
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDetection and beyond: challenges and advances in aptamer-based biosensors
Hyebin
Yoo†
a,
Hyesung
Jo†
a and
Seung Soo
Oh
 *ab
*ab
aDepartment of Materials Science and Engineering, Pohang University of Science Technology (POSTECH), Pohang, 37673, South Korea. E-mail: seungsoo@postech.ac.kr
bSchool of Interdisciplinary Bioscience and Bioengineering, Pohang University of Science and Technology (POSTECH), Pohang, 37673, South Korea
First published on 26th October 2020
Abstract
Beyond traditional needs of biosensors such as high sensitivity and selectivity for analyte detection, newly emerging requirements including a real-time detection ability and in-field applicability have been gradually emphasized to address clinical and environmental availability. Highly programmable, synthetic aptamers that can specifically recognize a broad range of targets have the potential to fulfill these requirements; cooperative binding to target molecules achieves a significant increase in sensitivity, and binding-induced structure-switching enables target detection even in complex mixtures. Due to the availability of chemical synthesis and functional modifications, these artificial ligand materials are easily installed in many devices, and the amenability to modularization allows the aptamer-based biosensors to diversify detectable targets and signaling processes. In this review, we highlight current progress in the development of aptamer-based, next-generation biosensors including new types of field-effect transistors, electrochemical detectors, and microfluidic devices. As the nucleic acid aptamers have been rapidly generated by various in vitro selection techniques, the use of the versatile nanostructures is expected to expand further to include in-field and real-time biosensors.
 Hyebin Yoo | Hyebin Yoo graduated summa cum laude with her BS in Materials Science and Engineering from Pohang University of Science and Technology (POSTECH), South Korea in 2018. She continues to pursue her PhD, supervised by Prof. Seung Soo Oh at POSTECH. She obtained several fellowships including a master's scholarship from the Kwanjeong Educational Foundation and a Global PhD Fellowship from the National Research Foundation of Korea. Her research interests focus on the synthesis of artificial molecular channels based on nucleic acid aptamers as well as the generation of high-performance biosensors using hydrogel ionic transistors. |
 Hyesung Jo | Hyesung Jo graduated magna cum laude with her BS in Materials Science and Engineering from Pohang University of Science and Technology (POSTECH), South Korea in 2018. She is pursuing her PhD under the supervision of Prof. Seung Soo Oh at POSTECH. Her research currently focuses on the development of novel aptamer-based site-specific bioconjugation technology that can contribute to molecular sensing and next-generation antibody–drug conjugates. |
 Seung Soo Oh | Dr Seung Soo Oh is the Il-Beom chaired associate professor in the Department of Materials Science and Engineering at POSTECH, South Korea. He graduated summa cum laude with his BS and MS in Materials Science and Engineering from Seoul National University. He received his PhD in Materials from UCSB in 2012 with Prof. Tom Soh, focusing on high-throughput generation of multi-functional aptamers. He continued his research career as a postdoc in Prof. Craig Hawker's lab for DNA–polymer hybrid synthesis. Between 2014 and 2016, he worked as a research fellow with Prof. Jack Szostak at Massachusetts General Hospital/Harvard Medical School for in vitro selection of ribozymes. His research interests include the synthesis of biomolecular materials capable of performing cellular functions for diagnostic and therapeutic applications. |
Introduction
Precise and rapid detection of bioanalytes is invaluable in our life. We are currently experiencing a global pandemic of COVID-19, an infectious disease caused by SARS-CoV-2 (Severe Acute Respiratory Syndrome Coronavirus 2), and the growing number of COVID-19 deaths reminds us of the significance of biosensors that enable diagnostic detection of specific targets such as coronavirus biomarkers.1 As the analytical devices convert the presence or concentrations of certain biological analytes to recognizable signals (e.g., electrical current, fluorescence, and color), we can easily and rapidly detect desired targets in many types of samples, including clinical or environmental specimens. Development of fast, accurate, and sensitive biosensors could achieve early diagnosis of diseases and contribute to prevention of the spread of infectious diseases by making us ready to provide optimum care for patients.In recent years, the scope of biosensing application has widened, and the economic impacts of biosensors have grown substantially. The rapid increase in the prevalence of chronic illnesses (e.g., cancer, diabetes, and asthma) has elicited the interest in individual healthcare and point-of-care (PoC) diagnostics and thereby accelerated the growth of biosensing research.2 The food industry can also benefit; in situ biosensors can monitor food quality and can forestall possible contamination by detecting pathogenic microorganisms and toxins.3 Many types of biosensors also have the uses in various other fields, including drug discovery, environmental monitoring, metabolic engineering, and even forensic science.4 The global biosensors market size is growing fast and is anticipated to expand from US$ 21.2 billion in 2019 to US$ 31.5 billion by 2024 at a compound annual growth rate of 8.3%.5
However, current biosensors often do not have sufficient sensitivity and selectivity in target detection. For example, early diagnosis of diseases requires exceptionally high sensitivity and selectivity because most biomarker proteins exist in blood as a trace amount (10 pg mL−1 to 1 ng mL−1), which is far below the attainable detection limit of current diagnostic sensors.6 Moreover, the low selectivity of biosensors can lead to an excessive number of false positives or false negatives during several diagnostic tests.7 Accordingly, only a few biosensors have been commercialized, e.g., glucose meters. Blood sugar levels are relatively easy to detect; in blood, the concentrations of glucose (700 to 1300 μg mL−1) are much higher than those of any other disease biomarkers,8 so glucose detection does not require large samples of blood. Even if desired biosensors are available, they often require time-consuming, multi-step processes and specialized instruments; these requirements impede the adoption of these biosensors in emerging applications such as homecare diagnostics and PoC.9
To address these challenges, the performance of biosensors, including sensitivity and specificity in target detection, must be further enhanced, and these needs emphasize the significance of reliable molecular recognition elements. Typically, biosensors consist of a biological recognition agent that binds with or recognizes specific bioanalytes, a signal transducer which transforms the analyte–receptor interaction into a measurable signal, and a signal readout that displays the transduced signal which must be easily recognized by users. The molecular recognition elements are usually bioreceptors such as antibodies, enzymes, nucleic acids, and even whole cells, and these elements mainly affect the limit of detection (LOD), target selectivity, and detection ranges. However, the inherent limitations of conventional bioreceptors have impeded the improvement of biosensor attributes.10 For example, protein-based bioreceptors such as monoclonal antibodies are limited by their structural complexity; they are difficult to be physically or chemically modified, so improving their binding affinity to target molecules is significantly challenging.11 Non-specific adsorption of the bioreceptors reduces the selectivity of biosensors, which can be a serious problem in target detection. The disadvantages of current biosensors are the consequences of the constrained capability of molecular recognition elements.
Use of nucleic acid aptamers may be a great way to overcome these limitations (Fig. 1). By Watson–Crick base pairing, nucleic acids such as DNAs and RNAs build thermodynamically favored nanostructures. Among the incredibly diverse forms of three-dimensional (3D) complex structures, a unique folding may be able to bind to a specific target with high affinity and specificity; this is the definition of ‘aptamer’ (Fig. 1, top).12 To generate the sequence-controllable biopolymers, a variety of in vitro selection techniques have been developed, and the in vitro selected aptamers can be tailored for various targets (e.g., proteins, hormones, metabolites, ions, and even whole cells).12
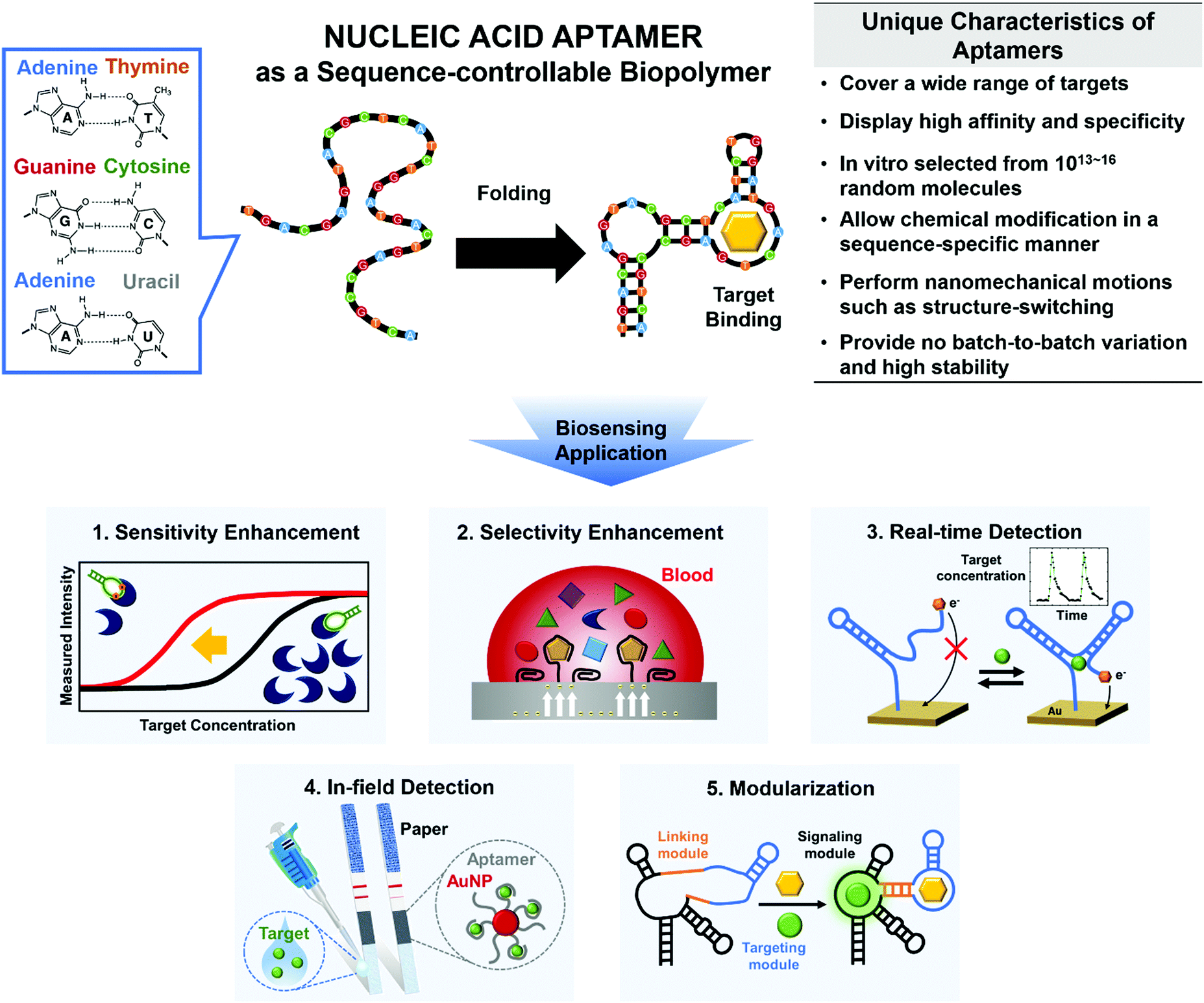 | ||
| Fig. 1 Main characteristics and functionalities of nucleic acid aptamers to overcome various limitations of conventional biosensors. Specific base pairing (top, left) folds the sequence-controllable biopolymers into thermodynamically-favored 3D nanostructures that enable molecular recognition (top, middle). The synthetic aptamers have unique features that can facilitate the development of next-generation biosensors (top, right). Here, we review technical advances in the development of aptamer-based biosensors, such as increases in sensitivity and selectivity, and actualization of newly emerging real-time and in-field detection applications, along with aptameric biosensors’ interesting properties, such as amenability to modularization (bottom). | ||
The feasibility of chemical synthesis gives the synthetic aptamers an edge over other bioreceptors. For instance, to improve the binding affinity to target molecules, the nucleobases and backbones within the aptamers can be readily modified in a sequence-specific manner, whereas monoclonal antibodies may require complex noncanonical protein translation before the site-specific chemical modification.13 The aptamers’ simple yet exponential self-amplification is also highly valuable to increase the sensitivity of biosensors, and this approach is not applicable to other bioreceptors.12 Not only the sensitivity but also the selectivity can be significantly improved; rather than mere binding-derived signals from conventional bioreceptors, the signals triggered by binding-induced structure-switching or catalytic activities can be produced by the versatile aptamers.14 These specific signals are expected only when the desired targets are present, so the aptamer-based biosensors have the potential to reduce false signals drastically.
Besides breaking the limits of sensitivity and selectivity, aptamers can be adapted to meet the evolving requirements of biosensors. For instance, continuous analysis of drug response is a requisite for personalized medicine to individually determine appropriate doses to achieve maximum efficacy.15 The rapid and reversible conformational change of aptamers can be exploited to attain the sensitivity and selectivity that are suitable for continuous real-time detection.15 Furthermore, an ability for in-field detection would enable daily healthcare monitoring or frequent environmental assessments.16 Aptamers can be easily modified to detect a wide range of specific targets and therefore provide an appropriate sensing tool for such in-field detectors while being physically and chemically robust, and competitively priced. In addition, the molecular sensors that use aptamers can be modular, and this trait may facilitate the development of multifunctional biosensors that are easy to use.
Aptamers can show the potential to fulfill these needs beyond a simple increase in binding strength. Focusing on how aptamers can address these technical challenges of biosensors, we specifically discuss the increases in sensitivity and selectivity of biosensors, and real-time and in-field detection of targets; then we present modular multifunctionalization and its application in next-generation biosensors (Fig. 1, bottom).
1. Increasing sensitivity
Increasing sensitivity in target detection is one of the most crucial challenges when developing biosensors.17 Diagnosis of diseases at their early stage requires an exceptionally low LOD because, at that stage, the biomarkers may exist at very low levels.18 Recently, several disease indicators including circulating tumor cells (CTCs) and exosomes have been reported to be effective for precise diagnosis, but their concentrations in blood are extremely low.18 Sample volumes may also be small; examples include sweat, tears, and saliva, and blood from infants and children,19 so their examination requires high sensitivity. The availability of ultra-sensitive biosensors would enable the evaluation of the non-invasive clinical samples, yielding painless diagnostic procedures.To increase the sensitivity of biosensors, many kinds of biosensing techniques20 and devices17,21,22 have been developed. For example, target-specific bioreceptors can be linked to the gate of field-effect transistor (FET). In response to target binding, the resulting ‘bio-FET’ amplifies the signal to convert small differences in electrostatic potential to large changes in electric current.21 However, such device-dependent signal amplification cannot fully meet the requirement of LOD in biosensing applications. To further increase the sensitivity, the bioreceptors should have improved affinity to targets; for this use, aptamers have demonstrated great potential for the development of ultra-sensitive biosensors (Table 1).
| Sensor type | Material | Target | Detection limit | Characteristic | Application | Ref. |
|---|---|---|---|---|---|---|
| Electrochemical sensor | DNA aptamer, DNA tetrahedron, and gold electrode | Protein (lysozyme) | 3.511 pM | Terminal fixation of aptamers | Allergenic protein detection in a real complex matrix | (Zhao et al., 2019) |
| Split DNA aptamer, gold electrode, and gold nanoparticle (AuNP) | Small molecule (ATP and cocaine) | Cocaine: 1 nM | AuNP modification with multiple split aptamers | Ultra-sensitive detection of small molecules | (Zhao et al., 2015) | |
| ATP: 5 nM | ||||||
| Fluorescence sensor | DNA aptamer, circular template, phi29 polymerase, and reduced graphene oxide (rGOx) | Small molecule (ATP), DNA, and protein (thrombin) | 10 pM | Single strand-specific DNA absorption of rGOx and isothermal signal amplification of RCA | Ultra-sensitive detection of wide-ranging targets in biological, medical, and environmental samples | (Liu et al., 2014) |
| DNA aptamer and DNA probe for HCR | Protein (tyrosine kinase-7) | 1 pM | Target binding-induced HCR on cellular surfaces | High-contrast fluorescence imaging and detection of tumor cells | (Wang et al., 2015) | |
| DNA aptamer and graphene | Protein (thrombin) | 31.3 pM | Graphene-dependent fluorescence quenching of dyes and desorption of G-quadruplex | Simple and highly sensitive FRET detection in blood | (Chang et al., 2010) | |
| DNA beacon and Exonuclease III | DNA | 20 aM | Fluorescence amplification by exonuclease III-aided target recycling | Extremely sensitive detection of target DNA by naked eyes | (Zuo et al., 2010) | |
| Field-effect transistor sensor | DNA aptamer, silicon nanowire, and Si substrate | Small-molecule (dopamine) | <10−11 M | Multiple-parallel-connection of silicon nanowires and target-induced electric field | Real-time cellular response monitoring of drug treatment or environmental alteration | (Li et al., 2013) |
| DNA aptamer, liquid ion, graphene, and polyethylene naphtalate (PEN) | Ion (Hg2+) | 10 pM | Liquid-ion gated field-effect and flexible graphene/PEN substrate | Real-time ion detection by flexible graphene-based aptasensor | (An et al., 2013) | |
| PNA probe, rGOx, and Si substrate | DNA | 100 fM | Strong hybridization between negatively charged DNA and netural PNA | Single nucleotide polymorphism (SNP) detection for PoC | (Cai et al., 2014) | |
1.1. Improvement of aptamer's binding affinity
The binding affinity of aptamers can be readily strengthened compared to other types of bioreceptors.23 High affinity indicates that bioreceptors can bind to targets even at small concentrations, and the improvement of this ability directly increases the detection sensitivity. To increase this binding affinity, several approaches have been developed; they include chemical modification, cooperative binding, and structural stabilization.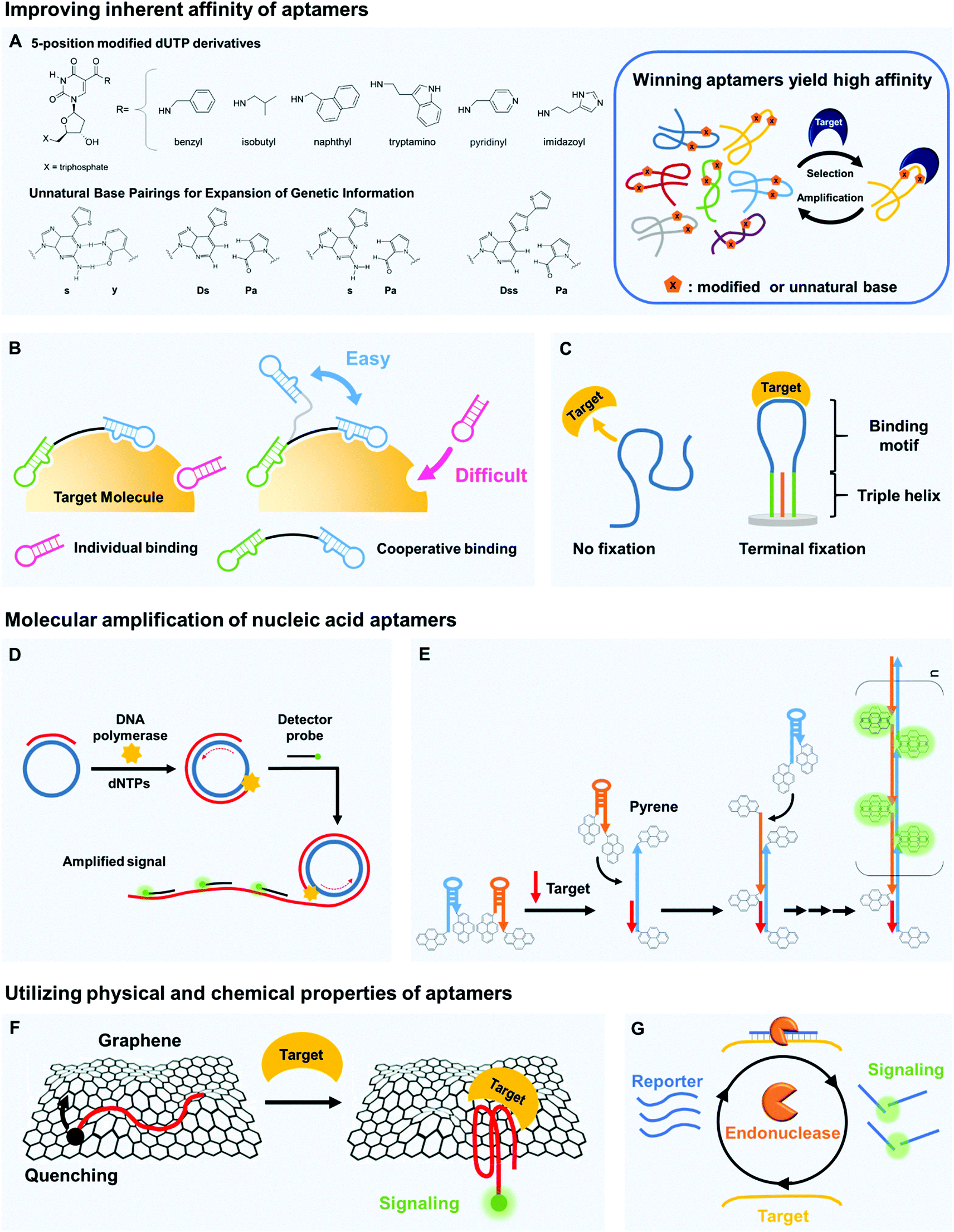 | ||
| Fig. 2 Aptamers can be key components of biosensors to increase sensitivity in target detection. (A) The inherent binding affinity of aptamers can be strengthened by chemical modification of nucleic acids, e.g., by attaching hydrophobic moieties to nucleotides and by adding non-canonical pairing bases. (B) Multivalency by cooperative binding of multiple aptamers and (C) terminal fixation of folding structures can be also effective to improve the target binding capability of aptamers. (D) Molecular amplification techniques such as rolling circle amplification (RCA) can be useful to develop ultra-sensitive biosensors by significantly increasing detectable signals. In RCA, primers are bound to circular templates; polymerases extend the primers to yield long single-stranded concatemers with tandem repeat structures, and the repeated hybridization of dye-labeled strands with tandem repeats produces amplified fluorescent signals. (E) Hybridization chain reaction (HCR) can also be used to increase sensitivity. In HCR, introduction of DNA targets can trigger a hybridization cascade of signaling probes such as pyrene-conjugated hairpin probes and thereby facilitate ultra-sensitive target detection. (F) Unique physical properties of nucleic acid aptamers can contribute to highly sensitive target detection. By π–π stacking, single-stranded nucleic acids bind well to graphene surfaces, whereas the target-bound aptamers are released due to folding in tertiary structures. This folding change of aptamers yields changes in fluorescent or electrical signals, which can be easily detected. (G) Target-bound aptamers are less vulnerable to nuclease digestion than their target-free forms, and this feature can be applied to signal accumulation; the exonuclease-based, enzyme-assisted target recycling (EATR) technique can significantly decrease the limit of detection by summing fluorescence signals. | ||
Natural nucleobases can be modified by attaching functional groups that tighten the binding of aptamers to their targets (Fig. 2A). Chemical linkage of aromatic or aliphatic functional groups to uridines by amide linkages can yield dUTP derivatives that are modified at the 5-position.26 The amide linkages contribute additional hydrogen bonding, so the aptamers that contain these modified uridines strongly interact with their target molecules. DNA modification chemistries have been exploited to prepare the dUTP derivatives and to confirm their compatibility with the polymerase chain reaction (PCR). Biological polymerases enabled incorporation of the modified uridine into DNA templates, so the aptamers to target human protein TNFRSF9 (Kd ∼ 5 nM) were successfully enriched by a PCR amplification process during Systematic Evolution of Ligands by EXponential enrichment (SELEX).27,28 The modification of dUTP has also been used in other strategies to strengthen the binding by aptamers. The Slow Off-rate Modified Aptamer (SOMAmer)29 is a new class of high-affinity aptamer that incorporates the uridine modified at the 5-position. Although the base-modified DNA library generally enhanced the efficacy of acquiring protein-binding aptamers with superior binding affinity, it remained unproven whether it could be applicable for generation of small-molecule-binding aptamers. Recently, there has been the first report that demonstrated improved binding properties of base-modified DNA aptamers capable of recognizing a small molecule target.30 A modified DNA library that included (E)-5-(2-(N-(2-(N6-adeninyl)ethyl))carbamylvinyl)-uracil bases yielded the high-affinity aptamers that bind to camptothecin, a potent antitumor drug (Kd ∼ 0.039 μM).
Chemical modifications can be further performed after the SELEX process to fine-tune the aptamer properties.31 For example, the SOMAmers initially selected to bind to platelet-derived growth factor B (PDGF-BB) were further optimized by substituting the original modification (in this case, the benzyl group) with 14 alternative moieties.32 By screening the best single substitutions, they could yield the modified aptamer that showed 5 times more improved binding to PDGF-BB than the unmodified aptamer. Similarly, the SOMAmers capable of binding to interleukin-6 (IL-6) were isolated and further optimized by substitution scans (2′-OMe and C3-spacer substitutions and beneficial 5-dU substitutions), leading to 10-fold enhanced IL-6 binding and 20-fold improved inhibition activity.33 However, the post-SELEX modification can hinder the binding characteristics of aptamers by altering their folding structures, which would be extremely difficult to predict.
Backbone replacement can also increase binding capability of aptamers. Several artificial DNA analogs with unnatural backbones have been reported, and among them, peptide nucleic acids (PNAs) are the best-known. Instead of phosphate-ribose as in DNA, PNAs have amide backbones of (N-(2-aminoethyl)glycine), which are electrically neutral; they are not repelled electrostatically by target DNAs or RNAs, so remarkably stable hybridization is possible.34 The superior binding affinity and chemical stability of PNAs has been exploited to detect DNA targets. For example, an ultra-sensitive biosensor used PNA probes on a reduced graphene oxide FET (rGO-FET) to sense specific DNAs with an LOD ∼ 100 fM, which is an order of magnitude lower than achieved by the biosensors that use complementary DNA.35
Encouraged by the success of the PNA-based DNA detector, researchers have considered developing PNA aptamers and relevant biosensors. The PNA that has the identical base sequence to that of well-known DNA aptamer may detect the same target,36 but a direct selection from a randomized PNA library, if available, would be a powerful way to create PNA aptamers.37 Xeno nucleic acids (XNAs) can also be used to generate target-specific aptamers; some of the XNAs that have been used include threose nucleic acid (TNA),38 P-alkyl phosphonate nucleic acid (phNA),39 and fluoro arabino nucleic acid (FANA).40In vitro selection techniques enabled the generation of TNA aptamers for thrombin that have a similar Kd to previously reported DNA and RNA aptamers, and phNA aptamers that bind to Streptavidin with comparable affinity to the same target DNA aptamer have been isolated. Importantly, in vitro selected FANA aptamers that bind HIV-1 reverse transcriptase showed a picomolar affinity, which is equivalent to that of the tightest binding DNA aptamer.40 These examples show that the XNAs can be excellent resources for the creation of high-affinity aptamers.
Directed evolution can generate optimal bivalent aptamers that have strong overall affinity.42 The main challenge in applying cooperative binding interactions is to rationally design an ideal scaffold that can place two different site-binding aptamers at the optimal position and orientation with respect to a target protein, especially when the folding structures of the aptamers are unknown. By employing an in vitro selection technique, the ideal scaffold of bivalent aptamers specific for thrombin was created; two different thrombin aptamers were connected with a randomized DNA linker (35 nt in length), and the optimal bivalent aptamer that showed an extremely high affinity to the target thrombin was isolated by microfluidic in vitro selection technology. The resulting divalent aptamer yielded Kd below 10 pM, which is >200 times higher affinity than monomeric aptamers.
To create such reagents that have bivalent affinity, aptamer pairs must be prepared in advance. However, their discovery is challenging because conventional selection methods were not intended to isolate aptamers that can recognize different binding sites. To address this challenge, the use of aptamer microarray was explored to efficiently identify new aptamers that did not share the same binding sites of human angiopoietin-2 (Ang2).43 After the identification of Ang2 aptamer pairs, an ultra-high-affinity bivalent aptamer was constructed by using a flexible linker to join the identified pairs; it could capture Ang2 at a Kd as low as 97 pM, which is more than 200 times higher affinity than monovalent aptamers for Ang2.
The distance between multiple linked aptamers may be optimized using a well-defined scaffold. DNA origami is complex nanoscale folding of DNA to create 2D and 3D shapes or scaffolds; using the DNA origami as a molecular pegboard, various sub-nanometer scale distances between different epitope-targeting aptamers can be examined with high accuracy.44 The distance between active sites in a bivalent binding significantly affected its sensitivity in target detection; an optimal spacing maximized the binding interaction of a bivalent thrombin aptamer. The resulting bivalent aptamer on the DNA origami had Kd ∼ 10 nM, which is about 10 times higher affinity than individual aptamers. Atomic force microscopy (AFM) measurements enabled to visualize the pincer-like strong grabbing of thrombin proteins by the bivalent aptamer on the DNA origami.
An intrinsic disorder-based generalizable strategy for the rational design of allosterically cooperative receptors was developed, and the cooperativity of two doxorubicin (DOX)-binding aptamers, originally noncooperative receptors, was explored.45 When the DOX aptamer was engineered to be split, a tandem repeat of one half of the aptamer was linked to a tandem repeat of the second half of the same aptamer by 30-base polythymine. As a result, an ideally cooperative behavior was observed for a two-site DOX receptor. The binding event of the first DOX led the two halves to associate together, forming both binding sites. It was relatively easy for the second DOX to be occupied in the other binding site, showing a cooperative response. By controlling the length of the polythymine linker, it was also possible to regulate the extent of cooperativity as well as a dynamic range.
Enthalpy-driven high-affinity aptamers have been developed using molecular crowding evolution.47 Aptamer screening in a molecular crowding environment (i.e., in blood plasma) both restricts the aptamer's degree of freedom and suppresses the entropic contribution to the binding interaction. Consequently, the binding events that occurred in this environment were mostly enthalpy-driven, so the isolation step predominantly yielded the aptamers that favored noncovalent yet specific binding. This method succeeded in the generation of aptamers that were specific for the tumor biomarker protein, EpCAM; they had 6.5 times higher binding affinity than those evolved using conventional SELEX and demonstrated outstanding accuracy in the detection of CTCs in blood samples.
1.2. Molecular amplification of nucleic acid aptamers
Nucleic acid amplification can benefit the development of highly sensitive biosensors. Nucleic acid aptamers are exponentially amplifiable using PCR; this is a powerful feature that cannot be easily matched by other affinity reagents. The PCR is widely used for diverse fields such as infectious disease diagnosis, clinical medicine, and forensic science, and simpler and easier amplification techniques are being developed.48 In particular, isothermal amplification does not require thermal cycling, so it is being evaluated as a strategy to achieve simple yet ultra-sensitive target detection.49 Here, we describe rolling circle amplification (RCA), hybridization chain reaction (HCR), and loop-mediated isothermal amplification (LAMP).RCA is an efficient isothermal technique that enables amplification of probe sequence by a factor of more than 109 (Fig. 2D). If a short primer binds to a circular template, DNA polymerases (e.g., ∅29 DNA polymerase) extend the primer to yield a long single-strand concatemer composed of tandem repeats. An ultra-sensitive biosensing tool that used the RCA and rGO substrates was developed.50 The rGOs can nonspecifically absorb single-stranded DNA aptamers flanked by primer regions, but if macromolecular targets such as proteins bind to the aptamers, the conformationally changed aptamers detach from the rGOs. If the flanking primer regions are simultaneously liberated to bind to circular DNA templates, long strands that contain repetitive sequence can be subsequently generated, which is easily detected using molecular beacons. This RCA-based biosensor achieved an LOD ∼ 10 pM, which is nearly two orders of magnitude lower than those of the biosensors without RCA amplification.
HCR is an efficient target-triggered amplification system (Fig. 2E). Two different DNA hairpin structures are rationally designed as a target DNA strand to initiate a hybridization cascade, and a long polymeric nanowire with repeated units can be formed by continuous cross-opening of DNA hairpins.51 The combination of the amplification capability of HCR and pyrene-conjugated hairpin probes yielded a highly sensitive DNA detection system.52 The pyrene moieties on hairpin probes are spatially separated in solution, so in the absence of the target, the only emission peak is that of single pyrene monomers. However, when the target DNA is introduced, it opens the hairpin structures to yield a long nicked double-helix in which the pyrene moiety from one probe becomes closely positioned with that from neighboring probes. As numerous excimers are produced, the LOD is in the femtomole range. This DNA targeting platform can be further upgraded as an aptamer-based biosensors because the HCR can also be triggered by a structure-switching aptamer. The real-time HCR activation and amplification was used for fluorescence imaging of tumor cells;53 when bound to targeted CEM cells, the structure of an sgc8 aptamer changed and thereby provoked HCR on the cell surface. These changes enabled sensitive fluorescence imaging and efficient therapy that targets tumors, and this aptamer-triggered HCR detected a membrane protein, tyrosine kinase-7 (PTK7), with an LOD ∼ 1 pM.
LAMP is a highly sensitive technique that uses four kinds of specially-constructed primers that recognize six distinct regions of target DNA.54 This technique can rapidly replicate a few copies of DNA to a tremendous number within an hour. A microfluidic electrochemical LAMP device could recognize as few as 16 copies of Salmonella genomic DNA and thereby demonstrated great potential in PoC detection of pathogen DNA.55 When combined with reverse transcription, the LAMP enabled sensitive, rapid detection of Zika viral RNA; 5 plaque-forming units (PFU) of the Zika virus could be readily detected by the naked eye.56 So far, LAMP has been only used for nucleic acid target detection, but the method has clear potential to increase the sensitivity of aptamer-based biosensors.
1.3. Utilizing physical and chemical properties of aptamers
The unique properties of nucleic acid aptamers can provide important hints in designing highly sensitive biosensors. Aptamers typically have lower molecular weight than monoclonal antibodies. Furthermore, nucleobases within the aptamers can have π–π stacking interactions that attract materials that bear aromatic ring moieties, so diverse carbon materials (e.g., graphene, carbon nanotubes) can be integrated with the aptamers to develop novel aptasensors. Another advantage of target-bound aptamers is that they may not be easily digested by nucleases that degrade nucleic acids. All of these properties can be valuable to increase the sensitivity of biosensors.The π–π stacking capability of nucleobases permits easy integration of aptamers with carbon materials (Fig. 2F). Graphene is known to be an excellent quencher of electronically-excited states of dyes, so combining graphene and dye-tagged aptamers is a promising approach to the development of sensitive biosensors. A graphene fluorescence resonance energy transfer (FRET) aptasensor for thrombin detection has extraordinarily high sensitivity even in blood serum;59 thrombin-specific aptamers labeled with FAM fluorophores were non-covalently assembled on graphene, which efficiently quenched the fluorescence of the dyes. In the presence of thrombin targets, the previously single-stranded aptamers that bound only weakly to the graphene formed quadruplex–thrombin complexes, so the FAM dyes moved away from the graphene, and fluorescence recovered. Due to the high quenching efficiency of graphene, the graphene aptasensor had significantly low background signals, providing excellent sensitivity relative to other types of aptamer-based assays, with an LOD as low as 31.3 pM. Other 2D materials can also be used; for example, single-layer 2D MoS2 nanosheets have been used for aptamer-based “capture-release” fluorescence detection of a malaria biomarker.60
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 hybridization between the beacon and the DNA target, the Exo III-based EATR strategy achieved an order of magnitude increase in the final fluorescence intensity.62 A prostate-specific antigen (PSA) has also been detected by combining an aptamer with the EATR technique; supported by terminal deoxynucleotidyl transferase and T7 exonuclease, the PSA aptasensor significantly improved the LOD to 0.043pg mL−1.63
:1 hybridization between the beacon and the DNA target, the Exo III-based EATR strategy achieved an order of magnitude increase in the final fluorescence intensity.62 A prostate-specific antigen (PSA) has also been detected by combining an aptamer with the EATR technique; supported by terminal deoxynucleotidyl transferase and T7 exonuclease, the PSA aptasensor significantly improved the LOD to 0.043pg mL−1.63
Amplified optical aptasensors for diverse small molecules were established by endonuclease-stimulated analyte regeneration.64 Small-molecule aptamers were modified to contain two duplex domains at each end, and the duplex domains were recognized by the endonucleases PvuII and HaeIII. As the aptamers were further modified to be split into two subunits, the duplex domains were disassembled without small molecule analytes. When aptamer–analyte complexes were formed, the duplex domains were regenerated and subsequently cleaved by endonucleases. Due to the cleavage, the aptamer–analyte complexes were destabilized, leading to the liberation of the analyte. The endonuclease-induced recycling of the analyte was readily combined with optical signal amplification techniques, which achieved much higher sensitivity than non-amplified aptamer assays in detecting adenosine triphosphate (LOD: 20 nM), vasopressin (LOD: 2 nM), and cocaine (LOD: 100 nM).
A general approach to sensitively detect small molecules using the EATR technique has been recently developed.65 To maximize a target-dependent response, cooperative binding split aptamers were employed for the development of EATR-amplified biosensors rather than single-domain split aptamers. Specifically, two aptamer domains were linked together by a duplexed C3 spacer abasic site that can be recognized and cleaved by Exo III. In the presence of targets, the two split aptamer domains were formed simultaneously, and the central C3 spacer abasic site was subsequently digested by Exo III. As a result, the unstable complex became disassembled, leading to the repetitive recycling of the small molecules and the split aptamer fragments. As a proof of concept, the researchers successfully detected dehydroisoandrosterone-3-sulfate with an LOD ∼ 1 μM in 50% urine, yielding 100-fold enhanced sensitivity compared to a non-EATR-based assay.
1.4. Use of aptamer's conformational change
Aptamers can be designed to undergo a conformational change upon binding to target molecules. Currently, numerous structure-switching aptamers have been applied to build advanced biosensors. Here, we describe their contributions to increasing sensitivity, by applying several interesting systems including electrochemical aptamer-based (E-AB) sensors, nanofluidic devices, and fluorescence sensors.E-AB sensors first demonstrated the application of aptamers’ conformational change to achieve sensitive detection without interference.66–68 E-AB sensors have been intensively studied to target diverse biomolecules,69–71 and some E-AB sensors have achieved high sensitivity even in undiluted whole blood.72–75 Compared to other affinity reagents, structure-switching aptamers can be highly useful for electrochemical detection; because of structural and functional limitations, other bioreceptors often fail to generate a measurable signal when they bind to a target. For instance, monoclonal antibodies do not alter their shapes in accordance with antigen binding, nor do they emit light or electrons. On the contrary, the aptamers that can be site-specifically labeled with redox tags change in conformation when they attach to targets. Due to these distinct advantages, the aptamers can easily fit into electrochemical devices and generate large signal changes induced by specific target binding.
ATP aptamer-based, DNA super-sandwich structures on solid-state nanochannels realized highly efficient gating.76 Densely packed DNA assemblies on the wall fully sealed the nanochannel, so the ion pathway was efficiently blocked (resistance on the gigaohm scale). In the presence of ATP, the conformation of aptamers changed, then the super-sandwich structures disassembled. As a result, the nanochannel opened, and a transmembrane ionic current was detected. This nanofluidic system had a high ON–OFF resistance ratio (up to 106); this result demonstrates the possibility of sensitive molecular biosensing.
Conformation-changing aptamers can also be actively combined with fluorophores to assemble sensitive fluorescent sensors. A new class of signaling aptamers, called forced intercalation (FIT) aptamers77 use a visco-sensitive quinoline blue dye as a surrogate of one specific base. When a target molecule binds to the modified aptamer probe, its conformation changes, so the quinoline blue intercalates between newly-formed base pairs. After the FIT, the fluorescence increased up to 20 times, which was 15 times higher signal-to-background ratio than obtained using a FRET-based probe. A light-up sensor that combines a partially oxidized G-quadruplex and a pyrene-modified guanine tract works by conformational change.78 A pyrene-tagged guanine repeat is thermodynamically favored to be a part of G-quadruplex compared to the oxidized guanine-containing strand. Therefore, the pyrene-modified guanine probe could participate to form a stable intermolecular G-quadruplex structure by substituting for the oxidized guanine tract; as a result, pyrenes stacked up and emitted a strong fluorescence. Several aptamers are known to fold into similar G-quadruplex structures upon target binding, so this report suggests that the pyrene-based intermolecular structure formation can be further used by aptamer-based biosensors to amplify signals.
1.5. Enhancing the amplification performance of devices
The synergy between aptamers and signaling devices can upgrade the target sensitivity of aptamer-based biosensors. Many device architectures and relevant materials enable excellent signal amplification, and their signaling strength can be boosted by integration with aptamers. Aptamers are often favored because of their small size, and the ability to perform nanomechanical motions gives them advantages over other bioreceptors.79 In this section, we describe several examples of aptasensors according to the type of amplifying systems including FETs and nanoparticle-based systems.Aptamers have been frequently chosen for fabrication of bio-FETs that enable hypersensitive detection of biomolecules such as proteins and small molecules.80 For example, an aptamer-based silicon nanowire (SiNW) FET can detect dopamine release from living PC12 cells.81 The SiNW-FET was sensitive by itself due to the large surface-to-volume ratio of SiNW; a substantial conductance change is induced by small variations of charge carriers at the nanowire surface. Furthermore, the incorporation of multiple dopamine aptamers connected in parallel greatly increased the sensitivity over a traditional single-channel SiNW-FET. The aptamers are much smaller than antibodies, so bound targets can be located extremely close to the surface of the FET sensor, within an electrical double layer; this proximity greatly intensifies the applied electric field. The SiNW FET biosensor had an LOD ∼ 10−11 M, and this LOD is much lower than any other existing dopamine biosensors which are not appropriate for clinical usage due to the lack of ability to detect extremely low levels of dopamine.81
Nanoparticles have been used in many kinds of biosensors, and aptamers can assist in increasing the sensitivity of the nanoparticle-based biosensors. Integration of gold nanoplasmonic particles (GNPs) with aptamers yielded a sensitive aptasensor for early diagnosis of cancer.82 On the surface of GNPs, negatively charged vascular endothelial growth factor-165 (VEGF165) aptamers were electrostatically attached by mediation of positively charged poly-L-lysine. Fluorescent probes were pre-attached to the aptamers and emitted surface-enhanced fluorescence (SEF) by interacting with GNPs. Upon binding to the VEGF165, the conformationally changed aptamers detached from the GNP surfaces; this process inactivated the SEF of the fluorescent probes. This tool enabled simple detection of the predominant biomarkers of cancer angiogenesis in a sample of only 10 μL. Further optimization of the sensor reduced the required sample volume to 170 pL.
2. Increasing selectivity
Selectivity is the ability to distinguish a specific analyte from a mixture of different molecules despite their structural and chemical similarity. Biosensors must detect only the desired substance, not others; for instance, if a biosensor does not have sufficient selectivity to discriminate cancer-specific biomarkers from other proteins, one may be misdiagnosed with cancer. However, conventional antibody-based biosensors cannot distinguish non-specific adsorption-derived signals from target-specific ones due to their mere binding-based mechanisms.70,83 Apart from the perspective of current limits, how do cells and our bodies accurately discriminate similar molecules? During complex yet precise cellular signaling processes and enzymatic reactions, relevant membrane receptors and enzyme proteins increase their target selectivity by using binding-induced structural change and stabilization of transition state.84–86 In this context, by mimicking cellular components, we can utilize binding-induced conformational change of aptamers and binding-activated catalytic reaction of aptazymes. Here, we introduce some interesting approaches that exploit such characteristics of aptamers to increase the selectivity of biosensors (Fig. 3).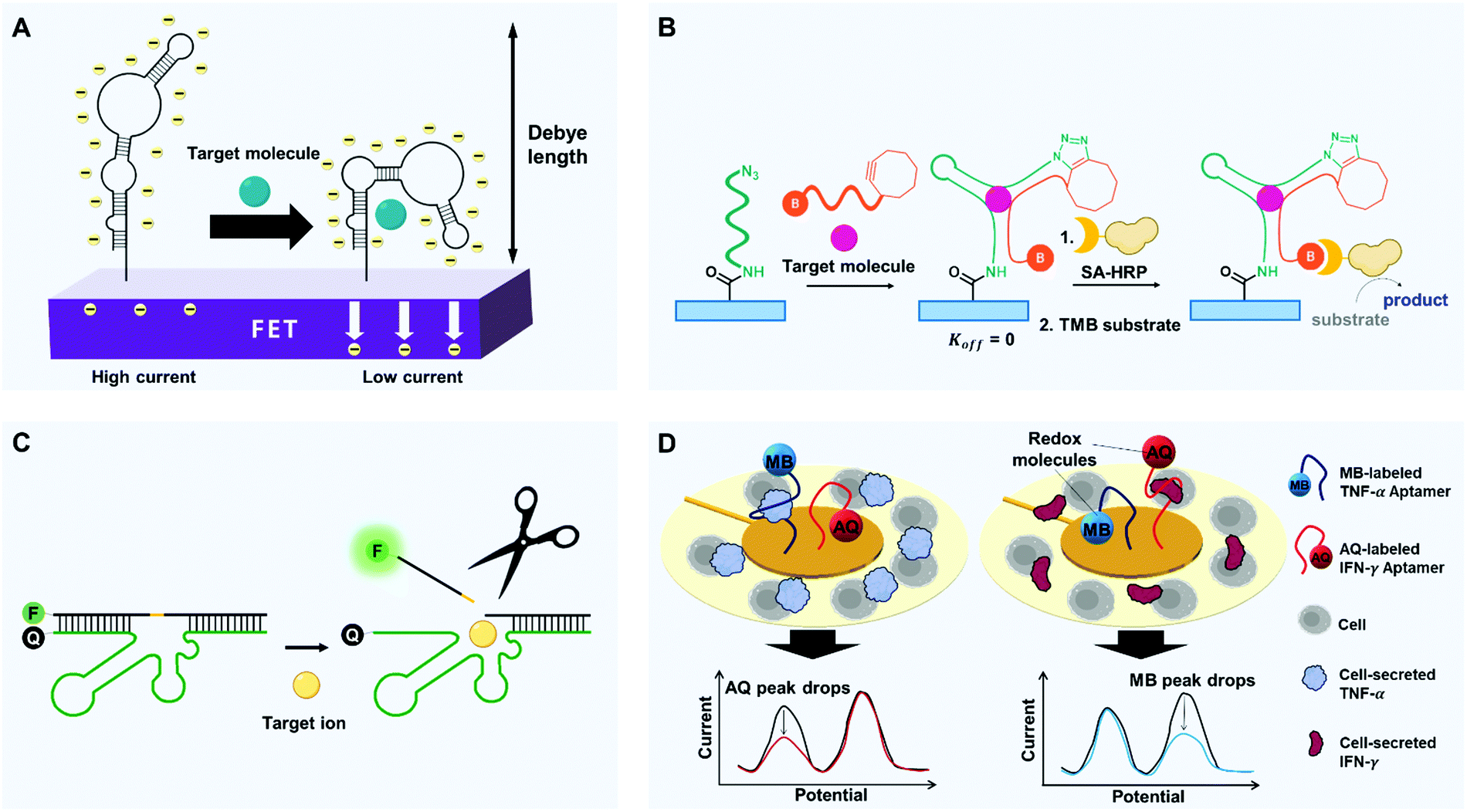 | ||
| Fig. 3 Diverse strategies with aptamers to improve selectivity of biosensors. (A) Binding-induced self-conformational change of aptamers. FETs modified with target-specific structure-switching aptamers enable selective electronic target detection. Within or near the Debye length, target-induced reorientations of stem-loop aptamers near semiconductor channels deplete the channels electrostatically and thereby decrease transconductance. (B) Binding-induced hetero-conformational change of split aptamers. Upon target binding, split aptamers can be covalently linked to each other by click chemistry. When biotinylated aptamer fragments recruit streptavidin-horseradish peroxidase (SA-HRP), chromogenic substrates such as TMB can be oxidated to emit detectable signals. (C) Binding-activated catalytic reaction. By target binding-induced cleavage of aptazymes, the release of fluorophore-linked fragments can be activated to emit highly target-specific fluorescence by reducing physisorption-derived signaling. (D) High selectivity-driven multiplexing. A multiplex analysis can be conducted by aptamers that are linked to redox molecules. Surrounded by cells, Au electrodes can be modified with different aptamers-redox reporter constructs. Binding to cytokines (TNF-α and IFN-γ) causes target-dependent conformation changes that decrease electron-transfer efficiency and thereby decrease the current. Redox molecules with different potential enable the simultaneous detection of multiple targets. | ||
2.1. Binding-induced conformational change
Cells manage to carry out and decode millions of signals in our bodies. During the cellular signaling process, each signaling molecule of extracellular environment binds to a specific membrane receptor. The ligand binding alters the conformation of the receptor protein, and this change triggers a post-signaling process.84,85 The receptor can bind only a restricted set of signal molecules, i.e., it has high selectivity. The high selectivity of the signaling process enables exchange of sophisticated signals at low error rates. Similarly, synthetic aptamers change their conformation only when they bind to targets, and this change of shape can produce a detectable signal (e.g., FRET or electric current).To detect small molecules under physiological conditions of high-ionic strength, FET arrays can be modified with DNA stem-loop aptamers.89 Nucleic acids have a backbone of negatively charged phosphodiesters, so ligand-induced stem-loop conformational rearrangements of aptamers, which bind to even low-charged or neutral targets, can cause signal transduction and amplification under biological conditions. The authors studied two types of aptamers: one type in which the stem-loops reorient toward the FETs after target binding, and one type in which they reorient away from the FETs. For the former group, substantial portions of the negatively charged backbones moved toward n-type semiconductor channels, which increased the electrostatic repulsion of charge carriers but decreased the transconductance. For the second group, the opposite response occurred. In both cases, the change in transconductance yielded a change in current, which was exploited to detect the target (Fig. 3A). These devices successfully detected charged or electroneutral targets (serotonin, dopamine, glucose, and sphingosine-1-phosphate) with great selectivity. For instance, the dopamine aptamer–FET distinctively responded to 10 nM dopamine but gave negligible signals upon exposure to 100 μM norepinephrine, serotonin, L-3,4-dihydroxyphenylalanine, or 3,4-dihydroxyphenylacetic acid.
A split aptamer inherently has a lower binding affinity than a full-length aptamer because the split aptamer's folded structure can be assembled only by binding to the target. However, this requirement also increases the selectivity by reducing the production of undesirable secondary structures, which might deliver false-positive or non-specific signals. This great selectivity of split aptamers has been exploited to develop biosensors, of which sensitivity is still high. In one case, an aptamer structure was divided into two fragments; one end of the first fragment was modified by redox labeling, and its other end was conjugated to a gold nanoparticle (AuNP) while the second fragment was bound by one end to the electrode.94 When a target was present, the two fragments self-assembled, so the redox molecule attached to the first fragment moved toward the electrode and enabled the target-specific current generation. This study was unique in that it conjugated a split aptamer fragment to AuNP instead of using free split aptamer strands. The AuNP conjugation strategy effectively eliminated background noise that can be generated otherwise by abundant free strands; as multiple redox-labeled fragments were attached to the AuNPs, local concentrations of split aptamers could always be high. Even with only 1/1000th of the number of probe strands, the same ones used in a previous report, cocaine could be ultra-sensitively detected in the concentration range of 1 nM to 4 mM; this was a sensitivity increase of 1000 to 100000 times compared to prior biosensors. Furthermore, no measurable change was observed when a different split aptamer was used for the reporter probe, even in the presence of 4 mM cocaine. This use of split cocaine-specific aptamer successfully prevented false signals that would be emitted by inappropriate self-folding.
Some strategies that use split aptamers provide a way to increase both selectivity and sensitivity, but they can only be applied to a closed system; this is a critical limitation. Even if the split aptamers are assembled to bind their target molecules, non-equilibrium events such as dilution and solution exchange, significantly interrupt their target binding. This response limits the use of split aptamers because many applications may require open systems that include buffer exchange or washing processes.93
This disadvantage of split aptamers can be overcome by locking or conjugating split aptamer pairs, once self-assembly has occurred. For example, an ATP split aptamer fragment was labeled with pyrene, and the fluorescent pyrene was also used for a host–guest interaction.95 The pyrene forms 1:2 or 1:1 host–guest complexes with γ-cyclodextrin (γ-CD); pyrene monomer and excimer display the maximal fluorescence at different wavelengths, 378 nm and 480 nm, respectively, and pyrene excimer–γ-CD complexes emit stronger fluorescence than pyrene monomer. In the presence of ATP targets, two DNA fragments assembled in proximity and formed pyrene excimer–γ-CD complex, which showed increased emission F480nm at 480 nm but decreased emission F378nm at 378 nm. This system achieved 8.2 times higher signal-to-background ratio compared to the non-labeled split aptamer and showed great selectivity among nucleotide triphosphates: F480nm/F378nm of 0.2 mM ATP, UTP, CTP, and GTP were 2.03, 0.22, 0.09, 0.16 respectively. This system also achieved quantitative detection of ATP in 50% human serum. Pyrene excimer has a long fluorescence lifetime and is therefore useful for time-resolved fluorescence monitoring. In the presence of ATP, the excimer emission intensity significantly increased over the full-time scale; the lifetime of the complex increased to 67.3 ns in the presence of 0.1 mM ATP.
Split aptamers can also be locked using click chemistry (Fig. 3B). One example is a split aptamer in which cyclooctyne and biotin are attached to opposite ends of one fragment, and azide is to one end of the other fragment.96 When the azide-labeled fragments were immobilized on a microplate, a test sample that was mixed with the doubly-labeled fragments was introduced into the well of microplate. As a result, the split aptamers formed the desired assembly to bind the targets, which yielded chemical ligation between the azide and the cyclooctyne moieties by copper-free click chemistry. The plates were washed, then streptavidin-modified horseradish peroxidase (HRP) and tetramethylbenzidine (TMB) substrates were added, and a colorimetric signal was generated only where chemical ligation had occurred. This use of chemical ligation was effective to make split aptamers applicable for open systems and showed excellent compatibility with an enzyme-linked immunosorbent assay (ELISA) assay. The system showed high selectivity for the cocaine target; structurally-similar metabolites, such as ecgonine, benzoylecgonine and ecgonine methyl ester did not elicit detectable signals.
2.2. Binding-activated catalytic reaction
Aptamers can also imitate enzymes, which perform catalytic reactions only on specific substrates. The target binding of enzymes is synchronized with the enzymatic reaction, so the conversion of substrates is extremely selective. Similarly, an aptazyme, which is a combination of aptamer and nucleozyme (e.g., ribozyme and DNAzyme), can generate selective signals only after a catalytic reaction stimulated by the formation of a complex with a specific molecule. This restriction increases the selectivity compared to conventional methods that mainly depend on the electrostatic binding of target receptors for detection, and several aptazymes have been used to increase the selectivity of biosensors.Sodium (Na+) is one of the most abundant metal ions in biological systems. As most conventional biosensors are suited for the detection of multivalent metal ion, monovalent Na+ has been regarded as a difficult target in biosensing applications. However, some researchers have overcome this problem by in vitro selection of a sodium-specific RNA-cleaving DNAzyme;97 when the DNAzyme assembled with the substrate, a partially-complementary DNA strand containing a ribonucleotide (rA), was exposed to Na+, the DNAzyme cleaved the rA-containing substrate at the ribonucleotide position (Fig. 3C). Importantly, among 23 metal ions, including Li+, K+, Rb+, Cs+, NH4+, and other divalent and trivalent ions, only Na+ caused a significant change in fluorescence. To exploit this mechanism for the intracellular sodium imaging, the researchers labeled each end of the substrate with a fluorophore and a quencher, and the internal ribonucleotide was photocaged to avoid cleavage during delivery of DNAzymes into cells. UV irradiation removed the UV-responsive caging group, and the delivered DNAzymes began to respond to Na+ within the cells. As a result, the intracellular distribution of sodium ions was successfully recorded by fluorescence mapping. This DNAzyme showed exceptionally high selectivity (>10000 times) for Na+ over 20 other metal ions.
DNA origami technology can also be used to achieve DNAzyme-based ion detection. A DNA origami scaffold was integrated with DNAzymes that were specific to metal ions; the DNAzymes generated nanoholes in the scaffold by programmed catalysis.98 The scaffold was designed to perform mechanical unlocking of the nanohole when a certain DNA strand was cleaved, which could be performed by the incorporated DNAzymes. When DNAzymes specific to Zn2+ or Pb2+ were incorporated into the DNA origami, the nanoholes formed within 10 min in response to target ions.99 The DNAzymes had high selectivity, so the nanoholes were only formed at the locations of target metal ions. This trait may enable the development of a unique and selective sensing platform; for instance, origami rafts with nanohole of distinct sizes could perform programmed unlocking of different-sized nanohole patterns, which could be used for multiplexed sensing.
Another enzyme-mimicking complex uses an aptamer as a binding module and a DNAzyme as a catalytic module.100 The aptamers can hold specific targets close, and the DNAzymes perform the catalytic reaction; the resulting construct can perform the efficient catalytic reaction of specific targets. The researchers combined a DNAzyme that mimics horseradish peroxidase with aptamers that were specific to dopamine or N-hydroxyl-L-arginine to stimulate the H2O2-mediated oxidation of organic substrates, similar to the action of real horseradish peroxidase. The complex had excellent selectivity, so it could even distinguish the chirality of dopamine, which suggests the possibility of binding-mediated catalysis. Another recent work introduced a method to achieve artificial photosynthesis by using electron acceptor/photosensitizer–aptamer conjugates.101 Furthermore, split aptamers were combined with split DNAzymes;102 the DNAzyme activation was synchronized with the split aptamer assembly.
2.3. High selectivity-driven multiplexing
These aptameric strategies can increase the selectivity of biosensors and suggest the possibility of multiplex analysis. Therapeutic responses and disease occurrences often involve the interplay of a variety of complex biological networks that involve many proteins, so multiplexing ability would fulfill some biosensing and clinical needs. Actualization of exceptional selectivity for various targets would enable the development of probes that could test for the presence of numerous diseases and biomarkers simultaneously.Sensitive, label-free, yet simple, multi small-molecule detection was achieved with aptamers by exploiting their degree of susceptibility to digestion. Binding of a small-molecule target to an aptamer can increase its resistance to digestion by an exonuclease. Use of exonucleases that digest only unbound aptamers achieved quantitative multiplex detection of cocaine and ATP individually or simultaneously.103 The survivorship of the probes could be assessed by using a fluorescent molecular beacon that bears a pair of fluorophore and quencher. Multiplex detection of cocaine and ATP yielded the LOD ∼ 15 μM for cocaine and ∼5 μM for ATP. These results demonstrate that expanded versions of multiplex assays can be easily achieved by simply adding other structure-switching aptamers and using fluorophores that have different wavelengths.
Electrochemical sensing platforms can be used to achieve multiplex detection. Electrochemical multi-analyte aptasensors with rapid and sensitive detection have been achieved by labeling aptamers with two redox reporters, anthraquinone (AQ) and methylene blue (MB), and anchoring the aptamers to an electrode104 (Fig. 3D). The redox reporters had redox peaks at −0.37 V (AQ) and −0.15 V (MB). Once a target was bound to the redox-labeled aptamer, the redox molecule oriented away from the electrode, so the current decreased. The concentration of the target could be measured by quantifying the decrease of current in each unique potential. The system achieved simultaneous detection of inflammatory cytokines IFN-γ and TNF-α, and the sensor showed great specificity over IL-12, IL-6, IL-10, and bovine serum albumin. An electrochemical multiplex assay using three distinct redox reporters has also been reported, and these results assure the potential and accuracy of this approach.105
Use of aptamers combined with AFM has been proposed as a distinctive strategy for multiplex recognition imaging.106 Two different aptamers were attached simultaneously to the end of the AFM tip by a click reaction using three-arm linkers. The small size and chemical stability of aptamers made them ideal candidates for use as recognition heads. Analysis of cognate proteins, VEGF-TNFα pair and thrombin–streptavidin pair, was conducted to quantify the specificity of detection. Use of the AFM tip labeled with the dual aptamers enabled analysis of multiple proteins immobilized on a surface, which provided an aptamer recognition image of the cognate proteins, i.e., detecting two proteins at the same time. However, when an individual image of each protein was needed, one type of aptamer on the AFM tip must be blocked to prevent interaction with its target. This method enabled simultaneous or separate viewing of two proteins at the molecular level and could detect proteins in only 2 to 3 μL of protein solution with a concentration of ∼2 ng mL−1.
3. Real-time detection
An ability of target detection in real time is invaluable for the next generation of biosensors. In this review, real-time detection covers methods that provide immediate and rapid analytical responses, referred to as near real-time detection, and importantly, methods that continuously measure the concentration changes of targets over time. Continuous detection of certain molecules in patients’ body fluids (e.g., blood, saliva, and tears) would enable tracking of their health or fitness. In particular, real-time monitoring of responses to therapeutic drugs would permit the development of personalized medicine. On-demand diagnosis would allow delivery of the right drug at the adequate dose at the right time and could contribute to maximizing drug efficacy while minimizing side effects.107 Continuous detection of signaling molecules such as chemokines could also give early warnings of microbial or viral infections and autoimmune flare-ups.107Most real-time biosensors monitor mechanical or physical signals such as heartbeat, temperature, and blood pressure; for instance, a pulse oximeter measures oxygen saturation by monitoring light absorption of blood. However, the detectors capable of measuring constantly changing levels of molecular signals that are highly relevant to health conditions are not common because typical bioreceptors like monoclonal antibodies are often not suitable for the use in real-time detection. The target binding of the bioreceptors must be synchronized with readable signals of biosensors, but signal conversion that exploits physisorption is severely hampered by nonspecific binding.
In contrast, aptamers can be manipulated to undergo a conformational change when they bind to a target; conversion of binding events to detectable signals can be selective and continuous. Here, we present key examples of real-time detection using the structure-switching aptamers, according to the types of targets.
3.1. Small molecules
To recognize small molecules, aptamers readily provide fittable binding pockets within their tertiary structures. This characteristic makes the aptamers much better small-molecule receptors compared to monoclonal antibodies. Aptamer discovery technologies have significantly advanced, and innovative in vitro selection techniques for structure-switching aptamers have been developed.108,109 These techniques do not require the target to be immobilized on a solid support; this is a great advantage for ligand generation of small molecules because chemical modification of the small targets is not easy and often causes structural and functional lapses. We introduce representative examples of the use of aptamers to monitor small molecules, in particular, drugs and toxins.Microfluidic Electrochemical Detector for In vivo Continuous monitoring (MEDIC) is a real-time tracking device that uses aptamers to detect therapeutic agents in living animals.15 As a proof of concept, MEDIC has been used to measure the concentration of DOX (Fig. 4A). As an E-AB sensor, MEDIC incorporated several technological advances to overcome the limitations of conventional electrochemical sensors for real-time detection. On the detection surface inside the microfluidic channel, conformation-changing aptamer probes were chemically attached, and redox molecules were further attached to the ends of the aptamers. When the aptamers bind to DOX, the degree of their conformation changes depends on DOX concentrations, which is converted to detectable redox currents in real time. To facilitate the conformational changes, the DOX aptamers were protected by a continuous-flow diffusion filter (CDF) that can prevent physical occlusion of the sensor surface by large interferents, such as blood cells. This work also demonstrated the electrochemical kinetic differential measurement (KDM) technique to self-correct the signal drift and to increase the signal-to-noise ratio. A MEDIC device connected to a living rat's bloodstream could continuously track the circulating DOX level and measure pharmacokinetics. The device achieved stable and quantitative detection for 4 h of continuous operation.
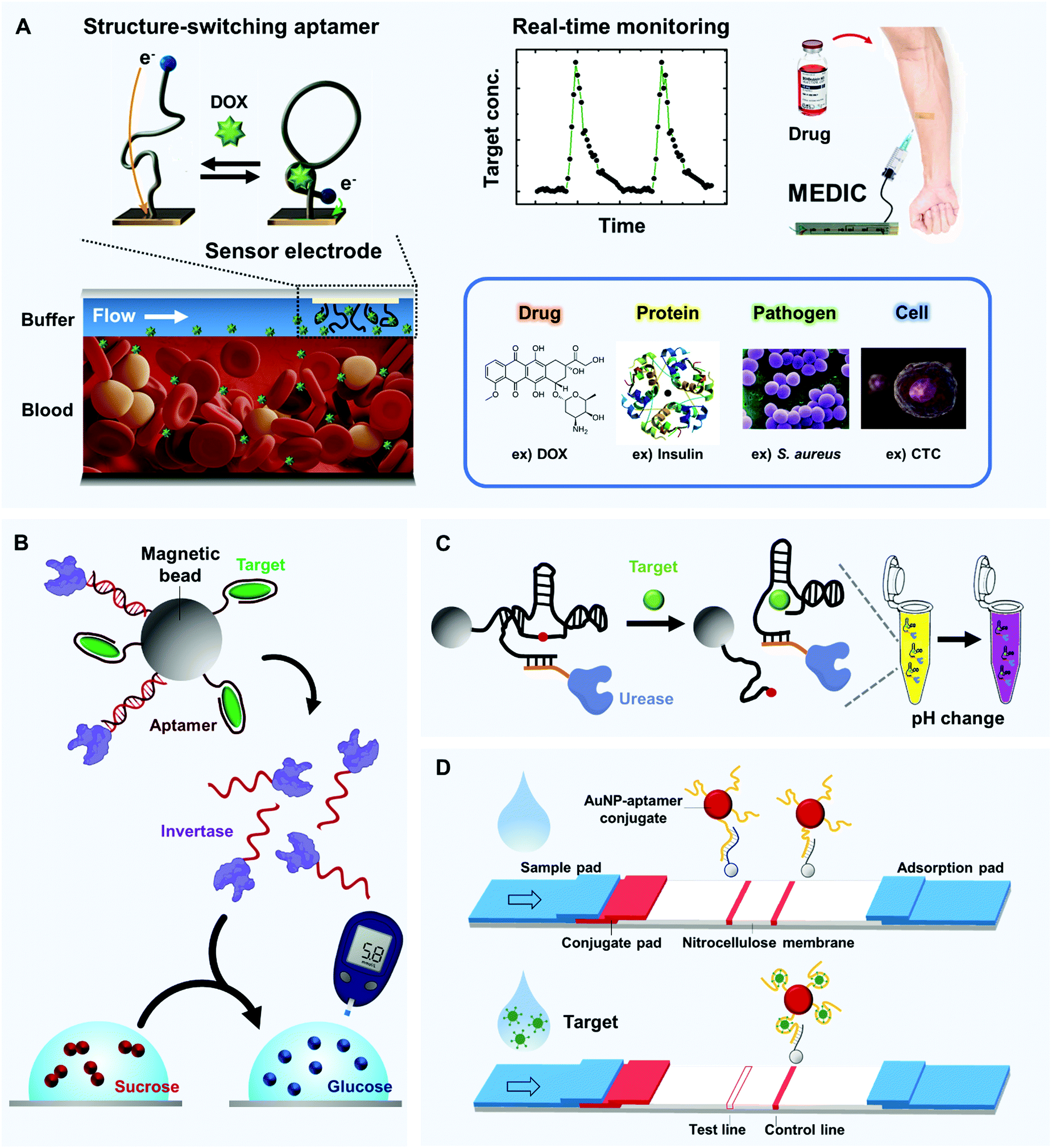 | ||
| Fig. 4 Real-time and in-field detection of aptamer-based biosensors. (A) Microfluidic Electrochemical Detector for In vivo Continuous monitoring (MEDIC) device that enables real-time detection of small molecules in undiluted blood. When structure-switching aptamers are installed inside the microfluidic channel, their DOX-dependent conformational change is synchronized with the detectable redox current change in real time (left). By connecting the device to a living subject's bloodstream, the circulating DOX level can be tracked (top, right); similar techniques can be applied to a diverse range of targets (bottom, right). (B) Glucometer-based sensor. Aptamers can collaborate with a well-established glucometer to detect various biomolecules; structure-switching aptamers can selectively release invertase-tagged DNAs, and glucose production can be easily detected by the portable glucometer. (C) Solution-based sensor. An aptazyme linked to urease that alters pH in the presence of urea can be linked to magnetic beads. In the presence of a target, the aptazyme self-cleaves, and the isolated aptazyme-urease complex alters the color of a solution that contains urea and litmus reagent. (D) Paper-based sensor that uses aptamer-linked AuNPs. Upon target binding, aptamers lose the ability to hybridize with their complementary strands which are site-specifically coated on the nitrocellulose membrane; this result is readily visualized by a lack of a red band at the test line. | ||
A newly developed E-AB sensor can realize the real-time and multi-hour measurement of DOX directly in the bodies of awake, even freely-moving animals.110 The sensors were encased in biocompatible polysulfone membranes to prevent fouling by blood cells. Similar E-AB sensor platforms have achieved real-time detection of antibiotics such as kanamycin, ampicillin,111 and anesthetics including cocaine.112 These successes demonstrate the potential utility of these devices in personalized medicine.
Aptamers have a huge competitive advantage in targeting small toxins. Traditionally, antibody production involves the injection of an antigen into an animal to induce the expression of specific antibodies; this approach is inviable when the antigen is fatal. However, aptamers can be discovered by in vitro selection, so toxicity is not a limiting factor; aptamers specific for biotoxins (e.g., okadaic acid) and neurotoxins (e.g., α-bungarotoxin, gonyautoxin 1/4 (GTX1/4)) have been generated.113–115 An aptamer-based optical biosensor exploited biolayer interferometry (BLI) to achieve real-time detection of GTX1/4, which is a potent marine neurotoxin that can accumulate in shellfish. The device showed good reproducibility and stability in GTX1/4 detection and yielded a target-specific response within 150 s.
3.2. Ions
Real-time monitoring of metal ions has become increasingly necessary because of their biological, environmental, and industrial significance. However, the generation of ion-binding reagents is difficult,116 and ion-induced selective signaling is exceptionally challenging. Use of aptamers may solve this problem, and several research groups have used metal ion-binding aptamers to construct real-time biosensors.117,118A FET-type flexible graphene aptasensor for real-time detection of mercury ions was developed by immobilizing mercury-specific aptamers to a surface composed of modified graphene.119 To achieve physical flexibility, the graphene was transferred onto a polyethylene naphthalate (PEN) film, which is a transparent plastic substrate. The mercury aptamers were anchored to the graphene surface by introducing 1,5-diaminonaphthalene (DAN) and glutaraldehyde (GA) as linkers to induce a Schiff-base reaction with 3′-amine and 5′-carboxylic acid-modified aptamers. When mercury ions specifically bound to the aptamers, an electrostatic change occurred at the device interface and yielded a change in electrical current. The aptasensor was two to three orders of magnitude more sensitive than electrochemical detection of mercury ions. It also has a very rapid response time of <1 s, which enabled a real-time detection of mercury ions in mussels.
A DNA device for reversible detection of lead ion was achieved by exploiting a turn-on of fluorescence in response to the target ion.120 In this device, the DNA underwent a structural change from a duplex to a G-quadruplex in the presence of lead ions. The G-quadruplex structure subsequently interacted with zinc protoporphyrin IX (ZnPPIX) to sharply increase its fluorescence intensity. The DNA aptamer could switch conformation reversibly, so the introduction of chelator, 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA), caused the destabilization of the G-quadruplex and thereby turned off the fluorescence of ZnPPIX. Therefore, this device that does not require any labeling or extra chemical modification of DNAs in fabrication can be used repeatedly for lead ion detection.
A rapid and label-free strategy to isolate aptamers for metal ions can be further utilized for real-time detection.121 The authors used a novel aptamer discovery method, called structure-switching particle display (SS-PD); it starts with a randomized pool of monoclonal DNA particles. Using water-in-oil droplets, they prepared the particles covered by specifically designed monoclonal DNAs that contain a central domain flanked by two random domains for metal ion binding. The central domain with predefined sequences was hybridized to a fluorescence-labeled complementary strand that could be released upon binding-induced structure-switching, reporting readable signals. Microparticles of structure-switching aptamers that responded to metal ions could be selectively screened using fluorescence-activated cell sorting (FACS). The SS-PD strategy was more efficient than conventional selection methods because it enabled the direct measurement of metal ion binding by FACS, without requiring any labels, solid support, or modifications. SS-PD successfully generated high-affinity aptamers for mercury ion and copper ion as a proof-of-concept. This technique is generally applicable to discovering high-quality structure-switching aptamers that target other metal ions, and the resulting aptamers would be valuable for real-time detection.
3.3. Proteins
PoC testing and clinical treatment require continuous measuring of diagnostically relevant protein biomarkers. Advanced methods for protein detection can also be practical in the biological research and food industry. Conventionally, proteins are detected using antibodies, but aptamers have decisive advantages over proteins, including resistance to denaturation and amenability to manipulation of binding characteristics by rational design.122 Aptamers can also be designed to bind to non-immunogenic or toxic proteins, which is a huge advantage. Importantly, synthetic aptamers can be readily modified with functional groups that allow covalent immobilization on a variety of novel materials; this ability can provide compatibility with numerous well-established devices. The aptamers that recognize proteins can be much more easily generated than those of small molecules or ions, and in principle, a specific aptamer can be developed for every protein target.122 Several approaches have been used to discover novel aptamers and to develop innovative biosensing platforms for real-time detection of proteins.An aptamer-functionalized molecular electronic device achieved the continuous detection of thrombin.123 The device used single-walled carbon nanotubes (SW-CNT) that were connected end-to-end by thrombin-specific aptamer linkers to leave nano-sized gaps among the SW-CNTs. When thrombin bound to the aptamer bridge, the G-quadruplex DNA structure became further stabilized and promoted tight π–π stacking within the G4 conformation. This base stacking yielded increased charge transport through the DNA aptamer linkers, so a sharp increase in electrical conductance was observed within 1 min. This device achieved reversible real-time detection of thrombin and was sensitive enough to detect single thrombin molecules.
Aptamer-functionalized porous silicon scaffolds have been used to develop interesting optical biosensors.124 Aptamers that were specific for polyhistidine tag (his-tag) were conjugated within the pores of nanostructured porous silicon (PSi), by using silanization and coupling chemistries. When the conjugated aptamers captured his-tagged proteins, the PSi's optical interference spectrum changed. These devices can emit stable readout signals continuously and therefore have the potential to achieve real-time detection of all kinds of proteins that have his-tags.
3.4. Pathogens
Whole organisms can also be detected by aptamer-based biosensors. The food industry, water quality control, and public health all require the detection of pathogens and viruses. If pathogenic bacteria in food cannot be identified in advance, it may lead to terrible consequences, posing a high risk to human health. Also, the precise detection of COVID-19 is now a critical task in the fight against ongoing global pandemics. A potentiometric biosensor has been developed to detect Staphylococcus aureus in the skin.125 The biosensor was composed of aptamers that function as biorecognition molecules and SW-CNTs that act as ion-to-electron potentiometric transducers, and the S. aureus-specific aptamers were attached to the SW-CNTs. The aptamer's binding to S. aureus induced a conformation change that clearly changed the surface charge of the SW-CNT layers. The change in charge was immediate, so real-time pathogen detection was possible.3.5. Circulating tumor cells
Several approaches that use aptamers have been developed for real-time detection of CTCs or tumor exosomes.126–130 CTCs are cancer cells that have been shed from a tumor into the bloodstream, and they have possible use as biomarkers of metastatic cancer. Tumor exosomes are extracellular vesicles with diameters of 30–100 nm, and they are actively involved in cancer progression and metastasis and therefore may also be useful as cancer biomarkers. However, the concentrations of CTCs and exosomes in blood are extremely low; for example, whole blood of cancer patients contains only 1–10 CTCs per mL, so detection is significantly challenging. However, highly sensitive aptamers that are specific to CTCs and exosomal proteins are being developed, so real-time detection should be achieved in the near future. This method would provide a useful tool for periodic monitoring of anticancer therapy effects and might contribute to effective early diagnosis of cancer.As an example of near real-time detection, a cancer cell-activatable aptamer-based one-step assay for the rapid detection of CTCs was demonstrated.131 A reporter probe consisted of an anti-CD30 RNA aptamer with fluorochrome and quencher at 5′- and 3′-ends, respectively. This probe interacted with specific surface biomarker proteins of cancer cells, triggering receptor-mediated endocytosis and internalization of probes into lysosomes. When the aptamer probes were degraded by lysosomal nucleases, fluorochrome was released from the nearby quencher, resulting in the emission of strong fluorescence signals. Using these aptamer probes, the researchers successfully identified the CTCs in the whole blood of the patients with lymphoma tumors. In comparison with antibody-based assays, the aptamer-based assay yielded much lower background noise; as the probe was optically silent in the absence of target cells, this assay did not require additional wash steps to remove the potential off-target signals, making the assay completed in a single-step within minutes.
4. In-field detection
The ability to immediately identify a desired target at the time and place of contact, i.e., in-field detection, has significant advantages in the identification of infectious diseases and environmental toxins. Most studies to actualize biosensors for in-field detection use antibodies. However, these devices have disadvantages such as low thermal stability, low tolerance for changes in pH, high price, and batch-to-batch deviation. In addition, most antibodies react only to proteins, so they are therefore not appropriate for the detection of small molecules and ions.Nucleic acid aptamers that have excellent thermal stability and pH tolerance are relatively inexpensive, so they are suitable for disposable biosensors. Moreover, the synthetic biopolymers do not show batch-to-batch variation, and in vitro selection methods generate high-quality aptamers customized for specific targets, in a range of categories, such as ions, proteins, lipids, and various small molecules. Various aptamers have been thus developed for many distinct signaling strategies to actualize in-field detection. These strategies include sensors that use glucometers, solutions, paper, and that exploit material properties (Fig. 4B).
4.1. Glucometer-based aptasensor
Several suggested approaches have combined the glucose-quantification capability of glucometers with the target-detecting capability of aptamers for in-field detection of molecules other than glucose. In general, they use a mechanism that transfers a signal from target biomolecules to glucose. Aptamers that are specific for the target molecules mediate the transition process and enable rapid target detection by commercial glucose sensors. This section introduces several strategies to expand the target range by using various aptamers and glucose meters.Aptamer-modified magnetic beads are combined with enzyme-tagged DNA strands that can hydrolyze sucrose into glucose, which converts the target-binding event to a detectable signal of glucose meter132 (Fig. 4B). To be specific, invertase (β-fructofuranosidase), an enzyme that catalyzes the hydrolysis of sucrose to fructose and glucose, was linked to a specific DNA that hybridized with an aptamer or aptazyme that had been immobilized on a magnetic bead. When the target was present, the aptamer bound to it, and the DNA-invertase conjugate was freely released from the magnetic bead (Fig. 4B, top). The magnetic beads were removed, followed by transferring the solution into a buffer that contained sucrose; the measured glucose transfer rates depended on target concentrations (Fig. 4B, bottom). Variants of the device obtained portable, low-cost and quantitative detection of many targets beyond glucose, including cocaine (LOD: 3.4 μM; detection range (DR): ∼0–500 μM), adenosine (LOD: 18 μM; DR: ∼0–1000 μM), interferon-γ (LOD: 2.6 nM; DR: ∼0–400 nM) and uranium (LOD: 9.1 nM; DR: ∼0–200 nM). This approach is easily expandable to many other targets because desired aptamers can be developed using SELEX technology.133
To produce glucose signals in response to non-glucose targets, another glucometer-based method utilized aptamers as crosslinkers to form a hydrogel wherein glucose-producing enzymes were trapped.134 To be compatible with glucose meters, this biosensor used glucoamylases that hydrolyze amylose, a polysaccharide made of α-D-glucose units, to a number of glucose molecules. When targeting molecules were present in the solution that contained amylose, the aptamers that crosslinked the polymer chains of hydrogel fell off to form aptamer-target complexes; as a result, the hydrogel collapsed and released glucoamylases. The released glucoamylases produced glucose that was detected by a glucometer. This method can be adapted to detect various targets, and the amplification of the glucose signal, by the use of polysaccharide substrate, provides detection sensitivity of a few micromoles.
4.2. Solution-based aptasensor
Aptamers can be coupled with enzymes or nanoparticles to trigger a color change of a sample for naked-eye detection that does not require battery power. For example, solution reagents that change color under certain conditions, such as litmus reagents or AuNPs, can be chosen for signaling target detection by the aptamer; this battery-free strategy can rapidly convert biomolecule-dependent signals to colorimetric signals. We present several examples of simple solution-based sensors.When an aptazyme is linked to an enzyme capable of modulating pH, a target-specific signal can be easily provided by a litmus reagent that changes color according to pH (Fig. 4C). Urease was coupled to an aptazyme that performs a target binding-induced cleavage activity, and the complex was attached to a magnetic bead, which was subsequently added to a litmus solution that included urea.135 Target molecules activated self-cleavage of the aptazymes and released the urease from the magnetic beads; the urease increased the pH by hydrolyzing the urea, and as a result, the color of the solution changed. A device that used EC1 DNAzyme detected Escherichia coli in a few minutes by using cheap and widely available pH-sensitive dyes or paper strips. This sensor can detect a desired target anytime and anywhere, at low cost, with no need for a power source or heavy detection instruments.
Nucleic acids interact uniquely with AuNPs; DNA has four types of nucleobases and a phosphate backbone, and these bases are strongly adsorbed to a gold surface; the adsorption energy decreases in the order of A > C > G > T ≫ phosphate.136,137 Adsorption requires protonated adenine and cytosine. They have comparably low pKa = 3.5 and 4.2, respectively, so the pH of physiological solution must drop to ∼3 before these bases are protonated.138 At this pH, the coordination interaction of nucleobases with AuNPs is strengthened, so they stably adsorb the DNA. When AuNPs are reduced using citrate, their surfaces collect chloride and citrate ligands and therefore become negatively charged. The addition of salt (>20 mM of NaCl) shortens the Debye length, so the AuNPs can approach each other closely enough that van der Waals force becomes significant. As a result, the AuNPs aggregate irreversibly, and solution color changes from red to blue. However, if DNAs are densely grafted to the surface of AuNPs, steric stabilization and increased electrostatic repulsion would stabilize the DNA-bound AuNPs. This colloid typically has quite high stability and can survive even in 1 M NaCl.
The intrinsic adhesion of aptamers to AuNPs actualizes the detection of target molecules by changes in color that result from AuNP aggregation. This method has been used to detect oxytetracycline (OTC), one of the most common antibacterial agents, by using an OTC-specific aptamer.139 When OTC was present in a red solution that included the aptamer-grafted AuNPs, the aptamers left the AuNP surface, then the AuNPs aggregated at high concentrations of NaCl, so the solution changed to purple. Such target-specific AuNP aggregation enables rapid detection of biotargets with few steps, and its production is so simple and cost-effective; the solution sensor can be made just by mixing aptamers with AuNPs. This work achieved qualitative sensing of OTC with an LOD ∼ 25 nM. Sensitive detection of other targets such as pesticides,140,141 arboviruses,142 exosome surface proteins,130 and organic synthetic compounds143 has also been achieved.
A sensor that uses luminescence as its signal can be simple to use in the field, and a combination of light-emitting nucleozymes and aptamers can be applied to chemiluminescent target detection. A biosensor that shines even without a light source would be useful for detection at night, or for military purposes that necessitate superior visibility.144 The luminescent sensor has two advantages. First, it has no background signal because it does not emit any light in the absence of targets; this trait increases its sensitivity. Second, it is a simple ON/OFF system. For a FRET system, if the Stokes shift is too small, the resolution becomes substandard. However, a luminescent sensor does not have this drawback.
One such sensor generates a chemiluminescent signal when the binding of the aptamer to the target induced the formation of active DNAzyme structures that mimic HRP.145 In the presence of target substances, the target binding of aptamers changed the complex of hemin and G-quadruplex DNAzyme to the active form, and subsequent luminol oxidation generated a luminescent signal at 420 nm. The sensor had a very low background signal and required no external light source; furthermore, if quantum dots (QDs) were attached to the ends of the DNA, the emission wavelength could be adjusted by chemiluminescence resonance energy transfer. This ability implies that multiplex detection can be achieved by introducing QDs of various sizes.
4.3. Paper-based aptasensor
To display the signal evoked by the detection of targets, some biosensors use paper, which has the advantages of being cheap and disposable. Generally, these sensors exploit the mechanism of strand displacement caused by target binding of aptamers. In an environment that includes the target, the aptamers undergo a conformational change and displace the partially complementary strands. Enzymes or AuNPs can be used to generate a detectable signal in response to the strand displacement. For instance, the target-specific strand displacement may trigger an enzyme chain reaction that can cause a color change or electron migration, or can disunite AuNP-tagged ssDNAs which produce a colorimetric signal when they accumulate on the paper. This section describes several examples of paper-based sensors that can measure various targets rapidly and inexpensively anywhere at any time.Toehold-mediated strand displacement (TMSD) is an enzyme-free molecular tool to replace one input nucleic acid strand with a specific output strand. A combination of bead-linked ssDNA/aptamer assembly and TMSD amplification successfully quantified adenosine signals on paper devices.146 When the adenosine target was present, the adenosine aptamer released the bead-linked strand, which hybridized with the complementary strand and eventually activated HCR. The resulting HCR product contained a number of biotinylated hybridized strands; it captured streptavidin that had been linked to glucose oxidase (GOx), and the attached GOx catalyzed the oxidation of pre-added glucose to produce an excess amount of H2O2. The H2O2 reacted with KMnO4 that had been embedded in paper, it changed from purple to colorless. This isothermal HCR process produced a colorimetric signal without the need for specialized instruments (e.g., thermocyclers), and microzones that were stained with different concentrations of KMnO4 enabled quantitative readout by the unaided eye.
A similar TMSD-activated paper device achieved in-field electrochemical detection of adenosine.147 A carbon electrode was coated on one side of paper, and [Fe(CN)6]3−, one of battery charging materials, and a bead-linked aptamer/GOx-tagged strand assembly were applied to the other side. Then, the paper was rolled up to bring the opposite sides into contact, and a solution that contained adenosine targets was dropped onto the inlet of the device. When the target substance reached the beads by diffusion, the aptamers linked to the beads induced TMSD in response to the adenosine. As a result, the separated GOx-tagged strands converted [Fe(CN)6]3− to [Fe(CN)6]4−, which charged a capacitor that was connected to the paper. When the capacitor is discharged, a short and amplified current flowed, which could be used for electrochemical detection of targets. This paper device used aptamers to charge the capacitor only when the target was present, and it transduced a biomolecular signal to an electronic signal, so it resembles a biobattery.
A strip-paper analysis has been developed using structure-switching aptamers and AuNPs that change colors upon accumulation.148,149 The sensor consists of four components: sample pad, conjugate pad, nitrocellulose membrane, and absorption pad (Fig. 4D). One design uses an aptamer that has a poly-A tail for the AuNP–aptamer conjugates. These conjugates (5 μL per strip) were prepared and added to the glass fiber membrane (conjugate pad), and two types of streptavidin-linked DNA probes, one with aptamer-complementary sequence and the other with poly-T sequence, were immobilized on the nitrocellulose membrane as test line and control line, respectively. When a droplet of solution was deposited on the sample pad, the solution diffused toward the absorption pad and carried AuNP–aptamer conjugates from the conjugation pad. If target molecules were present, the AuNP-conjugated aptamers bound to the targets; the AuNPs could not be captured by the aptamer-complementary strands on the test line, so the test line did not show any bands. However, the control line always captured the AuNP–aptamer complex regardless of the presence of the target, and this trait confirmed the sample movement. In this way, a red band caused by the formation of AuNP aggregates occurred only on the control line and indicated the existence of target molecules. These strip-paper sensors were used to detect ochratoxin A (OTA) in wine by applying a solution to the bottom of the strip. The strip had a visual LOD ∼ 1 ng mL−1 and great specificity against four different common toxins, zearalenone (ZEN), fumonisin B1 (FB1), deoxynivalenol (DON) and microcystin-LR (MC-LR) regardless of their concentrations. This simple and easy-to-use method has been applied to sensitively detect various toxins,150,151 ions,152 and bacteria.153
4.4. Material property-based aptasensor
Target-responsive changes in the physical properties of materials can be also used for aptamer-based in-field detection. For example, when a hydrogel is constructed using aptamer crosslinkers, target binding events can cause the crosslinkers to be cleaved or conformationally changed, so the material's mechanical or optical properties can change. These changes are easily noticeable in field, so they have been exploited in several sensing devices.Analyte-responsive DNA hydrogel sensors that employ aptamers and their complementary strands as crosslinkers have demonstrated interesting sol–gel transition in the presence of targets.154–156 If aptamers dehybridize with the complementary strands by binding targets, crosslinker cleavage can be induced. Typically, antibody encapsulation into hydrogels is difficult because proteins can be irreversibly denatured during hydrogel synthesis. On the contrary, synthetic aptamers are reversibly denatured and stable during synthesis, so they are readily used to prepare such sensing hydrogels. In the presence of targets, the aptamer-crosslinked hydrogels lose rigidity due to cleavage, i.e., dehybridization of crosslinkers. Therefore, a gel-to-solution transition occurs, which is easy to detect. Hydrogels that include aptamers can also be used to reversibly catch and release target materials in response to changes in pH or temperature.157 Moreover, the programmable 3D structure of hydrogels can provide matrices that immobilize nucleic acids without the need to add additional supports; this trait simplifies the structure of the sensor.
Use of liquid crystals (LCs) enables direct detection of aptamers’ conformation changes. One example is an LC sensor that uses an aqueous interface laden with octadecyltrimethylammoniumbromide (OTAB) surfactant, with structure-switching aptamers placed on the top of the interface.158 Without target molecules, the proximity of hydrophobic nucleobases to the hydrophobic LC perturbed the interfacial structure of the surfactant monolayer, so the LC assumed a planar configuration. When targets bound to the aptamers, their conformation changed immediately, so they developed partially-hybridized domains. As a result, the number of exposed hydrophobic nucleobases decreased, so the hydrophobicity of the aptamers decreased. This change allowed LC to return to the initial homeotropic state; the reorientation was easily observed by the change of optical property. For example, when arginine-specific aptamers were applied to the LC interface, the presence of arginine induced LC reorientation within 3 min. This sensor exploited the features of LCs to determine the presence of a target in field, without the need for any battery or complicated manipulation. However, not all aptamers can undergo these conformation changes, so applications of this sensor are limited.
5. Modularization
Modularity describes the degree to which individual components of a system can be separated and reassembled for easy manufacturing. Biosensors can be constructed using a modular approach because of the distinct functions of the components. Biosensors consist of three independent modules: one for detection, one for amplification, and one for signaling; they must be well-synchronized for the system to operate well. Modularization of biosensors can effectively reduce the time and costs for the development of sophisticated biosensors. Sometimes, a systematic correlation of complex functions among the modules leads to increased detection sensitivity that surpasses the fundamental limits of biosensors. The inherent modularity of nucleic acids means that aptamers, as a recognition module, can be combined with other functional nucleic acid modules to build a complex biosensor on a molecular level. In this part, we introduce modular strategies to diversify the targets using a single type of device, and a unique method to expand the detection range of sensors by strategic fusion of several modules.Many researchers have developed biosensing systems that exploit molecular modularization. One of the most representative examples is a molecular biosensor that uses Spinach, which is an RNA mimic of the green fluorescent protein.159,160 The fluorescent sensor was composed of a target-specific aptamer module and fluorescence-emitting Spinach module, which was destabilized if it had not bound to the target. When the target bound to the aptamer module, the aptamer reconfigured into an active form, which stabilized the interconnected folding structure of the Spinach module. This change activated the fluorescence of Spinach and allowed efficient imaging of the target. The target-induced structure stabilization successfully correlated the molecular recognition module (aptamer) and the signaling module (Spinach), so the system operated as a molecular biosensor. The biggest benefit of this system is that it can be adapted to diverse targets by simply changing the aptamer module.
A DNA-based beacon to detect antibodies and other proteins uses a mechanism that relies on cooperation between a structure module and a recognition module.161 The structure module was a stem-loop DNA scaffold, and the recognition modules could be either small molecules, polypeptides, or nucleic acids, which were conjugated to each end of the stem in the structure module. When target molecules bound to both of the recognition elements, a steric strain was imposed on the stem-loop structure, so the base-paired stem could not be maintained. For this reason, a fluorophore and quencher pair that were initially close together on the structure module became segregated, and as a result, the fluorescence emission intensified. As a proof of principle, platelet-derived growth factor (PDGF), a dimeric protein, was chosen, and PDGF-specific aptamers as recognition elements were installed at both stem ends of the structure module. The molecular device could detect the PDGF rapidly (<10 min) and selectively even in complex samples, such as blood serum. The device can be adapted to the detection of diverse macromolecular targets such as antibodies and proteins, by simply changing the recognition elements while retaining the stem-loop DNA structure.
A pH-independent DNA nanodevice called a Clensor was proposed to quantify chloride transport in organelles of living cells.162 The Clensor was composed of three components: one for sensing, one for targeting, and one for normalization. The targeting module included an aptamer that binds to a human transferrin receptor. The sensing module was a PNA labeled with a chloride-sensitive molecule, 10,10′-bis[3-carboxypropyl]-9,9′-biacridinium dinitrate (BAC). The normalizing module consisted of a DNA strand modified with chloride-insensitive Alexa 647 fluorophore (A647); this module mediated the connection of the other two modules by DNA hybridization that constructed a complete nanostructure. When the nanodevice bound to the transferrin receptor, cellular uptake occurred, which delivered the internalized nanodevice to organelles along the endolysosomal pathway. In the presence of intracellular chloride ions, BAC underwent collisional quenching of its fluorescence, whereas the fluorescence of Alexa 647 in the normalizing module remained constant regardless of the chloride level. Normalization of the signals enabled the quantification of chloride concentrations in the subcellular organelles of Drosophila melanogaster. The modularity of this nanodevice makes other aptamers easily incorporated in the molecular device, so it can detect chloride levels of specific subcellular compartments that are targeted by the incorporated aptamers.
A nucleic acid nanostructure can also be constructed using modular construction that exploits ligation. This approach has been used to realize electrochemical detection of a broad range of analytes by employing the concept of on-electrode ligation.163 The molecular sensor consisted of three DNA modules. The first was an anchor module that was immobilized on a gold electrode by a thiol group. The second module was a target-recognizing part that was composed of a DNA strand with an internal small molecule label. The third module was a signaling DNA modified with a methylene blue redox tag. T4 DNA ligase was used to enzymatically link these three modules to construct a complete probe directly on the surface of an electrode. Diffusion of tethered redox molecules was initially fast, but when target proteins were bound to the internally-labeled small molecules within the target-recognizing module, the diffusion rate decreased. The diffusion rates caused a change in current, so the concentration of proteins could be measured even in undiluted serum. The device could be modified by simple replacement of target-recognition modules; it was used successfully to quantify a wide range of proteins, including streptavidin, anti-digoxigenin, and anti-tacrolimus. Considering the high cost of long DNAs and the complexity of modification by commercial solid-phase synthesis, this modular construction would be highly advantageous.
The modularization strategy can contribute to the further improvement of biosensor sensitivity and specificity. The approach has been used to develop a dual-sensing DNA structure that has an exceptionally broad detection range.164 For ATP detection, the DNA nanostructure includes both an aptamer module and a DNAzyme module, which capture the same target but generate signals by different mechanisms. Within the modular construct, an ATP aptamer, which is designed to emit fluorescence at low (micromolar) ATP concentration, and a G-quadruplex-hemin DNAzyme, of which peroxidation activity is greatly enhanced at a high (millimolar) concentration of ATP were systematically integrated. The two functional modules were fused into one molecule, so their distinct dynamic ranges were also combined to achieve an ultra-broad detection range; it could detect ATP concentrations ranging from 1 μM to 500 mM; this range of more than 5 orders of magnitude is currently the widest dynamic range that has been achieved in a single biosensor.
Conclusions
To address current technical challenges in biosensing applications, we focused on the unique abilities of aptamers in this review. In particular, we highlighted various strategies wherein the nucleic acid biopolymers become key materials that surpass the sensitivity and selectivity of conventional biosensors. Moreover, we identified newly emerging demands including real-time detection, in-field detection, and structure modularity, and various studies that used aptamer-based sensors to meet these demands were introduced.Several limitations hinder commercialization of the aptamers and aptameric biosensors. First, nucleic acids are susceptible to in vivo environments, e.g., degradation by ubiquitous nucleases and aggregation with plasma proteins; this can be a critical impediment to applications in in vivo imaging and sensing.165 Second, the small size of aptamers sometimes can be disadvantageous; renal filtration and rapid clearance from the bloodstream can also impede their use in vivo.166 Third, the aptamer generation is not easy; current SELEX technologies that are usually time-consuming and labor-intensive have an unsatisfactory success rate in discovery. Advances in aptameric biosensing technology require the development of a fast and effective method to produce desired aptamers. In addressing these problems, we can consider potential solutions to generate advanced biosensors.
Incorporation of modified nucleotides can increase the stability of aptamers in vivo. This is a relatively easy strategy; chemically-engineered backbones, modified side chains, unnatural nucleotides, and 5′ or 3′ terminal caps, all reduce the susceptibility of nucleic acid aptamers to nucleases.167 For example, in ribonucleotides, 2′-OH moieties can be replaced by diverse chemical groups such as 2′-fluoro or 2′-O-methoxy motifs.168,169 Moreover, the native phosphodiester backbone can be replaced with boranophosphate or phosphorothioate residues, and a neutral backbone can be realized by using PNA.170 Locked nucleic acid (LNA) or XNA technology can increase the nuclease resistance and also promote binding characteristics.171 In addition, the 3′- or 5′-terminal of aptamers can be protected using additional hairpin structures.172
Large biocompatible nanomaterials, either synthetic or natural, can be easily conjugated to aptamers for their in vivo use; this approach would effectively reduce their renal filtration rate. Representatively, the weight of aptamers can increase to greater than the renal filtration threshold of 40 kDa by linking them to polyethylene glycol (PEG).173 This incorporation of PEG also increases the resistance of aptamers to nucleases. Furthermore, conjugation of aptamers with lipids can lead to the formation of aptamer-decorated liposomes,174 which have reduced the renal clearance rate and increased the binding affinity due to multivalent effects.
SELEX protocols should be improved to reduce the selection periods and increase the success rate of aptamer discovery. For instance, an initial pool of oligonucleotides could be optimized, rather than random. We also believe that every step of SELEX, including separation, amplification, sequencing, and characterization, should be further improved. For example, integration of microfluidic technology, high-throughput sequencing (HTS), and parallel microarray characterization has provided a highly efficient workflow for discovering aptamers that have high affinity.175–177 Furthermore, the use of cell SELEX and in vivo SELEX178 may permit the effective discovery of aptamers that bind to the native state of targets under in vivo physiological conditions. Aided by state-of-the-art instruments such as FACS, the efficiency and success rate of aptamer screening can be increased.179
In addition to target-binding aptamers, other functional nucleic acids, including nucleozymes, also have wide applicability to biosensors. Among catalytic RNA molecules, self-cleaving ribozymes that respond to ligand binding can be used as molecular switches in genetic circuits.180 When small molecules like metabolites bind to the ribozymes, autocatalytic cleavage is induced to regulate downstream gene expression.181 Moreover, DNAzymes are frequently combined with FRET technology and applied for quantitative detection of diverse metal ions.182 For instance, anchoring of DNAzyme-based sensors to the cell membrane enabled real-time monitoring of metal ions in the cellular microenvironment;183 this task is extremely difficult using other contemporary biotechniques. Furthermore, even hybrid molecules in which aptamers are integrated with other materials can be created to achieve complex molecular functions,184 which would be useful for biosensing applications. Also, we envision that the directed evolution of such hybrid-based affinity reagents could yield exceptionally high affinity and specificity.
Bioinformatics approaches have the possibility of reducing the time and cost to generate specific aptamers. Aptamers’ tertiary structures, binding affinity and specificity, thermodynamic characteristics, and target-relevant interactions can be predicted using in silico approaches, and numerous programs such as Rosetta, DOVIS, and CLADE are currently in use.185 Molecular dynamics simulations of molecular docking might also be useful to discover new aptamers,185 and the binding characteristics of existing aptamers can be further optimized by operating in silico screening and mutation analysis.186 Artificial intelligence (AI) and machine learning (ML) are increasingly applied to in silico aptamer design and screening; combined with other technologies such as HTS,187 AI- and ML-based big data analysis would provide new approaches to identification and characterization of aptamers.
Biosensors need not be inanimate. Microbes and living cells have been evaluated as biosensors that can operate without batteries.188 The cells can be genetically engineered to possess sensing functionalities, and the whole-cell biosensors are being studied concurrently with the rapid growth of synthetic biology. The cells are programmed to recognize the molecular entities among the cellular environments and can report the presence of molecules in a variety of ways, such as fluorescence emission or cellular suicide.181,189 If microorganism or cell sensors are created, their production would be inexpensive, and these living sensors have the potential to be applied for disease theranostics and environmental monitoring. These applications require precise control of the whole-cell biosensors, which can be realized by aptamers. Inside the cells, RNA aptamers can be incorporated into genetic circuits in the form of riboswitches, which undergo a conformational change when they bind to metabolites or secondary messengers, in the same way as structure-switching aptamers. The programmed action is synchronized with necessary gene activation and suppression, so the aptamer incorporation into cells would be a core technology to actualize living biosensors.
We believe that aptamers can be applied to various forms of biosensors to flexibly meet users’ needs, even unforeseen needs that may arise in the future. Use of the versatile nucleic acid biopolymers has enabled biosensors to surpass the limitations of existing biosensors and will contribute to breakthroughs in the field of biosensing. The rapid development of aptamer production technology and the next-generation devices will yield advanced aptamer-based biosensors that can address remaining technical challenges in detection.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NRF (National Research Foundation of Korea) Grant funded by the Korean Government (NRF-2017R1C1B3012050 and NRF-2019-Global PhD Fellowship Program).References
- F. Cui and H. S. Zhou, Biosens. Bioelectron., 2020, 165, 112349 CrossRef CAS.
- J. Kirsch, C. Siltanen, Q. Zhou, A. Revzin and A. Simonian, Chem. Soc. Rev., 2013, 42, 8733–8768 RSC.
- L. A. Terry, S. F. White and L. J. Tigwell, J. Agric. Food Chem., 2005, 53, 1309–1316 CrossRef CAS.
- P. Mehrotra, J. Oral Biol. Craniofac. Res., 2016, 6, 153–159 CrossRef.
- Markets and Markets, 2019.
- R. Schiess, B. Wollscheid and R. Aebersold, Mol. Oncol., 2009, 3, 33–44 CrossRef CAS.
- A. P. F. Turner, Chem. Soc. Rev., 2013, 42, 3184–3196 RSC.
- G. Danaei, M. M. Finucane, Y. Lu, G. M. Singh, M. J. Cowan, C. J. Paciorek, J. K. Lin, F. Farzadfar, Y.-H. Khang, G. A. Stevens, M. Rao, M. K. Ali, L. M. Riley, C. A. Robinson and M. Ezzati, Lancet, 2011, 378, 31–40 CrossRef CAS.
- M. L. Sin, K. E. Mach, P. K. Wong and J. C. Liao, Expert Rev. Mol. Diagn., 2014, 14, 225–244 CrossRef CAS.
- C. I. L. Justino, A. C. Freitas, R. Pereira, A. C. Duarte and T. A. P. Rocha Santos, TrAC, Trends Anal. Chem., 2015, 68, 2–17 CrossRef CAS.
- V. Thiviyanathan and D. G. Gorenstein, Proteomics: Clin. Appl., 2012, 6, 563–573 CAS.
- A. D. Keefe, S. Pai and A. Ellington, Nat. Rev. Drug Discovery, 2010, 9, 537–550 CrossRef CAS.
- R. Van Vught, R. J. Pieters and E. Breukink, Comput. Struct. Biotechnol. J., 2014, 9, e201402001 CrossRef.
- R. Nutiu and Y. Li, J. Am. Chem. Soc., 2003, 125, 4771–4778 CrossRef CAS.
- B. S. Ferguson, D. A. Hoggarth, D. Maliniak, K. Ploense, R. J. White, N. Woodward, K. Hsieh, A. J. Bonham, M. Eisenstein, T. E. Kippin, K. W. Plaxco and H. T. Soh, Sci. Transl. Med., 2013, 5, 213ra165 CrossRef.
- K. B. Yeh, H. Wood, M. Scullion, J. A. Russell, K. Parker, B. T. Gnade, A. R. Jones, C. Whittier and K. Mereish, mSphere, 2019, 4, e00695–19 CrossRef CAS.
- A. P. F. Turner, Science, 2000, 290, 1315–1317 CrossRef CAS.
- S. B. Nimse, M. D. Sonawane, K. S. Song and T. Kim, Analyst, 2016, 141, 740–755 RSC.
- A. Rentka, K. Koroskenyi, J. Harsfalvi, Z. Szekanecz, G. Szucs, P. Szodoray and A. Kemeny-Beke, Ann. Clin. Biochem., 2017, 54, 521–529 CAS.
- R. M. Lequin, Clin. Chem., 2005, 51, 2415–2418 CrossRef CAS.
- M. J. Schöning and A. Poghossian, Analyst, 2002, 127, 1137–1151 RSC.
- N. J. Ronkainen, H. B. Halsall and W. R. Heineman, Chem. Soc. Rev., 2010, 39, 1747–1763 RSC.
- H. Hasegawa, N. Savory, K. Abe and K. Ikebukuro, Molecules, 2016, 21, 421 CrossRef.
- A. K. Ogawa, Y. Wu, D. L. McMinn, J. Liu, P. G. Schultz and F. E. Romesberg, J. Am. Chem. Soc., 2000, 122, 3274–3287 CrossRef CAS.
- M. Kimoto, R. Yamashige, K. I. Matsunaga, S. Yokoyama and I. Hirao, Nat. Biotechnol., 2013, 31, 453–457 CrossRef CAS.
- J. D. Vaught, C. Bock, J. Carter, T. Fitzwater, M. Otis, D. Schneider, J. Rolando, S. Waugh, S. K. Wilcox and B. E. Eaton, J. Am. Chem. Soc., 2010, 132, 4141–4151 CrossRef CAS.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 CrossRef CAS.
- C. Tuerk and L. Gold, Science, 1990, 249, 505–510 CrossRef CAS.
- L. Gold, D. Ayers, J. Bertino, C. Bock, A. Bock, E. N. Brody, J. Carter, A. B. Dalby, B. E. Eaton, T. Fitzwater, D. Flather, A. Forbes, T. Foreman, C. Fowler, B. Gawande, M. Goss, M. Gunn, S. Gupta, D. Halladay, J. Heil, J. Heilig, B. Hicke, G. Husar, N. Janjic, T. Jarvis, S. Jennings, E. Katilius, T. R. Keeney, N. Kim, T. H. Koch, S. Kraemer, L. Kroiss, N. Le, D. Levine, W. Lindsey, B. Lollo, W. Mayfield, M. Mehan, R. Mehler, S. K. Nelson, M. Nelson, D. Nieuwlandt, M. Nikrad, U. Ochsner, R. M. Ostroff, M. Otis, T. Parker, S. Pietrasiewicz, D. I. Resnicow, J. Rohloff, G. Sanders, S. Sattin, D. Schneider, B. Singer, M. Stanton, A. Sterkel, A. Stewart, S. Stratford, J. D. Vaught, M. Vrkljan, J. J. Walker, M. Watrobka, S. Waugh, A. Weiss, S. K. Wilcox, A. Wolfson, S. K. Wolk, C. Zhang and D. Zichi, PLoS One, 2010, 5, e15004 CrossRef CAS.
- Y. Imaizumi, Y. Kasahara, H. Fujita, S. Kitadume, H. Ozaki, T. Endoh, M. Kuwahara and N. Sugimoto, J. Am. Chem. Soc., 2013, 135, 9412–9419 CrossRef CAS.
- J. P. Elskens, J. M. Elskens and A. Madder, Int. J. Mol. Sci., 2020, 21, 4522 CrossRef CAS.
- D. R. Davies, A. D. Gelinas, C. Zhang, J. C. Rohloff, J. D. Carter, D. O'Connell, S. M. Waugh, S. K. Wolk, W. S. Mayfield, A. B. Burgin, T. E. Edwards, L. J. Stewart, L. Gold, N. Janjic and T. C. Jarvis, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19971–19976 CrossRef CAS.
- S. Gupta, M. Hirota, S. M. Waugh, I. Murakami, T. Suzuki, M. Muraguchi, M. Shibamori, Y. Ishikawa, T. C. Jarvis, J. D. Carter, C. Zhang, B. Gawande, M. Vrkljan, N. Janjic and D. J. Schneider, J. Biol. Chem., 2014, 289, 8706–8719 CrossRef CAS.
- P. E. Nielsen, Chem. Biodivers., 2010, 7, 786–804 CrossRef CAS.
- B. Cai, S. Wang, L. Huang, Y. Ning, Z. Zhang and G. J. Zhang, ACS Nano, 2014, 8, 2632–2638 CrossRef CAS.
- E. J. Lee, H. K. Lim, Y. S. Cho and S. S. Hah, RSC Adv., 2013, 3, 5828–5831 RSC.
- Y. Brudno, M. E. Birnbaum, R. E. Kleiner and D. R. Liu, Nat. Chem. Biol., 2010, 6, 148–155 CrossRef CAS.
- H. Yu, S. Zhang and J. C. Chaput, Nat. Chem., 2012, 4, 183–187 CrossRef CAS.
- S. Arangundy-Franklin, A. I. Taylor, B. T. Porebski, V. Genna, S. Peak-Chew, A. Vaisman, R. Woodgate, M. Orozco and P. Holliger, Nat. Chem., 2019, 11, 533–542 CrossRef CAS.
- I. A. Ferreira-Bravo, C. Cozens, P. Holliger and J. J. DeStefano, Nucleic Acids Res., 2015, 43, 9587–9599 CAS.
- J. D. Badjić, A. Nelson, S. J. Cantrill, W. B. Turnbull and J. F. Stoddart, Acc. Chem. Res., 2005, 38, 723–732 CrossRef.
- K. M. Ahmad, Y. Xiao and H. Tom Soh, Nucleic Acids Res., 2012, 40, 11777–11783 CrossRef CAS.
- M. Cho, S. S. Oh, J. Nie, R. Stewart, M. J. Radeke, M. Eisenstein, P. J. Coffey, J. A. Thomson and H. T. Soh, Anal. Chem., 2015, 87, 821–828 CrossRef CAS.
- S. Rinker, Y. Ke, Y. Liu, R. Chhabra and H. Yan, Nat. Nanotechnol., 2008, 3, 418–422 CrossRef CAS.
- A. J. Simon, A. Vallée-Bélisle, F. Ricci and K. W. Plaxco, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 15048–15053 CrossRef CAS.
- L. Zhao, X. Qi, X. Yan, Y. Huang, X. Liang, L. Zhang, S. Wang and W. Tan, J. Am. Chem. Soc., 2019, 141, 17493–17497 CrossRef CAS.
- M. Huang, J. Song, P. Huang, X. Chen, W. Wang, Z. Zhu, Y. Song and C. Yang, Anal. Chem., 2019, 91, 10879–10886 CrossRef CAS.
- M. Fakruddin, K. S. B. Mannan, A. Chowdhury, R. M. Mazumdar, M. N. Hossain, S. Islam and M. A. Chowdhury, J. Pharm. BioAllied Sci., 2013, 5, 245–252 CrossRef.
- Y. Zhao, F. Chen, Q. Li, L. Wang and C. Fan, Chem. Rev., 2015, 115, 12491–12545 CrossRef CAS.
- M. Liu, J. Song, S. Shuang, C. Dong, J. D. Brennan and Y. Li, ACS Nano, 2014, 8, 5564–5573 CrossRef CAS.
- S. Bi, S. Yue and S. Zhang, Chem. Soc. Rev., 2017, 46, 4281–4298 RSC.
- J. Huang, Y. Wu, Y. Chen, Z. Zhu, X. Yang, C. J. Yang, K. Wang and W. Tan, Angew. Chem., Int. Ed., 2011, 50, 401–404 CrossRef CAS.
- Y. M. Wang, Z. Wu, S. J. Liu and X. Chu, Anal. Chem., 2015, 87, 6470–6474 CrossRef CAS.
- T. Notomi, H. Okayama, H. Masubuchi, T. Yonekawa, K. Watanabe, N. Amino and T. Hase, Nucleic Acids Res., 2000, 28, e63 CrossRef CAS.
- K. Hsieh, A. S. Patterson, B. S. Ferguson, K. W. Plaxco and H. T. Soh, Angew. Chem., Int. Ed., 2012, 51, 4896–4900 CrossRef CAS.
- J. Song, M. G. Mauk, B. A. Hackett, S. Cherry, H. H. Bau and C. Liu, Anal. Chem., 2016, 88, 7289–7294 CrossRef CAS.
- D. Rotem, L. Jayasinghe, M. Salichou and H. Bayley, J. Am. Chem. Soc., 2012, 134, 2781–2787 CrossRef CAS.
- R. Kawano, T. Osaki, H. Sasaki, M. Takinoue, S. Yoshizawa and S. Takeuchi, J. Am. Chem. Soc., 2011, 133, 8474–8477 CrossRef CAS.
- H. Chang, L. Tang, Y. Wang, J. Jiang and J. Li, Anal. Chem., 2010, 82, 2341–2346 CrossRef CAS.
- Kenry, A. Geldert, X. Zhang, H. Zhang and C. T. Lim, ACS Sens., 2016, 1, 1315–1321 CrossRef CAS.
- Y. V. Gerasimova and D. M. Kolpashchikov, Chem. Soc. Rev., 2014, 43, 6405–6438 RSC.
- X. Zuo, F. Xia, Y. Xiao and K. W. Plaxco, J. Am. Chem. Soc., 2010, 132, 1816–1818 CrossRef CAS.
- M. Chen, Z. Tang, C. Ma and Y. Yan, Sens. Actuators, B, 2020, 302, 127178 CrossRef CAS.
- C.-H. Lu, F. Wang and I. Willner, Chem. Sci., 2012, 3, 2616–2622 RSC.
- H. Yu, J. Canoura, B. Guntupalli, O. Alkhamis and Y. Xiao, Anal. Chem., 2018, 90, 1748–1758 CrossRef CAS.
- Y. Xiao, B. D. Piorek, K. W. Plaxco and A. J. Heeger, J. Am. Chem. Soc., 2005, 127, 17990–17991 CrossRef CAS.
- Y. Xiao, R. Y. Lai and K. W. Plaxco, Nat. Protoc., 2007, 2, 2875–2880 CrossRef CAS.
- R. J. White, N. Phares, A. A. Lubin, Y. Xiao and K. W. Plaxco, Langmuir, 2008, 24, 10513–10518 CrossRef CAS.
- B. R. Baker, R. Y. Lai, M. S. Wood, E. H. Doctor, A. J. Heeger and K. W. Plaxco, J. Am. Chem. Soc., 2006, 128, 3138–3139 CrossRef CAS.
- A. Vallée-Bélisle, F. Ricci, T. Uzawa, F. Xia and K. W. Plaxco, J. Am. Chem. Soc., 2012, 134, 15197–15200 CrossRef.
- Y. Liu, N. Tuleouva, E. Ramanculov and A. Revzin, Anal. Chem., 2010, 82, 8131–8136 CrossRef CAS.
- R. Y. Lai, K. W. Plaxco and A. J. Heeger, Anal. Chem., 2007, 79, 229–233 CrossRef CAS.
- Y. Xiao, A. A. Lubin, A. J. Heeger and K. W. Plaxco, Angew. Chem., Int. Ed., 2005, 44, 5456–5459 CrossRef CAS.
- H. Li, N. Arroyo-Currás, D. Kang, F. Ricci and K. W. Plaxco, J. Am. Chem. Soc., 2016, 138, 15809–15812 CrossRef CAS.
- H. Li, P. Dauphin-Ducharme, G. Ortega and K. W. Plaxco, J. Am. Chem. Soc., 2017, 139, 11207–11213 CrossRef CAS.
- Y. Jiang, N. Liu, W. Guo, F. Xia and L. Jiang, J. Am. Chem. Soc., 2012, 134, 15395–15401 CrossRef CAS.
- S. B. Ebrahimi, D. Samanta, H. F. Cheng, L. I. Nathan and C. A. Mirkin, J. Am. Chem. Soc., 2019, 141, 13744–13748 CrossRef CAS.
- S. Takahashi, K. T. Kim, P. Podbevšek, J. Plavec, B. H. Kim and N. Sugimoto, J. Am. Chem. Soc., 2018, 140, 5774–5783 CrossRef CAS.
- A. A. Lubin and K. W. Plaxco, Acc. Chem. Res., 2010, 43, 496–505 CrossRef CAS.
- T. Goda and Y. Miyahara, Biosens. Bioelectron., 2013, 45, 89–94 CrossRef CAS.
- B.-R. Li, Y.-J. Hsieh, Y.-X. Chen, Y.-T. Chung, C.-Y. Pan and Y.-T. Chen, J. Am. Chem. Soc., 2013, 135, 16034–16037 CrossRef CAS.
- H. Cho, E. C. Yeh, R. Sinha, T. A. Laurence, J. P. Bearinger and L. P. Lee, ACS Nano, 2012, 6, 7607–7614 CrossRef CAS.
- J. Y. Lichtenberg, Y. Ling and S. Kim, Sensors, 2019, 19, 2488 CrossRef CAS.
- C. Hoffmann, A. Zürn, M. Bünemann and M. J. Lohse, Br. J. Pharmacol., 2008, 153, S358–S366 CrossRef CAS.
- N. R. Latorraca, A. J. Venkatakrishnan and R. O. Dror, Chem. Rev., 2017, 117, 139–155 CrossRef CAS.
- G. Hou and Q. Cui, J. Am. Chem. Soc., 2013, 135, 10457–10469 CrossRef CAS.
- A. Vacic, J. M. Criscione, N. K. Rajan, E. Stern, T. M. Fahmy and M. A. Reed, J. Am. Chem. Soc., 2011, 133, 13886–13889 CrossRef CAS.
- P. S. Weiss, P. L. Trevor and M. J. Cardillo, J. Chem. Phys., 1989, 90, 5146–5153 CrossRef CAS.
- N. Nakatsuka, K. A. Yang, J. M. Abendroth, K. M. Cheung, X. Xu, H. Yang, C. Zhao, B. Zhu, Y. S. Rim, Y. Yang, P. S. Weiss, M. N. Stojanović and A. M. Andrews, Science, 2018, 362, 319–324 CrossRef CAS.
- D. E. Huizenga and J. W. Szostak, Biochemistry, 1995, 34, 656–665 CrossRef CAS.
- J. D. Klemm, S. L. Schreiber and G. R. Crabtree, Annu. Rev. Immunol., 1998, 16, 569–592 CrossRef CAS.
- C. H. Heldin, Cell, 1995, 80, 213–223 CrossRef CAS.
- M. Debiais, A. Lelievre, M. Smietana and S. Müller, Nucleic Acids Res., 2020, 48, 3400–3422 CrossRef.
- T. Zhao, R. Liu, X. Ding, J. Zhao, H. Yu, L. Wang, Q. Xu, X. Wang, X. Lou, M. He and Y. Xiao, Anal. Chem., 2015, 87, 7712–7719 CrossRef CAS.
- F. Jin, Y. Lian, J. Li, J. Zheng, Y. Hu, J. Liu, J. Huang and R. Yang, Anal. Chim. Acta, 2013, 799, 44–50 CrossRef CAS.
- A. K. Sharma, A. D. Kent and J. M. Heemstra, Anal. Chem., 2012, 84, 6104–6109 CrossRef CAS.
- S. F. Torabi, P. Wu, C. E. McGhee, L. Chen, K. Hwang, N. Zheng, J. Cheng and Y. Lu, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5903–5908 CrossRef CAS.
- J. Wang, L. Yue, Z. Li, J. Zhang, H. Tian and I. Willner, Nat. Commun., 2019, 10, 1–10 CrossRef.
- X.-B. Zhang, Z. Wang, H. Xing, Y. Xiang and Y. Lu, Anal. Chem., 2010, 82, 5005–5011 CrossRef CAS.
- E. Golub, H. B. Albada, W. C. Liao, Y. Biniuri and I. Willner, J. Am. Chem. Soc., 2016, 138, 164–172 CrossRef CAS.
- G. F. Luo, Y. Biniuri, W. H. Chen, E. Neumann, M. Fadeev, H. B. Marjault, A. Bedi, O. Gidron, R. Nechushtai, D. Stone, T. Happe and I. Willner, Nano Lett., 2019, 19, 6621–6628 CrossRef CAS.
- Y. Luo, H. Yu, O. Alkhamis, Y. Liu, X. Lou, B. Yu and Y. Xiao, Anal. Chem., 2019, 91, 7199–7207 CrossRef CAS.
- J. Canoura, Z. Wang, H. Yu, O. Alkhamis, F. Fu and Y. Xiao, J. Am. Chem. Soc., 2018, 140, 9961–9971 CrossRef CAS.
- Y. Liu, Y. Liu, Z. Matharu, A. Rahimian and A. Revzin, Biosens. Bioelectron., 2015, 64, 43–50 CrossRef CAS.
- D. Kang, R. J. White, F. Xia, X. Zuo, A. Vallée-Bélisle and K. W. Plaxco, NPG Asia Mater., 2012, 4, e1 CrossRef.
- S. Manna, S. Senapati, S. Lindsay and P. Zhang, J. Am. Chem. Soc., 2015, 137, 7415–7423 CrossRef CAS.
- K. W. Plaxco and H. T. Soh, Trends Biotechnol., 2011, 29, 1–5 CrossRef CAS.
- S. S. Oh, K. Plakos, Y. Xiao, M. Eisenstein and H. T. Soh, ACS Nano, 2013, 7, 9675–9683 CrossRef CAS.
- S. S. Oh, K. Plakos, X. Lou, Y. Xiao and H. T. Soh, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14053–14058 CrossRef CAS.
- N. Arroyo-Currás, J. Somerson, P. A. Vieira, K. L. Ploense, T. E. Kippin and K. W. Plaxco, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 645–650 CrossRef.
- Z. G. Yu and R. Y. Lai, Talanta, 2018, 176, 619–624 CrossRef CAS.
- J. S. Swensen, Y. Xiao, B. S. Ferguson, A. A. Lubin, R. Y. Lai, A. J. Heeger, K. W. Plaxco and H. T. Soh, J. Am. Chem. Soc., 2009, 131, 4262–4266 CrossRef CAS.
- S. Eissa, A. Ng, M. Siaj, A. C. Tavares and M. Zourob, Anal. Chem., 2013, 85, 11794–11801 CrossRef CAS.
- L. H. Lauridsen, H. A. Shamaileh, S. L. Edwards, E. Taran and R. N. Veedu, PLoS One, 2012, 7, e41702 CrossRef CAS.
- S. Gao, B. Hu, X. Zheng, Y. Cao, D. Liu, M. Sun, B. Jiao and L. Wang, Biosens. Bioelectron., 2016, 79, 938–944 CrossRef CAS.
- W. Zhou, R. Saran and J. Liu, Chem. Rev., 2017, 117, 8272–8325 CrossRef CAS.
- C. H. Chung, J. H. Kim, J. Jung and B. H. Chung, Biosens. Bioelectron., 2013, 41, 827–832 CrossRef CAS.
- F. Long, A. Zhu, H. Shi, H. Wang and J. Liu, Sci. Rep., 2013, 3, 1–7 Search PubMed.
- J. H. An, S. J. Park, O. S. Kwon, J. Bae and J. Jang, ACS Nano, 2013, 7, 10563–10571 CrossRef CAS.
- T. Li, S. Dong and E. Wang, J. Am. Chem. Soc., 2010, 132, 13156–13157 CrossRef CAS.
- H. Qu, A. T. Csordas, J. Wang, S. S. Oh, M. S. Eisenstein and H. T. Soh, ACS Nano, 2016, 10, 7558–7565 CrossRef CAS.
- B. Strehlitz, N. Nikolaus and R. Stoltenburg, Sensors, 2008, 8, 4296–4307 CrossRef CAS.
- S. Liu, X. Zhang, W. Luo, Z. Wang, X. Guo, M. L. Steigerwald and X. Fang, Angew. Chem., Int. Ed., 2011, 50, 2496–2502 CrossRef CAS.
- K. Urmann, J. G. Walter, T. Scheper and E. Segal, Anal. Chem., 2015, 87, 1999–2006 CrossRef CAS.
- G. A. Zelada-Guillén, J. L. Sebastián-Avila, P. Blondeau, J. Riu and F. X. Rius, Biosens. Bioelectron., 2012, 31, 226–232 CrossRef.
- B. Hong and Y. Zu, Theranostics, 2013, 3, 377–394 CrossRef CAS.
- Y. Song, Z. Zhu, Y. An, W. Zhang, H. Zhang, D. Liu, C. Yu, W. Duan and C. J. Yang, Anal. Chem., 2013, 85, 4141–4149 CrossRef CAS.
- F. Zheng, Y. Cheng, J. Wang, J. Lu, B. Zhang, Y. Zhao and Z. Gu, Adv. Mater., 2014, 26, 7333–7338 CrossRef CAS.
- H. Dong, H. Chen, J. Jiang, H. Zhang, C. Cai and Q. Shen, Anal. Chem., 2018, 90, 4507–4513 CrossRef CAS.
- Y. Jiang, M. Shi, Y. Liu, S. Wan, C. Cui, L. Zhang and W. Tan, Angew. Chem., Int. Ed., 2017, 56, 11916–11920 CrossRef CAS.
- Z. H. Zeng, C. H. Tung and Y. L. Zu, Mol. Ther.–Nucleic Acids, 2014, 3, 9 Search PubMed.
- Y. Xiang and Y. Lu, Nat. Chem., 2011, 3, 697–703 CrossRef CAS.
- C. Gu, T. Lan, H. Shi and Y. Lu, Anal. Chem., 2015, 87, 7676–7682 CrossRef CAS.
- L. Yan, Z. Zhu, Y. Zou, Y. Huang, D. Liu, S. Jia, D. Xu, M. Wu, Y. Zhou, S. Zhou and C. J. Yang, J. Am. Chem. Soc., 2013, 135, 3748–3751 CrossRef CAS.
- K. Tram, P. Kanda, B. J. Salena, S. Huan and Y. Li, Angew. Chem., Int. Ed., 2014, 53, 12799–12802 CrossRef CAS.
- J. J. Storhoff, R. Elghanian, C. A. Mirkin and R. L. Letsinger, Langmuir, 2002, 18, 6666–6670 CrossRef CAS.
- H. Kimura-Suda, D. Y. Petrovykh, M. J. Tarlov and L. J. Whitman, J. Am. Chem. Soc., 2003, 125, 9014–9015 CrossRef CAS.
- B. Liu and J. Liu, Anal. Methods, 2017, 9, 2633–2643 RSC.
- Y. S. Kim, J. H. Kim, I. A. Kim, S. J. Lee, J. Jurng and M. B. Gu, Biosens. Bioelectron., 2010, 26, 1644–1649 CrossRef CAS.
- P. Weerathunge, R. Ramanathan, R. Shukla, T. K. Sharma and V. Bansal, Anal. Chem., 2014, 86, 11937–11941 CrossRef CAS.
- W. Bai, C. Zhu, J. Liu, M. Yan, S. Yang and A. Chen, Environ. Toxicol. Chem., 2015, 34, 2244–2249 CrossRef CAS.
- A. Bosak, N. Saraf, A. Willenberg, M. W. C. Kwan, B. W. Alto, G. W. Jackson, R. H. Batchelor, T. D. Nguyen-Huu, V. Sankarapani, G. D. Parks, S. Seal and B. J. Willenberg, RSC Adv., 2019, 9, 23752–23763 RSC.
- K. V. Ragavan, L. S. Selvakumar and M. S. Thakur, Chem. Commun., 2013, 49, 5960–5962 RSC.
- V. C. Coffey, Opt. Photonics News, 2011, 22, 26–31 CrossRef CAS.
- R. Freeman, X. Liu and I. Willner, J. Am. Chem. Soc., 2011, 133, 11597–11604 CrossRef CAS.
- Y. Zhang, D. Gao, J. Fan, J. Nie, S. Le, W. Zhu, J. Yang and J. Li, Biosens. Bioelectron., 2016, 78, 538–546 CrossRef CAS.
- H. Liu, Y. Xiang, Y. Lu and R. M. Crooks, Angew. Chem., Int. Ed., 2012, 51, 6925–6928 CrossRef CAS.
- L. Wang, W. Ma, W. Chen, L. Liu, W. Ma, Y. Zhu, L. Xu, H. Kuang and C. Xu, Biosens. Bioelectron., 2011, 26, 3059–3062 CrossRef CAS.
- H. Xu, X. Mao, Q. Zeng, S. Wang, A.-N. Kawde and G. Liu, Anal. Chem., 2009, 81, 669–675 CrossRef CAS.
- L. Wang, W. Chen, W. Ma, L. Liu, W. Ma, Y. Zhao, Y. Zhu, L. Xu, H. Kuang and C. Xu, Chem. Commun., 2011, 47, 1574–1576 RSC.
- W. B. Shim, M. J. Kim, H. Mun and M. G. Kim, Biosens. Bioelectron., 2014, 62, 288–294 CrossRef CAS.
- J. Chen, S. Zhou and J. Wen, Anal. Chem., 2014, 86, 3108–3114 CrossRef CAS.
- Z. Fang, W. Wu, X. Lu and L. Zeng, Biosens. Bioelectron., 2014, 56, 192–197 CrossRef CAS.
- H. Yang, H. Liu, H. Kang and W. Tan, J. Am. Chem. Soc., 2008, 130, 6320–6321 CrossRef CAS.
- P. Song, D. Ye, X. Zuo, J. Li, J. Wang, H. Liu, M. T. Hwang, J. Chao, S. Su, L. Wang, J. Shi, L. Wang, W. Huang, R. Lal and C. Fan, Nano Lett., 2017, 17, 5193–5198 CrossRef CAS.
- Z. Zhu, C. Wu, H. Liu, Y. Zou, X. Zhang, H. Kang, C. J. Yang and W. Tan, Angew. Chem., Int. Ed., 2010, 49, 1052–1056 CrossRef CAS.
- L. Xu, L. Qiu, Y. Sheng, Y. Sun, L. Deng, X. Li, M. Bradley and R. Zhang, J. Mater. Chem. B, 2018, 6, 510–517 RSC.
- P. S. Noonan, R. H. Roberts and D. K. Schwartz, J. Am. Chem. Soc., 2013, 135, 5183–5189 CrossRef CAS.
- R. L. Strack, W. Song and S. R. Jaffrey, Nat. Protoc., 2014, 9, 146–155 CrossRef CAS.
- K. Höfer, L. V. Langejürgen and A. Jäschke, J. Am. Chem. Soc., 2013, 135, 13692–13694 CrossRef.
- S. Ranallo, M. Rossetti, K. W. Plaxco, A. Vallée-Bélisle and F. Ricci, Angew. Chem., Int. Ed., 2015, 54, 13214–13218 CrossRef CAS.
- S. Saha, V. Prakash, S. Halder, K. Chakraborty and Y. Krishnan, Nat. Nanotechnol., 2015, 10, 645–651 CrossRef CAS.
- S. Somasundaram and C. J. Easley, J. Am. Chem. Soc., 2019, 141, 11721–11726 CrossRef CAS.
- B. Kang, S. V. Park, H. T. Soh and S. S. Oh, ACS Sens., 2019, 4, 2802–2808 CrossRef CAS.
- M. A. Eckert and W. Zhao, Interface Focus, 2013, 3, 20130014 CrossRef.
- H. Sun and Y. Zu, Molecules, 2015, 20, 11959–11980 CrossRef CAS.
- S. Ni, H. Yao, L. Wang, J. Lu, F. Jiang, A. Lu and G. Zhang, Int. J. Mol. Sci., 2017, 18, 1683 CrossRef.
- R. Padilla and R. Sousa, Nucleic Acids Res., 1999, 27, 1561–1563 CrossRef CAS.
- J. Ruckman, L. S. Green, J. Beeson, S. Waugh, W. L. Gillette, D. D. Henninger, L. Claesson-Welsh and N. Janjić, J. Biol. Chem., 1998, 273, 20556–20567 CrossRef CAS.
- R. Pipkorn, M. Wiessler, W. Waldeck, U. Hennrich, K. Nokihara, M. Beining and K. Braun, Int. J. Med. Sci., 2012, 9, 1–10 CrossRef CAS.
- M. A. Campbell and J. Wengel, Chem. Soc. Rev., 2011, 40, 5680–5689 RSC.
- C. Kratschmer and M. Levy, Nucleic Acid Ther., 2017, 27, 335–344 CrossRef CAS.
- F. Odeh, H. Nsairat, W. Alshaer, M. A. Ismail, E. Esawi, B. Qaqish, A. Bawab and S. I. Ismail, Molecules, 2020, 25, 3 CrossRef CAS.
- A. N. Cadinoiu, D. M. Rata, L. I. Atanase, O. M. Daraba, D. Gherghel, G. Vochita and M. Popa, Polymers, 2019, 11, 1515 CrossRef CAS.
- M. Cho, Y. Xiao, J. Nie, R. Stewart, A. T. Csordas, S. S. Oh, J. A. Thomson and H. T. Soh, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 15373–15378 CrossRef CAS.
- M. Cho, S. S. Oh, J. Nie, R. Stewart, M. Eisenstein, J. Chambers, J. D. Marth, F. Walker, J. A. Thomson and H. T. Soh, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 18460–18465 CrossRef CAS.
- K. M. Ahmad, S. S. Oh, S. Kim, F. M. McClellen, Y. Xiao and H. T. Soh, PLoS One, 2011, 6, e27051 CrossRef CAS.
- K. Sefah, D. Shangguan, X. Xiong, M. B. O’Donoghue and W. Tan, Nat. Protoc., 2010, 5, 1169–1185 CrossRef CAS.
- G. Mayer, M. S. L. Ahmed, A. Dolf, E. Endl, P. A. Knolle and M. Famulok, Nat. Protoc., 2010, 5, 1993–2004 CrossRef CAS.
- A. R. Ferré-D’Amaré and W. G. Scott, Cold Spring Harbor Perspect. Biol., 2010, 2, a003574 Search PubMed.
- Y. Zhang, J. Wang, H. Cheng, Y. Sun, M. Liu, Z. Wu and R. Pei, Gene Ther., 2017, 24, 84–91 CrossRef CAS.
- H. K. Kim, I. Rasnik, J. Liu, T. Ha and Y. Lu, Nat. Chem. Biol., 2007, 3, 763–768 CrossRef CAS.
- L. Qiu, T. Zhang, J. Jiang, C. Wu, G. Zhu, M. You, X. Chen, L. Zhang, C. Cui, R. Yu and W. Tan, J. Am. Chem. Soc., 2014, 136, 13090–13093 CrossRef CAS.
- S. S. Oh, B. F. Lee, F. A. Leibfarth, M. Eisenstein, M. J. Robb, N. A. Lynd, C. J. Hawker and H. T. Soh, J. Am. Chem. Soc., 2014, 136, 15010–15015 CrossRef CAS.
- A. B. Kinghorn, L. A. Fraser, S. Liang, S. C. C. Shiu and J. A. Tanner, Int. J. Mol. Sci., 2017, 18, 2516 CrossRef.
- M. Darmostuk, S. Rimpelova, H. Gbelcova and T. Ruml, Biotechnol. Adv., 2014, 33, 1141–1161 CrossRef.
- S. Hoon, B. Zhou, K. D. Janda, S. Brenner and J. Scolnick, BioTechniques, 2011, 51, 413–416 CrossRef CAS.
- Q. Gui, T. Lawson, S. Shan, L. Yan and Y. Liu, Sensors, 2017, 17, 1623 CrossRef.
- B. Saltepe, E. Ş. Kehribar, S. S. Su Yirmibeşoǧlu and U. O. Şafak Şeker, ACS Sens., 2018, 3, 13–26 CrossRef CAS.
Footnote |
| † Equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2020 |