
Core hyper-cross-linked star polymers from block polymer micelle precursors†
Jongmin
Park
 a,
Stefan J. D.
Smith
bc,
Colin D.
Wood
b,
Xavier
Mulet
b and
Myungeun
Seo
*ad
a,
Stefan J. D.
Smith
bc,
Colin D.
Wood
b,
Xavier
Mulet
b and
Myungeun
Seo
*ad
aDepartment of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Korea. E-mail: seomyungeun@kaist.ac.kr
bCommonwealth Scientific and Industrial Research Organisation (CSIRO), Australia
cMonash Centre for Membrane Innovation (MCMI), Monash University, Australia
dKAIST Institute for Nanocentury, KAIST, Daejeon 34141, Korea
First published on 27th October 2020
Abstract
This study explores hyper-cross-linking of the cores of block copolymer micelles as a means to generate star polymers with a hyper-cross-linked core surrounded by linear corona arms. A solution of poly(methyl methacrylate)-b-polystyrene (MS) was prepared in acetonitrile to form micelles which were reacted with α,α′-dichloro-p-xylene in the presence of FeCl3 to produce core hyper-cross-linked star (CHS) polymers by selective cross-linking of the PS core. A kinetic investigation showed formation of high-molar mass species (>104 kg mol−1) within 1 h of reaction, which supported conversion of individual MS micelles into CHS polymers. We synthesized several CHS polymers by varying the PS core fractions from 20 to 53%. All the polymers possessed discrete spherical cores that were 19–60 nm in diameter and all were highly soluble in organic solvents retaining the CHS architecture. While permanent microporosity was not detected by gas sorption measurements, increased dye uptake of CHS polymer in solution suggests utility of CHS polymers as stable and solution-processible nanocontainers with accessible free volume in the core.
Introduction
Core cross-linked star (CCS) polymers consist of multiple polymer chains tethered to a densely cross-linked core.1,2 The mobile linear chains, typically referred to as “arms”, are responsible for interaction with external environments and make the CCS polymer processible in solution and melt states. The cross-linked core stabilizes the whole macromolecule through covalent bonding and provides a distinctive inner compartment that can be useful for encapsulation. Given these features, CCS polymers may be considered unimolecular micelles distinguishable from conventional block copolymer micelles, which are disintegrated into individual chains in a good solvent or upon dilution. Synthesis of CCS polymers with a functionalized core by covalent anchoring of functional groups has been demonstrated for applications such as catalysis, imaging, and storage of sensitive molecules that can be released in a controlled fashion.3–14CCS polymers have typically been synthesized via the “arm-first” approach including cross-linking polymerization from the end of the first polymer chain.1,2,15–20 Cross-linking of the core of block copolymer micelle can be an alternative route to attain a well-defined and discrete core. A block copolymer precursor consisting of solvophobic and solvophilic blocks is introduced into a selective solvent to form micelles.21 Cross-linking of the micellar core converts the micelle into the CCS polymer while retaining the morphology. As the self-assembly process allows for the morphological parameters to be tuned by adjusting the molar mass and composition of the precursor, CCS polymers can be obtained with controlled dimensions of the core and the whole macromolecule.
Core cross-linking requires orthogonal chemistry to polymerization that does not interfere with the synthesis of the block copolymer precursor. Hyper-cross-linking can be an effective way to cross-link polymers containing aromatic units. Friedel–Crafts alkylation forms methylene bridges between the aromatic rings and generates densely cross-linked polymer networks with substantial porosity.22–24 Unlike to conventional methodologies that require a reactive functionality on the core block typically installed by copolymerization, in hyper-cross-linking external carbocations can be used to link the native aromatic rings without specific functionality.
Hyper-cross-linking has recently been utilized for corona cross-linking of polystyrene (PS)-containing block copolymers to produce a stable cross-linked shell containing microporous void (<2 nm) with high thermal and chemical stability.25,26 While no examples of core cross-linking via hyper-cross-linking strategy have yet been reported, we envision that CCS polymers consisting of a hyper-cross-linked core will be an interesting platform considering utility of hyper-cross-linked materials for adsorption, separation, and catalysis.24,27 The processibility of the CCS architecture can be synergistically combined with the utility of the hyper-cross-linked compartment, to create solvent-dispersible and processible nanoobjects possessing a stable core with high loading capacity.28–31
Here we describe the straightforward synthesis of core hyper-cross-linked star (CHS) polymers by hyper-cross-linking of the core of poly(methyl methacrylate)-b-polystyrene (PMMA-b-PS) micelles. PMMA-b-PS with PS core and PMMA corona was prepared in acetonitrile (MeCN) which is a selective solvent to PMMA. Addition of α,α′-dichloro-p-xylene (p-DCX) as an external cross-linker and FeCl3 as a Lewis acid to the micellar solution yields the CHS polymer that has the parent micellar morphology. Characterization of the CHS polymer by 1H NMR spectroscopy and size exclusion chromatography (SEC) clearly indicates formation of ultrahigh molar mass species (>104 kg mol−1) containing the cross-linked PS core. The spherical morphology of the parent PMMA-b-PS micelle is successfully transferred into the CHS polymer with controllable size in the range of 50–200 nm. Increased thermal stability of the CHS polymer than PMMA-b-PS is evident. The CHS polymer retains the structures in a good solvent to PS such as chloroform and is processible into films, suggesting potential for use as membranes or membrane additives due to their natural dispersion of hyper-cross-linked cores.32 Higher dye uptake of CHS than the micelle precursor is consistent with larger accessible volume of the hyper-cross-linked core, suggesting its utility for catalytic and separation applications.
Results and discussion
A synthetic scheme to CHS polymer is depicted in Fig. 1. A solution of PMMA-b-PS and p-DCX as a cross-linker is prepared in MeCN. At 80 °C, FeCl3 is added to the mixture to initiate the hyper-cross-linking. The amount of p-DCX and FeCl3 was set to 4 equiv. relative to the styrene repeating units in PMMA-b-PS, following the literature protocols using the external cross-linker.33,34 The reaction is quenched by precipitating the mixture in HCl(aq.)/methanol. The resulting grey powder is further purified by Soxhlet extraction with methanol. The CHS polymer is denoted by Mn and the volume fraction of PS block (fPS) of the parent PMMA-b-PS (MS) determined using 1H NMR spectroscopy. For example, CHS(72.4, 0.42) indicates the CHS polymer synthesized from MS with Mn = 72.4 kg mol−1 and fPS = 0.42.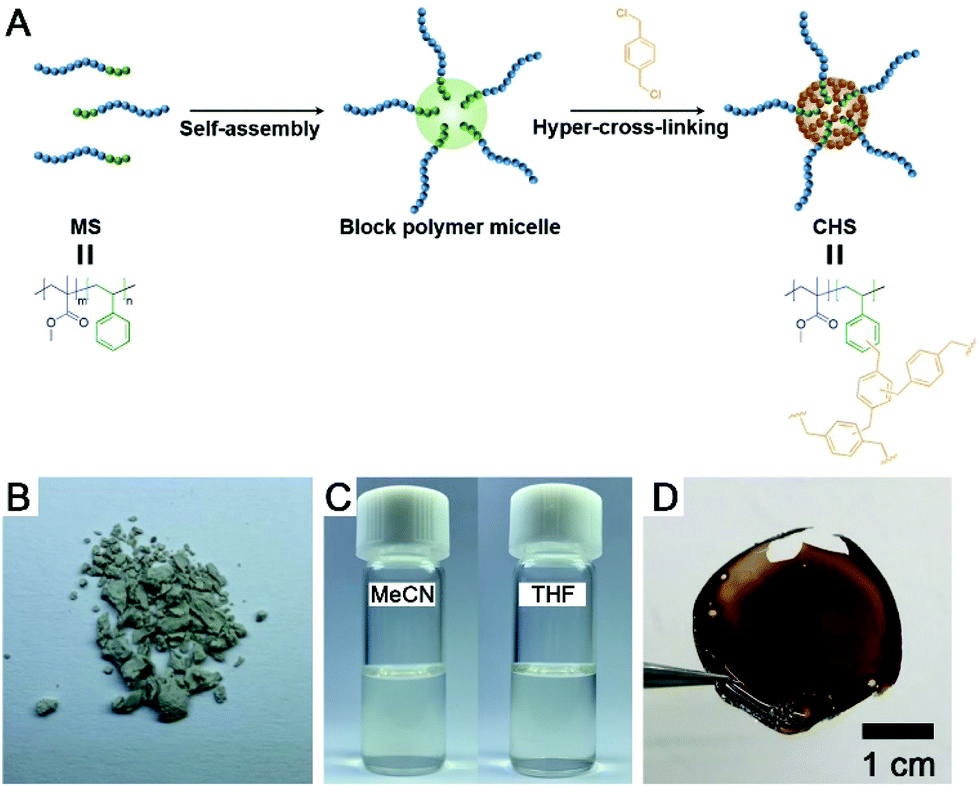 | ||
| Fig. 1 (A) Synthetic route to CHS polymer via hyper-cross-linking of PMMA-b-PS in MeCN. (B) Photo of CHS(69.2, 0.36) powder. (C) Photos of CHS(69.2, 0.36) solutions in MeCN (left) and THF (right). (D) Film of CHS(72.4, 0.42) obtained by casting from toluene solution. | ||
The CHS polymer is soluble in common organic solvents such as MeCN, acetone, chloroform, toluene, and DMF, indicating that the nanoobject is well solubilized by the PMMA chains. A representative 1H NMR spectrum of CHS(69.2, 0.36) in CDCl3 is shown in Fig. 2A, obtained after 24 h of reaction. Protons corresponding to the PMMA segment are clearly visible including the methoxy proton at 3.6 ppm. In contrast, peaks corresponding to the aromatic protons of the PS block at 6–7 ppm diminishes after reaction. Strong suppression of the PS signals is consistent with the formation of the hyper-cross-linked and solid-like polystyrenic core, whose protons are difficult to detect due to the decreased T2 relaxation time.
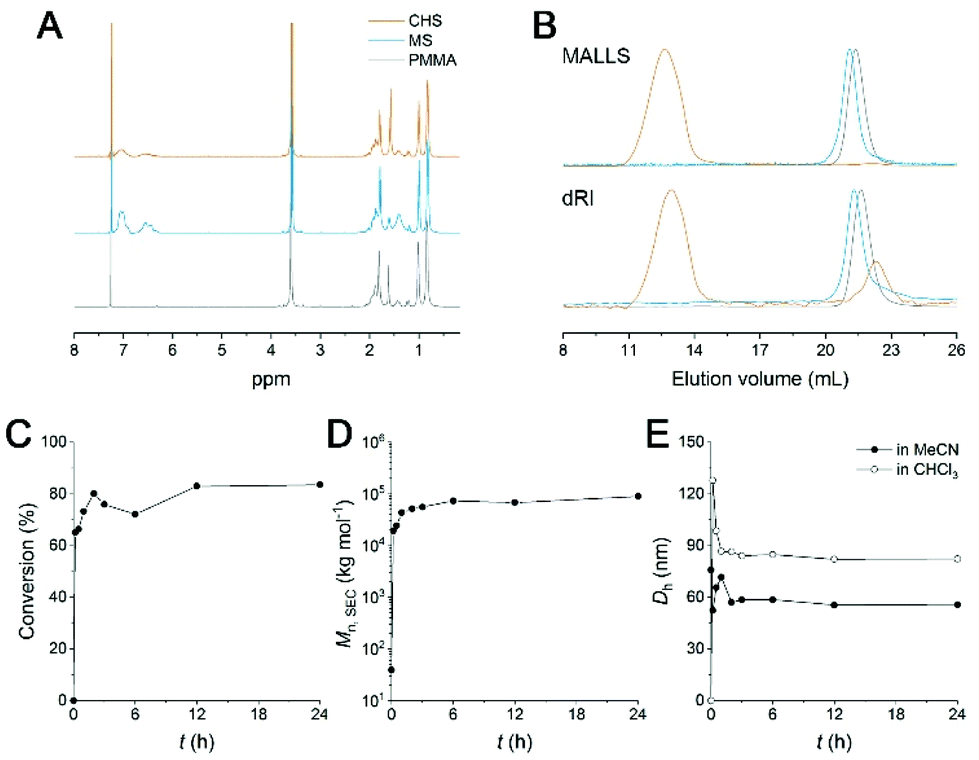 | ||
| Fig. 2 (A and B) Normalized 1H NMR spectra (A) and SEC traces (B) of CHS(69.2, 0.36) obtained after 24 h of reaction and its precursors. The spectra were normalized by the peak at 3.6 ppm corresponding to PMMA methoxy proton. The traces were also normalized by the main peak intensity. (C–E) Evolution of conversion (C), Mn,SEC (D), and Dh (E) as a function of reaction time. Dh was estimated by DLS analysis of the purified polymer solutions in MeCN (filled circle) and CHCl3 (open circle). | ||
A SEC trace of CHS(69.2, 0.36) compared to PMMA and MS is shown in Fig. 2B. In both the differential refractive index (dRI) and multi-angle light scattering (MALLS) detections, the CHS polymer is eluted very early, indicating its ultrahigh molar mass. In this particular example obtained after 24 h of reaction, Mn,SEC of 89![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 710 kg mol−1 was estimated by linear PMMA calibration standards. We note that the value is not reliable as it exceeds the upper limit of the SEC column used for the analysis (<10000 kg mol−1). The number of MS chains per CHS was estimated from the absolute molecular weight (Mw) determined by MALLS-SEC analysis (Fig. S1 and Table S1†). As a representative sample, Mw of MS(56.9, 0.24) and CHS(56.9, 0.24) was determined as 103 and 5972 kg mol−1, respectively. Assuming full incorporation of p-DCX and complete removal of HCl via Soxhlet extraction, we concluded 12 MS chains were covalently tied per hyper-cross-linked core.
710 kg mol−1 was estimated by linear PMMA calibration standards. We note that the value is not reliable as it exceeds the upper limit of the SEC column used for the analysis (<10000 kg mol−1). The number of MS chains per CHS was estimated from the absolute molecular weight (Mw) determined by MALLS-SEC analysis (Fig. S1 and Table S1†). As a representative sample, Mw of MS(56.9, 0.24) and CHS(56.9, 0.24) was determined as 103 and 5972 kg mol−1, respectively. Assuming full incorporation of p-DCX and complete removal of HCl via Soxhlet extraction, we concluded 12 MS chains were covalently tied per hyper-cross-linked core.
Kinetics of the hyper-cross-linking reaction was monitored by estimating the consumption of the MS precursor in the SEC analysis (Fig. 2E, S2, and Table S2†). At the constant sample concentration (1 mg ml−1), the amount of MS remaining in the reaction mixture was evaluated by the peak integral as a function of time. It seems that more than 73% of the chains are rapidly converted to high molar mass CHS polymer within 1 h (Fig. 2D). We also observed that the intensity of the PS aromatic protons decreased noticeably after 1 h, supporting formation of a densely cross-linked core (Fig. S2†). Then increase in both conversion and in molar mass becomes slower. This suggests that individual MS micelles are converted into CHS polymer, as the hyper-cross-linking reaction is confined to the miceller core. Steric hindrance may also hinder further core-core coupling. Mn,SEC reached a plateau after 6 h. 83.5% Conversion was achieved after 24 h. The hydrodynamic diameter (Dh) of CHS(69.2, 0.36) estimated by Cumulant fit of the dynamic light scattering (DLS) data also reached a constant value of ca. 56 nm after 3 h of reaction (Fig. 2E and S3†). This also suggests the intercore cross-linking is minimized. Compared to the MS micelle prior to the reaction, formation of the hyper-cross-linked core reduced Dh by 24%. The covalently joined CHS architecture retains its hydrodynamic size in CHCl3, which is a good solvent for PS and PMMA. While MS is homogeneously dissolved in CHCl3, CHS polymer produced by 10 min of reaction (65% conversion) shows a noticeable decay in the autocorrelation function. The size is initially large (Dh > 120 nm) and the distribution is broad, but the Dh value converges to 82 nm after 3 h of reaction with narrower polydispersity index (0.092). Somewhat larger dimension in CHCl3 than MeCN suggests the hyper-cross-linked core may swell more in CHCl3. The loosely cross-linked micelle in the initial stage is knitted more tightly as the reaction proceeds further, resulting in a smaller but well-defined hydrodynamic dimension. Overall, these data nicely support that CHS polymer was successfully generated with PMMA corona and hyper-cross-linked PS core.
Based on this success, we synthesized several CHS polymers by varying fPS, while keeping the molar mass of the PMMA block nearly identical. We set the reaction time to be 18 h to help ensure formation of a densely cross-linked network in the micellar core. The 1H NMR spectra and SEC traces of the parent MS and the corresponding CHS polymers are shown in Fig. S4 and S5.† The characteristic details are summarized in Table 1. We note that CHS(91.4, 0.53) could not be analyzed by SEC analysis, presumably because its hydrodynamic size was too large. We also conducted thermogravimetric analysis (TGA) of the CHS and parent MS polymer with fPS = 0.29 to confirm the presence of the hyper-cross-linked core (Fig. S6†). Ca. 30% of residue was observed from the CHS polymer sample above 420 °C, while the parent PMMA-b-PS was entirely decomposed by 410 °C. Considering the fPS of PMMA-b-PS used for the measurement was 0.29, we interpret the results to indicate a hyper-cross-linked core with enhanced thermal stability remains after decomposition of the PMMA segment. Carbonization may occur with further ramping temperature (a char yield of 5% was observed after 700 °C). We note that hyper-cross-linked polymers are well known precursors to microporous carbon.24,35
| Entrya | Conversionb (%) | M n,PMMA (kg mol−1) | M n,PS (kg mol−1) | M n,SEC (kg mol−1) | Đ | f PS | D h (nm) | d SAXS (nm) |
|---|---|---|---|---|---|---|---|---|
| a The numbers in parentheses indicate Mn,PS-b-PMMA (kg mol−1) and fPS. b Determined by CHCl3 SEC based on linear PMMA standards. We note that the values >104 kg mol−1 are not reliable as they exceed the upper molar mass limit of the column. c Determined by 1H NMR analysis in CDCl3. d Determined by DLS analysis in MeCN. e Determined by dSAXS = 2π/q* from bulk SAXS where q* is the position of the principal peak. | ||||||||
| MS(56.2, 0.20) | — | 45.9 | 10.3 | 46.4 | 1.52 | 0.20 | 40.9 | 25.8 |
| MS(56.9, 0.24) | — | 44.1 | 12.8 | 39.7 | 1.44 | 0.24 | 68.3 | 25.9 |
| MS(62.5, 0.29) | — | 45.9 | 16.6 | 50.6 | 1.40 | 0.29 | 111.4 | 28.0 |
| MS(72.4, 0.42) | — | 44.1 | 28.3 | 56.0 | 1.35 | 0.42 | 122.5 | 33.4 |
| MS(91.4, 0.53) | — | 45.9 | 45.5 | 60.8 | 1.55 | 0.53 | 336.0 | 58.2 |
| CHS(56.2, 0.20) | 86.0 | — | — | 1.12 × 104 | 2.71 | 0.20 | 50.2 | 25.5 |
| CHS(56.9, 0.24) | 84.4 | — | — | 6.57 × 104 | 2.35 | 0.24 | 56.6 | 23.0 |
| CHS(62.5, 0.29) | 77.3 | — | — | 1.85 × 104 | 2.55 | 0.29 | 62.5 | 27.2 |
| CHS(72.4, 0.42) | 92.5 | — | — | 1.52 × 105 | 1.47 | 0.42 | 81.9 | 30.2 |
| CHS(91.4, 0.53) | 91.2 | — | — | — | — | 0.53 | 184.2 | 62.6 |
Morphology of the CHS polymers in MeCN was examined by transmission electron microscopy (TEM), and compared to that of the parent MS (Fig. 3). Samples were prepared by dropping the solutions on a TEM grid and evaporating the solvent under ambient conditions, and then imaged without staining. In this case the polystyrene-based core appears dark.36,37 All of the MS block copolymers self-assembled into spherical micelles regardless of fPS. The diameter of the micellar core increased with increasing fPS (i.e., PS molar mass increase) from 23 to 63 nm. The CHS polymers retained their spherical morphology and well-dispersible nature of the parent MS micelles. This indicates the hyper-cross-linking reaction in the PS core transformed the micellar aggregate into the CHS polymer, while maintaining an intact PMMA corona. The core size did not noticeably vary after hyper-cross-linking.
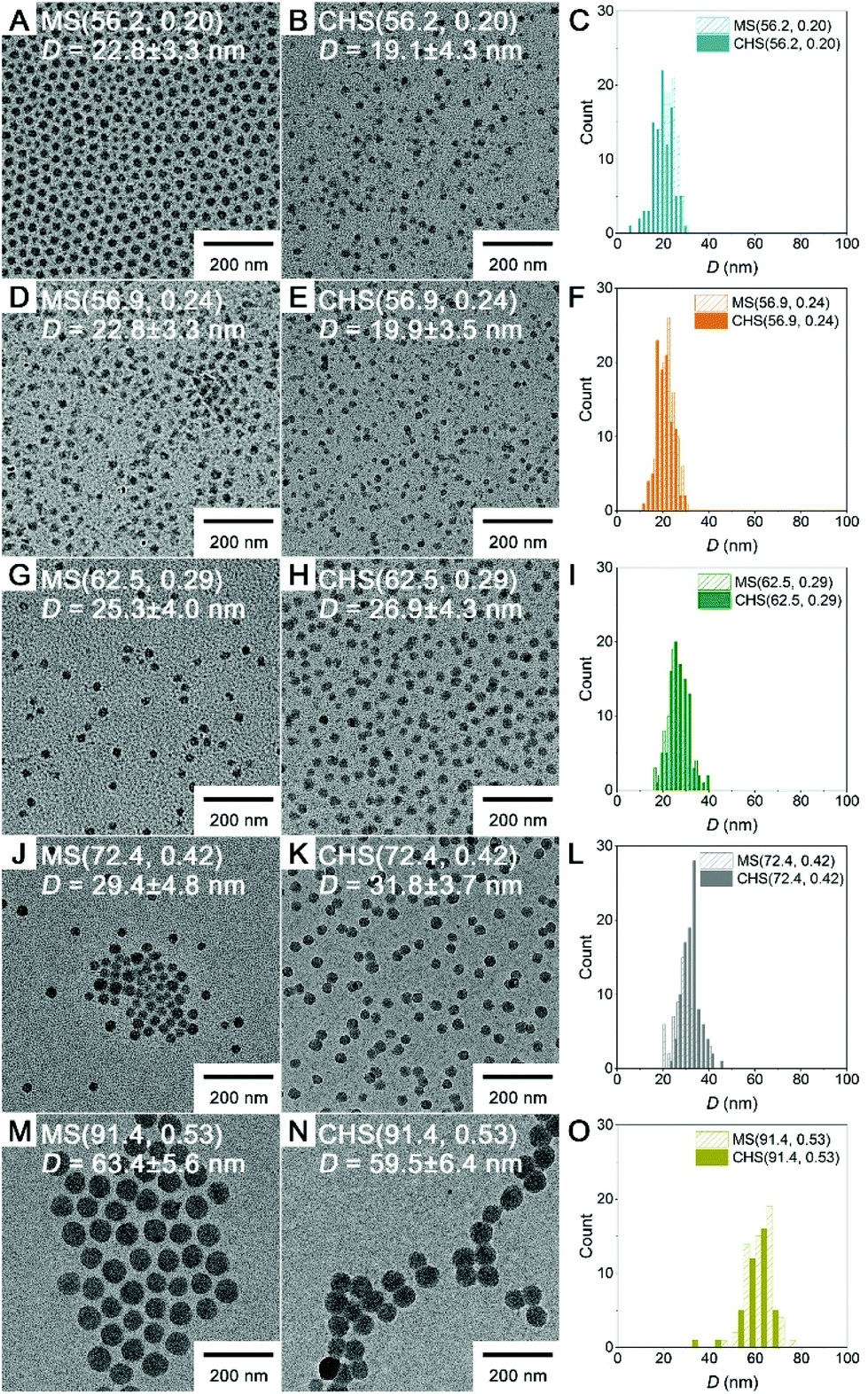 | ||
| Fig. 3 TEM images of MS (A, D, G, J and M) and CHS polymers (B, E, H, K and N) cast from the MeCN solution. Size distribution in diameter is shown in C, F, I, L, and O, and the average value is included in the image. | ||
Free-standing polymer films prepared by casting the MeCN solution were also analyzed by small-angle X-ray scattering (SAXS) (Fig. S7†). MS samples exhibited an intense but relatively broad principle scattering peak, which is consistent with disordered packing of spherical micelles. The CHS polymers showed similar scattering patterns while retaining the length scale. However, the principal scattering peak q* was much weaker and broader compared to MS. The lower degree of long-range order in the CHS polymers is attributed to the presence of a few core-core coupled species observed in the TEM images.
By DLS analysis, Dh of both the MS and CHS polymers in MeCN increased with fPS (Fig. 4 and S8†). Dh reduction after CHS formation was observed in most cases. By comparing the DLS data to those obtained at 60 °C and also in the presence of p-DCX, we suggest that the reduced micellar dimension at 80 °C than room temperature is arrested by hyper-cross-linking to result in the smaller Dh (Fig. S9 and Table S4†). The data also suggest slight swelling of the micellar core by preferential partitioning of p-DCX, which would facilitate hyper-cross-linking of the core. In CHCl3, the CHS polymers also showed Dhs comparable to MeCN.
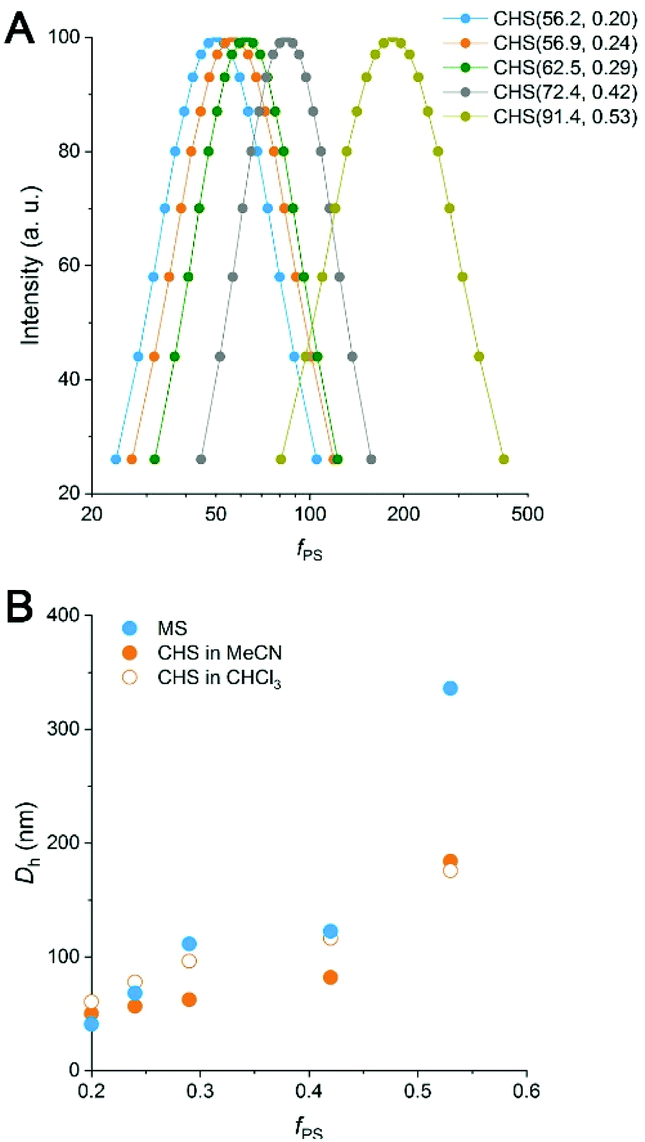 | ||
| Fig. 4 (A) Particle size distribution of CHS polymers obtained by DLS measurements in MeCN. (B) Dh of MS (blue) and CHS (orange) polymers in different solvents. Filled circle: MeCN, open circle: CHCl3. | ||
We attempted to further analyze the CHS morphology in MeCN by solution SAXS measurements. Preliminary results for CHS(56.9, 0.24) and CHS(72.4, 0.42) are shown in Fig. 5 with their parent MS. A distinct minimum was observed from all scattering data correlating to the presence of micelles. Its position did not move significantly by the hyper-cross-linking reaction, which is consistent with conversion of individual MS micelles into CHS conserving the length scale. Higher-order features were difficult to discern in the high q regime. The relatively broad scattering patterns qualitatively resemble that of a polymer star (soft sphere) rather than a spherical micelle (hard sphere).38,39 Interestingly, CHS(72.4, 0.42) exhibited a second-order minimum suggesting that potentially the hyper-cross-linked core becomes more sphere-like and facilitates more contiguous packing. Indeed, the hard sphere model fits the scattering profile of CHS(72.4, 0.42) much better than MS(72.4, 0.42). From the fitting, the radius of gyration (Rg) of CHS(72.4, 0.42) core was estimated to be 17.7 nm, which is comparable to the core size observed by TEM (D = 31.8 nm).
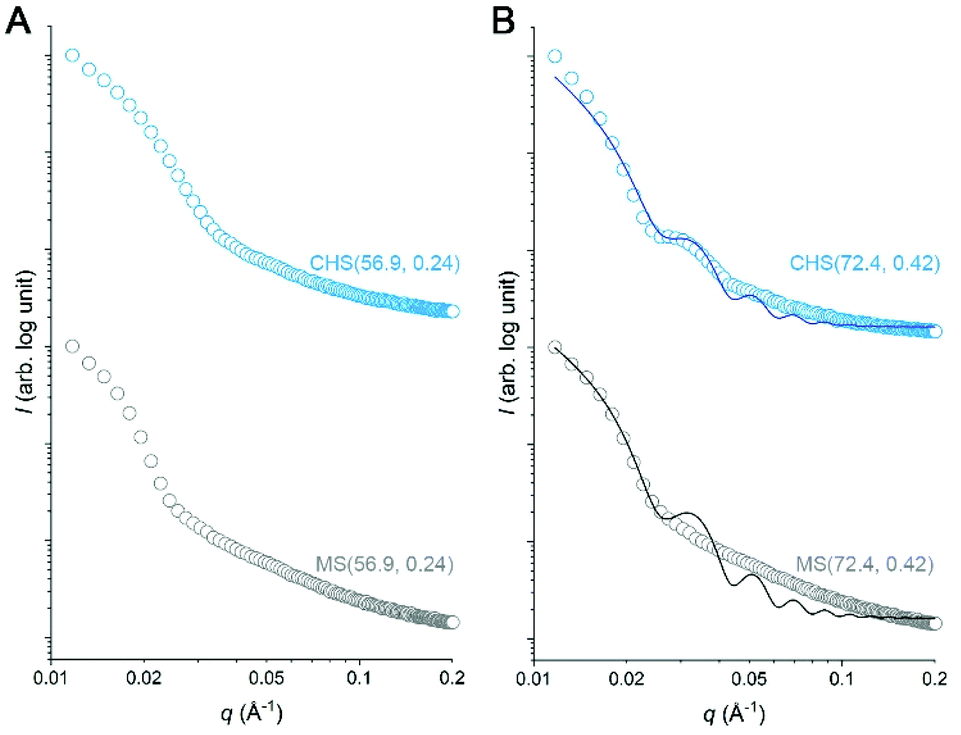 | ||
| Fig. 5 Solution SAXS profiles of CHS(56.9, 0.24) (A) and CHS(72.4, 0.42) (B) along with their parent MS. The data were obtained from the MeCN solutions at 2 mg ml−1 concentration. For the data shown in B, the scattering profiles were fitted with the hard-sphere model. | ||
N2 uptake by the CHS polymers was negligible, suggesting the core does not possess accessible permanent microporosity in the solid state (Fig. S10†). Low-pressure CO2 adsorption was also low, suggesting the PMMA corona may be hindering access to the microporous core. Other size-limited hyper-cross-linked polymers have also shown poor gas sorption at low pressures, suggesting that confined hyper-cross-linked cores are unable to form large pore volumes. Nonetheless, we observed that the hyper-cross-linked core has a higher adsorption capacity than the micellar core in the solution state as a stable nanocontainer.
Fig. 6A shows a solution of CHS(72.4, 0.42) in THF and MeCN encapsulating Nile red, which was selected as a model guest compound. We tested CHS(56.9, 0.24) and CHS(72.4, 0.42) to see the effect of the core volume on the Nile red uptake. The change in color of the MeCN solution upon addition of CHS is consistent with adsorption of Nile red in more nonpolar environments provided by the hyper-cross-linked core.40 Uptake of Nile red also occurs in THF as the CHS polymer stably retains the conformation, but the adsorption capacity (Q) in relatively nonpolar THF is much lower than MeCN (Fig. 6B, S11, and Table S3†). Based on the absolute molar mass determined by MALLS analysis, 41.8 and 9.8 Nile red molecules are adsorbed on average per individual CHS(56.9, 0.24) in MeCN and THF, respectively. The Q value obtained from the MeCN solution is comparable to those reported using other unimolecular micelles, where water was used as a solvent that strongly facilitates adsorption of hydrophobic Nile red.41,42 Thus this result supports that the hyper-cross-linked core in CHS provides large hydrophobic free volume that can effectively uptake hydrophobic guests even from organic solvents. Compared to the MS micelles in MeCN, CHS also exhibited higher amount of Nile red uptake. Interestingly, we observed noticeable increase in Dh for CHS(72.4, 0.42) after Nile red adsorption (Fig. S12†). For this CHS polymer with the relatively large core fraction, aggregates appear to form driven by increased hydrophobicity upon guest encapsulation. A similar transition has been reported in literature for a different unimolecular micelle system in water.41,42
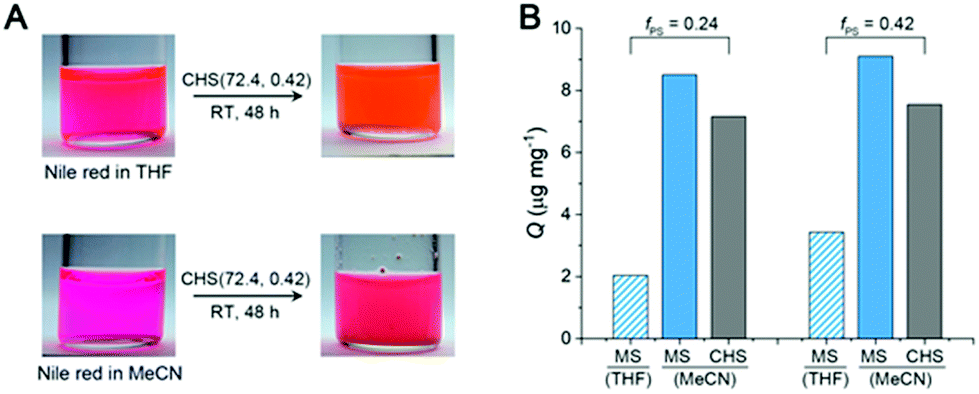 | ||
| Fig. 6 (A) Photos of Nile red solutions in THF and MeCN (left), and of the solution after adding CHS(72.4, 0.42) and stirring for 48 h (right). (B) Q of CHS(56.9, 0.24) and CHS(72.4, 0.42) in THF and MeCN obtained at the initial concentration of 100 μg mL−1. The Q values of the parent MS micelles in MeCN are also shown as a reference. | ||
Conclusions
Hyper-cross-linking was investigated as a synthetic method to produce core cross-linked star polymers, by selective cross-linking of the core of block copolymer micelles. Successful synthesis of the CHS polymer was achieved with PMMA-b-PS micelles using p-DCX as an external cross-linker. Because the PS block is confined within the micellar core, the reaction appears to convert individual micelles into CHS polymers. Polymer nanoobjects with high-molar mass (>104 kg mol−1) are produced, which possess a discrete spherical core surrounded by the corona arms. The CHS polymers can be synthesized in a wide range of PS core fractions (20–53%), are well soluble in organic solvents, and processible into free-standing films, while retaining its architecture covalently stabilized by core hyper-cross-linking. The free volume available in the core provides higher adsorption capacity for dye uptake in solution compared to the parent micelles. We believe that the solution-processible hyper-cross-linked polymer will be a useful platform as a stable nanocontainer for the solution-based catalytic and separation applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2019R1I1A2A01063804). Experiments at Pohang Accelerator Laboratory (PAL) were supported in part by Ministry of Science and ICT of Korea and POSTECH. The authors thank M. Munir Sadiq and Kristina Konstas for their assistance conducting gas adsorption experiments. Parts of this research was undertaken on the Small Angle/Wide Angle X-Ray Scattering (SAXS/WAXS) beamline at the Australian Synchrotron, part of ANSTO.References
- H. Gao and K. Matyjaszewski, Synthesis of functional polymers with controlled architecture by CRP of monomers in the presence of cross-linkers: From stars to gels, Prog. Polym. Sci., 2009, 34, 317–350 CrossRef CAS.
- A. Blencowe, J. F. Tan, T. K. Goh and G. G. Qiao, Core cross-linked star polymers via controlled radical polymerisation, Polymer, 2009, 50, 5–32 CrossRef CAS.
- B. Helms, S. J. Guillaudeu, Y. Xie, M. McMurdo, C. J. Hawker and J. M. J. Fréchet, One-pot reaction cascades using star polymers with core-confined catalysis, Angew. Chem., Int. Ed., 2005, 44, 6384–6387 CrossRef CAS.
- H. Gao and K. Matyjaszewski, Low-polydispersity star polymers with core functionality by cross-linking macromonomers using functional ATRP initiators, Macromolecules, 2007, 40, 399–401 CrossRef CAS.
- M. Spiniello, A. Blencowe and G. G. Qiao, Synthesis and characterization of fluorescently labeled core cross-linked star polymers, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2422–2432 CrossRef CAS.
- A. Blencowe, G. T. Kit, S. P. Best and G. G. Qiao, Synthesis of buckminsterfullerene C60 functionalised core cross-linked star polymers, Polymer, 2008, 49, 825–830 CrossRef CAS.
- Z. Zhang, P. Bilalis, H. Zhang, Y. Gnanou and N. Hadjichristidis, Core cross-linked multiarm star polymers with aggregation-induced emission and temperature responsive fluorescence characteristics, Macromolecules, 2017, 50, 4217–4226 CrossRef CAS.
- T. Terashima, M. Kamigaito, K.-Y. Baek, T. Ando and M. Sawamoto, Polymer catalysts from polymerization catalysts: direct encapsulation of metal catalyst into star polymer core during metal-catalyzed living radical polymerization, J. Am. Chem. Soc., 2003, 125, 5288–5289 CrossRef CAS.
- T. Terashima, M. Ouchi, T. Ando, M. Kamigaito and M. Sawamoto, Amphiphilic, thermosensitive ruthenium(II)-bearing star polymer catalysts: one-pot synthesis of PEG armed star polymers with ruthenium(II)-enclosed microgel cores via metal-catalyzed living radical polymerization, Macromolecules, 2007, 40, 3581–3588 CrossRef CAS.
- J. Dong, J. Tong, J. Luo and X. Liu, Gold nanoparticles for smart and recoverable catalyst using thermoresponsive core-crosslinked star polymer as the nanoreactor, Appl. Surf. Sci., 2020, 507, 144950–144960 CrossRef CAS.
- A. M. James, M. J. Derry, J. S. Train and R. Dawson, Dispersible microporous diblock copolymer nanoparticles via polymerisation-induced self-assembly, Polym. Chem., 2019, 10, 3879–3886 RSC.
- S. H. Yu, M. Patra, S. Ferrari, P. R. Garcia, N. A. Veldhuis, L. M. Kaminskas, B. Graham, J. F. Quinn, M. R. Whittaker, G. Gasser and T. P. Davis, Linker chemistry dictates the delivery of a phototoxic organometallic rhenium(I) complex to human cervical cancer cells from core crosslinked star polymer nanoparticles, J. Mater. Chem. B, 2018, 6, 7805–7810 RSC.
- D. Gu, K. Ladewig, M. Klimak, D. Haylock, K. M. McLean, A. J. O'Conor and G. G. Qiao, Amphiphilic core cross-linked star polymers as water-soluble, biocompatible and biodegradable unimolecular carriers for hydrophobic drugs, Polym. Chem., 2015, 6, 6475–6487 RSC.
- J. Liu, H. Duong, M. R. Whittaker, T. P. Davis and C. Boyer, Synthesis of functional core, star polymers via RAFT polymerization for drug delivery applications, Macromol. Rapid Commun., 2012, 33, 760–766 CrossRef CAS.
- J. Du and Y. Chen, PCL star polymer, PCL-PS heteroarm star polymer by ATRP, and core-carboxylated PS star polymer thereof, Macromolecules, 2004, 37, 3588–3594 CrossRef CAS.
- G. Zheng and C. Pan, Preparation of star polymers based on polystyrene or poly(styrene-b-N-isopropyl acrylamide) and divinylbenzene via reversible addition-fragmentation chain transfer polymerization, Polymer, 2005, 46, 2802–2810 CrossRef CAS.
- J. A. Syrett, D. M. Haddleton, M. R. Whittacker, T. P. Davis and C. Boyer, Functional, star polymeric molecular carriers, built from biodegradable microgel/nanogel cores, Chem. Commun., 2011, 47, 1449–1451 RSC.
- K. L. Thompson, J. M. A. Cockram, N. J. Warren, V. J. Cunningham, E. R. Jones, R. Verber and S. P. Armes, Are block copolymer worms more effective Pickering emulsifiers than block copolymer spheres?, Soft Matter, 2014, 10, 8615–8626 RSC.
- H. Ding, S. Park, M. Zhong, X. Pan, J. Pietrasik, C. J. Bettinger and K. Matyjaszewski, Facile arm-first synthesis of star block copolymers via ARGET ATRP with ppm amounts of catalyst, Macromolecules, 2016, 49, 6752–6760 CrossRef CAS.
- S. Qian, R. Liu, G. Han, K. Shi and W. Zhang, Star amphiphilic block copolymers: synthesis via polymerization-induced self-assembly and crosslinking within nanoparticles, and solution and interfacial properties, Polym. Chem., 2020, 11, 2532–2541 RSC.
- A. Blanazs, S. P. Armes and A. J. Ryan, Self-Assembled Block Copolymer Aggregates: From Micelles to Vesicles and their Biological Applications, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS.
- V. A. Davankov and M. P. Tsyurupa, Structure and properties of hypercrosslinked polystyrene – the first representative of a new class of polymer networks, React. Polym., 1990, 13, 27–42 CrossRef CAS.
- J.-H. Ahn, J.-E. Jang, C.-G. Oh, S.-K. Ihm, J. Cortez and D. C. Sherrington, Rapid generation and control of microporosity, bimodal pore size distribution, and surface area in Davankov-type hyper-cross-linked resins, Macromolecules, 2006, 39, 627–632 CrossRef CAS.
- L. Tan and B. Tan, Hypercrosslinked porous polymer materials: design, synthesis, and applications, Chem. Soc. Rev., 2017, 46, 3322–3356 RSC.
- Z. Li, D. Wu, X. Huang, J. Ma, H. Liu, Y. Liang, R. Fu and K. Matyjaszewski, Fabrication of novel polymeric and carbonaceous nanoscale networks by the union of self-assembly and hypercrosslinking, Energy Environ. Sci., 2014, 7, 3006–3012 RSC.
- T.-N. Gao, T. Wang, W. Wu, Y. Liu, Q. Huo, Z.-A. Qiao and S. Dai, Solvent-induced self-assembly strategy to synthesize well-defined hierarchically porous polymers, Adv. Mater., 2019, 31, 1806254–1806260 CrossRef.
- S. J. D. Smith, C. D. Wood, P. H. M. Feron, H. Mahdavi, R. J. Mulder, C. M. Doherty, M. R. Hill and X. Mulet, Hyper-cross-linked polymer liquids for gas purification, To be submitted Search PubMed.
- M. P. Tsyurupa, T. A. Mrachkovskaya, L. A. Maslova, G. I. Timofeeva, L. V. Dubrovina, E. F. Titova, V. A. Davankov and V. M. Menshov, Soluble intramolecularly hypercross-linked polystyrene, React. Polym., 1993, 19, 55–66 CrossRef CAS.
- Y. Yang, B. Tan and C. D. Wood, Solution-processable hypercrosslinked polymers by low cost strategies: a promising platform for gas storage and separation, J. Mater. Chem. A, 2016, 4, 15072–15080 RSC.
- W. Mai, B. Sun, L. Chen, F. Xu, H. Liu, Y. Liang, R. Fu, D. Wu and K. Matyjaszewski, Water-dispersible, responsive, and carbonizable hairy microporous polymeric nanospheres, J. Am. Chem. Soc., 2015, 137, 13256–13259 CrossRef CAS.
- Y. Xie, W. Huang, B. Zheng, S. Li, Q. Liu, Z. Chen, W. Mai, R. Fu and D. Wu, All-in-one porous polymer adsorbents with excellent environmental chemosensory responsivity, visual detectivity, superfast adsorption, and easy regeneration, Adv. Mater., 2019, 31, 1900104–1900112 CrossRef.
- R. Hou, R. O'Loughlin, J. Ackroyd, Q. Liu, C. M. Doherty, H. Wang, M. R. Hill and S. J. D. Smith, Greatly enhanced gas selectivity in mixed-matrix membranes through size-controlled hyper-cross-linked polymer additives, Ind. Eng. Chem. Res., 2020, 59, 13773–13782 CrossRef CAS.
- X. Yang, L. Tan, L. Xia, C. D. Wood and B. Tan, Hierarchical porous polystyrene monoliths from polyHIPE, Macromol. Rapid Commun., 2015, 36, 1553–1558 CrossRef CAS.
- J. Park, K. Kim and M. Seo, Hyper-cross-linked polymers with controlled multiscale porosity via polymerization-induced microphase separation within high internal phase emulsion, Chem. Commun., 2018, 54, 7908–7911 RSC.
- J.-S. M. Lee, M. E. Briggs, T. Hasell and A. I. Cooper, Hyperporous carbons from hypercrosslinked polymers, Adv. Mater., 2016, 28, 9804–9810 CrossRef CAS.
- N. Y. Ahn and M. Seo, Synthetic route-dependent intramolecular segregation in heteroarm core cross-linked star polymers as Janus-like nanoobjects, Polym. Chem., 2020, 11, 449–460 RSC.
- J. Park, N. Y. Ahn and M. Seo, Cross-linking polymerization-induced self-assembly to produce branched core cross-linked star block polymer micelles, Polym. Chem., 2020, 11, 4335–4343 RSC.
- T. J. Prosa, B. J. Bauer and E. J. Amis, From star to spheres: A SAXS analysis of dilute dendrimer solutions, Macromolecules, 2001, 34, 4897–4906 CrossRef CAS.
- J. Bang, S. Jain, Z. Li, T. P. Lodge, J. S. Pedersen, E. Kesselman and Y. Talmon, Sphere, cylinder, and vesicle nanoaggregates in poly(styrene-b-isoprene) diblock copolymer solutions, Macromolecules, 2006, 39, 1199–1208 CrossRef CAS.
- A. Yadigarli, Q. Song, S. I. Druzhinin and H. Schönherr, Probing of local polarity in poly(methyl methacrylate) with the charge transfer transition in Nile red, Beilstein J. Org. Chem., 2019, 15, 2552–2562 CrossRef CAS.
- O. G. Schramm, G. M. Pavlov, H. P. van Erp, M. A. R. Meier, R. Hoogenboom and U. S. Schubert, A versatile approach to unimolecular water-soluble carriers: ATRP of PEGMA with hydrophobic star-shaped polymeric core molecules as an alternative for PEGylation, Macromolecules, 2009, 42, 1808–1816 CrossRef CAS.
- J. Park, M. Moon, M. Seo, H. Choi and S. Y. Kim, Well-defined star-shaped rod–coil diblock copolymers as a new class of unimolecular micelles: encapsulation of guests and thermoresponsive phase transition, Macromolecules, 2010, 43, 8304–8313 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py01225d |
| This journal is © The Royal Society of Chemistry 2020 |