
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A chelate like no other: exploring the synthesis, coordination chemistry and applications of imidoyl amidine frameworks
Alexandros A.
Kitos
 ,
Niki
Mavragani
,
Muralee
Murugesu
and
Jaclyn L.
Brusso
*
,
Niki
Mavragani
,
Muralee
Murugesu
and
Jaclyn L.
Brusso
*
Department of Chemistry and Biomolecular Sciences, University of Ottawa, 10 Marie Curie, Ottawa, Ontario K1N 6N5, Canada. E-mail: jbrusso@uottawa.ca; m.murugesu@uottawa.ca
First published on 7th November 2020
Abstract
Since the remarkable work by Werner, the design and use of chelating ligands have played an essential part in coordination chemistry. While ligands such as 2,4-pentanedione (acac) and 1,5-diazapentadienyl (NacNac) have been established in the literature for over a century and their rich coordination chemistry is well known, 1,3,5-triazapentadiene ligands remain the significantly less explored members of this family. Also known as imidoyl amidine (ImAm), the coordination chemistry of this ligand framework has recently attracted a great deal of attention due to its multiple binding sites, ability to be incorporated into organic and inorganic materials, and diverse applicability (catalysis, biomolecular probes, materials science, etc.) making them a class of chelates like no other. The scope of this review is to provide a critical overview of the research progress on the chemistry of ImAm as well as complexation with different metal ions. General synthetic routes and new insights for the preparation of ImAm are discussed along with their use as starting materials or intermediates in organic and coordination chemistry in the development of materials that have found use in fields ranging from magnetic, conducting and luminescent materials to catalysis and biomedical applications. Concluding remarks including the limitations and perspectives of ImAm are discussed.
 Alexandros A. Kitos | Alexandros A. Kitos holds an MSc in Analytical Chemistry-Nanotechnology and a PhD in Chemistry from University of Patras, Greece. In August 2019, he joined the groups of Profs M. Murugesu and J. Brusso as a postdoctoral fellow at the University of Ottawa. His ongoing research explores enhancing the magnetic (SMMs, SCO, etc.), luminescence and gas sorption properties of hybrid materials. |
 Niki Mavragani | Niki Mavragani received her Bachelor and Master's degree in chemistry at the University of Patras, Greece. She is a PhD student since January 2019 under the supervision of Prof. M. Murugesu in the Department of Chemistry and Biomolecular Sciences, University of Ottawa. Her research field includes the design and synthesis of advanced radical-based coordination and organometallic complexes for magnetic applications. |
 Muralee Murugesu | Muralee Murugesu received his PhD from the University of Karlsruhe in 2002 under the supervision of Prof. A. K. Powell. He undertook postdoctoral stays at the University of Florida (2003–2005) with Prof. G. Christou, and jointly at the University of California, Berkeley and the University of California, San Francisco under the supervision of Prof. J. R. Long and the Nobel Laureate Prof. S. Pruissner (2005–2006). In 2006, he joined the University of Ottawa as an assistant professor and since 2015 he is a full professor. Prof. Murugesu's research focuses on the design and development of single-molecule magnets, metal–organic frameworks and high-energy materials. |
 Jaclyn L. Brusso | Jaclyn Brusso obtained her PhD from the University of Waterloo under the direction of R. T. Oakley in 2006. Following an NSERC Postdoctoral Fellowship with D. Perepichka at McGill University and then with S. Holdcroft's group at Simon Fraser University, Jaclyn joined the University of Ottawa in 2010. Jaclyn's research program focuses on the design and development of novel organic/inorganic based optical, magnetic and electronic materials. |
1. Introduction
Classical β-diketones or 1,3-diketones (R1COC(R2)COR1) represent one of the most valuable classes of ligands for metal ion chelation (Scheme 1). They have been studied for more than a century and their rich coordination chemistry is well explored along with their industrial applications.1 Substitution of the central α-carbon in R1COC(R2)COR1 for nitrogen affords the bis(carbonyl)amide ligand (bca; Scheme 1), which has been employed in coordination complexes for a variety of applications in materials chemistry including single-chain magnets, spin crossover (SCO) materials, metal–organic frameworks (MOFs) and luminescent complexes.2 Since the earliest development of the coordination chemistry of dinitrogen analogues of 1,3-dicarbonyl compounds (i.e., 1,5-diazapentadienyl, β-diketiminates or NacNac) by Bradley3 and Holm,4 a great deal of research has focused on the use of NacNac leading to it becoming one of the most employed monoanionic nitrogen-based ligands in the field (Scheme 1).5 Indeed, NacNac ligand frameworks have been much less explored compared to the R1COC(R2)COR1 analogues but their complexes show various applications in many fields.6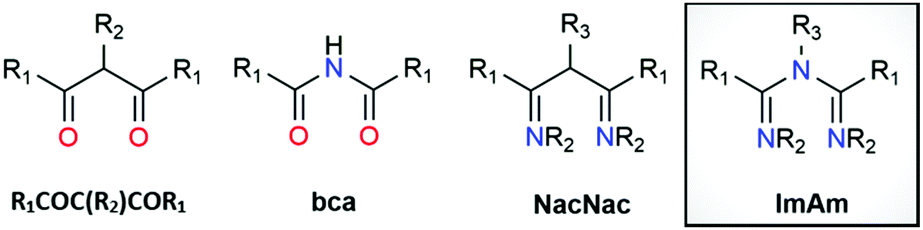 | ||
| Scheme 1 Structure of 1,3-diketones (R1COC(R2)COR1), bis(carbonyl)amide (bca), 1,3-diketoiminate (NacNac) and imidoyl amidine (ImAm). | ||
The less explored member of this family (compared to R1COC(R2)COR1, bca and NacNac) is the trinitrogen analogue of the 1,3-dicarbonyls, i.e., 1,3,5-triazapentadiene or imidoyl amidine (Scheme 1). While both terms are equally applied in literature, for the purposes of this article the latter (imidoyl amidine) will be used exclusively with the corresponding abbreviation ImAm. The presence of an additional N-donor atom (compared to NacNac) in ImAm has triggered the attention of coordination chemists. In addition to the N-N bidentate chelate coordination mode (also known as U-shape fashion) of NacNac, ImAm ligands can also coordinate in a tridentate fashion (W-shape) when donor atoms are incorporated onto the substituents at the β-carbon atoms or act as linkers between metal ions in their deprotonated/anionic form.7 The ability of these frameworks to adopt multiple coordination modes (vide infra) forming stable five- and six-membered chelate rings, can be exploited to efficiently target coordination to various metal ions including heterometallic systems. Moreover, the central N-atom can easily switch between its protonated and deprotonated form based on the pH conditions and act as an “on–off” switch, particularly attractive for luminescent applications.8 Computational studies of trinuclear heterometallic coordination complexes of FeII with ZnII, NiII, CuII or CoII, based on ImAm ligands with pyrazole functional groups, revealed that weak exchange interactions are expected to occur between the metal centers, undergoing a two-step SCO process.9 Another interesting feature of the ImAm framework is the ability to functionalize the carbon atoms (R1) and/or the nitrogen atoms (R2 and R3), leading to a vast array of organic molecules with desirable properties. Despite these advantages and the potential of ImAm ligands, complexes containing a pre-synthesized ImAm are rather rare compared to other 1,3-dicarbonyl derivatives and analogues. This can be attributed to the challenge associated with isolating non-coordinated ImAm frameworks, which until recently21 the synthesis was poorly developed.
Having established facile synthetic methodology for the isolation of ImAm chelates and their derivatives, enables one to tap into the potential of this versatile framework. To that end, the scope of the present review is to provide new insights into the preparation of ImAm along with its usage as building blocks in organic chemistry and employment as ligands in coordination chemistry. Our aim is to cover the recent research progress on the chemistry and applications of ImAm, excluding biguanidine and related derivatives as they have already been reviewed10 and will therefore not be addressed herein.
2. Brief introduction to the organic chemistry of the imidoyl amidine frameworks
2.1 Tautomerism of imidoyl amidines
Imidoyl amidines are multifunctional, nitrogen-rich molecules bearing formally fused amidine, imide and amine groups forming a characteristic unsaturated N–C–N–C–N chain. As such, ImAm can exist in two main tautomeric forms, namely 1,3,5-triaza-1,3-pentadienes (i.e., amino–imino; Scheme 2a) and 1,3,5-triaza-1,4-pentadienes (i.e., imino–imino; Scheme 2b), depending on the position of the double bond. Upon coordination of deprotonated ImAm ligands to a metal center, delocalization of the double bond system over the entire molecule occurs11 while the C–N bonds are usually localized in the neutral form.12 Based on the number of hydrogen atoms attached to the nitrogen atoms, ImAm can be classified as primary (three N–H groups), secondary (two N–H groups), tertiary (one N–H group) or quaternary (where no N–H groups are present).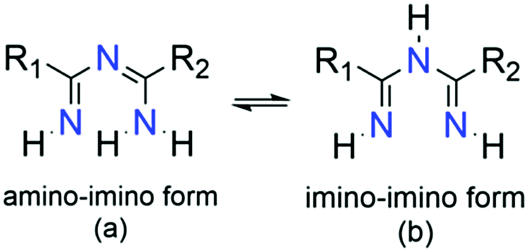 | ||
| Scheme 2 The 1,3,5-triaza-1,3-pentadienes or amino–imino (a) and 1,3,5-triaza-1,4-pentadienes or imino–imino (b) tautomeric forms of ImAm ligands. | ||
2.2 Synthetic approaches for the development of imidoyl amidines
The majority of literature references that contain ImAm involves their preparation through metal assisted reactions (vide infra) with only a few examples of the synthesis of such organic molecules without metal mediated transformations.13–21 Thus far, reported methods for the preparation of ImAm chelates mostly lead to heavily N-substituted ImAm with bulky substituents at the terminal nitrogen atoms and/or strong electron-acceptor functional groups at the carbon atoms, as shown in Scheme 3.13 Since these synthetic routes have already been discussed in detail by Kopylovich and Pombeiro,14 herein we will only focus on the main advantages and disadvantages of each of these procedures. For example, the preparation of non-coordinated ImAmvia the Pinner reaction15 (Scheme 3a) leads to rather low yields and a large number of by-products, making this a relatively inefficient preparative route. A more versatile and simplified method proposed by Ley and Muller16 (Scheme 3b) leads to relatively good yields and provides a pathway towards the isolation of a large number of ImAm (where R1, R2, R3 can be either phenyl groups or different substituted phenyl groups). As highlighted in Scheme 3c step I, N-imidoylimidoates can be obtained by the reaction of imidoate hydrochlorides with N-imidoyl chlorides under basic conditions and, upon further reaction with a primary or secondary amine (Scheme 3c step II), tertiary or quaternary ImAm frameworks can also be isolated. This was well demonstrated by Häger et al. for the synthesis of tertiary and quaternary ImAm ligands and their complexation with various transition metal ions as well as boron difluoride.13c Alternative synthetic routes involve the preparation of N-tiobenzoylbenzamidines proposed by Titherley and Hughes,17 which are then converted to the corresponding ImAm by the desulfurization reaction with an amine or amidine (Scheme 3d).15 Condensation of perfluorocarbon (or perchloro-) activated amidines with similarly activated nitriles (Scheme 3e, reaction I) or by the reaction of a perfluoroalkylnitrile with an excess of anhydrous ammonia (Scheme 3e, reaction II) affords halide substituted ImAm.18 The main disadvantage of these substituted ImAm frameworks is their rather unstable nature, which leads to cyclization thereby converting them to the corresponding triazines.19 An alternative procedure for the synthesis of ImAm containing perfluoro moieties involves the reaction of perfluoro-5-aza-4-nonene with primary amines (Scheme 3f).20 This synthetic route gives access not only to halide substituted ImAm but also to N-substituted ImAm, depending on the initial amine used for the synthesis.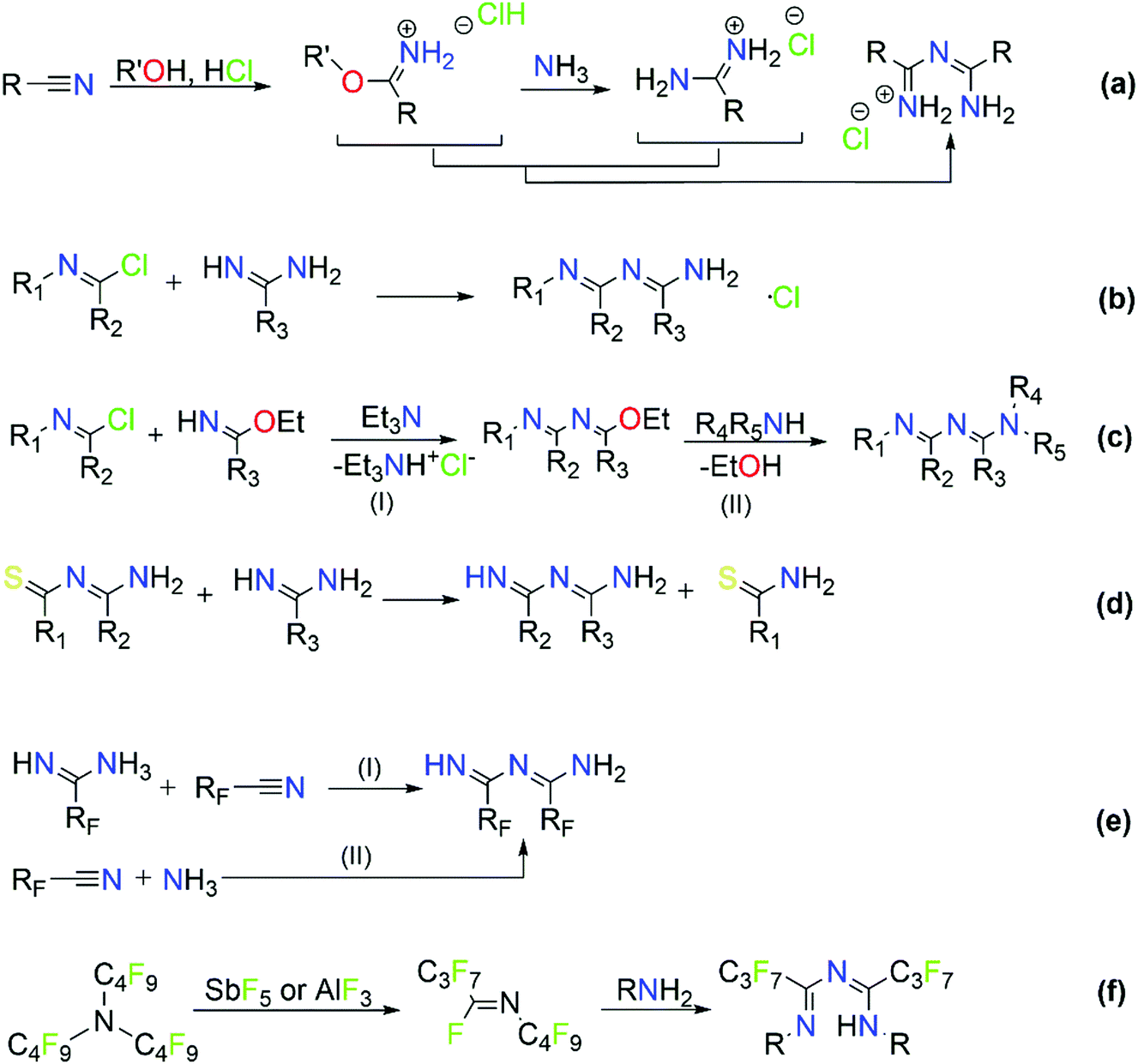 | ||
| Scheme 3 Common synthetic routes for the synthesis of non-coordinated ImAm frameworks. | ||
As mentioned, these preparative routes typically afford heavily N-substituted ImAm with bulky substituents at the terminal nitrogen atoms and/or strong electron-acceptor functionalities at the carbon atoms. Additionally, the overall yields reported are poor and, in most cases, the starting materials are not commercially available, thus adding more steps to their synthesis. Recently, we developed a high yielding one-pot metal free synthetic procedure for the preparation of carbon substituted ImAm.21 This procedure gave access to the isolation of N-2-pyridylimidoyl-2-pyridylamidine (Py2ImAm) and N-2-pyrimidylimidoyl-2-pyrimidylamidine (Pm2ImAm), starting from commercially available 2-pyridinecarbonitrile and 2-pyrimidinecarbonitrile, respectively (Scheme 4). The synthesis of both compounds involves the reaction of ammonia gas with the appropriately substituted nitriles. In the case of Py2ImAm, the reaction is carried out under increased pressure affording a 63% yield. The Pm2ImAm is isolated by allowing 2-pyrimidinecarbonitrile to react slowly with ammonia over a period of two weeks at room temperature and under atmospheric pressure resulting in a 76% yield. Encouragingly, the versatility of this procedure enables the preparation of various ImAm in high yields depending on the substituted nitriles in use. With that said, attempts to synthesize N-2-thienylimidoyl-2-thienylamidine (Th2ImAm) by reacting 2-cyanothiophene with NH3(g) led to negligible yields of the desired product regardless of the temperature or pressure conditions used.35d This was overcome by treating 2-thienylamidine with sodium hydride thereby strengthening the nucleophilicity of the amidine, which subsequently reacts with 2-cyanothiophene to form Th2ImAm in high yields and purity (Scheme 4).
 | ||
| Scheme 4 Synthesis of Py2ImAm, Pm2ImAm (top) and Th2ImAm (bottom). Reagents and conditions: (a) 2-cyanopyridine, acetonitrile (MeCN), 110 °C; (b) NH3, MeCN, RT; (c) (i) NaOMe, MeOH, RT; (ii) NH4Cl; (d) NaH, 2-thiophenecarbonitrile, DMSO, RT. | ||
2.3 Imidoyl amidines as building blocks in organic materials
Although their synthesis has not been widely investigated, ImAm moieties have attracted interest as building blocks in the development of new organic frameworks. For example, ImAm have been successfully used as intermediates in the formation of triazines19 and as starting materials for the synthesis of aminotriazines (Scheme 5).22 In the past few decades these two classes of compounds have been extensively studied in various fields, such as medicinal chemistry due to their excellent antitumor and anti-inflammatory activity,22,23 in coordination and materials chemistry24 as ligands for the preparation of Metal Organic Frameworks (MOFs) with significant CO2 capture capabilities,25 as well as in catalysis.26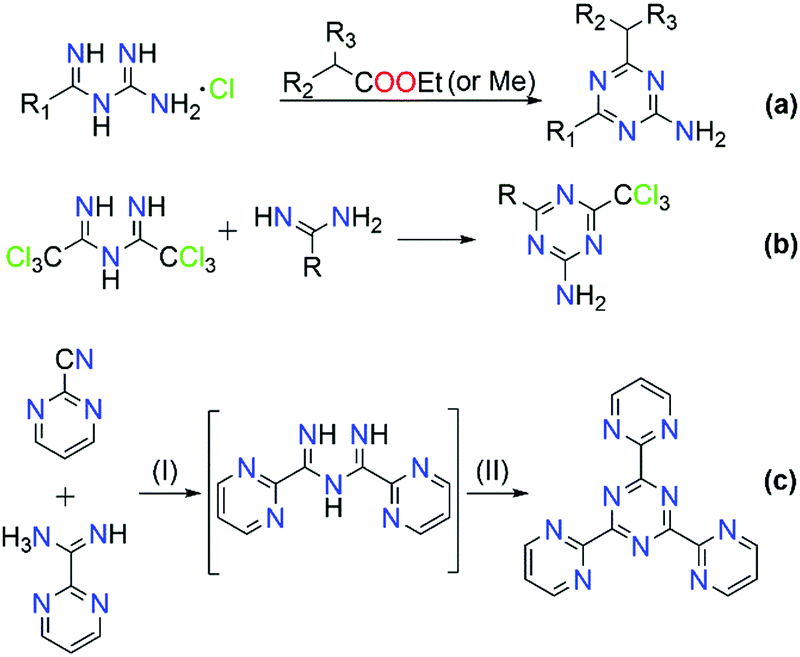 | ||
| Scheme 5 Synthesis of triazines and aminotriazines using ImAm as starting material. Reagents and conditions on (c): (I) NH3, acetonitrile (MeCN), 110 °C, 72 h; (II) MeCN, 110 °C, 120 h. | ||
Additionally, ImAm moieties have been successfully used as starting materials for the preparation of boratriazines (BTA). Complexation of ImAm with boron leads to a new class of materials, similar to those of boron dipyrromethene (BODIPY), which have been studied for their sharp fluorescence peaks,27 high thermal resistance,28 strong UV-absorption29 and excellent stability.29 Recently, the incorporation of boron into ImAm frameworks was reported, yielding compounds which may be considered as unfused analogues to the extensively studied BODIPY dyes.21b In Scheme 6, the general synthetic route for the synthesis of 2,2-difluoro-4,6-bis(2-pyridyl)-1,3-dihydro-1,3,5,2-triazaborinine (Py2F2BTA) and 2,2-difluoro-4,6-bis(2-pyrimidinyl)-1,3-dihydro-1,3,5,2-triazaborinine (Pm2F2BTA) is illustrated. Furthermore, the tridentate (NNN) coordination site in Py2F2BTA and Pm2F2BTA is expected to promote coordination through the chelating effect to a metal center.30a Taking advantage of the ligating properties of these boratriazines, a series of Py2F2BTA and Pm2F2BTA coordination complexes with iron and cobalt salts that exhibited interesting magnetic properties (i.e. strong ferromagnetic interactions and field induced Single-Molecule Magnet (SMM) behavior) were reported.30 As shown, it is possible to maintain the chelating ability of these organic ligands while tuning the physical properties of the construct. This approach may give access to interesting photocatalytic activity thanks to the photoactive features of the system and careful choice of the metal ion.
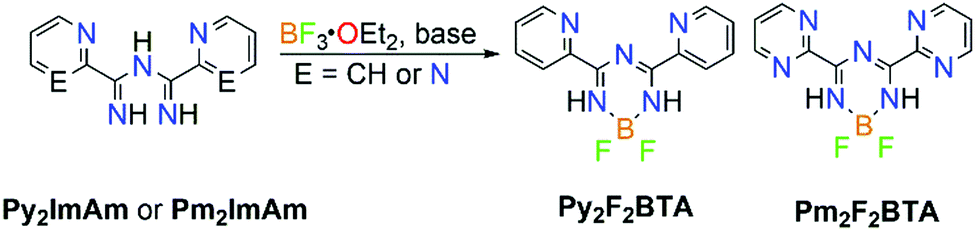 | ||
| Scheme 6 General synthetic route used for the synthesis of 2,2-difluoro-4,6-bis(2-pyridyl)-1,3-dihydro-1,3,5,2-triazaborinine (Py2F2BTA) and 2,2-difluoro-4,6-bis(2-pyrimidinyl)-1,3-dihydro-1,3,5,2-triazaborinine (Pm2F2BTA). Reagents and conditions: (Py2F2BTA) triethylamine (Et3N), tetrahydrofuran (THF), 60 °C; (Pm2F2BTA) Et3N, dichloromethane (DCM). | ||
Organic radicals represent another field in which ImAm have been employed as starting materials, in particular for the synthesis of thiatriazinyls (TTA).31 In 1984, Oakley and co-workers first isolated and structurally characterized 3,5-bis-(phenyl)-1,2,4,6-thiatriazinyl (Ph2TTA) triggering the scientific interest for the preparation of such molecules.31b Commonly, 1,2,4,6-thiatriazinyls can be prepared from the reaction of trichlorocyclotrithiazene with amidines32 or from the reaction of ImAm with excess SCl2.33 Boeré and coworkers proposed a general synthetic route for the synthesis of asymmetrical 5-aryl-3-trifluoromethyl-1,2,4,6-thiatriazinyls where ImAm treated with SCl2 affords S-chlorothiatriazines, which can then be reduced with Ph3Sb yielding the corresponding radicals (Scheme 7).34 Recently, we developed the synthetic methodology for the preparation of symmetrical TTA such as 3,5-dipyridylthiatriazinyl (Py2TTA) and bis-(2-thienyl)-1,2,4,6-thiatriazinyl (Th2TTA).35 In the case of Py2TTA, reaction of Py2ImAm with S2Cl2 affords the thiadiazolium dication, which upon treatment with Ph3Sb undergoes a two-electron reduction affording Py2TTAH. Upon oxidation of Py2TTAH with N-chlorosuccinimide (NCS), in the presence of base, the radical form of Py2TTA is isolated (Scheme 7). In their crystal packing, Py2TTA and Th2TTA form dimeric species via S⋯S contacts at ∼2.6 Å. Electron paramagnetic resonance (EPR) spectroscopic studies on symmetrical and asymmetrical TTA revealed that the unpaired spin density is localized on the heterocyclic TTA ring. Additionally, cyclic voltammetric studies suggested that there is a monomer–dimer equilibria in solution.34,35 Lastly, the coordination chemistry of Py2TTA has been explored, affording complexes with transition metal ions and lanthanides that exhibit interesting magnetic properties (i.e. SMM behavior), with the ligand in either the anionic or oxidized form.36
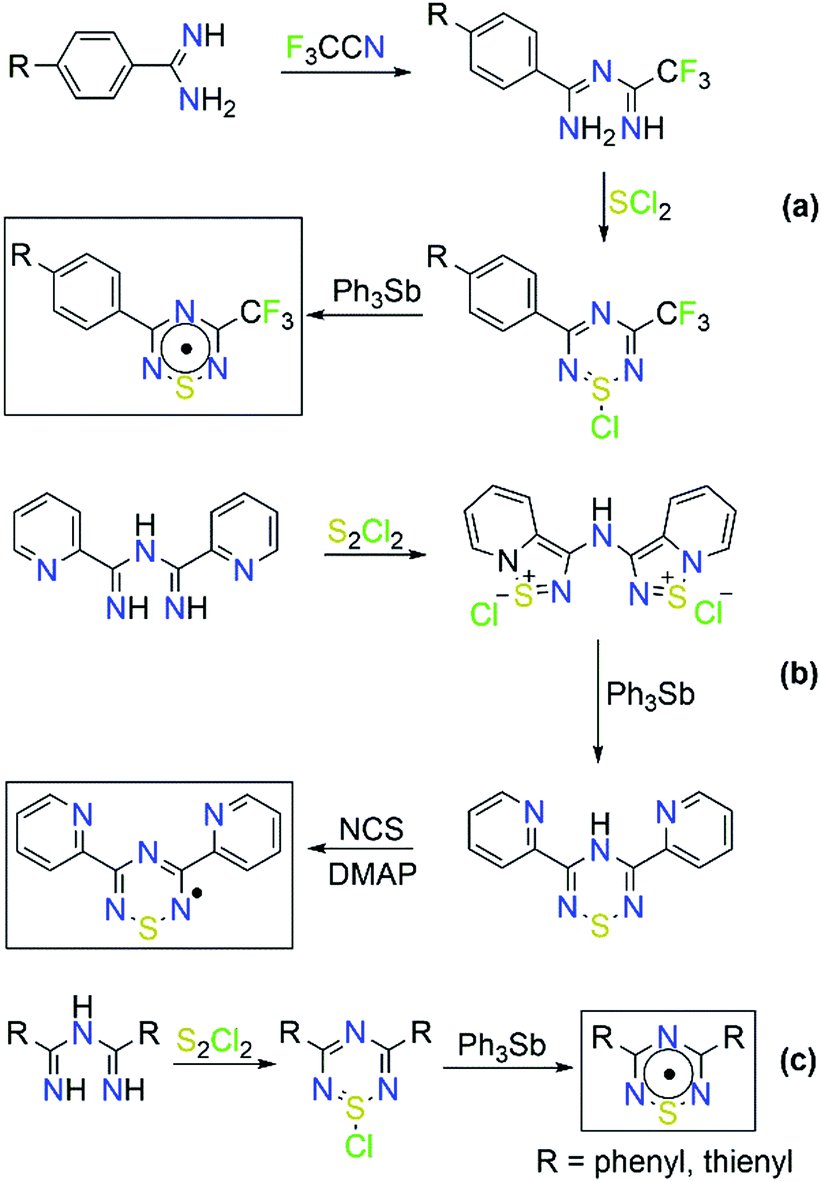 | ||
| Scheme 7 General synthetic routes for the synthesis of (a) asymmetrical 5-aryl-3-trifluoromethyl-1,2,4,6-thiatriazinyls, (b) symmetrical 3,5-dipyridylthiatriazinyl (Py2TTAH) and 3,5-bis-(phenyl)-1,2,4,6-thiatriazinyl (Ph2TTA), and (c) 3,5-bis-(2-thienyl)-1,2,4,6-thiatriazinyl (Th2TTA). | ||
2.4 Metal-assisted formation of imidoyl amidine ligands
From a historic perspective, the first ImAm complex was reported in 1984.37 The methylimidoyl-methylamidine ligand was generated in situ via a metal assisted transformation, when acetamidine was reacted in the presence of nickel metal ions. Subsequent work led to other metal assisted transformations to afford ImAm frameworks, such as the metal-mediated hydrolytic decomposition of oximes38a,b or triazines,38c,d the nucleophilic addition to dicyanamide,38e the template condensation of nitriles and/or amidines at a metal center,38f–i as well as in situ reactions of a metal ion and a nitrile under solvothermal conditions.38j In 2011, a review by Kopylovich and Pombeiro14 covered this topic in an excellent way. Thus, all these synthetic approaches will not be described herein in detail. In the following sections, coordination complexes and significant progress in the field of the last decade will be covered.3. The coordination chemistry of symmetrical imidoyl amidine chelates
While the coordination chemistry of ImAm is significantly influenced by the R1, R2 and R3 substituents, functionalization at the central nitrogen atom (i.e., R3) is rare with this site either protonated or involved in coordination to transition metal ions. The R1 and R2 groups, on the other hand, can either contribute to the coordination sphere of the metal ion by having donor atoms such as oxygen, nitrogen or sulfur, or not contribute at all. Furthermore, the presence of aryl-, aromatic or halogen-rich moieties are likely to stabilize discrete coordination complexes by forming stronger supramolecular networks (via H-bond, π–π interactions, halogen bonds, etc.). In general, there are three coordination modes that most commonly appear in the literature (Scheme 8). The U-shaped coordination mode allows for the ImAm ligand to act as a bidentate chelate forming a six-membered ring, which represents the most common coordination mode of ImAm based complexes and has yielded a large number of mononuclear complexes. On the other hand, ImAm can also act as ligands either in the W-shape (through the middle N atom) or terminal mode (through the peripheral N atom), leading mostly to mononuclear complexes. By utilizing both the U- and W-shape coordination modes, the syn–anti and syn–syn bridging motifs have led to the isolation of polynuclear complexes. The main difference between these two coordination modes is that the middle nitrogen atom in the case of the syn–syn bridging mode coordinates to two metal centers, while in the case of syn–anti, it binds to only one metal ion.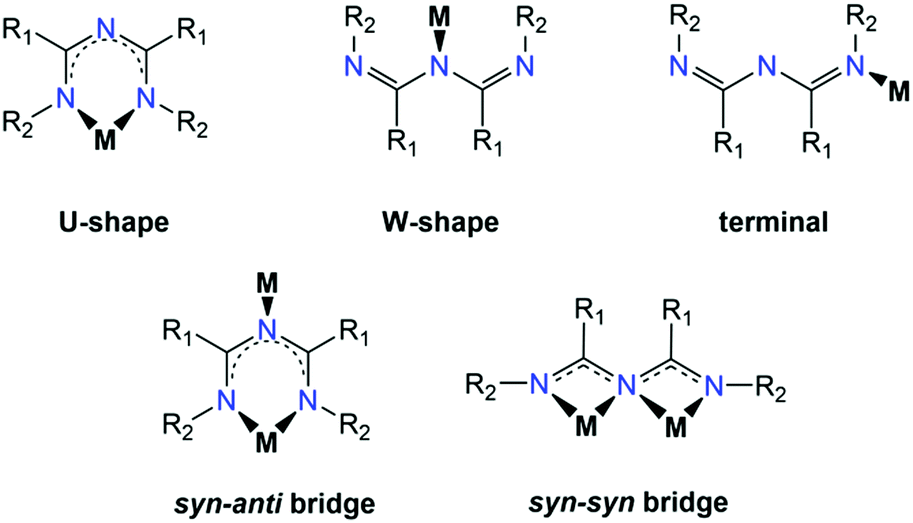 | ||
| Scheme 8 The crystallographically established coordination modes of ImAm. | ||
3.1 ImAm chelates without additional donor atoms in the substituents
In Scheme 9, ImAm chelates with no additional coordinating groups such as H, Me, Ph, CF3, C6F5, C3F7, etc. are presented. The synthesis of the dimethyl ImAm ligand (L1) was achieved in situ in the presence of a NiII metal center.39 Although deprotonation of the central N-atom can be relatively facile, depending on the reaction conditions, it is not always achieved. This was aptly illustrated in the work of Thiele et al. where they obtained the mononuclear cationic complex [NiII(L1)2]·Cl (1) in which one of the L1 ligands was neutral while the other was anionic (Fig. 1).39b By changing the metal source from NiCl2 to NiSO4 enabled isolation of a mononuclear NiII complex, where both ligands were in their neutral form.39b In both complexes, the ligand acts as a chelate stabilizing the 3d metal ion in a slightly distorted square planar geometry. The same group also reported a similar square planar mononuclear NiII complex with in situ preparation of L2 in which both ligands are deprotonated.39b | ||
| Scheme 9 Examples of ImAm ligands that do not possess additional donor atoms in the R1, R2 or R3 groups. | ||
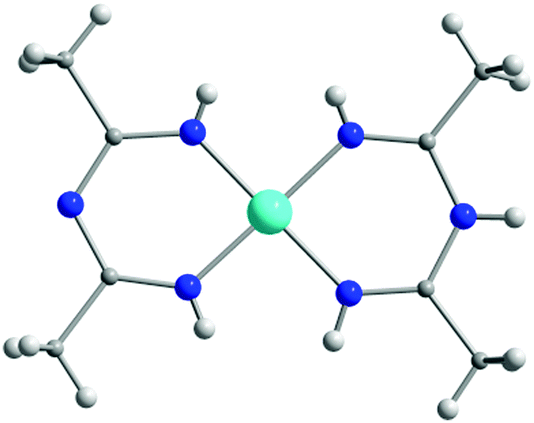 | ||
| Fig. 1 The molecular structure of the cationic complex 1,39b where the L1 ligand exists in both its neutral and anionic form. Lattice solvent molecules and counter ions have been omitted for clarity. Colour code: C: grey, H: white, N: blue, NiII: light blue. Reproduced using reported cif. | ||
Replacement of hydrogen as the R2 substituent by bulkier aryl or aromatic groups can lead to structurally interesting ImAm ligands such as L3, L4 and L5. Pernik et al. employed these bulky frameworks in the formation of mononuclear and dinuclear complexes of LiI, KI, CaII and MgII.40 In contrast to the previous examples, in this work the ligands were pre-synthesised prior to reactions with metal salts. In all these complexes, the deprotonated ligands act in the U-shaped coordination mode. In a similar manner, the H-atom of the R3 group can also be replaced, as demonstrated by Bolaño et al.41 They reported the in situ synthesis of the L6 ligand upon coordination to a RhIII center leading to the formation of the trication [(tbpy)2-RhIII(L6)][OTf]3 (2) (tbpy = 4,4′-di-tert-butyl-2,2′-bipyridine; OTf = CF3SO3−; Fig. 2), representing one of the few examples of ImAm-based complexes where the central N-atom is substituted. In 2, the RhIII metal center has a slightly distorted octahedral geometry with two tbpy and one L6 ligand completing the coordination sphere.
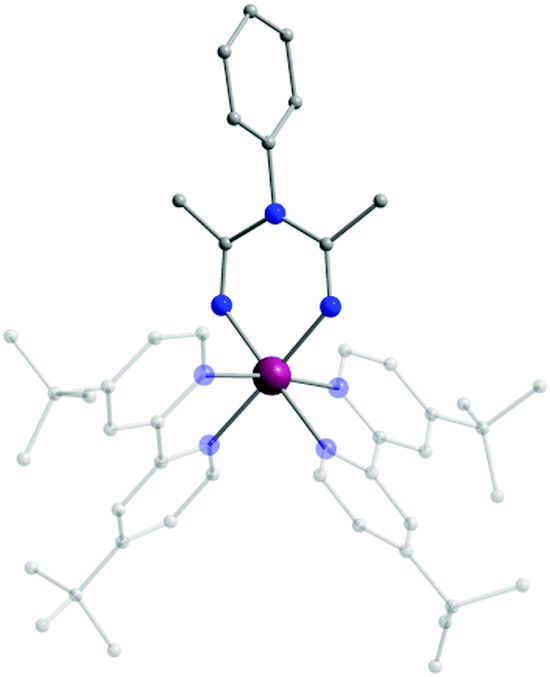 | ||
| Fig. 2 The molecular structure of the cationic complex 2.41 For clarity reasons H-atoms, disorder conformers and lattice counter ions have been omitted, as well as partial transparency has been employed. Colour code: C: grey, N: blue, RhIII: purple. Reproduced using reported cif. | ||
Replacement of the H-atoms in the methyl groups of L1 with chlorides affords the ligand L7, which was also prepared in situ as reported by Shixaliyev et al. where they isolated a family of mononuclear square planar complexes of the generalized type {MII[L7]2}·2(Me2SO), where M = NiII (3), CuII (4) or PdII (5) (Fig. 3).42 Strong H-bonds of the form N–H⋯O (where O belongs to a dimethylsulfoxide (Me2SO) lattice solvent molecule) as well as Cl⋯Cl halogen bonds create an elaborate 2D supramolecular network. Due to the chemical and thermal stability as well as enhanced solubility in halocarbons (i.e., dichloromethane, chloroform, chlorobenzene, etc.) that these halogenated ImAm complexes exhibit, the researchers explored the catalytic properties of similar mononuclear complexes of L7 of the general formula [MIII(L7)3]·x(Me2SO), where M = MnIII (6) with x = 1; FeIII (7) with x = 2; and CoIII (8) with x = 2.43 As in the previous study, 6–8 were prepared using a one pot synthetic procedure. In each complex, three L7 ligands are coordinated to the metal center in a bidentate fashion leading to a slightly distorted octahedral geometry about the metal ions. As previously observed, the H-atoms of the amine groups were engaged in strong H-bonding with a Me2SO lattice solvent molecule and halogen bonds lead to the formation of a supramolecular organization. These complexes were tested as catalyst precursors for the additive-free microwave (MW) assisted homogenous oxidation of secondary alcohols (such as 1-phenylethanol) to the corresponding ketones by aqueous tert-butylhydroperoxide (TBHP) or H2O2 without an added solvent. Although the MnIII and FeIII complexes showed notable catalytic properties, the best yields were achieved for the CoIII analogue.
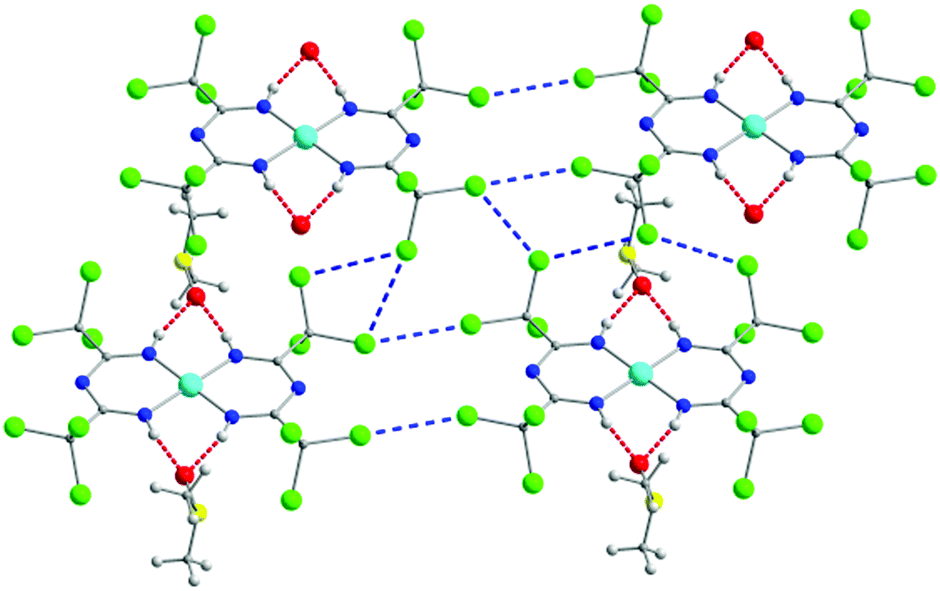 | ||
| Fig. 3 The 2D supramolecular organization of 3.42 Cl⋯Cl interactions are represented as dashed blue lines, while H-bonds as dashed red lines. Colour code: C: grey, N: blue, O: red, Cl: green, NiII: light blue. Reproduced using reported cif. | ||
The introduction of bulkier groups such as chlorobenzene (i.e., ligand L8) in the R1 groups was achieved by Kopylovich et al. in a one pot synthesis starting with halogenated benzonitriles in the presence of PdCl2.44 This led to the formation of the cationic [PdII(L8)2]2+ (with the ligand L8 being in the neutral form) and the neutral square planar PdII complex [PdII(L8)2] (9, where both ligands are in the monoanionic form).
The presence of halogen atoms in both the R1 and R2 groups of ImAm has inspired the synthesis of ligands with rich halogen content due to their potential catalytic applications.42–45 To that end, ligands such as L9, L10, L11 and L12 have been prepared and, upon reaction with CuI sources, have led to the isolation of mononuclear complexes. In the work of Ponduru et al., [CuI(L9)MeCN] (10) was isolated upon treatment of L9 with CuO in acetonitrile.45a In 10, the ligand acts in a chelating fashion in the U-shape coordination mode and the copper center adopts a trigonal planar geometry (Fig. 4). This complex was tested as a possible catalyst for the oxidation of anthracene, naphthalene and pyrene to the corresponding quinones by usage of H2O2 as an oxidant under mild conditions without the presence of an acid co-catalyst. Similar work was also published by the same group utilizing L10, L11 and L15 as ligands, which led to the isolation of analogous mononuclear complexes of CuI.45b
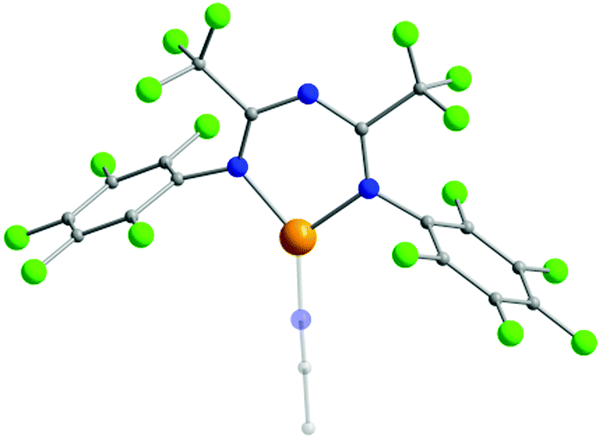 | ||
| Fig. 4 The molecular structure of the cationic complex 10.45a For clarity reasons H-atoms and disorder conformers have been omitted, as well as partial transparency has been employed. Colour code: C: grey, N: blue, F: green, CuI: orange. Reproduced using reported cif. | ||
Despite the prominence of U-shape chelating coordination mode that these ligands exhibit, other coordination modes have also been reported. In the work of Dias et al. a series of mononuclear CuI and AgI complexes were isolated.46 In [AgI(L9)(CNtBu)2] (11), the L9 ligand coordinates in a terminal W-shaped fashion through the central N-atom with the three-coordinate Ag metal center adopting a distorted trigonal planar geometry (Fig. 5a). Changing from L9 to L12, which possesses bulkier R1 substituents, leads to the formation of the isostructural AgI complex [AgI(L12)(CNtBu)2] (12). By addition of triphenylphosphine to Ag2O in the presence of L9 and L12, two isostructural complexes of AgI [AgI(L9)(PPh3)2] (13) and [AgI(L12)(PPh3)2] (14) were isolated. In these complexes both ligands act in a terminal fashion through one of the terminal N-atoms, while the three-coordinate AgI metal center is in a distorted trigonal planar geometry (Fig. 5b). Apart from these, they also reported a mononuclear CuI complex, [CuI(L9)(CNtBu)2] (15) with L9, where the ligand binds to the metal in a U-shape manner as previously illustrated. In 15, the metal center is four-coordinate in a slightly distorted tetrahedral geometry. This work highlights the versatility of ImAm ligands, which can bind to metal ions in several different manners. Additionally, computational analysis at the PM3 level of theory on the U- and W-shape coordination motifs has shown that these are the lowest energy conformations.46 Moreover, due to the almost evenly distributed electron density among the three N-atoms within the HOMO, these ligands are flexible enough to adapt relatively easily by altering their coordination mode in order to placate any steric-electronic demands of the coordination fragment.
 | ||
| Fig. 5 The molecular structure of the cationic complex 11 (a) and 14 (b).46 For clarity reasons H-atoms and disorder conformers molecules have been omitted, as well as partial transparency has been employed. Colour code: C: grey, N: blue, F: green, AgI: light blue. Reproduced using reported cifs. | ||
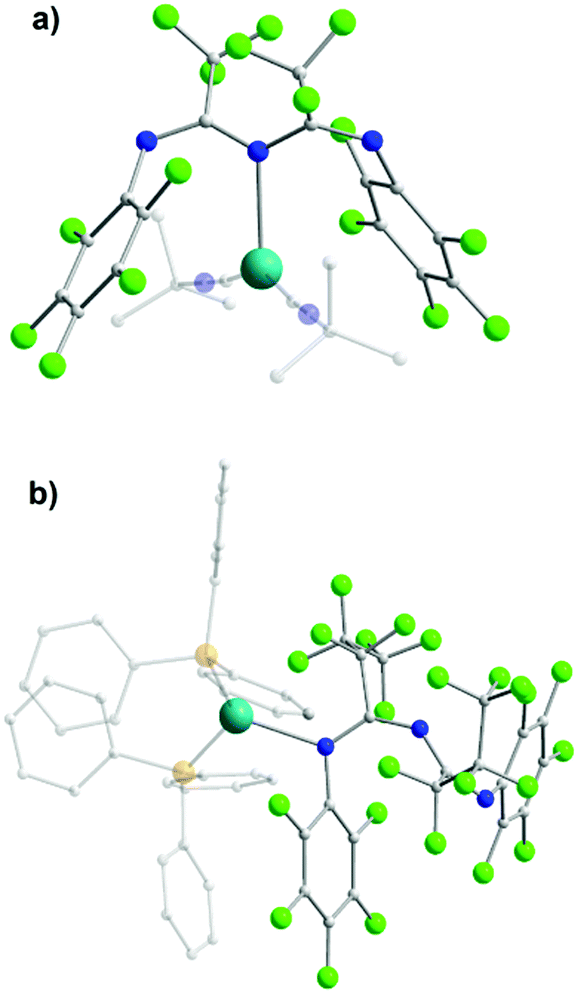
The coordination possibilities of ImAm ligands that bear non coordinating groups was fully explored by Bakthavachalam et al. in a series of LiI and AlIII complexes with L13.47 They illustrated that by employing various reaction conditions, mono- and dinuclear complexes of LiI and AlIII metal centers could be isolated (Scheme 10). Due to structural similarities only three of the complexes are discussed in detail. In complex [L13AlMe2(AlMe3)] (16) the L13 ligand acts as a syn–anti tridentate bridging moiety by utilizing all its donor atoms. This is the only example where the ImAm ligand acts as a syn–anti bridge when the R1 groups do not possess donor atoms. Additionally, by using LiI instead of AlIII, the authors were able to isolate the dinuclear LiI complex [(L13Li)2] (17) from Et2O. In 17, each LiI ion is bound to four N-atoms in a highly distorted tetrahedral geometry with both ligands adopting a syn–syn W-shape coordination motif, which stabilizes the two LiI metal centers. In contrast, using THF instead of Et2O a mononuclear LiI complex has been obtained, [L13Li(THF)3], where L13 coordinated in the terminal fashion through only one of the N atoms.
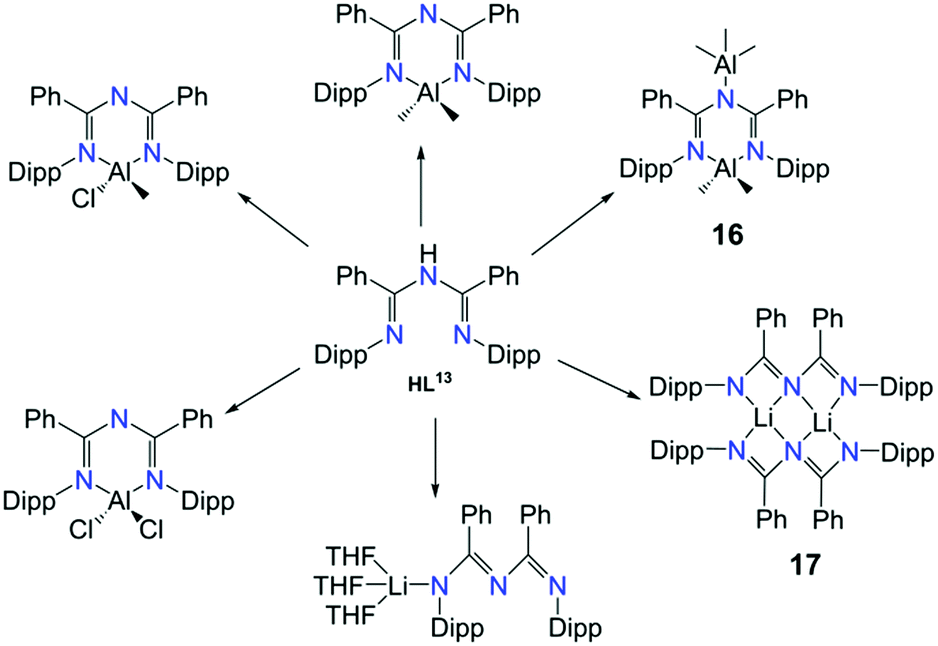 | ||
| Scheme 10 The synthesis of mononuclear and dinuclear complexes of AlIII and LiI with HL13.47 | ||
In an effort to explore the coordination chemistry of HL13 with 3d transition metal ions, Nareddula and co-workers focused their attention on the isolation of ZnII complexes.48a By reacting HL13 with ZnEt2 they were able to isolate the mononuclear complex [L13ZnEt] (18). Here, L13 is deprotonated and acts as a chelating (U-shaped) ligand, stabilizing the ZnII ion in a slightly distorted trigonal planar geometry. Interestingly, by changing the ZnII metal source to Zn(OAc)2 the authors were able to isolate a tetranuclear ZnII cluster [(L13)2Zn4O(OAc)4] (19) (Fig. 6). In this complex, the four ZnII centers are connected via a central μ4-O2− bridge. Two bridging acetato ligands also contribute to the coordination sphere of each metal center, while the L13 coordinates in a syn–syn W-shape manner, bridging two ZnII metal centers. This coordination motif suggests that ImAm ligands can also act as bridging moieties for the formation of high nuclearity complexes in 3d metal chemistry. Additionally, these complexes were also tested as efficient catalysts for the ring opening polymerization (ROP) of ε-caprolactone.48a
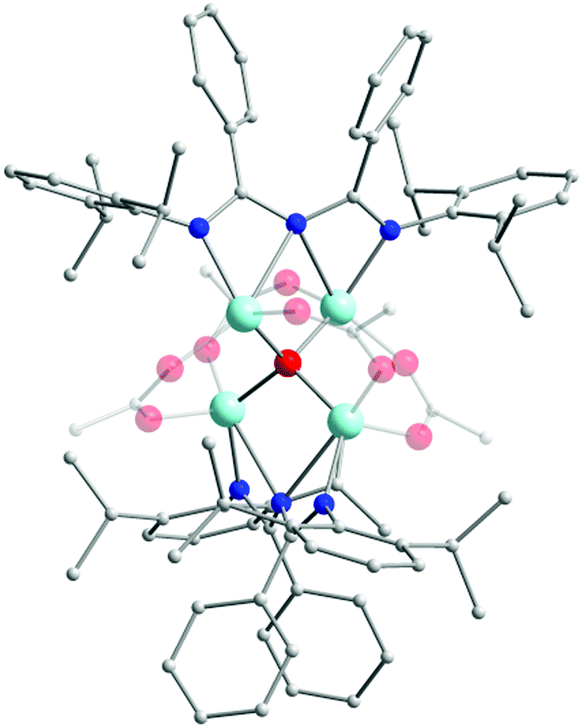 | ||
| Fig. 6 The molecular structure of the cationic complex 19.48a For clarity reasons H-atoms and disorder conformers have been omitted, as well as partial transparency has been employed. Colour code: C: grey, N: blue, O: red ZnII: pale blue. Reproduced using reported cif. | ||
A series of mononuclear and dinuclear complexes of ZnII was reported by Kulkarni et al. where reaction of L14, L15, L16 and L17 with ZnEt2 led to the isolation of a mononuclear coordination complex of the generalized formula [LxZnEt] (where x = 14, 15, 16 or 17).48b While these complexes are similar to 18, upon exposure to O2 for a limited time followed by recrystallization at specific temperatures, dimerization of the complexes containing L14, L16 and L17 occurred (Scheme 11). In particular, when L14 and L16 ligands were employed, dimerization was achieved through the formation of ethoxy bridges, while in the case of L17 the formation of ethylperoxy bridges was observed. In case of L15, when the corresponding complex was exposed to O2, a mixture of intractable products was obtained. This difference can be attributed to the nature of the R2 groups that bind to the terminal N-atoms of the ImAm framework. For example, the L17 ligand bears electron-donating groups making it more capable of stabilizing the zinc-ethylperoxy complex. In contrast, the halogen rich substituents in L14 and L16 are electron-withdrawing and therefore lead to the formation of the zinc-ethoxy complexes.
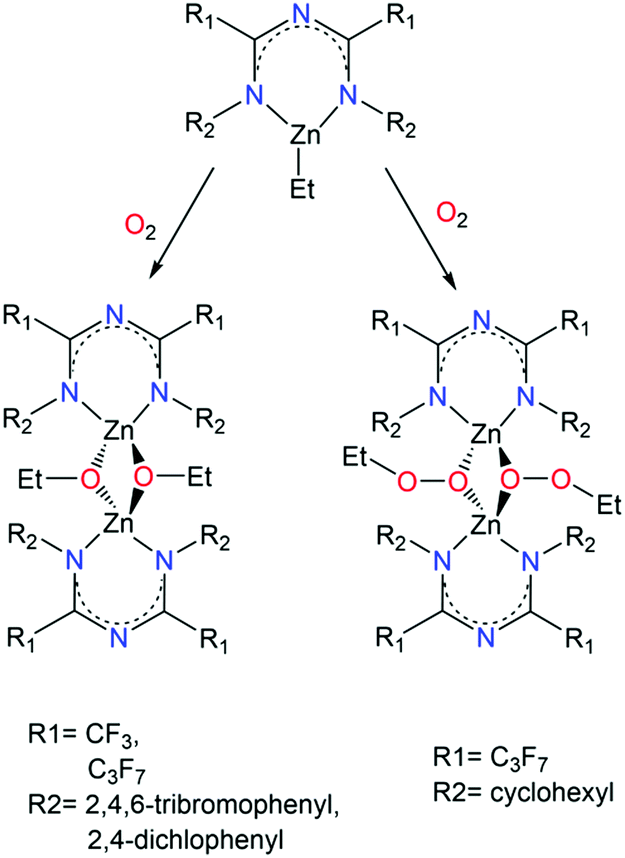 | ||
| Scheme 11 The synthesis of ZnII ethoxide and ethylperoxide dimers.48b | ||
Overall, the coordination chemistry of these ligands has not been fully explored. This may be attributed to the absence of donor atoms in the R1 and R2 groups and the energetically favored U-shape coordination mode, leaving little room for isolation of metal complexes with higher nuclearities. Nonetheless, polynuclearity can be achieved by the inclusion of donor atoms in the R1 and R2 moieties.
3.2 ImAm chelates containing additional donor atoms in the R1 positions
Apart from the three N-atoms of the N3C2 moiety, efforts to synthesize polydentate imidoyl amidine ligands have led to impressive results. Of particular relevance to the current review, the ligands presented in Scheme 12 will be discussed in turn all of which possess multiple sites for coordination.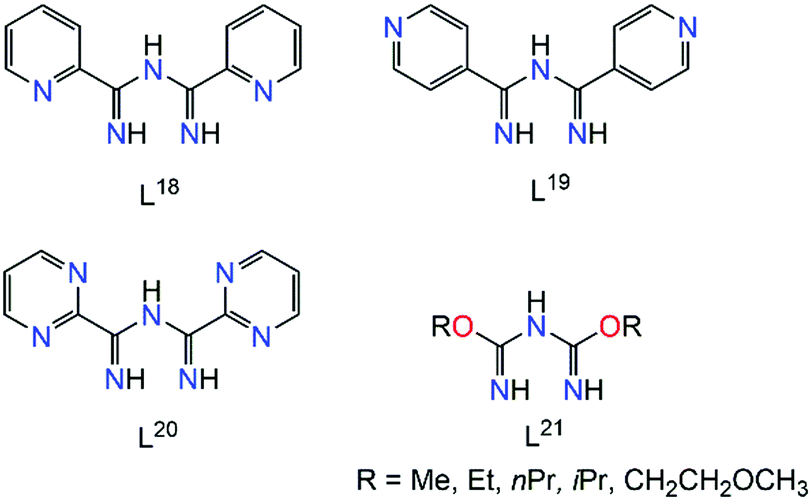 | ||
| Scheme 12 Examples of polydentate ImAm ligands with additional coordination groups in the R1 positions. | ||
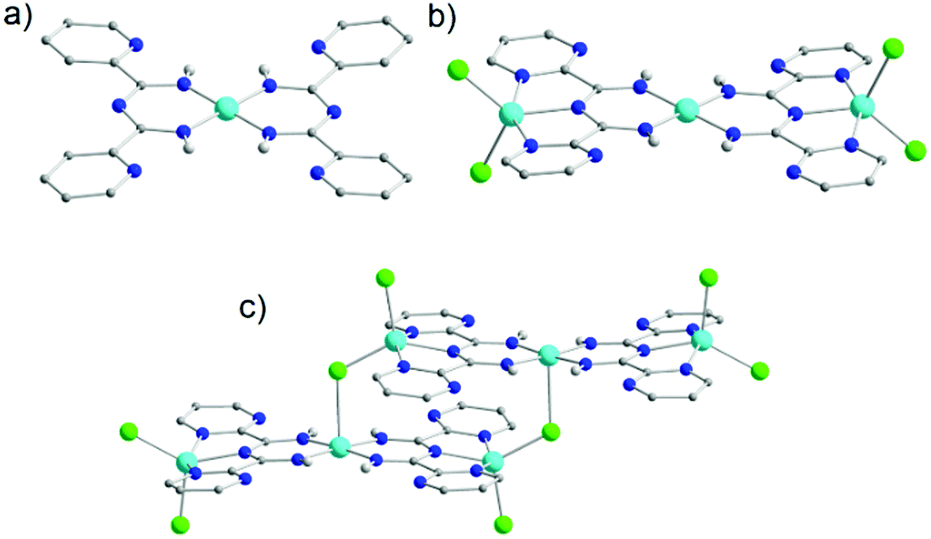
Due to the presence of two pyridyl substituents in L18, both bidentate and tridentate coordination sites are created. Coordination in a bidentate fashion has been well known since several groups managed to isolate complexes by the in situ synthesis of L18via metal-assisted transformations.49 Such a preparative manner that is reliant on the serendipitous assembly of the ligand framework does not enable full exploration of the capabilities of L18. On the other hand, having the ligand framework premade and then utilized in coordination chemistry provides better control over the synthetic process and overall yields. The importance of this was highlighted in the development of a series of mononuclear coordination complexes with MnIII, FeIII and CoIII under various synthetic conditions, which we recently reported.50 In particular, when reactions were carried out under basic or neutral conditions mononuclear complexes with a generalized molecular formula [MIII(L18)3] (where M = MnIII (20), FeIII (21) or CoIII (22)) (Fig. 7a) were isolated. In these complexes, the monoanionic ligand binds through its bidentate chelate in a U-shape fashion, stabilizing the trivalent metal ions in an octahedral molecular geometry. Conversely, under acidic conditions the tridentate coordination pocket of L18 could be accessed, leading to the isolation of the complexes [MII(L18)Cl2] (where M = MnII (23) or CoII (24)) and [FeIII(L18)Cl3] (25) in which the ligand was not deprotonated. The additional hydrogen atom on L18 in 23–25 is shared between the imino groups (50% occupancy due to the presence of a crystallographic C2 axis) effectively blocking the bidentate pocket, thus leading to coordination to the metal ions through the two pyridyl and the central amidine nitrogen atoms. In the MnII (23) and CoII (24) analogues metal oxidation was not observed, as was the case under basic conditions, leading to metal ions in which bivalent oxidation states are preserved. Two chloro terminal ligands complete their coordination spheres (Fig. 7b). In the case of the iron derivative 25, due to the trivalent iron starting material, FeIII has an octahedral molecular geometry with three chloro terminal groups completing its coordination sphere (Fig. 7c). Interestingly, neither the change of the transition metal nor the solvent seemed to affect the products isolated, although a change of the latter from MeCN to dichloromethane (DCM) affected the crystal packing.
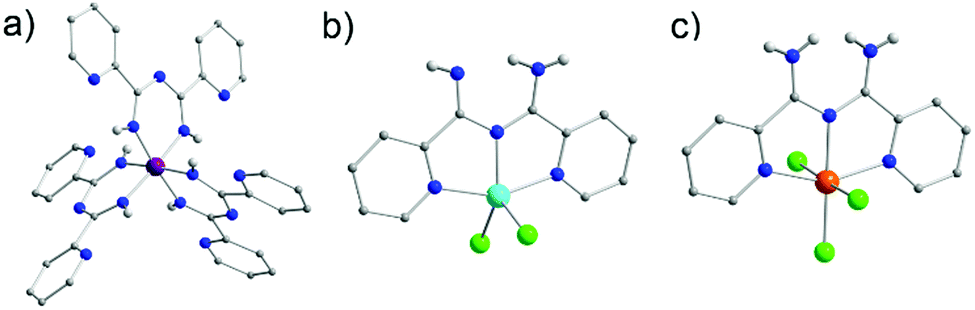 | ||
| Fig. 7 The molecular structure of the complex 20 (a), 24 (b) and 25 (c).50 For clarity reasons H-atoms and lattice solvent molecules have been partially omitted. Colour code: C: grey, N: blue, Cl: green, H: light grey, MnIII: purple, CoII: tale, FeIII: orange. Reproduced using reported cifs. | ||
Intrigued by these findings, we sought to isolate polynuclear coordination complexes with similar ligand frameworks, such as L20 where the pyridyl rings have been replaced by pyrimidyl groups.51 In this work, two coordination complexes of MnII/III and two coordination complexes of FeIII were isolated and their magnetic behaviour along with the magnetic behaviours of the previously isolated 20 and 21, were investigated (Scheme 13). The first complex isolated, [MnII2(μ-Cl)2(L20)2Cl2] (26), is a centrosymmetric dinuclear complex with two μ-Cl− the two MnII centers. The neutral ligand coordinates in a similar fashion as in the case of 23, through its tridentate binding pocket, while the slightly distorted octahedral coordination geometry of the MnII centers is completed by a terminal chloride. By changing to less acidic conditions, the tetranuclear complex [MnIIIMnII3(L20)3Cl6] (27) was isolated. In this complex, three deprotonated L20 ligands are bound to the central MnIII ion in a bidentate chelate fashion as in the case of complex 20. Each ligand is simultaneously coordinated through its tridentate pocket to another MnII metal center while two terminal chloro co-ligands complete its coordination sphere. By employing similar reaction conditions as in the case of 25 with FeIII, the mononuclear [FeIII(L20)Cl3] (28) was obtained. This shares structural resemblance with complex 25, since the neutral ligand is coordinating again in a mer-fashion, with three Cl− ions completing the coordination sphere of FeIII. The tetranuclear FeIII analogue of 27, [FeIII4(L20)3Cl9] (29), was achieved by changing the reaction solvent as well as the starting material of iron. Complexes 27 and 29 share structural similarities such as the coordination fashion of the deprotonated L20 and the coordination environment of the central metal ion. However, in the case of [FeIII4(L20)3Cl9] (29), all four iron ions are in their 3+ oxidation state and in order to satisfy charge balance considerations each of the peripheral FeIII ions is bound to three Cl− terminal ligands.
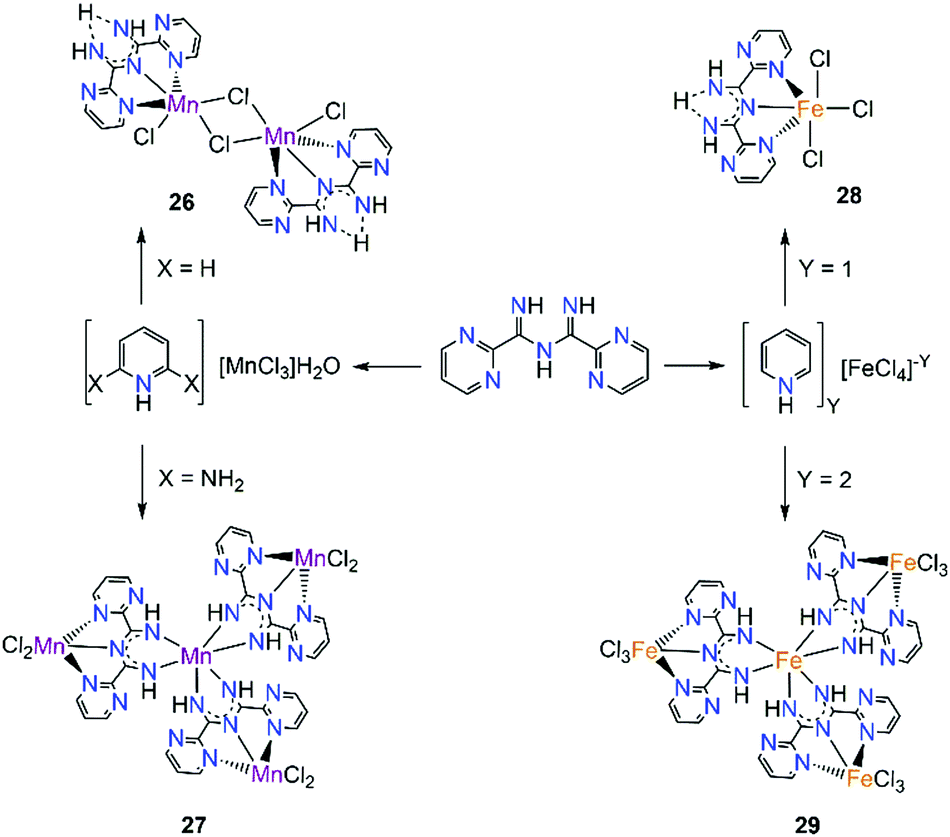 | ||
| Scheme 13 Synthesis of complexes 26, 27, 28 and 29 employing L20.51 | ||
Direct current (dc) magnetic susceptibility measurements performed on complexes 20, 21, 26, 27, 28 and 29 (Fig. 8) as well as Mössbaeur spectroscopy performed on 29 (Fig. 9), revealed that these ImAm ligands can stabilize both high and low spin configurations of these metal centers. For instance, in the case of 28 and 29 the central metal ions have a low-spin configuration due to their N-rich coordination environment provided by the ImAm ligands. On the contrary, the peripheral metal ions that also bind to Cl− adopt high-spin configurations. The incorporation of such ImAm frameworks that can promote both ferro- and antiferromagnetic interactions between the metal centers can prove to be most useful in the field of Molecular Magnetism, as such examples are rare. Therefore, synthetic strategies as those implemented in these cases can prove helpful in the targeted design of polynuclear species with different spin states configurations.
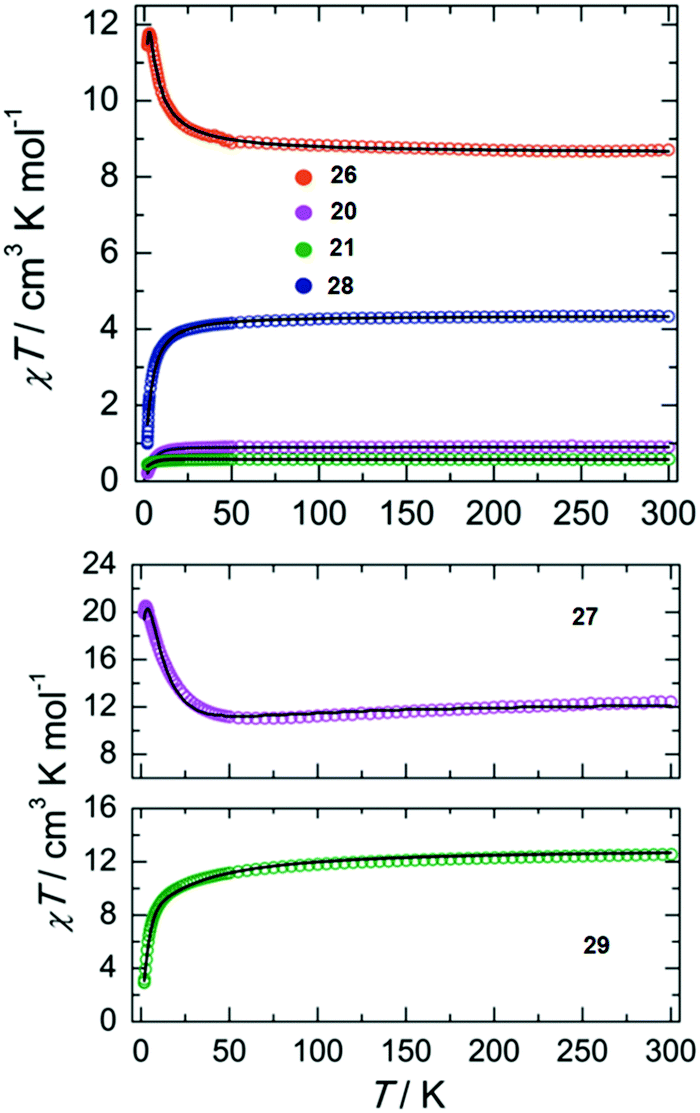 | ||
| Fig. 8 Top: Temperature dependence of the χT product of 20 (purple), 21 (green), 26 (orange) and 28 (blue) with χ being the molar magnetic susceptibility per molecule as defined by M/H. Bottom: Temperature dependence of the χT product of 27 (purple) and 29 (green) with χ being the molar magnetic susceptibility per molecule as defined by M/H. In both plots the black solid lines represent the best-fits to the data. Adapted with permission from ref. 51. Copyright 2019 Wiley. | ||
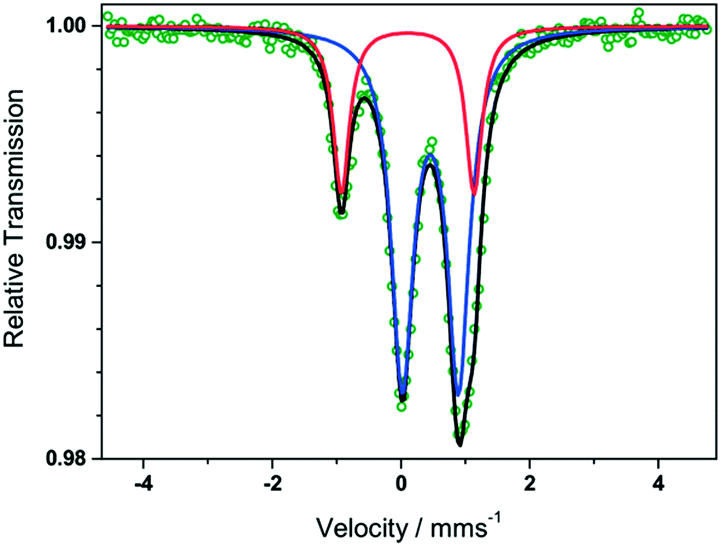 | ||
| Fig. 9 Zero-field Mössbauer spectrum of 29 recorded at 80 K; the collected experimental data is indicated as green circles. The blue and red lines correspond to the fit of outer high spin and the central low spin FeIII ions, respectively, showing that there are two different species of FeIII in the complex. The overall sum of the FeIII ions is represented by the black line. Adapted with permission from ref. 51. Copyright 2019 Wiley. | ||
Although these synthetic procedures have yielded impressive results, we wanted to explore the potential of the aforementioned ligands under inert conditions. Recently, both L18 and L20 were employed to isolate mononuclear as well as polynuclear CoII complexes, respectively.52 Under basic reaction conditions in an inert environment mononuclear [CoII(L18)2] (30) was obtained (Fig. 10a). In contrast to complex 22, the CoII has an almost ideal square planar geometry since both ligands coordinate in a bidentate fashion and the metal ion is in its 2+ oxidation state, as confirmed by both charge balance considerations and dc magnetic susceptibility measurements. Replacement of L18 with L20, led to the isolation of [CoII3(L20)2Cl4] (31) and [CoII6(μ-Cl)2(L20)4Cl6] (32) (Fig. 10b and c, respectively). Complex 31 is a linear trinuclear CoII complex, where both the bidentate and tridentate coordination pockets of each deprotonated L20 moiety is occupied by a CoII ion. The central CoII adopts a square planar geometry identical to that of 30. The two peripheral CoII ions have geometries similar to the one observed for complex 24, with three N-donor atoms of the L20 ligand and the two terminal Cl− atoms completing their coordination sphere. Interestingly, the degree of hydration of the reaction solvent (MeOH) is the key difference between isolating complex 31 and 32. Complex 32 can be described as two trinuclear moieties (similar to 31) bridged by two μ-Cl−. The asymmetric unit of 32 consists of three unique CoII centers. Within this unit, the two L20 ligands coordinate in the same manner as in the case of 31. However, the central CoII is pentacoordinate due to the bridging Cl−, resulting in a slightly distorted square pyramidal geometry.
 | ||
| Fig. 10 The molecular structures of complexes 30 (a), 31 (b) and 32 (c).52 For clarity reasons H-atoms and lattice solvent molecules have been omitted. Colour code: C: grey, N: blue, Cl: green, CoII: tale. Reproduced using reported cifs. | ||
Magnetic susceptibility measurements as well as magnetostructural correlations of the {CoII3} aggregates in 31 and 32 reveal a unique combination of low spin CoII (center) and high spin CoII (peripheral) configurations. Additionally, complex 32 exhibits SMM behavior (Fig. 11) under a small applied field, where Raman, quantum tunneling of the magnetization and direct process are responsible for the slow relaxation of the magnetization (Fig. 12).
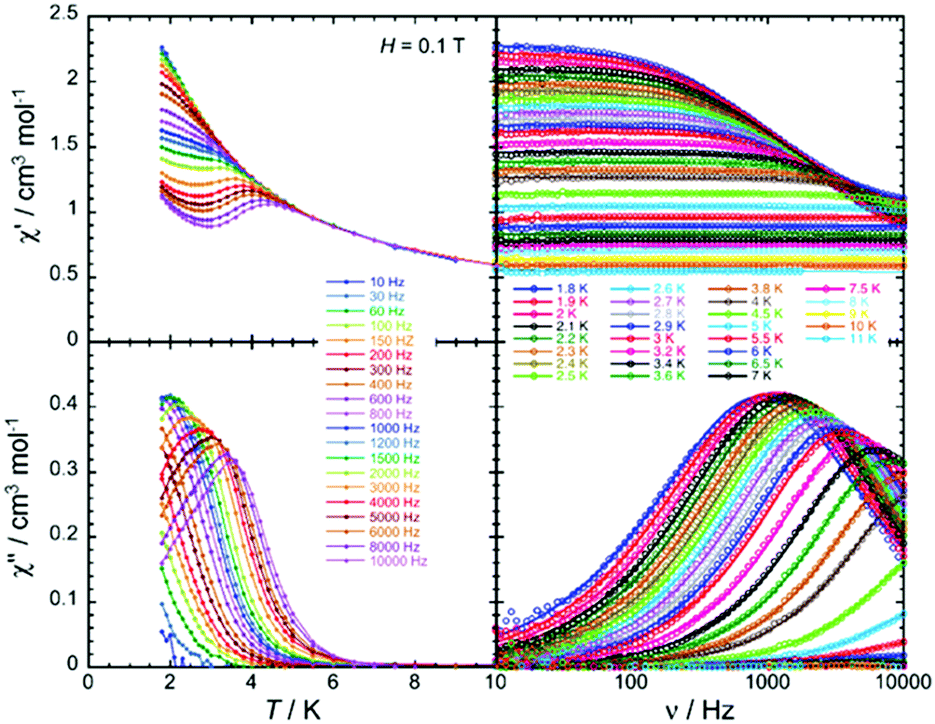 | ||
Fig. 11 Temperature (left) and ac frequency (right) dependences of the in-phase (χ′, top) and out-of-phase (χ′′, bottom) ac susceptibility in the temperature region of 1.85 to 10 K and between 10 and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Hz for 32 in a 0.1 T dc field. Solid lines serve, on the left plots as guides to the eye while on the right they show the generalized Debye fit of the ac data. Adapted with permission from ref. 52. Copyright 2020 Royal Society of Chemistry. 000 Hz for 32 in a 0.1 T dc field. Solid lines serve, on the left plots as guides to the eye while on the right they show the generalized Debye fit of the ac data. Adapted with permission from ref. 52. Copyright 2020 Royal Society of Chemistry. | ||
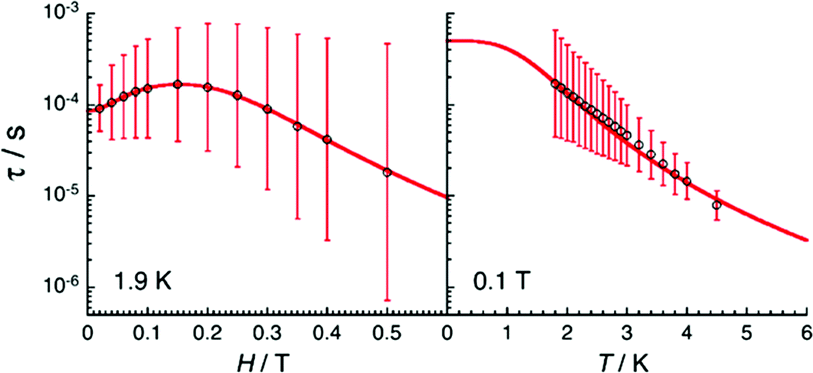 | ||
| Fig. 12 Dc-field (left) and temperature (right) dependences of the relaxation time for 32 estimated from the generalized Debye fits of the ac data shown in Fig. 10 collected at 1.9 K and under an 0.1 T applied field, respectively. The vertical solid bars are the estimated standard deviations of the relaxation time. The solid red line represents the best fit. Adapted with permission from ref. 52. Copyright 2020 Royal Society of Chemistry. | ||
Recently, Piers and co-workers presented two mononuclear NiII complexes with L18.53 The first was the neutral complex [NiII(L18)2] (33) in which the monoanionic bidentate chelate ligands stabilize the square planar NiII ion (Fig. 13a). The one e− reduction of 33 results in the formation of [K(crypt)][NiII(L18)2] (crypt = 2,2,2-cryptand) (34) in which the ImAm ligand is in its reduced form. As supported by density functional theory (DFT) calculations and electron paramagnetic resonance (EPR) spectra the radical species is delocalized almost exclusively onto the ligand framework (Fig. 13b and c, respectively). Controlled potential electrolysis revealed that reduced 33 can convert CO2 to CO, highlighting that the redox activity of this ligand framework can play a significant role in the reduction of CO2via the utilization of 3d metal complexes.
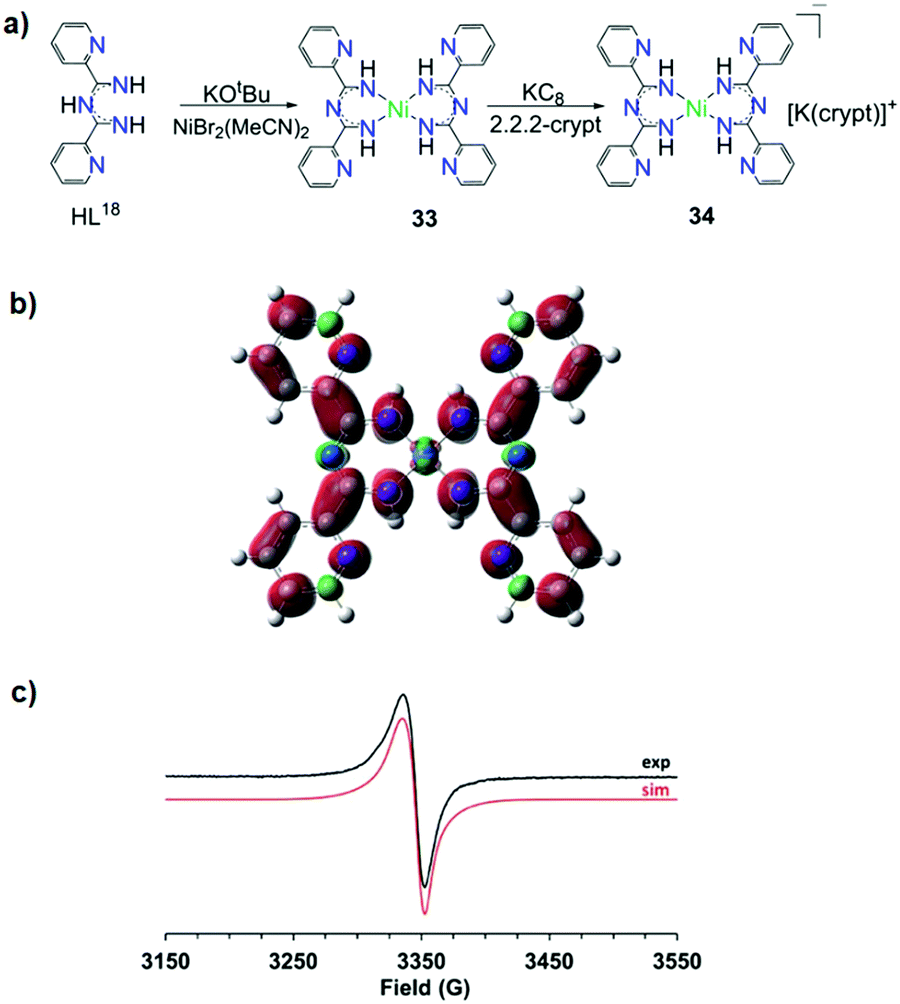 | ||
| Fig. 13 (a) The synthetic procedure that led to the isolation of complexes 33 and 34; (b) the Mulliken spin density plot of 34; (c) experimental (back) and simulated (red) X-band EPR spectra of 34. Reprinted with permission from ref. 53. Copyright 2019 Royal Society of Chemistry. | ||
By changing the pyridine N-atom from the ortho- to the para-position, L19 can be formed. Jin and co-workers reported a series of heterometallic cationic complexes of the generalized formula of {[Cp*2Rh2X2]4[MII(L19)2]2} (35–37) (where X = Cl, (μ-η4-C2O4), (μ-chloroanilic acid) and M = NiII or PdII).54 The authors isolated six heterometallic complexes (3d/4d and 4d/4d) following the generalized synthetic procedure outlined in Scheme 14. In all six cationic complexes, the L19 ligands coordinate in the same fashion, the only difference being the bridging co-ligand between the Rh metal centers or the exchange of NiII with PdII. Thus, for the purposes of this review article, only 36a will be discussed in detail. In this complex, each L19 ligand coordinates to the NiII metal centers through its bidentate U-shape site, stabilizing the NiII ions in a square planar geometry. Four coplanar N-pyridyl atoms in L19 coordinate with four {Cp*Rh} moieties, with each of the Cp*Rh moieties bridged through two chloride co-ligands with a second Cp*Rh moiety that has been stabilized in the “corners” of the L19 ligand. As a result, two Ni(L19)2 moieties stack in a parallel fashion, forming a double decker bridged by the {Cp*2Rh2Cl2}.
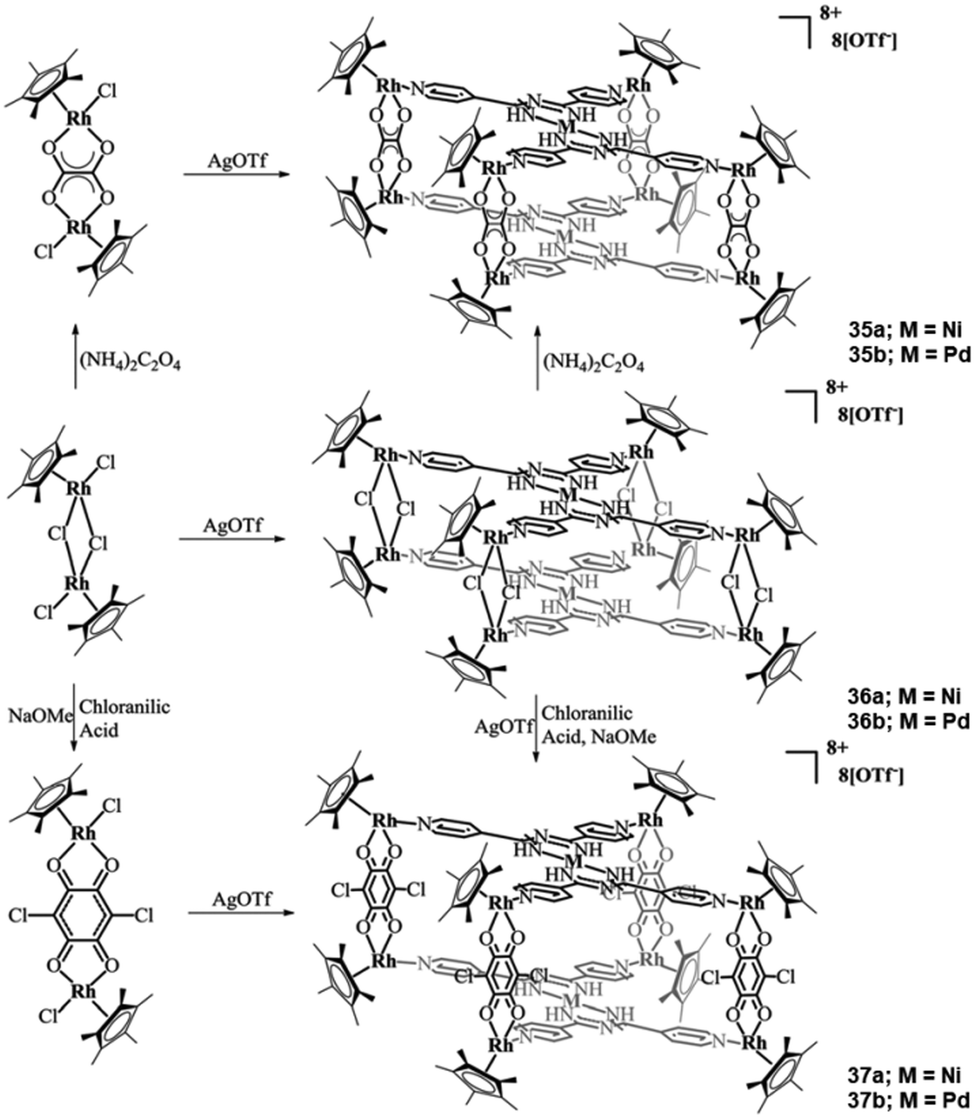 | ||
| Scheme 14 The synthetic routes that led to the isolation of the organometallic heterometallic complexes, where M = NiII or PdII. Adapted with permission from ref. 54. Copyright 2017 Royal Society of Chemistry. | ||
As evident from the aforementioned complexes, the introduction of functional groups with N-atoms has yielded impressive results based on stepwise inorganic syntheses. Attempts to incorporate other functional groups such as RO−, have led to the formation of a small family of mononuclear complexes. The main difference in this category of ligands, such as L21, is that these chelates are synthesized in situ, via a metal assisted pathway. In particular, the synthesis of these 2,4-alkoxy-1,3,5-triazapentadienes is based on the reaction of sodium dicyanamide with alcohols in the presence of a transition metal source, leading to mononuclear complexes of CuII55a–c or NiII55d with a generalized formula [MII(L21)2] (where M = CuII or NiII). Efforts to understand the mechanisms behind this ligand formation, such as the one given in Scheme 15, have been made by several groups.55a,b In these complexes, the ligand acts in the chelate, U-shape, coordination mode with the divalent transition metal ion adopting a square planar geometry. In the case of NiII, by employing pyrazole-based co-ligands, dinuclear and trinuclear NiII complexes were isolated with the ligand coordinating in the same fashion. The synthesis of these ligands has only been achieved in situ and efforts to utilize the alkoxy groups as possible coordination sites remain an ongoing challenge given there is no control over this synthetic approach.
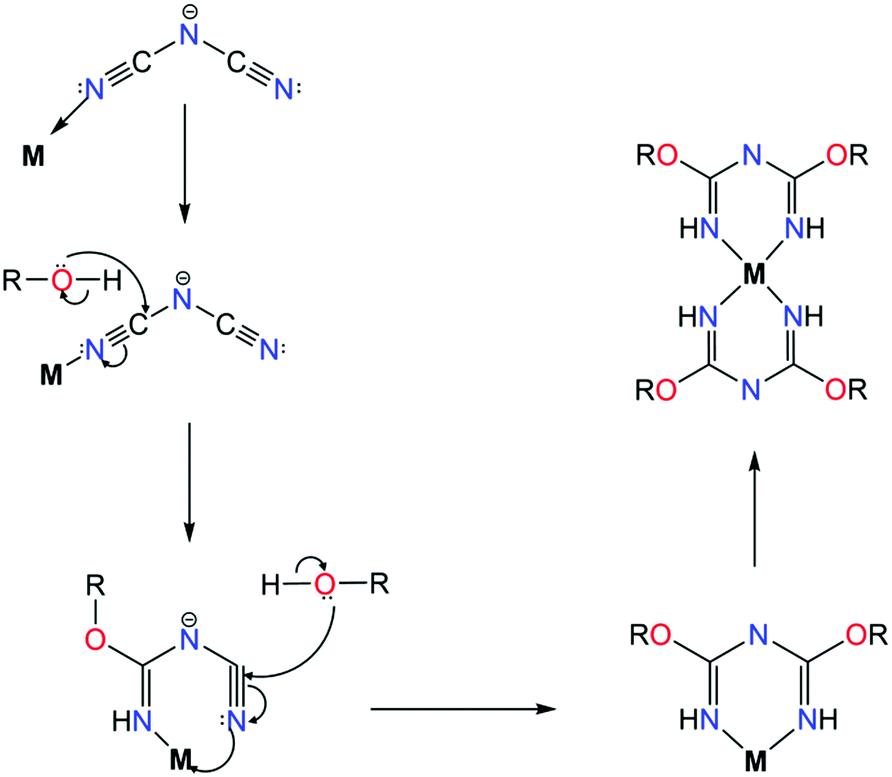 | ||
| Scheme 15 The proposed mechanism for the in situ synthesis of 2,4-alkoxy-1,3,5-triazapentadiene ligands, where M = CuII or NiII and R = Me, Et, nPr, iPr or CH2CH2OCH3.55b | ||
ImAm ligands can provide promising complexes for catalytic applications like their β-diketone analogues.55c For this reason, some of the 2,4-alkoxy-1,3,5-triazapentadienato CuII complexes have been tested as efficient catalysts for the nitroxyl radical mediated mild aerobic oxidation of alcohols to the corresponding aldehydes and for the solvent-free microwave-assisted synthesis of ketones from secondary alcohols with tert-butylhydroperoxide as oxidant.55a In addition to that, other ImAm based complexes have been studied as catalysts for the mild oxidation of alkanes with H2O2 as an oxidant, pyridine as promoting agent and cyclohexane as a main model substrate.55c
4. The coordination chemistry of asymmetrical imidoyl amidine chelates
As evident from the aforementioned examples, the presence of different substituents plays a key role in the coordination chemistry of ImAm based ligands. By introducing different R groups, desired structural motifs can be targeted. For instance, such frameworks can have an R1 group, such as pyridine, that stabilize metal ions through chelating coordination and a different R1′ substituent, such as hydroxyl, that can bridge multiple metal centers. Moreover, apart from an additional coordination site, the central N-atom can also take part in facile acid/base equilibria, providing pH sensing properties to complexes.56 This can prove to be important for future application of such complexes that exhibit luminescence properties. In Scheme 16 the ligands that will be discussed in this section are presented.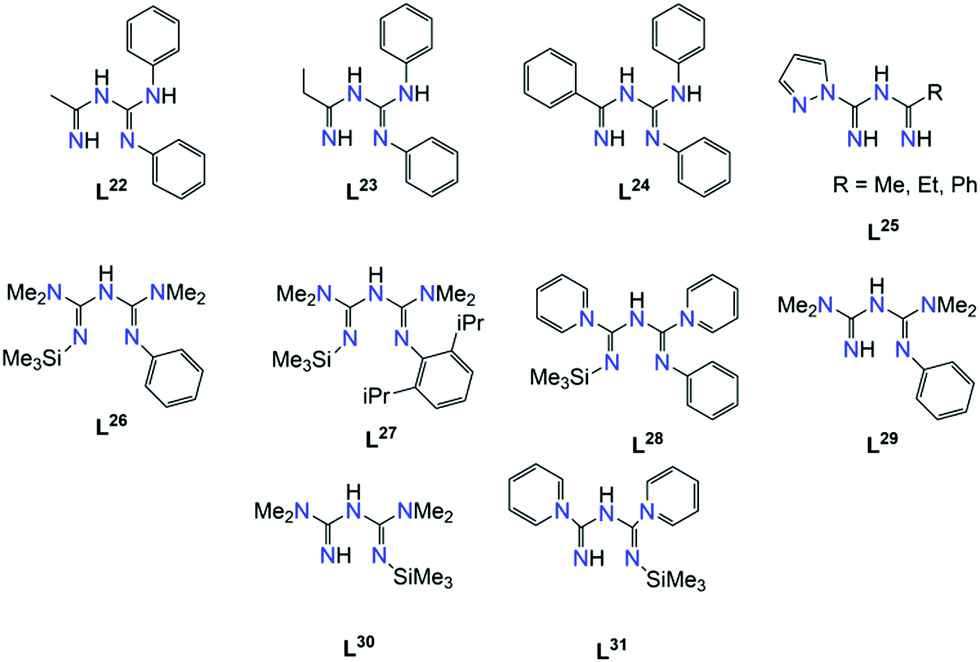 | ||
| Scheme 16 The asymmetrical ImAm chelates that will be discussed in this section. | ||
In an effort to explore the luminescent properties of asymmetric ImAm ligands with PtII, Eliseev et al. synthesized a series of mononuclear PtII complexes [PtII(dpa)(Lx)][OTf] (38) (where x = 22, 23, 24; dpa = 2,2′-dipyridylamine; OTf = SO3CF3).56 Due to the fact that in all three cationic complexes the ligands act in the same coordination mode and since all three share structural similarities, only 38a will be discussed with L22 as ligand. As shown in Fig. 14 (top), the L22 ligand, which was synthesized in situ, is in its monoanionic form, coordinating in the chelate, U-shape, coordination fashion. Four N-atoms (two from the ImAm ligand and two from the dpa co-ligand) complete the coordination sphere of the PtII ion leading to a slightly distorted square planar molecular geometry. Surprisingly, these complexes showed intense emission in the solid state (Fig. 14, bottom), but they were non-emissive in solution. This can be explained by the square planar configuration of PtII in which an unoccupied d orbital is located perpendicular to the plane. Theoretically, the empty orbital can be subject to coordination by electron donors of the lattice solvent molecules, which leads to dominant non-radiative deactivation pathways.
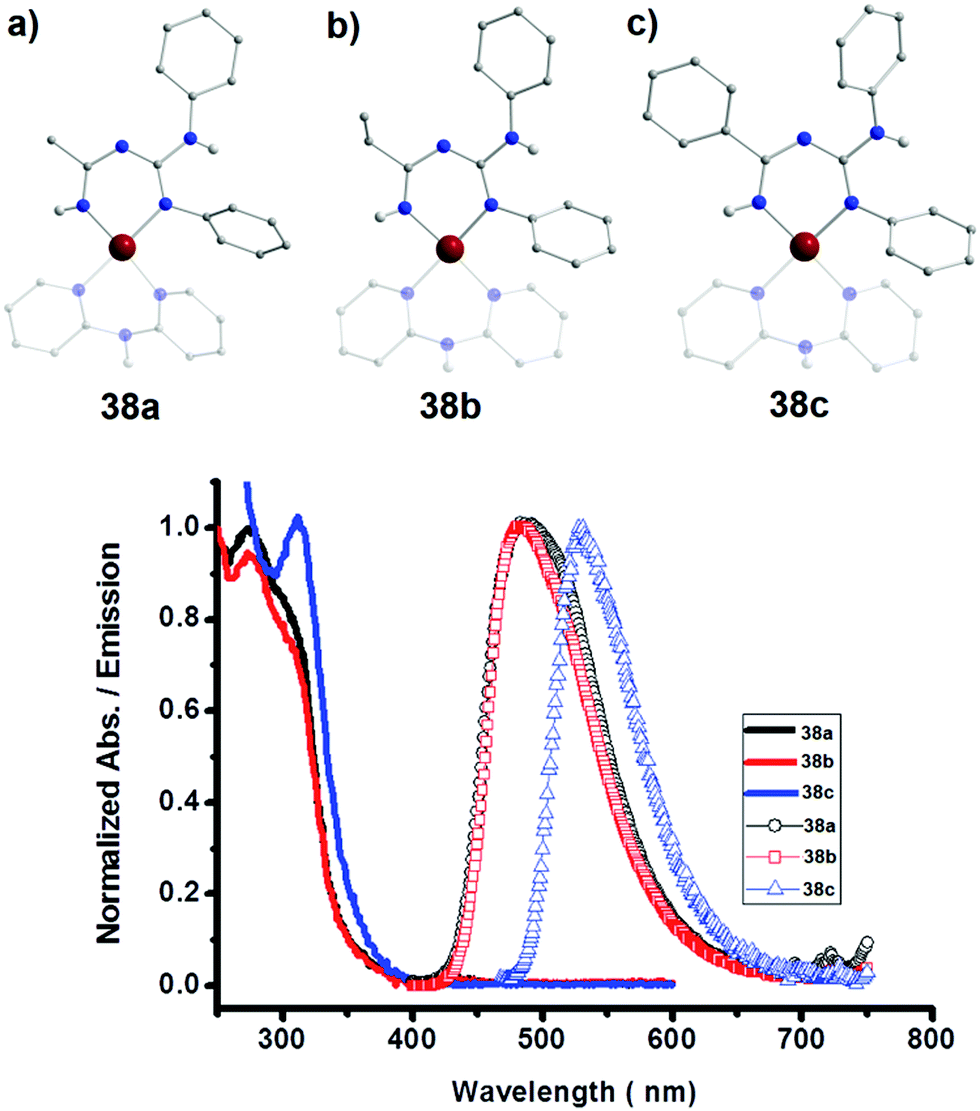 | ||
| Fig. 14 Top: Molecular structures of 38a (a), 38b (b) and 38c (c). For clarity reasons only the H-atoms of the nitrogen are shown, lattice solvent molecules and counter ions have been omitted, as well as partial transparency has been employed. Colour code: C: grey, N: blue, H: white, PtII: dark red. Reproduced using reported cifs. Bottom: Absorption in CH2Cl2 (solid lines) and the normalized solid-state emission spectra of 38a, 38b and 38c at room temperature (icons with solid lines). Adapted with permission from ref. 56. Copyright 2014 Wiley. | ||
Since the emissive states of PtII complexes can be affected dramatically by the bulkiness of the substituents on the ImAm ligands, Tanaka et al. synthesized a series of PtII based complexes with L25.57 The introduction of the pyrazole substituent is key, for the careful tuning of the basicity of pyrazolate ligands and enables control over the structure of mononuclear PtII complexes. The general synthetic procedure that led to the isolation of these complexes is given in Scheme 17. The ligand is generated in situ, in a nitrile solution with a pyrazole derivative bearing the amidine group in the presence of the metal salt. Formation of this ligand is achieved by the coupling of the coordinated nitriles with the pyrazolecarboxamidine. The final products of these reactions, depending on the nitrile in use, have a generalized formula of [PtII(bpy)(L25R)]X (39R) (where bpy = 2,2′-bipyrimidine, R = Me (a), Et (b) or Ph (c) and X = BF4−, PF6−). Different recrystallization methods led to two different crystal phases; type I, which exhibits yellow luminescence and type II, which exhibits yellow-green luminescence. In Fig. 15 (top), the solid state emission spectra of these complexes is given.
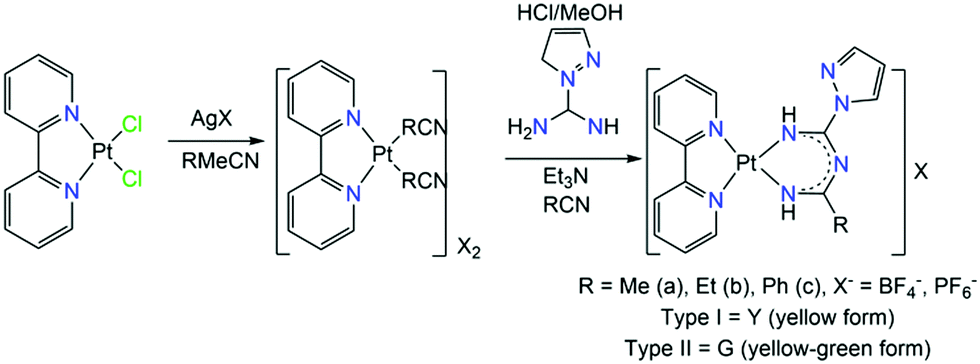 | ||
| Scheme 17 Synthesis of [PtII(bpy)(L25R)]X (39R) (where bpy = 2,2′-bipyrimidine, R = Me (a), Et (b) or Ph (c) and X = BF4−, PF6−).57 | ||
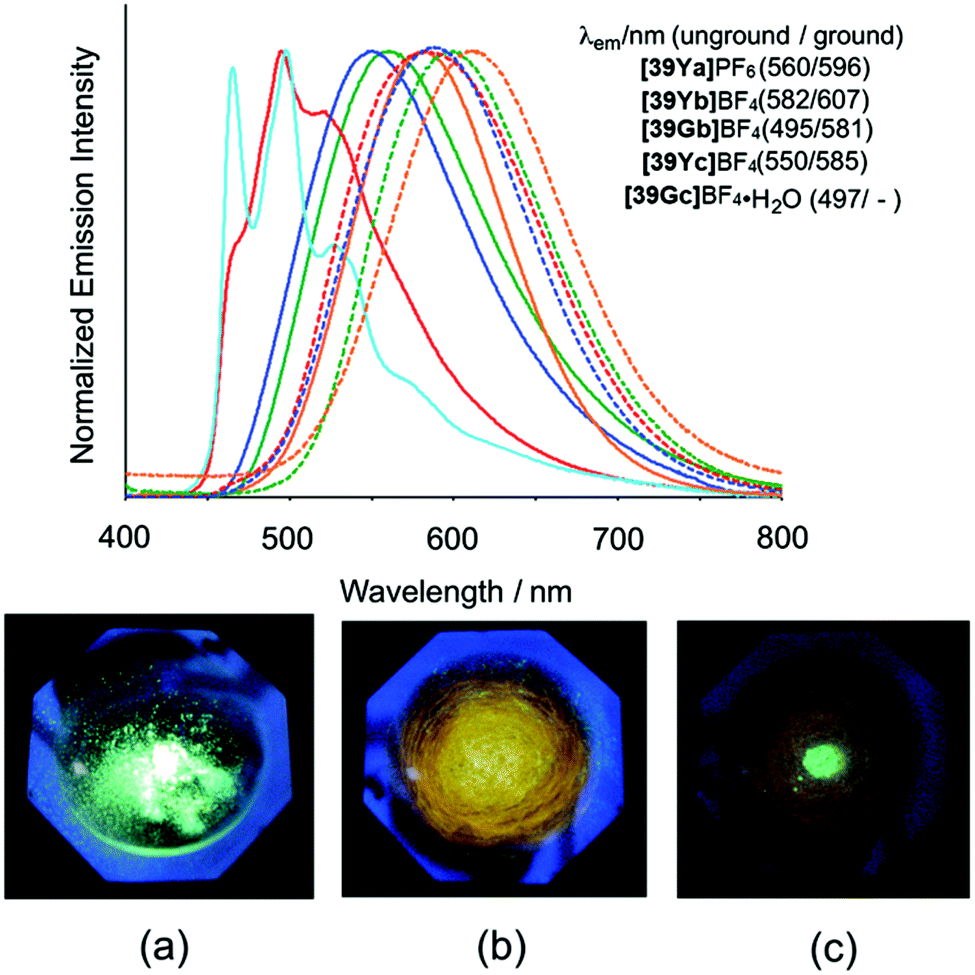 | ||
| Fig. 15 Top: Normalized emission spectra of the PtII based complexes in the solid state at 295 K (λex = 355 nm). Solid and dashed lines correspond to the spectra of unground and ground samples, respectively. Bottom: Photographic images of [39Gb]BF4 in response to mechanical grinding under UV light irradiation (365 nm): (a) unground sample, (b) completely ground sample, (c) ground sample with a drop of acetonitrile. Reprinted with permission from ref. 57. Copyright 2014 Royal Society of Chemistry. | ||
Based on time-dependent density functional theory (TD-DFT) calculations, it was found that the lowest energy absorption bands of these complexes can be assigned to a combination of ligand-to-ligand charge-transfer (LLCT) and metal-to-ligand charge transfer (MLCT) transitions. These findings highlight the prosperity of aromatic ImAm ligands in the development of complexes for optical applications. Interestingly, these complexes also exhibited unique mechanochromic behavior. As shown in Fig. 15 (bottom), the luminescence of the unground sample dramatically changes from green to yellow-orange after it has been completely ground. However, upon addition of a drop of acetonitrile the luminescence becomes bright again but is slightly different from the original luminescence. This difference can be assigned to the formation of a new crystalline phase of the material caused by the addition of the solvent. These interesting luminescence properties may originate from the one-dimensional stacking of the highly planar PtII complexes, where the bpy and ImAm ligands stack alternately.
Replacement of the NHPh groups by NMe2 substituents in the R1 positions of the ImAm framework, as well as the addition of SiMe3 in place of the H-atom at one of the R2 positions, afford the ligand L26. A slight modification of this ligand can also lead to L27, in which 2,6-diisopropylphenyl (Dipp) is introduced on the amide N-atom instead of a Ph group. In the work by Zhou et al. these ligands were synthesized in situ during the formation of mononuclear and dinuclear complexes of MgII.58 Although both ligand frameworks are rather similar, small changes in the synthetic conditions led to different coordination motifs for each ligand, altering the nuclearity of the isolated complex. In the mononuclear complex [MgII(L26)2] (40), the ligand stabilizes the MgII center in a distorted tetrahedral geometry by acting as a chelate terminal ligand (U-shape fashion) (Fig. 16a). On the contrary, in the dinuclear [MgII2Br2(L27)2] (41), each MgII ion is bound to four N-atoms in a highly distorted square pyramidal geometry (a terminal bromo co-ligand completes the coordination sphere of each metal center) with both ligands adopting a syn–syn W-shape coordination motif, stabilizing the two MgII metal centers (Fig. 16b).
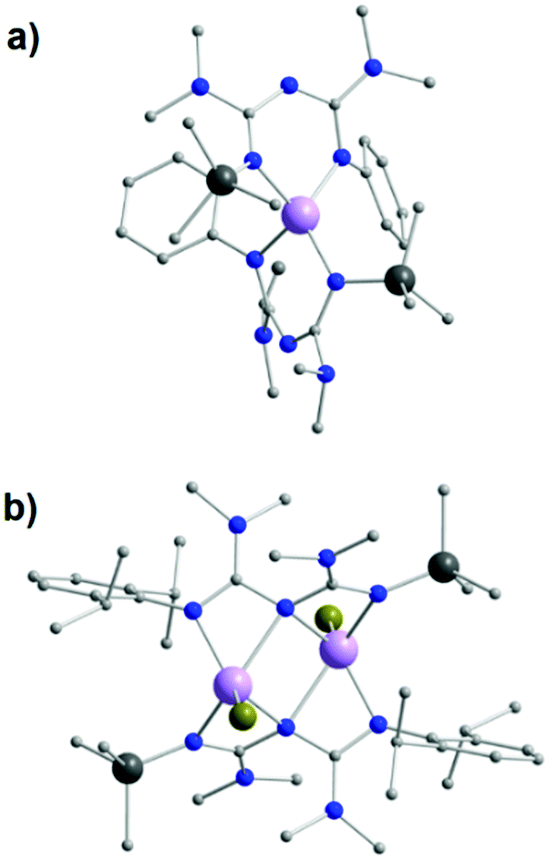 | ||
| Fig. 16 The molecular structure of the complex 40 (a) and 41 (b).58 For clarity reasons H-atoms and have been omitted. Colour code: C: grey, N: blue, Br: dark yellow, Si: dark grey, MgII: mauve. Reproduced using reported cifs. | ||
Apart from alkaline earth metals, the coordination chemistry of these ligands has also been explored with transition metals. Mononuclear complexes of CuII have been isolated with L26: [CuII(L26)2] (42) and L27: [CuIICl(L27)] (43) and [CuII(PPh3)(L27)] (44).58a,b Both ligands coordinate on the metal center in a U-shape fashion, stabilizing the metal center in a square planar geometry in 42 and a trigonal planar geometry in 43 and 44. The same group has also isolated the ZnII analogue of 42, which was demonstrated to possess catalytic properties for the ring-opening polymerization (ROP) of racemic-lactide.58c
By replacing the NMe2 groups with pyridine, L28 can be formed. The obvious similarities to L26 allows for a similar coordination behavior in the transition metal chemistry. Mononuclear complexes of this ligand with a generalized formula [MII(L28)2] (where M = ZnII,58c FeII59 and CoII59) have been isolated. In all these cases, the ligands act as terminal chelates stabilizing the metal centers in a slightly distorted square planar geometry, with the ZnII analogue acting as a catalyst for the ROP of racemic-lactide.
The growing interest in asymmetric ImAm chelates and their ability to form stable complexes with transition metal ions, led Zhou and co-workers in studying the chemistry of L26, L27 and L28 in TiIV complexes.60 These complexes might also exhibit promising activities towards olefin polymerization. Starting from nitriles, the in situ synthesis of these ligands led to the isolation of a family of mononuclear TiIV complexes with the general formula [TiIVCl(Lx)2] (where x = 26, 28 or 29, respectively). In all three complexes the five-coordinate TiIV ion bonds to one chloro co-ligand and four nitrogen atoms from two ImAm ligands. Both ligands adopt the U-shape conformation, while the coordination geometry of the metal center can best be described as distorted trigonal bipyramidal. Surprisingly, one of the Ti–N bonds is significant shorter (average length 1.751(3) Å) than the other three distances (average length 2.075(6) Å), indicating that one ligand is in its dianionic form while the other is monoanionic (Scheme 18). Due to the potential activity towards olefin polymerization, the catalytic behavior of the TiIV complexes with L28 and L29 towards ethylene polymerization with methylaluminoxane (MAO) as co-catalyst was investigated, demonstrating both complexes were active in ethylene polymerization.
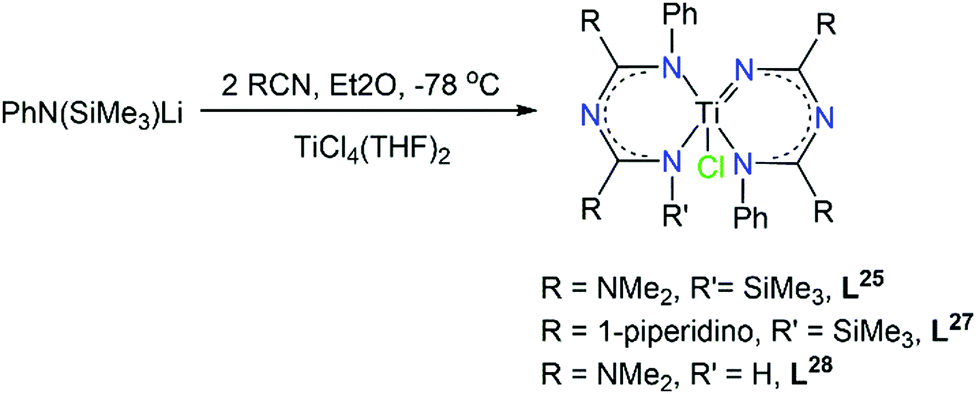 | ||
| Scheme 18 Synthesis of TiIV complexes with L26, L28 and L29 as ligands.60 | ||
The catalytic activity that the aforementioned complexes have shown inspired Guo et al. to explore the chemistry and catalytic activity of asymmetric ImAm ligands with SnII.61 For this purpose, several ligands such as L26, L28, L29 and L30 have been employed. These ligands were synthesized in situ from nitriles and by slight changes of the reaction conditions a family of mononuclear, dinuclear and tetranuclear complexes has been isolated. In Scheme 19, the synthesis of these complexes is given. In the mononuclear complexes [SnII(L26)2] (45) and [SnII(L28)2] (46), two monoanionic L26 ligands are coordinated to the SnII metal center. Both L26 are coordinated to the metal center in the U-shape coordination mode.61a In the mononuclear [SnIICl(L29)] (47), the monoanionic L29 coordinates in a similar fashion.61a However, in 48 two different coordination motifs are observed, as one of the L26 adopts a U-shaped conformation while the other a twisted W-shaped conformation. In this W-shaped mode, the N3C2 framework forms a twisted zigzag conformation.61b Furthermore, higher nuclearities were obtained with L30 and L31. In the dinuclear [SnII2(L30)4] (49), dimerization is achieved through the unsubstituted N of the N3C2 framework of two dianionic L30 moieties. The other two monoanionic L30 act as terminal ligands to each SnII in a U-shape conformation.61b By replacement of the NMe2 groups with 1-piperidine, L31 leads to the formation of the tetramer [SnII4(L31)4] (50). Here, each dianionic L31 bridges two SnII ions through its unsubstituted N-atom.61a Lastly, the catalytic activities of complexes 45, 47, 48 and 49 were tested. Complexes 45 and 47 were found to be efficient catalysts for the catalytic addition of phenylamine to N,N′-diisopropylcarbodiimide affording guanide,61a while complexes 48 and 49 could be used as pre-catalysts in the addition of aniline or 2,6-diisopropylaniline to N,N′-diisopropylcarbodiimide.61b
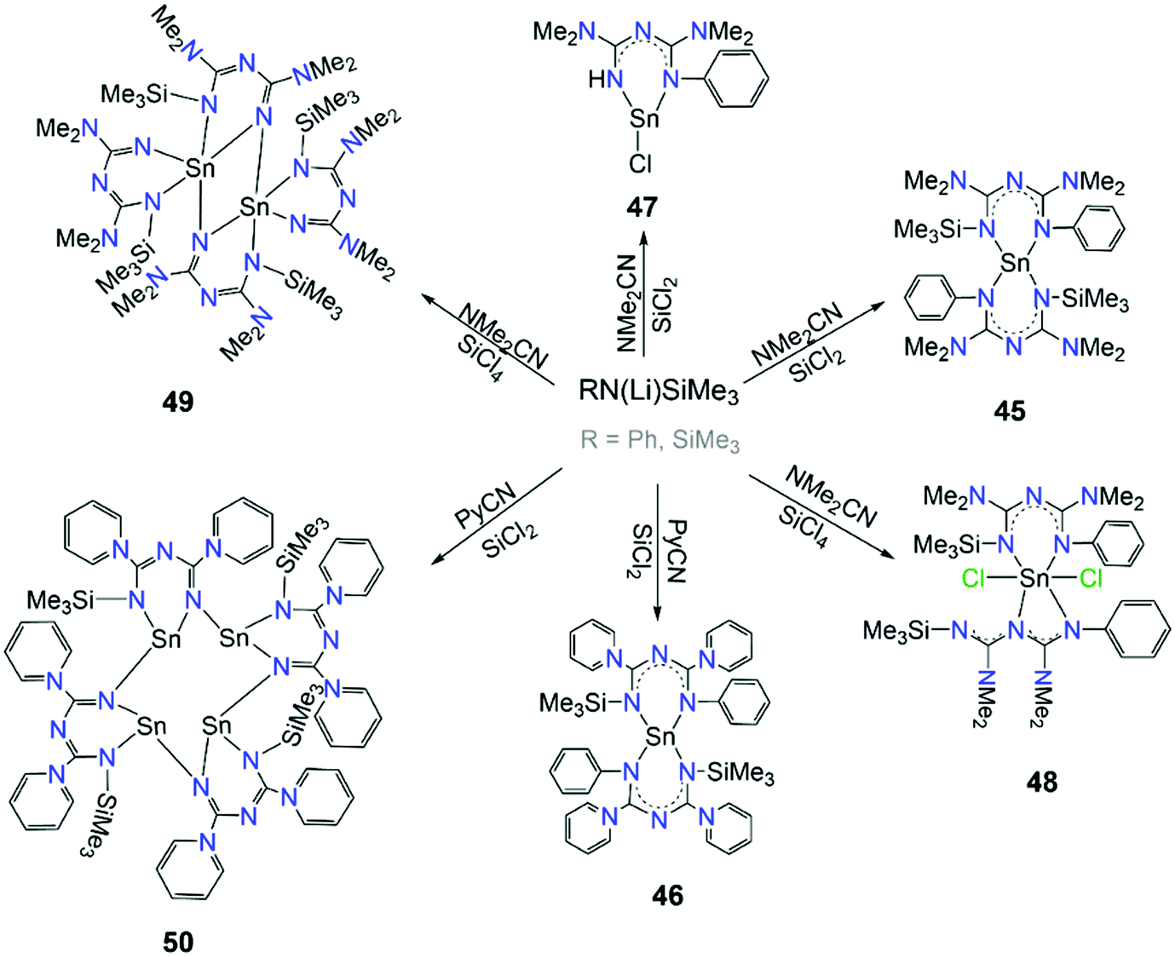 | ||
| Scheme 19 Synthetic routes for the preparation of complexes 45–49.61 | ||
5. Conclusions, limitations and perspectives
From the results presented herein, it is clear that imidoyl amidines represent a versatile class of organic ligands for the construction of new mono- or polynuclear coordination complexes exhibiting a variety of coordination modes, coordination geometries and associated properties. The limited number of ImAm coordination complexes as well as their synthetic routes reported to date, reveals only the surface of this field has been scratched and deserves further exploration. Despite the Pinner synthetic procedure and other proposed synthetic routes by Muller, Häger, Hughes and their coworkers in the literature, the synthesis of ImAm chelates has not yet been fully explored and, therefore, holds great potential. In particular, these methods generally afford heavily N-substituted ImAm with bulky substituents at the terminal nitrogen atoms and/or strong electron-acceptor functionalities at the carbon atoms. Additionally, the overall yields reported are poor and the starting materials often are not commercially available, thereby adding more steps to their synthesis and limiting their further development. Another synthetic approach involves the use of metal mediated transformations, affording ImAm-based coordination complexes; however, this leaves little room for control during preparation of the ImAm framework. With that said, the recent report on an efficient, high yielding, one-pot synthesis of ImAm21 provides an avenue for creating a vast library of chelates with untapped potential. This therefore opens new perspectives in fields ranging from magnetic and luminescent materials to catalysis and biomedical applications.Creative attention to both the organic and coordination chemistry of ImAm ligands can lead to the design of new ImAm frameworks and their metallo-assemblies. As an example, Py2ImAm affords two distinct coordination pockets; a tridentate chelating group akin to terpyridine along with a bidentate chelating mode reminiscent of NacNac. These two coordination pockets afford stable five and six membered coordination rings, respectively, thus can be exploited to efficiently target coordination to various metal ions including mixed-metal systems. This unique ligand elegantly demonstrates the combination of two prolific ligand frameworks into a single chelate, which represents just one of the many ImAm to be recently explored. Overall, this review aims to highlight the versatility and vast potential of ImAm, a chelate like no other.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the University of Ottawa and the National Sciences and Engineering Council of Canada.Notes and references
- (a) C. Pettinari, F. Marchetti and A. Drozdov, β-Diketones and Related Ligands, in Comprehensive Coordination Chemistry, Elsevier, Amsterdam, 2nd edn, 2004, pp. 97–115 Search PubMed; (b) G. Aromi, P. Gamez and J. Reedijk, Coord. Chem. Rev., 2008, 252, 964–989 CrossRef CAS; (c) A. J. Brock, J. K. Clegg, F. Li and L. F. Lindoy, Coord. Chem. Rev., 2018, 375, 106–133 CrossRef.
- (a) T. Kajiwara, M. Nakano, Y. Kaneko, S. Takaishi, T. Ito, M. Yamashita, A. Igashira-Kamiyama, H. Nojiri, Y. Ono and N. Kojima, J. Am. Chem. Soc., 2005, 127, 10150–10151 CrossRef CAS; (b) A. M. Madalan, K. Bernot, F. Pointillart, M. Andruh and A. Caneschi, Eur. J. Inorg. Chem., 2007, 5533–5540 CrossRef CAS; (c) S. Wocadlo, W. Massa and J.-V. Folgado, Inorg. Chim. Acta, 1993, 207, 199–206 CrossRef CAS; (d) A. Kamiyama, T. Noguchi, T. Kajiwara and T. Ito, Angew. Chem., Int. Ed., 2000, 39, 3130–3132 CrossRef CAS; (e) M. G. Cowan, R. G. Miller, P. D. Southon, J. R. Price, O. Yazaydin, J. R. Lane, C. J. Kepert and S. Brooker, Inorg. Chem., 2014, 53, 12076–12083 CrossRef CAS; (f) S. Zebret, N. Dupont, G. Bernardinelli and J. Hamacek, Chem. – Eur. J., 2009, 15, 3355–3358 CrossRef CAS; (g) S. Zebret, E. Torres, E. Terreno, L. Guénée, C. Senatore and J. Hamacek, Dalton Trans., 2011, 40, 4284–4290 RSC.
- (a) R. Bonnett, D. C. Bradley and K. J. Fisher, Chem. Commun., 1968, 886–887 RSC; (b) R. Bonnett, D. C. Bradley, K. J. Fisher and I. F. Rendall, J. Chem. Soc. A, 1971, 1622–1627 RSC.
- J. E. Parks and R. H. Holm, Inorg. Chem., 1968, 7, 1408–1416 CrossRef CAS.
- L. Bourget-Merle, M. F. Lappert and J. R. Severn, Chem. Rev., 2002, 102, 3031–3066 CrossRef CAS.
- (a) Y.-C. Tsai, Coord. Chem. Rev., 2012, 256, 722–758 CrossRef CAS; (b) S. P. Sarish, S. Nembenna, S. Nagendran and H. W. Roesky, Acc. Chem. Res., 2011, 44, 157–170 CrossRef CAS; (c) M. Asay, C. Jones and M. Driess, Chem. Rev., 2011, 111, 354–396 CrossRef CAS; (d) F. T. Edelmann, Coord. Chem. Rev., 2015, 284, 124–205 CrossRef CAS; (e) F. T. Edelmann, Coord. Chem. Rev., 2016, 318, 29–130 CrossRef CAS; (f) W. E. Piers and D. J. H. Emslie, Coord. Chem. Rev., 2002, 233–234, 131–155 CrossRef CAS; (g) A. A. Mohamed, Coord. Chem. Rev., 2010, 254, 1918–1947 CrossRef CAS; (h) V. T. Annibale and D. Song, RSC Adv., 2013, 3, 11432–11449 RSC; (i) D. Zhu and P. H. M. Budzelaar, Dalton Trans., 2013, 42, 11343–11354 RSC; (j) S. J. Malthus, S. A. Cameron and S. Brooker, Coord. Chem. Rev., 2016, 316, 125–161 CrossRef CAS.
- (a) M. N. Kopylovich, A. J. L. Pombeiro, A. Fischer, L. Kloo and V. Y. Kukushkin, Inorg. Chem., 2003, 42, 7239–7248 CrossRef CAS; (b) M. N. Kopylovich, M. Haukka, A. M. Kirillov, V. Y. Kukushkin and A. J. L. Pombeiro, Chem. – Eur. J., 2007, 13, 786–791 CrossRef CAS; (c) N. A. Bokach, T. V. Kuznetsova, S. A. Simanova, M. Haukka, A. J. L. Pombeiro and V. Y. Kukushkin, Inorg. Chem., 2005, 44, 5152–5160 CrossRef CAS; (d) P. V. Gushchin, M. R. Tyan, N. A. Bokach, M. D. Revenco, M. Haukka, M.-J. Wang, C.-H. Lai, P.-T. Chou and V. Y. Kukushkin, Inorg. Chem., 2008, 47, 11487–11500 CrossRef CAS; (e) X. M. Guo, H. D. Guo, L. S. Fu, L. D. Carlos, R. A. S. Ferreira, L. N. Sun, R. P. Deng and H. J. Zhang, J. Phys. Chem., 2009, 113, 12538–12545 CAS.
- G. H. Sarova, N. A. Bokach, A. A. Fedorov, M. N. Berberan-Santos, V. Y. Kukushkin, M. Haukka, J. J. R. Fraústo-da-Silva and A. J. L. Pombeiro, Dalton Trans., 2006, 3798–3805 RSC.
- A. A. Starikova, Chem. Pap., 2018, 72, 821–828 CrossRef CAS.
- F. Kurzer, Chem. Rev., 1956, 56, 96–197 CrossRef.
- J. Baker, M. Kilner, M. M. Mahmoud and S. C. Wallwork, J. Chem. Soc., Dalton Trans., 1989, 837–841 RSC.
- N. Heße, R. Fröhlich, I. Humelnicu and E.-U. Würthwein, Eur. J. Inorg. Chem., 2005, 2189–2197 CrossRef.
- (a) N. Heße, R. Fröhlich, B. Wibbeling and E.-U. Würthwein, Eur. J. Org. Chem., 2006, 3923–3937 CrossRef; (b) H. J. Breslin, M. J. Kukla, R. W. Tuman, M. C. Rebarchak and C. R. Bowden, J. Med. Chem., 1993, 36, 1597–1603 CrossRef CAS; (c) I. Häger, R. Fröhlich and E.-U. Würthwein, Eur. J. Inorg. Chem., 2009, 2415–2428 CrossRef; (d) N. C. Aust, A. Beckmann, R. Deters, R. Krämer, L. Terfloth, S. Warzeska and E.-U. Würthwein, Eur. J. Inorg. Chem., 1999, 1189–1192 CrossRef CAS; (e) H. Behrens, R. Fröhlich and E.-U. Würthwein, Eur. J. Org. Chem., 2005, 3891–3899 CrossRef CAS; (f) M. Buhmann, E.-U. Würthwein, M. H. Möller and U. Rodewald, Angew. Chem., Int. Ed. Engl., 1994, 33, 2337–2339 CrossRef; (g) A. R. Siedle, R. J. Webb, M. Brostrom, R. A. Newmark, F. E. Behr and V. G. Young, Organometallics, 2004, 23, 2281–2286 CrossRef CAS; (h) H. V. R. Dias and S. Singh, Inorg. Chem., 2004, 43, 7396–7402 CrossRef CAS; (i) H. V. R. Dias, S. Singh and J. A. Flores, Inorg. Chem., 2006, 45, 8859–8861 CrossRef CAS.
- M. N. Kopylovich and A. J. L. Pombeiro, Coord. Chem. Rev., 2011, 255, 339–355 CrossRef CAS.
- D. A. Peak, J. Chem. Soc., 1952, 215–226 RSC.
- H. Ley and F. Müller, Ber. Dtsch. Chem. Ges., 1907, 40, 2950–2958 CrossRef.
- A. W. Titherley and E. G. Hughes, J. Chem. Soc., 1911, 99, 1493–1510 RSC.
- H. C. Brown and P. D. Schuman, J. Org. Chem., 1963, 28, 1122–1127 CrossRef CAS.
- (a) F. C. Schaefer, I. Hechenbleikner, G. A. Peters and V. P. Wystrach, J. Am. Chem. Soc., 1959, 81, 1466–1470 CrossRef CAS; (b) D. A. Safin, N. A. Tumanov, A. A. Leitch, J. L. Brusso, Y. Filinchuk and M. Murugesu, CrystEngComm, 2015, 17, 2190–2195 RSC.
- A. R. Siedle, R. J. Webb, F. E. Behr, R. A. Newmark, D. A. Weil, K. Erickson, R. Naujok, M. Brostrom, M. Mueller, S.-H. Chou and V. G. Young, Inorg. Chem., 2003, 42, 932–934 CrossRef CAS.
- (a) A. A. Leitch, I. Korobkov, A. Assoud and J. L. Brusso, Chem. Commun., 2014, 50, 4934–4936 RSC; (b) M. Yousaf, N. J. Yutronkie, R. Castañeda, J. A. Klein and J. Brusso, New J. Chem., 2017, 41, 12218–12224 RSC.
- S. Hayashi, M. Furukawa, Y. Fujino and S. Yoshimatsu, Chem. Pharm. Bull., 1969, 17, 329–334 CrossRef CAS.
- (a) Z. Brzozowski, F. Saczewski and M. Gdaniec, Eur. J. Med. Chem., 2000, 35, 1053–1064 CrossRef CAS; (b) L. Marín-Ocampo, L. A. Veloza, R. Abonia and J. C. Sepúlveda-Arias, Eur. J. Med. Chem., 2019, 162, 435–447 CrossRef; (c) S. Cascioferro, B. Parrino, V. Spano, A. Carbone, A. Montalbano, P. Barraja, P. Diana and G. Cirrincione, Eur. J. Med. Chem., 2017, 142, 328–375 CrossRef CAS.
- (a) M. Liu, L. Guo, S. Jin and B. Tan, J. Mater. Chem. A, 2019, 7, 5153–5172 RSC; (b) C. Krishnaraj, H. S. Jena, K. Leus and P. Van Der Voort, Green Chem., 2020, 22, 1038–1071 RSC.
- H. Wang, D. Jiang, D. Huang, G. Zeng, P. Xu, C. Lai, M. Chen, M. Cheng, C. Zhang and Z. Wanga, J. Mater. Chem. A, 2019, 7, 22848–22870 RSC.
- J. Artz, ChemCatChem, 2018, 10, 1753–1771 CrossRef CAS.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS.
- F. Li, S. I. Yang, Y. Ciringh, J. Seth, C. H. Martin, D. L. Singh, D. Kim, R. R. Birge, D. F. Bocian, D. Holten and J. S. Lindsey, J. Am. Chem. Soc., 1998, 120, 10001–10017 CrossRef CAS.
- J. Bañuelos, Chem. Rec., 2016, 16, 335–348 CrossRef.
- (a) J. M. Savard, R. Castañeda, B. Gabidullin and J. L. Brusso, Polyhedron, 2019, 162, 8–13 CrossRef CAS; (b) J. M. Savard, G. Brunet, A. Srinivasan, M. Murugesu and J. L. Brusso, Dalton Trans., 2018, 47, 14875–14879 RSC.
- (a) C. P. Constantinides and P. A. Koutentis, Adv. Heterocycl. Chem., 2016, 119, 173–207 CrossRef CAS; (b) P. J. Hayes, R. T. Oakley, A. Cordes and W. T. Pennington, J. Am. Chem. Soc., 1985, 107, 1346–1351 CrossRef CAS.
- P. J. Hayes, R. T. Oakley, A. W. Cordes and W. T. Pennington, J. Am. Chem. Soc., 1985, 107, 1346–1351 CrossRef CAS.
- R. T. Oakley, R. W. Reed, A. W. Cordes, S. L. Craig and J. B. Graham, J. Am. Chem. Soc., 1987, 109, 7745–7749 CrossRef CAS.
- R. T. Boeré, T. L. Roemmele and X. Yun, Inorg. Chem., 2011, 50, 5123–5136 CrossRef.
- (a) A. A. Leitch, I. Korobkov, A. Assoud and J. L. Brusso, Chem. Commun., 2014, 50, 4934–4936 RSC; (b) E. Kleisath, N. J. Yutronkie, I. Korobkov, B. M. Gabidullin and J. L. Brusso, New J. Chem., 2016, 40, 4472–4479 RSC; (c) N. J. Yutronkie, P. Tami, S. Singh, E. Kleisath, B. M. Gabidullin, R. Davis and J. L. Brusso, New J. Chem., 2017, 41, 2268–2276 RSC; (d) N. J. Yutronkie, A. A. Leitch, I. Korobkov and J. L. Brusso, Cryst. Growth Des., 2015, 15, 2524–2532 CrossRef CAS.
- (a) K. L. M. Harriman, A. A. Leitch, S. A. Stoian, F. Habib, J. L. Kneebone, S. I. Gorelsky, I. Korobkov, S. Desgreniers, M. L. Neidig, S. Hill, M. Murugesu and J. L. Brusso, Dalton Trans., 2015, 44, 10516–10523 RSC; (b) N. J. Yutronkie, I. A. Kühne, I. Korobkov, J. L. Brusso and M. Murugesu, Chem. Commun., 2016, 52, 677–680 RSC; (c) K. L. Harriman, I. A. Kühne, A. A. Leitch, I. Korobkov, R. Clérac, M. Murugesu and J. L. Brusso, Inorg. Chem., 2016, 55, 5375–5383 CrossRef CAS.
- R. Norrestam, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1984, 40, 955–957 CrossRef.
- For example, see: (a) V. Duros, H. Sartzi, S. J. Teat, Y. Sanakis, O. Roubeau and S. P. Perlepes, Inorg. Chem. Commun., 2014, 50, 117–212 CrossRef CAS; (b) M. M. Turnbull, M. Y. Wei and R. D. Willett, J. Coord. Chem., 1995, 35, 11–17 CrossRef CAS; (c) M. N. Kopylovich, J. Lasri, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Dalton Trans., 2009, 3074–3084 RSC; (d) T. Kajiwara, A. Kamiyama and T. Ito, Chem. Commun., 2002, 1256–1257 RSC; (e) M.-L. Tong, Y.-M. Wu, Y.-X. Tong, X.-M. Chen, H.-C. Chang and S. Kitagawa, Eur. J. Inorg. Chem., 2003, 2385–2388 CrossRef CAS; (f) D. R. Aris, J. Barker, P. R. Phillips, N. W. Alcock and M. G. H. Wallbridge, J. Chem. Soc., Dalton Trans., 1997, 909–910 RSC; (g) C. R. G. Calona, A. S. Filatov, B. Pedro and E. V. Rybak-Akimova, Organometallics, 2019, 38, 2512–2522 CrossRef; (h) W. J. Bland, R. D. W. Kemmitt, I. W. Nowell and D. R. Russell, Chem. Commun., 1968, 1065–1066 RSC; (i) M. N. Kopylovich, A. M. Kirillov, E. A. Tronova, M. Haukka, V. Y. Kukushkin and A. J. L. Pombeiro, Eur. J. Inorg. Chem., 2010, 2425–2432 CrossRef CAS; (j) J.-P. Zhang, Y.-Y. Lin, X.-C. Huang and X.-M. Chen, J. Am. Chem. Soc., 2005, 127, 5495–5506 CrossRef CAS.
- (a) Y.-Q. Xie, J.-J. Li, Y. Guo, Y.-M. Zhang and T.-B. Wei, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2013, 69, m3 CrossRef CAS; (b) G. Thiele, B. Wagner and S. Dehnen, Eur. J. Inorg. Chem., 2015, 5329–5334 CrossRef CAS.
- I. Pernik, B. J. Maitland, A. Strasch and C. Jones, Can. J. Chem., 2018, 96, 513–521 CrossRef CAS.
- T. Bolaño, T. B. Gunnoe and M. Sabat, Dalton Trans., 2013, 42, 347–350 RSC.
- (a) N. Q. Shixaliyev, A. M. Maharramov, A. V. Gurbanov, N. V. Gurbanova, V. G. Nenajdenko, V. M. Muzalevskiy, K. T. Mahmudov and M. N. Kopylovich, J. Mol. Struct., 2013, 1041, 213–218 CrossRef CAS; (b) N. G. Shikhaliyev, A. M. Maharramov, V. M. Muzalevskiy, V. G. Nenajdenko and V. N. Khrustalev, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, m1220–m1221 CrossRef CAS.
- N. Q. Shixaliyev, A. V. Gurbanov, A. M. Maharramov, K. T. Mahmudov, M. N. Kopylovich, L. M. D. R. S. Martins, V. M. Muzalevskiy, V. G. Nenajdenko and A. J. L. Pombeiro, New J. Chem., 2014, 38, 4807–4815 RSC.
- M. N. Kopylovich, J. Lasri, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Eur. J. Inorg. Chem., 2011, 377–383 CrossRef CAS.
- (a) T. T. Ponduru, C. Qiu, J. X. Mao, A. Leghissa, J. Smuts, K. A. Schug and H. V. R. Dias, New J. Chem., 2018, 42, 19442–19449 RSC; (b) V. A. K. Adriraju, J. A. Flore, M. Yousufuddin and H. V. R. Dias, Organometallics, 2012, 31, 7926–7932 CrossRef.
- H. V. R. Dias, J. A. Flores, M. Pellei, B. Morresi, G. G. Lobbia, S. Singh, Y. Kobayashi, M. Yousufuddin and C. Santini, Dalton Trans., 2011, 40, 8569–8580 RSC.
- K. Bakthavachalam and N. Dastagiri Reddy, Organometallics, 2013, 32, 3174–3184 CrossRef CAS.
- (a) K. Bakthavachalam, B. Raghavendra and N. Dastagiri Reddy, Appl. Organomet. Chem., 2017, 31, e3833 CrossRef; (b) N. V. Kulkarni, A. Das, S. G. Ridlen, E. Maxfield, V. A. K. Adiraju, M. Yousufuddin and H. V. R. Dias, Dalton Trans., 2016, 45, 4896–4906 RSC.
- (a) T. Kajiwara and T. Ito, Chem. Commun., 2002, 1256–1257 RSC; (b) M. N. Kopylovich, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Dalton Trans., 2009, 3074–3084 RSC; (c) V. Duros, H. Sartzi, S. J. Teat, Y. Sanakis, O. Roubeau and S. P. Perlepes, Inorg. Chem. Commun., 2014, 50, 117–121 CrossRef CAS.
- R. Castañeda, A. Hollingshead, B. Gabidullin and J. L. Brusso, Cryst. Growth Des., 2017, 17, 6572–6578 CrossRef.
- R. Castañeda, K. L. M. Harriman, J. W. L. Wong, B. Gabidullin, M. Murugesu and J. L. Brusso, Eur. J. Inorg. Chem., 2019, 963–972 CrossRef.
- R. Castañeda, M. Rouzières, R. Clérac and J. L. Brusso, J. Mater. Chem. C, 2020, 8, 4401–4407 RSC.
- Z. Dubrawski, J. Heidebrecht, B. M. P. Lombardi, A. S. Hyla, J. Willkomm, C. L. Radford, J.-B. Lin, G. C. Welch, S. Ponnurangam, R. Roesler, D. E. Prokopchuk and W. E. Piers, Sustainable Energy Fuels, 2019, 3, 1172–1181 RSC.
- B.-B. Guo, Y.-J. Lin and G.-X. Jin, Dalton Trans., 2017, 46, 8190–8197 RSC.
- (a) M. N. Kopylovich, Y. Y. Karabach, M. D. C. G. da Silva, P. J. Figiel, J. Lasri and A. J. L. Pombeiro, Chem. – Eur. J., 2012, 18, 899–914 CrossRef CAS; (b) S. Jana, S. Khan, A. Bauzá, A. Frontera and S. Chattopadhyay, J. Mol. Struct., 2017, 1127, 355–360 CrossRef CAS; (c) O. V. Nesterova, M. N. Kopylovich and D. S. Nesterov, Inorganics, 2019, 7, 82 CrossRef CAS; (d) E. V. Andrusenko, E. V. Kabin, A. S. Novikov, N. A. Bokach, G. L. Starova and V. Y. Kukushkin, New J. Chem., 2017, 41, 316–325 RSC.
- I. I. Eliseev, P. V. Gushchin, Y.-A. Chen, P.-T. Chou, M. Haukka, G. L. Starova and V. Y. Kukushkin, Eur. J. Inorg. Chem., 2014, 4101–4108 CrossRef CAS.
- T. Tanaka, R. Nouchi, Y. Nakao, Y. Arikawa and K. Umakoshi, RSC Adv., 2014, 4, 62186–62189 RSC.
- (a) Q. Xie, H. Tong and M. Zhou, Inorg. Chem. Commun., 2014, 44, 37–40 CrossRef CAS; (b) F. Li, L. Yan, H. Tong and M. Zhou, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2015, 71, m54 CrossRef CAS; (c) D. Tian, Q. Xie, L. Yan, H. Tong and M. Zhou, Inorg. Chem. Commun., 2015, 58, 35–38 CrossRef CAS.
- F. Li, X. Qiao, M. Wang, M. Zhou, H. Tong, D. Guo and D. Liu, Polyhedron, 2013, 52, 639–644 CrossRef.
- X. Wang, Z. Guo, H. Tong and M. Zhou, J. Organomet. Chem., 2017, 828, 10–15 CrossRef CAS.
- (a) Z. Guo, F. Liu, H. Tong, J. Chao, H. Wang and M. Zhou, Polyhedron, 2018, 151, 273–278 CrossRef CAS; (b) Z. Guo, D. Tian, Q. Yang, H. Tong, Y. Wang and M. Zhou, Polyhedron, 2019, 166, 162–165 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |