
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLanthanide MOFs for inducing molecular chirality of achiral stilbazolium with strong circularly polarized luminescence and efficient energy transfer for color tuning†
Min
Zeng‡
ac,
Ang
Ren‡
ac,
Wubin
Wu
ac,
Yongsheng
Zhao
 *ac,
Chuanlang
Zhan
*abc and
Jiannian
Yao
*ac
*ac,
Chuanlang
Zhan
*abc and
Jiannian
Yao
*ac
aBeijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Photochemistry, Institute of Chemistry, Chinese Academy of Sciences, Beijing, 100190, China. E-mail: clzhan@iccas.ac.cn; yszhao@iccas.ac.cn; jnyao@iccas.ac.cn
bKey Laboratory of Excitonic Materials Chemistry and Devices (EMC&D), College of Chemistry and Environmental Science, Inner Mongolia Normal University, Huhehot 010022, China
cUniversity of Chinese Academy of Sciences, Beijing, 100049, China
First published on 4th August 2020
Abstract
We present herein an innovative host–guest method to achieve induced molecular chirality from an achiral stilbazolium dye (DSM). The host–guest system is exquisitely designed by encapsulating the dye molecule in the molecule-sized chiral channel of homochiral lanthanide metal–organic frameworks (P-(+)/M-(−)-TbBTC), in which the P- or M-configuration of the dye is unidirectionally generated via a spatial confinement effect of the MOF and solidified by the dangling water molecules in the channel. Induced chirality of DSM is characterized by solid-state circularly polarized luminescence (CPL) and micro-area polarized emission of DSM@TbTBC, both excited with 514 nm light. A luminescence dissymmetry factor of 10−3 is obtained and the photoluminescence quantum yield (PLQY) of the encapsulated DSM in DSM@TbTBC is ∼10%, which is close to the PLQY value of DSM in dilute dichloromethane. Color-tuning from green to red is achieved, owing to efficient energy transfer (up to 56%) from Ln3+ to the dye. Therefore, this study for the first time exhibits an elegant host–guest system that shows induced strong CPL emission and enables efficient energy transfer from the host chiral Ln-MOF to the achiral guest DSM with the emission color tuned from green to red.
Introduction
Circularly polarized luminescence (CPL) has attracted more and more interest in recent years owing to potential applications such as in photoelectric devices,1 nonlinear optics,2,3 CPL sensors,4 biological probes,5 and organic light-emitting diodes (OLEDs).6,7 CPL is the differential emission intensity of chiral luminescent species upon excitation by the right- or left circularly polarized light. It is deeply associated with the structures of excited states of chiral luminescent species.8 For applications using CPL as probing light sources, CPL emissions with high quantum yields are prerequisite. It thus requires not only high luminescence quantum efficiencies (ϕlum) but also large luminescence dissymmetry factors (glum). glum is defined by the coupling of transition moments (μ, m, and Θ) summed over all possible excited states,9 where μ, m, and Θ are the electric transition dipole, magnetic transition dipole, and electric quadrupole operators, respectively.10 Generally, large glum and high ϕlum cannot be simultaneously obtained from a single chiral luminescent species because there are few substances found to have large |μ| and large |m| at the same time. For example, chiral organic molecules, polymers, and transition-metal complexes have high ϕlum values, while generally having small glum values (10−5 to 10−3) due to the negligible |m|.11–14 In contrast, chiral lanthanide complexes have high values of glum (10−2 to 0.5)15 due to the existence of a magnetic transition dipole, while they commonly suffer from low ϕlum because of the forbidden electric transition dipole.16 Self-assembly of chiral or achiral luminescent organic molecules into chiral aggregates can lead to amplification of the glum values to some degree, normally in the order of 10−5 to 10−3.17–19 A glum value of 10−1 was observed from self-assembled nematic liquid crystalline polymers20 and emissive charge-transfer complexes.21 Unfortunately, the CPL seriously suffers from the aggregation-caused quenching (ACQ) effect.22–24 Fluorophores exhibiting aggregation-induced emission (AIE) could result in enhanced luminescence performance in the condensed phase.25–27 Nevertheless, AIE emitters normally require complicated organic synthesis. Again, they also suffer from lower glum.Metal–organic frameworks (MOFs) which have a highly ordered crystalline porous structure are self-assembled by metal ions and organic ligands.28,29 The size, shape (including the chiral one), and surface composition of the pores can be easily adjusted by changing the size and structure of organic ligands and altering the type of metal ions.30–32 Hence, MOFs with a pore confinement effect (with the pore size matching well with the size of organic fluorophores) are the most suitable candidates for minimizing the ACQ of fluorophores.33–36 Chiral MOFs have been recently emerging as potential hosts for chiral synthesis,37 separation38,39 and transcription.40 A recent case based on MOFs showed that the glum value was increased from 10−4 to 10−3 and simultaneously the ϕlum enhanced from 0.3 to 0.4 by reorganizing a chiral luminescent binaphthyldiamine-imidazole based emitter on the skeleton of a zeolitic imidazolate framework,41 indicating that MOFs can be candidates for CPL materials.
Here, we present an innovative supramolecular approach to report an exquisitely designed CPL host–guest system with the encapsulated achiral organic molecule showing enhanced CPL emission having a glum value of 10−3 and a ϕlum value of ∼10%. This host–guest system is achieved by encapsulating (4-p-(dimethylamino)styryl)-1-methylpyridinium iodide (DSM) into the chiral pores of homochiral lanthanide MOFs, P-(+)/M-(−)-TbBTC, having a glum value of 10−3 (Fig. 1). DSM is selected as the guest because of its high luminescence efficiency in its molecular state (ϕlum, ∼10%), and most importantly, it is a linear achiral organic dye (Fig. 1a) that is chosen herein as a model achiral molecule to create induced molecular chirality by following the present concept (Fig. 1). Colour-tuning from green to red is achieved by changing the loading contents of DSM in the MOFs from 0.09% to 1%, and is due to the efficient energy transfer efficiency from Ln3+ to the dye.
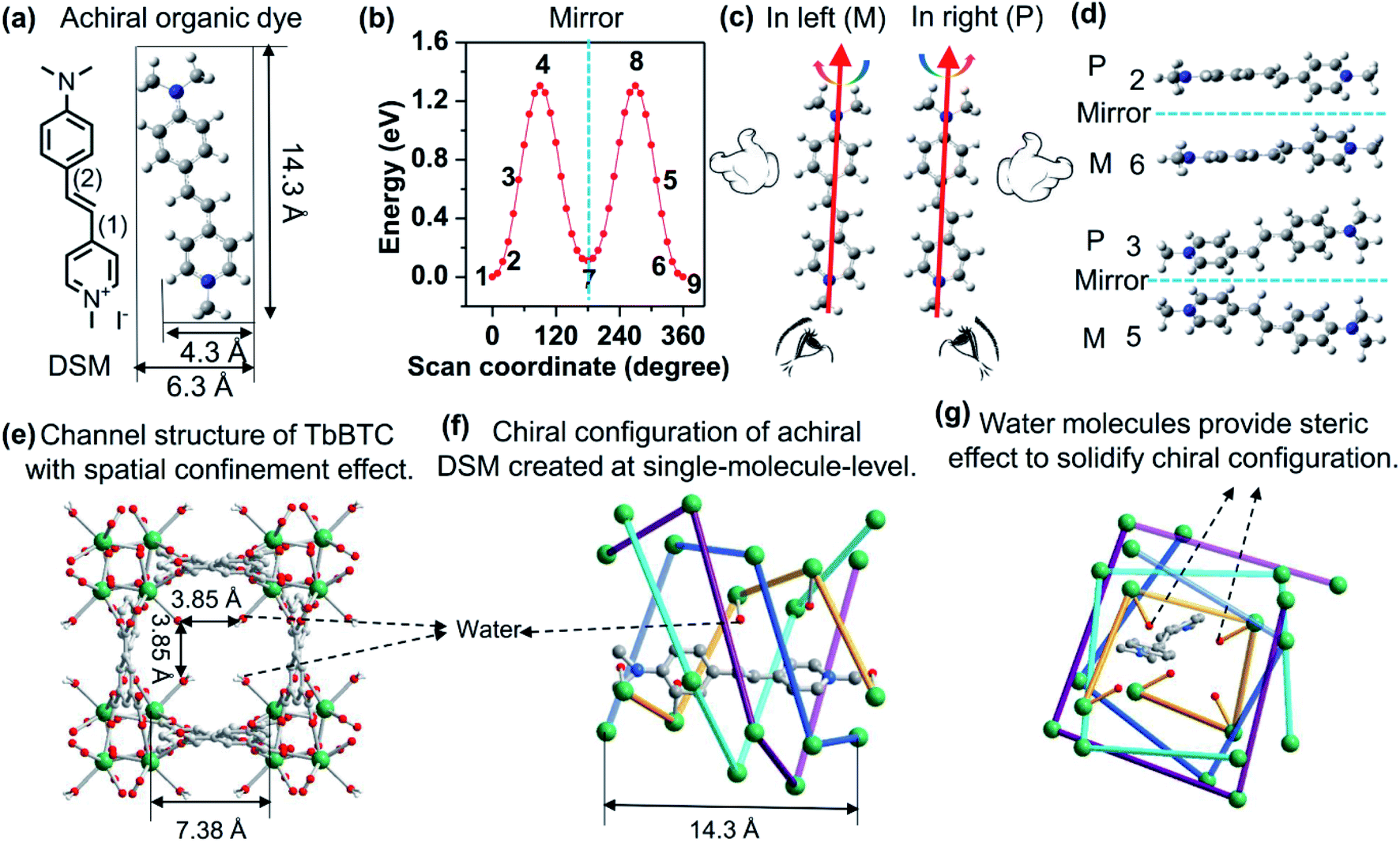 | ||
| Fig. 1 (a) Molecular structure and optimal conformation of DSM. (b) Potential energy surface scan of DSM around single bonds (1) and (2) simultaneously. (c) Diagram of rotation of a DSM molecule in the left or in right. (d) Examples of four enantiomorphic configurations of achiral DSM created by rotation around single bonds (1) and (2) simultaneously: the configurations labelled with 1, 4, 7, 8, and 9 in (b) are achiral. The chirality of ‘P’ or ‘M’ is defined as twisting of the phenyl plane relative to the pyridinium plane right (<90°) or left (<90°) along the dipole vector of the DSM conjugation, shown in (c). (e) View of dimensions of the 1D channel of right-hand P-(+)-TbBTC-a along the c axis. (f and g) Schematic views of molecule-state encapsulation of DSM in the chiral channel of right-hand P-(+)-TbBTC along the b and c axis, respectively. The dangling water molecules suspended in the chiral channel are coordinated with the Tb3+ ions and aligned into the single helix labelled by a yellowish orange color. | ||
Results and discussion
Density functional theory (DFT, Gaussian 09 software, B3LYP) calculations demonstrate the energy requirements of rotation of a DSM molecule along carbon–carbon bonds (1) and (2) simultaneously (Fig. 1b), which suggests that about 0.025 eV is required for a DSM molecule to twist 10° (between the phenyl and pyridinium planes). The energy at room temperature (RT, 298 K) is 0.025 eV calculated via kRT. This indicates that DSM molecules can be slightly twisted to a degree such as below 10° at RT. Normally, DSM molecules randomly rotate both in the left and right (Fig. 1c). Speculatively, if we can unidirect the rotation either left or right and then fix the left or right twist, we can create enantiomorphic configurations that are mirror images of each other, for examples, those shown in Fig. 1b and d. The width of a DSM molecule is 4.3 Å. To create a chiral channel with the channel size matched with the width of DSM, we select Tb3+ and 1,3,5-benzenetricarboxylic acid (BTC) and synthesize TbBTC. The exquisitely detailed one-dimensional (1D) chiral channel of TbBTC (Fig. 1e–g) not only encapsulates DSM at the molecular state but also induces unidirectional rotation (P- or M-) on a DSM molecule. Again, the dangling water molecules coordinated with Tb3+, which are positioned into the channel, provide spatial steric effects to solidify the geometrical configuration made after unidirectional rotation. As a result, enantiomorphic configurations made from achiral DSM molecules are created and molecule-state chirality is observed.TbBTC was obtained through self-assembly of Tb3+ and fully deprotonated achiral ligand BTC. It crystallized as a mixture of tetragonal chiral space groups P4122 [TbBTC-a, CCDC no. 1960864; TbBTC-d, CCDC no. 1966281] and P4322 [TbBTC-b, CCDC no. 1960863; TbBTC-c, CCDC no. 1966280] (Table 1). Their crystallographic data are displayed in Tables S1–S8.† The racemic mixture of TbBTC was separated into right-hand P-(+)-TbBTC (-a, and -b) and left-hand M-(−)-TbBTC (-c and -d) with (S)-(+)-2-amino-1-butanol and (R)-(−)-2-amino-1-butanol as chiral dopants (Fig. S1†). P-(+)-TbBTC-a and M-(−)-TbBTC-c constitute an enantiomorphic space-group pair; their fundamental building unit consists of one Tb(III) ion, one deprotonated BTC3− and one water. P-(+)-TbBTC-b and M-(−)-TbBTC-d constitute another enantiomorphic space-group pair; their asymmetric unit consists of three Tb(III) ions, three deprotonated BTC3− and three water molecules. H3BTC in these four MOFs has the same coordination mode: it is fully deprotonated and bidentate coordinated with six Tb3+ ions. All four TbBTCs feature 3D porous structures with 1D chiral channels of 3.85 × 3.85 Å2 along the c-axis (Fig. 1e, f, S1 and S2†). Simulated powder X-ray diffraction (PXRD) patterns of these four crystals are nearly identical to each other (Fig. S3†), suggesting that they have similar spatial structures. The alignment of Tb3+ ions leads to chiral assemblies with right-hand helices in TbBTC-a and TbBTC-b; in contrast, in TbBTC-c and TbBTC-d, the alignment of Tb3+ ions results in left-hand helices (Fig. 1e, f, S1 and S4†). The chiral channel is assembled by four single helices of aligned Tb3+; each single-helix repeat unit contains four Tb3+ ions, featuring as an i + 4 helix. The length of per repeat unit of helix, between i and i + 4 Tb3+ is ∼14.3 Å, nearly equaling to the length of DSM. The water molecules oriented into the chiral channel are all coordinated with Tb3+ ions that are aligned into one single-helix (Fig. 1f and g).
Here, DSM is introduced into the chiral channel of TbBTC by post-synthesis. The PXRD pattern of DSM@TbBTC is nearly identical to that of TbBTC (Fig. S5†), indicating that the loading of DSM does not destroy the crystal structure of TbBTC. Elemental analysis of C, H, and N indicates that the ratio of TbBTC to DSM is 15.7, which corresponds to ∼0.5 DSM molecule per MOF unit cell (containing one pore) for TbBTC-a and -c, and ∼1.5 DSM molecules per MOF unit cell (containing three pores) for TbBTC-b and -d. Fig. 1f and g show a representative structure of encapsulation of one DSM molecule in one chiral channel of TbBTC-a.
As shown in Fig. 2a, the absorption and emission peaks of DSM in DSM@TbBTC are located at 507 and 615 nm, respectively. In solutions, the absorption and emission peaks of DSM are changed with solvent polarity (Fig. 2b). As the solvents change from CHCl3 to DMF, the emission peak demonstrates a large redshift of 40 nm: 592 nm for CHCl3, 615 nm for CH2Cl2, 628 nm for acetone, and 632 nm for DMF. In contrast, the absorption peak illustrates a distinct blueshift: 505 nm for CHCl3, 507 nm for CH2Cl2, 472 nm for acetone, and 468 nm for DMF. DSM shows identical peak positions (absorption and fluorescence) in DSM@TbBTC and dilute CH2Cl2, indicating that the polarity of the chiral pore of neutral TbBTC is similar to that of CH2Cl2.
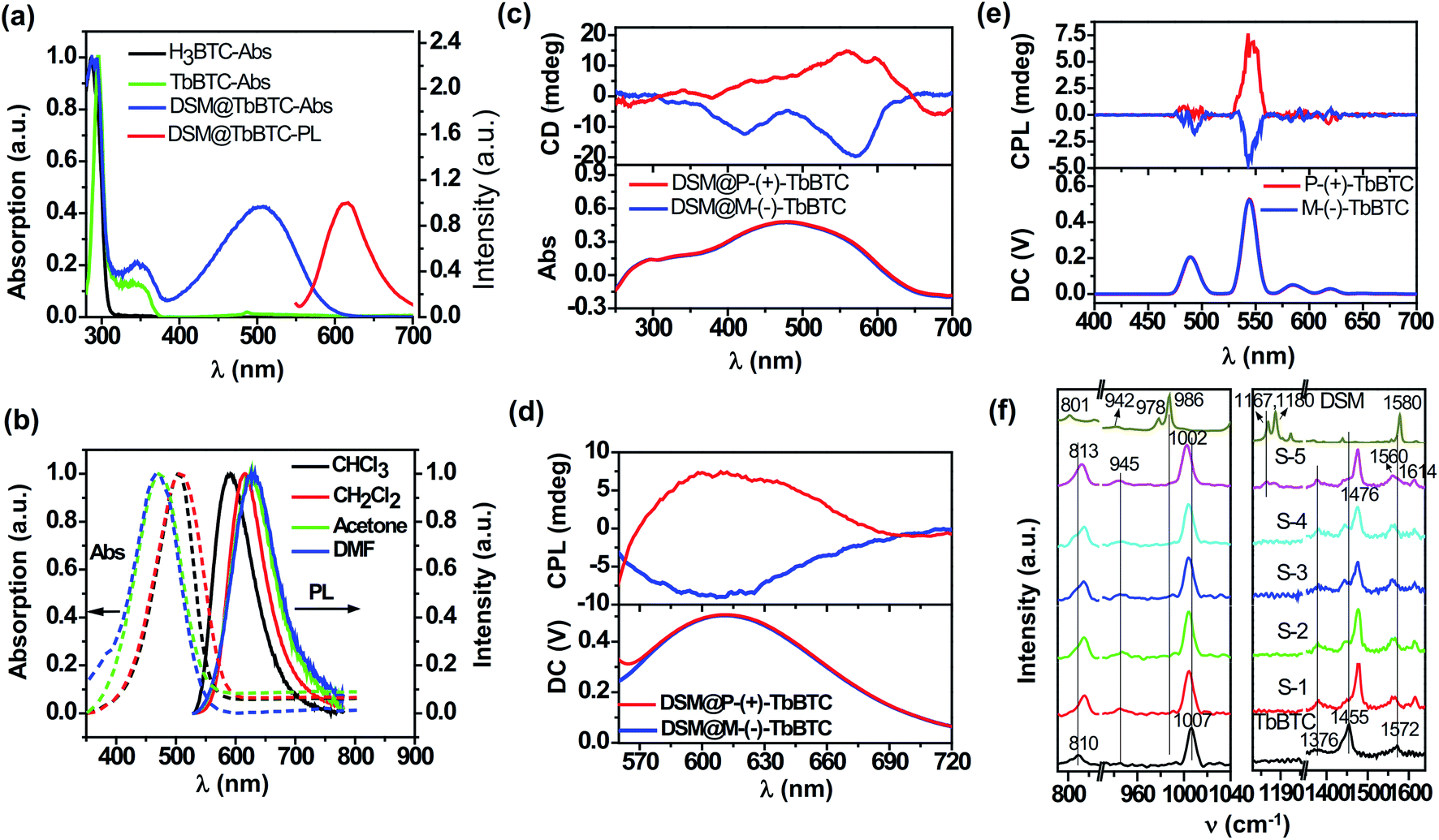 | ||
| Fig. 2 (a) Solid-state adsorption spectra of H3BTC, TbBTC, and DSM@TbBTC, and the solid-state PL spectra of DSM in DSM@TbBTC under 514 nm excitation. (b) Normalized absorption (Abs) and PL spectra of DSM in different solvents. (c and d) Solid-state CD (c) and CPL (d) spectra of DSM@TbBTC, respectively. CD spectra were measured via the refractive mode and CPL spectra were measured under 514 nm excitation. (e) CPL spectra of TbBTC under 290 nm excitation. Both CD and CPL spectra were measured with the powders of DSM@TbBTC and TbBTC, respectively. (f) Powder Raman spectra of TbBTC, DSM and five samples of DSM@TbBTC (S-1 to S-5 with different loading contents of DSM, 0.09%, 0.16%, 0.21%, 0.6% and 1%, respectively). Raman spectra were obtained under 785 nm laser excitation. | ||
The photoluminescence quantum yield (PLQY) of DSM is 10.6% in dilute CH2Cl2 solution and 1.12% measured from DSM powder, both excited at 514 nm. Under 514 nm excitation, the PLQY of DSM in DSM@TbBTC is 9.6% which is close to the PLQY in dilute CH2Cl2 solution, evidence indicating single-molecule encapsulation of DSM in TbBTC.
DSM is an achiral molecule. No chiral optical properties can be seen in DSM solutions and powder. MOF-5 features 1D achiral channels with sizes of 10.7 × 10.7 Å2.42 The length of DSM is 14.3 Å and its size is 4.3 Å. When DSM is encapsulated in the achiral channel of MOF-5, it would rotate randomly left or right. No solid-state CPL signals were detected (Fig. S6†). In contrast, TbBTC features 1D chiral channels. The size is 7.38 × 7.38 Å2, which is just larger than the size of DSM. Again, the dangling water molecules in the chiral channel further reduce the pore size to 3.85 × 3.85 Å2, which will avoid ππ-stacking of DSM molecules inside the chiral channel.
When encapsulating DSM in P-(+)-TbBTC, a broad positive circular dichroism (CD) (Fig. 2c) and CPL signal (peaking around 615 nm, Fig. 2d) are detected. Oppositely, on encapsulation of DSM in M-(−)-TbBTC, a negative CD and CPL signal (around 615 nm) are observed. For P-(+)-TbBTC and M-(−)-TbBTC, positive and negative CPL signals are seen at 544 nm, upon 290 nm excitation (Fig. 2e). The glum was calculated from equation glum = 2(IL − IR)/(IL + IR),43IL and IR are the luminescence intensities of the left- and right-handed CPL, respectively. Here, the glum factor is 2.1 × 10−3 for P-(+)-TbBTC and −1.3 × 10−3 for M-(−)-TbBTC. For DSM@P-(+)-TbBTC and DSM@M-(−)-TbBTC, glum values are 2.1 × 10−3 and −2.5 × 10−3, respectively. First, the glum values of DSM CPL in DSM@TbBTC are close to the values of TbBTC, the Ln-MOF. Second, the glum values of DSM CPL in DSM@TbBTC are comparable to the values for pure organic assembled materials.25,44,45
The micro-area CPL of DSM@P-(+)-TbBTC was measured by the combination of a λ/4 plate and a linear polarizer with an experimental setup shown in Fig. 3a. After being filtered by a band-pass filter (514 ± 12.5 nm), a continuous 514 nm laser excited the single crystal microrod and the emitted red light was seen at the same position on the crystal microrod (Fig. S7†). The red light passed through the 550 nm long-pass filter and λ/4 waveplate. Subsequently, the PL intensities can be recorded by rotating the linear polarizer by 30°. As shown in Fig. 3b and c, by using a λ/4 waveplate, CPL can change into linearly polarized light, while no change occurred in the unpolarized light. Therefore, it can be predicted that the DSM@P-(+)-TbBTC will show circular polarization features depending on the changed PL intensities by rotating the linear polarizer from 0° to 360°.46 Upon 514 nm excitation, the emission intensities of both the ±45° linear polarized light exhibit a maximum value at 615 nm (Fig. 3d and e). It was observed that, as the polarization angle θ changes from 0° to 360°, the PL intensities of the ±45° linear polarized light change in the opposite direction. e.g. when the polarization angle θ changes from 0° to 90°, the PL intensities of the −45° linear polarized light decreased and +45° linear polarized light increased gradually. The maximum emission intensity values for the −45° linear polarized light occurred at about 180° and 360° and the maximum emission intensity for the +45° linear polarized light occurred at about 90° and 270°. From the same one single-crystal microrod of DSM@P-(+)-TbBTC, the polarization directions of the −45° linear polarized light and +45° linear polarized light are nearly perpendicular (Fig. 3f and g), which again indicates that achiral DSM shows CPL activity when encapsulated into the chiral pores of TbBTC.47
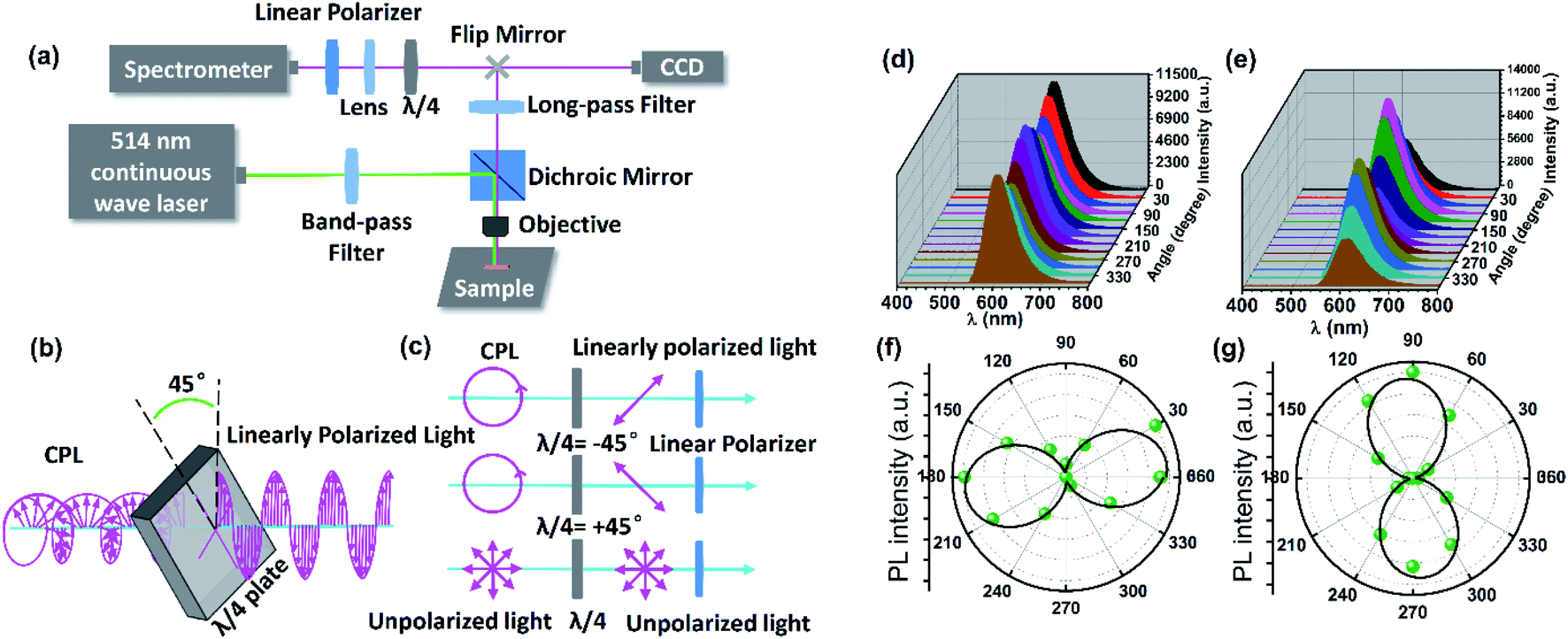 | ||
| Fig. 3 (a) Schematic illustration of the experimental setup for measurements of micro-area CPL of single-crystal microrods of DSM@P-(+)-TbBTC. (b) Illustrative representation of the conversion between CPL and linearly polarized light through a λ/4 waveplate. (c) Evaluation of the circular polarization light. (d and e) Polarized emission spectra from one single-crystal microrod of DSM@P-(+)-TbBTC under 514 nm excitation at changed angles (0–360°), angles of the λ/4 waveplate = −45° (d) and +45° (e). (f and g) Maximum emission intensity of the same DSM@P-(+)-TbBTC microrod at different polarizer rotation angles, angles of the λ/4 waveplate = −45° (f) and +45° (g). | ||
In order to obtain information about the configuration of the DSM molecule inside the chiral pore of Tb-BTC, we tried to grow single crystals of DSM@TbBTC. Luckily; we obtained a single crystal of the DSM@P-(+)-TbBTC structure with encapsulation of DSM. Unfortunately, the structure of the encapsulated DSM cannot be obtained, due to the severe disorder of the DSM structure. Nevertheless, the structure of P-(+)-TbBTC is obtained by following PLATON/SQUEEZE to remove the DSM molecule from the structure. The data are shown in Tables S9 and S10.† The structure of P-(+)-TbBTC of DSM@TbBTC is nearly identical to the structure of P-(+)-TbBTC (-a) before encapsulating DSM. Specifically, the dangling water molecules are clearly seen on P-(+)-TbBTC of DSM@TbBTC, indicating that the coordinated water molecules remain right at their positions after encapsulation of DSM.
We also obtained a single crystal structure of DSM (Tables S11 and S12†), which is identical to the one previously reported (CCDC no. 1904261).48 The crystal structure shows that there is an ∼10° twist between the phenyl plane and the pyridinium plane (Fig. 4a). There are four DSM molecules in a cell unit. Two are paired in largely displaced edge-to-π antiparallel mode. Along the dipole vector, going from pyridinium to phenyl of the conjugation system, two of them are twisted in the right and another two in the left (Fig. 4b). As shown in Fig. 4c, one DSM molecule (twisted in the right, P) is surrounded by eight antiparallel DSM molecules, four of them (twisted in the right, P) at the phenyl end and another four (twisted in the left, M) at the pyridinium end, which is most likely the reason for induction of the chiral configuration of DSM. Again, the ‘four-P’ and ‘four-M’ molecules are differently oriented, relative to the phenyl and pyridinium planes, which also induce different intermolecular interactions with the phenyl or pyridinium unit; hence, this may be another reason for the induction of the chiral configuration of DSM. That is to say that sequential packing of the alternative ‘four-P’ and ‘four-M’ forms a channel encapsulating the DSM molecule (Fig. 4d). Although the channel is formed alternately by four right-twisted and four left-twisted DSM molecules, which is different from the configuration of the chiral channel of TbBTC, the configuration of the DSM molecule inside the channel formed by DSM molecules can be helpful for us to speculate about the DSM conformation inside the chiral channel of TbBTC. Both the left- and right-twisted configurations are seen in the same crystal of DSM; no chiral optical signals can be detected from the DSM single crystal.
 | ||
| Fig. 4 (a) The P and M configurations of the DSM molecule derived from a single crystal of DSM, which shows that there is an ∼10° twist between the phenyl plane and the pyridinium plane. The configurations of P and M are indicated along the dipole vector of the π conjugation. (b) A unit cell of the DSM single crystal. (c and d) Packing of nine DSM molecules showing one DSM is surrounded by four antiparallel right-twisted (P) DSM molecules at its phenyl end and another four antiparallel left-twisted (M) DSM molecules at the pyridinium end; alternate packing of the four-P and four-M DSM molecules forms a channel-like environment encapsulating one DSM molecule inside. | ||
The Raman spectra of TbBTC, DSM and DSM@TbBTC powders were obtained (Fig. 2f) to give spectral evidence for the conformation changes of organic ligands in TbBTC after encapsulation of DSM. For TbBTC, the bands located at ∼810 cm−1 and ∼1007 cm−1 can be ascribed to the out-of-plane CH bending vibration and the ring-breathing mode of benzene, respectively. The bands located at ∼1376 cm−1 and ∼1455 cm−1 can be ascribed to the symmetric C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations of the carboxylic group, and the asymmetric CO stretching vibration appears at ∼1572 cm−1. The separation (Δ) between the two peaks of νasym(COO) and νsym(COO) is in accordance with the coordination mode of the carboxylic group.49 After encapsulation of DSM forming DSM@TbBTC (S-5), the ∼810 cm−1 signal is shifted to ∼813 cm−1 and the ∼1007 cm−1 to ∼1002 cm−1. Meanwhile, the ∼1455 cm−1 symmetric and ∼1572 cm−1 asymmetric stretching vibrations of the carboxylic group are both separated: the former one into ∼1455 cm−1 and ∼1476 cm−1; the latter one into ∼1560 cm−1 and ∼1614 cm−1, respectively. Shifting of ligand benzene signals and separation of ligand CO signals are most likely due to the conformation variations of the ligands after encapsulation of DSM.
O stretching vibrations of the carboxylic group, and the asymmetric CO stretching vibration appears at ∼1572 cm−1. The separation (Δ) between the two peaks of νasym(COO) and νsym(COO) is in accordance with the coordination mode of the carboxylic group.49 After encapsulation of DSM forming DSM@TbBTC (S-5), the ∼810 cm−1 signal is shifted to ∼813 cm−1 and the ∼1007 cm−1 to ∼1002 cm−1. Meanwhile, the ∼1455 cm−1 symmetric and ∼1572 cm−1 asymmetric stretching vibrations of the carboxylic group are both separated: the former one into ∼1455 cm−1 and ∼1476 cm−1; the latter one into ∼1560 cm−1 and ∼1614 cm−1, respectively. Shifting of ligand benzene signals and separation of ligand CO signals are most likely due to the conformation variations of the ligands after encapsulation of DSM.
For DSM in deuterated chloromethane (Fig. S8†), the band located at ∼1577 cm−1 is attributed to the CC stretching vibration. The bands located at ∼1167 cm−1 and ∼1180 cm−1 are attributed to deformation vibration of the –CH3 of the N-dimethylamino group.50 The band at ∼984 cm−1 can be assigned to a mode consisting of the in-phase ethylenic CH wag.51 The out-of-plane CH bending vibration is seen at ∼810 cm−1 as weak signals. For DSM powder, the CC stretching vibration is slightly shifted to ∼1580 cm−1; the in-phase ethylenic CH wag is seen as two bands at ∼978 cm−1 and ∼986 cm−1, respectively, and the out-of-plane CH bending vibration is seen at ∼801 cm−1 as a single signal. The shifting, especially, the occurrence of the 978 cm−1 signal demonstrates the twisting of the central CC bond in relation to the covalent benzene and pyridinium units when DSM molecules are shifted from solution to the solid powder. This is well consistent with the observation of the twisted conformation of DSM between the benzene and the pyridinium units, torsion around the central CC bond, in the single crystal structure of DSM (Fig. 4). When DSM is encapsulated into the chiral pore of TbBTC, the signals of the deformation vibration of the –CH3 of the N,N-dimethylamino group are detected, albeit weakly, this indicates encapsulation of DSM. The weak Raman signals of DSM detected from DSM@TbBTC are consistent with the low content of DSM relative to TbBTC (about 1%). After encapsulation of DSM, the signals of the deformation vibration of the –CH3 of the N,N-dimethylamino group are slightly shifted to ∼1164 cm−1 and ∼1177 cm−1 and the relative intensity between those two bands is reversed. The spectral changes suggest a slight difference in conformational torsion of the N,N-dimethylamino group in DSM powders and in the chiral pore of TbBTC.
TbBTC emits green light, while DSM emits red light, and both of them show high PLQY. Again, the emission spectrum of Tb3+ is overlapped with the absorption spectrum of DSM (Fig. S9†). We therefore conducted the color tuning through the energy transfer from Tb3+ to the encapsulated DSM by loading different amounts of DSM inside the chiral pores of TbBTC. Energy levels between energy donor Tb3+ and energy acceptor DSM are matched (Fig. 5a). The HOMO–LUMO gap on a 10° twisted DSM is 2.48 eV, calculated with DFT. The excited state level (5D4) of Tb3+ is 2.55 eV, which is higher than the S1 state of DSM. The fluorescence of DSM in DSM@TbBTC is enhanced significantly in comparison to that in DSM–TbBTC [a thoroughly ground mixture of DSM and TbBTC], and meanwhile, the emission of TbBTC in DSM@TbBTC at 487 and 544 nm is significantly reduced (Fig. S10†), featuring efficient energy transfer from Tb3+ to DSM. By varying dye concentrations encapsulated in TbBTC, five samples of DSM@TbBTC-X (X = 1, 2, 3, 4 and 5) were obtained. The loading content (wt/wt) of DSM was determined by elemental analysis to be 0.09%, 0.16%, 0.21%, 0.6% and 1%, respectively (Table S13†). PLQYs of DSM@TbBTC-X (X = 1–5) are 23.84, 16.91, 17.58, 17.04 and 14.75%. The PLQY of TbBTC is 41.38%, excited at 355 nm. With the increase of DSM content, the main emission intensities of Tb3+ at 487 and 544 nm reduced and the intensity of DSM at 615 nm increased accordingly (Fig. 5b). TbBTC shows monoexponential fluorescence decay and the lifetime is 1.37 ms (Fig. 5c and Table S14†). Each sample of DSM@TbBTC-X (X = 1–5) exhibits biexponential fluorescence decay and the average emission lifetimes are 1.15, 0.99, 0.92, 0.71 and 0.6 ms, respectively. The shortening of lifetime values upon loading DSM into the channels of TbBTC is attributed to the energy transfer from Tb3+ to DSM. The energy transfer efficiency between Tb3+ and DSM is calculated from the equation Ø = 1 − τDA/τD, τDA and τD are the lifetime values of the energy donor in the presence and absence of the energy acceptor. Ø in DSM@TbBTC-X (X = 1–5) is 16.1, 27.7, 32.8, 48.2 and 55.7%, respectively. The CIE coordinates of TbBTC and DSM@TbBTC-X (X = 1–5) are (0.27, 0.56), (0.37, 0.51), (0.45, 0.45), (0.51, 0.42), (0.59, 0.37) and (0.61, 0.37), respectively. The corresponding photographs show that with the increase of the content of DSM, the photoluminescence color can be gradually tuned from the green to red region (Fig. 5d).
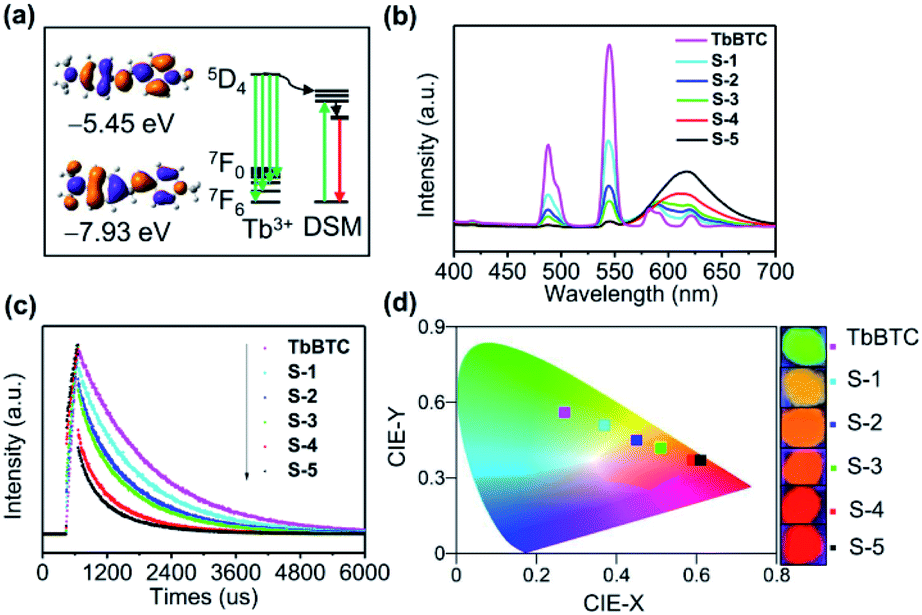 | ||
| Fig. 5 (a) Calculated HOMO and LUMO levels of DSM and the diagram of energy transfer from Tb3+ to DSM. (b) Solid-state emission spectra, (c) time-resolved emission decay curves (λem = 544 nm) and (d) CIE chromaticity coordinates and colors of powders of neat TbBTC and five DSM@TbBTC samples: S1–S5. Fluorescence spectra were obtained under 355 nm excitation. | ||
Conclusions
In summary, we present an exquisite method to create molecular chirality from an achiral organic molecule DSM. We designed and synthesized an elegant chiral channel that enables achievement of the following three aspects – encapsulation of DSM at the molecule-state, unidirectional twist of DSM into either P- or M-configuration to create symmetry breaking, and solidification of the asymmetry made after unidirectional rotation – at the same time and by the same chiral channel. Strong CPL emission with a glum factor of 10−3 and a ϕlum value of ∼10% is characterized by solid-state CPL and micro-area polarized emission of DSM@TbBTC. Controllable color-tuning in the green and red regions was achieved in DSM@TbBTC on account of energy level matching between Tb3+ and the symmetry-broken DSM. Our study reveals that molecular chirality with strong CPL emission may be created from an achiral molecule if we can unidirect the symmetry breaking and fix the resulting asymmetry. Again, this study provides valuable information for understanding the structural origins of supramolecular chirality of an achiral molecule.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. Z. and M. Z. acknowledge financial support from the National Natural Science Foundation of China (NSFC, Grant no. 21327805).Notes and references
- Y. Yang, R. C. da Costa, M. J. Fuchter and A. J. Campbell, Nat. Photonics, 2013, 7, 634–638 CrossRef CAS.
- C. Wang, T. Zhang and W. Lin, Chem. Rev., 2012, 112, 1084–1104 CrossRef CAS PubMed.
- N. J. Long, Angew. Chem., Int. Ed., 1995, 34, 21–38 CrossRef CAS.
- F. Song, G. Wei, X. Jiang, F. Li, C. Zhu and Y. Cheng, Chem. Commun., 2013, 49, 5772 RSC.
- M. Seitz, E. G. Moore, A. J. Ingram, G. Muller and K. N. Raymond, J. Am. Chem. Soc., 2007, 129, 15468–15470 CrossRef CAS PubMed.
- Z.-G. Wu, H.-B. Han, Z.-P. Yan, X.-F. Luo, Y. Wang, Y.-X. Zheng, J.-L. Zuo and Y. Pan, Adv. Mater., 2019, 31, 1900524 CrossRef PubMed.
- N. Sharma, E. Spuling, C. M. Mattern, W. Li, O. Fuhr, Y. Tsuchiya, C. Adachi, S. Braese, I. D. W. Samuel and E. Zysman-Colman, Chem. Sci., 2019, 10, 6689–6696 RSC.
- K. Dhbaibi, L. Favereau, M. Srebro-Hooper, C. Quinton, N. Vanthuyne, L. Arrico, T. Roisnel, B. Jamoussi, C. Poriel, C. Cabanetos, J. Autschbach and J. Crassous, Chem. Sci., 2020, 11, 567–576 RSC.
- M. Krykunov and J. Autschbach, J. Chem. Phys., 2006, 125, 034102 CrossRef PubMed.
- K. Claborn, C. Isborn, W. Kaminsky and B. Kahr, Angew. Chem., Int. Ed., 2008, 47, 5706–5717 CrossRef CAS PubMed.
- B. A. S. Jose, S. Matsushita and K. Akagi, J. Am. Chem. Soc., 2012, 134, 19795–19807 CrossRef PubMed.
- B. A. San Jose, J. Yan and K. Akagi, Angew. Chem., Int. Ed., 2014, 53, 10641–10644 CrossRef CAS PubMed.
- R. Aoki, R. Toyoda, J. F. Koegel, R. Sakamoto, J. Kumar, Y. Kitagawa, K. Harano, T. Kawai and H. Nishihara, J. Am. Chem. Soc., 2017, 139, 16024–16027 CrossRef CAS PubMed.
- N. Hellou, M. Srebro-Hooper, L. Favereau, F. Zinna, E. Caytan, L. Toupet, V. Dorcet, M. Jean, N. Vanthuyne, J. A. G. Williams, L. Di Bari, J. Autschbach and J. Crassous, Angew. Chem., Int. Ed., 2017, 56, 8236–8239 CrossRef CAS PubMed.
- Y. Zhou, H. Li, T. Zhu, T. Gao and P. Yan, J. Am. Chem. Soc., 2019, 141, 19634–19643 CrossRef CAS PubMed.
- J. L. Lunkley, D. Shirotani, K. Yamanar, S. Kaizaki and G. Muller, J. Am. Chem. Soc., 2008, 130, 13814–13815 CrossRef CAS PubMed.
- J. Kumar, T. Nakashima and T. Kawai, J. Phys. Chem. Lett., 2015, 6, 3445–3452 CrossRef CAS PubMed.
- G. Longhi, E. Castiglioni, J. Koshoubu, G. Mazzeo and S. Abbate, Chirality, 2016, 28, 696–707 CrossRef CAS PubMed.
- C. Dee, F. Zinna, W. R. Kitzmann, G. Pescitelli, K. Heinze, L. Di Bari and M. Seitz, Chem. Commun., 2019, 55, 13078–13081 RSC.
- B. A. San Jose, S. Matsushita and K. Akagi, J. Am. Chem. Soc., 2012, 134, 19795–19807 CrossRef CAS PubMed.
- J. Han, D. Yang, X. Jin, Y. Jiang, M. Liu and P. Duan, Angew. Chem., Int. Ed., 2019, 58, 7013–7019 CrossRef CAS PubMed.
- H. Song, H. Zhu, Z. Huang, Y. Zhang, W. Zhao, J. Liu, Q. Chen, C. Yin, L. Xing, Z. Peng, P. Liao, Y. Wang, Y. Wang and K. Wu, ACS Nano, 2019, 13, 7202–7208 CrossRef CAS PubMed.
- Y. Yang, Y. Zhang and Z. Wei, Adv. Mater., 2013, 25, 6039–6049 CrossRef CAS PubMed.
- P. Xing and Y. Zhao, Acc. Chem. Res., 2018, 51, 2324–2334 CrossRef CAS PubMed.
- J. Roose, B. Z. Tang and K. S. Wong, Small, 2016, 12, 6495–6512 CrossRef CAS PubMed.
- H. Shang, Z. Ding, Y. Shen, B. Yang, M. Liu and S. Jiang, Chem. Sci., 2020, 11, 2169–2174 RSC.
- Y. Li, K. Liu, X. Li, Y. Quan and Y. Cheng, Chem. Commun., 2020, 56, 1117–1120 RSC.
- A. P. Katsoulidis, D. Antypov, G. F. S. Whitehead, E. J. Carrington, D. J. Adams, N. G. Berry, G. R. Darling, M. S. Dyer and M. J. Rosseinsky, Nature, 2019, 565, 213–217 CrossRef CAS PubMed.
- J.-D. Xiao and H.-L. Jiang, Acc. Chem. Res., 2019, 52, 356–366 CrossRef CAS PubMed.
- J. Lyu, X. Zhang, K.-i. Otake, X. Wang, P. Li, Z. Li, Z. Chen, Y. Zhang, M. C. Wasson, Y. Yang, P. Bai, X. Guo, T. Islamoglu and O. K. Farha, Chem. Sci., 2019, 10, 1186–1192 RSC.
- D. Yu, Q. Shao, Q. Song, J. Cui, Y. Zhang, B. Wu, L. Ge, Y. Wang, Y. Zhang, Y. Qin, R. Vajtai, P. M. Ajayan, H. Wang, T. Xu and Y. Wu, Nat. Commun., 2020, 11, 927 CrossRef CAS PubMed.
- W. Zhao, G. Li and Z. Tang, Nano Today, 2019, 27, 178–197 CrossRef CAS.
- Z. Wang, C.-Y. Zhu, J.-T. Mo, P.-Y. Fu, Y.-W. Zhao, S.-Y. Yin, J.-J. Jiang, M. Pan and C.-Y. Su, Angew. Chem., Int. Ed., 2019, 58, 9752–9757 CrossRef CAS PubMed.
- X.-G. Yang, X.-M. Lu, Z.-M. Zhai, Y. Zhao, X.-Y. Liu, L.-F. Ma and S.-Q. Zang, Chem. Commun., 2019, 55, 11099–11102 RSC.
- Q. Li, Z.-L. Fan, D.-X. Xue, Y.-F. Zhang, Z.-H. Zhang, Q. Wang, H.-M. Sun, Z. Gao and J. Bai, J. Mater. Chem. A, 2018, 6, 2148–2156 RSC.
- X.-Y. Liu, K. Xing, Y. Li, C.-K. Tsung and J. Li, J. Am. Chem. Soc., 2019, 141, 14807–14813 CrossRef CAS PubMed.
- P. Wu, C. He, J. Wang, X. Peng, X. Li, Y. An and C. Duan, J. Am. Chem. Soc., 2012, 134, 14991–14999 CrossRef CAS PubMed.
- J. Y. Chan, H. Zhang, Y. Nolvachai, Y. Hu, H. Zhu, M. Forsyth, Q. Gu, D. E. Hoke, X. Zhang, P. J. Marriot and H. Wang, Angew. Chem., Int. Ed., 2018, 57, 17130–17134 CrossRef CAS PubMed.
- S. Das, S. Xu, T. Ben and S. Qiu, Angew. Chem., Int. Ed., 2018, 57, 8629–8633 CrossRef CAS PubMed.
- T. Kitao, Y. Nagasaka, M. Karasawa, T. Eguchi, N. Kimizuka, K. Ishii, T. Yamada and T. Uemura, J. Am. Chem. Soc., 2019, 141, 19565–19569 CrossRef CAS PubMed.
- T. Zhao, J. Han, X. Jin, Y. Liu, M. Liu and P. Duan, Angew. Chem., Int. Ed., 2019, 58, 4978–4982 CrossRef CAS PubMed.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS.
- J. P. Riehl and F. S. Richardson, Chem. Rev., 1986, 86, 1–16 CrossRef CAS.
- J. Ma, Y. Wang, X. Li, L. Yang, Y. Quan and Y. Chen, Polymer, 2018, 143, 184–189 CrossRef CAS.
- E. M. Sanchez-Carnerero, F. Moreno, B. L. Maroto, A. R. Agarrabeitia, M. J. Ortiz, B. G. Vo, G. Muller and S. de la Moya, J. Am. Chem. Soc., 2014, 136, 3346–3349 CrossRef CAS PubMed.
- S. Furumi, Chem. Rec., 2010, 10, 394–408 CAS.
- J. Zhao, T. Zhang, X. Y. Dong, M. E. Sun, C. Zhang, X. Li, Y. S. Zhao and S. Q. Zang, J. Am. Chem. Soc., 2019, 141, 15755–15760 CrossRef CAS PubMed.
- L. Guo, C. Li, H. Shang, R. Zhang, X. Li, Q. Lu, X. Cheng, Z. Liu, J. Z. Sun and X. Yu, Chem. Sci., 2020, 11, 661–670 RSC.
- R. Luo, H. Xu, H.-X. Gu, X. Wang, Y. Xu, X. Shen, W. Bao and D.-R. Zhu, CrystEngComm, 2014, 16, 784–796 RSC.
- M. Todorova, R. Bakalska and T. Kolev, Spectrochim. Acta, Part A, 2013, 108, 211–222 CrossRef CAS PubMed.
- K. Furuya, K. Kawato, H. Yokoyama, A. Sakamoto and M. Tasumi, J. Phys. Chem. A, 2003, 107, 8251–8258 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1960863, 1960864, 1966280 and 1966281. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc02856h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |