DOI:
10.1039/D0SC02641G
(Edge Article)
Chem. Sci., 2020,
11, 7789-7799
Metal-chelating benzothiazole multifunctional compounds for the modulation and 64Cu PET imaging of Aβ aggregation†
Received
12th May 2020
, Accepted 7th July 2020
First published on 9th July 2020
Abstract
While Alzheimer's Disease (AD) is the most common neurodegenerative disease, there is still a dearth of efficient therapeutic and diagnostic agents for this disorder. Reported herein are a series of new multifunctional compounds (MFCs) with appreciable affinity for amyloid aggregates that can be potentially used for both the modulation of Aβ aggregation and its toxicity, as well as positron emission tomography (PET) imaging of Aβ aggregates. Firstly, among the six compounds tested HYR-16 is shown to be capable to reroute the toxic Cu-mediated Aβ oligomerization into the formation of less toxic amyloid fibrils. In addition, HYR-16 can also alleviate the formation of reactive oxygen species (ROS) caused by Cu2+ ions through Fenton-like reactions. Secondly, these MFCs can be easily converted to PET imaging agents by pre-chelation with the 64Cu radioisotope, and the Cu complexes of HYR-4 and HYR-17 exhibit good fluorescent staining and radiolabeling of amyloid plaques both in vitro and ex vivo. Importantly, the 64Cu-labeled HYR-17 is shown to have a significant brain uptake of up to 0.99 ± 0.04 %ID per g. Overall, by evaluating the various properties of these MFCs valuable structure–activity relationships were obtained that should aid the design of improved therapeutic and diagnostic agents for AD.
Introduction
Alzheimer's disease (AD) is the most common neurodegenerative disease, associated with loss of memory and cognitive decline.1 An estimated 5.8 million Americans of all ages are living with Alzheimer's dementia currently.2 The presence of amyloid plaques and neurofibrillary tangles in the brain is the hallmark of AD.3 Amyloid β (Aβ) peptides, the main component of amyloid plaques,4–7 are formed from the cleavage of amyloid precursor protein (APP) by β- and γ-secretases. The main alloforms of Aβ are Aβ40 and Aβ42, containing 40 and 42 amino acids, respectively.8 Even though Aβ40 is present in the deposits in larger amounts, Aβ42 exhibits higher neurotoxicity and aggregates more easily.9–12 In the past two decades, soluble Aβ oligomers have been found to be the most toxic form among all Aβ species13–19 through their interactions with membrane and synaptic receptors18,20 that influence intracellular systems21 and affect neurotransmission,22,23 leading to neurodegeneration.
In addition, the amyloid deposits contain uncommonly high concentrations of metal ions such as Fe2+, Cu2+ and Zn2+,24,25 and it has been found that these metal ions promote the formation of neurotoxic Aβ aggregates.26–29 Cu and Fe ions can also cause the formation of reactive oxygen species (ROS), which exacerbates Aβ toxicity.25,30–32 Previously, we have reported that Cu2+ ions can slow down Aβ fibrillization and stabilize Aβ oligomers,33,34 and thus small molecules that can inhibit the interaction between metal ions and Aβ peptides, can be used as potential therapeutic compounds for AD.35–38
Moreover, the development of novel diagnostic agents is essential for the prevention and treatment of AD. Recently, several positron emission tomography (PET) compounds have been approved by FDA and can be used to visualize amyloid plaques in AD patients. However, these radiolabeled agents are employing short-lived radionuclides, such as 11C and 18F (t1/2 = 20.4 min and 109.8 min,39–45 respectively), thus limiting their widespread use. Thus, the development of longer-lived radiolabeled compounds is essential for further expanding the use of PET imaging in healthcare, and diagnostic agents employing longer-lived radionuclides such as 64Cu (t1/2 = 12.7 h, β+ = 17%, β− = 39%, EC = 43%, Emax = 0.656 MeV) are viewed as optimal PET imaging agents.46,47
We have previously reported first-generation multifunctional compounds (MFCs) that contain Aβ binding and metal chelating fragments33,48 and these MFCs were found to have the ability to modulate the aggregation of Aβ42 species, although some led to the formation of neurotoxic soluble Aβ42 oligomers.33,48 Moreover, some MFCs could be converted into PET imaging agents through pre-chelation with 64Cu and have with MFCs, PET imaging agents could be obtained for the diagnosis of Alzheimer's disease.49,50 Herein, we report a series of second-generation multifunctional compounds (MFCs) containing metal-binding and Aβ-interacting fragments that also exhibit additional key properties (Fig. 1). In contrast to the first-generation MFCs, which were shown to promote the formation of neurotoxic oligomeric Aβ species,33,48 these MFCs are capable of re-routing the neurotoxic metal-stabilized Aβ oligomers into less toxic aggregates, while also decreasing the formation of ROS. Moreover, these MFCs can be easily converted into PET imaging agents by chelation with the 64Cu radionuclide, and ex vivo labeling studies using AD mouse brain sections reveal that the MFCs and their Cu complexes can clearly label the amyloid plaques. In addition, biodistribution studies show that the 64Cu-radiolabeled compounds cross the blood–brain barrier (BBB) efficiently and thus should be able to act as potential therapeutic or imaging agents in vivo. Finally, structure–activity relationship (SAR) studies suggest that the presence of a monomethylamine group leads to increased specificity for binding to the Aβ aggregates, while the introduction of a pyridyl group is essential for modulating the neurotoxicity of metal–Aβ species. Most importantly, repositioning of the hydroxyl group and the metal-chelating azamacrocycle on the benzothiazole ring in HYR-17 has a dramatic effect on improving the brain uptake of the corresponding 64Cu complex, which could be a useful design approach for the development of improved PET imaging agents.
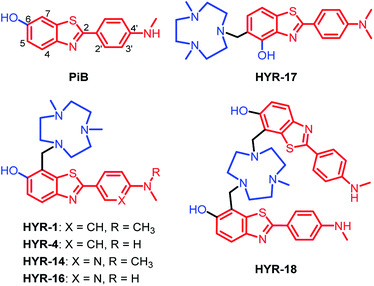 |
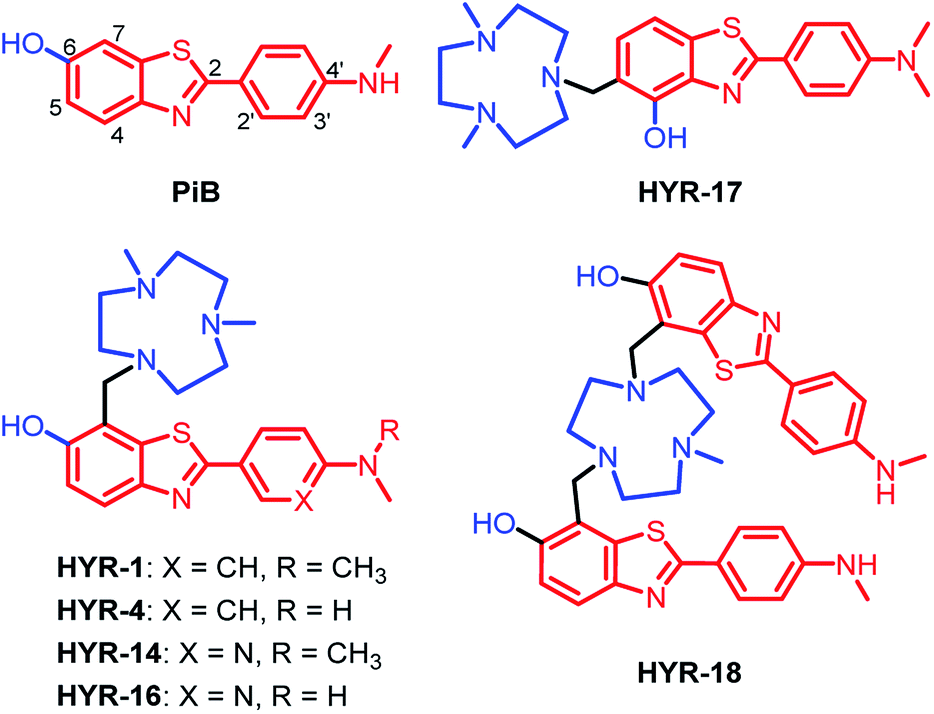
| | Fig. 1 Structures of Pittsburgh compound B (PiB) and the developed multifunctional compounds (MFCs) HYR-1, -4, -14, -16, -17 and -18. The metal-binding and Aβ-interacting fragments are shown in blue and red, respectively. The numbering of the different positions on the aromatic rings is shown only for the first structure. | |
Results and discussion
Design, synthesis and characterization of MFCs
Following up on our previous results, we set out to develop second-generation MFCs aimed to better alleviate the metal-induced Aβ toxicity, while also obtaining valuable structure–activity relationships (SAR) that should give us insight into the design of improved therapeutic and imaging agents for AD.
Previously, 2-aryl-benzothiazole derivatives have been found to have appreciable Aβ binding affinity and fluorescence properties,33,48 and thus we continued to use such molecular frameworks. Inspired by the structure of Pittsburgh compound B (PiB, Fig. 1), a widely-used amyloid-binding compound with high Aβ binding affinity,39 we introduced a hydroxyl group in the 6-position of the benzothiazole aromatic ring (Fig. 1). In addition, dimethylamino and monomethylamino groups were introduced at the 4′ position of the phenyl ring, since such functional groups are present in many Aβ binding compounds such as Thioflavin T (ThT) and Florbetapir.43,51 Then, we have introduced a 1,4-dimethyl-1,4,7-triazacyclononane (tacn) metal-chelating group attached to the benzothiazole aromatic ring in order to generate a MFC that can modulate the metal–Aβ interactions. Importantly, previous studies have shown that the tacn-phenolate metal-chelating fragment can bind more tightly to Cu versus other metal ions such as Zn and Fe.52–54 Accordingly, HYR-1 and HYR-4 were developed, and we have also designed HYR-14 and HYR-16 as 3′-pyridyl analogues in order to probe the effect of a hydrogen-bond acceptor pyridyl group vs. a phenyl group.43,51 Moreover, it was previously found that a PiB derivative with a hydroxyl group in the 4-position of benzothiazole framework performed similarly as PiB in Aβ binding and bio-distribution studies,55 and thus we have designed HYR-17, which contains a 4-hydroxyl substituent and has the tacn azamacrocycle connected to the 5 position of the benzothiazole ring (Fig. 1). Finally, the MFC HYR-18 that contains two 2-aryl-benzothiazole fragments attached to one tacn azamacrocycle was also synthesized, in order to probe whether additional Aβ binding fragments will improve the affinity for Aβ aggregates.
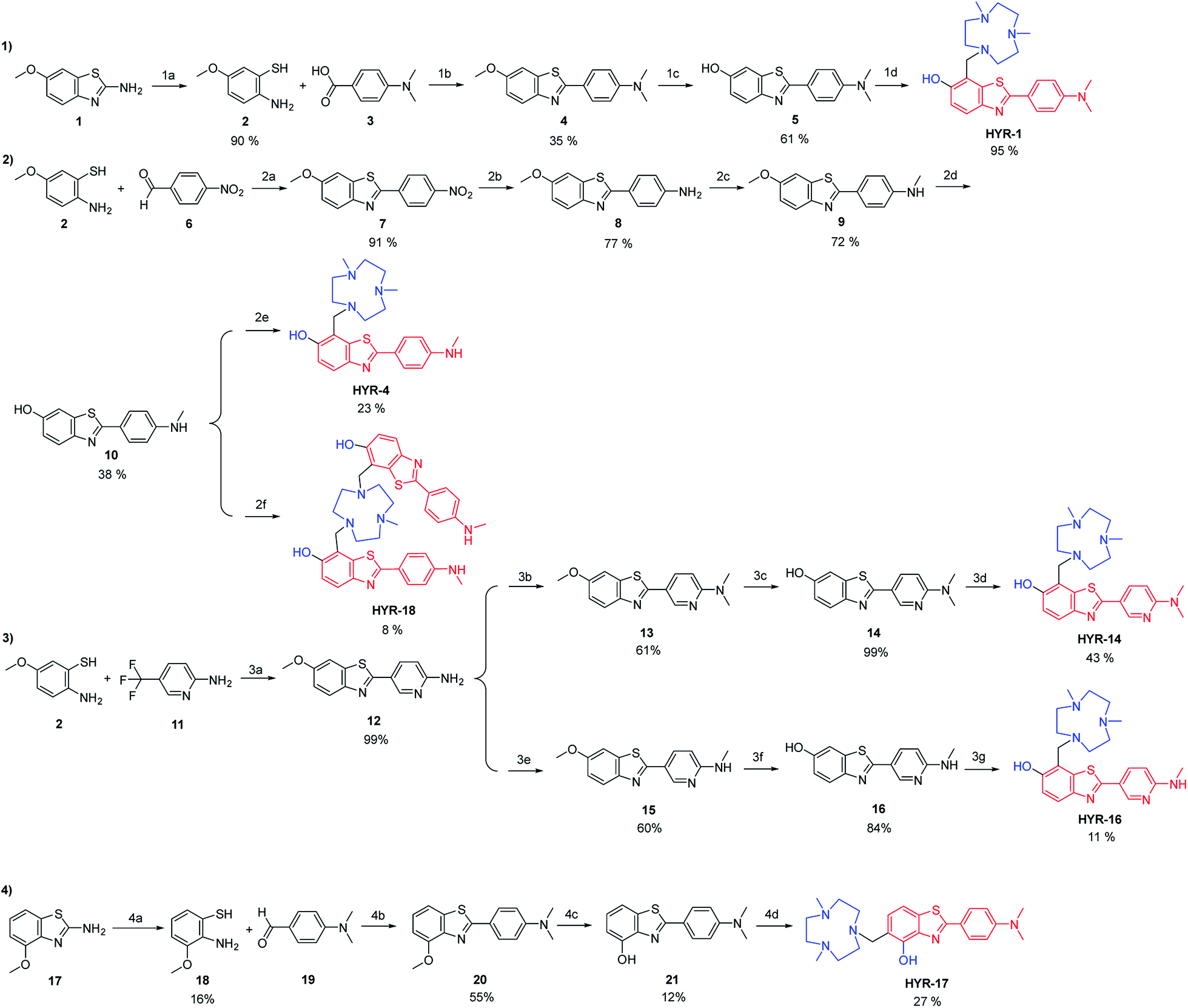
The synthesis of the MFCs HYR-1, -4, -14, -16, -17 and -18 follows a stepwise sequence of steps that typically includes an oxidative cyclization step between a 2-amino-methoxybenzenethiol derivative and a benzaldehyde or benzoic acid derivative to generate the 2-aryl-benzothiazole fragments (Scheme 1). In cases where the 2-amino-methoxy-benzothiazole precursor was readily available, hydrolysis under basic conditions afforded the 2-amino-methoxybenzenethiol starting materials, while for the pyridyl derivatives 2-amino-5-(trifluoromethyl)pyridine was employed in the oxidative cyclization reaction. Reduction of the nitro group to the aniline derivative using tin(II) chloride (if needed) and subsequent N-monomethylation using paraformaldehyde and sodium borohydride or N-dimethylation using paraformaldehyde and sodium cyanoborohydride generated the 2-(4′-aniline-aryl)-benzothiazole derivatives. Deprotection of the methoxy group using boron tribromide afforded the hydroxyl-benzothiazole derivatives, while the last synthetic step for all compounds is the Mannich reaction with paraformaldehyde and 2,4-dimethyl-1,4,7-triazacyclononane under reflux to generate the targeted MFCs (Scheme 1).
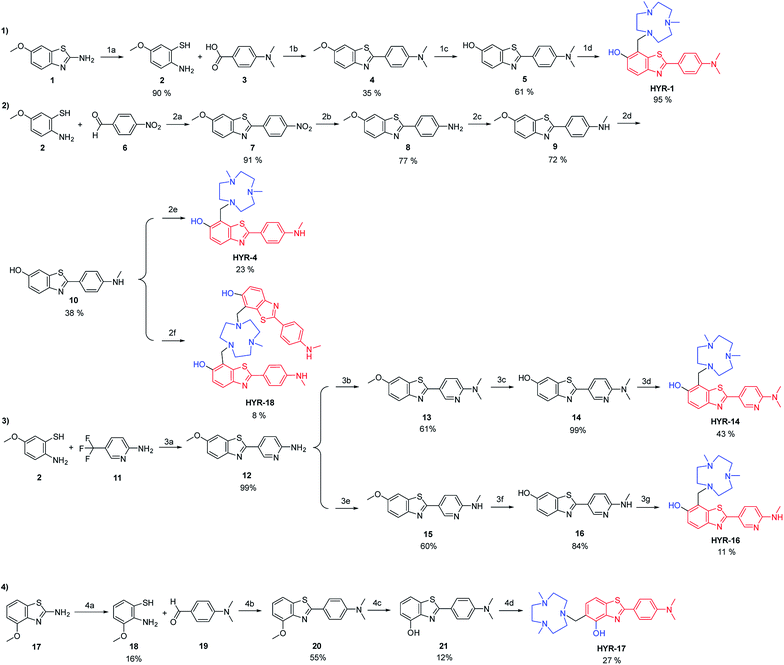 |
| | Scheme 1 Synthesis of the investigated MFCs HYR-1, -4, -14, -16, -17 and -18. The metal-binding and Aβ-interacting fragments are shown in blue and red, respectively. Reagents and conditions: (1a) KOH, H2O, ethylene glycol, reflux, 48 h; (1b) DMSO, 170 °C, 30 min; (1c) BBr3, DCM, rt, 24 h; (1d) Me2HTACN, (CH2O)n, MeCN, reflux, 24 h; (2a) DMSO, 125 °C, overnight; (2b) SnCl2, EtOH, conc. HCl, reflux, 3 h; (2c) (i) (CH2O)n, NaOMe, MeOH, reflux, 2 h; (ii) NaBH4, 0 °C to rt, 1 h; (2d) BBr3, DCM, rt, 24 h; (2e) Me2HTACN, (CH2O)n, MeCN, reflux, 24 h; (2f) MeH2TACN, (CH2O)n, MeCN, reflux, 24 h; (3a) NaOH (1 M), 90 °C, 3 h; (3b) (CH2O)n, NaBH3CN, acetic acid, rt, overnight; (3c) BBr3, DCM, rt, 24 h; (3d) Me2HTACN, (CH2O)n, MeCN, reflux, 24 h; (3e) (i) (CH2O)n, NaOMe, MeOH, reflux, 2 h; (ii) NaBH4, reflux, 1 h; (3f) BBr3, DCM, rt, 24 h; (3g) Me2HTACN, (CH2O)n, MeCN, reflux, 24 h; (4a) KOH, H2O, ethylene glycol, reflux, 48 h; (4b) DMSO, 125 °C, 30 min; (4c) BBr3, DCM, rt, 24 h; (4d) Me2HTACN, (CH2O)n, MeCN, reflux, 24 h. | |
ThT fluorescence competition assays
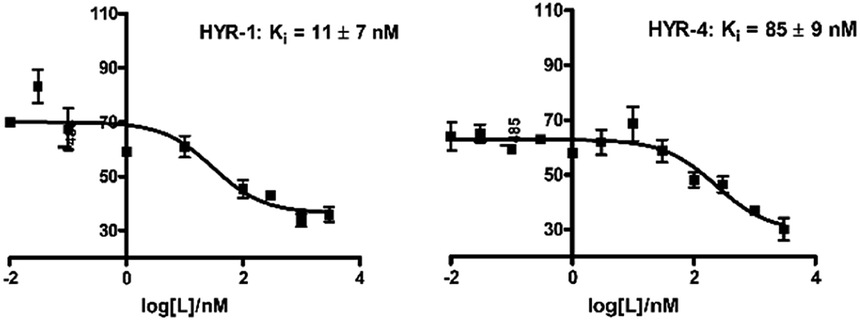
In order to measure the binding affinity of MFCs toward amyloid fibrils, ThT fluorescence competition assays were performed. The Aβ40 peptide was used in these experiments, since it is known that Aβ40 forms well-defined amyloid fibrils.56,57 Excitingly, HYR-1 and HYR-4 exhibit nanomolar affinities for the Aβ40 fibrils with Ki values of 11 ± 7 nM and 85 ± 9 nM, respectively, indicating that the MFCs can replace ThT efficiently and bind tightly to the Aβ40 fibrils (Fig. 2). However, for the other MFCs it was difficult to obtain reproducible Ki values, likely due to the larger structural differences between these MFCs and ThT and thus the inability to compete with ThT for the same binding site(s) on the amyloid fibrils and that not lead to an appreciable decrease in ThT fluorescence. In addition, we consider that probing the affinity of the MFCs toward native amyloid plaques by performing ex vivo binding studies with transgenic AD mice brain sections should provide more physiologically relevant results and also rule out any non-specific binding (see below).
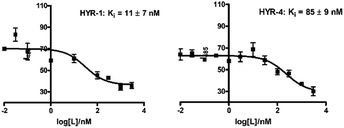 |
| | Fig. 2 ThT fluorescence competition assays for MFCs HYR-1 and HYR-4 with ThT-bound Aβ40 fibrils ([Aβ] = 5 μM, [ThT] = 2 μM). | |
Fluorescence imaging of amyloid plaques in 5×FAD mice brain sections
Brain sections collected from 8 month-old 5×FAD transgenic mice were employed in these ex vivo Aβ binding studies. The transgenic 5×FAD mice overexpress mutant forms of the amyloid precursor protein (APP) and presenilin 1, develop amyloid plaque deposits at an younger age, and show progressive cognitive impairment similar to that found in AD in humans.58
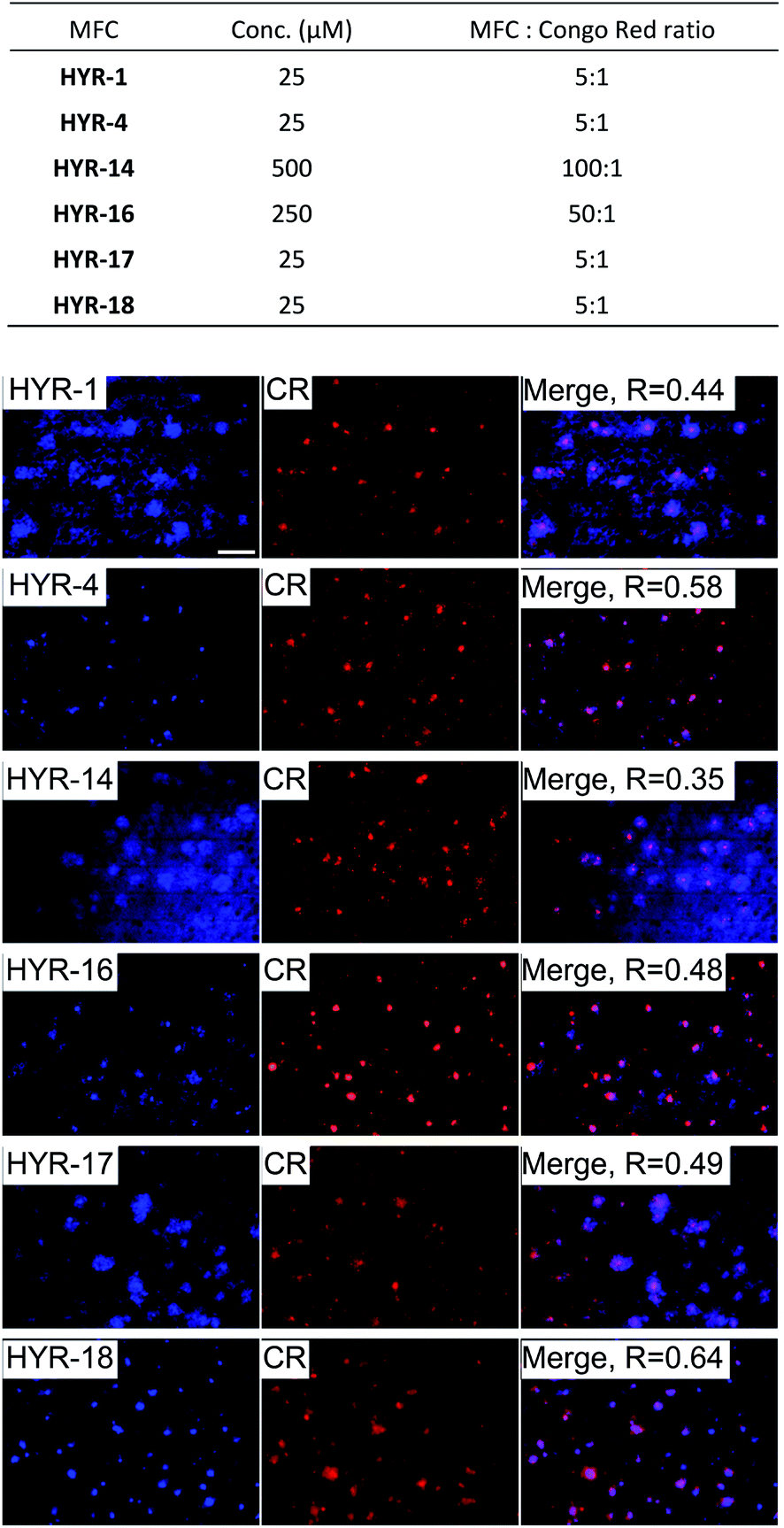
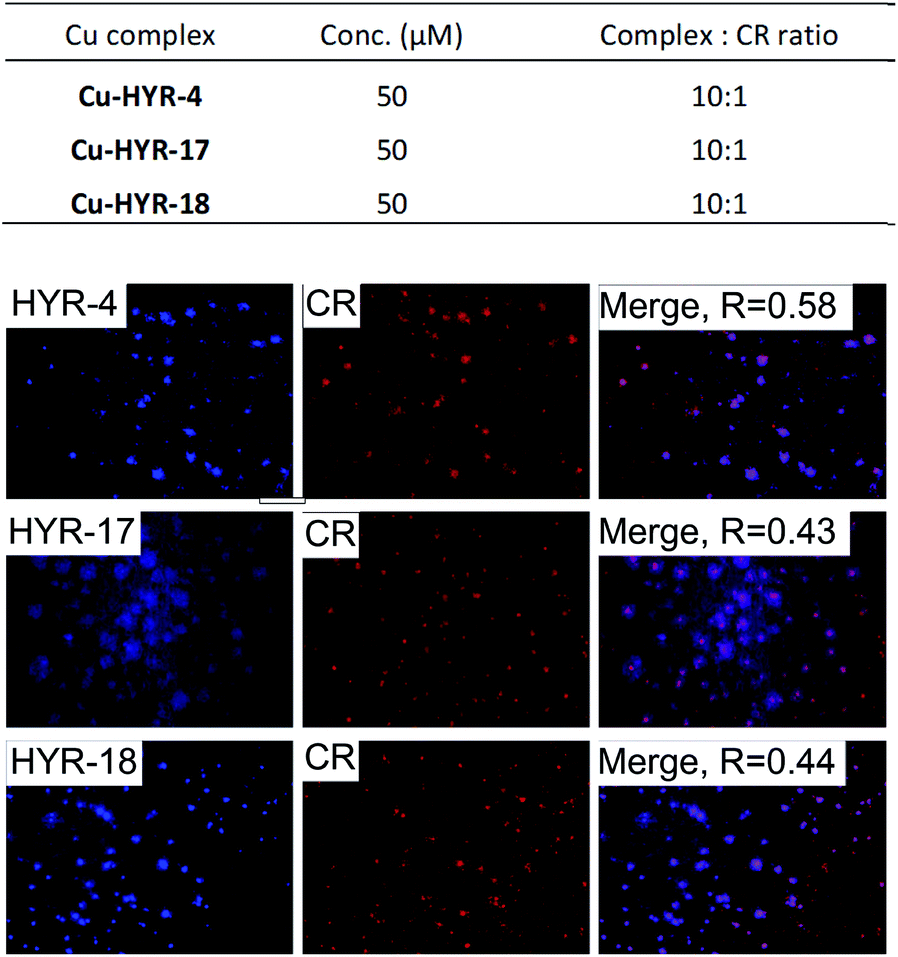
Interestingly, incubation of the 5×FAD mouse brain sections with the different MFCs for 1 hour reveals significant fluorescent staining of the amyloid plaques, as confirmed by co-staining with Congo Red (CR), a well-known amyloid-binding fluorescent dye (Fig. 3). Among the MFCs, HYR-1, -4, -17, and -18 show significant amyloid staining at low concentration (25 μM), suggesting an appreciable Aβ binding affinity. The most specific amyloid-binding MFCs are HYR-4 and HYR-18 (and HYR-17 to a slightly lesser extent), which bind to the dense core of the amyloid plaques and thus show the best colocalization with CR. By contrast, HYR-1 seems to label other endogenous proteins, both in AD and WT brain sections (Fig. 3 and S5,† respectively). Also, HYR-1 can label other regions in wild type mice brain section (Fig. S5†), which means its specificity is extremely low. By comparison, the pyridyl-containing MFCs HYR-14 and HYR-16 exhibit appreciable amyloid plaque staining only at high concentrations (250–500 μM), suggesting that their Aβ binding affinity is somewhat reduced and thus these two compounds were not employed in radiolabeling studies (see below).
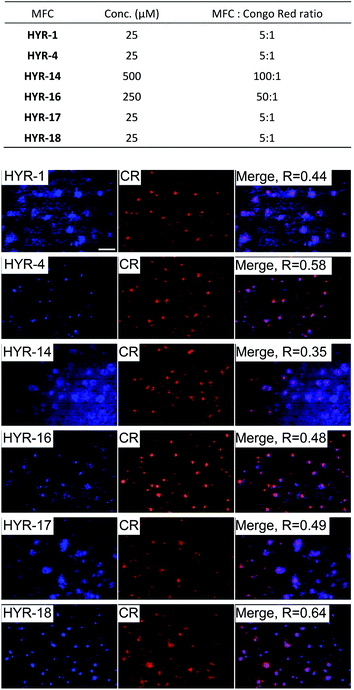 |
| | Fig. 3 Fluorescence microscopy images of 5×FAD mice brain sections incubated with multifunctional compounds HYR-1, -4, -14, -16, -17 and -18 (left panels), Congo Red (CR, middle panels), and merged images (right panels, with the Pearson's coefficient R shown). Scale bar: 100 μm. The concentration of MFC and MFC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Congo Red ratio used are listed at the top. :Congo Red ratio used are listed at the top. | |
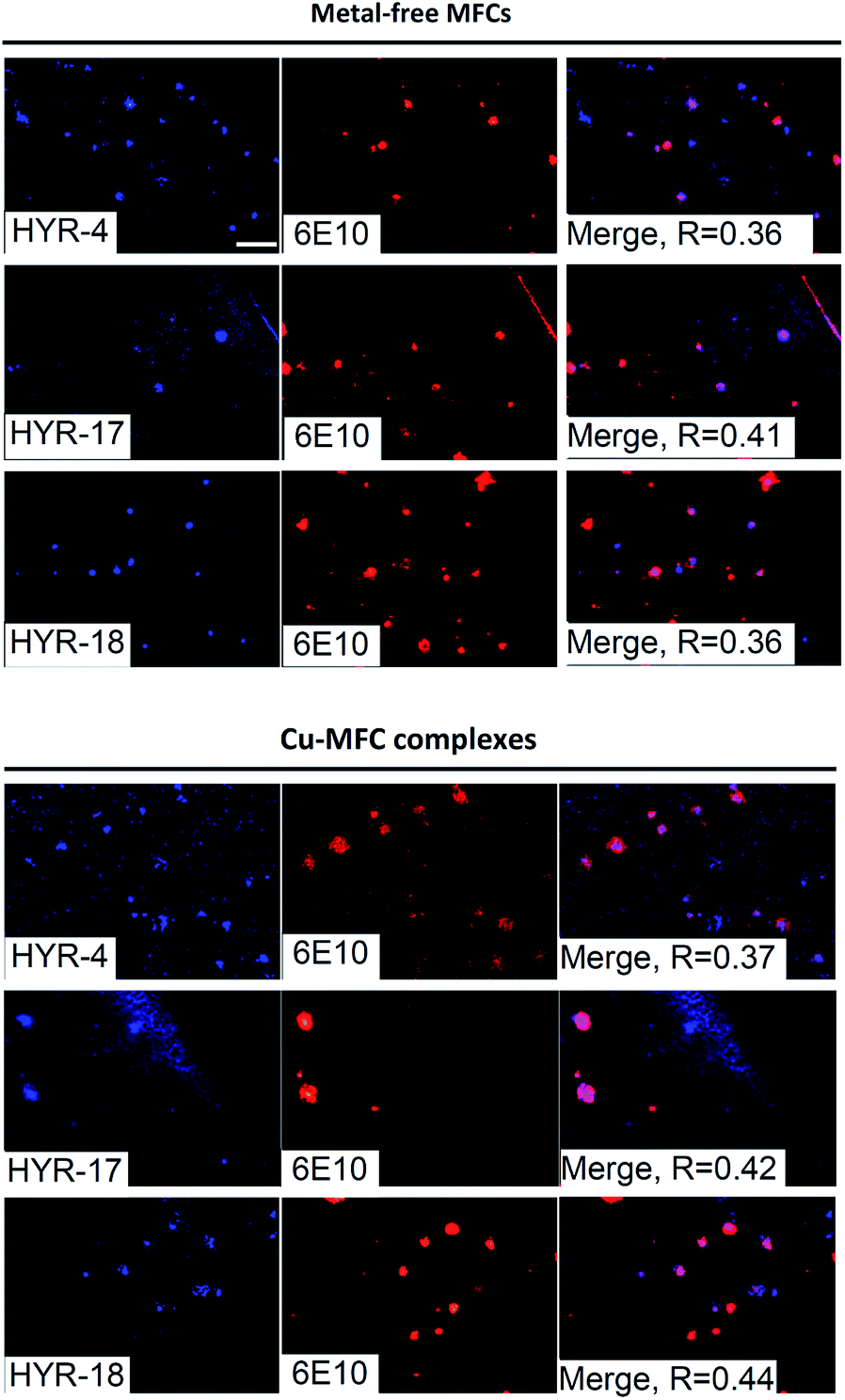
We have also probed the ability of the Cu2+ complexes of HYR-4, -17 and -18 to label the amyloid plaques in 5×FAD mice brain sections (Fig. 4). In this case, higher ligand to Congo Red ratios were used since the Cu2+ ions lead to some fluorescence quenching for our MFCs (Fig. S5 and S6†). However, the Cu2+ complexes of HYR-4, -17 and -18 can still label the amyloid plaques efficiently (Fig. 3), with the complexes of HYR-4 and -18 showing higher specificity compared with the Cu-HYR-17 complex. Finally, the fluorescently labeled CF594-6E10 antibody – which binds to a wide range of Aβ species, was employed to confirm that our MFCs and their Cu2+ complexes are able to specifically label the Aβ42 species (Fig. 5). It is important to note that we have previously determined the stability constants for the Cu2+ complex of another 1,4,7-triazacyclononane-phenolate ligand, which was shown to bind Cu2+ very tightly.49,50 Since the structural changes for the different MFCs described herein are remote from the Cu-binding site, we envision that the stability constants for the corresponding Cu2+ complexes will not vary significantly.
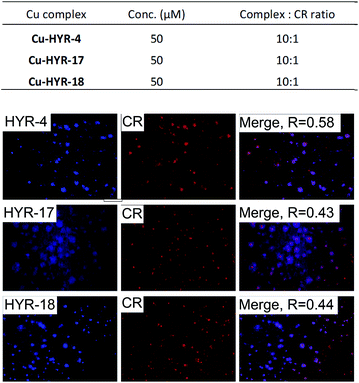 |
| | Fig. 4 Fluorescence microscopy images of 5×FAD mice brain sections incubated with Cu2+ complexes of HYR-4, -17 and -18 (left panels), Congo Red (middle panels), and merged images (right panels). Scale bar: 100 μm. | |
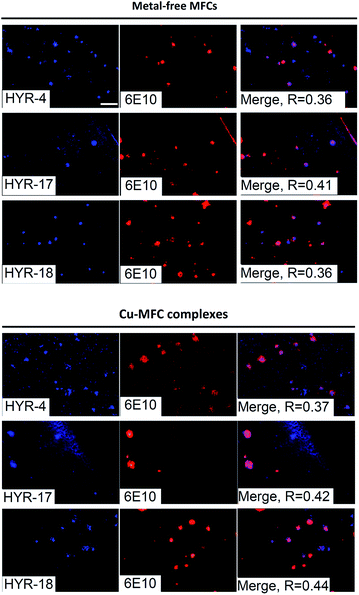 |
| | Fig. 5 Fluorescence microscopy images of 5×FAD mice brain sections incubated with MFCs HYR-4, -17 and -18 or their Cu(II) complexes (left panels), CF594-6E10 antibody (middle panels), and merged images (right panels). Scale bar: 100 μm. | |
Modulation of metal-free and metal-induced Aβ aggregation
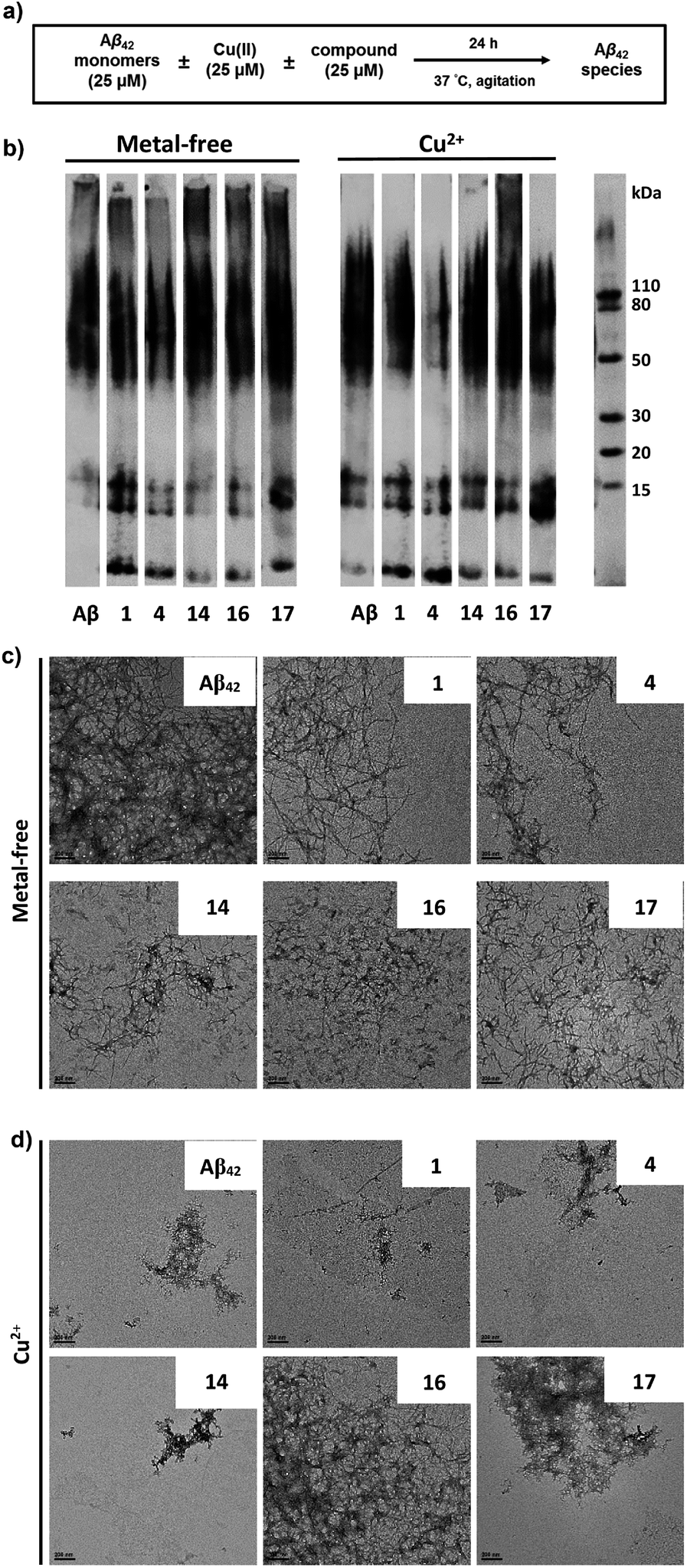
The ability of these MFCs to modulate the aggregation of Aβ42 was then explored, both in the absence or presence of Cu2+ ions. The Aβ42 peptide was used since it was shown to form neurotoxic soluble Aβ42 oligomers.12,13,18 Freshly prepared monomeric Aβ42 solutions were treated with MFCs, Cu2+, or both, and incubated for 24 hours at 37 °C, and the resulting samples were analysed by native gel electrophoresis/western blot analysis and transmission electron microscopy (TEM, Fig. 6). The former analysis method reveals the presence of smaller, soluble Aβ aggregates and their MW distribution, while the latter method provides the characterization of the larger, insoluble Aβ aggregates that cannot penetrate the gel, and thus allowing us to visualize all Aβ42 aggregates of various types and sizes.
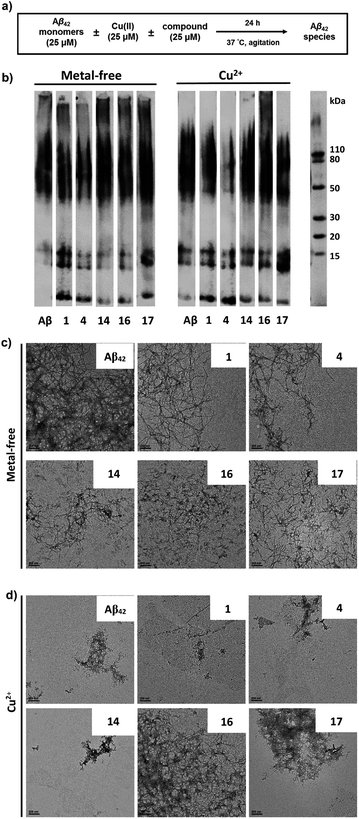 |
| | Fig. 6 Effects of the compounds on Cu2+-free and Cu2+-induced Aβ42 aggregation. (a) Scheme of the inhibition experiment: freshly prepared Aβ42 (25 μM) in the presence or absence of Cu2+ (25 μM) and with or without MFCs (25 μM) was incubated at 37 °C for 24 h with constant agitation. (b) Native gel/western blot analysis of the resulting Aβ42 species using the 6E10 anti-Aβ antibody. (c) Representative TEM images of the Aβ42 aggregates upon incubation with or without MFCs (scale bar, 200 nm). (d) Representative TEM images of the Aβ42 aggregates upon incubation with Cu2+ and with or without MFCs (scale bar, 200 nm). | |
While the aggregation of Aβ42 in the absence of Cu2+ ions leads to well-defined Aβ42 fibrils – as confirmed by TEM (Fig. 6c), the Aβ42 aggregation in the presence of Cu2+ ions generates a limited amount of Aβ fibrils (Fig. 6d), and native gel/western blotting shows that the aggregation of Aβ42 with Cu2+ ions yields mostly soluble Aβ42 oligomers of various sizes (Fig. 6b). These results are consistent with our previous studies which suggest that Cu2+ can stabilize the soluble Aβ42 oligomers and slow down the Aβ42 aggregation.33,59
All MFCs HYR-1, -4, -14, -16 and -17 did not seem to significantly inhibit the Aβ42 aggregation in the absence of Cu2+, even though some morphological changes were observed for the Aβ42 fibrils (Fig. 6b and c). Interestingly, the presence of HYR-16 had a dramatic effect on the Cu2+-mediated oligomerization of Aβ42 and promoted the formation of larger Aβ42 aggregates, as observed by TEM (Fig. 6d). Moreover, native gel/western blotting analysis reveals the presence of large, insoluble Aβ42 aggregates at the top of the gel, which were not observed for the Aβ42 aggregation in presence of only Cu2+ ions. Thus, HYR-16 is expected to control the neurotoxicity of Aβ42 species by accelerating the aggregation of toxic Aβ42 oligomers into nontoxic Aβ42 aggregates. By comparison, the other MFCs do not show a dramatic effect on Cu2+-mediated Aβ42 aggregation (Fig. 6b, left panels).
Cytotoxicity of MFCs and modulation of Aβ42 neurotoxicity
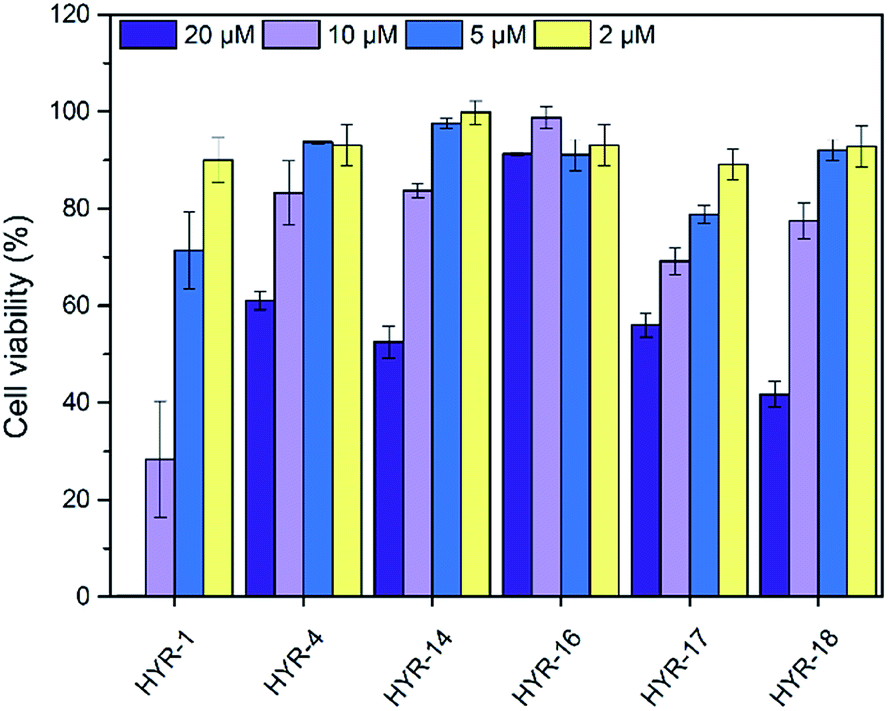
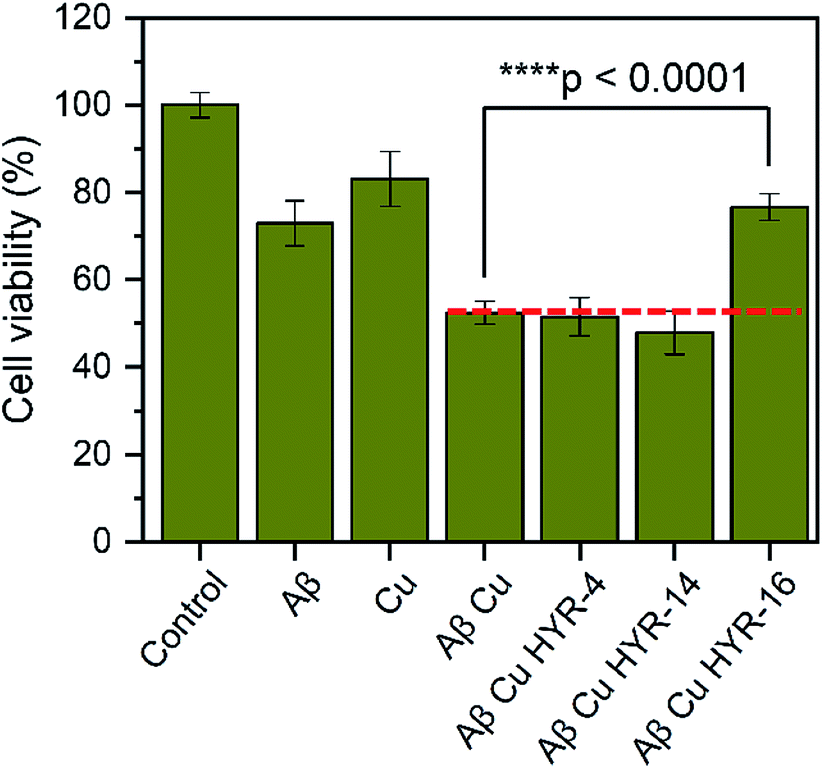
The neurotoxicity of the MFCs and their ability to alleviate the Cu-induced Aβ42 toxicity was performed using mouse neuroblastoma (N2a) cells. First, we examined the toxicity of all MFCs at various concentrations ranging from 2 to 20 μM (Fig. 7). Among the different MFCs, HYR-4, -14 and -16 exhibit no appreciable cell toxicity (>80% cell viability) up to 10 μM concentration, with HYR-16 showing no cell toxicity up to 20 μM concentration, and thus these three MFCs were employed in Aβ42-induced cytotoxicity studies. As we have shown before,33,34 the presence of both Cu2+ ions and Aβ42 leads to pronounced cell toxicity (∼50% cell viability), likely due to the formation of neurotoxic Aβ42 oligomers (Fig. 8). Interestingly, HYR-16 can significantly alleviate the toxicity of Cu-stabilized Aβ42 oligomers and increase the cell viability up to 80% vs. a 1% DMSO control, which is consistent with in vitro results that HYR-16 can accelerate the aggregation of toxic Aβ42 oligomers into nontoxic Aβ42 aggregates. By comparison, HYR-4 and HYR-14 do not seem to significantly reduce the neurotoxicity of the Cu-Aβ42 species (Fig. 8), suggesting that the 2-monomethylamino-pyridyl fragment found in HYR-16 is needed for the efficient alleviation of Aβ42 oligomer neurotoxicity.
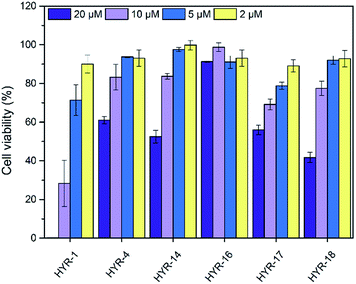 |
| | Fig. 7 Toxicity of MFCs at various concentrations in N2a cells, reported vs. a 1% DMSO control. The error bars indicate the standard deviations based on at least five samples per group. | |
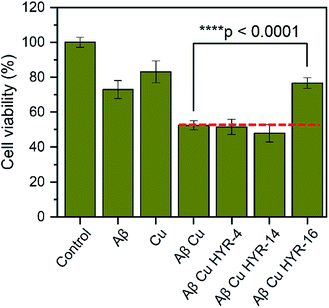 |
| | Fig. 8 The effect of MFCs on Cu2+-induced Aβ42 cytotoxicity, reported vs. a 1% DMSO control. N2a cells were treated with Aβ42 (20 μM), CuCl2 (20 μM), and MFCs (10 μM) and incubated for 40 h at 37 °C. The error bars indicate the standard deviations based on at least three samples per group, and the statistical analysis was evaluated according to one-way ANOVA (****p < 0.0001). | |
Antioxidant properties
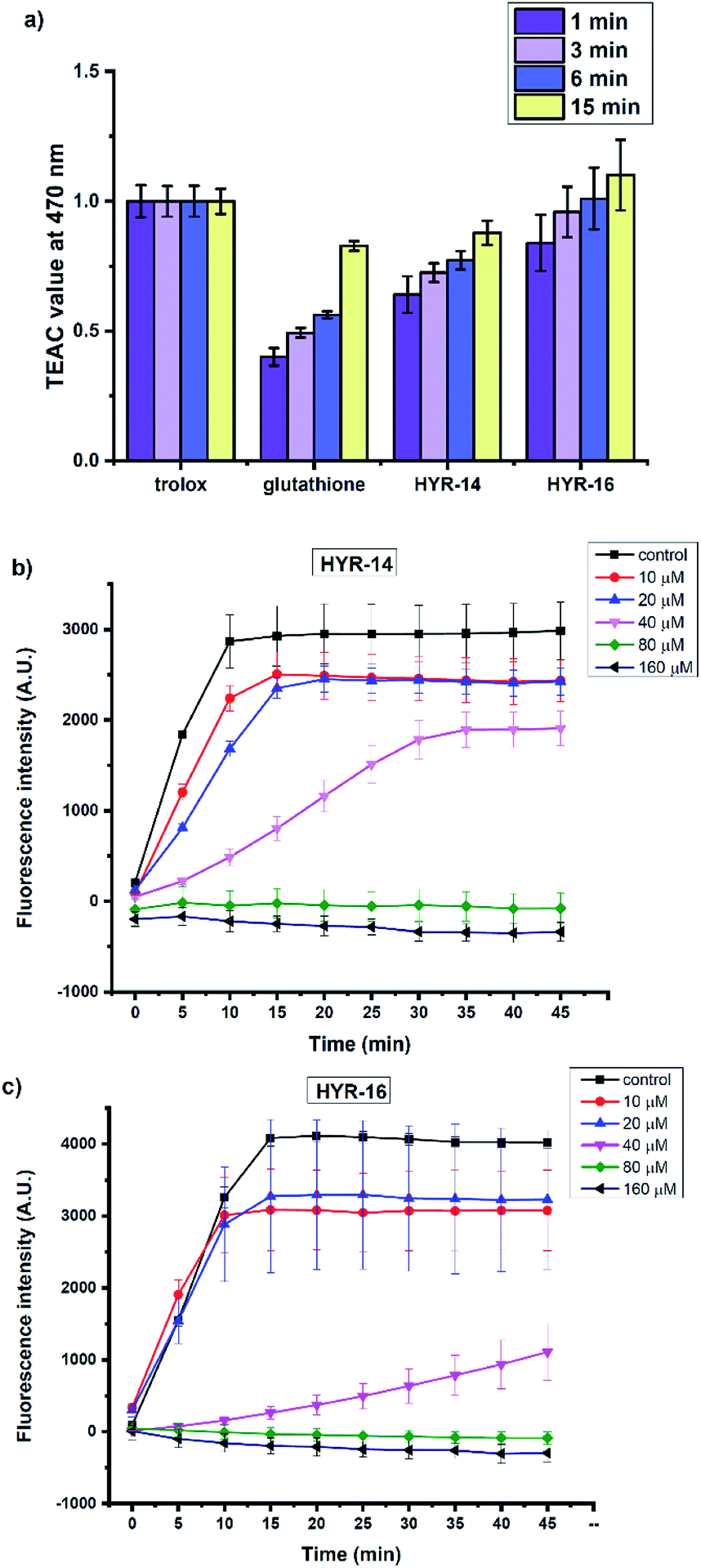
Several previous reports have shown that the Cu2+ ions can interact with various Aβ species and lead to formation of reactive oxygen species (ROS) such as H2O2 and hydroxyl radicals (OH˙).31,60 In this regard, the antioxidant capacity of the pyridine derivatives HYR-14 and HYR-16 – the least toxic MFCs investigated herein, was first evaluated via the Trolox equivalent antioxidant capacity (TEAC) assay.61 Both HYR-14 and HYR-16 showed better performance on scavenging the free radical ABTS+˙ (ABTS = 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid)) than glutathione – a well-known antioxidant, at all the selected time points (1–15 min, Fig. 9a), with HYR-16 showing an antioxidant capacity similar to Trolox.
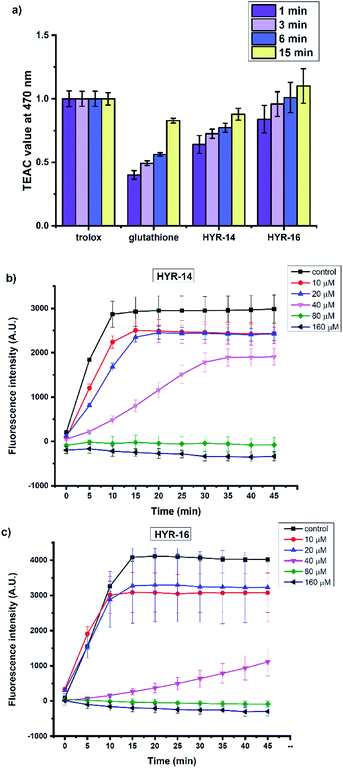 |
| | Fig. 9 (a) The antioxidant activity of Trolox, glutathione, HYR-14, and HYR-16 (25–100 μM) at 1, 3, 6, and 15 minutes assessed by TEAC assay. The TEAC values of glutathione, HYR-14, and HYR-16 were normalized to the Trolox activity. The inhibitory activity of (b) HYR-14 and (c) HYR-16 toward Cu-mediated hydroxyl radical generation, evaluated via the CCA antioxidant assay. Conditions: [CCA] = 100 μM; [CuSO4] = 40 μM; [ascorbic acid] = 400 μM; [HYR-14/HYR-16] = 0–160 μM; λex = 395 nm; λex = 450 nm. All experiments were performed in triplicate and the error bars indicate the standard deviation for each measurement. | |
In addition, the ability of HYR-14 and HYR-16 to quench the Cu-induced hydroxyl radical (OH˙) generation was measured by using coumarin-3-carboxylic acid (CCA) antioxidant assay.62 In this assay, the Cu2+ ions are reduced by ascorbic acid to Cu+ ions that then react with O2 to produce OH˙. The non-fluorescent CCA reacts with OH˙ to produce the fluorescent 7-hydroxycoumarin-3-carboxylic acid (CCA-OH), and CCA-OH formation can be monitored to evaluate the efficacy of ligand binding to Cu and inhibition of OH˙ generation. At 80 μM, both of HYR-14 and HYR-16 show a strong Cu2+ binding affinity (corresponding to a 2:1 MFC:Cu ratio) and limit the generation of OH˙ (Fig. 9b and c), while HYR-16 is able to limit the generation of OH˙ even at 40 μM (corresponding to a 1:1 MFC:Cu ratio). Overall, these studies reveal that HYR-16 is the most potent anti-oxidant among the investigated MFCs, and along with its ability to efficiently destabilize the neurotoxic soluble Aβ42 oligomers strongly suggest that HYR-16 may potentially exhibit therapeutic properties in animal studies.
Radiolabeling and log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) Doct value determination
Doct value determination
One crucial factor for developing imaging agents for neurodegenerative diseases is that they should be able to effectively cross the blood–brain barrier (BBB). To determine the hydrophobicity of the radiolabeled compounds, the octanol/PBS partition coefficient values logDoct were determined for the 64Cu complexes of HYR-1, -4, -14, -16, -17 and -18. The obtained logDoct values for the 64Cu-radiolabeled complexes HYR-1, -4, -17 and -18 are in the range of 1.08–1.30 (Table 1), which supports their potential ability to cross the BBB, since logD values between 0.9 and 2.5 are considered optimal.63 In contrast, the 64Cu complexes of HYR-14 and -16 exhibit logDoct values of ∼0.6, suggesting that 2-pyridyl-benzothiazole derivatives may be too hydrophilic to cross the BBB. Thus, only the MFCs HYR-1, -4, -17 and -18 were employed in the subsequent radiochemistry studies.
Table 1 Molecular weights for MFCs and logDoct values for the corresponding 64Cu-labeled complexes
| Ligand |
MW (g mol−1) |
logDoct |
|
HYR-1
|
439.6 |
1.08 ± 0.11 |
|
HYR-4
|
425.6 |
1.29 ± 0.19 |
|
HYR-14
|
440.6 |
0.58 ± 0.05 |
|
HYR-16
|
426.6 |
0.56 ± 0.17 |
|
HYR-17
|
439.6 |
1.30 ± 0.15 |
|
HYR-18
|
679.9 |
1.09 ± 0.11 |
Ex vivo autoradiography studies
Ex vivo autoradiography studies using brain sections of transgenic 5×FAD mice were also performed to determine the specific binding to the amyloid plaques of the 64Cu-labeled HYR-1, -4, -17 and -18. The brain sections were stained, washed, and radioimaged as described in the experimental section. When compared with WT brain sections that show a limited background intensity (Fig. 10, first row), the 64Cu-labeled complexes of HYR-4, -17 and -18 show an increased autoradiography intensity for the 5×FAD mouse brain sections (Fig. 10, second row). The 64Cu-labeled HYR-1 also exhibits non-specific binding as appreciable autoradiography intensity is observed for WT brain sections, which is consistent with the brain section fluorescent imaging studies described above. Finally, the specific binding to amyloid plaques of the 64Cu-labeled MFCs was confirmed by blocking with the non-radioactive blocking agent B1 (Fig. S6†), which led to a markedly decreased autoradiography intensity (Fig. 10, third row). Overall, these autoradiography results strongly suggest that the 64Cu-labeled MFCs HYR-4, -17 and -18 have the necessary amyloid-binding specificity to be used as imaging agents in vivo. The MFC HYR-18, the bis-(2-phenylbenzothiazole) analogue of HYR-4, exhibits a slightly lower logDoct value than HYR-4, and it was also perceived to have a too large MW for an efficient brain uptake. Since HYR-4 already exhibits a promising logDoct value and specific binding to amyloid plaques, we decided to not include HYR-18 in the in vivo biodistribution studies.
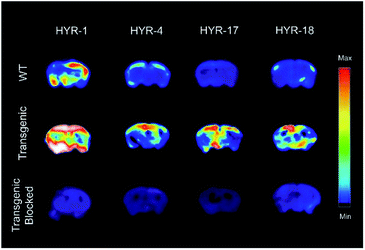 |
| | Fig. 10 Autoradiography images of 5×FAD mice brain sections with the absence and presence with a known Aβ blocking agent (B1). | |
Biodistribution studies
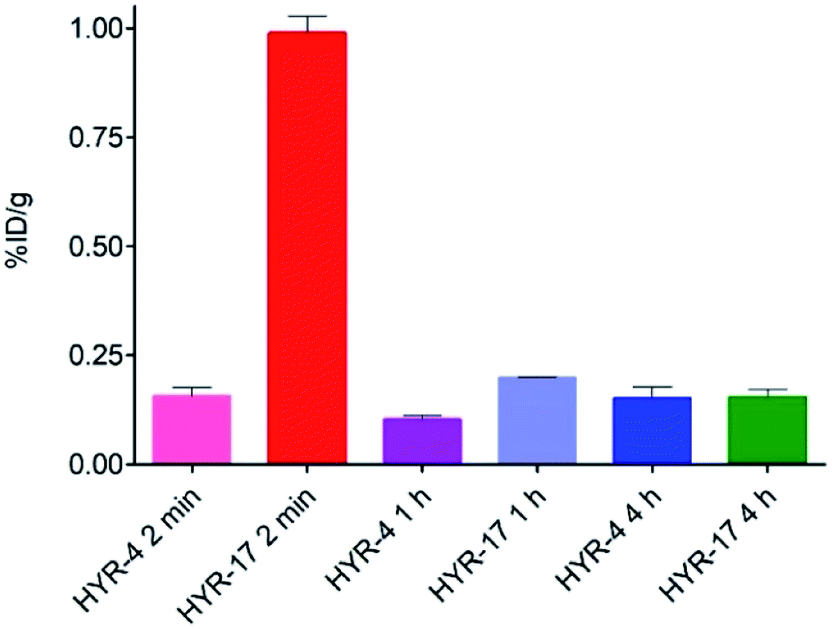
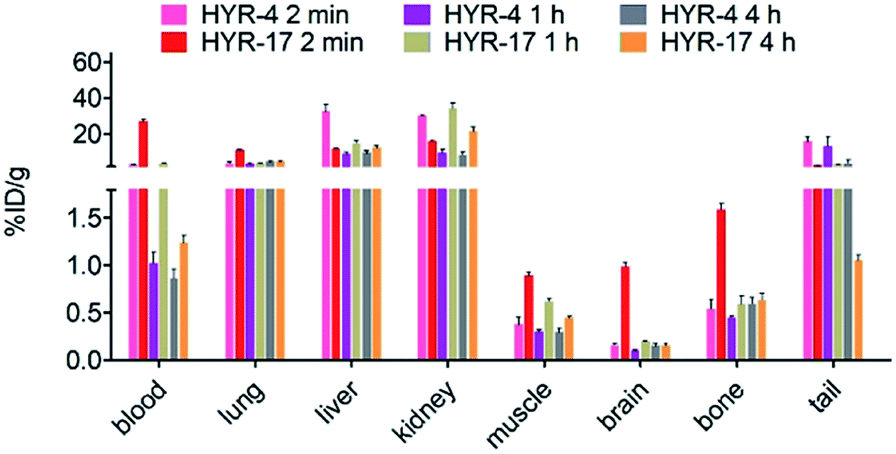
Encouraged by the promising in vitro radiolabeling and amyloid-binding studies, in vivo biodistribution experiments were performed to probe the uptake of 64Cu-labelled HYR-4 and -17 complexes using normal CD-1 mice. The retention and accumulation of the 64Cu-labeled complexes in selected organs was evaluated at 2, 60, and 240 minutes after tracer administration. Excitingly, 64Cu-HYR-17 shows an appreciable brain uptake of 0.99 ± 0.04 % injected dose per gram (%ID per g) at 2 min post injection, which drops to 0.20 ± 0.01 %ID per g at 60 min (Fig. 11 and Table 2). By comparison, 64Cu-HYR-4 shows a relatively low brain uptake of 0.16 ± 0.02 %ID per g at 2 min post injection, although 64Cu-HYR-4 shows a similar blood uptake to that of 64Cu-HYR-17 (Fig. 12), and suggesting that the structure of HYR-17 containing the 4-hydroxyl substituent and tacn azamacrocycle connected to the 5 position of the benzothiazole ring should lead to improved brain uptake properties for the corresponding 64Cu-labeled complexes. Overall, these biodistribution studies strongly suggest that the 64Cu-HYR-17 complex can efficiently cross the BBB and thus could serve as a PET imaging agent for the detection of Aβ aggregates in vivo. Importantly, the rapid clearance from the brain of WT mice suggest that these radiolabeled MFCs do not release 64Cu in the brain to an appreciable extent, and thus should not lead to a significant background PET signal in WT or healthy controls.
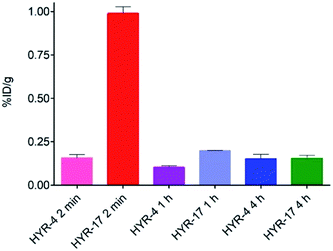 |
| | Fig. 11 Brain uptake (% injected dose per gram, %ID per g) results from the biodistribution studies in CD-1 mice, at 2, 60, and 240 min post injection. | |
Table 2 Overall biodistribution results of 64Cu-Labeled HYR-4 and HYR-17 for the three time points evaluated (2, 60, and 240 min; % injected dose/gram, mean ± SEM)
| Organ |
HYR-4
|
HYR-17
|
| 2 min |
60 min |
240 min |
2 min |
60 min |
240 min |
| Blood |
2.85 ± 0.52 |
1.02 ± 0.12 |
0.86 ± 0.10 |
26.82 ± 1.59 |
3.70 ± 0.06 |
1.24 ± 0.08 |
| Lung |
3.77 ± 0.88 |
3.36 ± 0.25 |
4.47 ± 0.49 |
11.02 ± 0.52 |
3.65 ± 0.16 |
4.47 ± 0.49 |
| Liver |
32.48 ± 3.94 |
8.84 ± 0.98 |
9.57 ± 1.19 |
11.50 ± 0.69 |
14.79 ± 1.71 |
12.16 ± 1.28 |
| Kidney |
30.27 ± 0.44 |
9.39 ± 2.02 |
8.24 ± 1.95 |
15.96 ± 0.81 |
34.17 ± 2.87 |
21.40 ± 2.27 |
| Muscle |
0.37 ± 0.08 |
0.30 ± 0.02 |
0.30 ± 0.04 |
0.89 ± 0.04 |
0.62 ± 0.02 |
0.45 ± 0.01 |
|
Brain
|
0.16 ± 0.02
|
0.10 ± 0.01
|
0.15 ± 0.03
|
0.99 ± 0.04
|
0.20 ± 0.00
|
0.15 ± 0.02
|
| Bone |
0.54 ± 0.11 |
0.45 ± 0.01 |
0.59 ± 0.07 |
1.59 ± 0.07 |
0.59 ± 0.09 |
0.64 ± 0.07 |
| Tail |
15.91 ± 2.84 |
12.8 ± 5.38 |
3.69 ± 1.92 |
2.53 ± 0.16 |
3.20 ± 0.29 |
1.05 ± 0.05 |
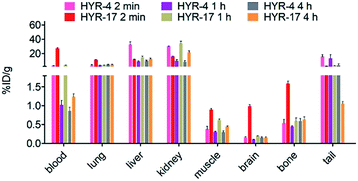 |
| | Fig. 12 Various organs uptake (%ID per g) of 64Cu-labeled HYR-4 and HYR-17 from the biodistribution studies in CD-1 mice, at 2, 60, and 240 min post injection. | |
Conclusions
Herein we report several metal-chelating benzothiazole multifunctional compounds (MFCs) and investigate their various biochemical, in vitro, and in vivo properties to bind to various Aβ species, modulate Aβ aggregation and its neurotoxicity, and potentially act as 64Cu PET imaging agents for in vivo detection of Aβ aggregates. During these studies, we have obtained important structure–activity relationships (SAR) that will guide us to develop improved MFCs as potential therapeutic or diagnostic agents for AD. Firstly, when comparing HYR-4vs.HYR-1 and HYR-16vs.HYR-14, we can conclude that the compounds containing a monomethylamino vs. a dimethylamino group exhibit an increased specificity for the Aβ aggregates as well as reduced cytotoxicity. Secondly, the introduction of a pyridyl group in MFCs such as HYR-14 and HYR-16 dramatically reduces their cytotoxicity and improves their antioxidant properties. Taken together, the MFC HYR-16 containing the 2-monomethylamino-pyridyl fragment is most effective at alleviating the Aβ42 oligomer neurotoxicity and modulating Aβ aggregation. Thirdly, all investigated MFCs can be efficiently radiolabeled with the 64Cu radioisotope, and the 64Cu complexes of HYR-4, HYR-17, and HYR-18 exhibit acceptable lipophilicity and specific radiolabeling of amyloid plaques ex vivo.
Interestingly, the position of the hydroxyl group and the metal-chelating tacn azamacrocycle on the benzothiazole ring has a dramatic effect on the BBB permeability of these 64Cu-labelled MFCs: the 64Cu complex of HYR-17 – which contains the 4-hydroxyl substituent and tacn azamacrocycle connected to the 5 position of the benzothiazole ring, exhibits the highest brain uptake. Finally, our results suggest that employing HYR-18, the bis-(2-phenylbenzothiazole) analogue of HYR-4, does not improve the lipophilicity or specificity for amyloid plaques, while unnecessarily increasing the MW and thus possibly limiting its BBB permeability.
Overall, these detailed studies suggest that HYR-16 is the most effective MFC at alleviating the neurotoxicity of soluble Aβ oligomers, and thus it lends promise to the use of HYR-16 and its second-generation derivatives in future animal studies to evaluate their therapeutic properties. Moreover, HYR-17 exhibits the largest brain uptake and ex vivo specificity for native amyloid plaques, and derivatives of this MFC with similar positioning of the benzothiazole ring substituents will be used as lead compounds for microPET imaging studies in WT vs. AD transgenic mice, toward the development of improved 64Cu PET imaging agents for AD diagnosis.
Ethical statement
All animal studies were performed in strict accordance with the NIH guidelines for the care and use of laboratory animals (NIH Publication No. 85-23 Rev. 1985) and were in compliance with the Guidelines for Care and Use of Research Animals established by the Division of Comparative Medicine and the Animal Studies Committee of Washington University in St. Louis. The animal protocol #20190073 including all the animal studies performed herein was reviewed and approved by the Institutional Animal Care and Use Committee of Washington University in St. Louis.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
L. M. M. acknowledges research funding from NIH (R01GM114588), the Alzheimer’s Association (NIRG 12-259199), and the Washington University Knight Alzheimer’s Disease Research Center (NIH P50AG05681). The authors would like to thank the small animal imaging facility at Washington University School of Medicine for excellent technical assistance, Cedric Mpoy for valuable assistance with animal studies, and the Isotope Production Group at Washington University for on time weekly production of 64Cu.
Notes and references
- K. P. Kepp, Chem. Rev., 2012, 112, 5193–5239 CrossRef CAS PubMed.
-
Alzheimer's Association, Alzheimer's Dementia, 2019, vol. 15, pp. 321–387 Search PubMed.
- P. Sweeney, H. Park, M. Baumann, J. Dunlop, J. Frydman, R. Kopito, A. McCampbell, G. Leblanc, A. Venkateswaran, A. Nurmi and R. Hodgson, Transl. Neurodegener., 2017, 6, 6 CrossRef PubMed.
- M. Citron, Nat. Rev. Drug Discovery, 2010, 9, 387–398 CrossRef CAS PubMed.
- F. M. LaFerla, K. N. Green and S. Oddo, Nat. Rev. Neurosci., 2007, 8, 499–509 CrossRef CAS PubMed.
- E. Karran, M. Mercken and B. D. Strooper, Nat. Rev. Drug Discovery, 2011, 10, 698–712 CrossRef CAS PubMed.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2017, 86, 27–68 CrossRef CAS PubMed.
- M. P. Mattson, Nature, 2004, 430, 631–639 CrossRef CAS PubMed.
- E. McGowan, F. Pickford, J. Kim, L. Onstead, J. Eriksen, C. Yu, L. Skipper, M. P. Murphy, J. Beard, P. Das, K. Jansen, M. DeLucia, W. L. Lin, G. Dolios, R. Wang, C. B. Eckman, D. W. Dickson, M. Hutton, J. Hardy and T. Golde, Neuron, 2005, 47, 191–199 CrossRef CAS PubMed.
- J. Kim, L. Onstead, S. Randle, R. Price, L. Smithson, C. Zwizinski, D. W. Dickson, T. Golde and E. McGowan, J. Neurosci., 2007, 27, 627–633 CrossRef CAS PubMed.
- I. Kuperstein, K. Broersen, I. Benilova, J. Rozenski, W. Jonckheere, M. Debulpaep, A. Vandersteen, I. Segers-Nolten, K. Van Der Werf, V. Subramaniam, D. Braeken, G. Callewaert, C. Bartic, R. D'Hooge, I. C. Martins, F. Rousseau, J. Schymkowitz and B. De Strooper, EMBO J., 2010, 29, 3408–3420 CrossRef CAS PubMed.
- K. Pauwels, T. L. Williams, K. L. Morris, W. Jonckheere, A. Vandersteen, G. Kelly, J. Schymkowitz, F. Rousseau, A. Pastore, L. C. Serpell and K. Broersen, J. Biol. Chem., 2012, 287, 5650–5660 CrossRef CAS PubMed.
- Y. S. Gong, L. Chang, K. L. Viola, P. N. Lacor, M. P. Lambert, C. E. Finch, G. A. Krafft and W. L. Klein, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10417–10422 CrossRef CAS PubMed.
- W. L. Klein, G. A. Krafft and C. E. Finch, Trends Neurosci., 2001, 24, 219–224 CrossRef CAS PubMed.
- M. P. Lambert, A. K. Barlow, B. A. Chromy, C. Edwards, R. Freed, M. Liosatos, T. E. Morgan, I. Rozovsky, B. Trommer, K. L. Viola, P. Wals, C. Zhang, C. E. Finch, G. A. Krafft and W. L. Klein, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6448–6453 CrossRef CAS PubMed.
- A. I. Bush, Curr. Opin. Chem. Biol., 2000, 4, 184–191 CrossRef CAS PubMed.
- I. Benilova, E. Karran and B. De Strooper, Nat. Neurosci., 2012, 15, 349–357 CrossRef CAS PubMed.
- S. J. C. Lee, E. Nam, H. J. Lee, M. G. Savelieff and M. H. Lim, Chem. Soc. Rev., 2017, 46, 310–323 RSC.
- M. D. Kirkitadze, G. Bitan and D. B. Teplow, J. Neurosci. Res., 2002, 69, 567–577 CrossRef CAS PubMed.
- J. W. Um, H. B. Nygaard, J. K. Heiss, M. A. Kostylev, M. Stagi, A. Vortmeyer, T. Wisniewski, E. C. Gunther and S. M. Strittmatter, Nat. Neurosci., 2012, 15, 1227–U1285 CrossRef CAS PubMed.
- T. Umeda, T. Tomiyama, N. Sakama, S. Tanaka, M. P. Lambert, W. L. Klein and H. Mori, J. Neurosci. Res., 2011, 89, 1031–1042 CrossRef CAS PubMed.
- S. Lesne, M. T. Koh, L. Kotilinek, R. Kayed, C. G. Glabe, A. Yang, M. Gallagher and K. H. Ashe, Nature, 2006, 440, 352–357 CrossRef CAS PubMed.
- D. M. Walsh and D. J. Selkoe, J. Neurochem., 2007, 101, 1172–1184 CrossRef CAS PubMed.
- M. A. Lovell, J. D. Robertson, W. J. Teesdale, J. L. Campbell and W. R. Markesbery, J. Neurol. Sci., 1998, 158, 47–52 CrossRef CAS PubMed.
- K. J. Barnham, C. L. Masters and A. I. Bush, Nat. Rev. Drug Discovery, 2004, 3, 205–214 CrossRef CAS PubMed.
- H. Kozlowski, A. Janicka-Klos, J. Brasun, E. Gaggelli, D. Valensin and G. Valensin, Coord. Chem. Rev., 2009, 253, 2665–2685 CrossRef CAS.
- P. Faller, ChemBioChem, 2009, 10, 2837–2845 CrossRef CAS PubMed.
- P. Faller and C. Hureau, Dalton Trans., 2009, 1080–1094 RSC.
- P. Faller, C. Hureau and G. La Penna, Acc. Chem. Res., 2014, 47, 2252–2259 CrossRef CAS PubMed.
- L. E. Scott and C. Orvig, Chem. Rev., 2009, 109, 4885–4910 CrossRef CAS PubMed.
- C. Hureau and P. Faller, Biochimie, 2009, 91, 1212–1217 CrossRef CAS PubMed.
- C. Cheignon, M. Tomas, D. Bonnefont-Rousselot, P. Faller, C. Hureau and F. Collin, Redox Biol., 2018, 14, 450–464 CrossRef CAS PubMed.
- A. K. Sharma, S. T. Pavlova, J. Kim, D. Finkelstein, N. J. Hawco, N. P. Rath, J. Kim and L. M. Mirica, J. Am. Chem. Soc., 2012, 134, 6625–6636 CrossRef CAS PubMed.
- A. K. Sharma, S. T. Pavlova, J. Kim, J. Kim and L. M. Mirica, Metallomics, 2013, 5, 1529–1536 RSC.
- M. G. Savelieff, A. S. DeToma, J. S. Derrick and M. H. Lim, Acc. Chem. Res., 2014, 47, 2475–2482 CrossRef CAS PubMed.
- L. M. F. Gomes, A. Mahammed, K. E. Prosser, J. R. Smith, M. A. Silverman, C. J. Walsby, Z. Gross and T. Storr, Chem. Sci., 2019, 10, 1634–1643 RSC.
- S. Ayala, P. Genevaux, C. Hureau and P. Faller, ACS Chem. Neurosci., 2019, 10, 3366–3374 CrossRef CAS PubMed.
- M. G. Savelieff, G. Nam, J. Kang, H. J. Lee, M. Lee and M. H. Lim, Chem. Rev., 2019, 119, 1221–1322 CrossRef CAS PubMed.
- W. E. Klunk, H. Engler, A. Nordberg, Y. M. Wang, G. Blomqvist, D. P. Holt, M. Bergstrom, I. Savitcheva, G. F. Huang, S. Estrada, B. Ausen, M. L. Debnath, J. Barletta, J. C. Price, J. Sandell, B. J. Lopresti, A. Wall, P. Koivisto, G. Antoni, C. A. Mathis and B. Langstrom, Ann. Neurol., 2004, 55, 306–319 CrossRef CAS PubMed.
- A. Nordberg, Curr. Opin. Neurol., 2007, 20, 398–402 CrossRef CAS PubMed.
- H. A. Archer, P. Edison, D. J. Brooks, J. Barnes, C. Frost, T. Yeatman, N. C. Fox and M. N. Rossor, Ann. Neurol., 2006, 60, 145–147 CrossRef PubMed.
- S. R. Choi, G. Golding, Z. Zhuang, W. Zhang, N. Lim, F. Hefti, T. E. Benedum, M. R. Kilbourn, D. Skovronsky and H. F. Kung, J. Nucl. Med., 2009, 50, 1887–1894 CrossRef CAS PubMed.
- H. F. Kung, S. R. Choi, W. Qu, W. Zhang and D. Skovronsky, J. Med. Chem., 2009, 53, 933–941 CrossRef PubMed.
- M. A. Mintun, G. N. LaRossa, Y. I. Sheline, C. S. Dence, S. Y. Lee, R. H. Mach, W. E. Klunk, C. A. Mathis, S. T. DeKosky and J. C. Morris, Neurology, 2006, 67, 446–452 CrossRef CAS PubMed.
- K. Serdons, C. Terwinghe, P. Vermaelen, K. Van Laere, H. Kung, L. Mortelmans, G. Bormans and A. Verbruggen, J. Med. Chem., 2009, 52, 1428–1437 CrossRef CAS PubMed.
- J. L. Hickey, S. Lim, D. J. Hayne, B. M. Paterson, J. M. White, V. L. Villemagne, P. Roselt, D. Binns, C. Cullinane, C. M. Jeffery, R. I. Price, K. J. Barnham and P. S. Donnelly, J. Am. Chem. Soc., 2013, 135, 16120–16132 CrossRef CAS PubMed.
- M. Roger, L. M. P. Lima, M. Frindel, C. Platas-Iglesias, J.-F. Gestin, R. Delgado, V. Patinec and R. Tripier, Inorg. Chem., 2013, 52, 5246–5259 CrossRef CAS PubMed.
- A. K. Sharma, J. Kim, J. T. Prior, N. J. Hawco, N. P. Rath, J. Kim and L. M. Mirica, Inorg. Chem., 2014, 53, 11367–11376 CrossRef CAS PubMed.
- N. Bandara, A. K. Sharma, S. Krieger, J. W. Schultz, B. H. Han, B. E. Rogers and L. M. Mirica, J. Am. Chem. Soc., 2017, 139, 12550–12558 CrossRef CAS PubMed.
- A. K. Sharma, J. W. Schultz, J. T. Prior, N. P. Rath and L. M. Mirica, Inorg. Chem., 2017, 56, 13801–13814 CrossRef CAS PubMed.
- S. R. Choi, J. A. Schneider, D. A. Bennett, T. G. Beach, B. J. Bedell, S. P. Zehntner, M. J. Krautkramer, H. F. Kung, D. M. Skovronsky, F. Hefti and C. M. Clark, Alzheimer Dis. Assoc. Disord., 2012, 26, 8–16 CrossRef CAS PubMed.
- E. Arturoni, C. Bazzicalupi, A. Bencini, C. Caltagirone, A. Danesi, A. Garau, C. Giorgi, V. Lippolis and B. Valtancoli, Inorg. Chem., 2008, 47, 6551–6563 CrossRef CAS PubMed.
- M. Roger, L. M. Lima, M. Frindel, C. Platas-Iglesias, J.-F. o. Gestin, R. Delgado, V. r. Patinec and R. l. Tripier, Inorg. Chem., 2013, 52, 5246–5259 CrossRef CAS PubMed.
- E. M. Snyder, D. Asik, S. M. Abozeid, A. Burgio, G. Bateman, S. G. Turowski, J. A. Spernyak and J. R. Morrow, Angew. Chem., Int. Ed., 2020, 132, 2435–2440 CrossRef.
- K. Serdons, T. Verduyckt, D. Vanderghinste, P. Borghgraef, J. Cleynhens, F. Van Leuven, H. Kung, G. Bormans and A. Verbruggen, Eur. J. Med. Chem., 2009, 44, 1415–1426 CrossRef CAS PubMed.
- W. E. Klunk, Y. Wang, G.-f. Huang, M. L. Debnath, D. P. Holt and C. A. Mathis, Life Sci., 2001, 69, 1471–1484 CrossRef CAS PubMed.
- A. Lockhart, L. Ye, D. B. Judd, A. T. Merritt, P. N. Lowe, J. L. Morgenstern, G. Z. Hong, A. D. Gee and J. Brown, J. Biol. Chem., 2005, 280, 7677–7684 CrossRef CAS PubMed.
- H. Oakley, S. L. Cole, S. Logan, E. Maus, P. Shao, J. Craft, A. Guillozet-Bongaarts, M. Ohno, J. Disterhoft, L. Van Eldik, R. Berry and R. Vassar, J. Neurosci., 2006, 26, 10129–10140 CrossRef CAS PubMed.
- Y. Zhang, D. L. Rempel, J. Zhang, A. K. Sharma, L. M. Mirica and M. L. Gross, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 14604–14609 CrossRef CAS PubMed.
- M. A. Smith, P. L. Harris, L. M. Sayre and G. Perry, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 9866–9868 CrossRef CAS PubMed.
- R. Re, N. Pellegrini, A. Proteggente, A. Pannala, M. Yang and C. Rice-Evans, Free Radical Biol. Med., 1999, 26, 1231–1237 CrossRef CAS PubMed.
- Y. Manevich, K. D. Held and J. E. Biaglow, Radiat. Res., 1997, 148, 580–591 CrossRef CAS PubMed.
- D. D. Dischino, M. J. Welch, M. R. Kilbourn and M. E. Raichle, J. Nucl. Med., 1983, 24, 1030–1038 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section and Fig. S1–S8. See DOI: 10.1039/d0sc02641g |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Hong-Jun
Cho
a,
Nilantha
Bandara
a,
Hong-Jun
Cho
a,
Nilantha
Bandara