
Graphene-induced growth of Co3O4 nanoplates with modulable oxygen vacancies for improved OER properties†
Lei
Qi
ab,
Mei
Wang
ab and
Xinheng
Li
 *a
*a
aThe State Key Laboratory for Oxo Synthesis and Selective Oxidation, Suzhou Research Institute of LICP, Lanzhou Institute of Chemical Physics (LICP), Chinese Academy of Sciences, Lanzhou, 730000, China. E-mail: xinhengli@licp.cas.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 28th April 2021
Abstract
Transition metal oxide/hydroxide is intensively studied for the oxygen evolution reaction (OER). Herein, the graphene-induced growth of Co3O4 nanoplates with modulable oxygen vacancies via a hydrothermal treatment is reported. With the increase of reaction time before the formation of Co(OH)2, the oxygen vacancies and conductivity of Co3O4 nanoplates continued to increase resulting in dramatically enhanced OER performances. An ultralow overpotential of 354 mV at a current density of 100 mA cm−2 and a Tafel slope as low as 63.24 mV dec−1 in 1 M KOH solution were obtained, superior to those of most reported oxides and RuO2.
Introduction
Owing to the depletion of fossil fuels and the increase of the greenhouse gases, energy conversion processes and storage devices, such as regenerative fuel cells, electrolysis cells, and metal–air batteries, have attracted great attention in the past decades.1–3 Among them, water splitting via photo electrocatalytic or electrocatalytic reaction is promising for producing reliable clean energy.4–6 The water splitting process consists of two parts: the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER). However, the OER often has a sluggish kinetics thus requiring high overpotentials, which will result in high energy consumption.5,7–11 One of the challenges is to develop excellent electrocatalysts using earth-abundant materials, with high stability and prominent catalytic activity. At present, noble-metal oxides (such as IrO2 and RuO2) are commercially used as OER catalysts, but their scarcity and high cost limit their applications. In recent decades, transition metal oxide/hydroxide has attracted great interests for the OER.Transition metal-based compounds, such as sulfides, hydroxides, oxides, and phosphides, have been reported for the OER owing to their tunable electronic structures and abundant active sites. For example, Co3O4, an inexpensive earth-abundant material, has been considered to be promising for the OER process because of its high electrical conductivity, adjustable structure, and oxygen defects.12–16 Generally, oxygen defects (such as oxygen vacancies and interstitials) are found to be vitally affecting the material properties such as electronic structure,17 conductivity,10 and intrinsic catalytic activity.18,19 Graphene has been intensively and extensively studied due to its excellent conductivity. It can also be used as a template to form two dimensional nanomaterials. Herein, we report the graphene-induced growth of Co3O4 nanoplates (denoted as g-Co3O4) via a hydrothermal treatment (HT) of pre-formed Co3O4, where the oxygen vacancies are modulated by the reduction time until Co(OH)2 was formed. Note that graphene oxide was reduced during the hydrothermal process. Graphene assisted the formation of Co3O4 nanoplates, by contrast to the formation of bulky materials without graphene. The OER activity was evaluated over the as-obtained g-Co3O4 electrocatalysts. And oxygen defects and conductivity were analyzed with respect to the HT time.
Results and discussion
Fig. 1 shows the schematic diagram of the formation of g-Co3O4 nanoplates via a facile hydrothermal reaction. The morphology and particle size distribution diagram with respect to different reaction times were characterized by scanning electron microscopy (SEM). As shown in Fig. 2a and S1,† pristine Co3O4 has sphere-like morphology with an average size of 60 nm. The SEM images of g-Co3O4 are shown in Fig. 2b–e. The TEM image (Fig. S2†) of the home-made graphene shows clear wrinkles indicating an ultrathin film. When the HT time was 3 h, triangular nanoplates formed with an average lamellar size of ca. 0.17 μm and were uniformly coated by gauze-like graphene. Fig. 2c shows a further change of the material morphology as the reaction proceeded. Hexagonal nanoplates formed after 6 h, where graphene behaved like glue between the nanoplates. It can be clearly observed that the lamellar size increased fivefold to ca. 0.82 μm. After 6 h, the morphology of hexagonal nanoplates did not change much but its size gradually increased. As control experiments, the pristine Co3O4 samples were treated by the HT process without graphene, as shown in Fig. S3–S6,† which exhibited bulky materials instead of any nanoplate morphology. Therefore, we can come to a short conclusion that as a template, graphene assisted the formation of Co3O4 nanoplates.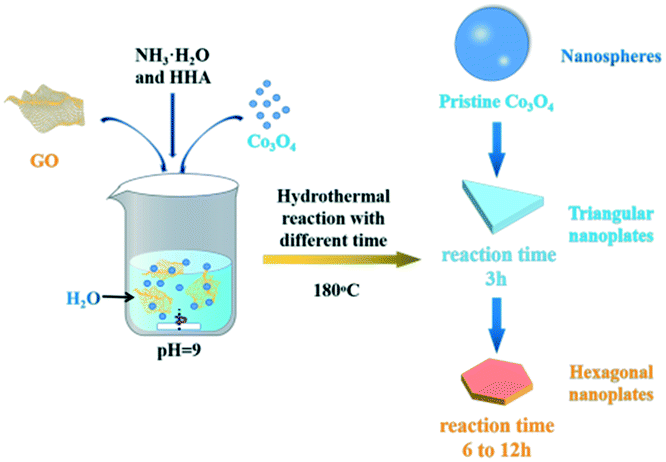 | ||
| Fig. 1 Schematic illustration of the preparation of g-Co3O4 nanoplates via a hydrothermal treatment. | ||
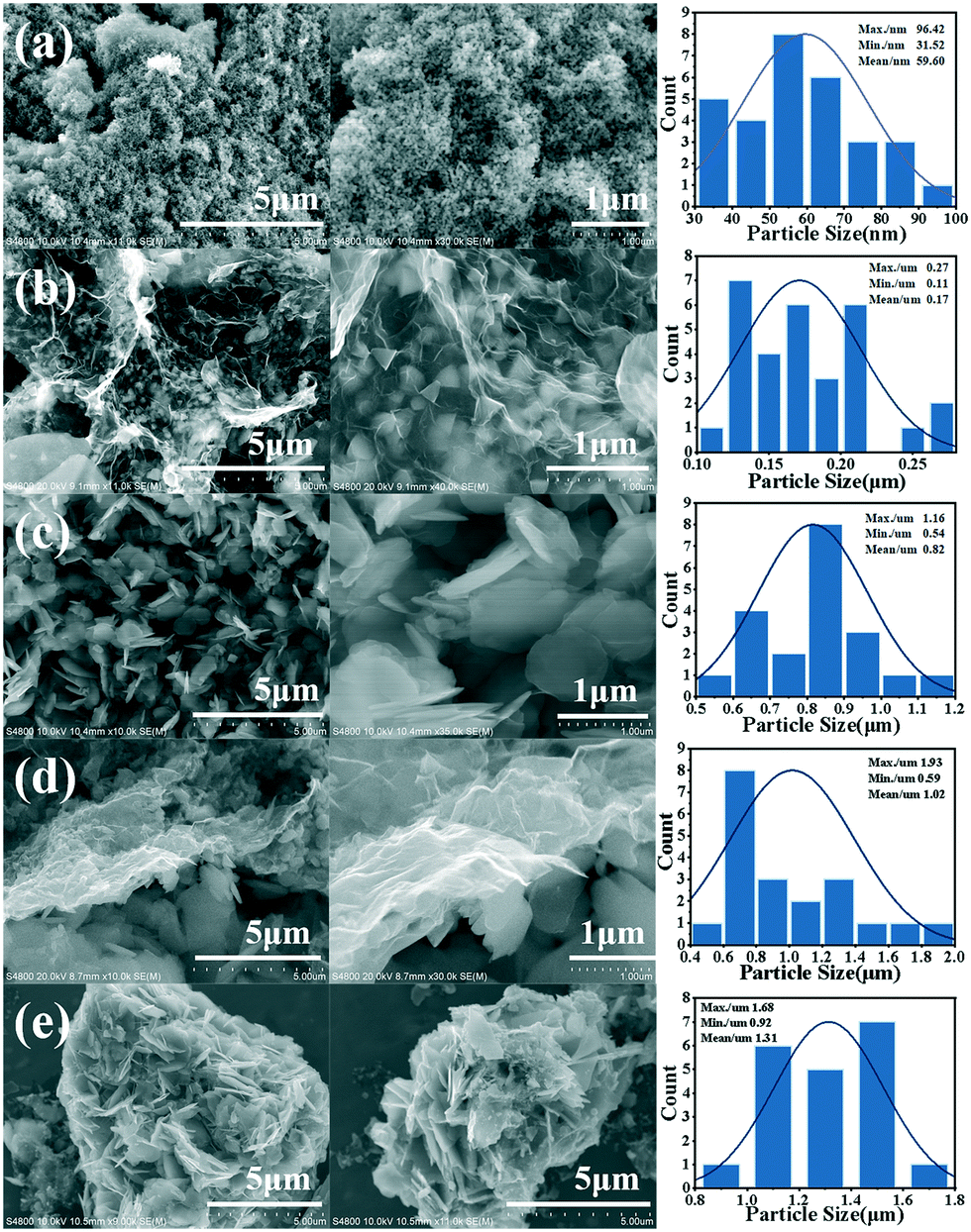 | ||
| Fig. 2 SEM images and particle size distribution diagrams of pristine Co3O4 (a) and g-Co(OH)2 with different hydrothermal reaction times: (b) 3 h; (c) 6 h; (d) 9 h and (e) 12 h. | ||
The as-obtained samples of g-Co3O4 were characterized by XRD, as shown in Fig. 3a. The peaks at 19.00, 31.28, 36.88, 44.88, 55.68 and 59.38 and 65.28° can be attributed to the (111), (220), (311), (400), (422), (511) and (440) planes of Co3O4 (PDF no. 43-1003). It can be found that the pristine Co3O4 has a cubic crystal structure, and its cell parameters are: a = b = c = 8.084. As for g-Co3O4 3 h and 6 h, it could be clearly observed that the characteristic peak intensity of Co3O4 decreased significantly. This proved that the crystal structure of the material had started to transition from a cubic structure of Co3O4 to a hexagonal structure of Co(OH)2. In other words, oxygen vacancies appeared in g-Co3O4 after 3 h due to reduction reaction. As for the sample of g-Co3O4 12 h, the diffraction peak of the material is consistent with that of Co(OH)2 with a hexagonal structure. And the cell parameters are a = b =3.183, c =4.652. So, g-Co3O4 12 h is considered as Co(OH)2, and the remaining samples are Co3O4 with adjustable oxygen vacancies. Note that there is no obvious graphene signal in the catalyst, suggesting that the graphene content is very low. The existence of oxygen vacancies over the as-obtained samples were further confirmed by X-ray photoelectron spectroscopy (XPS). The pristine Co3O4 exhibits two subpeaks. The one at 529.8 eV corresponds to lattice O arising from the Co–O bond, and the other at 531.4 eV corresponds to oxygen vacancies. As the HT time increased until 9 h, the peak intensities and areas of the oxygen vacancies increased. From 9 h to 12 h, the formation of Co(OH)2 made the oxygen defects disappear, corroborated by the Co 2p spectra as shown in Fig. S7.† The peaks at 779.8 eV and 781.3 eV for Co3O4, g-Co3O4 3 h and g-Co3O4 6 h correspond to Co3+ and Co2+, respectively. As the HT reaction time proceeded to 9 h and 12 h, only Co2+ was observed.
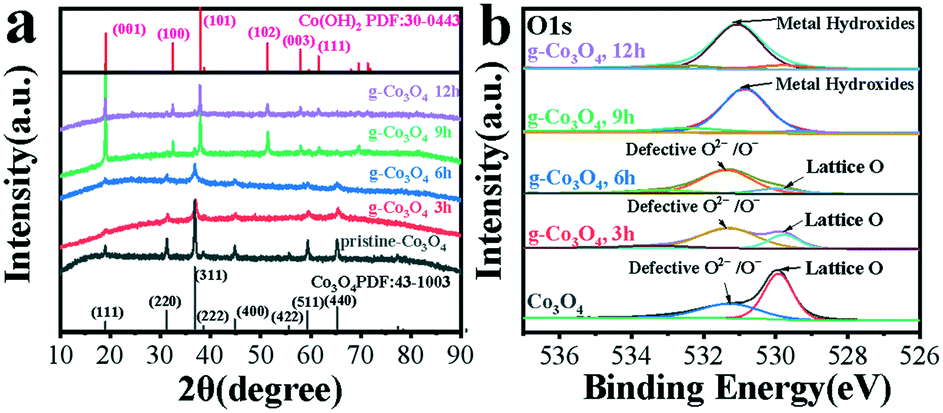 | ||
| Fig. 3 (a) The XRD patterns and (b) O1s spectra of pristine Co3O4 (black) and g-Co3O4 with different hydrothermal reaction times: (red) 3 h; (blue) 6 h; (green) 9 h and (purple) 12 h. | ||
OER performances over pristine Co3O4 and g-Co3O4 were evaluated by cyclic voltammetry (CV) and linear sweep voltammetry (LSV). Fig. S8 and S9† show the CV curves of the g-Co3O4 and control experiments, as well as the LSV curves of the control experiments. It is proved that the active species was CoO(OH). By contrast, the onset potential to form CoOOH decreased from 0.308 V to 0.272 V, and the overpotential decreased by 20 mV at a current density of 100 mA cm−2 over the g-Co3O4 6 h electrocatalyst. Fig. 4 shows that the onset potentials to form CoOOH were only 0.256 V and 0.239 V, respectively, over the g-Co3O4 3 h and g-Co3O4 6 h samples. And their overpotentials were 36 mV and 71 mV at 100 mA cm−2, respectively, much lower than those over pristine Co3O4, g-Co3O4 9 h and g-Co3O4 12 h.
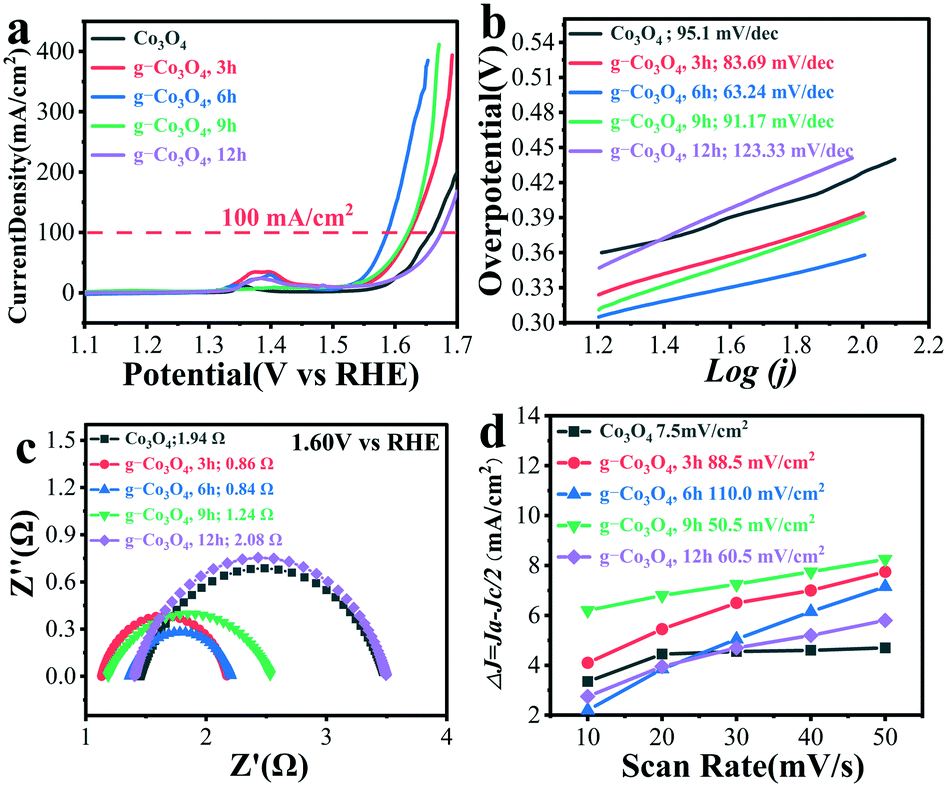 | ||
| Fig. 4 (a) LSV curves of pristine Co3O4 (black) and g-Co3O4 with different hydrothermal reaction times: (red) 3 h; (blue) 6 h; (green) 9 h and (purple) 12 h. (b) Related Tafel slope. (c) and (d) Nyquist plots and double layer capacitance of the as-prepared catalysts. | ||
The Tafel slope was employed to confirm the kinetic process of electron migration in the OER process. As shown in Fig. 4b, Co3O4 with a complete crystal structure exhibited a Tafel slope of 95.1 mV dec−1. Electrochemical impedance spectroscopy (EIS) was then carried out to further probe the transfer kinetics of charge carriers (Fig. S9c† and Fig. 4c). Pristine Co3O4 show a small charge transfer resistance (Rct) of 4.89 Ohm at a potential of 1.60 V vs. RHE. As the HT time increased, the Rct significantly decreased. The as-prepared g-Co3O4 6 h exhibited an Rct value as low as 0.84 Ω, corroborating the fast kinetics of water oxidation. The double-layer capacitance test further demonstrates that as the HT time increased, the electrochemical surface area (ECSA) increased. Combining with the above-mentioned results, we can conclude that the increase of oxygen vacancies and conductivity largely improved the catalytic activity.
Conclusions
In summary, the graphene-induced growth of Co3O4 nanoplates with modulable oxygen vacancies via a hydrothermal treatment was reported. With the increase of the hydrothermal reaction time before the formation of Co(OH)2, the oxygen vacancies and conductivity of Co3O4 increased, which resulted in greatly improved OER performances. An ultralow overpotential of 354 mV at a current density of 100 mA cm−2 and a Tafel slope as low as 63.24 mV dec−1 in 1 M KOH solution were obtained, superior to those of most reported oxides and RuO2. The enhanced activity can be attributed to the nanoplate morphology, abundant oxygen vacancies and improved conductivity. Our strategy for the rational design of non-precious metal catalysts with superior OER activity can be applied to other systems, which may find ways for practical applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21872157 and 21573263), the National Key Research and Development Program of China from Ministry of Science and Technology of China (2016YFE0105700), and the Natural Science Foundation of Jiangsu Province (BK20191198).References
- T. Chao, X. Luo, W. Chen, B. Jiang, J. Ge, Y. Lin, G. Wu, X. Wang, Y. Hu, Z. Zhuang, Y. Wu, X. Hong and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 16047–16051 Search PubMed.
- C. C. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 Search PubMed.
- M. S. Dresselhaus and I. L. Thomas, Nature, 2001, 414, 332–337 Search PubMed.
- E. Cui, G. Hou, X. Chen, M. Xie, F. Zhang, Y. Deng, Y. Wu and X. Yang, Langmuir, 2021, 37, 894–907 Search PubMed.
- Q. Hu, X. Huang, Z. Wang, G. Li, Z. Han, H. Yang, P. Liao, X. Ren, Q. Zhang, J. Liu and C. He, Small, 2020, 16, e2002210 Search PubMed.
- H. Yan, Y. Xie, A. Wu, Z. Cai, L. Wang, C. Tian, X. Zhang and H. Fu, Adv. Mater., 2019, 31, e1901174 Search PubMed.
- Z. Zhang, H. Zhang, Y. R. Yao, J. J. Wang, H. Guo, Y. D. Deng and X. P. Han, ChemSusChem, 2021, 14, 1659–1673 Search PubMed.
- Z. H. Zang, X. W. Wang, X. Li, Q. L. Zhao, L. L. Li, X. J. Yang, X. F. Yu, X. H. Zhang and Z. M. Lu, ACS Appl. Mater. Interfaces, 2021, 13, 9865–9874 Search PubMed.
- D. Wu, K. Kusada, S. Yoshioka, T. Yamamoto, T. Toriyama, S. Matsumura, Y. Chen, O. Seo, J. Kim, C. Song, S. Hiroi, O. Sakata, T. Ina, S. Kawaguchi, Y. Kubota, H. Kobayashi and H. Kitagawa, Nat. Commun., 2021, 12, 1145 Search PubMed.
- Z. Wei, W. C. Wang, W. L. Li, X. Q. Bai, J. F. Zhao, E. C. M. Tse, D. L. Phillips and Y. F. Zhu, Angew. Chem., Int. Ed., 2021, 60, 8236–8242 Search PubMed.
- P. Plate, C. Hohn, U. Bloeck, P. Bogdanoff, S. Fiechter, F. F. Abdi, R. van de Krol and A. C. Bronneberg, ACS Appl. Mater. Interfaces, 2021, 13, 2428–2436 Search PubMed.
- N. F. Yu, W. Huang, K. L. Bao, H. Chen, K. Hu, Y. Zhang, Q. H. Huang, Y. Zhu and Y. P. Wu, Dalton Trans., 2021, 50, 2093–2101 Search PubMed.
- Y. Wu, Z. Xiao, Z. Jin, X. Li and Y. Chen, J. Colloid Interface Sci., 2021, 590, 321–329 Search PubMed.
- J. X. Flores-Lasluisa, F. Huerta, D. Cazorla-Amoros and E. Morallon, Nanomaterials, 2020, 10, 22 Search PubMed.
- X. Yang, J. Chen, Y. Chen, P. Feng, H. Lai, J. Li and X. Luo, Nano-Micro Lett., 2018, 10, 15 Search PubMed.
- G. Zhang, J. Yang, H. Wang, H. Chen, J. Yang and F. Pan, ACS Appl. Mater. Interfaces, 2017, 9, 16159–16167 Search PubMed.
- A. Karmakar, K. Karthick, S. Kumaravel, S. S. Sankar and S. Kundu, Inorg. Chem., 2021, 60, 2023–2036 Search PubMed.
- J. Dai, N. Shao, S. Zhang, Z. Zhao, Y. Long, S. Zhao, S. Li, C. Zhao, Z. Zhang and W. Liu, ACS Appl. Mater. Interfaces, 2021, 13, 7259–7267 Search PubMed.
- H. Liu, H. Fu, Y. Liu, X. Chen, K. Yu and L. Wang, Chemosphere, 2021, 272, 129534 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ce00255d |
| This journal is © The Royal Society of Chemistry 2021 |