
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ synthesis of polyynes in a polymer matrix via pulsed laser ablation in a liquid
Sonia
Peggiani
 a,
Anna
Facibeni
a,
Alberto
Milani
a,
Chiara
Castiglioni
b,
Valeria
Russo
a,
Andrea
Li Bassi
a and
Carlo S.
Casari
*a
a,
Anna
Facibeni
a,
Alberto
Milani
a,
Chiara
Castiglioni
b,
Valeria
Russo
a,
Andrea
Li Bassi
a and
Carlo S.
Casari
*a
aDepartment of Energy, Politecnico di Milano, Via Ponzio 34/3, 20133 Milan, Italy. E-mail: carlo.casari@polimi.it
bDepartment of Chemistry, Materials, and Chemical Engineering “Giulio Natta”, Politecnico di Milano, Piazza Leonardo da Vinci 32, 20133 Milan, Italy
First published on 14th August 2020
Abstract
Polyynes are finite chains formed by sp-hybridized carbon atoms with alternating single and triple bonds and display intriguing electronic and optical properties. Pulsed laser ablation in liquid (PLAL) is a well assessed technique for the physical synthesis of hydrogen-capped polyynes in solution; however, their limited stability prevents further exploitation in materials for different applications. In this work, polyynes in poly(vinyl alcohol) (PVA) were produced in a single-step PLAL process by ablating graphite directly in an aqueous solution of PVA which, as a participating medium for PLAL, is shown to favour the formation of polyynes. The addition of Ag colloids to the aqueous PVA/polyynes solution allowed surface-enhanced Raman spectroscopy (SERS) measurements, carried out both on liquid samples and on free-standing nanocomposites, obtained after solvent evaporation. A non-trivial behaviour of the polymer matrix structure as a function of the PVA concentration is revealed showing that an intimate blend of polyynes and Ag nanoparticles with the polymer can be achieved. We demonstrate that polyynes in the nanocomposite remain stable for at least 11 months, whereas the corresponding Ag/PVA/polyynes solution displayed a strong polyyne decomposition already after 3 weeks. These results pave the way to further characterization of the properties of polyyne-based films and materials.
Introduction
Past achievements in the research on carbyne and linear sp-carbon chains have outlined great potential for their huge effective area, the largest Young's modulus, high electron mobility and thermal conductivity.1–5 A few proof of concept demonstrations of possible applications have been reported only recently, as in the case of optical imaging of biomolecules and field effect transistor realization.6,7 Finite sp-carbon linear chains with single–triple bond alternation, also called polyynes, display semiconducting optoelectronic properties modulated by the length (i.e. number of carbon atoms) and by the terminations (atom or molecular group).8 Polyynes can be synthesized by physical methods potentially scalable to mass production, for example, submerged arc discharge in liquid (SADL) and pulsed laser ablation in liquid (PLAL). In both cases, the most exploited liquid media are organic solvents,9–15 whereas the use of water is less explored in the literature.16–18 However, the use of water in SADL was recently shown to achieve polyynes of up to 16 carbon atoms or to obtain polyynes by collecting and condensing the gas generated by arc discharge.19,20To fully take advantage of the properties of polyynes, the availability of a stable material under ambient conditions is required. The stability of sp-carbon atomic chains is not a trivial issue.21 In fact, hydrogen-capped polyynes, typically synthesized using physical methods such as SADL and PLAL, show poor stability against cross-linking, especially when the solvent is removed.22–24 Different strategies have been developed so far to avoid degradation, for instance encapsulation of long sp-carbon wires inside double- or multi-walled carbon nanotubes,25–27 or surrounding the polyynes by a rotaxane macrocycle28 or immersing them in an ionic liquid.29 In general, bulky groups at the end of the sp-carbon chains are known to act as spacers preventing cross-linking reactions,30 for example, sp2 clusters,31 graphene edges32 or phenyl groups.8,33–35 Another very promising technique to stabilize carbon chains is to embed them in solid matrices, such as solid Ag nanoparticle assemblies,23 SiO2 dried gels36 and poly(vinyl alcohol) (PVA).37–39 PVA is widely exploited in the formation of carbon or metal-based composites because it is a low-cost, chemically stable material, and is soluble in water with good film formation properties.40,41 A few studies have addressed the encapsulation of polyynes in such a polymer to form a nanocomposite by simply adding PVA to a solution of an already formed aggregate of polyynes and Ag or Au nanoparticles.37,38 In this case, the aggregation of polyynes with metal nanoparticles could prevent the complete blending of polyynes, metal nanoparticles and polymer. In addition, those studies involved polyyne synthesis in organic solvents and a subsequent heating process to dissolve PVA pellets in the polyyne/metal solution. In view of possible future applications, further investigations are needed to deepen the understanding of the structure of the composite by optimizing the blending of all the components and to improve simple and fast production methods without employing toxic solvents and a heating process which degrades polyynes.21,42
Thus, some still open questions regard the possibility of producing a nanocomposite by directly synthesizing polyynes in an aqueous polymer solution and how this process can affect the polymer matrix and the final degree of blending of polyynes and Ag particles with the polymer.
In this study, we address these questions by reporting a one-step and simple method for in situ synthesis of polyynes in PVA by employing laser ablation of graphite in aqueous solutions of PVA. We show a two-fold advantage of this process: on one side is the use of non-toxic liquids as solvents for nanocomposite formation and on the other, there is no need for any further process involving heating or dilution to obtain the solution of PVA and polyynes. The participating role of aqueous solutions of PVA at different concentrations as a solvent for PLAL is explored by using UV-Vis and surface-enhanced Raman spectroscopy (SERS). A non-trivial behaviour of the polymer matrix structure is observed as a function of the PVA concentration showing that an intimate blend of polyynes and Ag with the polymer can be achieved using the in situ synthesis of polyynes in aqueous PVA. After adding Ag colloids to the aqueous solution of PVA with polyynes, the solution was left to dry, forming a free-standing film. SERS analysis in different areas of the film was performed to investigate the uniformity of polyynes dispersion and to evaluate the connection between morphology and the SERS enhancement. Finally, a comparison between the stability of Ag/PVA/polyynes solution and the corresponding free-standing film is given, confirming the prolonged stability of polyynes embedded in the polymer and the practicality of the method presented in this work for development of polyyne-based nanocomposites in view of future applications.
Experimental procedures
Polyynes were synthesized by ablating a graphite target in liquid media by means of a ns-pulsed laser. Ablation was performed using a Nd:YAG laser (2nd harmonic, λ = 532 nm) with a pulse duration of 5–7 ns and repetition rate of 10 Hz (Quantel Q-smart 850). The laser beam was focused from the top side of the vial. The estimated energy fluence reaching the target was 5.2 J cm−2 and the spot size at the target was 0.14 cm. The ablation time was 15 minutes in all the experiments, except in one case where it was 30 minutes meant for studying the viscosity role of PVA during the ablation, as described later in the Results section. The solvent was prepared by dissolving poly(vinyl alcohol) (PVA) granules (Fluka, MW 130![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, degree of polymerization N ∼ 2700) in deionized water Milli-Q (0.055 μS) at 373 K and at different concentrations, namely 0.03, 0.5, 1, 3, 10 wt%. We hereafter name the solutions as PVAx%. A graphite target 2 mm thick and 8 mm in diameter with a purity of 99.99% (Testbourne Ltd) was placed at the bottom of a glass vial containing 10 ml of solution. To perform surface-enhanced Raman spectroscopy (SERS), we varied the ratio of polyynes in aqueous PVA solution (indicated by PVA/polyynes) with respect to silver nanoparticles to obtain the highest SERS intensity of polyynes. We achieved the optimum for a volume ratio of 1:2 (1 for PVA/polyynes, 2 for Ag colloid). We refer to the SERS-active solution as Ag/PVA/polyynes solution. The Ag nanoparticles, employed in this work, were prepared using the Lee–Meisel method,43 reaching a concentration of 10−3 M and a plasmonic peak centred at 413 nm. Their medium size of 60–80 nm was estimated thanks to the morphological characterization using a field emission scanning electron microscope (FEG-SEM, Zeiss Supra 40). The drop-cast solution of Ag/PVA/polyynes was left to dry in a small plastic container at room temperature. After 24 hours, we detached the solid Ag/PVA/polyynes nanocomposite material, and we squared it adding a graduated scale to perform localized SERS analyses. The step by step procedure for the formation of free-standing films is reported in Table 1.
000, degree of polymerization N ∼ 2700) in deionized water Milli-Q (0.055 μS) at 373 K and at different concentrations, namely 0.03, 0.5, 1, 3, 10 wt%. We hereafter name the solutions as PVAx%. A graphite target 2 mm thick and 8 mm in diameter with a purity of 99.99% (Testbourne Ltd) was placed at the bottom of a glass vial containing 10 ml of solution. To perform surface-enhanced Raman spectroscopy (SERS), we varied the ratio of polyynes in aqueous PVA solution (indicated by PVA/polyynes) with respect to silver nanoparticles to obtain the highest SERS intensity of polyynes. We achieved the optimum for a volume ratio of 1:2 (1 for PVA/polyynes, 2 for Ag colloid). We refer to the SERS-active solution as Ag/PVA/polyynes solution. The Ag nanoparticles, employed in this work, were prepared using the Lee–Meisel method,43 reaching a concentration of 10−3 M and a plasmonic peak centred at 413 nm. Their medium size of 60–80 nm was estimated thanks to the morphological characterization using a field emission scanning electron microscope (FEG-SEM, Zeiss Supra 40). The drop-cast solution of Ag/PVA/polyynes was left to dry in a small plastic container at room temperature. After 24 hours, we detached the solid Ag/PVA/polyynes nanocomposite material, and we squared it adding a graduated scale to perform localized SERS analyses. The step by step procedure for the formation of free-standing films is reported in Table 1.
| Step | Description | Sample name |
|---|---|---|
| 1 | Preparation of solvents for PLAL, i.e. PVA at different concentrations in water wt%: x = 0.03, 0.5, 1, 3, 10. | PVAx% |
| 2 | PLAL of graphite in aqueous PVA at different concentrations (PVA/polyynes solution). | L-x% |
| 3 | Addition of Ag colloid in a volume ratio of 2:1 with respect to PVA/polyynes solution (Ag/PVA/polyynes solution). |
L-x%_Ag |
| 4 | Drop casting and solvent evaporation for 24 hours at room temperature (Ag/PVA/polyynes nanocomposite). | S-x%_Ag |
To check the repeatability of the results each SERS measurement on liquid and solid samples was repeated at least three times.
UV-Vis absorption spectra of PVA/polyynes liquid samples were obtained on a Shimadzu UV-1800 UV/Visible scanning spectrophotometer (190–1100 nm). SERS measurements were carried out using a Renishaw InVia micro-Raman spectrometer with an argon ion laser (λ = 514.5 nm) and a spectral resolution of about 3 cm−1. SERS spectra of both Ag/PVA/polyynes solutions and the corresponding nanocomposites were taken using an objective of 50× and a laser power of 0.13 and 0.013 mW on the liquid and solid samples, respectively. SERS maps of Ag/PVA1%/polyynes films (S-1%_Ag) were obtained on a Renishaw InVia micro-Raman spectrometer equipped with an automatic translator x–y–z of micrometric resolution and a diode-pumped solid-state laser (λ = 532 nm), using a laser power of 0.35 mW. The laser powers used in these experiments do not cause any laser-induced degradation to the material.
Results
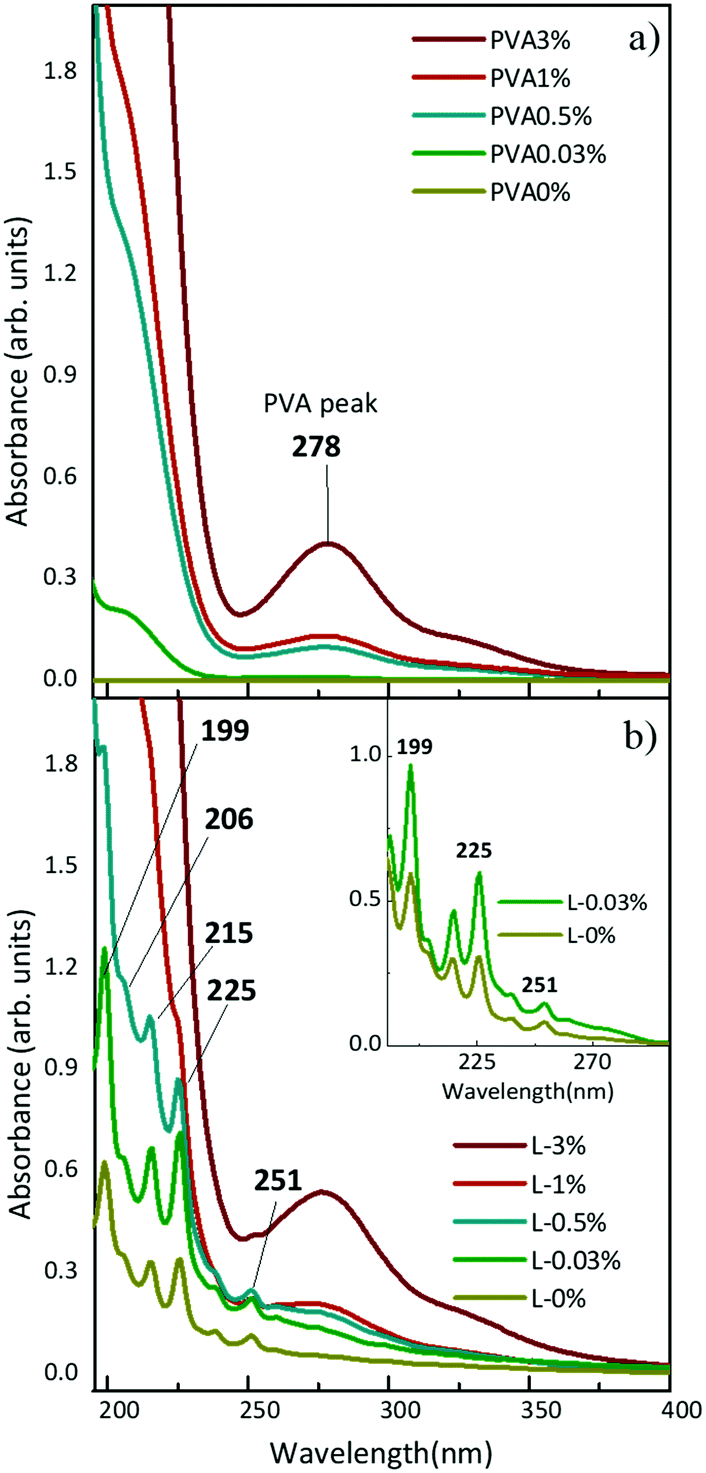
In situ synthesis of hydrogen-capped polyynes in a polymer was achieved by laser ablation of a solid graphite target in aqueous solutions of PVA. The UV-Vis absorption spectra of solutions with different concentrations of PVA in water (e.g. 0.03, 0.5, 1, 3, 10 wt%) before and after ablation of graphite are shown in Fig. 1. We refer to the PVA/polyynes solutions by L-0.03%, L-0.5%, L-1%, L-3%, L-10%, and to the ablation in water by L-0% (see Table 1 for the sample name). Pure PVA is transparent in the visible range and it has an absorption peak at 278 nm, related to the π → π* transition of the carbonyl groups (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), associated with the ethylene unsaturation (CC) of the type –(CHCH)2CO−, typical of end groups, as reported in ref. 44. By increasing the concentration of PVA in water, a consequent growth of the absorption peak at 278 nm is observed. After ablation, the UV-Vis spectra indicate the presence of polyynes via the characteristic vibronic peaks at 199, 206, 215, 225 and 251 nm. These features are related to H-terminated polyynes, which for simplicity we indicate as Cn, with n (i.e. the number of carbon atoms) ranging from 6 to 10 in agreement with previous studies.19,45,46 Longer polyynes may be present but are hardly detectable due to their low concentration, which results in a weak signal also partially covered by the PVA peak. In fact, for PVA concentration exceeding 0.5 wt%, the strong PVA peak makes it difficult to identify the presence of polyyne.
O), associated with the ethylene unsaturation (CC) of the type –(CHCH)2CO−, typical of end groups, as reported in ref. 44. By increasing the concentration of PVA in water, a consequent growth of the absorption peak at 278 nm is observed. After ablation, the UV-Vis spectra indicate the presence of polyynes via the characteristic vibronic peaks at 199, 206, 215, 225 and 251 nm. These features are related to H-terminated polyynes, which for simplicity we indicate as Cn, with n (i.e. the number of carbon atoms) ranging from 6 to 10 in agreement with previous studies.19,45,46 Longer polyynes may be present but are hardly detectable due to their low concentration, which results in a weak signal also partially covered by the PVA peak. In fact, for PVA concentration exceeding 0.5 wt%, the strong PVA peak makes it difficult to identify the presence of polyyne.
 | ||
| Fig. 1 UV-Vis spectra of PVA solutions at different concentrations in water (a) before and (b) after ablation. Inset: Comparison of spectra of polyynes solution with and without PVA0.03%. Spectra are obtained after subtraction from the corresponding background spectra, respectively PVA0.03% and PVA0%. | ||
By comparing the spectrum of L-0.03% with that of L-0% after subtraction of the corresponding PVA background (see the inset of Fig. 1), we observe a higher absorption of polyynes synthesized in PVA0.03%. Using the peak–valley difference in the absorbance of the polyynes 0–0 band of the 1∑u+ ← X1∑g+ transition, we register an increase of a factor of 2.3 both for C6 (at 199 nm) and C8 (at 225 nm). By means of UV-Vis absorption measurements we concluded that aqueous PVA, employed as a PLAL solvent, favours the formation of polyynes with respect to pure water because it may help the sp-carbon chain formation by providing further carbon and hydrogen atoms46 and by changing the viscosity of the solution, as discussed below.
Since the PVA UV-Vis signal does not allow us to directly evaluate the effect of PVA on the polyyne yield at all concentrations, we adopted Raman spectroscopy, whose polyyne fingerprint is quite different from that of PVA. In fact, the sp-carbon Raman-active mode, which is related to the different collective stretching vibrations of sp-hybridized C–C, C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bonds,47 is in the 1800–2300 cm−1 range and the intense Raman band of the PVA pellet closer to polyynes region is at about 2910 cm−1, which refers to the symmetrical stretching vibrational mode νs(CH2). It also shows two shoulders, one at 2851 cm−1 related to weak intensity stretching mode ν(CH) and one at 2934 cm−1 related to medium intensity asymmetrical stretching mode νa(CH2).48 Such features, typical of the semi-crystalline polymer, can change due to structural disorder, as outlined later in this section.
C bonds,47 is in the 1800–2300 cm−1 range and the intense Raman band of the PVA pellet closer to polyynes region is at about 2910 cm−1, which refers to the symmetrical stretching vibrational mode νs(CH2). It also shows two shoulders, one at 2851 cm−1 related to weak intensity stretching mode ν(CH) and one at 2934 cm−1 related to medium intensity asymmetrical stretching mode νa(CH2).48 Such features, typical of the semi-crystalline polymer, can change due to structural disorder, as outlined later in this section.
Here we specifically performed surface-enhanced Raman spectroscopy (SERS) to overcome problems in weak Raman signals due to the low polyynes concentration. To obtain SERS-active liquid samples, we added the same amount of chemically synthesized Ag colloids to all the PVA/polyynes solutions (denoted by L-x%_Ag). Then, we drop-cast the liquid specimens on a substrate, and we let them dry at room temperature under stationary conditions to obtain the solid nanocomposites (indicated by S-x%_Ag, with x being the PVA concentration ranging from 0.5% to 10%). Considering that we added the same quantity of silver colloid into all the solutions, the SERS spectra can be compared to get qualitative information on the presence of polyynes.
The SERS spectra of liquid samples (L-0.03%_Ag, L-0.5%_Ag, L-1%_Ag, L-3%_Ag, L-10%_Ag) are presented in Fig. 2a. These spectra are compared to the spectrum of the sample in pure water without PVA (L-0%_Ag) and to the one without Ag colloids composed by PVA1% used as a background reference (“L background”).
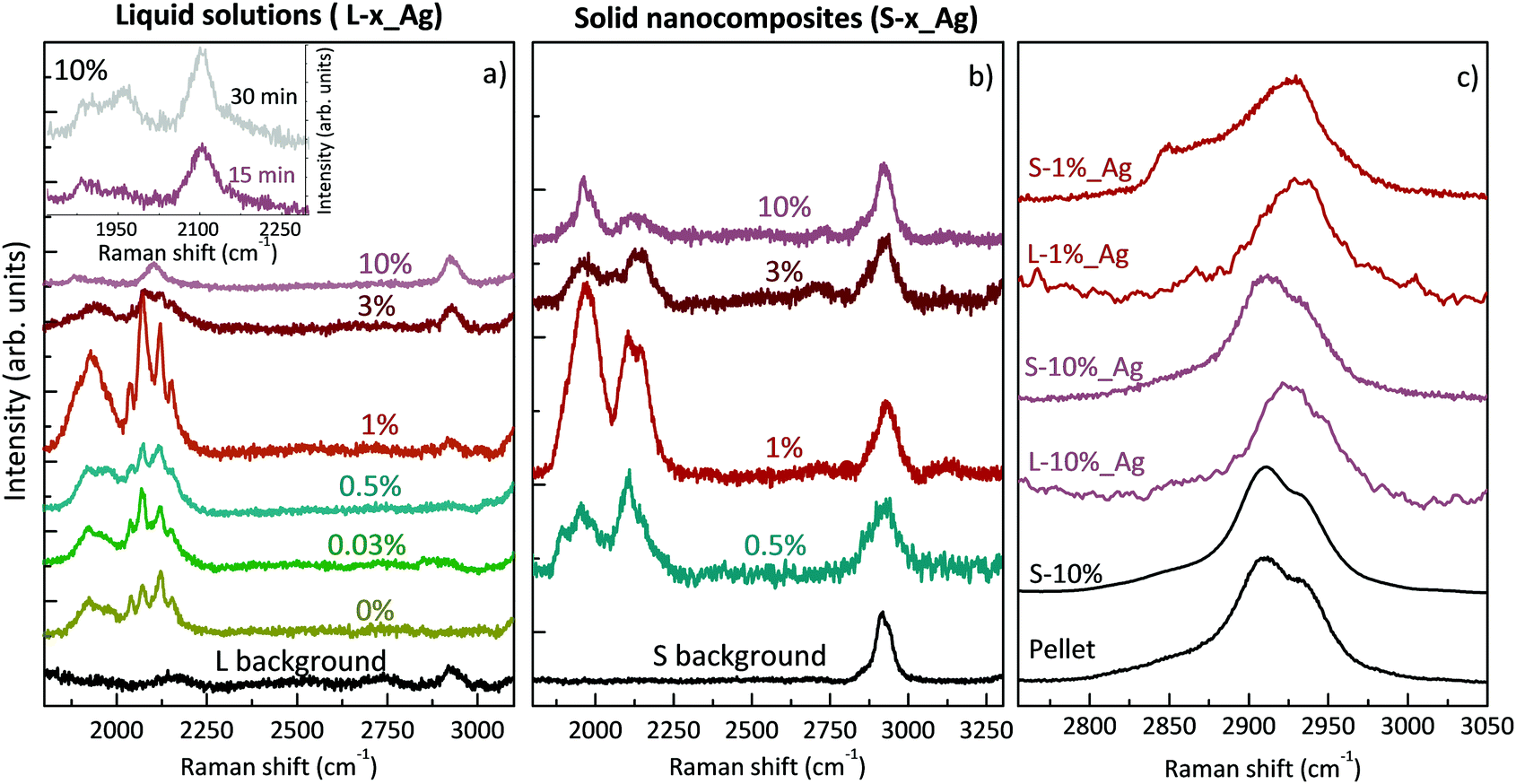 | ||
| Fig. 2 (a) SERS spectra of Ag/PVA/polyynes solutions at different concentrations. In the black line, a spectrum of liquid PVA1% before ablation (L background). Inset: Ag/PVA10%/polyynes (L-10%_Ag) solution spectra after 15 and 30 minutes of ablation. (b) SERS spectra of Ag/PVA/polyynes nanocomposites; for all samples the measurements were taken at the centre. In black, the free-standing film of solidified PVA1% without polyynes and Ag colloid (S background). (c) SERS spectra zoomed on the stretching vibrational mode of PVA in different phases. | ||
In all the spectra of liquid samples (Fig. 2a) the CH band of PVA at about 2900 cm−1 increases consistently with the concentration of the polymer and two main bands are visible in the sp-carbon spectral region (1800–2200 cm−1). One band at 2000–2200 cm−1 is due to collective CC stretching modes of the sp-carbon chain (effective conjugation coordinate (ECC) mode),49 and the other one at 1800–2000 cm−1 can be associated with the interaction between polyynes and silver nanoparticles that can modify the electronic structure of the chains causing an equalization of the bond lengths, in agreement with our previous studies.8,35,50 The bands at 2000–2200 cm−1 for all the samples, except L-10%_Ag, are characterized by four features, which refer to polyynes of different lengths. Raman peaks of longer polyynes are expected at lower wavenumbers, in agreement with molecular modelling and experiments reported in previous studies.49–51 In SERS experiments, the spectra are affected by the solvent and by the presence of metal nanoparticles; hence further studies of size-selected polyynes in the Ag/PVA matrix are needed to assign each band to polyynes of a specific length.
In Ag/PVA/polyynes solutions up to the PVA concentration of 1 wt% (i.e. L-0.03%_Ag, L-0.5%_Ag, L-1%_Ag), SERS features of sp-carbon are more intense with respect to samples synthesized in water (L-0%_Ag), in agreement with UV-Vis data already presented. Exceeding that concentration, as in the case of L-3%_Ag and L-10%_Ag, the SERS spectra show weaker and less resolved sp-carbon bands than in L-0%_Ag. These results indicate a non-trivial role of PVA that can be possibly explained considering the effects of liquids of different viscosities on polyyne formation. In some studies, decalin was employed as a solvent, considering that the reactive species, i.e. C2 radicals, hardly diffuse in viscous solvents and are strongly confined to the plasma region. As a consequence, polyynes keep on growing in length up to C30, before being terminated by hydrogen atoms.11,12 In the last-mentioned studies, they used a mixture of two different conformations of decalin, cis-decalin with a viscosity at 25 °C of 3.042 mPa s and the trans-decalin with a value of 1.948 mPa s, giving an average viscosity of 2.50 mPa s, which is close to that of PVA1% (∼2.53 mPa s at 25 °C).52 Consequently, viscous media, as decalin and PVA1%, can help plasma confinement and thus the synthesis of sp-carbon species. However, we observe a decrease of the sp-carbon signal intensity when exceeding the 1% PVA concentration. When the viscosity is too high, the overall yield of polyynes can be hindered by the solvent. In fact, the already formed end-capped chains, due to their low mobility in the viscous liquid, remain in the plasma region, and they may be decomposed and thermally degraded. Another effect that may decrease the polyyne formation yield in highly viscous liquids is the slower release of the microbubbles created during the ablation which can partially shield the laser beam.53,54 In order to observe whether we can improve polyyne signal in highly viscous solvents (PVA > 1%) by increasing the ablation time, we ablated graphite for a longer time (30 minutes instead of 15 minutes) in a solution of PVA10% (∼25.91 mPa s at 25 °C),55 which is the maximum concentration of PVA that can dissolve in water.56 Increasing the ablation time, only a slight increment of the intensity of polyyne SERS signal is recorded (see inset of Fig. 2) but always in the presence of one broad band without the features observed in the other cases. Another contributing factor to explain the different SERS polyyne signals relies on the fact that highly concentrated PVA may surround Ag nanoparticles and polyynes, preventing the direct adsorption of polyynes on Ag colloids and hence leading to the formation of hot spots. Several different effects, all related to the concentration of PVA, can indeed play a role in the non-trivial behaviour observed.
The SERS spectra of solid nanocomposites resulting from the solvent evaporation of the solutions with polyynes, silver colloids and PVA in water (i.e. L-x%_Ag) are shown in Fig. 2b. It was not possible to prepare a free-standing film starting from a PVA concentration below 0.5% (i.e. from sample L-0.03%_Ag) due to the limited concentration of PVA. All the spectra of the nanocomposites are characterized by the CH band of the PVA at about 2900 cm−1 showing the same intensity and two main broad bands in the polyyne region, as in the liquid case, displaying the fact that polyynes survive in the solvent evaporation process. The same intensity of the CH band of PVA observed in all the solid samples and in the reference without Ag colloids indicates that, differently from the polyyne signal, PVA does not give substantial SERS enhancement.
SERS features in the sp-carbon region are less resolved in the solid sample with respect to the liquid case and the SERS enhancement trend is in agreement with what happens in solution, also showing that S-1%_Ag has the highest SERS enhancement as compared to the spectra of the other free-standing films. To investigate the differences between the liquid and solid SERS analyses, we now focus on liquid and solid samples with PVA1% (i.e. L-1%_Ag and S-1%_Ag). The region over 2000 cm−1 is characterized by four resolved narrow bands in L-1%_Ag and by a broader band in S-1%_Ag whereas the signals below 2000 cm−1 appear as broad bands in both cases. The relative intensity of the SERS signal of over and under 2000 cm−1 is opposite in L-1%_Ag and S_1%_Ag.
All the differences between the two samples can be caused by the different SERS effects in liquids and solids.23,37,57 In liquids, polyynes can diffuse and be adsorbed more easily on Ag nanoparticles leading to creation of hot spots which can be very sensitive to the specific polyyne lengths. Conversely, in a solid phase, polyynes and Ag colloids are immobilized in the PVA matrix, and the different chemical environments of polyynes in proximity to metal nanoparticles may lead to a broad band, as observed in a previous work.35 Moreover, the complexes formed by strong polyyne–Ag interactions, which contribute to the band at a lower wavenumber,50 are more preserved by the solid polymeric matrix, justifying the higher intensity of this band with respect to the one above 2000 cm−1 in the solid sample. Further studies are needed to analyse the complex physical and chemical phenomena involved in the SERS mechanism, which is still not completely understood. We underline that, differently from previous studies in which PVA was added to a polyyne–metal nanoparticle aggregate, Ag colloids are added to the polyyne–PVA liquid leading to a possibly different blending when the solid nanocomposite is formed, as discussed in the following.
To better understand how the structure of the polymer changes in the presence of silver nanoparticles and polyynes, we compare the CH stretching band in the case of a PVA pellet, a PVA film drop cast from water, aqueous solutions with Ag colloids and polyynes and the corresponding solid nanocomposites (see Fig. 2c). The PVA band is clearly visible and is of the same intensity in all the solid samples, including the reference, thus indicating that the polymer does not undergo relevant degradation by the laser in the ablation process. We observe no differences between the PVA band in the pellet and in the film obtained from the aqueous solution of PVA10%, both showing the main peak located at 2910 cm−1, a shoulder at 2934 cm−1 and a weak shoulder at 2851 cm−1. Looking at L-10%_Ag, we notice that the main peak shifts up to 2925 cm−1 but when the solution is dried, its position goes back to that of the pellet. Also in the case of the aqueous solution of L-1%_Ag, the PVA peak moves to 2933 cm−1, but in this case it shows a small shift to 2928 cm−1 when the composite is formed; moreover, the shoulder at lower wavenumber becomes more visible while the other one at higher wavenumber is barely evident. It is known in the literature that PVA band changes its position from a solid pellet or cast film (2910, 2911 cm−1), to an aqueous solution (2918 cm−1) due to structural disorder.58 The anomalously high peak wavenumber of the co-deposited L-1%_Ag film suggests that the presence of Ag colloid and polyynes could limit the capability of the polymer to form ordered crystalline phases during the solvent evaporation process. Interestingly, if we deposit from a solution where PVA concentration in water is low, e.g. PVA1%, we get a film (S-1%_Ag) showing a CH stretching band position very close to that of PVA in water, namely to the wavenumber typical of conformationally disordered polymer chains. This indicates that polyynes, Ag nanoparticles and the polymer form an intimate blend in the solid phase obtained. Instead, in the case of a higher concentration of PVA as in S-10%_Ag, the PVA band wavenumber matches that of the pellet, which suggests that segregation of PVA crystalline domains occurs during the solvent evaporation. This effect can reduce the blending between all the components and encapsulation of polyynes. The PVA concentration is thus fundamental to achieve full blending and to avoid possible segregation of polyynes and Ag colloids occurring when PVA forms crystalline domains.
The free-standing films of a few cm size obtained after solvent evaporation of polyynes in PVA1% (Fig. 3a) are characterized by a non-complete homogeneous distribution of silver nanoparticles which, during solvent evaporation, accumulate preferentially along the original drop edge due to the coffee ring effect. Such a phenomenon can become visible to the naked eye, because of the colour difference between the centre and the edge of the sample. SEM images confirmed a higher concentration of Ag colloids at the film edge with respect to the centre, characterized by fewer but locally well dispersed nanoparticles (see Fig. 3b and c). To investigate the effect of the inhomogeneity of the Ag nanoparticles in the sample, we mapped its SERS response along a line from the centre to the border, as drawn in Fig. 3a. All the spectra have been normalized with respect to the PVA band. The polyyne signal is always present and its intensity decreases on moving towards the edge of the film where Ag nanoparticles are more concentrated, as shown in Fig. 3d. To evaluate this effect, we plot the ratio of the areas of the whole polyyne signal between 1800 and 2200 cm−1 and the PVA peak as a function of the distance from the centre to the border of the sample (see the inset of Fig. 3d).
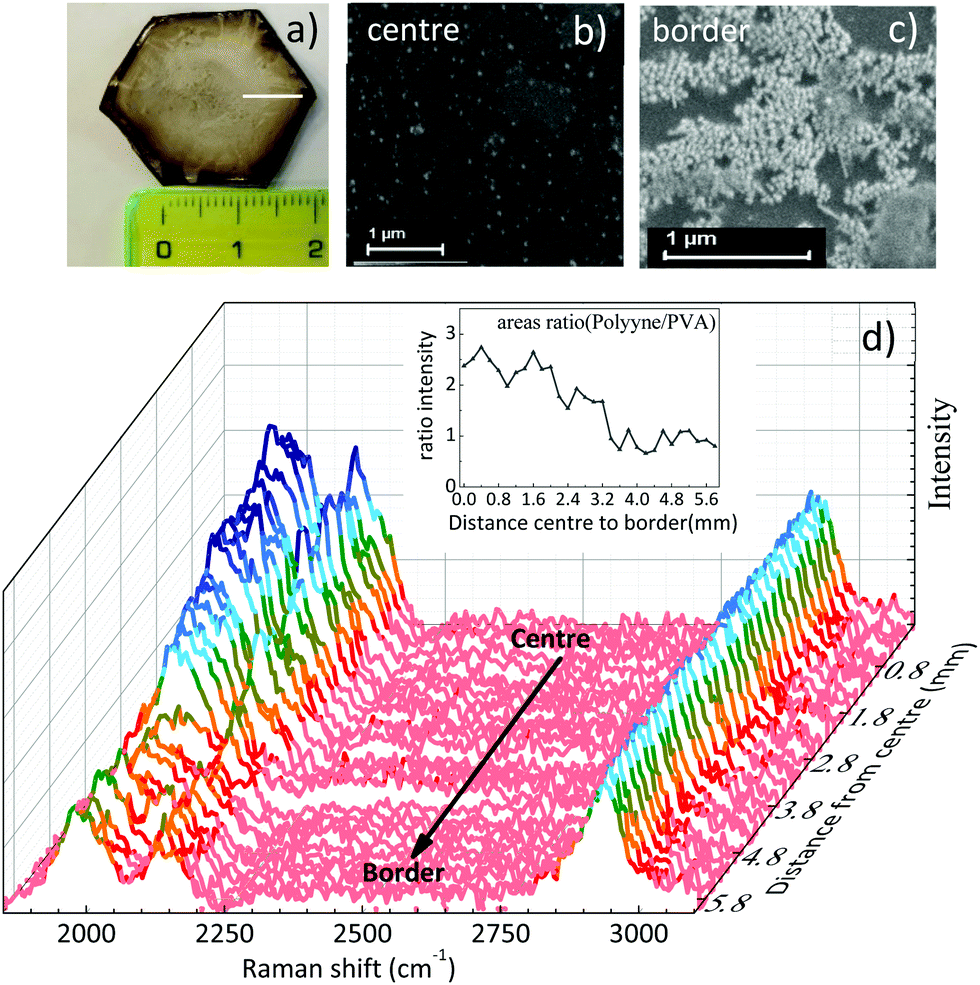 | ||
| Fig. 3 (a) Free-standing film of Ag/PVA1%/polyynes (S-1%_Ag), (b) SEM image of the centre of the sample, (c) SEM image of the border of the film, (d) normalized SERS spectra of the free-standing film from the centre to the border with a step size of 200 μm following the white line drawn in (a). Inset: ratio between the area of the polyynes bands to the area of the PVA peak. | ||
The SERS signal of polyynes (1800–2250 cm−1) lowers at the edge of the sample where the concentration of silver colloids is higher. If we assume a uniform distribution of polyynes, overabundant Ag nanoparticles may absorb the incident laser used for Raman reducing the sampling volume or may occupy the active sites for SERS effects in place of sp-carbon chains leading to an overall lower polyyne signal at the edge than in the centre.59 This result highlights how hard it is to obtain a good SERS enhancement because of its dependence on several factors including the concentration of Ag nanoparticles and the position of plasmonic peak, which in turn depends on the nanoparticle size and aggregation.35 Notwithstanding the slight non-uniformity of Ag colloids distribution over several mm scale attributed to the coffee-ring effect during solvent evaporation, we do not observe any effect related to the possible segregation of Ag colloids and polyynes with respect to PVA, in agreement with the behaviour of the Raman CH band of PVA discussed above.
Once the films were prepared and the morphologically homogeneous areas with the higher SERS enhancement were selected (i.e. the centre of the sample), we studied the stability of polyynes immobilized in the polymer matrix. We performed SERS analysis of the liquid and solid samples at different times starting from the as-prepared sample. Fig. 4 reports the comparison between the SERS time evolution of the liquid solution of the Ag colloid and PVA1% (L-1%_Ag) and the corresponding solid nanocomposite (S-1%_Ag). Spectra are normalized with respect to the peak of PVA at 2910 cm−1. In the case of the liquid, the well-defined polyyne features of the SERS spectrum are replaced by two broad bands after just 1 week and, after 2 months, no features are visible in the sp-carbon spectral region. After 3 weeks, the decomposition of polyynes in Ag/PVA solution, evaluated considering the integrated area of the SERS signal in sp-carbon range, is about 60%, which is anyway strongly reduced with respect to the case of polyynes in the aqueous Ag colloid, whose area decrease after 3 weeks is 95% (as reported in ref. 19). In the spectra of solid nanocomposites, we observe after 1 week a decrease of the intensity of the polyynic SERS bands of the order of 50% but then, the spectrum does not substantially change up to 11 months. Our results agree with the few studies addressing the stability of polyynes in PVA. Okada et al. showed that Ag–polyynes aggregates in PVA are stable for a month.38 An and co-workers observed the SERS signal from Au/polyynes/PVA films for up to 6 months.37 In our work, we indicate that stability of polyynes in liquid media is improved in the presence of PVA, even if only slightly, because the reactions between sp-carbon chains are partially inhibited by the presence of the polymer. However, sp-carbon chains are not completely immobilized in PVA solution and the mechanism of crosslinking does not stop completely. In the case of solid nanocomposites, the stabilizing effect is instead quite clear and effective. In fact, once polyynes are encapsulated in the PVA matrix, they are somehow protected, and they are stable for a prolonged time.
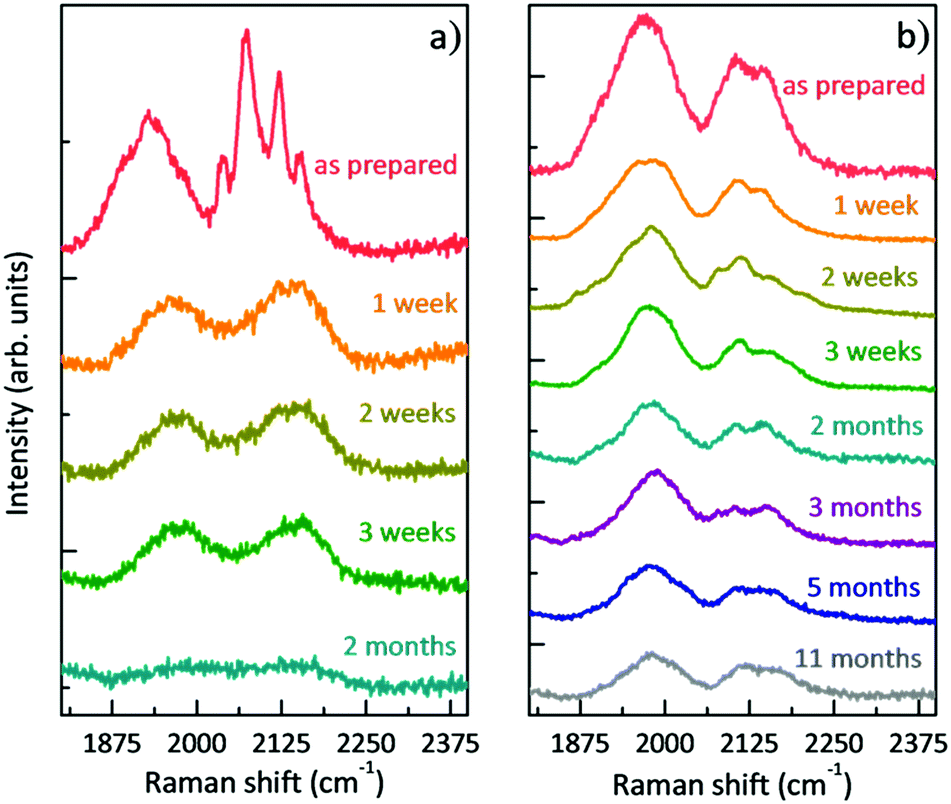 | ||
| Fig. 4 Evolution in time of the SERS spectra: (a) polyynes solution of PVA1% with Ag colloid (L-1%_Ag) and (b) free-standing nanocomposite of Ag/PVA1%/polyynes (S-1%_Ag). | ||
Conclusions
In this work, we presented the ablation of graphite directly in a pure aqueous solution of PVA as an in situ method to directly form polyynes in a polymer matrix. Our method does not require the use of organic solvents and does not need the dilution of polyynes by the subsequent addition of the polymer in the pristine solutions or their exposure to the heating stage necessary when PVA granules are directly dissolved in the solution containing sp-carbon chains. The preparation of different PVA/polyynes solutions varying the PVA concentration in water allowed us to elucidate the non-trivial behaviour of the polymer in the composite formation. After the addition of the Ag colloid to aqueous solution of PVA/polyynes and its drying, we obtained a polyyne-based free-standing film. The nanocomposite formed in this way is characterized by a non-complete crystallization of the PVA domains revealing an intimate blending of the components.The presence of PVA in the aqueous solution is shown to favour polyynes formation with respect to ablation in pure water. A combined SEM and SERS map analysis on free-standing films allowed us to individuate the area of the nanocomposite with a homogeneous morphology and the highest SERS intensity, where it was possible to perform a stability study. Polyynes embedded in Ag nanoparticles and PVA are still stable after 11 months. In this way, our simple method of synthesis of hydrogen-capped polyynes in PVA and the enhanced stability in the polymer matrix is promising for future optical and mechanical characterization of polyyne-based films.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program ERC-Consolidator Grant (ERC CoG 2016 EspLORE grant agreement no. 724610, website: http://www.esplore.polimi.it).References
- C. S. Casari, M. Tommasini, R. R. Tykwinski and A. Milani, Nanoscale, 2016, 8, 4414–4435 RSC.
- C. S. Casari and A. Milani, MRS Commun., 2018, 8, 207–219 CrossRef CAS.
- P. B. Sorokin, H. Lee, L. Y. Antipina, A. K. Singh and B. I. Yakobson, Nano Lett., 2011, 2660–2665, DOI:10.1021/nl200721v.
- Z. Zanolli, G. Onida and J. C. Charlier, ACS Nano, 2010, 4, 5174–5180 CrossRef CAS.
- V. I. Artyukhov, M. Liu and B. I. Yakobson, Nano Lett., 2014, 14, 4224–4229 CrossRef CAS.
- F. Hu, C. Zeng, R. Long, Y. Miao, L. Wei and Q. Xu, et al. , Nat. Mater., 2018, 15, 194–200 CAS.
- A. D. Scaccabarozzi, A. Milani, S. Peggiani, S. Pecorario, B. Sun and R. R. Tykwinski, et al. , J. Phys. Chem. Lett., 2020, 11, 1970–1974 CrossRef CAS.
- A. Milani, M. Tommasini, V. Barbieri, A. Lucotti, V. Russo and F. Cataldo, et al. , J. Phys. Chem., 2017, 121, 10562–10570 CAS.
- M. Tsuji, T. Tsuji, S. Kuboyama, S. Yoon, Y. Korai and T. Tsujimoto, et al. , Chem. Phys. Lett., 2002, 355, 101–108 CrossRef CAS.
- H. Tabata, M. Fujii, S. Hayashi, T. Doi and T. Wakabayashi, Carbon, 2006, 44, 3168–3176 CrossRef CAS.
- K. Inoue, R. Matsutani, T. Sanada and K. Kojima, Carbon, 2010, 48, 4197–4214 CrossRef.
- R. Matsutani, K. Inoue, T. Sanada, N. Wada and K. Kojima, J. Photochem. Photobiol., A, 2012, 240, 1–4 CrossRef CAS.
- R. Matsutani, T. Kakimoto, H. Tanaka and K. Kojima, Carbon, 2011, 49, 77–81 CrossRef CAS.
- R. Matsutani, T. Kakimoto, K. Wada, T. Sanada, H. Tanaka and K. Kojima, et al. , Carbon, 2008, 46, 1091–1109 CrossRef.
- K. S. Khashan and M. H. Mohsin, Iraqi J. Phys., 2013, 11, 37–47 Search PubMed.
- F. Cataldo, Carbon, 2004, 42, 129–142 CrossRef CAS.
- G. Compagnini, V. Mita, L. D’Urso, R. S. Cataliotti and O. Puglisi, J. Raman Spectrosc., 2008, 39, 177–181 CrossRef CAS.
- S. K. Shin, J. K. Song and S. M. Park, Appl. Surf. Sci., 2011, 257, 5156–5158 CrossRef CAS.
- S. Peggiani, A. Senis, A. Facibeni, A. Milani, P. Serafini and G. Cerrato, et al. , Chem. Phys. Lett., 2020, 740, 137054–137061 CrossRef CAS.
- H. Li, Y. Wu, Y. Zhang, T. Zhu, T. Maruyama and Y. Liu, et al. , Chem. Phys., 2020, 535 DOI:10.1016/j.chemphys.2020.110804.
- C. S. Casari, A. Li Bassi, L. Ravagnan, F. Siviero, C. Lenardi and P. Piseri, et al. , Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 075422–075427 CrossRef.
- F. Cataldo, Fullerenes, Nanotubes, Carbon Nanostruct., 2007, 15, 155–166 CrossRef CAS.
- C. S. Casari, V. Russo, A. Li Bassi, C. E. Bottani, F. Cataldo and A. Lucotti, et al. , Appl. Phys. Lett., 2007, 90, 1–3 CrossRef.
- G. Compagnini and S. Scalese, in Laser Ablation in Liquids: Principles and Applications in the Preparation of Nanomaterials, ed. G. Yang, Pan Stanford Publishing Pte. Ltd, 2012 Search PubMed.
- L. Shi, P. Rohringer, K. Suenaga, Y. Niimi, J. Kotakoski and J. C. Meyer, et al. , Nat. Mater., 2016, 15, 634–640 CrossRef CAS.
- Y. Zhang, J. Zhao, Y. Fang, Y. Liu and X. Zhao, Nanoscale, 2018, 10, 17824–17833 RSC.
- D. Nishide, H. Dohi, T. Wakabayashi, E. Nishibori, S. Aoyagi and M. Ishida, et al. , Chem. Phys. Lett., 2006, 428, 356–360 CrossRef CAS.
- L. D. Movsisyan, M. Franz, F. Hampel, A. L. Thompson, R. R. Tykwinski and H. L. Anderson, J. Am. Chem. Soc., 2016, 138, 1366–1376 CrossRef CAS.
- L. G. Bettini, F. Della Foglia, P. Piseri and P. Milani, Nanotechnology, 2016, 27, 1–6 CrossRef.
- S. Szafert and J. A. Gladysz, Chem. Rev., 2003, 103(11), 4175–4206 CrossRef CAS.
- L. Ravagnan, F. Siviero, C. Lenardi, P. Piseri, E. Barborini and P. Milani, Phys. Rev. Lett., 2002, 89, 28–31 CrossRef.
- R. Rivelino, R. B. dos Santos and F. de Brito Mota, J. Phys. Chem. C, 2010, 114, 16367–16372 CrossRef CAS.
- R. R. Tykwinski, W. Chalifoux, S. Eisler, A. Lucotti, M. Tommasini, D. Fazzi, M. Del Zoppo and G. Zerbi, Pure Appl. Chem., 2010, 82, 891–904 CAS.
- F. Cataldo, O. Ursini, A. Milani and C. S. Casari, Carbon, 2017, 126, 232–240 CrossRef.
- A. Milani, A. Lucotti, V. Russo, M. Tommasini, F. Cataldo and A. Li Bassi, et al. , J. Phys. Chem. C, 2011, 115, 12836–12843 CrossRef CAS.
- R. Matsutani, F. Ozaki, R. Yamamoto, T. Sanada, Y. Okada and K. Kojima, Carbon, 2009, 47, 1659–1663 CrossRef CAS.
- K. An, G. Wei, G. Qi, L. Sheng, L. Yu and W. Ren, et al. , Chem. Phys. Lett., 2015, 637, 71–76 CrossRef CAS.
- S. Okada, M. Fujii and S. Hayashi, Carbon, 2011, 49, 4704–4709 CrossRef CAS.
- R. Sata, H. Suzuki, N. Ueno, Y. Morisawa, M. Hatanaka and T. Wakabayashi, Chin. J. Chem. Phys., 2019, 32(2), 175–181 CrossRef CAS.
- S. Mallakpoura and A. N. Ezhieh, Carbohydr. Polym., 2017, 166, 377–386 CrossRef.
- S. Pandey, S. K. Pandey, V. Parashar, G. K. Mehrotra and A. C. Pandey, J. Mater. Chem., 2011, 21, 17154–17159 RSC.
- S. K. Shin and S. M. Park, Bull. Korean Chem. Soc., 2012, 33, 597–601 CrossRef CAS.
- P. C. Lee and D. J. Meisel, J. Phys. Chem., 1982, 86, 3391–3395 CrossRef CAS.
- W. H. Eisa and A. A. Shabaka, React. Funct. Polym., 2013, 73, 1510–1516 CrossRef CAS.
- F. Cataldo, Polyynes: synthesis, properties and applications, CRS Press, 2005 Search PubMed.
- M. Tsuji, S. Kuboyama, T. Matsuzaki and T. Tsuji, Carbon, 2003, 41, 2141–2148 CrossRef CAS.
- A. Milani, M. Tommasini, V. Russo, A. Li Bassi, A. Lucotti and F. Cataldo, et al. , Beilstein J. Nanotechnol., 2015, 6, 480–491 CrossRef.
- T. F. Cooney, L. Wang, S. K. Sharma, R. W. Gauldie and A. J. Montana, J. Polym. Sci., Part B: Polym. Phys., 1994, 32, 1163–1174 CrossRef CAS.
- M. Tommasini, D. Fazzi, A. Milani, M. Del Zoppo, C. Castiglioni and G. Zerbi, J. Phys. Chem. A, 2007, 111, 11645–11651 CrossRef CAS.
- A. Lucotti, M. Tommasini, M. Del Zoppo, C. Castiglioni, G. Zerbi and F. Cataldo, et al. , Chem. Phys. Lett., 2006, 78–82, DOI:10.1016/j.cplett.2005.10.016.
- C. Castiglioni, M. Tommasini and G. Zerbi, Philos. Trans. R. Soc., A, 2004, 362, 2425–2459 CrossRef CAS.
- M. Mohsen-Nia and H. Modarress, J. Adhes. Sci. Technol., 2006, 20, 1273–1280 CrossRef CAS.
- M.-R. Kalus, N. Bärsch, R. Streubel, E. Gökce, S. Barcikowski and B. Gökcea, Phys. Chem. Chem. Phys., 2017, 19, 7112–7123 RSC.
- T. Hupfeld, G. Laurens, S. Merabia, S. Barcikowski, B. Gökce and D. Amans, J. Appl. Phys., 2020, 127(4), 044306 CrossRef CAS.
- Y. Wang and Y. L. Hsieh, J. Membr. Sci., 2008, 309, 73–81 CrossRef CAS.
- M. Perfetti, N. Gallucci, I. Russo Krauss, A. Radulescu, S. Pasini and O. Holderer, et al. , Macromolecules, 2020, 53(3), 852–861 CrossRef CAS.
- A. Milani, V. Barbieri, A. Facibeni, V. Russo, A. Li Bassi and A. Lucotti, et al. , Sci. Rep., 2019, 9, 1648 CrossRef CAS.
- Y. A. Badr, M. Abd El-Kader and R. M. Khafagy, J. Appl. Polym. Sci., 2004, 92, 1984–1992 CrossRef CAS.
- Q. He, A. Zhao, L. Li, H. Sun, D. Wang and H. Guo, New J. Chem., 2017, 41, 1582–1590 RSC.
| This journal is © The Royal Society of Chemistry 2020 |