
Highly enhanced UV-vis-NIR light harvesting and photoelectric conversion of a pyrene MOF by encapsulation of the D–π–A cyanine dye†
Xiao-Gang
Yang
 a,
Jian-Hua
Qin
*a,
Ya-Dan
Huang
a,
Zhi-Min
Zhai
a,
Lu-Fang
Ma
*a and
Dongpeng
Yan
*bc
a,
Jian-Hua
Qin
*a,
Ya-Dan
Huang
a,
Zhi-Min
Zhai
a,
Lu-Fang
Ma
*a and
Dongpeng
Yan
*bc
aCollege of Chemistry and Chemical Engineering, Luoyang Normal University, Henan Province Function-Oriented Porous Materials Key Laboratory, Luoyang 471934, P. R. China. E-mail: mazhuxp@126.co.m
bCollege of Chemistry, Beijing Normal University, Beijing Key Laboratory of Energy Conversion and Storage Materials, Beijing 100875, P. R. China. E-mail: yandp@bnu.edu.cn
cCollege of Chemistry, Zhengzhou University, Zhengzhou 450001, P. R. China
First published on 2nd November 2020
Abstract
The development of photofunctional metal–organic frameworks (MOFs) has received increasing attention owing to their adjustable molecular assembly modes and broad optoelectronic applications. However, efficient wide-range solar energy utilization and conversion of such materials remain scarce. Herein, an anionic pyrene-based MOF [(HOEtMIm)2][Mn3(TBAPy)2(μ2-OH2)2(H2O)2] (1) (H4TBAPy = 1,3,6,8-tetrakis(p-benzoic acid)-pyrene) was synthesized under ionothermal conditions. Through a facile ion exchange process, D–π–A cation dye cyanine (DMP) was successfully encapsulated into nanochannels of MOFs, forming molecular level heterojunctions of a DMP@1 donor–acceptor system. The emission color of 1 was largely modulated from cyan to orange through Förster resonance energy transfer as confirmed by both experiments and theoretical calculations. Moreover, the obtained DMP@1 shows a distinct light absorption from the UV/visible to the NIR region, and extremely high luminescence polarization anisotropy (0.97) due to the regular orientation and orderly arrangement of the linear DMP into the MOF host. Photoelectric measurements show that the photocurrent of DMP@1 is at least 15 times higher than that of 1 under monochromatic light.
Introduction
The rational design and controlled synthesis of photosensitizers for efficient light harvesting have attracted great attention in the fields of photovoltaics, artificial photosynthesis and photocatalysis.1,2 Pyrene and its derivatives with the four-ring-fused planar electron-enriched skeleton have been widely exploited as important photoelectric units in photochemistry and photoelectronics.3 It is well recognized that the optoelectronic performance of pyrene is highly dependent on the interchromophoric interactions and stacking modes.4 In this sense, metal–organic frameworks (MOFs), a fascinating class of crystalline porous materials,5–7 can be regarded as an excellent platform to arrange and control organic chromophores orderly at the molecular level. The intrinsic long-range order of such materials provides new opportunities for diverse applications such as light harvesting, photovoltaic devices, and so on.8 Over the past few years, some prominent advancements have been achieved by adjusting the interactions and aggregation state of pyrene-based chromophores in the MOF matrix.1b,4,9–12 For example, a 2D layered porous network with reversible dimensional adjustment has been reported, and the emissive properties are significantly altered by changes in non-convalent interactions upon the structural transformation.10b Besides the framework topology and the linker concentration, Stylianou's group found that the interchromophoric interactions and fluorescence emission of a pyrene-based MOF can also be affected by the external temperature.4a Very recently, Deria et al. have shown that the 1D pores of the NU-1000 MOF can be anchored by the porphyrin zinc(II) pigment for an efficient light-harvesting system.12 Despite these great efforts, the high efficiency of solar energy utilization and conversion based on photoactive MOFs remain scarce, particularly for the pyrene-based systems.1b,13In general, two key factors towards high light-harvesting need to be considered: photon absorption and charge carrier separation efficiency.12 In this context, the use of well-defined porous structures to encapsulate diverse chromophores supplies an attractive platform for the adjustment of host–guest interactions, photosensitization efficiency and photoelectric performances. Herein, a pyrene-tetralactic acid linker 1,3,6,8-tetrakis(p-benzoic acid)-pyrene (H4TBAPy) was selected to construct an anionic cluster-based MOF, [(HOEtMIm)2][Mn3(TBAPy)2(μ2-OH2)2(H2O)2] (1), by the use of an ionothermal synthesis with the aid of ionic liquid 1-(2-hydroxyethyl)-3-methylimidazolium chloride ([HOEtMIm]Cl). The MOF absorbs strongly in the 300–450 nm region but weakly in the visible region. By the incorporation of the cyanine cation dye into anionic MOF channels through a facile ion exchange process, the obtained host–guest MOF shows a distinct light absorption from the UV/visible light to NIR region. Cyanine cation 2-[4-(dimethylamino)styryl]-1-methylpyridinium (DMP) was selected as the guest molecule, since it is a typical type of D–π–A compound that possesses excellent two-photon absorption and electrochemical properties.14 In DMP, the styryl π-conjugated system is modified by an electron-deficient methylpyridinium and an electron-rich dimethylamino group. Thus, it features asymmetrical charge distribution, easily polarized electron cloud, facilitating intramolecular charge transfer. Typically, it tends to generate aggregation-caused quenching in the solid state and twisted intramolecular charge transfer in strong polar organic solvents.15 Benefitting from both confinement and isolation effects, the robust pore of MOFs can endow the encapsulated cyanine dye with preferential geometries, and thus minimize the above unfavorable factors.16 In the host–guest system, weak interactions (electrostatic and hydrogen bonding) between donor (D) and acceptor (A) molecular entities afford long-range order electron transfer channels, facilitating the suppression of charge recombination.8 As a result, the synergistic effect of the TBAPy antenna chromophore and cyanine energy acceptor in DMP@1 leads to highly enhanced photo-current responses and photoelectric conversion performance. Therefore, this work provides an effective way to construct a new type of pyrene based MOF host–guest for efficient light harvesting and photoelectric applications.
Results and discussion
Pale yellow block crystals of the title MOF can be obtained with high quality and large scale under ionothermal conditions. Herein, the [HOEtMIm]Cl ionic liquid not only serves as the solvent and structure-directing agent that facilitates crystal growth by adjusting the reaction kinetics, but also enters the resulting structure as charge compensating unit. Single crystal X-ray analysis indicates that the TBAPy ligand in 1 acts as a μ6-bridging, linking six Mn(II) ions through one μ2-η1:η1, one μ2-η1:η2 and two μ1-η1:η0 coordination modes for four carboxylate groups, respectively (Fig. 1a). Mn1, Mn2 and Mn1A are connected together by two μ2-H2O and eight carboxylate groups of eight TBAPy ligands to form a linear [Mn3(CO2)8(μ2-H2O)2] trinuclear Mn(II) cluster, as shown in Fig. 1b and Fig. S1a (ESI†). These {Mn3} clusters are linked by pairs of TBAPy ligands, extending along the bc plane to form an infinite 2D anionic double layer (Fig. 1c and Fig. S1b, ESI†). Topologically, the {Mn3} clusters can be considered as 8-coordinated nodes and TBAPy ligands as 4-coordinated nodes. Therefore, the layered structure of 1 can be viewed as a rare example of the (4,8)-connected network (Fig. 1d and Fig. S2, ESI†). Along the a-axis, the individual layers are held together, generating a 3D porous network by O–H⋯O hydrogen bonds between the terminal coordinated water molecules and carboxylate groups (Fig. 1e and Fig. S3a, b, ESI†). The effective free volume of 1 was calculated by PLATON analysis as 40.6% of the crystal volume (1015 out of the 2503 Å3 unit cell volume), which are occupied by heavily disordered [HOEtMIm]+ cations as counter charges.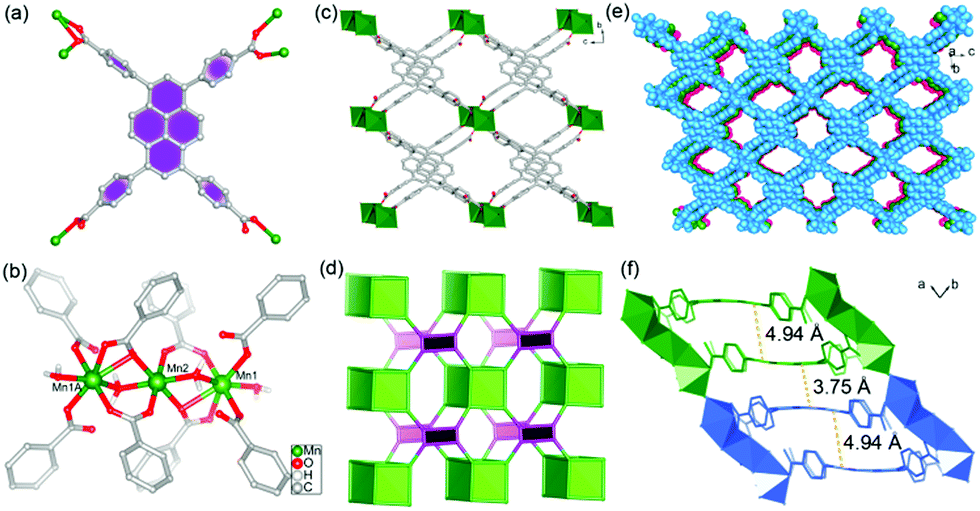 | ||
| Fig. 1 (a) The coordination modes of the TBAPy ligand in 1. (b) View of the trinuclear Mn(II) cluster in 1. Symmetry codes: a 1 − x, 2 − y, −z. (c) View of the 2D anionic double layer structure of 1 along the a direction. (d) Schematic representation of the (4,8)-connected network. (e) Spacing filling view of the 3D stacking structure along the a direction. (f) Distance between pyrene cores for inter- and intra-layers. (Layers show different colors for clarity.) | ||
It is worth noting that the TBAPy ligand has a seriously twisted conformation between the central pyrene core and four benzoate motifs, showing dihedral angles of 56.03, 60.45, 66.34 and 80.45°, respectively (Fig. S3c, ESI†). These values are distinctly different from the trinuclear Mg(II)4b and mononuclear Zn(II)13 based MOFs, which exhibit dihedral angles in the range of 49.83–88.32 and 57.83–75.33°, respectively. The diversities of molecular conformations arise from the coordinated metal ions. Meanwhile, the central pyrene core also exhibits slightly concave–convex arrangement. Concave pyrene cores within a 2D double layer face each other owing to the structure-directing effect of rigid {Mn3} clusters, which results in a large offset packing of TBAPy with a pyrene core distance of 4.94 Å. However, the small displacement between convex pyrene core inter-layers leads to overlap of the π systems with an inter-planar distance of 3.75 Å, suggesting the existence of π⋯π interactions in 1 (Fig. 1f). These results indicate that the formation of the {Mn3} cluster plays a significant role in arranging the packing style of TBAPy chromophores, in which the twisted conformation of TBAPy is in favor of electron–hole separation during the charge-transfer process.
To evaluate the photophysical properties, steady-state and time-resolved photoluminescence (PL) spectra of the pristine H4TBAPy and Mn cluster-based MOF 1 in the solid state were measured at room temperature. Excited at 380 nm, the solid-state PL spectrum of pristine H4TBAPy displays a major peak at 549 nm (Fig. 2a). In contrast, the hybrid system of 1 shows a distinct fluorescence emission, and possesses a shorter wavelength of 475 nm (λex = 335 nm), with a large blue shift of about 74 nm in comparison with the pristine H4TBAPy (Fig. 2b). Thus, the emission color of H4TBAPy can be extensively tuned from green to cyan upon being linked with Mn-clusters as shown in the insets. The large blue-shift is assigned to the fact that the strong electron-withdrawing effect of the Mn2+ cations can decrease the electron density of TBAPy in the MOF structure, and the spatial isolation effect can also reduce the molecular aggregation of the ligands to some extent. Such an observation is similar to the tetraphenylethylene based MOF.17 In addition, the time-resolved fluorescence decay gives a fluorescence lifetime of 1.51 ns for pure H4TBAPy and 3.43 ns for 1 (Fig. 2c and d). The prolonged lifetime is attributable to the fact that the TBAPy molecules are tightly fixed between {Mn3} clusters in the rigid MOF matrix through strong coordination interactions, which suppresses the torsional relaxation, the thermal vibration and the nonradiative relaxation process.
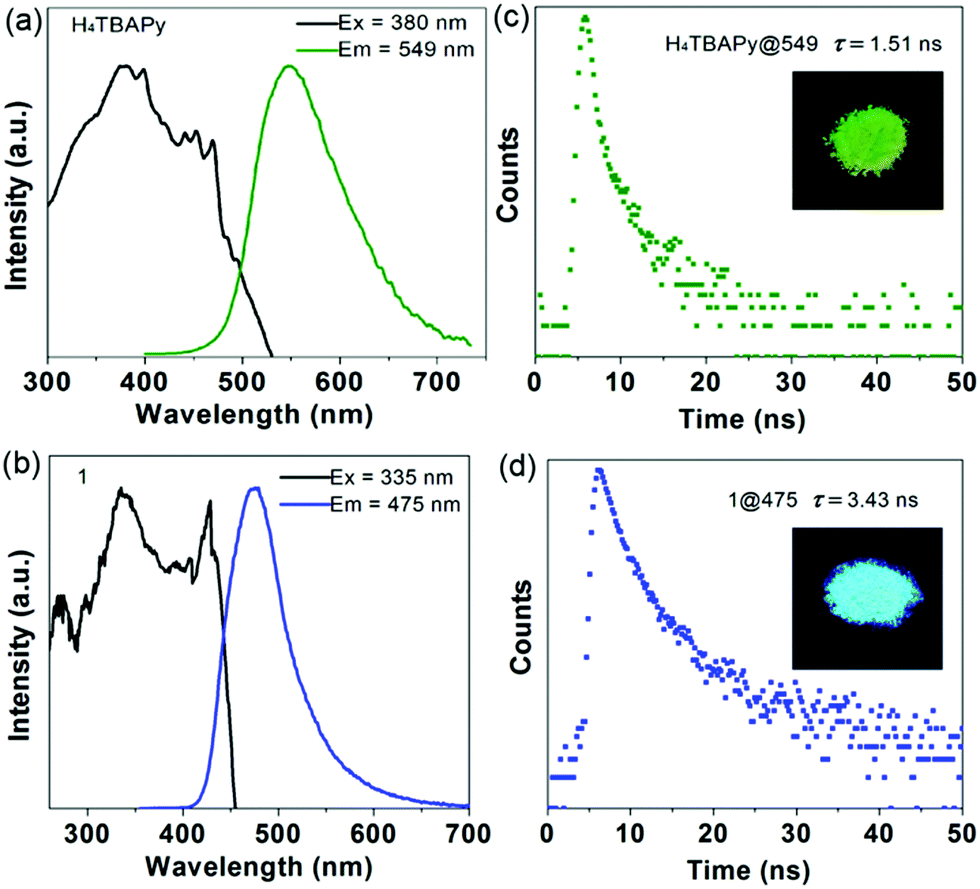 | ||
| Fig. 2 Normalized excitation and emission spectra of free H4TBAPy (a) and 1 (b) in the solid state. Fluorescence decay curves of free H4TBAPy (c) and 1 (d) measured at room temperature. The insets show the crystal samples of free H4TBAPy and 1 under UV (365 nm) light radiation. | ||
The temperature-dependent PL spectra were conducted in the range of 293 to 20 K to detect the inter-chromophoric interactions in 1. It is observed that the characteristic emission bands of the excimer increase obviously upon the decrease of the temperature from the ambient conditions to 80 K. Further cooling to 20 K, a mixture of monomer and excimer emission (Fig. S4a, ESI†) is observed.4b We reason that the excimer fluorescence of 1 comes from the interactions of coupled H4TBAPy dimers between adjacent layers. However, the emergence of monomer emission at low temperature may be the result of the fact that the fixed ionic liquid [HOEtMIm] in the framework of 1 can reduce such intermolecular interactions obviously. Moreover, the temperature-dependent PL at 475 nm shows a linear correlation between the intensity and temperature ranging from 200 to 20 K (Fig. S4b, ESI†), demonstrating the high degree of adjustment on interchromophoric interaction in 1 as a function of temperature, which can potentially serve as a temperature sensor.
To obtain the information on the electronic structure and energy level characteristics of the above photoactive materials, density functional theory (DFT) calculations were further performed (Fig. 3). In the monomer state of the free H4TBAPy, it is observed that the electronic isodensity surfaces of the HOMO up to HOMO−3 are exclusively distributed on the pyrene cores, and the LUMO and LUMO+1 are distributed on the whole H4TBAPy molecule. However, the LUMO+2 to LUMO+4 absolutely appear on the benzoate motifs (Fig. S5, ESI†). As for the H4TBAPy dimer in 1 (Fig. S6, ESI†), large overlap of π-stacking between the H4TBAPy dimer leads to stronger intermolecular coupling, which affords the distribution of electronic isodensity surfaces transfer from one excited pyrene core to another one, forming an excimer. Moreover, the delocalized π electrons tend to migrate outside benzoate groups (LUMO+2 to LUMO+4).
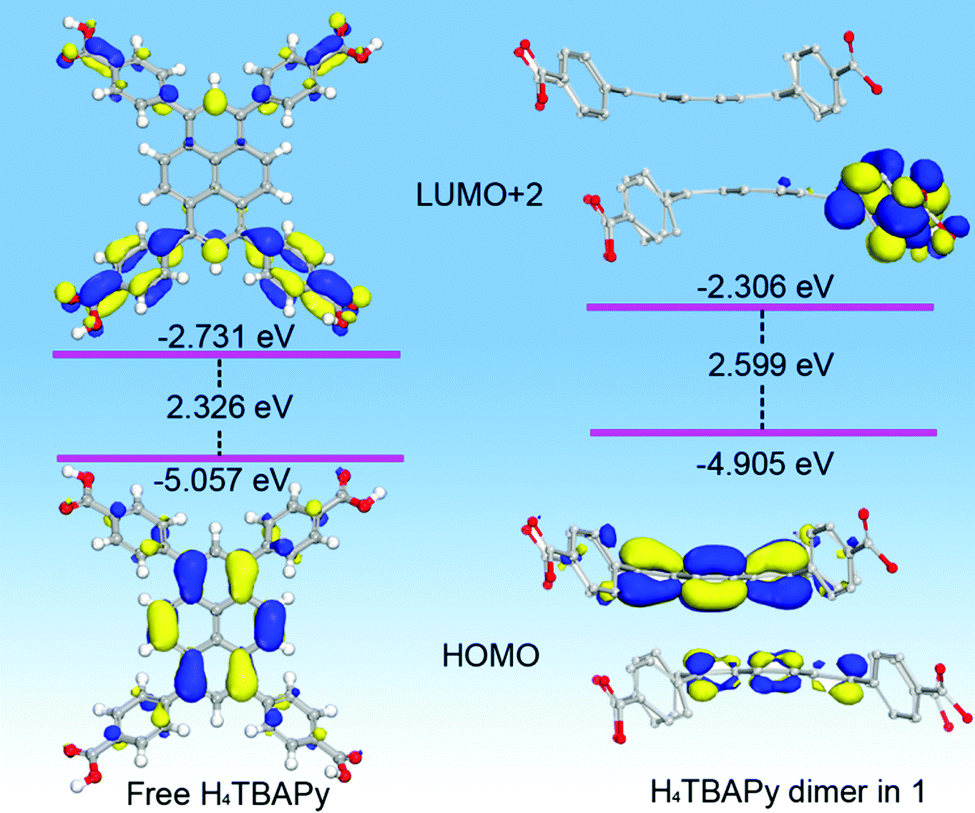 | ||
| Fig. 3 View of selected HOMOs, LUMOs and relevant energy level for the DFT optimized structure of free H4TBAPy and H4TBAPy dimers in 1. | ||
The good separation of molecular orbitals stems from the seriously twisted conformation of H4TBAPy. Contrastively, the free H4TBAPy exhibits a relatively small dihedral angle ranging from 47.62 to 48.29° between the central pyrene core and four benzoate motifs (Fig. S7, ESI†). The larger degree of distortion for the H4TBAPy molecule breaks down the delocalized conjugate system, increasing the energy gap between the HOMO and LUMO, which is in accordance with the blue-shift of fluorescence emission in experiment. Energy level analysis reveals that the fluorescence emission peaked at 549 nm (2.259 eV) for free H4TBAPy and at 475 nm (2.610 eV) for 1 can be assigned to HOMO → LUMO+2 transitions, which bear an energy gap of 2.326 and 2.599 eV, respectively. During the transition process, the electronic isodensity surfaces completely move from the pyrene core to benzoate motifs. Therefore, the photoexcitation and emission for the free H4TBAPy and 1 occur within and/or between the twisty molecules during the fluorescence process. The complete separation of molecular orbitals facilitates to suppress electron–hole recombination in the MOF for long lifetime emission.18
An essential property for any effective photosensitizer is efficient light harvesting in the visible to near-IR region.19 However, most current MOFs including the title MOF show a broad band gap, which cannot meet this requirement. Considering the anionic type framework of 1, a suitable cationic dye was selected to extend its light absorption range through ion exchange. It is observed that the absorption band of the DMP acceptor strongly overlaps with the fluorescence emission of the MOF donor (Fig. S8, ESI†), suggesting that potential Förster resonance energy transfer (FRET) can take place. The ion exchange (Fig. 4a) was conducted by immersing the as-synthesized crystal sample of 1 in EtOH solution containing DMP (3 × 10−4 mg L−1) for 3 days in the dark at room temperature, and then the color of 1 changed from pale yellow to red (Fig. S9, ESI†). Furthermore, thermogravimetric curves and FT-IR spectra suggest the efficient encapsulation of the DMP dyes (Fig. S10, ESI†). The PXRD patterns (Fig. S11, ESI†) and SEM images (Fig. S12, ESI†) indicate that 1 largely retains its phase purity and morphology after the incorporation of DMP. The ratio of DMP to the TBAPy ligand in DMP@1 was calculated as about 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 based on the 1H-NMR measurements (Fig. S13, ESI†). The UV-vis-NIR absorption spectra (Fig. 4b) show that the free H4TBAPy has a broad absorption ranging from 300 to 550 nm. However, 1 exhibits distinctly narrow absorption ranging up to 480 nm. The blue-shift of absorption compared to the free H4TBAPy results from the extension of the energy gap for 1. In sharp comparison, host–guest DMP@1 broadens the light absorption from UV to visible-light and even the NIR region, implying high enhancement of solar light-harvesting. It is noted that the new peak centred at 536 nm (Fig. S14, ESI†) stems from the charge-transfer transition between the MOF host and the DMP cationic guest.
:9 based on the 1H-NMR measurements (Fig. S13, ESI†). The UV-vis-NIR absorption spectra (Fig. 4b) show that the free H4TBAPy has a broad absorption ranging from 300 to 550 nm. However, 1 exhibits distinctly narrow absorption ranging up to 480 nm. The blue-shift of absorption compared to the free H4TBAPy results from the extension of the energy gap for 1. In sharp comparison, host–guest DMP@1 broadens the light absorption from UV to visible-light and even the NIR region, implying high enhancement of solar light-harvesting. It is noted that the new peak centred at 536 nm (Fig. S14, ESI†) stems from the charge-transfer transition between the MOF host and the DMP cationic guest.
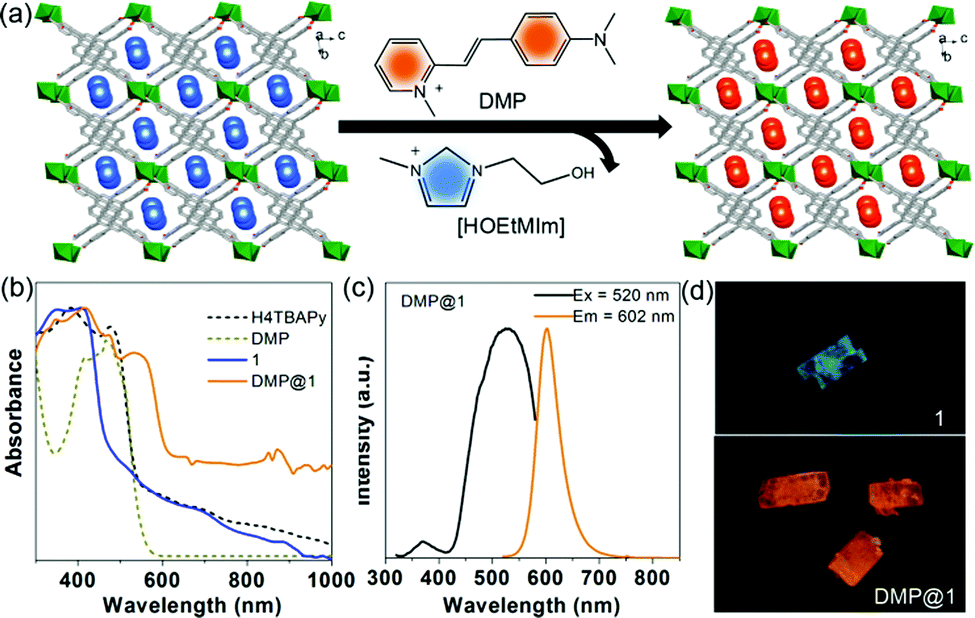 | ||
| Fig. 4 (a) Schematic diagram of the ion exchange experiment by the incorporation of DMP cations. (b) The UV-vis-NIR absorption of free 1 H4TBAPy, DMP, 1 and DMP@1. (c) Normalized excitation and emission spectra of DMP@1 in the solid state. (d) Optical microscope characterization of bulk crystals of 1 and DMP@1 under UV (365 nm) light radiation. | ||
Excited at 335 nm, it presents an extremely weak monomer fluorescence emission (450 nm) of the 1 host and a strong fluorescence peak (590 nm) from the DMP guest (Fig. S15, ESI†), implying that efficient FRET between host–guest components occurs in DMP@1. Moreover, it also reveals that the DMP guests can completely eliminate the coupled interactions between TBAPy dimers and thus lead to the deformation of excimer fluorescence. Upon excitation at 520 nm, the emission maxima of DMP@1 is at 602 nm, vastly red-shifted about 127 nm compared to the pristine MOF (Fig. 4c). The fluorescence decay curve gives a long lifetime value of 7.38 ns (Fig. S16, ESI†). Under light irradiation (365 nm) on the single crystals of 1 and DMP@1, the uniform emission color from cyan to orange can be observed before and after DMP encapsulation, further confirming that the guest molecules are highly dispersed and confined within the MOF nanochannels (Fig. 4d).15
To further understand the emission color change induced by the integration of DMP guests, DFT calculations were further carried out on the idealized model of DMP@1 (Fig. 5). It can be observed that the HOMO-2 is mainly populated on the pyrene core. Owing to high electron affinity of the pyrene core, the intermolecular polarization occurs between the pyrene core and DMP guest, instead of the benzoate substituents within the TBAPy molecule. As a result, the HOMO−1 mainly appears on the styryl and dimethylamino groups. The HOMO and LUMO are mainly dominated by carboxyl groups in TBAPy and methylpyridinium in DMP, respectively. Different from those of the reported dye@MOF system exhibiting direct charge transfer from the host to the guest,20 DMP@1 in this work presents a distinct and reiterative charge transfer process between host and guest systems rooting from the special D–π–A structure of DMP and seriously twisted conformation of TBAPy. Energy level analysis shows that the incorporation of DMP highly reduces the band gap, and the transitions of HOMO−2 → LUMO with a band gap of 1.933 eV corresponds to the fluorescence emission peaked at 602 nm for DMP@1. The calculation results confirm efficient charge transfer between the host and guest, and the DMP cationic dye largely influences the frontier orbital distributions in the host–guest system, and thus tunes the fluorescence emission in a wide range.
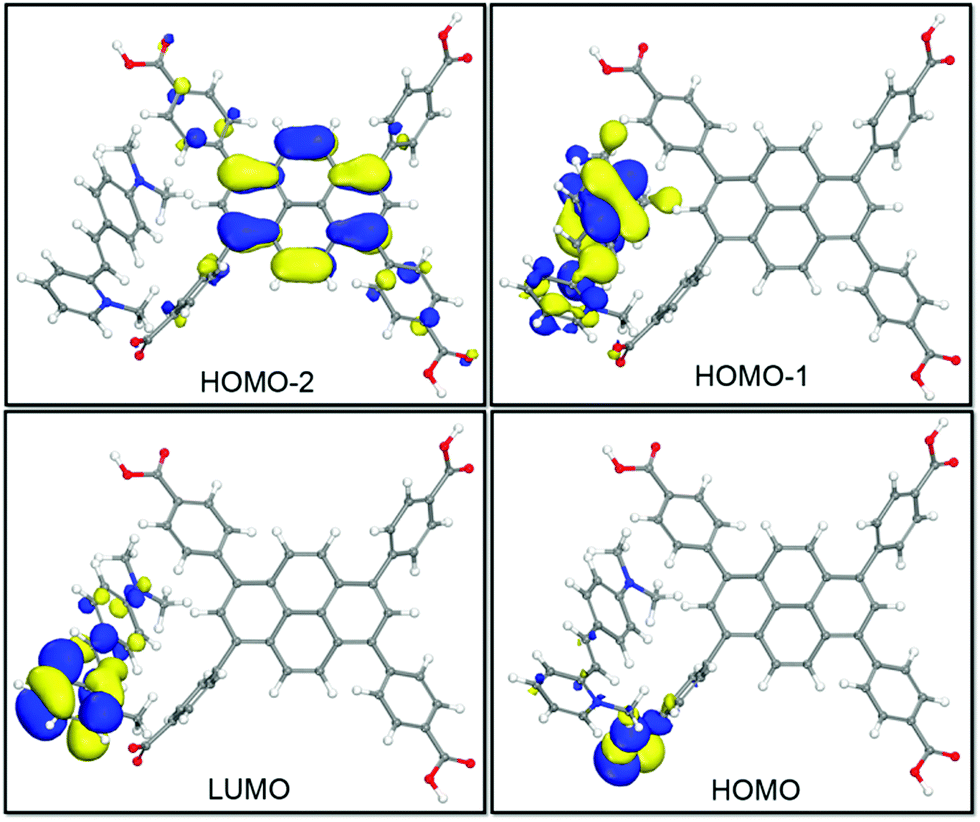 | ||
| Fig. 5 View of selected HOMOs and LUMOs for the DFT optimized structure of DMP@1. | ||
Inspired by highly ordered arrangement of DMP cations encapsulated in the MOF host, angle-dependent polarized fluorescence spectra of DMP@1 were measured by rotating the polarizer at different polarization angles. Fig. 6a and b show the relationship between the fluorescence intensity of DMP@1 and the polarization angle θ. It exhibits the maximum and minimum values at 0/180° and 90/270°, respectively, suggesting that the DMP molecules are aligned preferentially and parallel to the a direction of the MOF crystal. In comparison with the as-made 1, the PXRD of DMP@1 shows strong diffraction peak at the (02![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) crystal plane owing to the encapsulation of DMP molecules (Fig. S11, ESI†). The dihedral angle between (02) and (100) platelets was calculated as 84.25° (Fig. S17, ESI†). Thus, the accurate orientation of encapsulated DMP within MOF channels is a 84.25° dihedral angle between the liner DMP molecule and the (100) platelet. According to equations Rd = Imax/Imin and ρ = (Rd − 1)/(Rd + 1),6d DMP@1 shows a polarization anisotropy ρ of 0.97. This value is higher than the state-of-the-art low-dimensional systems (such as the lanthanide MOF6d and organic–inorganic hybrid perovskite18a). The high anisotropy can be related to the ordered arrangement of 1D D–π–A linear structures of cyanine molecules in the host. Remarkably, polarized emission switching between “on” and “off” states can be controlled by rotating the polarization angle θ from 180° to 0° (Fig. 6c) owing to the polarization-sensitive character of DMP@1. Such on–off switching is instantaneous and can be readily repeated several times during the rotation process (Fig. 6d). The above results reveal that the incorporation of DMP cations into the MOF rigid matrix affords a uniformly ordered structure for potential optical displays and information switches.
) crystal plane owing to the encapsulation of DMP molecules (Fig. S11, ESI†). The dihedral angle between (02) and (100) platelets was calculated as 84.25° (Fig. S17, ESI†). Thus, the accurate orientation of encapsulated DMP within MOF channels is a 84.25° dihedral angle between the liner DMP molecule and the (100) platelet. According to equations Rd = Imax/Imin and ρ = (Rd − 1)/(Rd + 1),6d DMP@1 shows a polarization anisotropy ρ of 0.97. This value is higher than the state-of-the-art low-dimensional systems (such as the lanthanide MOF6d and organic–inorganic hybrid perovskite18a). The high anisotropy can be related to the ordered arrangement of 1D D–π–A linear structures of cyanine molecules in the host. Remarkably, polarized emission switching between “on” and “off” states can be controlled by rotating the polarization angle θ from 180° to 0° (Fig. 6c) owing to the polarization-sensitive character of DMP@1. Such on–off switching is instantaneous and can be readily repeated several times during the rotation process (Fig. 6d). The above results reveal that the incorporation of DMP cations into the MOF rigid matrix affords a uniformly ordered structure for potential optical displays and information switches.
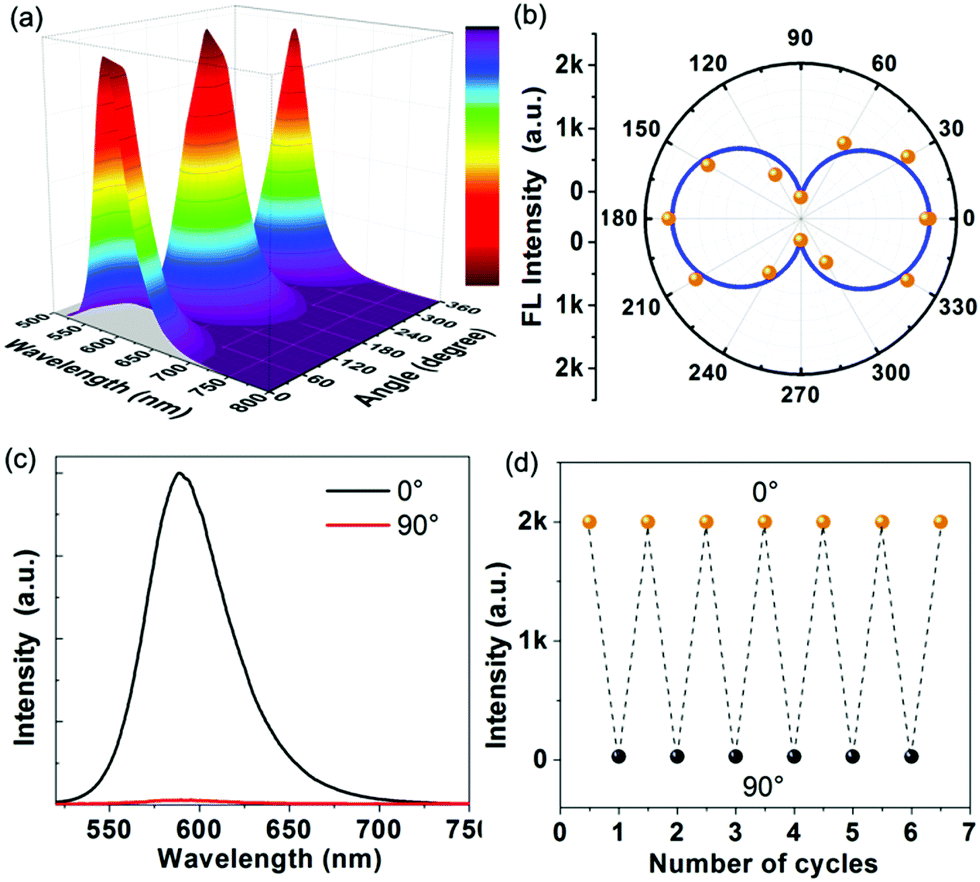 | ||
| Fig. 6 3D view (a) and polar coordinates (b): representation of the fluorescence intensity of DMP@1 as a function of the polarizer rotation angle. (c) Polarized fluorescence spectra of DMP@1 recorded at a polarization angle θ of 0° and 90°. (d) Reversible variation of the fluorescence intensity of DMP@1 at 0° and 90°. | ||
Encouraged by the efficient light-harvesting capability, the photoelectric performance was tested in 0.5 M Na2SO4 aqueous solution in a standard three-electrode system with 1 and DMP@1 nanoparticle (Fig. 7a and b) modified indium tin oxide (ITO) substrates as working electrodes. The transient current density–time curve (Fig. 7c) reveals that both 1 and DMP@1 exhibit quick transient photocurrent response under sudden illumination. The photocurrent response is steady and reproducible during several on–off cycles of illumination. In comparison, DMP@1 generates an average maximum photocurrent (Ilight) of about 4.8 μA cm−2, which is 6 times higher than that of 1 (0.8 μA cm−2). The average minimum current (Idark) in the dark state is 0.03 and 0.06 μA cm−2, respectively. Herein, the average on–off ratio values (Ilight/Idark) are calculated as 13 for 1 and 160 for DMP@1. This value is 4 times higher than that of the Zn(II)-TBAPy MOF13 in aqueous solution and 6 times higher than that of the lead iodine-based perovskite monocrystalline wire.2d The higher photocurrent response and on/off ratio indicate efficient photogeneration electron–hole separation of DMP@1.
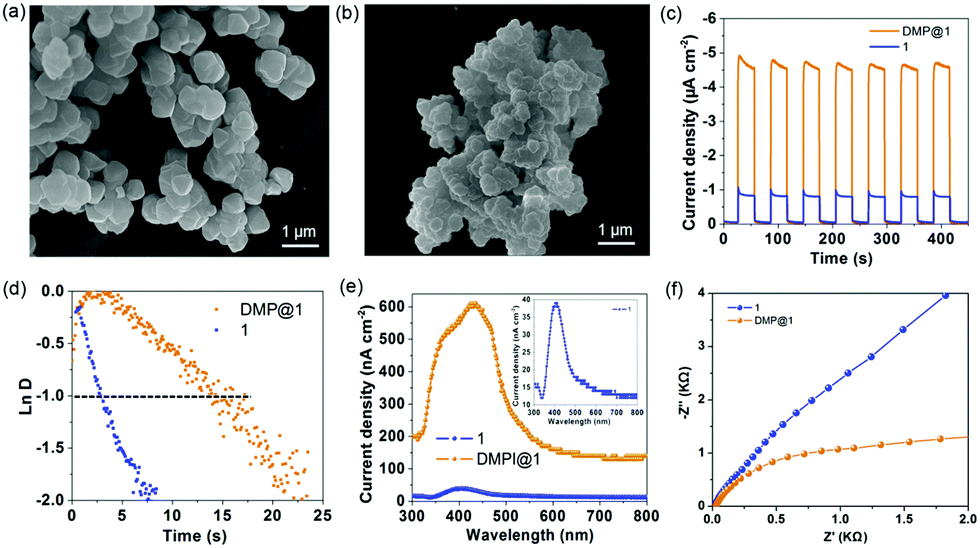 | ||
| Fig. 7 SEM images of 1 (a) and DMP@1 (b) nanoparticles used for photoelectric experiments. Transient photocurrent response (c) and normalized plots of the photocurrent–time dependence (d) for 1 and DMP@1 measured in a 0.5 M Na2SO4 aqueous solution under xenon lamp irradiation. (e) Wavelength dependent photocurrent response of 1 and DMP@1 without bias potential. (f) Electrochemical impedance spectroscopy (EIS). Nyquist plots of 1 and DMP@1 measured at a bias potential of −0.5 V vs. Ag/AgCl. | ||
To further evaluate the charge recombination behaviour of the photoelectrodes, the transient time constant (τ) is determined by the relationship between lnD and time when lnD = −1 (Fig. 7d). LnD was calculated using the equation lnD = (It − Ist)/(Iin − Ist), where It, Ist, and Iin are the time-dependent photocurrent, the steady-state photocurrent, and the initial photocurrent, respectively (Fig. S18, ESI†). The average τ values are estimated to be 2.8 and 14.8 s for 1 and DMP@1, respectively, indicating a slow charge recombination process for DMP@1. The wavelength dependent photocurrent response (Fig. 7e) demonstrates that the photocurrent response of DMP@1 spans 300–800 nm, which is a much wider range than that of 1 (350–500 nm). Illuminated with monochromatic 429 nm visible light, DMP@1 generates a maximum photocurrent of 608 nA cm−2, which is at least 15 times higher than that of 1 (39 nA cm−2) irradiated by 403 nm monochromatic light. Electrochemical impedance spectroscopy (EIS) analysis suggests that the charge transfer resistance of DMP@1 is obviously lower than that of 1 (Fig. 7f), indicating the fast charge transfer rate of DMP@1 under bias potential. Therefore, the confinement effects of the MOF host at the molecular level provide long-range order channels for efficient light harvesting, energy/charge transfer, and dissociation of photogenerated excitons.
Conclusions
In summary, we have developed an anionic MOF (1) based on linear trinuclear Mn(II) clusters and 1,3,6,8-tetrakis(p-benzoate)-pyrene photoactive linkers. The inter-chromophoric interactions and band gap of 1 can be tuned to a large extent by the incorporation of D–π–A cation dye cyanine (DMP) through a facile ion exchange process. As a result, the light-harvesting range of 1 can be largely extended from the visible-light to NIR region. Experiments and theoretical calculations reveal efficient energy/charge transfer from the pyrene core to DMP owing to strong overlap of the absorption band of DMP with the emission peak of 1, and confinement effects of the MOF host. The molecular-scale order of donor–acceptor integrated in the highly uniform DMP@1 heterojunction vastly promotes polarized fluorescence, charge mobility and energy transfer rates for excellent photo-electron conversion. This work not only provides an effective ionothermal route to develop a host–guest cluster-based MOF with a tunable band gap, enhanced UV-vis-NIR light harvesting and extremely high polarized emission by encapsulation of D–π–A cyanine, but also gives deep insights into the underlying host–guest energy transfer mechanisms of the orderly organized systems for applications in future artificial photosynthesis and optoelectronic materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21971100, U1504202, 21771021, and 21771097), the Project of Central Plains Science and Technology Innovation Leading Talents of Henan Province (No. 204200510001), and the Key Scientific Research Projects of Higher Education of Henan Province (No. 20A150005).Notes and references
- (a) T. Zhang and W. Lin, Chem. Soc. Rev., 2014, 43, 5982–5993 RSC; (b) Y. Zhang, J. Pang, J. Li, X. Yang, M. Feng, P. Cai and H.-C. Zhou, Chem. Sci., 2019, 10, 8455–8460 RSC.
- (a) S. Wang, D. Zhang, X. Zhang, D. Yu, X. Jiang, Z. Wang, M. Cao, Y. Xia and H. Liu, Sustainable Energy Fuels, 2019, 3, 529–538 RSC; (b) W. Lu, Y. Zhang, J. Zhang and P. Xu, Ind. Eng. Chem. Res., 2020, 59, 5536–5545 CrossRef CAS; (c) P. Xu, W. Lu, J. Zhang and L. Zhang, ACS Sustainable Chem. Eng., 2020, 8, 12366–12377 CrossRef CAS; (d) L. Wang, Y. Huang, A. Waleed, K. Wu, C. Lin, Z. Wang, G. Zheng, Z. Fan, J. Sun, H. Zhou, L.-D. Sun and C.-H. Yan, ACS Appl. Mater. Interfaces, 2017, 9, 25985–25994 CrossRef CAS.
- L. Stegbauer, S. Zech, G. Savasci, T. Banerjee, F. Podjaski, K. Schwinghammer, C. Ochsenfeld and B. V. Lotsch, Adv. Energy Mater., 2018, 8, 1703278 CrossRef.
- (a) Q. Yin, P. Zhao, R.-J. Sa, G.-C. Chen, J. Lü, T.-F. Liu and R. Cao, Angew. Chem., Int. Ed., 2018, 57, 7691–7696 CrossRef CAS; (b) A. Gładysiak, T. N. Nguyen, R. Bounds, A. Zacharia, G. Itskos, J. A. Reimer and K. C. Stylianou, Chem. Sci., 2019, 10, 6140–6148 RSC.
- (a) Z. Chen, P. Li, R. Anderson, X. Wang, X. Zhang, L. Robison, L. R. Redfern, S. Moribe, T. Islamoglu, D. A. Gómez-Gualdrón, T. Yildirim, J. F. Stoddart and O. K. Farha, Science, 2020, 368, 297–303 CrossRef CAS; (b) J. G. Jia, J. S. Feng, X. D. Huang, S. S. Bao and L. M. Zheng, Chem. Commun., 2019, 55, 2825–2828 RSC; (c) Y. X. Tan, F. Wang and J. Zhang, Chem. Soc. Rev., 2018, 47, 2130–2144 RSC; (d) Y. P. Wu, J. W. Tian, S. Liu, B. Li, J. Zhao, L. F. Ma, D. S. Li, Y. Q. Lan and X. Bu, Angew. Chem., Int. Ed., 2019, 58, 12185–12189 CrossRef CAS; (e) M. Ding, R. W. Flaig, H. L. Jiang and O. M. Yaghi, Chem. Soc. Rev., 2019, 48, 2783–2828 RSC.
- (a) X. G. Yang, X. M. Lu, Z. M. Zhai, Y. Zhao, X. Y. Liu, L. F. Ma and S. Q. Zang, Chem. Commun., 2019, 55, 11099–11102 RSC; (b) X. G. Yang and D. Yan, Chem. Sci., 2016, 7, 4519–4526 RSC; (c) T. F. Chen, S.-Y. Han, Z. P. Wang, H. Gao, L.-Y. Wang, Y.-H. Deng, C. Q. Wan, Y. Tian, Q. Wang, G. Wang and G. S. Li, Appl. Catal., B, 2019, 259, 118047 CrossRef CAS; (d) X. Yang, X. Lin, Y. Zhao, Y. S. Zhao and D. Yan, Angew. Chem., Int. Ed., 2017, 56, 7853–7857 CrossRef CAS; (e) Y.-C. Qiu, S. Yuan, X.-X. Li, D.-Y. Du, C. Wang, J.-S. Qin, H. F. Drake, Y.-Q. Lan, L. Jiang and H.-C. Zhou, J. Am. Chem. Soc., 2019, 141, 13841–13848 CrossRef CAS.
- (a) J. Zhu, W. A. Maza and A. J. Morris, J. Photochem. Photobiol., A, 2017, 344, 64–77 CrossRef CAS; (b) G. Lan, Y. Y. Zhu, S. S. Veroneau, Z. Xu, D. Micheroni and W. Lin, J. Am. Chem. Soc., 2018, 140, 5326–5329 CrossRef CAS; (c) P. Chinapang, M. Okamura, T. Itoh, M. Kondo and S. Masaoka, Chem. Commun., 2018, 54, 1174–1177 RSC; (d) H. Yang, M. Zhao, J. Zhang, J. Ma, P. Wu, W. Liu and L. Wen, J. Mater. Chem. A, 2019, 7, 20742–20749 RSC; (e) J.-S. Qin, S. Yuan, L. Zhang, B. Li, D.-Y. Du, N. Huang, W. Guan, H. F. Drake, J. Pang, Y.-Q. Lan, A. Alsalme and H.-C. Zhou, J. Am. Chem. Soc., 2019, 141, 2054–2060 CrossRef CAS.
- (a) Y. Zeng, Z. Fu, H. Chen, C. Liu, S. Liao and J. Dai, Chem. Commun., 2012, 48, 8114–8116 RSC; (b) R. Haldar, L. Heinke and C. Wöll, Adv. Mater., 2019, 1905227 Search PubMed.
- (a) P. Deria, J. Yu, T. Smith and R. P. Balaraman, J. Am. Chem. Soc., 2017, 139, 5973–5983 CrossRef CAS; (b) A. V. Wyk, T. Smith, J. Park and P. Deria, J. Am. Chem. Soc., 2018, 140, 2756–2760 CrossRef; (c) J. Yu, J. H. Park, A. V. Wyk, G. Rumbles and P. Deria, J. Am. Chem. Soc., 2018, 140, 10488–10496 CrossRef CAS.
- (a) K. C. Stylianou, R. Heck, S. Y. Chong, J. Bacsa, J. T. A. Jones, Y. Z. Khimyak, D. Bradshaw and M. J. Rosseinsky, J. Am. Chem. Soc., 2010, 132, 4119–4130 CrossRef CAS; (b) K. C. Stylianou, J. Rabone, S. Y. Chong, R. Heck, J. Armstrong, P. V. Wiper, K. E. Jelfs, S. Zlatogorsky, J. Bacsa, A. G. McLennan, C. P. Ireland, Y. Z. Khimyak, K. M. Thomas, D. Bradshaw and M. J. Rosseinsky, J. Am. Chem. Soc., 2012, 134, 20466–20478 CrossRef CAS.
- (a) C.-W. Kung, T. C. Wang, J. E. Mondloch, D. Fairen-Jimenez, D. M. Gardner, W. Bury, J. M. Klingsporn, J. C. Barnes, R. Van Duyne, J. F. Stoddart, M. R. Wasielewski, O. K. Farha and J. T. Hupp, Chem. Mater., 2013, 25, 5012–5017 CrossRef CAS; (b) A. V. Vinogradov, H. Zaake-Hertling, A. S. Drozdov, P. Lönnecke, G. A. Seisenbaeva, V. G. Kessler, V. V. Vinogradov and E. Hey-Hawkins, Chem. Commun., 2015, 51, 17764–17767 RSC; (c) P. Chandrasekhar, A. Mukhopadhyay, G. Savitha and J. Narasimha Moorthy, Chem. Sci., 2016, 7, 3085–3091 RSC; (d) V. A. Milichko, S. V. Makarov, A. V. Yulin, A. V. Vinogradov, A. A. Krasilin, E. Ushakova, V. P. Dzyuba, E. Hey-Hawkins, E. A. Pidko and P. A. Belov, Adv. Mater., 2017, 29, 1606034 CrossRef.
- X. Li, J. Yu, D. J. Gosztola, H. C. Fry and P. Deria, J. Am. Chem. Soc., 2019, 141, 16849–16857 CrossRef CAS.
- J. H. Qin, Y. D. Huang, Y. Zhao, X. G. Yang, F. F. Li, C. Wang and L. F. Ma, Inorg. Chem., 2019, 58, 15013–15016 CrossRef CAS.
- H. He, E. Ma, Y. Cui, J. Yu, Y. Yang, T. Song, C. D. Wu, X. Chen, B. Chen and G. Qian, Nat. Commun., 2016, 7, 11087 CrossRef CAS.
- Y. Wei, H. Dong, C. Wei, W. Zhang, Y. Yan and Y. S. Zhao, Adv. Mater., 2016, 28, 7424–7429 CrossRef CAS.
- I. H. Choi, S. B. Yoon, S. Huh, S. J. Kim and Y. Kim, Sci. Rep., 2018, 8, 9838 CrossRef.
- Z. Wei, Z.-Y. Gu, R. K. Arvapally, Y.-P. Chen, R. N. McDougald, Jr., J. F. Ivy, A. A. Yakovenko, D. Feng, M. A. Omary and H.-C. Zhou, J. Am. Chem. Soc., 2014, 136, 8269–8276 CrossRef CAS.
- (a) X. G. Yang, L. F. Ma and D. P. Yan, Chem. Sci., 2019, 10, 4567–4572 RSC; (b) X. G. Yang, Z. M. Zhai, X. M. Lu, L. F. Ma and D. Yan, ACS Cent. Sci., 2020, 6, 1169–1178 CrossRef CAS.
- M. E. Foster, J. D. Azoulay, B. M. Wong and M. D. Allendorf, Chem. Sci., 2014, 5, 2081–2090 RSC.
- K. Sivula, R. Zboril, F. L. Formal, R. Robert, A. Weidenkaff, J. Tucek, J. Frydrych and M. Grätzel, J. Am. Chem. Soc., 2010, 132, 7436–7444 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1998196. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0tc04292g |
| This journal is © The Royal Society of Chemistry 2020 |