reactions are compatible with non-ethereal solvents, although the use of Lewis donors such as PMDETA can facilitate the formation of more reactive monomeric structures.18,19 DFT calculations on this type of co-complexation have revealed that in many cases the formation of the relevant heterobimetallic complexes is thermodynamically driven.18,19
Homoleptic bimetallics can also be used as precursors for more complex heteroleptic species. For example, commercially available LiAlH4 reacts in THF with two equivalents of the hexamethyldisilazane [HMDS = N(SiMe3)2] to form [(THF)3LiAlH2(HMDS)2] (4) with subsequent elimination of hydrogen (Scheme 1d). Lithium aluminate 4 can effectively catalyse hydroboration of aldehydes and ketones (vide infra).20
Maximising cooperative effects
Through the isolation and structural elucidation of key reaction intermediates in stoichiometric transformations it has been deduced that in several cases the close proximity of the two metals in these heterobimetallic systems is crucial in order to maximise cooperative effects. Bimetallic cooperativity involving alkali metals can have different subject transforming outcomes. For example, Goicoechea and Aldridge pioneered the anionic aluminium(I) nucleophile in [K{Al(NON)}]2 (5) [NON = 4,5-bis(2,6-diisopropylanilido)-2,7-di-tert-butyl-9,9-dimethylxan-thene], where potassium plays a key stabilising role through K⋯π interactions (Fig. 1).21 This is remarkable since organoaluminium complexes are usually electrophiles not nucleophiles, however 5 is able to act as a nucleophile towards C–X and M–X bonds and it reacts with benzene via formal C–H addition to give a phenyl hydride potassium aluminate with aluminium in the oxidation state 3+.21 Interestingly, hinting at the key role played by potassium in these reactions, when 5 is reacted with benzene in the presence of [2.2.2]-cryptand, which can sequester the potassium cation, selective oxidative C–C bond activation of benzene is observed instead.22 Related to this work Coles has shown that the products of the activation of 1,3,5,7-cyclooctatetraene (COT) by potassium aluminyl K[Al(NONAr)] (NONAr = [O(SiMe2NAr)2]2−; Ar = 2,6-iPr2C6H3) are completely different if the reaction is carried out in the presence of 18-crown-6, reinforcing the idea that the alkali-metal is not just an spectator in these unique type of activations.23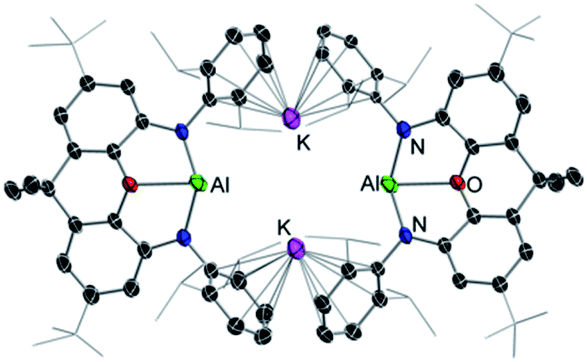 | ||
| Fig. 1 Molecular structure of potassium aluminyl [K{Al(NON)}]2 (5). | ||
Another striking example of K/Al cooperativity has been shown by Harder for a related potassium-stabilised anionic Al(I) complex which selectively activates two C–H bonds in benzene via oxidative addition at the positions 1 and 4.24 In this case encapsulation of K by a cryptand ligand also influences reactivity towards benzene, promoting a single C–H bond activation process. In this case, DFT calculations provide mechanistic insights on how K and Al(I) cooperate, with the coordination of benzene to the K cations being key to promote the 1,4-substituted double-C–H activation product.24
Alkali-metal effects have also proved to be very relevant in alkali-metal mediated magnesiation (AMMMg) of aromatic substrates. For instance, potassium magnesiate [KMg(TMP)2Bu] (6) deprotonates a molar equivalent of naphthalene to produce inverse crown complex [{KMg(TMP)2(C10H7)}6] (7) which in the crystal displays a 24-membered {KNMgN}6 ring with 6 naphthalene molecules hosted in its core which have been selectively magnesiated at their C2 position (Fig. 2a).25 Contrastingly when the sodium congener of 6, [NaMg(TMP)2Bu] (8), is reacted with naphthalene its 1,4-regioselective dimagnesiation is observed affording [{Na4Mg2(TMP)4(TMP*)2}(1,4-C10H6)] (9) (where TMP* is 2,2,6-trimethyl-1,2,3,4-tetrahydropyridide, a demethylated variant of TMP) (9) (Fig. 2b). While 9 also adopts an inverse crown structure, it is remarkably different to 7, made up by a 12-atom (NaNNaNMgN)2 ring, which hosts in its core the di-magnesiated molecule of naphthalene. While intuitively potassium magnesiate 6 could be expected to be more reactive than its sodium analog 8, the former only induces mono-metallation of naphthalene whereas the latter promotes selective dimetallation. Reflecting the key role played by the alkali-metal in these AMMMg reactions, this unexpected reactivity has been rationalised considering that by forming a larger ring size product, potassium can maximise its π interactions with the metallated naphthalene rings (Fig. 2a), a scenario that would not be possible if the Na atoms were replaced by K in 9.
 | ||
| Fig. 2 Molecular structures of alkali-metal magnesiates [{KMg(TMP)2(C10H7)}6] (7) (LHS) and [{Na4Mg2(TMP)4(TMP*)2}(1,4-C10H6)] (9) (RHS) resulting from AMMMg of naphthalene by [MMg(TMP)2Bu] (M = K, 6; M = Na, 8). | ||
The use of Lewis donors or presence of donor solvents should also be taken into account as their ability to coordinate to the alkali-metal can greatly affect the reactivity of the mixed-metal reagent. In some cases the addition of a donor additive such as TMEDA can have a deactivating effect as shown by Mulvey for the direct zincation of benzene by [NaZn(TMP)tBu2] (10) which in the absence of TMEDA induces the two-fold deprotonation of benzene at its positions 1 and 4; whereas if its TMEDA-solvated version [(TMEDA)NaZn(TMP)tBu2] (11) is employed only monozincation can be achieved.26,27 However, it should also be noted that on other occasions, the addition of the donor molecule is key in order to facilitate the metallation, as shown for the reaction of [LiZn(TMP)tBu2] (12) that forms a coordination adduct with N,N-diisopropyl benzamide with the carboxamide coordinated to lithium via its O atoms. While this zincate complex is stable at room temperature, addition of TMEDA induces metallation of the benzamide affording the relevant ortho-zincation product.28 In addition, it should also be noted that for many of these bimetallic systems, when a large excess of a donor is employed (or a Lewis donor with high denticity such as a crown ether), formation of solvent-separated species will be favoured, which in some cases hinders metal–metal’ cooperation and inhibits cooperative behaviours.17
Applications in homogenous catalysis
Alkali-metal/group 2 metal partnerships
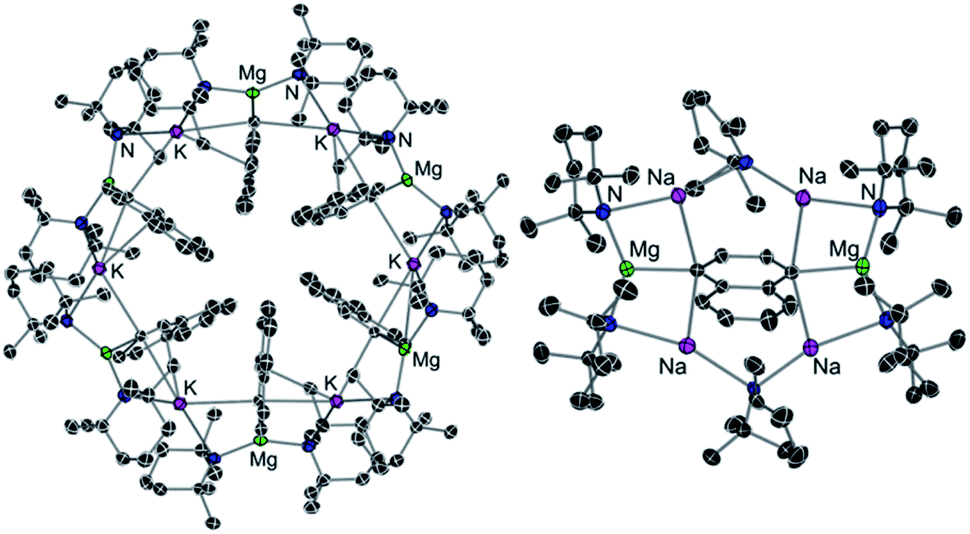
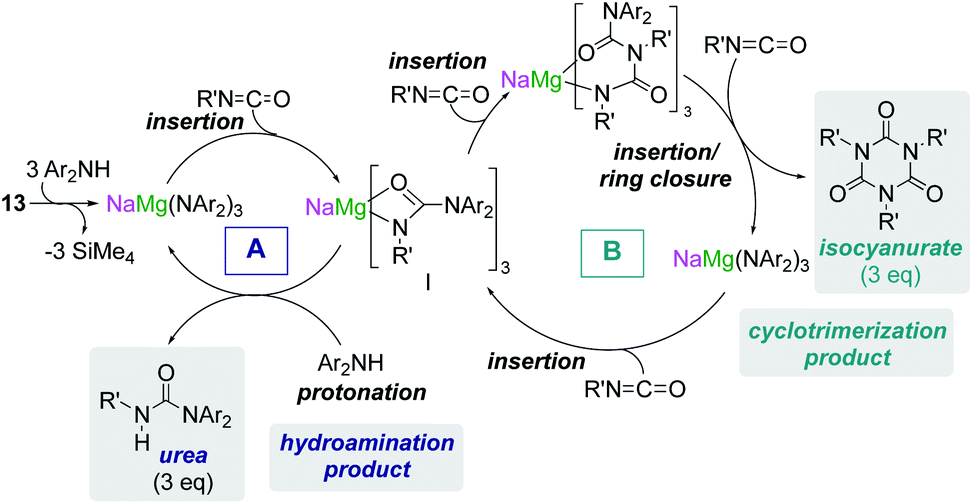
Some initial studies assessing the catalytic potential of s-block bimetallic reagents have focussed on hydroamination reactions. Generally described as the addition of an N–H fragment across an unsaturated C–X bond, hydroaminations constitute a powerful atom-efficient and waste-minimized method to access synthetically relevant organic molecules such as amines, N-heterocycles, ureas and guanidines to mention just a few. While several catalytic transition-metal systems based on rhodium, ruthenium, titanium or palladium have been developed, pioneering work by Hill and Harder has revealed that alkaline earth metal (particularly calcium and strontium) complexes can efficiently catalyse these processes, emerging as a cheaper and more sustainable alternative.4 Operating under mild reaction conditions, the catalytic activity of these systems is enhanced with increasing ionic radius of the group 2 metal, with magnesium notably displaying poor reactivity for hydroamination processes.5 Mechanistically s-block metal-mediated hydroaminations are thought to occur via the formation of a metal amide intermediate that in turn can undergo insertion into the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) X (X = O, N, C) bond of the unsaturated organic substrate (alkenes, isocyanates, carbodiimides, etc.).4 In these single-metal systems this substrate activation occurs by forming electrostatic interactions with the group 2 metal, which leads to the polarization of the π-electron density and facilitates the nucleophilic attack by the amide group. The limited poor reactivity of magnesium compounds to catalyse these processes can be overcome by forming alkali-metal magnesiate systems. Thus, research by our group has demonstrated that using mixed-metal homoleptic [NaMg(CH2SiMe3)3] (13) as a pre-catalyst for hydroamination of aliphatic isocyanates (RNCO) it is possible to access a wide range of ureas under mild reaction conditions,29 offering greater yield and a broader substrate scope than when using Ca or Ba catalysis.30 Unique cooperativity arises because Na can act as a Lewis acid allowing activation of the unsaturated organic substrate (isocyanate) facilitating the intramolecular nucleophilic attack by the highly nucleophilic Lewis basic tris(amide) magnesiate anion (cycle A in Scheme 2 and Fig. 3). Stoichiometric studies reacting sodium alkyl magnesiate with an excess of amine led to the isolation of [(THF)NaMg(NPh2)3(THF)] (14) (Fig. 3) which is catalytically competent and proposed to be the active species in the catalytic cycles depicted in Scheme 2.
X (X = O, N, C) bond of the unsaturated organic substrate (alkenes, isocyanates, carbodiimides, etc.).4 In these single-metal systems this substrate activation occurs by forming electrostatic interactions with the group 2 metal, which leads to the polarization of the π-electron density and facilitates the nucleophilic attack by the amide group. The limited poor reactivity of magnesium compounds to catalyse these processes can be overcome by forming alkali-metal magnesiate systems. Thus, research by our group has demonstrated that using mixed-metal homoleptic [NaMg(CH2SiMe3)3] (13) as a pre-catalyst for hydroamination of aliphatic isocyanates (RNCO) it is possible to access a wide range of ureas under mild reaction conditions,29 offering greater yield and a broader substrate scope than when using Ca or Ba catalysis.30 Unique cooperativity arises because Na can act as a Lewis acid allowing activation of the unsaturated organic substrate (isocyanate) facilitating the intramolecular nucleophilic attack by the highly nucleophilic Lewis basic tris(amide) magnesiate anion (cycle A in Scheme 2 and Fig. 3). Stoichiometric studies reacting sodium alkyl magnesiate with an excess of amine led to the isolation of [(THF)NaMg(NPh2)3(THF)] (14) (Fig. 3) which is catalytically competent and proposed to be the active species in the catalytic cycles depicted in Scheme 2.
 | ||
| Scheme 2 Proposed catalytic cycle using sodium magnesiate [[NaMg(CH2SiMe3)3] (13) as a pre-catalyst for hydroamination of isocyanates (cycle A) and trimerization of isocyanates (cycle B). | ||
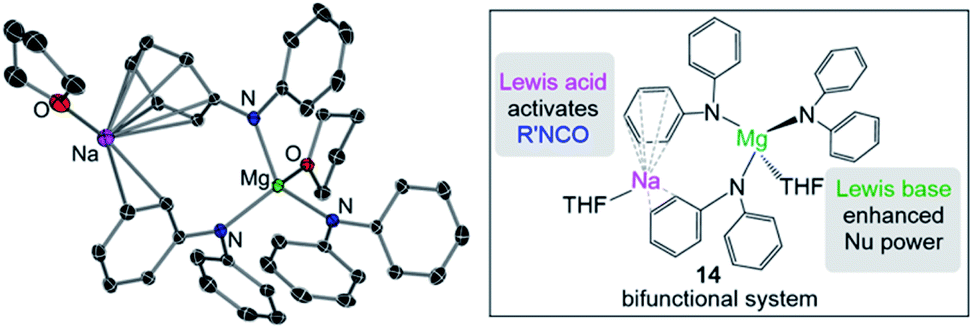 | ||
| Fig. 3 Molecular structures of alkali-metal magnesiate [(THF)NaMg(NPh2)3(THF)] (14). | ||
In 14 Mg bonds to all three amido N atoms; whereas Na π-engages with two Ph groups and one THF molecule. These distinct bonding modes have been proposed to maximise chemical cooperativity, enhancing the nucleophilicity of the {Mg(NPh2)3}− fragment as well as priming Na for coordination of isocyanate (Fig. 3). Interestingly using aromatic isocyanates triggers a second catalytic cycle, to produce the relevant isocyanurate (cycle B, Scheme 2) as a result of the selective trimerization of the aryl isocyanate. Thus, the tris(ureido) intermediate I (which is common to both catalytic cycles A and B) undergoes further insertion with two further equivalents of the aryl isocyanate followed by ring closure forming the sodium trisamidomagnesiate [NaMg(NAr2)3] that can in turn react with ArNCO to regenerate I. This reaction sequence contrasts with that of catalytic cycle A (Scheme 2) where I undergoes protonation by excess amine. The selectivity of these processes is remarkable since in each case only hydroamination or trimerization is seen, with none forming a mixture of both possible products.
We have also reported applications of mixed Na/Mg system 13 as an efficient pre-catalyst in hydroamination and hydrophosphination of carbodiimides. These proceed by a similar reaction pathway as that described for isocyanates, although in this case sodium seems to take a back seat in the catalytic cycle, with all processes taking place in the coordination sphere of Mg, suggesting that for these specific substrates the enhanced catalytic activity of the sodium magnesiate can be rationalised in terms of its anionic activation.31
Showcasing the power of these bimetallic partnerships, alkali–metal magnesiates can also catalyse the hydroamination of more challenging substrates such as alkynes and styrenes.32 This reactivity contrasts markedly with the lack of catalytic ability of Mg(CH2SiMe3)2, which even after 24 h at 80 °C fails to promote hydroamination of diphenylacetylene by piperidine (Table 1, entry 1).32 On the other hand, sodium magnesiate 13 enables formation of hydroamination product 15 although harsh reaction conditions are required (18 h, 80 °C) and limited E/Z selectivity is accomplished (entry 2, Table 1). Interestingly, reflecting the relevance of the nucleophilicity of the mixed-metal species, higher order (formally dianionic) magnesiates ([(D)2MI2Mg(CH2SiMe3)4] MI = Na, K; D = Lewis donor) have proven to be more efficient and powerful than lower order 13, capable of promoting hydroamination even while operating at room temperature. Highlighting the key role of the alkali-metal, potassium magnesiate 3 mediates the smooth hydroamination of diphenyl acetylene affording 15 in quantitative yields after just 3 h with greater E/Z selectivity than when using sodium analogue [(TMEDA)2Na2Mg(CH2SiMe3)4](16) which affords 15 in a modest 28% yield (entries 3 and 4, Table 1).
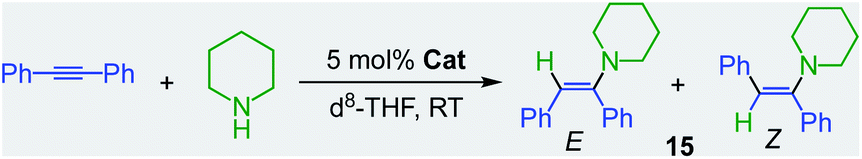
This conspicuous alkali-metal effect has been attributed to the greater ability of potassium to engage diphenyl acetylene (probably by forming π-electrostatic interactions as those discussed for 7), bringing it into the proximity of the nucleophilic {Mg(amide)4}2− dianion and priming it for insertion. A comparison of the catalytic performance of 3 with other heavier alkali-earth single-metal systems, shows that its performance in terms of E/Z selectivity and conversion is comparable to that described by Hill for [Sr{CH(SiMe3)2}2(THF)2], although for the latter, higher temperatures are required (2 h, 60 °C);33 whereas 3 operates at room temperature. Though 3 can also promote hydroamination of styrene with several amines, this method is not compatible with less nucleophilic aromatic amines. This limitation can be overcome by using potassium calciates as demonstrated by Westerhausen for the hydroamination of diphenylbutadiyne with a variety of anilines (Scheme 3a).34 Illustrating the cooperative effect of the ate compound as a catalyst, no reaction product is observed when adding diphenylamine to 1,4-diphenylbutadiyne in the presence of Ca(NPh2)2 or KNPh2. However, when the higher order bimetallic calciate [K2Ca(NPh2)4] (17) is used as a catalyst the hydroamination product 18 is formed in NMR yields exceeding 90% (Z isomer 40%, isolated yield) (Scheme 3a).34 Interestingly in the case of primary anilines, [K2Ca{N(H)Dipp}4] (19) promoted the attack on the two triple bonds of diphenylbutadiyne in a cascade reaction that resulted in either N-aryl-2,5-diphenylpyrroles or polycyclic compounds as 20 (Scheme 3b) depending on the substitution pattern in the parent aniline.35
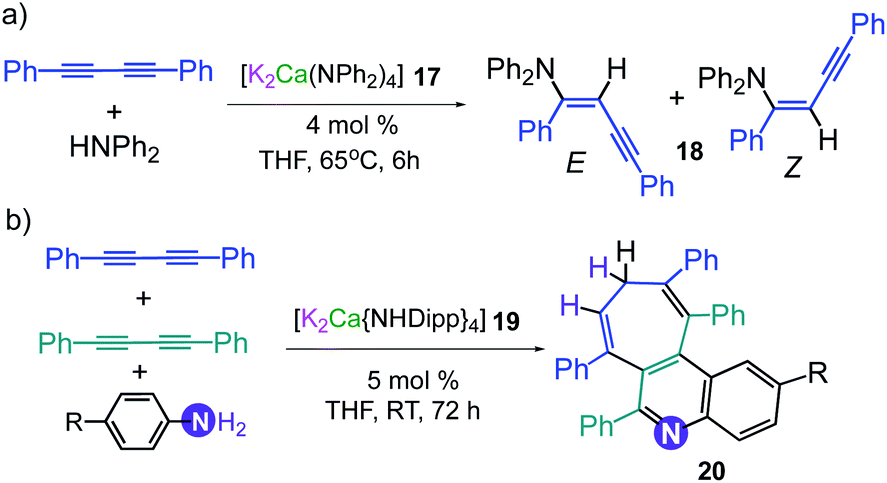 | ||
| Scheme 3 Catalytic hydroamination of diphenylbutadiyne using (a) [K2Ca(NPh2)4] (17) and [K2Ca{N(H)Dipp}4] (19) as pre-catalysts. | ||
Expanding further the applications of alkali-metal magnesiates in catalysis we recently reported their use in intramolecular hydroalkoxylation of alkynols, which provides access to a wide range of oxygen-containing heterocycles.36 Higher-order potassium magnesiate 3 enables the cyclisation of 4-pentynol (Scheme 4), disclosing a cooperativity which helps to overcome the inherent challenges of this type of transformation: namely OH activation and coordination to, and then addition across, a C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond. Each metal plays a vital role enabling a unique type of substrate activation that is not possible in conventional single-metal systems. To explain, coordination of the CC bond to potassium enables further activation of this unsaturated moiety and brings it into close proximity to the dianionically activated {Mg(OR)4}2− ate, which is significantly more nucleophilic than a neutral Mg(OR)2 species. Interestingly, reflecting the key role of donor additives in order to finely tune the efficiency of the bimetallic systems, 3 has to be paired with 18-crown-6. Since kinetic studies have revealed an inhibition effect of substrate on the catalyst, by formation of a coordination adduct which requires dissociation prior to the cyclisation step, the beneficial effect of 18-crown-6 has been attributed to its ability to minimise the coordination of additional substrate molecules to the catalyst.36
C bond. Each metal plays a vital role enabling a unique type of substrate activation that is not possible in conventional single-metal systems. To explain, coordination of the CC bond to potassium enables further activation of this unsaturated moiety and brings it into close proximity to the dianionically activated {Mg(OR)4}2− ate, which is significantly more nucleophilic than a neutral Mg(OR)2 species. Interestingly, reflecting the key role of donor additives in order to finely tune the efficiency of the bimetallic systems, 3 has to be paired with 18-crown-6. Since kinetic studies have revealed an inhibition effect of substrate on the catalyst, by formation of a coordination adduct which requires dissociation prior to the cyclisation step, the beneficial effect of 18-crown-6 has been attributed to its ability to minimise the coordination of additional substrate molecules to the catalyst.36
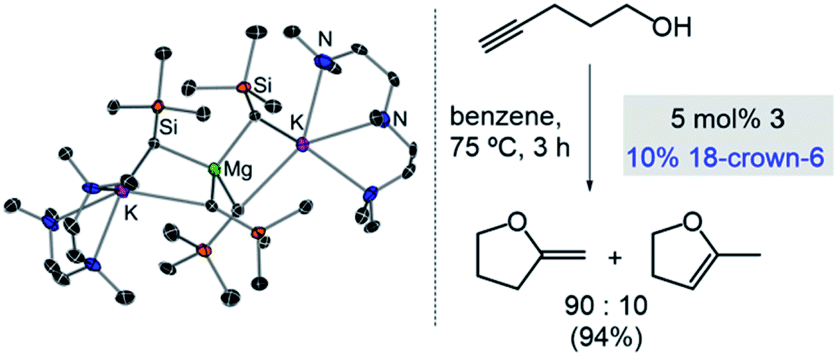 | ||
| Scheme 4 Molecular structures of higher-order potassium magnesiate [(PMDETA)2K2Mg(CH2SiMe3)4] (3) and its application as a pre-catalyst for cyclisation of alkynols. | ||
Alkali-metal/aluminium partnerships
Turning to other more sustainable catalytic systems, alkali-metal aluminates, including the classical commercially available reagent LiAlH4, have already shown substantial promise for hydrogenation, hydroboration and hydrophosphination reactions. In a seminal example, Harder has demonstrated that LiAlH4 can catalytically reduce imines to amines at 85 °C, using low H2 pressures and low catalyst loads without the requirement of organic solvents.37 Stoichiometric studies of LiAlH4 with the imine PhC(H)NtBu combined with DFT calculations suggest that the active catalytic species (II in Fig. 4) is generated in situ by the sequential insertion of the imine into the Al–H bonds of the catalyst. This proposed intermediate II has been trapped and isolated, and structurally defined by X-ray crystallography as the PMDETA adduct [(PMDETA)LiAlH2{NtBu(Bz)}2] (21) (Bz = CH2Ph, Fig. 4), with Al and Li being connected by a hydride bridge. II can in turn undergo insertion with a third equivalent of imine, furnishing [LiAlH{NtBu(Bz)}3] which in turn can react with H2 to generate the amine product PhCH2NHtBu (22) and regenerate II. Indirectly supporting a cooperative mechanism, neither LiH or AlH3 can promote these hydrogenation reactions on their own under substoichiometric conditions. Furthermore, an important alkali-metal effect is observed with NaAlH4 being a noticeably less effective catalyst than its lithium congener.37 More recently the same group has reported that alkaline-earth metal [M(AlH4)2(THF)x] (M = Mg, Ca, Sr; x = 4 or 5) which can also catalyse the hydrogenation of imines offering a greater substrate scope than LiAlH4.38 Reflecting the key role of the higher polarity metal in these transformations, the salt [Bu4N][AlH4] showed no catalytic activity.
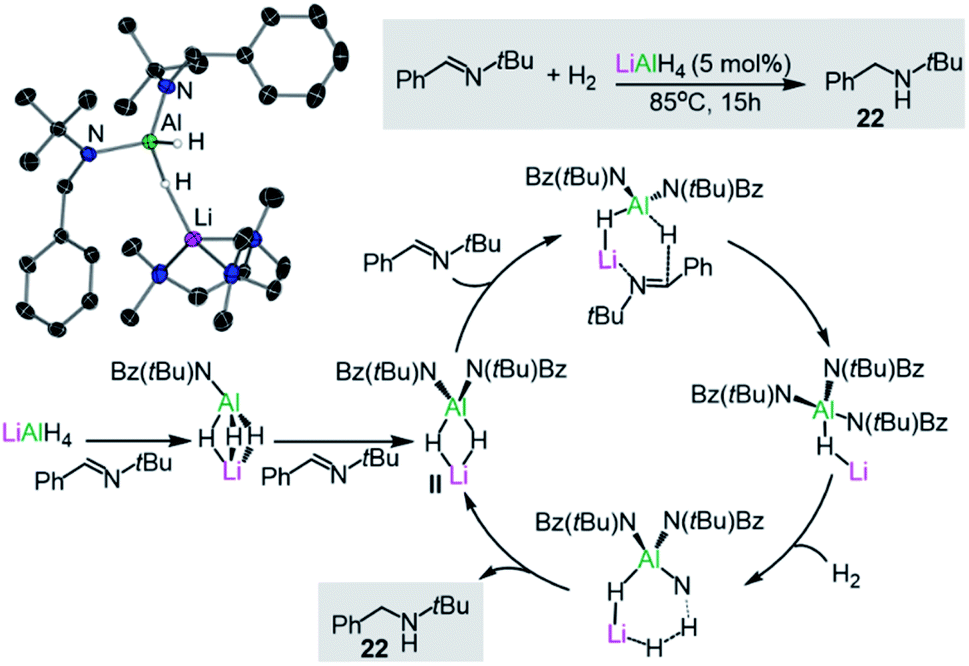 | ||
| Fig. 4 Molecular structure of [(PMDETA)LiAlH2{NtBu(Bz)}2] (21) and the catalytic cycle proposed for the hydrogenation of imines by LiAlH4. | ||
Experimental and computational studies have provided further insights, supporting a cooperative bimetallic mechanism with each metal playing an important role for the success of the hydrogenation process.37b The rate-determining step is the hydrogenolysis of [LiHAl{N(tBu)Bz}3] (Fig. 4) and in agreement with experimental findings, replacing Li for Na (or K) and Al for B (or Ga) led to higher calculated energy barriers.37b
In parallel to these findings Cowley and Thomas have reported the efficient hydroboration of alkenes with HBpin using LiAlH4 as a precatalyst.39 While LiAlH4 showed greater catalytic activity than its single metal components, the role of cooperative behaviour was not examined. Reactions were thought to occur via an initial hydroalumination, followed by σ-bond metathesis exchanging Al and B to regenerate the aluminium hydride reactive species, although the possibility of an alternative mechanism involving a borane or borohydride species cannot be unequivocally ruled out. Related to the latter interpretation, a particularly insightful study by Thomas has revealed that many of the recent hydroboration processes with HBpin reported in the literature that were thought to be catalysed by nucleophiles such as nBuLi or Bu2Mg are actually catalysed by BH3, and the role of the polar organometallic reagents is not genuinely catalytic but more promotes the formation of BH3.40 More recently, An has shown that commercially available lithium aluminate [LiAliBu2(H)(OtBu)] can also catalyse the hydroboration of alkenes under similar conditions to those described by Thomas and Cowley for LiAlH4 (neat, 110 °C, 2–3 h).41
Shedding light on the mechanisms operating in lithium-aluminate catalysed hydroborations, Mulvey has trapped and structurally characterised key reaction intermediates involved in the hydroboration of ketones and aldehydes with HBpin using [(THF)3LiAlH2(HMDS)2] (4). In this study hydroborations take place at room temperature using low loadings of 4 (Fig. 5). Stoichiometric studies of 4 with two equivalents of benzaldehyde led to the isolation of hydrometallated product [(THF)2LiAl(OCH2Ph)2(HMDS)2] (23) (Fig. 5) which is also catalytically competent.20b
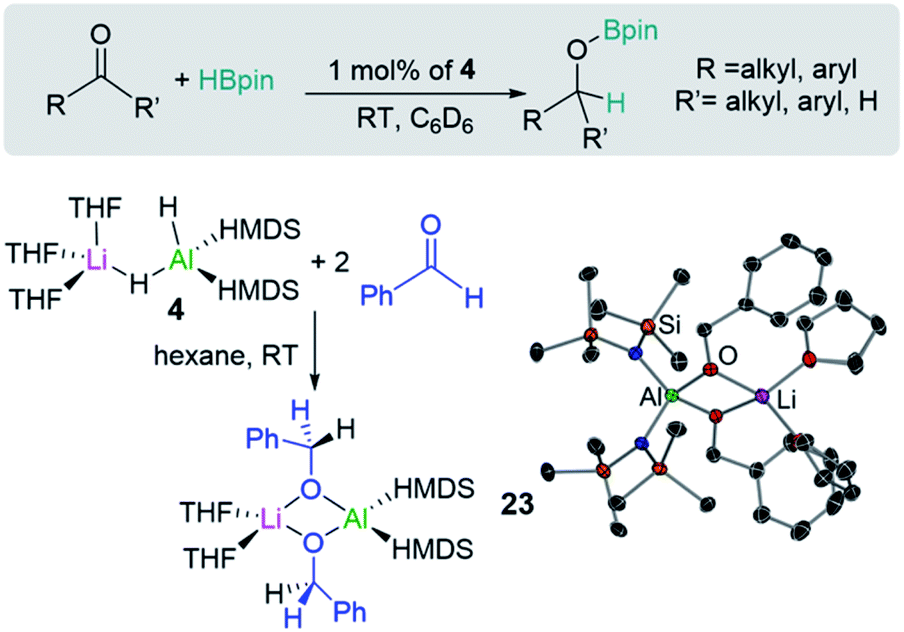 | ||
| Fig. 5 Catalytic hydroboration of aldehydes and ketones with HBPin using lithium aluminate [(THF)3LiAlH2(HMDS)2] (4) as a pre-catalyst and synthesis and molecular structure of [(THF)2LiAl(OCH2Ph)2(HMDS)2] (23). | ||
In order to assess the effect of Lewis donors as additives, the new family of lithium aluminates 24–28 derived from 4 was prepared and structurally characterised (Fig. 6). Assessing their catalytic ability for the hydroboration of acetophenone revealed that those complexes with non-labile chelating donors such as 26, which contains tridentate ligand PMDETA, were significantly less effective than 4.20 These findings are consistent with the catalysts operating through bimetallic cooperation with the hydride transfer being executed by Al, but the initial substrate coordination occurring at Li, at rates dependant on the relative lability of the donor ligands.
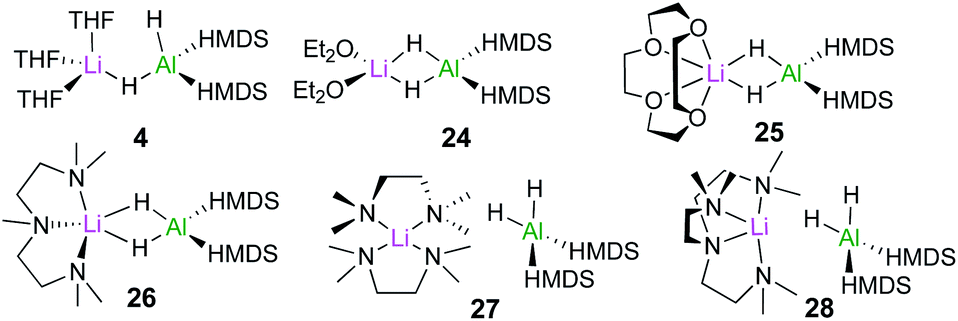 | ||
| Fig. 6 Structures of lithium aluminates [(D)xLiAlH2(HMDS)2] (4, 24–28) (x = 2 for THF, OEt2, TMEDA; x = 1 for 12-c-4, PMDETA and Me6-Tren). | ||
Lithium aluminate [iBu2Al(TMP)(H)Li] (29) resulting from the co-complexation of DIBAL(H) with LiTMP in hexane can also catalyse the hydroboration of ketones and aldehydes and imines.20b,42 A comparison of the catalytic ability of 29 in these reactions with those of neutral aluminium species [AliBu2(TMP)], [Al(H)(HMDS)2] and [AlHiBu2] showed that the lithium aluminate offers higher conversions in shorter timescales.42 This has been attributed to the higher degree of polarisation of key reaction intermediates induced by the presence of two distinct metals of different electropositivity.
Building on these insights, hydroboration of ketones and aldehydes by lithium aluminates can be envisaged to work by initial coordination of the substrate to the Li atom (see Scheme 5 for hydroboration of acetophenone catalysed by 29). This step can be hindered in the presence of chelating donors such as PMDETA.43 Coordination will activate the CO of the substrate towards insertion into the Al–H bond of the aluminate anion, followed by trans-elementation with HBpin to afford hydroborated product and regenerate an active lithium aluminate hydride catalyst. Lithium aluminate 29 also catalyses the hydroboration of phenylacetylene (2.5 mol% of 29, 70 °C, 18 h) although this process has been proposed to proceed via an alternative mechanism were initially 29 deprotonates the organic substrate using its basic TMP amide group.43
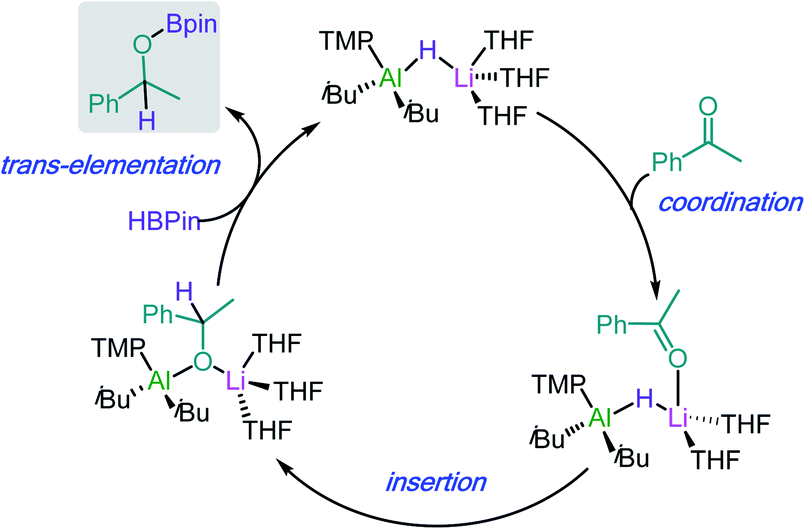 | ||
| Scheme 5 Proposed catalytic cycle for the hydroboration of acetophenone by lithium aluminate [iBu2Al(TMP)(H)Li] (29). | ||
Other catalytic applications noted for lithium aluminates include the use of LiAlH4 in dehydrocoupling of amine borane Me2NHBH3 although long reaction times are required (16 h, 65 °C).44 Through NMR reaction monitoring and X-ray crystallographic studies, Wright has uncovered the complexity of these processes which involve a sequence of deprotonation, B–N bond formation and B–N bond cleavage steps.44
Broadening the scope of applications of lithium aluminates in catalysis, Mulvey has also identified that [LiAl(H)iBu3] (30) is an efficient pre-catalyst for hydrophosphination of alkynes, alkenes and carbodiimides.45 The active catalytic species in this process was thought to be the structurally defined lithium phosphidoaluminate [iBu3Al(PPh2)Li(THF)3] (31) which forms by reacting [iBu3AlHLi]2 (30) and the secondary phosphine HPPh2 at room temperature in the presence of THF (Fig. 7).45 The authors envisage lithium aluminate 31 as a co-complex of LiPPh2 and iBu3Al/THF, with the two metals connected by a PPh2 bridge. Running the reaction in deuterated toluene, 31 catalyses hydrophosphination of terminal and internal alkynes with HPPh2 at 110 °C to afford the anti-Markovnikov addition products with variable levels of E/Z selectivities. Interestingly, the addition of Lewis donors such TMEDA as additives can increase the E/Z selectivity (19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 compared to 10:1 when using neat toluene).
:1 compared to 10:1 when using neat toluene).
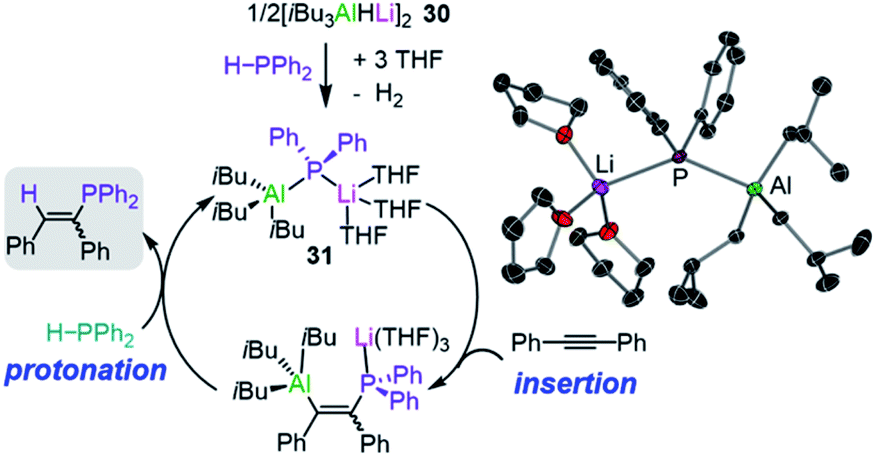 | ||
| Fig. 7 Proposed catalytic cycle for the hydrophosphination of diphenylacetylene by lithium aluminate [iBu3Al(PPh2)Li(THF)3] (31) and molecular structure of 31. | ||
Mechanistic insights have been gained using kinetic and computational studies which support the catalytic cycle depicted in Fig. 7 where pre-catalyst 30 reacts with HPPh2 to form 31 followed by insertion of the alkyne into the Al–P bond and then protonolysis with a second equivalent of HPPh2 to generate the vinylphosphine product and regenerate 31 (Fig. 7). Lithium is proposed to act as a Lewis acid to coordinate the substrate and allow the aluminate to engage in the hydrophosphination process; whereas attempts to catalyse these reactions with the single metal systems iBu3Al and iBu2AlH resulted in low conversion and low selectivities.45
Other heterobimetallic partnerships
As mentioned above alkali-metal zincates have found widespread applications in stoichiometric processes, in particular in deprotonative metallation processes, employing bimetallic combinations that contain kinetically activated amide groups such as TMP or HMDS.7 Thus for example the tris(amido) potassium zincate [KZn(HMDS)3] reacts with toluene at room temperature to form the benzyl product [KZn(HMDS)2(CH2Ph)] (32) resulting from the direct zincation of toluene. Significantly, neither KHMDS or Zn(HMDS)2 on their own can metallate the arene.46 Interestingly 32 can catalyse benzylic C–H bond addition of diarylmethanes to styrenes and conjugated dienes with good yields and chemoselectivity without the need of transition-metal catalysis or the use of external oxidants (Scheme 6).47 Representing a practical, atom economical process, the reactions involved are proposed to operate via initial zincation of diphenylmethane to afford the active species [KZn(HMDS)2(CHPh2)] (III) (Scheme 6) which in turn undergoes addition to styrene, with the latter probably activated by coordinating to potassium as described above for the hydroamination of styrene mediated by potassium magnesiate 3.32 Protonation of the insertion intermediate with diphenylmethane releases the alkylation product 33 and regenerates active species III (Scheme 6). To gain further mechanistic insights KIE values were measured, finding values that are consistent with the cleavage of the benzylic C–H bond being the rate determining step in these cycles.47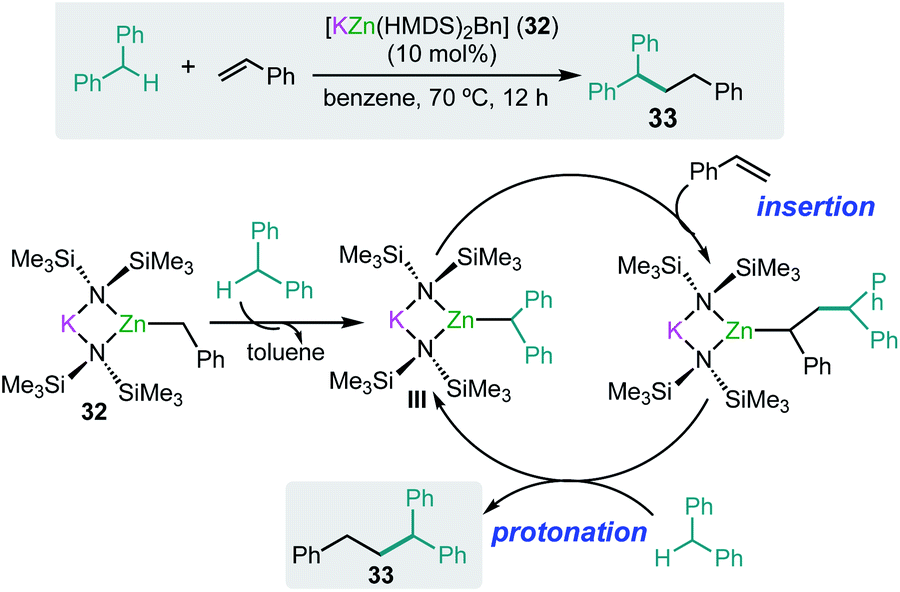 | ||
| Scheme 6 Potassium-zincate [KZn(HMDS)2(CH2Ph)] (32) catalyzed benzylic C–H bond addition of diphenylmethanes to styrene. | ||
Seeking the development of novel s-block bimetallic strategies to enable C–C bond forming processes while operating in catalytic regimes, Kobayashi has also shown that toluene and other related alkyl arenes can undergo addition to imines and stilbene derivatives48 using catalytic amounts of the LINK base previously developed by O'Shea. This base is made up of a mixed-alkali-metal combination of KOtBu and LiTMP.49 While the true constitution of this bimetallic combination is still not known with any certainty, its reactivity clearly has a cooperative origin since neither KOtBu or LiTMP on their own promote these processes. Hinting at the importance of coordination effects, the presence of high-denticity, Lewis donor PMDETA enables the smooth alkylation of stilbene (Scheme 7). This activating effect has been attributed to the ability of PMDETA to coordinate to the benzylic intermediate (resulting from the deprotonation of toluene by the LINK base), favouring the formation of a smaller more carbanionic molecule that will be expected to be more kinetically labile towards addition to the CC bond.48
 | ||
| Scheme 7 Coupling of toluene and stilbene using LINK base as a catalyst. | ||
Applications polymerization reactions
Another area related to catalysis in which main group heterobimetallic complexes have already shown excellent potential is polymerization processes. Some examples include ring-opening polymerization (ROP) of oxygenated cyclic molecules such as lactides50,51 and epoxides52 as well as CO2/epoxide copolymerisation.53–55 Thus, Mosquera has prepared a series of heteroleptic alkali-metal aluminates [MIAlMe2{2,6-(MeO)2C6H3O}2]n (MI = Li, 34a; Na, 34b; and K, 34c) which are active towards ROP of L-lactide revealing an alkali-metal effect with lithium aluminate 34a being the most active catalyst, offering narrower polydispersity values than the single metal [AlMe2{2,6-(MeO)2C6H3O}]2.50 The same group has also prepared lithium magnesiate [LiMg{OR}3]2 [OR = O(C10H6)(NNPh)] (35) derived from 1-phenylazo-2-naphtol which exhibits a dimeric structure which is retained in benzene solution. 35 can efficiently polymerize rac-lactide in the presence of benzyl alcohol as co-initiator, offering an activity intermediate between those witnessed for its homometallic components but greater stereoselectivity (Fig. 8).
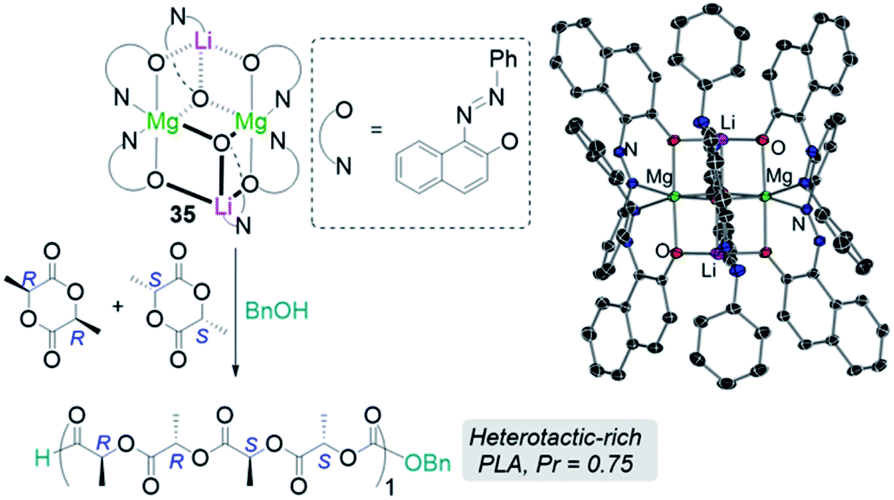 | ||
| Fig. 8 Polymerisation of rac-lactide using complex 3 and benzyl alcohol as initiators and molecular structure of lithium mangesiate [LiMg{OR}3]2 [OR = O(C10H6)(NNPh)] (35). | ||
Showcasing different reactivity profiles to single-metal aluminium initiators, alkali-metal aluminates [MIAlMe2(OR)4] (MI = Li, 36a; Na, 36b; and K, 36c; OR = 2,6-bis(diphenylmethyl)-4-tert-butylphenoxide) polymerize difunctional glycidyl methacrylate (GMA) via vinyl polymerization52 whereas [AlMe2(OR)2] promotes the selective polymerization of the oxirane group in GMA via ROP (Fig. 9).56 The acrylate polymerization mediated by alkali-metal aluminates 36 is likely to occur via an anionic mechanism (Fig. 9) contrasting with more common approaches for GMA polymerization using RAFT or radical mechanism.
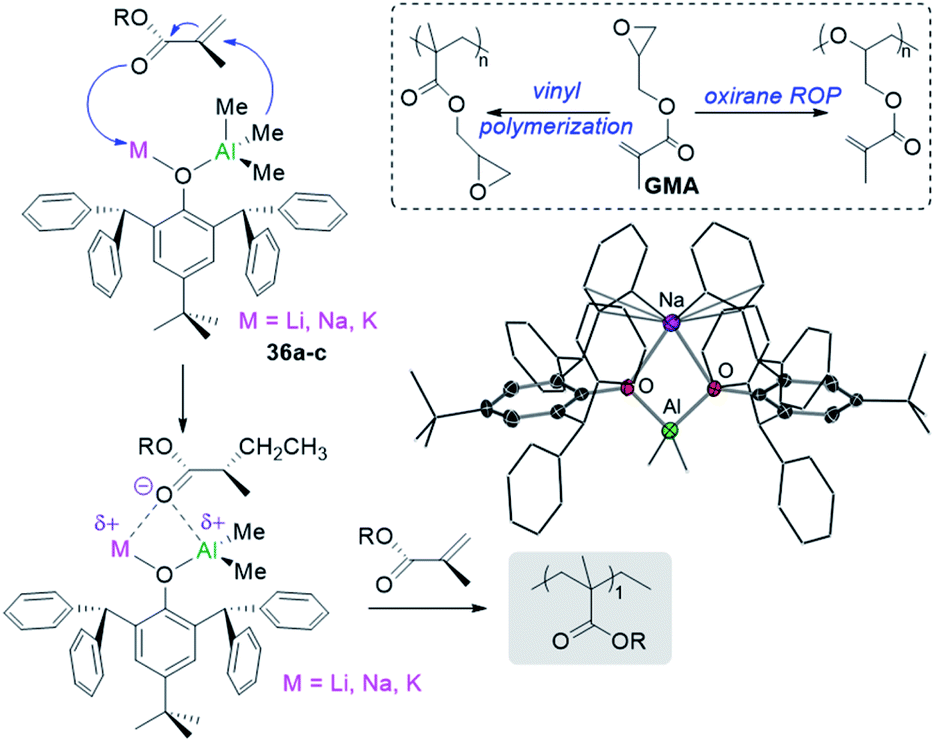 | ||
| Fig. 9 Polymerization of GMA using alkali-metal alumminates MIAlMe2(OR)4] (MI = Li, 36a; Na, 36b; and K, 36c; OR = 2,6-bis(diphenylmethyl)-4-tert-butylphenoxide) and molecular structure of 36b. | ||
Seminal work by Williams has also demonstrated the ability of cooperative bimetallics for epoxide and CO2/anhydride ring-opening copolymerizations (ROCOP).53–55 Key for the success of this approach was to synthesize Zn/Mg heterodinuclear complex 37 where both metals are coordinated by a symmetrical macrocyclic ligand (Fig. 10). Switching on chemical cooperativity between Mg and Zn, 37 offers significantly higher activity than either the relevant di-magnesium or di-zinc complexes or than mixtures of them.51 Mechanistic investigations are consistent with a chain shuttling copolymerization pathway, with Mg and Zn adopting distinct roles in the catalytic cycle, with the growing polymeric chain shuttling between both metals (Fig. 10).54 Interestingly in the rate determining step (RDS) Mg coordinates the epoxide which undergoes addition from the carbonate group coordinated to Zn, forming an alkoxide which binds to Mg and that in turn inserts rapidly CO2 to give the zinc carbonate intermediate (Fig. 10).54
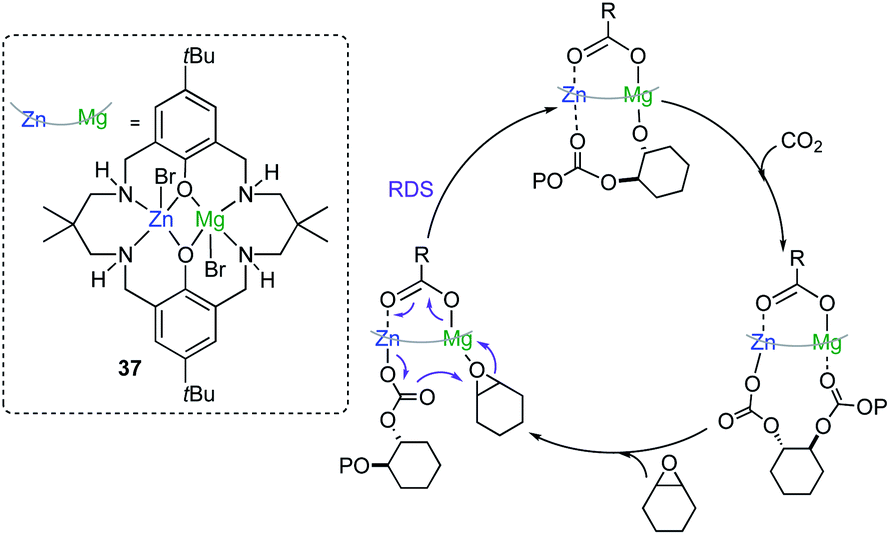 | ||
| Fig. 10 Proposed mechanism for CO2/epoxide copolymerization by hetrobimetallic catalyst 37. | ||
Expanding the boundaries outside of main group bimetallics William has recently extend the scope of these polymerization approaches to bimetallics pairs of Mg/Co(II) finding an even better reactivity and higher selectivity for these copolymerization processes than when using 37 which has been rationalised in terms of the synergy created between the two metals, with Mg favouring the epoxide coordination while Co(II) accelerates the carbonate attack.57
While this Perspective focusses on main group heterobimetallic systems, it should be noted that mixed-metal systems combining Al with a group 4 metal such as Ti or Zr have also played a pivotal role in alkene polymerization.58 It is now well established that group 4 metallocenes are activated by a group 13 co-catalyst, such as MAO (methylaluminoxane).59 Seminal work by Roesky has shown that heterobimetallic Al/Zr complexes can catalyse ethene polymerisation, however despite the presence of Al in these systems, the use of MAO is still required.60 Although it should be noted that significantly reduced amounts of this cocatalyst are needed for activity. Harder has also prepared several oxo-bridge heterobimetallic Al/Zr complexes such as [(TBB)Al(THF)OZr(Me)Cp*2] (38) (TBB = 3,3′,5,5′-tetra-tBu-2,2′-biphenolato) and [(DIPPnacnac)Al(Me)OZr(THF)Cp2]+[B(C6F5)4]− (39) (DIPPnacnac = HC[(Me)CN(2,6-iPr2-C6H3)]2) (Fig. 11).61 Interestingly while 38 polymerises ethene in the presence of iBu3Al as a co-catalyst, cationic complex 39 is inert towards these reaction conditions. Mechanistic studies suggest that the addition of this co-catalyst (or MAO) induces the cleavage of the Zr–O bond, forming [Cp*2ZrMe]+ which is the active catalyst for the polymerisation of ethene with the ease of activation being dependant of the ability of the [(TBB)Al(THF)O]− in 38 to act as a good leaving group.61 These reactivity patterns show that while both metals Al and Zr have an influence on the catalytic activity of these systems, the catalytic process can actually take place in just one metal centre.
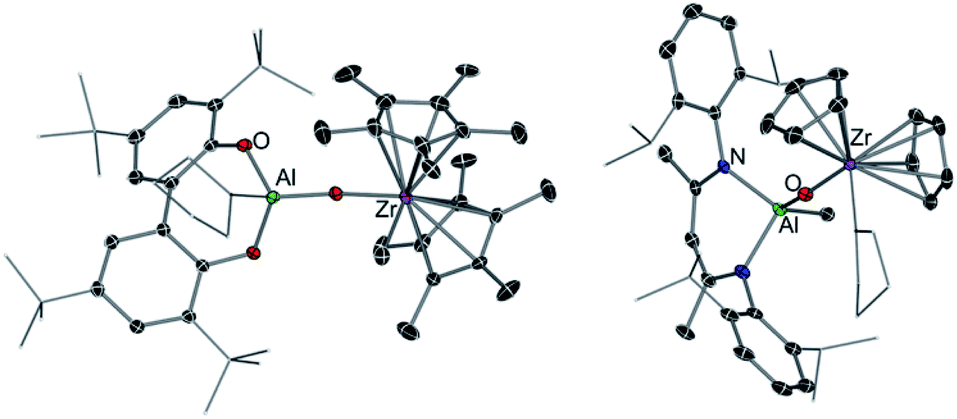 | ||
| Fig. 11 Molecular structure of [(TBB)Al(THF)OZr(Me)Cp2*] (38) (LHS) and structure of cation present in [(DIPPnacnac)Al(Me)OZr(THF)Cp2]+[B(C6F5)4]− (39) (RHS). | ||
Conclusions and outlook
Equipped with metal–metal′ cooperativity, s/p-block heterobimetallic systems are growing in stature as important additions to the main-group catalyst toolbox. Combining, in most cases, an alkali-metal with a lower electropositive metal such as Mg, Al or Zn, these tools offer synergic reactivities and selectivities, which arise from their unique combination of: (i) a low polarity metal ate anion with enhanced nucleophilic/basic power compared to that of a neutral metal complex; and (ii) a built-in Lewis acid, the alkali-metal, which can coordinate the organic substrate, activating it and bringing it into close proximity of the ate anionic fragment of the catalyst. This scenario is a feature of these mixed-metal systems that is unavailable to their single-metal components. Judicious choice of anionic ligands, the alkali-metal and the presence (or absence) of Lewis donors such as chelating TMEDA or PMDETA make possible to finely tune the catalytic behaviour of these heterobimetallic systems. Accordingly, they offer a growing range of opportunities, being able to catalyse processes from hydroelementation to C–C bond formation through to polymerization reactions. Interestingly, while the use of main group bimetallic partnerships for cooperative catalysis is an emerging new development, significant mechanistic understanding has been gained by the combination of kinetic and computational studies with the trapping and characterization of key reaction intermediates. Looking forward, it is anticipated that these pioneering studies should provide a solid foundation to stimulate more innovations in this evolving new branch of sustainable catalysis, including the discovery of new transformations as well as expanding substrate scope and functional group tolerance for some of the reactions that have already being uncovered. Transition metal catalysts still lead the way in homogeneous catalysis by a long distance, but with these main group advances catching the eye of many early career researchers looking for new, challenging research directions, the gap may be significantly reduced over the next few years.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC (EP/S020837/1) and the University of Bern for their financial support, as well as Rab Mulvey (University of Strathclyde) for many stimulating discussions on this exciting, evolving field. We also thank Leonie Bole for her assistance with the graphic material.Notes and references
- (a) J. Clayden, Organolithiums: Selectivity for Synthesis, Elsevier, 1st edn, 2002, vol. 23 Search PubMed; (b) Organomagnesium Compounds, ed. Z. Rappaport and I. Marek, Wiley, 2nd edn, 2008 Search PubMed; (c) Organolithium Compounds, ed. Z. Rappaport and I. Marek, Wiley, 2nd edn, 2004 Search PubMed; (d) Handbook of Functionalized Organometallics: Applications in Synthesis, ed. P. Knochel, Wiley, 1st edn, 2005 Search PubMed.
- Organometallics in Synthesis—Third Manual, ed. M. Schlosser, John Wiley & Sons, 2013 Search PubMed.
- Early Main Group Metal Catalysis, ed. S. Harder, Wiley-VHC, 2020 Search PubMed.
- (a) S. Harder, Chem. Rev., 2010, 110, 3852 CrossRef CAS; (b) A. G. M. Barrett, M. R. Crimmin and M. S. Hill, Proc. R. Soc. A, 2010, 466, 927 CrossRef CAS; (c) M. S. Hill, D. Liptrot and C. Weetman, Chem. Soc. Rev., 2016, 45, 972 RSC; (d) Y. Sarazin and J. F. Carpentier, Chem. Rec., 2016, 16, 2482 CrossRef CAS; (e) R. Rochat, M. J. Lopez, H. Tsurugi and K. Mashima, ChemCatChem, 2016, 8, 10 CrossRef CAS.
- (a) C. Brinkmann, A. G. M. Barrett, M. S. Hill and P. A. Procopiou, J. Am. Chem. Soc., 2012, 134, 2193 CrossRef CAS; (b) A. G. M. Barrett, C. Brinkmann, M. R. Crimmin, M. S. Hill and P. A. Procopiou, J. Am. Chem. Soc., 2009, 131, 12906 CrossRef CAS; (c) B. Liu, T. Rosinel, J. F. Carpentier and Y. Sarazin, Angew. Chem., Int. Ed., 2012, 51, 4943 CrossRef CAS.
- J. Penafiel, L. Maron and S. Harder, Angew. Chem., Int. Ed., 2015, 54, 201 CrossRef CAS.
- For an authoritative comprehensive review in the area see: S. D. Robertson, M. Uzelac and R. E. Mulvey, Chem. Rev., 2019, 119, 8332 CrossRef CAS.
- (a) A. Harrison-Marchand and F. Mongin, Chem. Rev., 2013, 113, 7470 CrossRef CAS; (b) F. Mongin and A. Harrison-Marchand, Chem. Rev., 2013, 113, 7563 CrossRef CAS; (c) D. Tilly, F. Chevallier, F. Mongin and P. C. Gros, Chem. Rev., 2014, 114, 1207 CrossRef CAS; (d) M. Balkenhohl and P. Knochel, Chem.–Eur. J., 2019, 26, 3688 CrossRef; (e) D. S. Ziegler, B. Wei and P. Knochel, Chem.–Eur. J., 2018, 25, 2695 CrossRef.
- A. Krasovskiy and P. Knochel, Angew. Chem., Int. Ed., 2004, 43, 3333 CrossRef CAS.
- (a) E. Hevia and R. E. Mulvey, Angew. Chem., Int. Ed., 2011, 50, 6448 CrossRef CAS; (b) R. L. Y. Bao, R. Zhao and L. Shi, Chem. Commun., 2015, 51, 6884 RSC.
- (a) D. R. Armstrong, W. Clegg, S. H. Dale, E. Hevia, L. M. Hogg, G. W. Honeyman and R. E. Mulvey, Angew. Chem., Int. Ed., 2006, 45, 3775 CrossRef CAS; (b) A. J. Martinez-Martinez, A. R. Kennedy, R. E. Mulvey and C. T. O'Hara, Science, 2014, 346, 834 CrossRef CAS.
- T. Furuyama, M. Yonehara, S. Arimoto, M. Kobayashi, Y. Matsumoto and M. Uchiyama, Chem.–Eur. J., 2008, 14, 10348 CrossRef CAS.
- M. Dell'Aera, F. M. Perna, P. Vitale, A. Altomare, A. Palmieri, L. C. H. Maddock, L. J. Bole, A. R. Kennedy, E. Hevia and V. Capriati, Chem.–Eur. J., 2020, 26, 8742 CrossRef.
- P. Garcia-Alvarez, D. V. Graham, E. Hevia, A. R. Kennedy, J. Klett, R. E. Mulvey, C. T. O'Hara and S. Weatherstone, Angew. Chem., Int. Ed., 2008, 47, 8079 CrossRef CAS.
- E. Hevia and R. E. Mulvey, Angew. Chem., Int. Ed., 2011, 50, 6448 CrossRef CAS.
- A. Krasovskiy, V. Krasovskaya and P. Knochel, Angew. Chem., Int. Ed., 2006, 45, 2958 CrossRef CAS.
- S. E. Baillie, T. Bluemke, W. Clegg, A. R. Kenedy, J. Klett, L. Russo, M. de Tullio and E. Hevia, Chem. Commun., 2014, 50, 12859 RSC.
- D. R. Armstrong, H. S. Emmerson, A. Hernán-Gómez, A. R. Kennedy and E. Hevia, Dalton Trans., 2014, 43, 14229 RSC.
- D. R. Armstrong, E. Brammer, T. Cadenbach, E. Hevia and A. R. Kennedy, Organometallics, 2013, 32, 480 CrossRef CAS.
- (a) A. Heine and D. Stalke, Angew. Chem., Int. Ed., 1992, 31, 854 CrossRef; (b) V. A. Pollard, S. A. Orr, R. McLellan, A. R. Kennedy, E. Hevia and R. E. Mulvey, Chem. Commun., 2018, 54, 1233 RSC.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Nature, 2018, 557, 92 CrossRef CAS.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, J. Am. Chem. Soc., 2019, 141, 1100 CrossRef.
- R. J. Schwamm, M. D. Anker, M. Lein and M. P. Coles, Angew. Chem., Int. Ed., 2019, 58, 1489 CrossRef CAS.
- S. Grams, J. Eyselein, J. Langer, C. Färber and S. Harder, Angew. Chem., Int. Ed., 2020, 59, 15982 CAS.
- A. J. Martinez-Martinez, D. R. Armstrong, B. Conway, B. J. Fleming, J. Klett, A. R. Kennedy, R. E. Mulvey, S. D. Robertson and C. T. O'Hara, Chem. Sci., 2014, 5, 771 RSC.
- D. R. Armstrong, W. Clegg, S. H. Dale, D. Graham, E. Hevia, L. M. Hogg, G. W. Honeyman, A. R. Kennedy and R. E. Mulvey, Chem. Commun., 2007, 598 RSC.
- P. C. Andrikopoulos, D. R. Armstrong, H. R. L. Barley, W. Clegg, S. H. Dale, E. Hevia, G. W. Honeyman, A. R. Kennedy and R. E. Mulvey, J. Am. Chem. Soc., 2005, 127, 6184 CrossRef CAS.
- W. Clegg, S. H. Dale, R. W. Harrington, E. Hevia, G. W. Honeyman and R. E. Mulvey, Angew. Chem., Int. Ed., 2006, 45, 2374 CrossRef CAS.
- A. Hernán-Gόmez, T. D. Bradley, A. R. Kennedy, Z. Livingstone, S. D. Robertson and E. Hevia, Chem. Commun., 2013, 49, 8659 RSC.
- A. G. M. Barrett, T. C. Boorman, M. R. Crimmin, M. S. Hill, G. Kociok-Köhn and P. A. Procopiou, Chem. Commun., 2008, 5206 RSC.
- M. De Tullio, A. Hernán-Gómez, Z. Livingstone, W. Clegg, A. R. Kennedy, R. W. Harrington, A. Antiñolo, A. Martínez, F. Carrillo-Hermosilla and E. Hevia, Chem.–Eur. J., 2016, 22, 17646 CrossRef CAS.
- L. Davin, A. Hernán-Gómez, C. McLaughlin, A. R. Kennedy, R. McLellan and E. Hevia, Dalton Trans., 2019, 48, 8122 RSC.
- C. Brinkmann, A. G. M. Barrett, M. S. Hill and P. A. Procopiou, J. Am. Chem. Soc., 2012, 134, 2193 CrossRef CAS.
- (a) C. Glock, H. Görls and M. Westerhausen, Chem. Commun., 2012, 48, 7094 RSC; (b) C. Glock, F. M. Younis, S. Ziemann, H. Görls, W. Imhof, S. Krieck and M. Westerhausen, Organometallics, 2013, 32, 2649 CrossRef CAS; (c) F. M. Younis, S. Krieck, H. Görls and M. Westerhausen, Dalton Trans., 2016, 45, 6241 RSC.
- F. M. Younis, S. Krieck, H. Görls and M. Westerhausen, Organometallics, 2015, 34, 3577 CrossRef CAS.
- M. Fairley, L. Davin, A. Hernan-Gomez, A. R. Kennedy, C. T. O'Hara, J. Garcia-Alvarez and E. Hevia, Chem. Sci., 2019, 10, 5821 RSC.
- (a) H. Elsen, C. Färber, G. Ballmann and S. Harder, Angew. Chem., Int. Ed., 2018, 57, 7156 CrossRef CAS; (b) H. Elsen, J. Langer, M. Wiesinger and S. Harder, Chem.–Eur. J., 2020 DOI:10.1002/chem.202003862.
- H. Elsen, J. Langer, M. Wiesinger and S. Harder, Organometallics, 2020 DOI:10.1021/acs.organomet.0c00226.
- A. Bismuto, M. J. Cowley and S. P. Thomas, ACS Catal., 2018, 8, 2001 CrossRef CAS.
- A. D. Bage, T. A. Hunt and S. P. Thomas, Org. Lett., 2020, 22, 4107 CrossRef CAS.
- A. K. Jaladi, W. K. Shin and D. K. An, RSC Adv., 2019, 9, 26483 RSC.
- V. A. Pollard, M. A. Fuentes, A. R. Kennedy, R. McLellan and R. E. Mulvey, Angew. Chem., Int. Ed., 2018, 57, 10651 CrossRef CAS.
- L. E. Lemmerz, R. McLellan, N. R. Judge, A. R. Kennedy, S. A. Orr, M. Uzelac, E. Hevia, S. D. Robertson, J. Okuda and R. E. Mulvey, Chem.–Eur. J., 2018, 24, 9940 CrossRef CAS.
- R. J. Less, H. R. Simmonds, S. B. Dane and D. S. Wright, Dalton Trans., 2013, 42, 6337 RSC.
- V. A. Pollard, A. Young, R. McLellan, A. R. Kennedy, T. Tuttle and R. E. Mulvey, Angew. Chem., Int. Ed., 2019, 58, 12291 CrossRef CAS.
- W. Clegg, G. C. Forbes, A. R. Kennedy, R. E. Mulvey and S. T. Liddle, Chem. Commun., 2003, 406 RSC.
- Y. F. Liu, D. D. Zhai, X. Y. Zhang and B. T. Guan, Angew. Chem., Int. Ed., 2018, 57, 8245 CrossRef CAS.
- Y. Yamashita, H. Suzuki, I. Sato, T. Hirata and S. Kobayashi, Angew. Chem., Int. Ed., 2018, 57, 6896 CrossRef CAS.
- (a) P. Fleming and D. F. O'Shea, J. Am. Chem. Soc., 2011, 133, 1698 CrossRef CAS; (b) M. Blangetti, P. Fleming and D. F. O'Shea, J. Org. Chem., 2012, 77, 2870 CrossRef CAS; (c) A. Manvar, P. Fleming and D. F. O'Shea, J. Org. Chem., 2015, 80, 8727 CrossRef CAS.
- M. T. Muñoz, T. Cuenca and M. E. G. Mosquera, Dalton Trans., 2014, 43, 14377 RSC.
- C. Gallegos, V. Tabernero, F. M. García-Valle, M. E. G. Mosquera, T. Cuenca and J. Cano, Organometallics, 2013, 32, 6624 CrossRef CAS.
- M. Palenzuela, M. T. Muñoz, J. F. Vega, A. Gutierrez-Rodriguez, T. Cuenca and M. E. G. Mosquera, Dalton Trans., 2019, 48, 6435 RSC.
- J. A. Garden, P. K. Saini and C. K. Williams, J. Am. Chem. Soc., 2015, 137, 15078 CrossRef CAS.
- G. Trott, J. A. Garden and C. K. Williams, Chem. Sci., 2019, 10, 4618 RSC.
- A. C. Deacy, C. B. Durr, J. A. Garden, A. J. P. White and C. K. Williams, Inorg. Chem., 2018, 57, 15575 CrossRef CAS.
- M. T. Muñoz, M. Palenzuela, T. Cuenca and M. E. G. Mosquera, ChemCatChem, 2018, 10, 936 CrossRef.
- A. C. Deacy, A. F. R. Kilpatrick, A. Regoutz and C. K. Williams, Nat. Chem., 2020, 12, 372 CrossRef CAS.
- (a) M. Bochmann, Organometallics, 2010, 29, 4711 CrossRef CAS; (b) M. C. Baier, M. A. Zuiderveld and S. Mecking, Angew. Chem., Int. Ed., 2014, 53, 9722 CrossRef CAS; (c) S. K. Manda and H. W. Roesky, Acc. Chem. Rev., 2009, 42, 248 Search PubMed.
- H. S. Zijlstra and S. Harder, Eur. J. Inorg. Chem., 2015, 19 CrossRef CAS.
- (a) G. Bai, S. Singh, H. W. Roesky, M. Noltemeyer and H. G. Schmidt, J. Am. Chem. Soc., 2005, 127, 3449 CrossRef CAS; (b) Y. Yang, T. Schulz, M. John, Z. Yang, V. M. Jiménez-Pérez, H. W. Roesky, P. M. Gurubasavaraj, D. Stalke and H. Ye, Organometallics, 2008, 27, 769 CrossRef CAS.
- (a) C. Boulho, H. S. Zijlstra, A. Hofmann, P. H. M. Budzelaar and S. Harder, Chem.–Eur. J., 2016, 22, 17450 CrossRef CAS; (b) C. Boulho, H. S. Zijlstra and S. Harder, Eur. J. Inorg. Chem., 2015, 2132 CrossRef CAS.
Footnote |
| † Dedicated to Prof. Alan Welch on the occasion of his retirement from Heriot–Watt University. |
| This journal is © The Royal Society of Chemistry 2021 |