
AB11O16(OH)2 (A = K and Cs): interpenetrating 2D layers with large birefringence†
Zhen
Chen‡
ab,
Jingyu
Guo‡
ab,
Shujuan
Han
 a,
Hao
Zeng
a,
Zhihua
Yang
a and
Shilie
Pan
*a
a,
Hao
Zeng
a,
Zhihua
Yang
a and
Shilie
Pan
*a
aCAS Key Laboratory of Functional Materials and Devices for Special Environments, Xinjiang Technical Institute of Physics & Chemistry, CAS, Xinjiang Key Laboratory of Electronic Information Materials and Devices, 40-1 South Beijing Road, Urumqi 830011, China. E-mail: slpan@ms.xjb.ac.cn; Fax: +86 991 3838957; Tel: +86 991 3674558
bCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, China
First published on 20th November 2020
Abstract
Two new isostructural alkali metal hydroxyborates, AB11O16(OH)2 (A = K and Cs), with two-dimensional (2D) interpenetrating [B11O18(OH)2]∞ layers have been successfully obtained. The calculated birefringences of KB11O16(OH)2 and CsB11O16(OH)2 are 0.104 and 0.100 at 546 nm, respectively. Moreover, KB11O16(OH)2 exhibits a short DUV cutoff edge (195 nm).
Birefringent materials have the capability to modulate polarized light, so they are expected to be used as polarization devices, which are essential in photoelectric modulators, photoelectric switches, optical isolators and coherent systems.1–4 In addition, birefringence is also one of the pivotal optical parameters for nonlinear optical (NLO) crystals. Furthermore, moderate birefringence enables the crystal to achieve phase matching at a specific wavelength.5–7 Borates, in particular, can be regarded as an excellent candidate for exploring birefringent materials due to various coordination patterns of B (BO3 or BO4), which can further be integrated via sharing oxygen atoms to form isolated groups, one-dimensional (1D) chains, 2D layers or 3D networks.8,9 As is known to all, borates with coplanar arrays of isolated [BO3]3−, [B2O5]4−, or [B3O6]3− groups might produce large birefringence,10–14 such as α-BaB2O4 (α-BBO, 0.122 at 546 nm),2 Li2Na2B2O5 (0.095 at 532 nm),15 and Ba2Ca(B3O6)2 (0.125 at 546 nm).16 Particularly, a 1D chain structure polymerized by coplanar [BO3]3− is expected to have a larger average anisotropic polarizability, generating a large birefringence, for example, Ca(BO2)2 (0.136 at 532 nm).17 In addition, the stereochemical activity lone pair (SCALP) cations also make a remarkable contribution to their huge birefringence except for the [BO3]3− anion group,18–22 such as α-BiB3O6,23 SbB3O6,24 and Sn2B5O9Cl.25 However, applications of the SCALP-containing borates in the ultraviolet (UV) region are limited due to the red shift of the cutoff edge.
How to design UV birefringent materials? In crystal engineering, it has been proved to be an effective way to obtain new crystals based on a well-known structural template for ion substitution or co-substitution.26–28 Through the chemical substitution strategy, a new crystal Sn2B5O9Cl with a large birefringence gain (16 times that of its isostructural Ba2B5O9Cl) has been synthesized after the substitution of Ba2+ with Sn2+;25,29 Na3Ba2(B3O6)2F (NBBF) can be structurally considered as a derivative from co-substituting the Ba2+ atoms in α-BBO with 3Na+ and F− atoms in NBBF;30 K7MIIRE2(B5O10)3 (MII = Ca, Sr, Ba, K/RE0.5; RE = Y, Lu, Gd)31 are derived from simultaneously replacing the Ba2+ groups in β-BBO with multiple rare-earth, alkali, and alkaline-earth metals. Recently, research has shown that introducing a fluorine atom into a BO4 tetrahedron forms BOF groups (BO3F, BO2F2, and BOF3),32 which are beneficial to improve the birefringence of borates due to their large polarizability anisotropy. In addition, hydroxyl (OH−) and fluorine (F−) have similar electronic compositions; therefore, hydroxyborates may show similar optical properties to fluorooxoborates if the arrangement of the functional active units is reasonable.33 Prior to this work, we obtained NH4B11O16(OH)2,34 which shows a short cutoff edge and large birefringence. In this work, guided by the above design strategy, we used alkali metals (K and Cs) to replace NH4, and finally two new crystals KB11O16(OH)2 (KBOH) and CsB11O16(OH)2 (CBOH) exhibiting relatively large birefringence and short UV cutoff edges were acquired in a closed system. Herein, characterization results on the optical and thermal properties, as well as theoretical calculation, were also presented.
The polycrystalline sample of KBOH was prepared through a solid-state reaction in a sealed system (Experimental section in the ESI†). The purity of the polycrystalline sample was confirmed by powder X-ray diffraction, and the experimental pattern agrees well with the calculated result, except for an impurity peak (Fig. S1 in the ESI†).
KBOH and CBOH belong to the centrosymmetric space group C2/c (no. 15) in the monoclinic system, and are isostructural to AB11O16(OH)2 (A = NH4, Rb).34,35 Therefore, KBOH is selected as a representative to be discussed in detail. The lattice parameters of the crystal are a = 14.163(17) Å, b = 10.101(12) Å, c = 10.664(12) Å, β = 106.827(15)° and Z = 4 (Table S1 in the ESI†). In the asymmetric unit, there are one, six, nine and one crystallographically unique K, B, O, and H atoms, respectively (Table S2 in the ESI†). It exhibits a 3D framework composed of KO8, BO3, BO4 and BO2(OH) groups. The B(1)O2(OH), B(3)O3 and B(6)O3 units are linked by sharing vertex O atoms to form a 3-membered ring (3-MRI), B3O5(OH) unit. The B(2)O3, B(4)O3 and B(5)O4 units are connected with each other by sharing vertex O atoms building 3-MRII, which is further linked to another 3-MRII sharing B(5)O4 to form a B5O10 double-loop. Then one [B5O10] double-loop and two [B3O5(OH)] single-rings form the fundamental building block (FBB) by sharing oxygen atoms, which is [B11O18(OH)2] (Fig. 1a). Finally, the FBBs are linked together to construct two-dimensional (2D) interpenetrating [B11O18(OH)2]∞ layers, which are separated by charge balancing K+ cations (Fig. 1b).
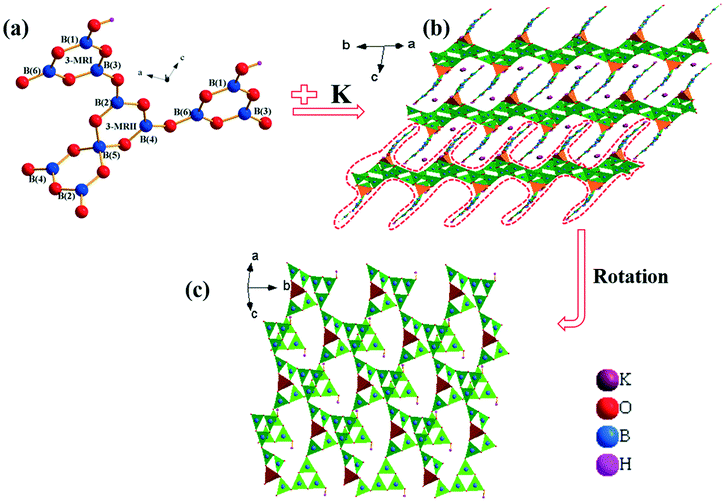 | ||
| Fig. 1 (a) The [B11O18(OH)2] group; (b) the crystal structure of KBOH; (c) the unique single-layered anionic arrangement from other direction (brown tetrahedron, [BO4]5− groups; green triangle, [BO3]3− groups). | ||
With respect to the B atoms, in the structure (Fig. 1a), the B(1), B(2), B(3), B(4) and B(6) atoms are coordinated by three O atoms, forming BO3 triangles with B–O bond lengths falling in a reasonable range (Table S3 in the ESI†). Further, the B(5) atoms are bonded to four O atoms forming BO4 tetrahedra with the B–O bond lengths ranging from 1.459(3) to 1.474(3) Å. All bond lengths are suitable, consistent with those observed in other borates. The K cations are bonded to eight O atoms, forming distorted KO8 polyhedra (Fig. S2 in the ESI†), which are isolated in the structure. The K–O bond lengths are in the range of 2.701(4)–3.098(4) Å (Table S3†).
To the best of our knowledge, sixteen hydroxyl potassium borate compounds have been reported besides the title compound. Table S4 in the ESI† summarizes the formula, space group, B–O anion framework, and cutoff edge for these compounds. It can be seen that, except for K2(B5O8(OH))(H2O)2 and the title compound, almost all hydroxyl potassium borates exhibit isolated anion groups and 1D chains, which is associated with the ratio of potassium to boron and the presence of hydroxyl groups in their structures. In general, introducing hydroxyl into borates could avoid the appearance of terminal oxygen atoms (the terminal oxygen atoms are connected to H) and reduce the dimensions of the B–O framework. In K2(B10O14(OH)4)(H2O), KB5O7(OH)2(H2O), K2(B6O9(OH)2), and KB5O7(OH)2, the FBBs contain two terminal oxygen atoms and are connected together through a common vertex to form a 1D chain. In addition, the FBBs [B5O10(OH)2] of K4B10O15(OH)4 form a 1D chain by sharing five terminal oxygen atoms. The FBBs of K2(B5O8(OH))(H2O)2 and the title compound are B5O10(OH) and B11O18(OH)2, respectively, containing four terminal oxygen atoms, which can be bonded with other ones through sharing oxygen atoms to form a higher-dimensional anionic framework. According to our conclusion, KBOH is the first compound with two interpenetrating layers among all the hydroxyl potassium borates. We also summarized hydroxyl cesium borates in Table S5 in the ESI,† and there are six cases in addition to the compound in this paper. Cs(B(OH)4)(H2O)2, Cs2(B4O5(OH)4)(H2O)3, Cs2(B12(OH)12)(H2O)2, and Cs2(B12(OH)12)(H2O)2 contain isolated B–O frameworks. In contrast, Cs0.4(H3O)0.6B3O5, CsB7O10(OH)2 and CBOH exhibit 2D B–O–H layers in their structures.
The IR spectrum of KBOH is presented in Fig. S3,† and the peaks are assigned by referring to ref. 34, 36, and 37. The peaks at 3315, 3228 and 1435 cm−1 are attributed to the stretching vibrations of the hydroxyl group O–H. Two obvious absorption peaks located at 1321 and 1278 cm−1 are caused by the asymmetric stretching in the BO3 groups. The peak at 1136 cm−1 stems from the in-plane bending of B–O–H. The absorption peak of the asymmetric stretching of the BO3 groups is located at 1056 cm−1. The absorption bands around 921, 885 and 810 cm−1 arise from the symmetric stretching of BO3. The main bands near 773 and 678 cm−1 originate from the out-of-plane bending mode of the B–O bond in BO3. Moreover, the crest below 600 cm−1 characterizes the bending pattern of the BO3 units.
As shown in Fig. 2, it can be found that the transmittance of KBOH is 19.1% at 195 nm, indicating that it can be used in the DUV area.
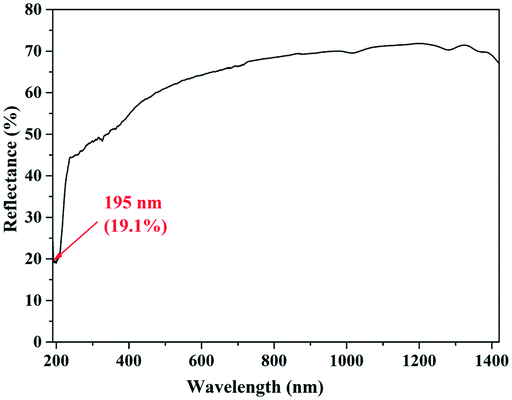 | ||
| Fig. 2 UV-vis-NIR optical spectrum of KBOH. | ||
In order to elucidate the mechanism between the optical properties and the structure of KBOH and CBOH, the first-principles calculations were performed based on density functional theory (DFT). The calculated band gaps using GGA (Fig. 3) are 6.04 and 5.85 eV with the direct band gap for KBOH (a) and CBOH (b), respectively. Simultaneously, the HSE06 method calculations show that KBOH and CBOH have large band gaps of 7.47 and 7.19 eV, respectively (Fig. S4 in the ESI†).
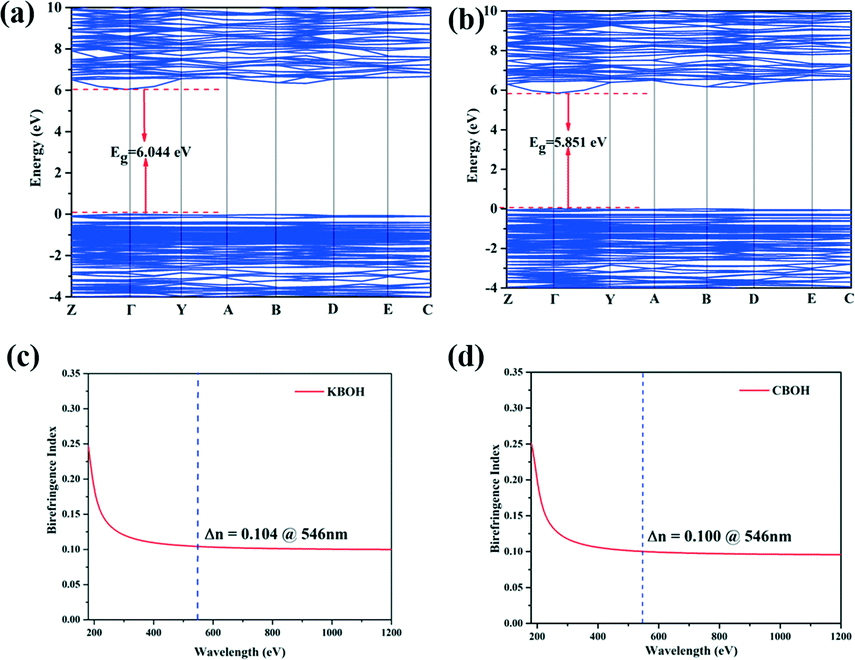 | ||
| Fig. 3 (a) The band structure of KBOH; (b) the band structure of CBOH; (c) the birefringence calculation of KBOH; (d) the birefringence calculation of CBOH. | ||
To understand the composition and origin of the electronic structures, the partial density of states (PDOS) for the compounds was calculated (Fig. 4). We select KBOH as a representative to be described in detail. At the top of the valence band, from −9 eV to the Fermi level, it is mainly composed of O-p orbitals slightly hybridized with B-p and H-s orbitals. The conduction bands from 6.2 eV to 14 eV are mainly contributed by B-p and B-s orbitals with slight contribution from O-p, H-s and K-p orbitals. The electron distribution near the Fermi level implies that the O-p orbitals slightly hybridized with the B-p orbital occupy the top area of the valence bands and the B-p orbital occupies the bottom region of the conduction bands. Therefore, the B–O bond in the title compounds plays a prominent role in the electronic structure and optical properties.
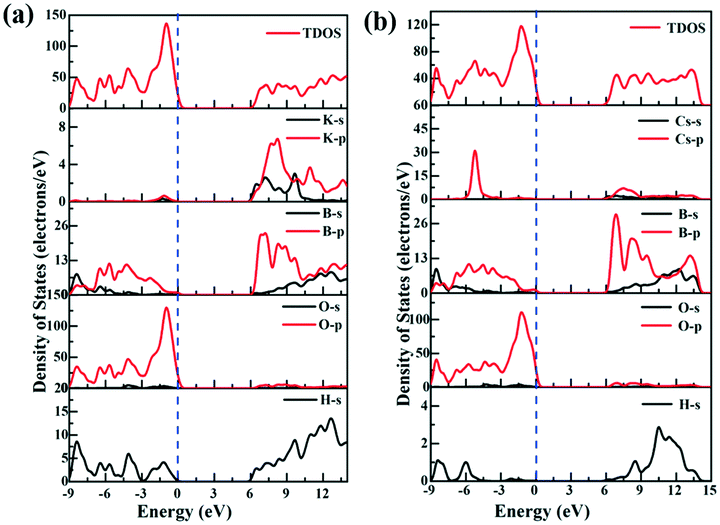 | ||
| Fig. 4 (a) Density of states (DOS) of KBOH; (b) density of states (DOS) of CBOH. | ||
As shown in Fig. 3c and d, the calculated birefringences of KBOH and CBOH are 0.104 and 0.100 at 546 nm by the first-principles methods. We measured the birefringence of KBOH on a polarizing microscope (ZEISS Axio Scope. A1). The maximum interference color is observed with the KBOH crystal under cross-polarized light and the thickness of the measured film is 12.50 μm (Fig. S5 in the ESI†). According to eqn (S2) in the ESI,† the retardation (R) value is 1.22 μm, and the birefringence of KBOH is 0.098 at the wavelength of 546 nm, indicating its potential application as a DUV birefringent material. Similarly, the calculated data fit the experimental ones well, so the accuracy of the calculation is also guaranteed well. Unfortunately, the measured birefringence value of CBOH is much smaller than the calculated value, and we speculate that the growth dissociates from the spindle direction.
Since the refractive index changes with the direction of light propagation in the crystal, birefringence will occur.25 In order to explain the source of birefringence, this work uses the electron localization function (ELF) to investigate the relevant performance of this structure. The ELF introduced by Becke and Edgecombe is one way to check the localization;38 they introduced the ELF range of [0, 1], in which a value close to 1 means that electrons are ‘well-localized’. The bonding behaviors in a crystal can be nicely illustrated by the ELF. As presented in Fig. S6,† it shows the strong hybridization between B and O atoms, which can be attributed to the covalent bond interaction between B and O atoms. Moreover, the bonding behavior interaction between the elements is inseparable from the electronic transition. Therefore, the main contributor to birefringence is directly related to the strong covalent interaction between B and O atoms.
Conclusions
In summary, two new hydroxyl borates KBOH and CBOH were successfully synthesized in a sealed system. The experimental birefringence (0.098 at 546 nm) of KBOH has been measured and agrees well with the calculated value. In addition, KBOH exhibits a short UV cutoff edge (195 nm), indicating that it can be used as an excellent material in the DUV region. The large birefringence is mainly due to the strong covalent interaction between O and B atoms. This work enriches the diversity of borates.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was completed with the help of the Shanghai Cooperation Organization Science and Technology Partnership Program (2020E01019), the Xinjiang Outstanding Young Talents in Science and Technology (2018Q004), the National Natural Science Foundation of China (U2003306, 61835014, and 51972336), and the Tianshan Innovation Team Program (2018D14001).Notes and references
- G. G. Kang, Q. F. Tan and G. F. Jin, Opt. Commun., 2010, 283, 4531–4535 CrossRef CAS.
- X. H. Meng, F. Liang, W. L. Yin, Z. S. Lin and M. J. Xia, New J. Chem., 2019, 43, 9766–9770 RSC.
- M. F. Weber, C. A. Stover, L. R. Gilbert, T. J. Nevitt and A. J. Ouderkirk, Science, 2000, 287, 2451–2456 CrossRef CAS PubMed.
- (a) P. Hlubina, D. Ciprian and L. Knyblová, Opt. Commun., 2006, 260, 535–541 CrossRef CAS; (b) S. Ghosh, W. H. Wang, F. M. Mendoza, R. C. Myers, X. Li, N. Samarth, A. C. Gossard and D. D. Awschalom, Nat. Mater., 2006, 5, 261–264 CrossRef CAS PubMed.
- (a) M. Mutailipu, M. Zhang, H. P. Wu, Z. H. Yang, Y. H. Shen, J. L. Sun and S. L. Pan, Nat. Commun., 2018, 9, 3089–3098 CrossRef PubMed; (b) Q. Huang, L. J. Liu, X. Y. Wang, R. K. Li and C. T. Chen, Inorg. Chem., 2016, 55, 12496–12499 CrossRef CAS PubMed; (c) K. M. Ok, Acc. Chem. Res., 2016, 49, 2774–2785 CrossRef CAS.
- (a) R. K. Li and Y. Y. Ma, CrystEngComm, 2012, 14, 5421–5424 RSC; (b) Z. Z. Zhang, Y. Wang, H. Li, Z. H. Yang and S. L. Pan, Inorg. Chem. Front., 2019, 6, 546–549 RSC; (c) W. G. Zhang, H. W. Yu, H. P. Wu and P. S. Halasyamani, Chem. Mater., 2017, 29, 2655–2668 CrossRef CAS.
- (a) L. Wu, S. Patankar, T. Morimoto, N. L. Nair, E. Thewalt, A. Little, J. G. Analytis, J. E. Moore and J. Orenstein, Nat. Phys., 2017, 13, 350–355 Search PubMed; (b) Y. C. Wu, T. Sasaki, N. Nakai, A. Yokotani, H. G. Tang and C. T. Chen, Appl. Phys. Lett., 1993, 62, 2614–2615 CrossRef CAS.
- J. S. Knyrim, P. Becker, D. Johrendt and H. Huppertz, Angew. Chem., Int. Ed., 2006, 45, 8239–8241 CrossRef CAS PubMed.
- (a) J. H. Huang, C. C. Jin, P. L. Xu, P. F. Gong, Z. S. Lin, J. W. Cheng and G. Y. Yang, Inorg. Chem., 2019, 58, 1755–1758 CrossRef CAS PubMed; (b) J. Chen, C. L. Hu, F. F. Mao, B. P. Yang, X. H. Zhang and J. G. Mao, Angew. Chem., Int. Ed., 2019, 58, 11666–11669 CrossRef CAS PubMed.
- C. T. Chen, Y. B. Wang, B. C. Wu, K. C. Wu, W. L. Zeng and L. H. Yu, Nature, 1995, 373, 322–324 CrossRef CAS.
- (a) F. L. Qin and R. K. Li, J. Cryst. Growth, 2011, 318, 642–644 CrossRef CAS; (b) Y. C. Hao, X. Xu, F. Kong, J. L. Songa and J. G. Mao, CrystEngComm, 2014, 16, 7689 RSC.
- (a) C. T. Chen, Y. C. Wu, A. D. Jiang, B. C. Wu, G. M. You, R. K. Li and S. J. Lin, J. Opt. Soc. Am. B, 1989, 6, 616–621 CrossRef CAS; (b) W. D. Cheng, H. Zhang, F. K. Zheng and J. T. Chen, Chem. Mater., 2000, 12, 3591–3594 CrossRef CAS.
- (a) B. C. Wu, D. Y. Tang, N. Ye and C. T. Chen, Opt. Mater., 1996, 5, 105–109 CrossRef CAS; (b) J. M. Zhao, H. K. Liu, X. D. Zhang, B. B. Zhang and Y. Wang, CrystEngComm, 2020, 22, 6495–6501 RSC.
- H. W. Yu, H. P. Wu, S. L. Pan, Z. H. Yang, X. L. Hou, X. Su, Q. Jing, K. R. Poeppelmeier and J. M. Rondinelli, J. Am. Chem. Soc., 2014, 136, 1264–1267 CrossRef CAS.
- M. Zhang, D. H. An, C. Hu, X. L. Chen, Z. H. Yang and S. L. Pan, J. Am. Chem. Soc., 2019, 141, 3258–3264 CrossRef CAS PubMed.
- Z. Jia, N. N. Zhang, Y. Y. Ma, L. W. Zhao, M. J. Xia and R. K. Li, Cryst. Growth Des., 2017, 17, 558–562 CrossRef CAS.
- X. L. Chen, B. B. Zhang, F. F. Zhang, Y. Wang, M. Zhang, Z. H. Yang, K. R. Poeppelmeier and S. L. Pan, J. Am. Chem. Soc., 2018, 140, 16311–16319 CrossRef CAS PubMed.
- (a) S. J. Han, M. Miriding, A. Tudi, Z. H. Yang and S. L. Pan, Chem. Mater., 2020, 5, 2172–2179 CrossRef; (b) K. M. Ok and P. S. Halasyamani, Angew. Chem., Int. Ed., 2004, 43, 5489–5491 CrossRef CAS; (c) X. L. Cao, F. Kong, C. L. Hu, X. Xu and J. G. Mao, Inorg. Chem., 2014, 53, 8816–8824 CrossRef CAS PubMed.
- M. Mutailipu, M. Zhang, B. B. Zhang, L. Y. Wang, Z. H. Yang, X. Zhou and S. L. Pan, Angew. Chem., Int. Ed., 2018, 57, 6095–6099 CrossRef CAS PubMed.
- H. Y. Li, H. P. Wu, X. Su, H. W. Yu, S. L. Pan, Z. H. Yang, Y. Lu, J. Han and K. R. Poeppelmeier, J. Mater. Chem. C, 2014, 2, 1704–1710 RSC.
- Q. Jing, Z. H. Yang, S. L. Pan and D. F. Xue, Phys. Chem. Chem. Phys., 2015, 17, 21968–21973 RSC.
- K. Wu, B. B. Zhang, Z. H. Yang and S. L. Pan, J. Am. Chem. Soc., 2017, 139, 14885–14888 CrossRef CAS PubMed.
- R. Fröhlich, L. Bohatẏ and J. Liebertz, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1984, 40, 343–344 CrossRef.
- Y. C. Liu, X. M. Liu, S. Liu, Q. G. Ding, Y. G. Li, L. N. Li, S. G. Zhao, Z. S. Lin, J. H. Luo and M. C. Hong, Angew. Chem., 2020, 59, 7793–7796 CrossRef CAS PubMed.
- J. Y. Guo, A. Tudi, S. J. Han, Z. H. Yang and S. L. Pan, Angew. Chem., Int. Ed., 2019, 58, 17675–17678 CrossRef CAS PubMed.
- Z. G. Xia and K. R. Poeppelmeier, Acc. Chem. Res., 2017, 50, 1222–1230 CrossRef CAS PubMed.
- J. Zhou, Z. G. Xia, M. S. Molokeev, X. W. Zhang, D. S. Peng and Q. L. Liu, J. Mater. Chem. A, 2017, 5, 15031–15037 RSC.
- (a) F. Li, Z. G. Xia, Y. Gong, L. Gu and Q. L. Liu, J. Mater. Chem. C, 2017, 5, 9281–9287 RSC; (b) R. Gautier, X. Y. Li, Z. G. Xia and F. Massuyeau, J. Am. Chem. Soc., 2017, 139, 1436–1439 CrossRef CAS PubMed.
- O. Ferro, S. Merlino, S. A. Vinogradova, D. Y. Pushcharovsky and O. V. Dimitrova, J. Alloys Compd., 2000, 305, 63–71 CrossRef CAS.
- H. Zhang, M. Zhang, S. L. Pan, Z. H. Yang, Z. Wang, Q. Bian, X. L. Hou, H. W. Yu, F. F. Zhang, K. Wu, F. Yang, Q. J. Peng, X. Y. Xu, K. B. Chang and R. P. Poeppelmeier, Cryst. Growth Des., 2015, 15, 523–529 CrossRef CAS.
- M. Mutailipu, Z. Q. Xie, X. Su, M. Zhang, Y. Wang, Z. H. Yang, M. R. S. A. Janjua and S. L. Pan, J. Am. Chem. Soc., 2017, 139, 18397–18405 CrossRef CAS PubMed.
- B. B. Zhang, G. Q. Shi, Z. H. Yang, F. F. Zhang and S. L. Pan, Angew. Chem., Int. Ed., 2017, 56, 3916–3919 CrossRef CAS.
- P. F. Gong, L. Kang and Z. S. Lin, J. Am. Chem. Soc., 2020, 142, 15157–15163 CrossRef CAS PubMed.
- C. M. Huang, F. F. Zhang, B. B. Zhang, Z. H. Yang and S. L. Pan, New J. Chem., 2018, 42, 12091–12097 RSC.
- S. S. Hu, S. W. Li and Z. Su, ChemistrySelect, 2019, 4, 3446–3449 CrossRef CAS.
- (a) Z. H. Miao, Y. Yang, Z. L. Wei, Z. H. Yang and S. L. Pan, Dalton Trans., 2020, 49, 1292–1299 RSC; (b) S. C. Wang, N. Ye and G. H. Zou, CrystEngComm, 2014, 16, 3971–3976 RSC.
- Y. Wang, J. Han, A. Tudi, Z. Z. Zhang, Z. H. Yang and S. L. Pan, CrystEngComm, 2019, 21, 6072–6079 RSC.
- D. Becke and K. E. Edgecombe, J. Chem. Phys., 1990, 92, 5397–5403 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC numbers 2023510 and 2023511 for KB11O16(OH)2 and CsB11O16(OH)2, respectively; crystallographic information file (CIF) for KB11O16(OH)2 and CsB11O16(OH)2; experimental section, optical characterization and theoretical calculations; tables of crystallographic data, selected bond lengths and angles, and the hydroxyl borates; experimental and calculated XRD patterns of KB11O16(OH)2, the coordination environments of K, and the electron localization function of KB11O16(OH)2. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ce01569e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |