
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExohedral functionalization vs. core expansion of siliconoids with Group 9 metals: catalytic activity in alkene isomerization†
Nadine E.
Poitiers
 ,
Luisa
Giarrana
,
Volker
Huch
,
Michael
Zimmer
and
David
Scheschkewitz
*
,
Luisa
Giarrana
,
Volker
Huch
,
Michael
Zimmer
and
David
Scheschkewitz
*
Krupp Chair of General and Inorganic Chemistry, Saarland University, D-66123 Saarbrücken, Germany. E-mail: scheschkewitz@mx.uni-saarland.de
First published on 2nd July 2020
Abstract
Taking advantage of pendant tetrylene side-arms, stable unsaturated Si6 silicon clusters (siliconoids) with the benzpolarene motif (the energetic counterpart of benzene in silicon chemistry) are successfully employed as ligands towards Group 9 metals. The pronounced σ-donating properties of the tetrylene moieties allow for sequential oxidative addition and reductive elimination events without complete dissociation of the ligand at any stage. In this manner, either covalently linked or core-expanded metallasiliconoids are obtained. [Rh(CO)2Cl]2 inserts into an endohedral Si–Si bond of the silylene-functionalized hexasilabenzpolarene leading to an unprecedented coordination sphere of the Rh centre with five silicon atoms in the initial product, which is subsequentially converted to a simpler derivative under reconstruction of the Si6 benzpolarene motif. In the case of [Ir(cod)Cl]2 (cod = 1,5-cyclooctadiene) a similar Si–Si insertion leads to the contraction of the Si6 cluster core with concomitant transfer of a chlorine atom to a silicon vertex generating an exohedral chlorosilyl group. Metallasiliconoids are employed in the isomerization of terminal alkenes to 2-alkenes as a catalytic benchmark reaction, which proceeds with competitive selectivities and reaction rates in the case of iridium complexes.
Introduction
The control of the reactivity of transition metal centres is a pivotal aspect of homogenous catalysis and therefore the development of novel ligands for transition metals is one of the priorities of organometallic chemistry. Unsaturated silicon compounds with the two major sub-categories of silylenes1 and disilenes1a,2 are typically characterized by a surplus of electrons and are therefore inherently stronger σ-donors than the corresponding carbon species, while retaining π-acceptor properties in some cases due to their unsaturated nature.3 First applications in homogeneous catalysis include C–H borylation,4 reduction of organic amides,5 Sonogashira and Heck cross-coupling reactions6 and hydrosilylation of ketones.7Recently, a third widely occurring sub-category was introduced into the class of stable unsaturated silicon species, the so-called siliconoids, partially unsubstituted neutral silicon clusters.8 Despite their unsaturated nature, applications of siliconoids as ligands towards transition metals are virtually unexplored. With our report on anionically functionalized Si6 derivatives,9 however, a conceptual link towards (poly)anionic, completely unsubstituted deltahedral Zintl anions of silicon10 was established, which in fact exhibit a rich chemistry towards transition metals: silicides and their heavier congeners of germanium and tin have frequently been employed as extraordinarily electron-rich ligands towards transition metal centres,11 and Zintl anions of Group 14 elements heavier than silicon can be converted to transition metal-centred derivatives M@Enx−.12 In all reported cases, the negative charges of the polyanionic precursors are (at least partially) retained in the transition metal-containing products with often adverse consequences for their solubility and stability, limiting their application, e.g. in homogeneous catalysis. Siliconoids with their stabilizing shell of organic ligands and high solubility due to their charge-neutrality appear to be the logical choice to overcome both limitations.
The first transition metal-substituted siliconoids, ligato-hexasilabenzpolarenes Si6–Zr(Cp)2Cl and Si6–Hf(Cp)2Cl with the covalently attached metallocene moiety were disclosed just recently.13 In contrast, the application of siliconoids as direct charge-neutral ligands towards transition metals has remained unsuccessful so far although the introduction of a silylene side-arm allowed for the coordination to Fe(CO)4 moieties in the periphery of the hexasilabenzpolarene motif.13 Now we show that by using the Group 9 metals rhodium and iridium a much larger variety of unprecedented coordination modes to siliconoids can be realized. We demonstrate that covalent bonding modes between the metal and the uncompromised hexasilabenzpolarene scaffold are related to the endohedral incorporation of the metal centre into the siliconoid cluster in a reversible manner. Finally, with the isomerization of alkenes we provide a first proof-of-principle for the application of the thus prepared soluble transition metal/silicon hybrid clusters as homogenous catalysts.
Results and discussion
Synthesis of iridium complexes
Treatment of tetrylene-functionalized siliconoids 1a–c with 0.5 equivalents of bis[(1,5-cyclooctadiene)iridium(I) chloride] affords the tetrylene-Si6 iridium complexes 2a–c in an NMR spectroscopically quantitative manner (Scheme 1). Complexes 2a–c were fully characterized by X-ray diffraction on single crystals, elemental analysis, and multinuclear NMR spectroscopy.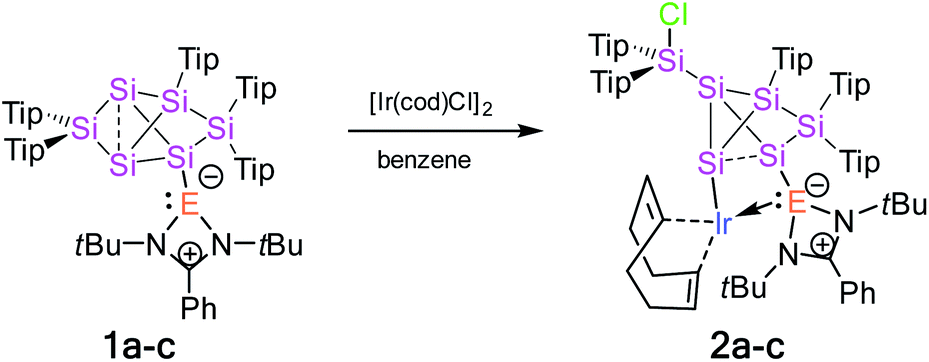 | ||
| Scheme 1 Synthesis of tetrylene-Si6 iridium complexes 2a–c from tetrylene-functionalized Si6 siliconoids 1a–c (2a: E = Si, 2b: E = Ge, and 2c: E = Sn). | ||
The 29Si NMR spectrum of 2a–c shows six sharp resonances in a much narrower range than usual for hexasilabenzpolarenes (+175.4 to −279.0 ppm),9 which provides a first hint at the rearrangement of the cluster scaffold and the ensuing loss of the spherical aromaticity and the associated magnetically induced cluster current. On the basis of the 2D 29Si/1H correlation NMR spectrum, the signals at 56.7 ppm (2a), 52.6 ppm (2b) and 50.2 ppm (2c) are assigned to the endohedral SiTip2 moieties. The signals at 13.3 (2a), 12.8 (2b) and 12.2 ppm (2c) are very close in chemical shift to that of the exocyclic silicon atom in chlorosilyltricyclo[2.1.0.02,5]pentasilane (5.8 ppm)14 and were therefore tentatively attributed to the extrusion of one SiTip2 unit from the cluster core of 2a–c. The resonances at −38.4 (2a), −41.5 (2b) and −42.1 ppm (2c) are assigned to the SiTip vertices. Most remaining 29Si signals are observed at a higher field (see ESI†) with the exception of one distinct resonance of 2a at 33.4 ppm, which is apparently due to the pendant silylene centre (Table 1).
| Siliconoid | δ 29Si2 [ppm] | δ 29Si5 [ppm] | δ 29Si6 [ppm] | δ 29Si3 [ppm] | δ 29Si4 [ppm] | δ 29Si7 [ppm] | δ 29Si2 solid [ppm] | δ 29Si6 solid [ppm] | δ 29Si7 solid [ppm] |
|---|---|---|---|---|---|---|---|---|---|
| 2a | 13.3 | 55.3 | −38.4 | −128.2 | −126.8 | 32.9 | 11.5 | −41.6 | 32.9 |
| 2b | 12.8 | 52.6 | −41.6 | −90.8 | −121.7 | — | 12.3 | −41.5 | — |
| 2c | 12.2 | 50.6 | −42.1 | −103.8 | −120.2 | — | 10.2 | −42.2 | — |
| 3 | 158.8 | −58.3 | 58.3 | 165.7 | — | 108.7 | 157.3 | 54.1 | 107.7 |
| 4 | 162.6 | 17.6 | 21.8 | — | −9.0 | 48.2 | 161.8 | 14.5 | 42.7 |
Single crystals of 2a–c were obtained by crystallization from hexane in 68% (2a), 61% (2b) and 63% (2c) yield and the tricyclic structures of the siliconoid–iridium complexes were confirmed by X-ray diffraction in the solid state (Fig. 1). As anticipated on the basis of NMR data, the privo-vertex has been extruded from the cluster core due to the additional bond to the chlorine atom transferred from the iridium centre, which in turn is not only coordinated by the pendant tetrylene moiety but also by the former nudo-vertex of the benzpolarene starting material. The structural model of 2c could not be refined in a satisfactory manner; the following discussion thus focuses on 2a,b. The Ir–E bond lengths (E = Si, Ge, Sn; 2a: Si3–Ir: 2.320(1) Å and Si7–Ir: 2.334(1) Å; 2b: Si3–Ir: 2.3517(7) Å and Ge–Ir: 2.4113(3) Å) are those of single bonds.16 The slightly longer distance between the pendant tetrylene and the iridium centre may be explained by the dative vs. covalent bonding situation. The endohedral bond distance between the iridium-bonded vertex and that bearing the tetrylene moiety (Si3–Si4 2a: 2.549(2) Å, 2b: 2.565(1) Å) is considerably elongated in comparison to those in the aforementioned chlorosilyltricyclo[2.1.0.02,5]pentasilane (2.356 Å)14 and a dianionic Si5 cluster with the same tricyclic scaffold (2.3822 Å).15
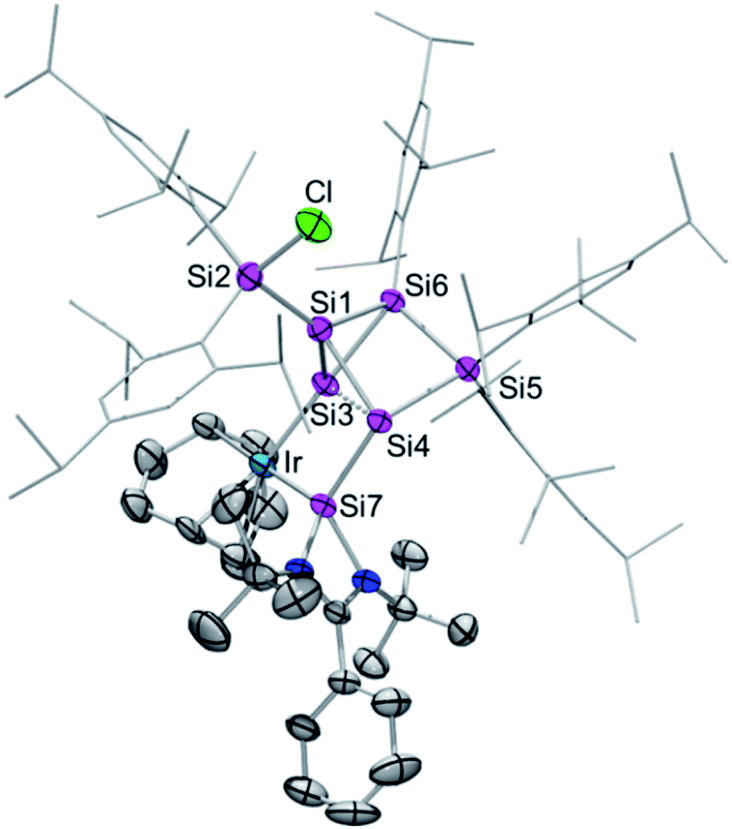 | ||
| Fig. 1 Representative molecular structure of silylene-functionalized siliconoid iridium complex 2a in the solid state. Hydrogen atoms are omitted for clarity. Thermal ellipsoids are set at 50% probability. For structures of 2b,c see ESI.† Selected bond lengths [Å] and angles [°]: 2a: Ir–Si7 2.320(1), Ir–Si3 2.334(1), Si1–Si3 2.305(2), Si1–Si4 2.313(2), Si1–Si6 2.364(2), Si1–Si2 2.369 (2), Si3–Si6 2.353(1), Si3–Si4 2.548(2), Si3–Si7 2.764(2), Si4–Si5 2.391(2), Si4–Si7 2.402(2), Si7–N2 1.857(3), Si7–N1 1.884(3), Cl–Si2–Si1 99.03(6), Si1–Si3–Ir 124.02(5), Ir–Si3–Si6 174.93(6), Ir–Si3–Si4 105.99(5), Ir–Si3–Si7 53.33(3), and Si7–Si4–Si3 67.81(4); 2b: Ir–Si3 2.3517(7), Ir–Ge 2.4113(3), Ge–Si4 2.4444(8), Ge–Si3 2.8481(8), Si1–Si3 2.296(1), Si1–Si4 2.302(1), Si1–Si2 2.356(1), Si1–Si6 2.375(1), Si3–Si6 2.339(1), Si3–Si4 2.565(1), Si4–Si5 2.389(1), Si5–Si6 2.368(1), Ge–N2 1.995(2), Ge–N1 2.005(2); Cl–Si2–Si1 100.91(4), Ir–Si3–Ge 54.243(17), Ir–Si3–Si4 106.90(3), Si6–Si3–Ir 173.61(4), Si1–Si3–Ir 124.75(4), Ir–Ge–Si3 52.322(16), Ir–Ge–Si4 108.95(2), Si3–Ir–Ge 73.436(19), and Ge–Si4–Si3 69.25(3). | ||
This is probably a consequence of the back and forth electron transfer between iridium and cluster orbitals, but may also indicate a propellane-like bonding situation as suggested by the hemispheroidality of Si4. On the other hand, Si3 does not fulfill the criterion of hemispheroidality,8a but rather adopts a very near planar-tetracoordinate coordination environment instead (2a: ϕ(Si3) = −0.0154 Å, ϕ(Si4) = +0.7559 Å; 2b: ϕ(Si3) = −0.0060 Å, ϕ(Si4) = +0.7276 Å). The bond lengths between the bridgehead silicon atoms Si1 and Si3 in 2a,b (2a: 2.305(2) Å, 2b: 2.2962(2) Å) are now at the short end of the usual range for silicon single bonds as also observed in the chlorosilyltricyclo[2.1.0.02,5]pentasilane (2.312 Å).14 The 29Si CP/MAS spectra of 2a–c show very similar signals to those in C6D6 solution thus confirming the integrity of the coordination modes upon solvation (the stannylene–Si6 iridium complex 2c shows a double set of signals due to two crystallographically independent molecules in the asymmetric unit; see ESI†). The longest wavelength absorption bands in the UV/vis spectra at λmax = 576 nm (2a), 580 nm (2b), 592 nm (2c) are strongly red-shifted compared to previously reported ligato-substituted siliconoids (λmax = 364 to 521 nm).9,13
Synthesis of rhodium complexes
The reaction of 1 equivalent of bis[(1,5-cyclooctadiene)rhodium(I) chloride] with 1a–c led to a complicated mixture of products in all cases, presumably due to competing oxidative addition and reductive elimination reactions. In one crystallization attempt of the product mixture from 1b (E = Ge) a few red-brownish crystals were collected and then investigated by X-ray diffraction showing the same motif as observed in 2a–c (see ESI†). In the anticipation that it might react in a similar manner despite the differing ligand set, we considered the rhodium(I) dicarbonyl chloride dimer [Rh(CO)2Cl]2 as an alternative. The reactions of 1a–c with 1 equivalent of [Rh(CO)2Cl]2, however, led to uniform conversion only in the case of 1a and inseparable mixtures of products for 1b,c according to NMR spectra. In addition, 1H NMR monitoring of the reaction mixture revealed the rearrangement of the initial product overnight in the case of 1a (Scheme 2).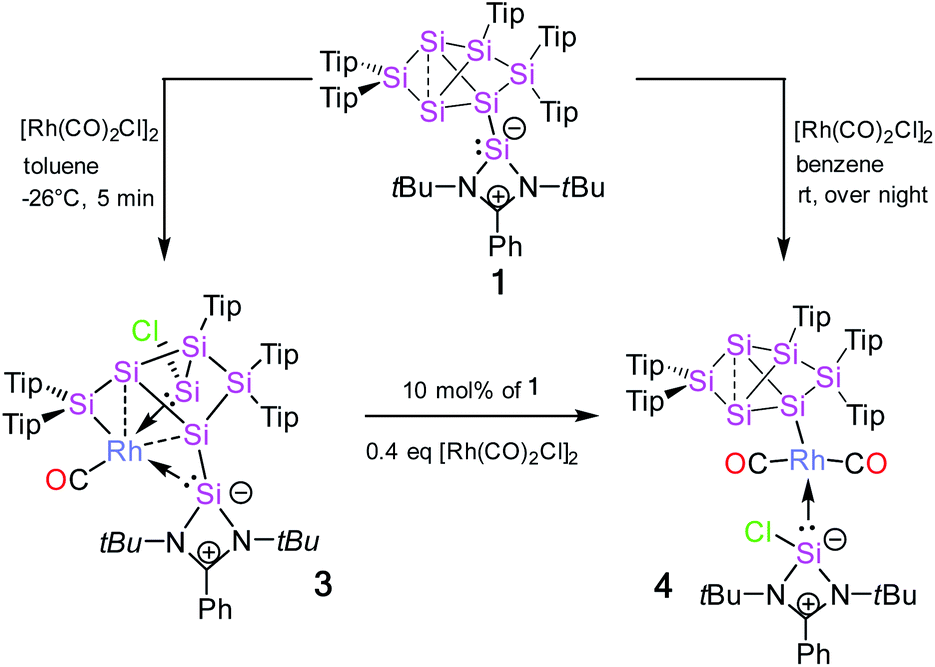 | ||
| Scheme 2 Synthesis of Si7 rhodium complexes 3 and 4 from silylene-functionalized Si6 siliconoid 1. | ||
Treatment of 1a with 1 equivalent of [Rh(CO)2Cl]2 in toluene followed by cooling to −26 °C after three minutes of stirring at ambient temperature yields dark red crystals of the primary product 3 after storage for 2 to 3 h in 56% crystalline yield. Conversely, the secondary product 4 is obtained as red-brownish crystals in 63% yield by crystallization from hexane after stirring the reaction mixture overnight. Both 3 and 4 were fully characterized by multinuclear NMR spectroscopy and X-ray diffraction on single crystals (Fig. 2 and 3).
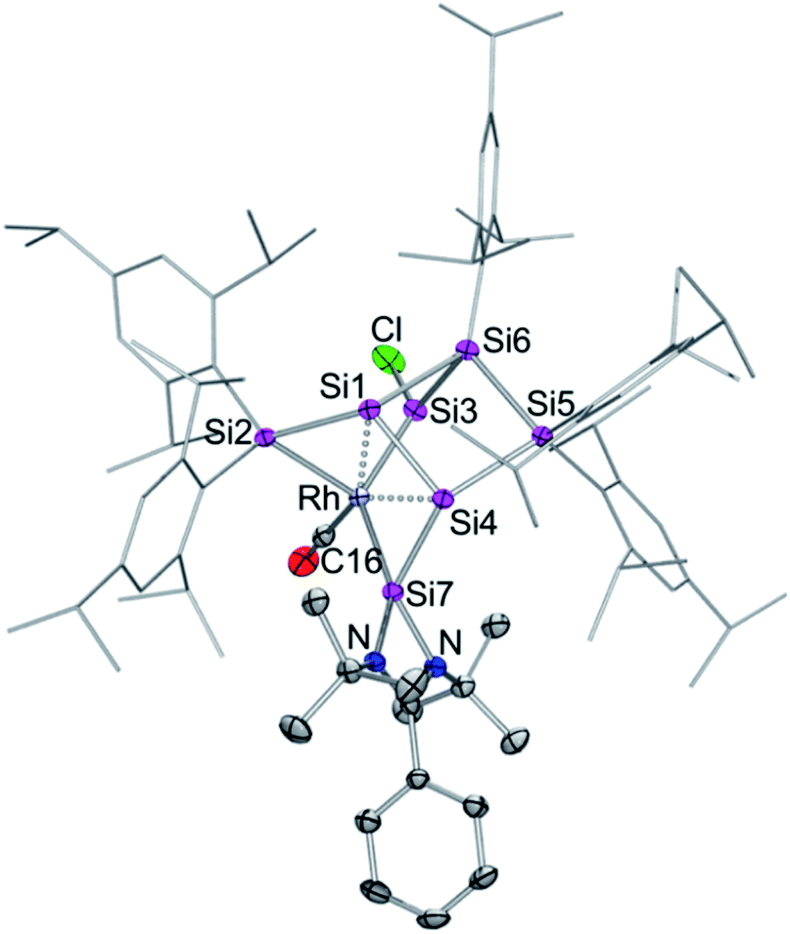 | ||
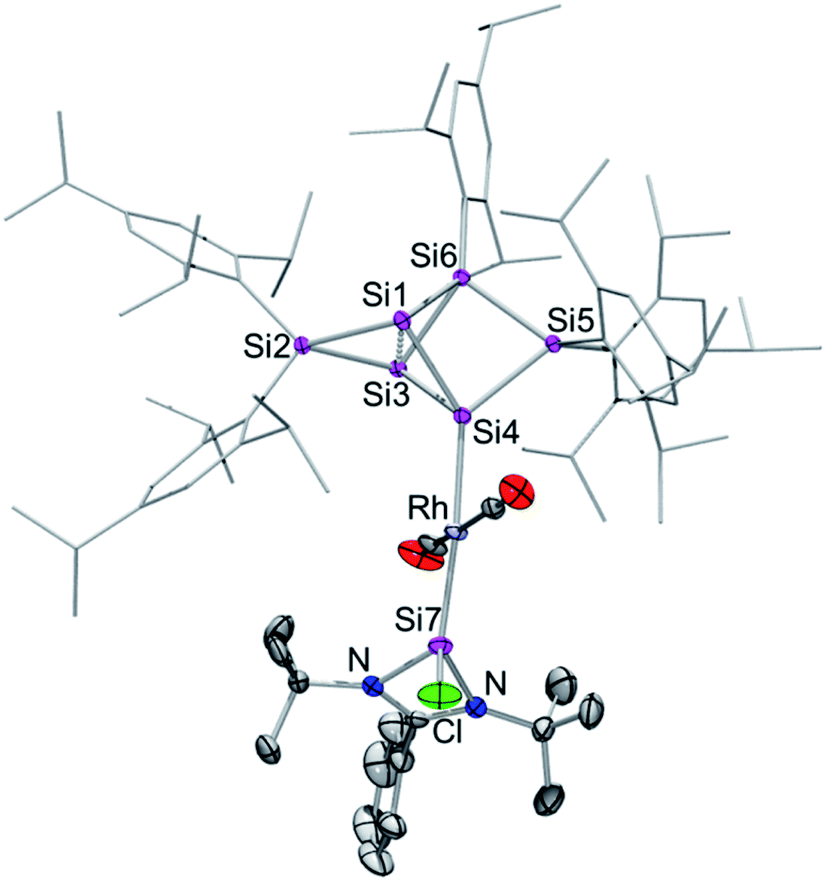
| Fig. 2 Molecular structure of silylene-functionalized siliconoid rhodium complex 3 in the solid state. Hydrogen atoms are omitted for clarity. Thermal ellipsoids are set at 50% probability. Selected bond lengths [Å] and angles [°]: Rh–Si3 2.2455(7), Rh–Si7 2.3104(7), Rh–Si5 2.3872(8), Rh–Si2 2.5936(7), Rh–Si1 2.5965(7), Si3–Cl 2.102(1), Si1–Si2 2.279(1), Si1–Si5 2.373(1), Si1–Si4 2.499(1), Si2–Si7 2.254(1), Si2–Si6 2.333(1), Si3–Si4 2.308(1), Si4–Si6 2.401(1), Si7–N1 1.837(2), Si7–N2 1.822(2), Si3–Rh–Si7 132.05(3), Si3–Rh–Si5 97.85(3), Si7–Rh–Si5 116.303, Si3–Rh–Si2 87.62(2), Si7–Rh–Si2 54.36(2), Si5–Rh–Si2 102.19(2), Si3–Rh–Si1 72.23(2), Si7–Rh–Si1 98.48(2), Si5–Rh–Si1 56.69(2), Si4–Si1–Rh 94.98(3), Si7–Si2–Rh 56.40(2), Si1–Si2–Rh 64.02(3), and Si6–Si2–Rh 115.58(3). | ||
 | ||
| Fig. 3 Molecular structure of silylene-functionalized siliconoid rhodium complex 4 in the solid state. Hydrogen atoms are omitted for clarity. Thermal ellipsoids are set at 50% probability. Selected bond lengths [Å] and angles [°]: Rh–Si7 2.305(1), Rh–Si4 2.398(1), Si3–Si6 2.342(1), Si2–Si3 2.352(1), Si3–Si4 2.386(1), Si1–Si2 2.619(1), Si1–Si4 2.336(1), Si1–Si6 2.351(1), Si1–Si2 2.392(1), Si1–Si3 2.619(1), Si4–Si5 2.384(1), Si5–Si6 2.371(1), Si7–N1 1.826(3), Si7–N2 1.831(3), Si7–Cl 2.077(2), Si7–Rh–Si4 172.63(4), Si3–Si4–Rh 131.22(5), Si5–Si4–Rh 132.67(5), Si1–Si4–Rh 124.01(5), and Si4–Si1–Si2 97.15(5). | ||
In contrast to the iridium complexes 2a–c, the rhodium centre of 3 is fully incorporated into the core structure under expansion to a 7-vertex motif. Only one of the CO ligands is retained in 3 completing the distorted trigonal-pyramidal coordination sphere at rhodium in an apical position (C16–Rh–Si2 102.586(4)°, C16–Rh–Si3 104.881(5)°, C16–Rh–Si7 45.489(4)°, centre(Si1–Si4)–Rh–Si2 79.149(4)°, centre(Si1–Si4)–Rh–Si3 78.869(4)°, centre(Si1–Si4)–Rh–Si7 75.997(4)°, C16–Rh–centre(Si1–Si4) 175.493(6)°, and Si7–Rh–Si3 132.095(5)°). The geometric parameter τ = (β − α)/60 is commonly used for pentacoordinate complexes as an index of the degree of the trigonality in trigonal-bipyramidal and square-planar pyramidal structural motifs.17 With the two largest angles α and β of 3 (β: C16–Rh–centre(Si1–Si4) 175.493(6)°; α: Si7–Rh–Si3 132.095(5)°) the angular parameter is τ = 0.72 in line with a distorted trigonal-bipyramidal coordination sphere of Rh. The chlorine atom is shifted to one of the former nudo-vertices so that the extrusion of the privo-SiTip2 moiety as in 2a–c is avoided in this case. Intriguingly, the chloro-substituted silicon vertex (Si3) is conferred a significant silylene character: the corresponding bond distance to rhodium (Si3–Rh 2.2455(7) Å) is considerably shorter than that of the amidinato silylene moiety, which binds to rhodium at a distance (Si7–Rh 2.3104(7) Å) similar to those reported for other complexes with this motif.5,16e The former privo-vertex binds to the Rh centre at a distance (Si2–Rh 2.3872(8) Å) in line with the covalent radii of silicon and rhodium. Si2, Si3 and Si7 bind in equatorial positions and thus form the base of the trigonal-bipyramidal coordination environment at Rh.
The Si1–Si4 bond is unusually short (2.279(1) Å) and occupies the remaining apical positions at Rh at relatively long distances (Si4–Rh 2.5936(7) Å and Si1–Rh 2.5965(7) Å). Both Si1 and Si4 exhibit a hemispheroidal coordination environment with hemispheroidalities of ϕ(Si1) = +0.8003 Å and ϕ(Si4) = +0.4764 Å.8a The degree of the metallacyclopropane character of this coordinating interaction according to the Dewar–Chatt–Duncanson model18 is difficult to estimate due to the complexity of the bonding situation as Si1–Si4, albeit shorter than a usual single bond, is heavily involved in cluster bonding.
The 29Si NMR spectrum in C6D6 is consistent with the bonding situation as discussed on the basis of the solid state structure. Four of the seven resonances are split into doublets by the coupling to the 103Rh nucleus suggesting the coordination of rhodium being uncompromised by solvation. The signals are, however, not as broadly dispersed as typically observed for Si6 siliconoids.9,13 The former privo-vertex Si2 gives rise to a low-field 29Si NMR signal, albeit it splits into a doublet at 158.8 ppm with a coupling constant of J29(Si,103Rh) = 41.0 Hz. The silylene character of the former nudo-vertex Si3, now bearing the chloro substituent, results in the significant deshielding of the corresponding 29Si NMR signal at 165.7 ppm. The interaction with the rhodium centre is reflected in the coupling constant of J29Si,103Rh = 53.4 Hz. The 103Rh coupling of the two hemispheroidally coordinated vertices Si1 and Si4 is too small to resolve resulting in singlets at −140.2 and −122.1 ppm. Although a discussion of the magnitude of experimental coupling constants is next to impossible in polycyclic systems such as 3 the absence of detectable coupling is in line with a predominant π-character of the coordination to rhodium.3a The assignment is backed by the absence of cross-peaks to Tip groups in the 2D 29Si/1H correlation. The doublet at 108.7 ppm (J29Si,103Rh = 59.6 Hz) is assigned to the N-heterocyclic silylene moiety (Si7) based on the observation of a cross-peak to the t-butyl groups. In notable contrast, the signal of the not directly Rh-bonded Si6 at 58.3 ppm (assigned on the basis of a cross-peak to one Tip substituent) is split into a doublet with J29Si,103Rh = 14.3 Hz.
Surprisingly, after the rearrangement of Rh(I) complex 3 to 4, the diagnostic wide dispersion of 29Si NMR shifts is again observed, which suggested the re-establishment of an uncompromised benzpolarene9,13 scaffold. Besides the characteristic highfield resonances for the nudo-vertices Si1 and Si3 at −256.1 and −258.3 ppm, the 29Si NMR signal of the tetracoordinate privo-vertex Si2 appears at the typical low field at 162.6 ppm. The doublet in the 29Si NMR at 48.2 ppm with J29Si,103Rh = 84.5 Hz is attributed to the N-heterocyclic silylene moiety on the basis of a cross-peak to the t-butyl groups in the 2D 29Si/1H correlation and the large coupling indicative of pronounced s-orbital contributions. In contrast, the doublet at −9.0 ppm with J29Si,103Rh = 31.5 Hz suggests a covalent bond to the 103Rh nucleus. Due to the absence of a cross-peak to a Tip group in the 2D 29Si/1H correlation NMR spectrum, it can be attributed to the ligato-vertex Si4. The remaining 29Si NMR chemical shifts are located in the usual range of saturated silicon atoms; only one of the signals showing a small coupling to the Rh centre (21.8 ppm; J29Si,103Rh = 8.5 Hz). The anticipated structure of 4 as an uncompromised benzpolarene scaffold covalently attached to rhodium was confirmed by X-ray diffraction in the solid state (Fig. 3).
The rhodium centre of 4 exhibits a typical square-planar coordination environment, with the hexasilabenzpolarene moiety indeed connected through the ligato-position Si4. Astonishingly, not only has the Si6 moiety been reinstated during the isomerization from 3, but the amidinato silylene – now disconnected from the siliconoid and coordinated to the rhodium centre in a trans-fashion – has reacquired its chloro-substituent as well. The coordination at the rhodium centre is completed by two CO ligands, which requires the “come-back” of the initially dissociated CO molecule. The Si7–Rh bond length of 2.305(1) Å is in line with the reported donor–acceptor bond length of Si–Rh complexes.5,16e Interestingly, it is significantly shorter than the covalent Si4–Rh bond length of 2.398(1) Å in the same molecule. The distance between the bridgehead silicon atoms (Si1–Si3 2.6188(4) Å) is similar to that in previously reported ligato-substituted Si6 siliconoids.9,13 The longest wavelength absorption bands are observed at λmax = 461 nm (3) and 466 nm (4) and thus are slightly blue-shifted compared to the ligato-metalated siliconoids Zr and Hf (Zr: 521 nm, Hf: 497 nm).13 The Rh complexes 3 and 4 exhibit IR characteristics of rhodium carbonyl complexes19 with CO stretching modes at ν = 1978 cm−1 (3) and 1951, 1949 cm−1 (4).
Mechanistic considerations
Due to the flexibility of the coordination environments, reactions involving transition metals generally proceed through multiple steps and, consequently, the mechanisms are often complicated, especially when backbone structures are reconstructed such as in the present case through a sequence of cleavage and formation of Si–Si bonds. Although, a computational treatment of such mechanisms is well out of reach to us due to the anticipated complexity of the potential energy surfaces and the large size of the involved molecules, we propose a plausible mechanism for the Ir and Rh structures based on the structurally characterized products (Chart 1).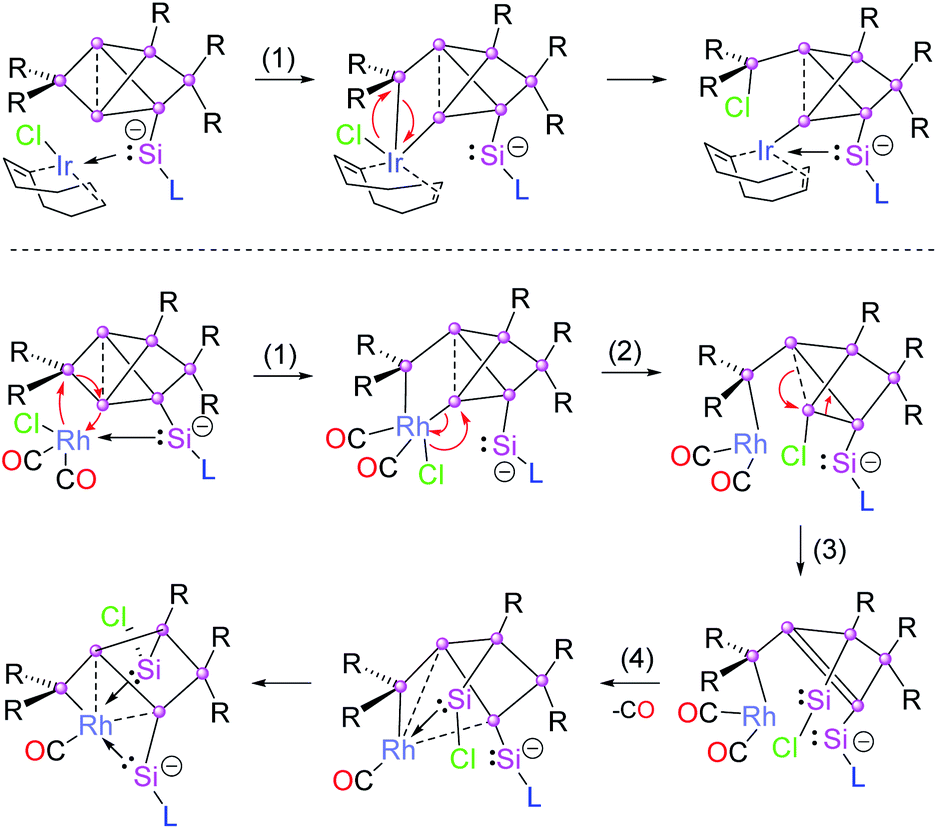 | ||
| Chart 1 Proposed mechanism of the formation of 2a (top) and 3 (bottom). | ||
All reactions are likely initiated by the straightforward coordination of the pendant silylene ligand to the metal. For the subsequent rearrangements, we suggest the oxidative addition to the Si2–Si3 single bond as the common first step (step 1 in Chart 1). From the second step onwards, however, the isomerizations proceed through distinct pathways for the rhodium and iridium species. In the case of 2a, the oxidative addition is directly followed by reductive elimination of the chloro group and the SiTip2 moiety from the Ir centre resulting in the formation of the exohedral chlorosilyl group in the final product 2a (and by extension 2b,c). In contrast, in the case of the primary rhodium product 3 the chlorine migrates to Si3 (step 2). In step 3, the formation of the chlorosilylene Si3 is suggested, while a formal double bond between Si1–Si4 is formed as a consequence. In the last step, the final product 3 is formed by elimination of a CO unit enforced by the coordination of the Si1–Si4 bond and the silylenes (Si3 and Si7) to the Rh centre. The question of the re-establishment of the intact hexasilabenzpolarene scaffold from intermediate 3 to yield the final product 4 is even more daunting as it requires the return of the previously eliminated CO ligand into the coordination sphere of rhodium. Notably, a solution of crystallized 3 turned out to be inert towards exposure to CO atmosphere as well as the addition of excess [(CO)2RhCl]2. We therefore assumed that the formation of 4 can only be attained by a reactive species formed in situ during the reaction of 1a with [(CO)2RhCl]2. Indeed, treatment of a solution of isolated crystals of 3 with 10 mol% of silylene-substituted siliconoid 1a and 0.4 equivalents of additional [(CO)2RhCl]2 results in the uniform conversion of isolated 3 to 4 in the course of 24 h. We speculate that either an extremely short-lived intermediate of monomeric [(CO)3RhCl]20 or a heterodimer not unlike the one reported by Braunschweig et al.21 might be responsible for the CO delivery.
Alkene isomerization catalysis
Despite the plethora of synthetic methods available for the introduction of C–C double bonds, the regioisomerization of an existing C–C double bond is a viable alternative.22 The so-called alkene isomerization, however, results in mixtures of (E) and (Z)-alkenes in many cases23 or further migration along a saturated carbon chain. The selective transformation of terminal alkenes to 2-alkenes has thus attracted considerable interest.24 We anticipated that electron-rich siliconoid ligands might fulfill two functions in a homogenous catalyst for alkene isomerization: (a) acting as an electron reservoir and thus facilitating oxidative addition reactions and (b) providing sufficient steric bulk to improve the selectivity regarding the number of positions the C–C double bond migrates. We therefore probed the isomerization of terminal alkenes to 2-alkenes in the presence of catalytic quantities of complexes 2a–c, 3 and 4.As rapidly indicated by first preliminary tests, no solvent is required for the catalytic activity of 2a–c, 3 and 4 and therefore all runs were carried out with the neat substrate, an attractive feature from both ecologic and economic perspectives. Allyltrimethylsilane and 1-hexene were used as neat substrates on an NMR scale using a C6D6 capillary as the locking signal; yields were calculated from 1H NMR integrations (Table 2). Fig. 4 shows the spectroscopic conversion to 2-trans-hexene using 0.05 mol% (2a), 0.1 mol% (2b, 2c) of the catalysts at room temperature as a function of time. The side chain migration to 3-trans-hexene is disregarded in the plots due to the overlap of the chemical shifts, and estimated to be <10% from the spectroscopic data. Blind tests without catalyst or in the presence of [(cod)IrCl]2 or [(CO)2RhCl]2 led to no detectable conversion under identical conditions. The reaction can therefore easily be quenched after the formation of 2-trans-hexene is complete by simple addition of water leading to the hydrolysis of the catalyst. The catalytic performance is best in the case of 2a (E = Si, TOF = 119 h−1) with catalyst loadings as low as 0.05 mol% and strongly decreases from 2b (E = Ge, TOF = 13.5 h−1) to 2c (E = Sn, TOF = 7.5 h−1) both requiring double catalyst loads. Preliminary results show a much lower catalytic activity of the rhodium complexes 3 and 4 which was therefore not investigated in detail (see ESI†). The isomerization of allyltrimethylsilane to 2-E/Z-vinyltrimethylsilane proceeds significantly slower even with the more active 2a–c and requires higher temperatures as well as one order of magnitude larger amounts of catalyst (0.8 mol%, 60 °C, Fig. 5, ESI†).
| Catalyst | Substrate | Cat. loading [mol%] | Time [h] | Temperature [°C] | Σ conversion [%] | E-isomer [%] | Z-isomer [%] | TON | TOF [h−1] |
|---|---|---|---|---|---|---|---|---|---|
| 2a | 1-Hexene | 0.05 | 16 | 25 | 95 | — | — | 1902.7 | 118.9 |
| 2b | 1-Hexene | 0.1 | 72 | 25 | 97 | — | — | 968.8 | 13.5 |
| 2c | 1-Hexene | 0.1 | 100 | 25 | 75 | — | — | 748.4 | 7.5 |
| 2a | Allyl-SiMe3 | 0.8 | 26 | 60 | 94 | 77 | 17 | 117.4 | 4.5 |
| 2b | Allyl-SiMe3 | 0.8 | 30 | 60 | 89 | 73 | 16 | 111.6 | 3.7 |
| 2c | Allyl-SiMe3 | 0.8 | 30 | 60 | 78 | 63 | 15 | 97.4 | 3.2 |
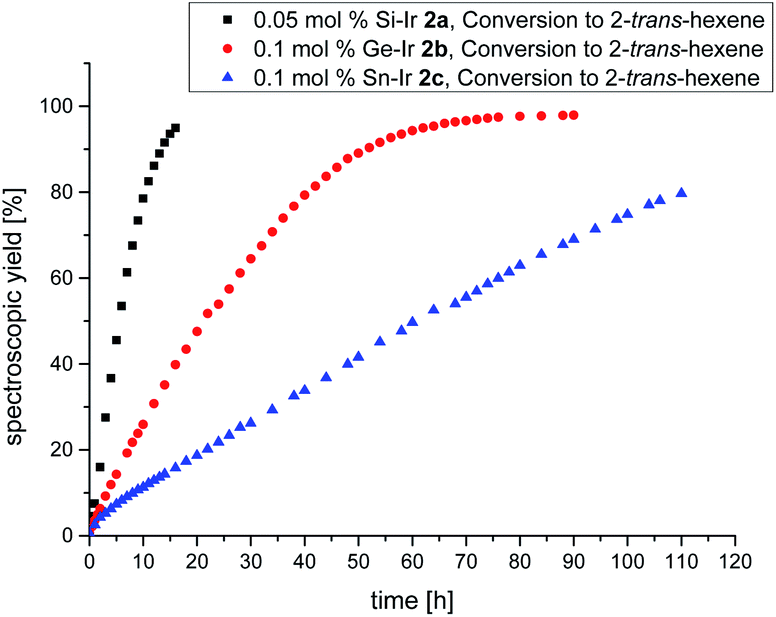 | ||
| Fig. 4 Plot of the spectroscopically determined conversion to 2-trans-hexene. Black = Si–Ir 2a (0.05 mol%), red = Ge–Ir 2b (0.1 mol%), and blue = Sn–Ir 2c (0.1 mol%). | ||
It should be noted, however, that the reaction at room temperature is probably slowed down even further due to the moderate solubility of the crystalline samples of the catalysts 2a–c in neat allyltrimethylsilane. This phenomenon is manifest in an extended induction period of approximately 5 h (Fig. 5) after which the spectroscopic yield increases much faster due to rapid dissolution in the mixture of allyltrimethylsilane and the isomerization products.
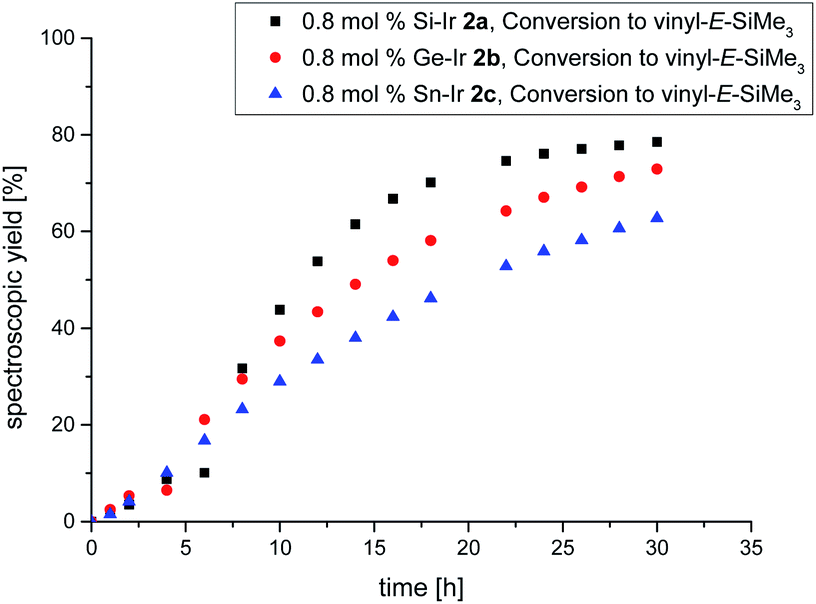 | ||
| Fig. 5 Plot of the spectroscopically determined conversion to E-vinyltrimethylsilane 60 °C using the 0.8 mol% catalyst. Black = Si–Ir 2a, red = Ge–Ir 2b, blue = Sn–Ir 2c. | ||
The driving force of the alkene isomerization is the higher thermodynamic stability of internal alkenes.25 As suggested by the negative blind tests with the siliconoid-free precursors, the intramolecular hydrogen migration is supported through the extremely electron-rich hexasilabenzpolarene scaffold.
Conclusion
In conclusion, with the Group 9 metal complexes 2a–c and 3 we reported the first siliconoids with endohedral incorporation of transition metals. As demonstrated by the isomerization of 3 to the 4 with complete reconstitution of the uncompromised benzpolarene scaffold, a temporary change in the coordination mode of these ligands is possible in principle. All isolated complexes show catalytic activity in the isomerization of alkenes with the best (2a) reaching competitive selectivity at satisfactory conversion rates in the case of 1-hexene.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding by the Deutsche Forschungsgemeinschaft (DFG SCHE906/4-1 and 4-2) is gratefully acknowledged.Notes and references
- Recent reviews:
(a) E. Rivard, Chem. Soc. Rev., 2016, 45, 989–1003 RSC
; (b) B. Blom and M. Driess, Functional Molecular Silicon Compounds II, in Structure and Bonding, ed. D. Scheschkewitz, 2013, vol. 156, pp. 85–123 Search PubMed
- Recent reviews:
(a) A. Rammo and D. Scheschkewitz, Chem.–Eur. J., 2018, 24, 6866–6885 CrossRef CAS PubMed
- Recent reviews:
(a) S. Ishida and T. Iwamoto, Coord. Chem. Rev., 2016, 314, 34–63 CrossRef CAS
-
(a) A. Brück, D. Gallego, W. Wang, E. Irran, M. Driess and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 11478–11482 CrossRef PubMed
- M. Stoelzel, C. Präsang, B. Blom and M. Driess, Aust. J. Chem., 2013, 66, 1163–1170 CrossRef CAS
-
(a) D. Gallego, A. Brück, E. Irran, F. Meier, M. Kaupp, M. Driess and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 15617–15626 CrossRef CAS PubMed
- B. Blom, S. Enthaler, S. Inoue, E. Irran and M. Driess, J. Am. Chem. Soc., 2013, 135, 6703–6713 CrossRef CAS PubMed
- Reviews:
(a) Y. Heider and D. Scheschkewitz, Dalton Trans., 2018, 47, 7104–7112 RSC
-
(a) P. Willmes, K. Leszczyńska, Y. Heider, K. Abersfelder, M. Zimmer, V. Huch and D. Scheschkewitz, Angew. Chem., Int. Ed., 2016, 55, 2907–2910 CrossRef CAS PubMed
-
(a) S. Scharfe, F. Kraus, S. Stegmaier, A. Schier and T. F. Fässler, Angew. Chem., Int. Ed., 2011, 50, 3630–3670 CrossRef CAS PubMed
- Recent review: R. J. Wilson, B. Weinert and S. Dehnen, Dalton Trans., 2018, 47, 14861–14869 RSC
- Recent review:
(a) S. D. Hoffmann and T. F. Fässler, Angew. Chem., Int. Ed., 2004, 43, 6242–6247 CrossRef PubMed
- N. E. Poitiers, L. Giarrana, K. I. Leszczyńska, V. Huch, M. Zimmer and D. Scheschkewitz, Angew. Chem., Int. Ed., 2020, 59, 8532–8536 CrossRef CAS PubMed
- K. Abersfelder, A. Russell, H. S. Rzepa, A. J. P. White, R. Haycock and D. Scheschkewitz, J. Am. Chem. Soc., 2012, 134, 16008–16016 CrossRef CAS PubMed
- Y. Heider, P. Willmes, V. Huch, M. Zimmer and D. Scheschkewitz, J. Am. Chem. Soc., 2019, 141(49), 19498–19504 CrossRef CAS PubMed
-
(a) M. Kilian, H. Wadepohl and L. H. Gade, Organometallics, 2008, 27(4), 524–533 CrossRef CAS
- A. W. Addison and T. N. Rao, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC
-
(a) M. J. S. Dewar, Bull. Soc. Chim. Fr., 1951, 18, C71 Search PubMed
- D. K. Dutta and M. M. Singh, Transition Met. Chem., 1979, 4, 230–234 CAS
- M. P. Keyes and K. L. Watters, J. Catal., 1986, 100, 477–481 CrossRef CAS
- K. Radacki, M. Forster and H. Braunschweig, Angew. Chem., Int. Ed., 2006, 45, 2132–2134 CrossRef PubMed
- G.-J. Boons, A. Burton and S. Isles, Chem. Commun., 1996, 141–142 RSC
- G. Chahboun, C. E. Petrisor, E. Gómez-Bengoa, E. Royo and T. Cuenca, Eur. J. Inorg. Chem., 2009, 1514–1520 CrossRef CAS
-
(a) R. H. Grubbs, Tetrahedron, 2004, 60, 7117–7140 CrossRef CAS
- D. B. Dahl, C. Davies, R. Hyden, M. L. Kirova and W. G. Lloyd, J. Mol. Catal. A: Chem., 1997, 123, 91–101 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2000911–2000916. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc02861d |
| This journal is © The Royal Society of Chemistry 2020 |