
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Donor and acceptor engineering for BINOL based AIEgens with enhanced fluorescence performance†
Pei-Long
Lu
,
Kun
Li
 *,
Lei
Shi
,
Xin
Liu
,
Mei-Lin
Feng
,
Hui-Zi
He
,
Hui
Yang
and
Xiao-Qi
Yu
*
*,
Lei
Shi
,
Xin
Liu
,
Mei-Lin
Feng
,
Hui-Zi
He
,
Hui
Yang
and
Xiao-Qi
Yu
*
Key Laboratory of Green Chemistry and Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu, 610064, China. E-mail: kli@scu.edu.cn; xqyu@scu.edu.cn
First published on 21st March 2020
Abstract
Organic AIEgens have been widely applied in multiple areas. However, most of them were designed based on AIE core skeletons. It's still a formidable challenge to design efficient AIEgens from simple ACQ fluorophores. Herein, we provided and perfected a strategy for developing new AIEgens by donor and acceptor engineering. By this strategy, we have synthesized two series of organic AIEgens based on a core skeleton of BINOL. The effect of steric hindrance and the electronic effect of the substituent on the optical properties have been discussed. Notably, the introduction of a donor and acceptor with large steric hindrance achieved fluorophores 2b and 2c with efficient solid emission, a high AIE effect factor and a large Stokes shift; typically, 2b exhibited a good performance in bioimaging. Meanwhile, the effect of the balance between the electron donor and acceptor should be paid attention. This AIEgen design strategy serves as new inspiration for turning ACQ skeletons to AIEgens.
Introduction
Organic fluorophores have great prospects in materials, biological and environmental sciences due to the multiple advantages of easy processing, a wide range of sources, structural diversity, tunable optical properties, etc.1 But traditional fluorophores have been problematic in practice because of aggregation-caused quenching (ACQ), which is caused by inherent strong intermolecular π–π stacking interactions consuming the excitation energy.2 Since the emergence of the concept of aggregation-induced emission (AIE), fluorescence materials based on AIE characteristics have attracted much attention due to the straightforward solution to the problem of ACQ, because avoiding the rebarbative ACQ phenomenon could obtain organic fluorophores with superior optical properties. So AIEgens have been widely used in optoelectronic and biological areas.3 Not only that, some of them have been recognized as advanced functional materials in high performance OLEDs,4 sensing and imaging,5 super resolution imaging,6 specific cancer cell imaging,7 theranostics, etc.8 Therefore, it's of great significance to develop more AIE molecules and continue this great work.As we know, there are two common ways to achieve AIE fluorophores. One is to modify the existing AIE structures, donor (D) and acceptor (A) as well as other functional groups, which could improve the fluorescence performance9 or realize multiple applications.10 Another one is to conjugate an AIE molecule with ACQ luminophores, which is an effective way to transform ACQ luminophores to AIE luminogens.11 The Tang group have obtained a few FR/NIR AIE fluorophores through this way.12 Admittedly, many excellent AIE fluorophores have been designed by these two methods, but the development of AIE fluorophores also was limited by expanding on a known AIE structure. Therefore, it is urgent and meaningful to find a new and common way to design AIE fluorophores. In order to break the limitation of inherent thinking, we decided to abandon those AIE structures such as TPE but focus on ACQ structures. We want to convert large numbers of ACQ luminophores into AIE luminescence. It's of great significance and challenging work.
Decoding the working mechanism of AIE has important fundamental significance to provide design guidance for the development of new AIE systems for new practical applications.13 Despite some controversy, restriction of intramolecular motions RIM has been generally recognized as central to the AIE working mechanism, not only in conventional AIEgens but also in some nonconventional systems, such as clusteration-triggered emission (CTE)14 and suppression of Kasha's rule.15 Most AIEgens show highly twisted propeller-like structures compared to conventional ACQ dyes, which suggests that the twisted structure plays a critical role in the AIE phenomenon.13 In solution, a highly twisted structure exhibits weak emission or is completely non-luminescent due to the free intramolecular motions; conversely intense emission will be observed in the aggregation state due to the RIM. These phenomena and mechanisms guide people on how to construct AIE fluorophores.
In the last few years, various AIE fluorophores have been reported which were based on various ACQ luminophores. People modified large steric groups on ACQ structures to get twisted structures, so as to convert their fluorescence behavior to AIE. The earliest case was reported by Diau, Hsu and co-workers. When the α-olefinic hydrogen atoms of DSB were replaced by methyl groups to generate methylated derivative PPB, the solid fluorescence behavior changed from ACQ to AIE.16 The crystal structure revealed that the strong π–π stacking interactions of DSB consumed the excitation energy and weakened the light emission, and the strong π–π stacking interactions of PPB were avoided due to the steric hindrance of the two methyl groups,17 and this made PPB highly emissive in the solid state by the deactivated RIR process. Someone used pyrene to modify fluorophores to convert fluorescence behavior due to its large steric hindrance and high luminous efficiency.18 Recently, P. K. Iyer's group modified a long alkyl chain to develop a smart AIEgenic nanostructure from a conventional ACQ-prone moiety.19 Tang's group used bulky aromatic groups successfully resulting in a twisted molecular conformation, which converted the fluorescence behavior from ACQ to AIE, and the increasing of the steric hindrance greatly improved the fluorescence quantum yield.20 These results tell us we can get an AIE molecule based on an ACQ group, and the mechanism was to get a twisted structure just the same as before.
These phenomena and conclusions inspired us very well. In 2018, our group provided a strategy for developing new AIE fluorophores based on BINOL derivative (S)-[10,100-bibenzo[g]chromene]-2,20-dione by decorating with diverse electronic and steric effects.21 Unfortunately, only one of the four compounds (BIN-COP) has the AIE effect. The quantum yield (QY) of BIN-COP in the solid state was only 0.97%, and the AIE effect numerical parameter (αAIE) of BIN-COP was just 1.31%. In terms of fluorescence properties, it was far from what we expected. The αAIE factor is another important parameter in addition to the QY, which can be quantified by the extent of emission enhancement from the solution state to the aggregation state;11 it's defined as below: αAIE = ϕF,solid/ϕF,solution. This definition provides us with an important design idea to improve the AIE effect: reducing the quantum yield in the solution state while increasing the quantum yield of the solid state.
Tang's group has effectively raised the AIE effect by reducing the quantum yield in solution.22 When enhancing the electron donor effect, molecular rotation was sped up to serve as nonradiative pathways to deactivate the excited states in the solution, and such rotational motions could be restricted in the aggregation state. In addition, color tunable AIEgens were obtained. The result also can be induced by enhancing the electron acceptor.23 There was also comprehensive regulation of the two, and the emission in solution was completely quenched.24 And there was another phenomenon – when the electronic effect was enhanced to a certain extent, the fluorescence will be quenched.25 From these research achievements, we have summarized and provided a systematic AIE fluorescence design strategy based on modifiable ACQ fluorescence: modifying several structures with large steric hindrance and a suitable electronic effect. It is conceivable that you can get some good results through our strategy: an ACQ to AIE transformation and enhanced fluorescence performance, which could be concretely described as: a better AIE effect, a larger solid quantum yield, larger Stokes shifts and tunable emission.
In this study, we have developed eight fluorophores with different AIE properties by our strategy (Scheme 1). As expected, with the change of electronic properties and steric hindrance, we have successfully obtained several fluorophores with much higher efficiency emission and a much better AIE effect than BIN-COP. Meanwhile, color tunable fluorophores with a large Stokes shift of BINOL derivatives were obtained, which initiated and developed color tunable solid luminescent materials of BINOL derivatives. In particular, the probe was applied in cell imaging.
 | ||
| Scheme 1 Design of molecular fluorophores. | ||
Results and discussion
Donor and acceptor engineering on the molecular fluorophores
Our design strategy mainly includes two aspects, steric hindrance and electronic effects of substituent groups. As shown in Scheme 1, we built a D–π–A structure to investigate our strategy, which could create an organic fluorophore with tunable optical properties.26 We chose methoxyphenyl as an electron donor which has a steric hindrance effect. We chose acetyl, benzoyl, methyl allyl dinitrile and aryl allyl dinitrile as electron acceptors which have different steric hindrance and electronic effects. The nitrile group has been frequently utilized in the design of AIE fluorophores due to its steric hindrance and electronic effects,27 which can be readily appreciated from the data shown by the Ma group.28 Obviously, for the electron donating effect: methoxyphenyl > H, for the electron accepting effect: methyl allyl dinitrile > aryl allyl dinitrile > acetyl- > benzoyl-, and for steric hindrance: aryl allyl dinitrile > methyl allyl dinitrile > benzoyl- > acetyl-, methoxyphenyl > H. To investigate donor and acceptor engineering, we made a subtle modification in the electron acceptor at position 2 (R2/R3); acetyl- and methyl allyl dinitrile were used as the acceptor to afford the fluorophores 1a and 1b, and methyl allyl dinitrile was used as the donor at position 6 (R1) conjugated with 1a and 1b to afford the fluorophores 1c and 1d, respectively. To further investigate the steric hindrance of the substituent group, aryl substituents were used on the basis of 1a–1d to afford the fluorophores 2a, 2b, 2c and 2d, respectively (Scheme 1).We could draw conclusions as defined below: the electron push–pull effect of these molecular fluorophores: BIN-COM < 1a < 1b < 1c < 1d (or BIN-COP < 2a < 2b < 2c < 2d); BIN-COP ∼ 2d < BIN-COM ∼ 1d, and the steric hindrance of these substituents: BIN-COM < 1a < 1b < 2a < 2b; BIN-COP < 2a < 2b; 1c < 2c < 1d < 2d. Preparation details and physical characterization of the BINOL(S) dialdehyde and BIN-COM and BIN-COP were given in the reported literature.21 Methoxyphenyl was connected with BINOL(S) dialdehyde through a Suzuki coupling reaction. Compounds BIN-COM, BIN-COP, 1c, and 2c were readily achieved by using a cyclization reaction between BINOL(S) dialdehyde derivatives and appropriate esters, and the dinitrile substituent compounds 1a, 1b, 1d, 2a, 2b and 2d were readily achieved by a one-step condensation reaction between ketones and malononitrile. All the compounds were easily purified by column chromatography or recrystallization with a reasonable yield. All of the target products were characterized by 1H NMR, 13C NMR, and high resolution mass spectrometry. The synthesis details are presented in the ESI.†
Frontier molecular orbitals and crystal structures
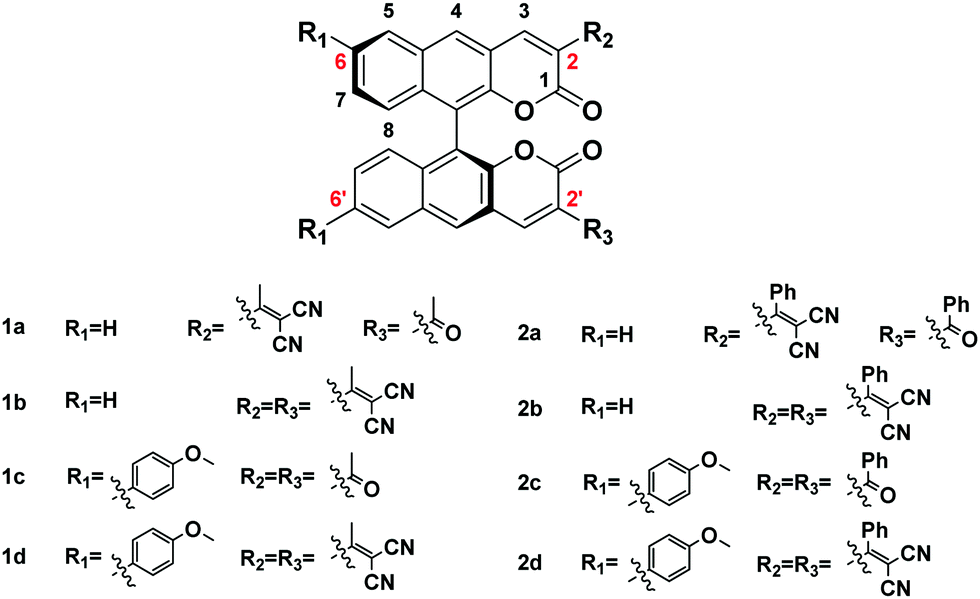
To better understand the electronic transitions of these fluorophores, density functional theory calculations were performed. The ground state geometries of all compounds were optimized using DFT with the B3LYP hybrid functional at the basis-set level of 6-31G*. As shown in Fig. 1, the calculation results showed that the majority of the electron cloud distribution of the highest occupied molecular orbital (HOMO) was very similar for each compound, which was located on the BINOL moiety and the electron-donating moieties. While the electron cloud of the lowest unoccupied molecular orbital (LUMO) was spread primarily over the coumarin and –C(X)R framework and the electron-accepting group, the LUMO was more delocalized over the electron acceptor group with increasing accepting effect. It demonstrated the existence of intramolecular charge transfer (ICT) character for these compounds and was consistent with the experimental results of solvent effects.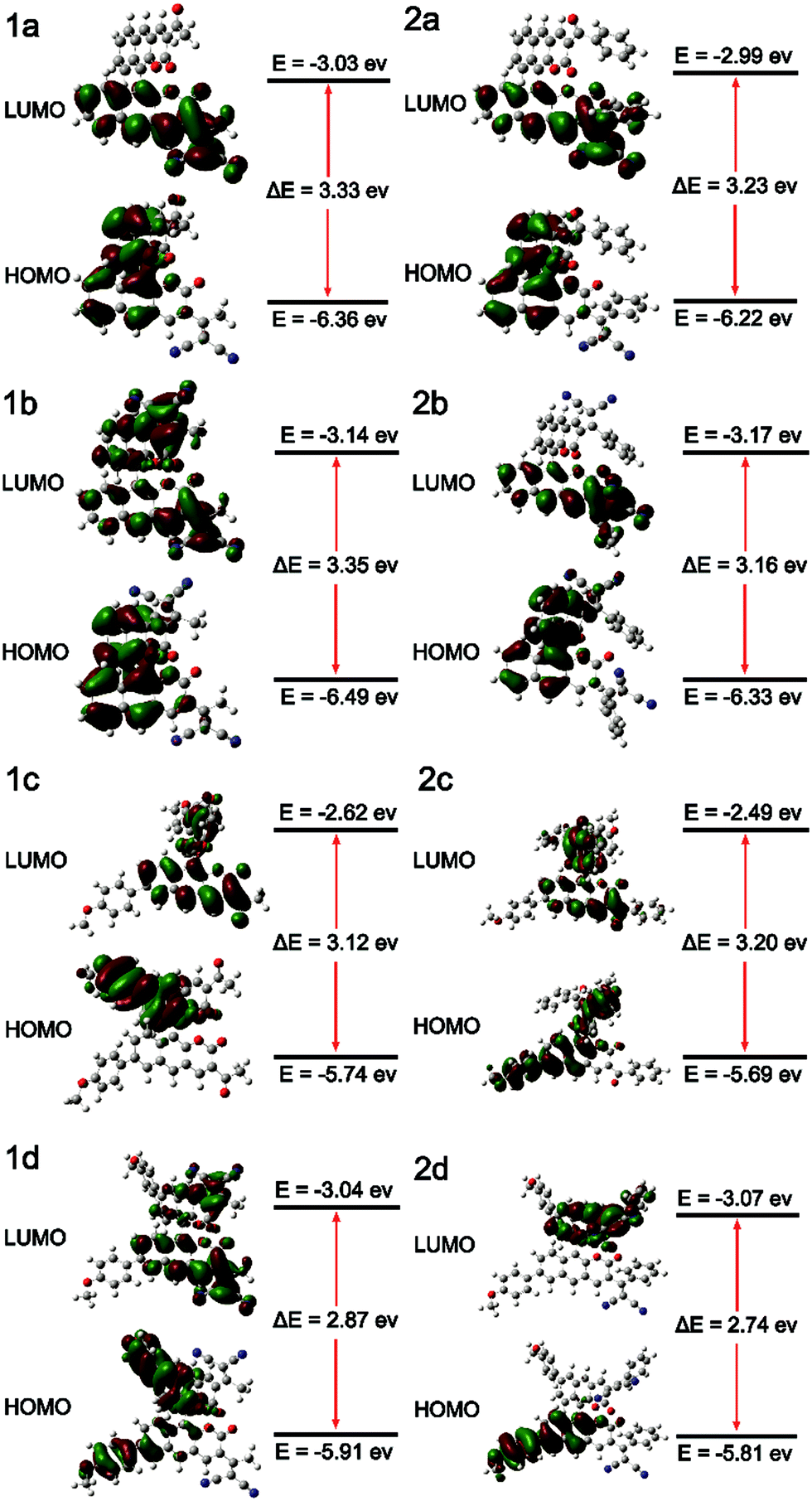 | ||
| Fig. 1 Molecular orbital amplitude plots of the HOMO and LUMO energy levels of fluorophores 1a–2d calculated by using the B3LYP/6-31G(d,p) basis set. | ||
The energy levels of the HOMO rise obviously with increasing electron donating effect, and the energy levels of the LUMO decrease with increasing electron accepting effect. It showed that 1d and 2d have a lower energy gap (ΔE) from the HOMO to the LUMO than other molecules due to the D–π–A structure; the ΔE values of 1d and 2d were 2.87 eV and 2.74 eV, respectively. With the decrease of the energy gap, the larger the red shift of the emission. It suggests that 1d and 2d have larger Stokes shifts, which is consistent with the experimental results. The substantial shifting of electron clouds indicates the occurrence of twisted intramolecular charge transfer (TICT) in these AIEgens upon excitation, which is responsible for their large Stokes shifts and redshift in DMSO.22 Through comparing the ΔE values of series 1 and 2, it's discovered that: 1a < 2a, 1b < 2b, 1c > 2c, and 1d < 2d. The results of 2a, 2b, and 2d were different from 2c due to the conjugation length of aryl allyl dinitrile. We can see the reason from the electron clouds; as shown in Fig. 1. 2c, the weak electron accepting effect of acyl- could only involve a small part of the benzene ring in ICT, and the weaker electron accepting effect of the substituent group led to this special result: ΔE1c > ΔE2c. Comparing the ΔE values with BIN-COP, all of the eight molecular fluorophores decrease significantly, which suggests a red emission peak.
To obtain the underlying reasons for these significant substituent groups with different steric hindrance and electronic effect dependent emission behavior, inspection of single crystals was performed. The single crystal structures of 2b, 1c, and 2c are shown in Fig. 2 and Fig. S1 (ESI†), and the distances between the adjacent molecules are shown in Table S1 (ESI†). According to our previous reports,21BIN-COM has an ACQ effect; such a phenomenon results from the rare steric hindrance, resulting in a short distance between the two adjacent planes, and leading to encountering strong and consecutive intermolecular π–π stacking interactions, and resulting in weakening of the fluorescence in the solid state. In contrast, BIN-COP shows a twisted molecular structure. The torsion angle between the central benzocoumarin planes is 62.9°, and the torsion angle between the central benzocoumarin plane and the R group is 47.5°, and the distance between the central benzocoumarin planes of two adjacent molecules is 4.828 Å, which can avoid fluorescence quenching in either the solid state or aggregation state by avoiding intermolecular π–π interactions. Compared with BIN-COP, the substituent groups of 2b are aryl allyl dinitrile, which has a larger steric hindrance. As shown in Fig. 2A, the torsion angle between the central benzocoumarin planes is 102.0°, and the torsion angles of the dinitrile group with the central benzocoumarin plane and phenyl are 67.3° and 39.8°, respectively. As a result, 2b is a more distorted structure than BIN-COP, which apparently resulted from steric congestion between aryl allyl dinitrile and neighboring phenyl rings. As shown in Fig. 2B, 2b doesn’t have two parallel planes like BIN-COP, and the distance of two adjacent molecules is 4.207 Å (>4.076 Å). Tang's group reported that there was no typical π–π stacking interactions in the 4.076 Å distance,29 and Xu's group observed that the ACQ phenomenon existed in bis(4-carbazol phenyl)sulfone, whose distance between two adjacent phenyls was just 3.70 Å, and a strong AIE phenomenon was noted in bis(4-phenothiazine phenyl)sulfone when the steric hindrance was enhanced.30 The above data fully illustrate that aryl allyl dinitrile can distort the molecular structure even more, and change the way that the molecules pack, which results in eliminating intermolecular π–π interactions completely, and then enhancing their emission efficiency in the solid state and aggregation state as we expected.
 | ||
| Fig. 2 Single-crystal structures of (A) 2b, (C) 1c and (E) 2c and the molecular packing of (B) 2b, (D) 1c and (F) 2c. Carbon, hydrogen, nitrogen and oxygen atoms are shown in grey, green, blue, and red, respectively. The single-crystal data can be found in the CCDC, the deposition numbers of 2b, 1c and 2c are 1907970, 1907963 and 1907968, respectively. | ||
As shown in Fig. 2C, crystal 1c adopted a slightly twisted conformation; the dihedral angle between the central benzocoumarin planes is 65.5°, and the torsion angles between the central benzocoumarin planes with the R1 and R2 groups are 34.6° and 17.6°, respectively. Careful evaluation of the details of the crystal packing indicates that the perpendicular distance and the center distance between two adjacent planes are 3.944 Å and 3.851–4.217 Å, respectively (Fig. 2D). It suggests that 1c reduces the intermolecular π–π stacking interactions and results in obvious enhancing of the fluorescence in the solid state. It is a pity that we did not get the single crystal structure of BIN-COM, but we can speculate that 1c is more distorted than BIN-COM in the solid state because of the methoxyphenyl group. Combined with the previous theories about αAIE, we can see the underlying reason for the hugely different fluorescence behaviors between BIN-COM and 1c. As shown in Fig. 2E, the dihedral angle between the central benzocoumarin planes is 109.9°, much larger than 1c, and the torsion angles between the central benzocoumarin planes with the R1 and R2 groups are 78.8° and 33.1°, respectively, which shows a more contorted conformation than BIN-COP and 1c because of the steric hindrance between the central benzocoumarin planes and benzoyl and methoxyphenyl. The perpendicular distance and center distance between the two adjacent phenyl groups are 4.156 Å and 4.823 (4.013) Å, respectively (Fig. 2F). As a result, 2c has more efficient solid fluorescence by avoiding the typical π–π stacking interactions. The difference in the fluorescence behavior between BIN-COM and BIN-COP could also be inferred by comparing the crystal structures of 1c and 2c. The data also prove that aromatic groups are helpful for the building of AIE molecules. Additionally, multiple intermolecular C–H⋯O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯H and CO⋯π interactions were found throughout the crystal structures of 2b, 1c and 2c (Fig. 3B, D and F), which could be helpful to rigidify the molecular conformation and restrain the nonradiative pathways, so as to hence enhance their emission efficiency in the solid state. Therefore, we can draw a conclusion that aromatic groups with nitrogen and oxygen can effectively enhance the efficiency of aggregation induced luminescence.
O⋯H and CO⋯π interactions were found throughout the crystal structures of 2b, 1c and 2c (Fig. 3B, D and F), which could be helpful to rigidify the molecular conformation and restrain the nonradiative pathways, so as to hence enhance their emission efficiency in the solid state. Therefore, we can draw a conclusion that aromatic groups with nitrogen and oxygen can effectively enhance the efficiency of aggregation induced luminescence.
 | ||
| Fig. 3 (A) Effect of subtle structure modification on solid state emission. (B) Absorption and emission spectra of 2b in different solutions. (C) Stokes shift (Δλ) of all compounds in the solid and aggregation state (90 vol% water fractions in DMSO/water). (D) Normalized fluorescence spectra of 1a–1d and 2a–2d in the solid state, λex = 370 nm, and photographs taken under irradiation with 365 nm UV light. (E) Solid fluorescence spectra of all compounds plotted on a CIE 1931 chromaticity diagram. | ||
Stokes shift study of the fluorophores
The absorption of all compounds in the solution and the aggregation state are shown in Fig. S2 and S3 and Table S2 (ESI†); each compound has a higher absorption peak appearing at around λ = 295 nm, which is attributed to a π–π* transition, and a weaker absorption ranging from λ = 340 nm to 380 nm. As shown in Fig. S4 and S5 and Table S3 (ESI†), all the fluorophores show remarkable solvent effects. Typically, as shown in Fig. 3B (or Fig. S4.7, ESI†), the emission peaks of 2b (or 2c) have a bathochromic shift from 525 to 582 nm (or 537 to 608 nm) with increasing solvent polarity from toluene to DMSO, and thus it can be seen that a significant ICT transition exists in these fluorophores. In addition, the discovery is consistent with the theoretical calculation results.All the fluorophores have large Stokes shifts ranging from 164 nm to 267 nm. The large Stokes shifts of all the fluorophores can be visualized in Fig. 3C. We investigate the effects of the electron donor and acceptor on the emission peak (λem). The emission peaks of all fluorophores in different states are shown in Table 1. When the substituent group of R2/R3 alters from acetyl- to methyl allyl dinitrile (or from benzoyl- to aryl allyl dinitrile), and the substituent group of R1 alters to methoxyphenyl, the corresponding compounds 1a–2d exhibit a long wavelength emission peak, which is assigned to the ICT transition from the different types of electron-donating group to the electron-accepting group. As shown in Fig. 3A, we further investigated the effect of substituents on solid state emission. With the increase of the electron-donor to electron-acceptor effect, λem of 1a–1d in the solid state presented a red-shift from 534 nm, 538 nm and 570 nm to 601 nm, respectively. By changing the aromatic group substituent, the corresponding compounds 2a (λem-solid = 538 nm), 2b (λem-solid = 541 nm), 2c (λem-solid = 560) and 2d (λem-solid = 609 nm) show similar red shifted emission. λem in the aggregation and solution state also presented a red-shift of 1a–1d (544 nm to 612 nm in the aggregation state) or 2a–2d (549 nm to 624 nm in the aggregation state), and it shows a red shift of the aromatic group substituent from series 1 to series 2 except the series c, which could be explained by the discussion result in the second section. All these results are consistent with the theoretical calculations of the frontier molecular orbitals. As a result, color tunable solid fluorophores with large Stokes shifts have been achieved by varying the type of donor and acceptor (Fig. 3D). The CIE chromaticity diagrams (Fig. 3E and Table S4, ESI†) demonstrate the same tunable emission from CIE yellow green (0.352, 0.5753) to CIE orange red (0.5743, 0.4236).
| Compound | λ abs [nm] | ε [M−1 cm−1] | λ em [nm] | Quantum yield (%) | ||||
|---|---|---|---|---|---|---|---|---|
| Solnb | Aggrec | Solidd | ϕ F | ϕ F | ϕ F | |||
| a Absorption maximum in DMSO. b Emission maximum of the solution state in DMSO. c Emission maximum of the aggregation state in H2O. d Emission maximum in the solid state. Fluorescence quantum yield determined by a calibrated integrating sphere. | ||||||||
| 1a | 340 | 5.24 × 104 | 562 | 544 | 534 | ∼0.1 | 0.58 | 3.1 |
| 1b | 349 | 4.21 × 104 | 574 | 550 | 538 | ∼0.1 | 3.23 | 2.55 |
| 1c | 346 | 3.29 × 104 | 605 | 588 | 570 | 0 | 0.3 | 2.93 |
| 1d | 379 | 1.35 × 104 | — | 612 | 601 | 0 | ∼0.1 | 0.16 |
| 2a | 354 | 6.20 × 104 | 579 | 551 | 538 | ∼0.1 | 3.58 | 4.43 |
| 2b | 354 | 6.22 × 104 | 582 | 553 | 541 | ∼0.1 | 5.41 | 6.3 |
| 2c | 340 | 4.80 × 104 | 608 | 580 | 560 | 0 | 3.46 | 6.3 |
| 2d | 353 | 5.14 × 104 | — | 624 | 609 | 0 | ∼0.1 | 0.15 |
It is noted that the emission peaks of 1a–2d were different in DMSO and the aggregation (in 90% water fraction) and the solid state, such as compound 2c (608 nm, 580 nm, and 560 nm, respectively). This phenomenon is often seen in AIE fluorophores with strong D–π–A structures.31 In the highly polar solvent DMSO, the emission peak of 2c (608 nm) is red-shifted compared to the solid state emission peak (560 nm) due to the TICT effect. While in 90 vol% water fraction DMSO–water mixtures, the formation of aggregates increases the hydrophobicity of the local environment and excludes the polar solvent molecules, so the emission was blue-shifted to 580 nm.
Optical property study of the fluorophores
The absorption and photoluminescence properties of all compounds in solution and the aggregation and the solid state are shown in Table 1, as well as Table S2 and S3 (ESI†). All fluorophores are almost non-emissive in DMSO; the QY of 1a, 1b, 2a and 2b is about 0.1%, and the QY of 1c, 1d, 2c and 2d is almost 0%, which should be explained by the twisted intramolecular charge transfer (TICT) effect in the solution state due to the D–π–A structures.32 All fluorophores show efficient emission in the aggregation and the solid state. Typically, the QYs of 2b and 2c are both 6.3% in the solid state, and are 5.41% and 3.46% in the aggregation state, respectively. We further investigate the effect of the steric hindrance of the substituents on the fluorescence performance. As we expected, when the substituent group alters from acetyl- (BIN-COM) to methyl allyl dinitrile (1b), the fluorescence behavior changes from the ACQ-effect to AIE-active. When we modified methoxyphenyl at the 6/6′ positions of BIN-COM to get 1c, the fluorescence behavior changes from the ACQ-effect to AIE-active, too. Not only that, the fluorescence performance is enhanced in the solid state (QYBIN-COM = 0.98%, QY1b = 2.55%, QY1c = 2.93%). When the substituent group alters from methyl allyl dinitrile (1b) and acetyl- (1c) to aryl allyl dinitrile and benzoyl-, the corresponding compounds 2b and 2c show higher emission; the quantum yields of them in the solid state (QYs) are both 6.3%. The fluorescence performance was further improved. Obviously, 2b is much better than BIN-COP (QY = 0.97%) due to the substituent group change from benzoyl- to aryl allyl dinitrile. These results are attributed to the increase of the steric hindrance and conjugation length; the more distorted structure enhances the solid emission efficiency. But when we further increase the steric hindrance to 1d and 2d, the emission becomes weak rapidly (QY1d = 0.16%, QY2d = 0.15%). In the solid state, the quenched emission of 1d and 2d should be ascribed to the exciton coupling between each molecule due to the large electron density overlap by the enhanced electron donating–accepting effect.33To better understand the steric hindrance of the substituents, we have studied two monosubstituted products 1a and 2a. The QY of 1a and 2a in the solid state is 3.1% and 4.43%, respectively. Both of them have good improvements. Compared to 1a, the substituent of 1b has larger steric hindrance, but the stronger electron accepting effect could lead to open radiative pathways to deactivate the excited states, and cause a quantum yield reduction. It's clear that aryl substituents have a more positive effect on the fluorescence emission efficiency; therefore 2b has a much better QY. These phenomena of 1d, 2d, and 1b show that while strengthening the steric hindrance attention should also be paid to the balance of push–pull electronic strength to avoid strong excition coupling in the solid state. We also calculated αAIE to verify the influence of the substituent groups on the emission behavior of the compounds (Fig. 4E). It's clear that the ratio of the fluorophores was greater than BIN-COP and BIN-COM; extraordinarily, the αAIE of 2c is more than 63. Therefore, the introduction of an electron donating group and enhancement of the electron accepting effect successfully reverse the trend of the quantum yields between the solution and solid phases. The results show that our strategy provides an extremely powerful method to create two fluorophores (2b and 2c) with efficient solid emission, a remarkable AIE effect and a large Stokes shift.
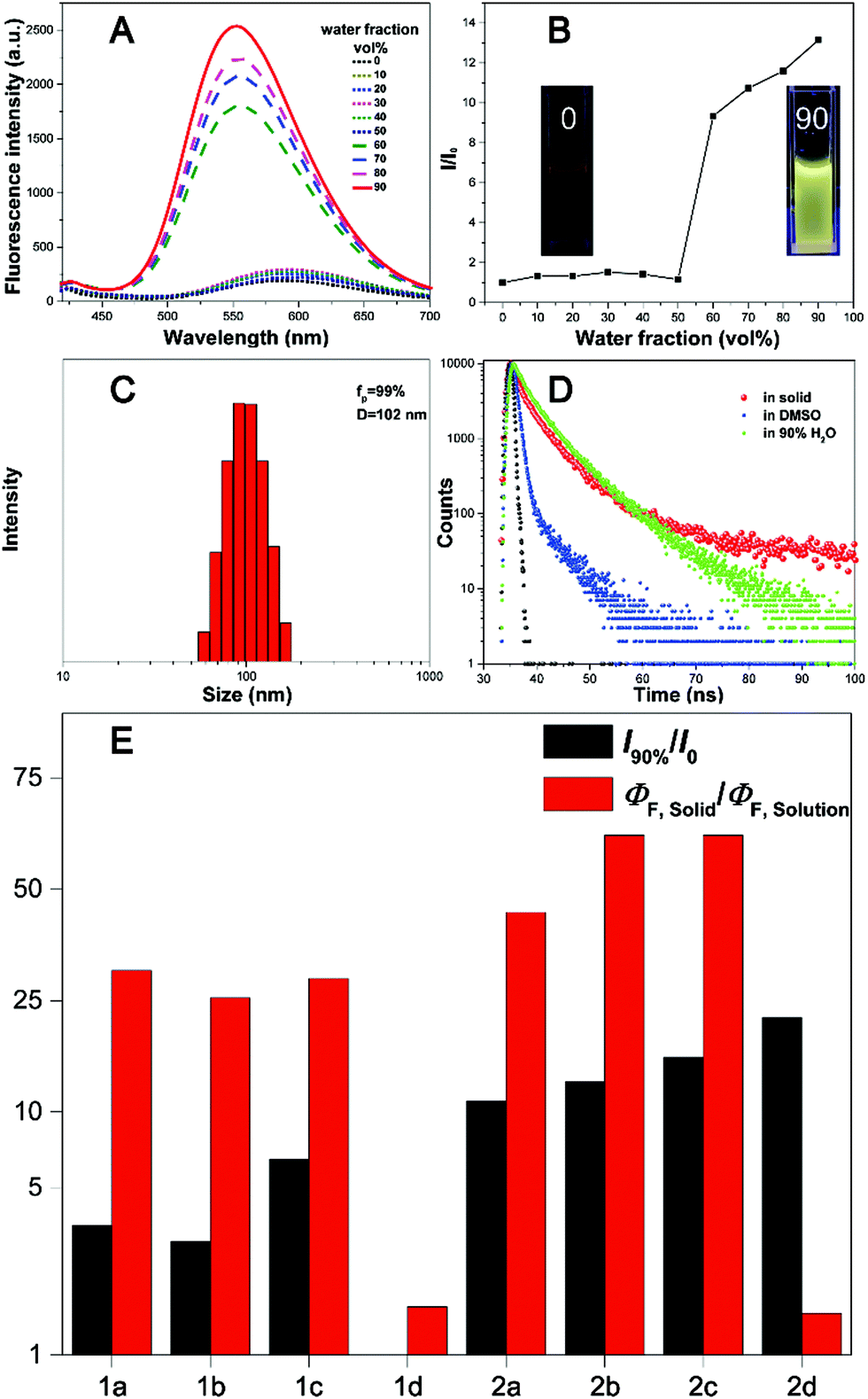 | ||
| Fig. 4 (A) Fluorescence spectra of 2b in DMSO/water with different water fractions. (B) The I/I0 ratios of 2b in DMSO/water with different water fractions. (C) Particle size distribution of 2b in a DMSO/water mixture with an fP value of 99%. (D) Emission decay of 2b in DMSO, H2O, and the solid state. (E) Ratio of the quantum yields for the solid and solution states of all compounds, and the ratio of the fluorescence intensity in the aggregation and solution states. | ||
AIE properties and decay effect study of the fluorophores
All the fluorophores exhibit remarkable AIE properties. We measured the AIE properties using DMSO and water (Fig. S6 ESI,†Fig. 4A and B), which served as a good and poor solvent, respectively. Typically, while 2b and 2c are almost non-emissive in DMSO, as the water fractions increases from 0 to 90%, the fluorescence intensities increase; such a phenomenon could be attributed to aggregation of the molecules in the mixed solution, activation of the RIM process and demonstration of AIE characteristics. As shown in Fig. 4E, we calculated the ratio of the fluorescence intensity in the aggregation and solution states. Typically, the fluorescence intensities of 2b and 2d in the aggregation state (90 vol% water fractions) are about 13.5 and 25 times greater than those in pure DMSO, respectively. The particle size for 1a–2d in the mixture with water fractions of 99% has been studied by using dynamic light scattering (DLS) (Fig. S7, ESI†). For example, in Fig. 4C the particle size of 2b is 102 nm, confirming the formation of nano aggregates.The fluorescence lifetime (τ) values of fluorophores 1a–2d are listed in Table S5 (ESI†). As shown in Fig. S8 (ESI†) and Fig. 4D, we can intuitively see the changes of the fluorescence lifetime in the solution, aggregated and solid states, which is significantly increased from the dissolved state to the aggregated state. To verify the photoemission dynamics, the radiative transition rate constant (Kr) and the non-radiative transition rate constant (Knr) were calculated using the fluorescence lifetimes and the quantum yields (Tables S6 and S7, ESI†). Taking compound 2b as an example, Knr (1.44 × 109) is remarkably greater than Kr (1.44 × 106) in solution. However, Knr decreases to 3.76 × 108 while Kr enhances to 2.53 × 107 in the solid state. Such results suggest that restraining the non-radiative decay and exciting the radiative decay are the major ways to improve the quantum yield in the solid state.
Cell imaging of HepG2
The high fluorescence QY and large Stokes shift of 2b can benefit biological imaging; the Stokes shift of 2b in PBS buffer is 206 nm, which is highly desirable for bioimaging application by avoiding autofluorescence, and the unique AIE property could avoid the ACQ effect of traditional organic fluorescent probes. We found that 2b can mix with oleic acid in PBS buffer to form nano-aggregates OA–2b, which improved the water-solubility of 2b. As shown in Fig. S9A (ESI†), OA–2b has a smaller particle size in PBS buffer than 2b, indicating that OA–2b can be better dispersed in PBS buffer, which suggests efficient emission based on AIE (Fig. S9B, ESI†). Oleic acid is often used as an assistant in lipid droplet (LD) application of biological imaging because it can enter the LDs and make them larger; we have imaged this process with OA–2b in live HepG2 cells by confocal lasing scanning microscopy (CLSM). As shown in Fig. 5, within 1 hour, most 2b gathered outside the cell membrane due to the hydrophobicity, while a little OA–2b entered the cell. In 2 hours, large amounts of OA–2b entered the cells and the emission of OA–2b was observed easily. As time went on, the amount of OA–2b in LDs gradually increased; after 4 hours, it achieved good imaging of HepG2 cells, and we can observe by the naked eye that the particles of LDs enlarged by about 2 times when incubated for 6 hours. We can see the enlarged inset details which are marked by the red ellipse in Fig. S10 (ESI†). The whole imaging process is detailed in Fig. 5S. The other data with different incubation times are presented in Fig. S11 (ESI†). After incubation with OA–2b for 6 hours, a commercially available LD-imaging agent BODIPY 493/503 was used to co-stain the HepG2 cells. To avoid optical crosstalk, we excited the OA–2b and BODIPY 493/503 at 405 and 488 nm, respectively. We collected the emission of 2b from 535 to 640 nm, and BODIPY 493/503 from 480 to 520 nm, respectively. As shown in Fig. 5U, the result verified the functionality of oleic acid. Additionally, autofluorescence was completely avoided under this condition. As shown in Fig. S12 (ESI†), the cell viability experiments indicated that 2b is suitable for biological applications with low cytotoxicity.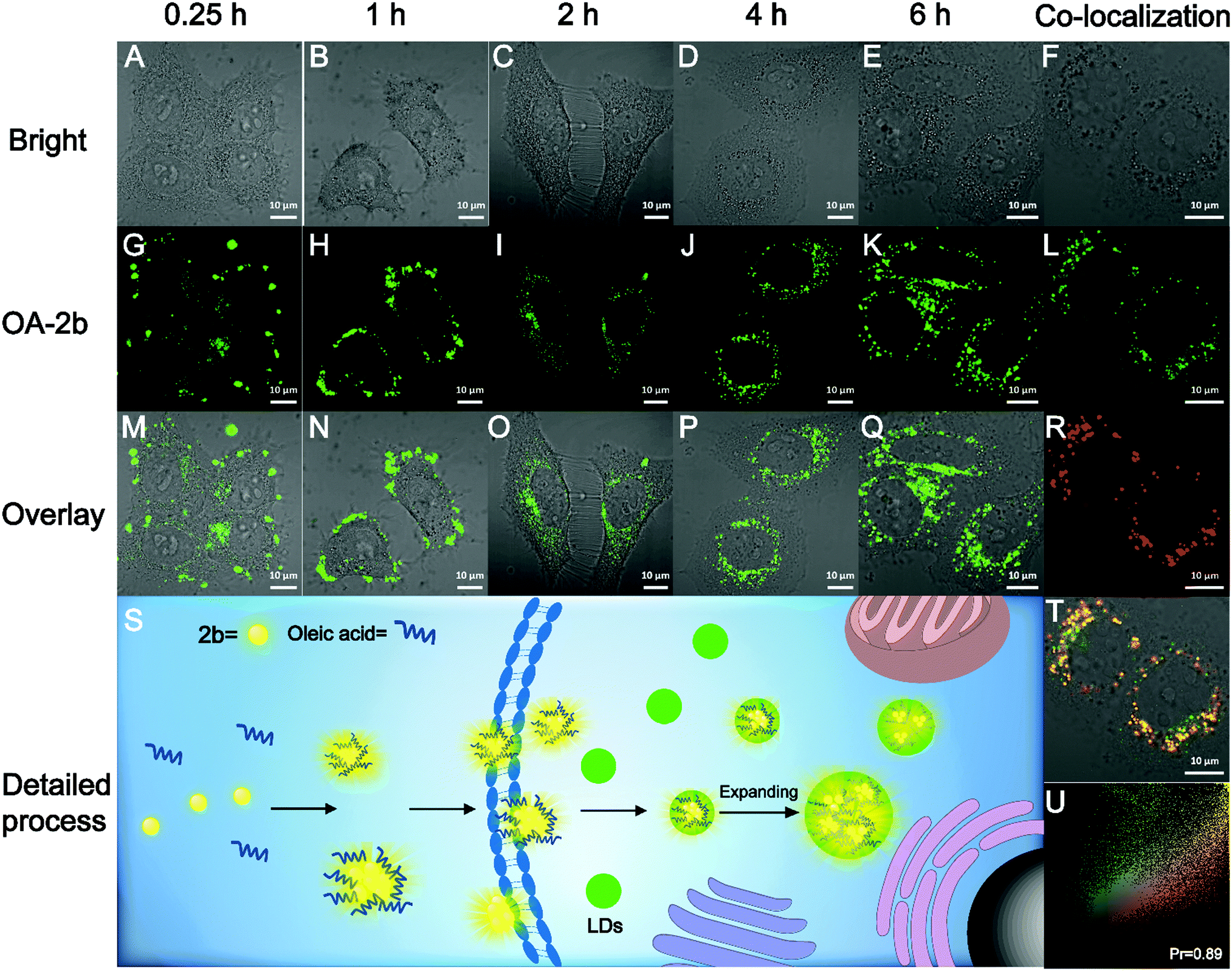 | ||
| Fig. 5 Live HepG2 cells incubated with OA–2b for different times: (S) detailed process of LD imaging. Live HepG2 cells incubated with OA–2b for 6 hours and BODIPY 493/503 for 15 min: (L) the 2b channel, (R) the BODIPY 493/503 channel, (X) the overlay of (L) and (R), and (Z) the co-localization. 2b channel: λex = 405 nm and λem = 535–640 nm; BODIPY 493/503 channel: λex = 488 nm and λem = 480–520 nm. | ||
Conclusions
In this work, we have provided a donor and acceptor engineering strategy to change the fluorescence behavior and enhance the fluorescence performance with aggregation-induced emission. By enlarging steric hindrance it's able to enhance the fluorescence emission efficiency in the solid state by a twisted molecular conformation and to avoid the typical π–π stacking interactions. In addition, by adjusting the electron accepting group and electron donating group we can reverse the trend of the quantum yields between the solution and solid phases. We applied this strategy to the creation of new AIEgens based on BINOL. We have obtained two series of color tunable AIE-active fluorophores by subtle structure modification. Their optical properties and several single crystal packing structures have been systematically investigated. The results indicate that methoxyphenyl, benzoyl and aryl allyl dinitrile are efficient substituents for AIE fluorophores, especially aryl allyl dinitrile. Finally, we have obtained two representative fluorophores 2b and 2c with large Stokes shift and high quantum yield, and 2b was successfully applied to HepG2 cell imaging very well. We believe our strategy provides a systematic and efficient way to build novel organic solid fluorophores from simple ACQ fluorophores. From fundamental and practical viewpoints, it will promote the rapid development of AIE fluorescence systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21877082). We also thank the comprehensive Training Platform of Specialized Laboratory, College of Chemistry, Sichuan University for sample analysis.Notes and references
- P. Shen, Z. Zhuang, Z. Zhao and B. Z. Tang, J. Mater. Chem. C, 2018, 6, 11835 RSC.
- (a) L. Hu, S. Yu, Y. Wang, X. Yu and L. Pu, Org. Lett., 2017, 19, 3779 CrossRef CAS PubMed; (b) Y. Wang, L. Hu, F. Zhao, S. Yu, J. Tian, D. Shi, X. Wang, X. Yu and L. Pu, Chem. – Eur. J., 2017, 23, 17678 CrossRef CAS PubMed.
- (a) Y. Dong, B. Xu, J. Zhang, X. Tan, L. Wang, J. Chen, H. Lv, S. Wen, B. Li, L. Ye, B. Zou and W. Tian, Angew. Chem., Int. Ed., 2012, 124, 10940 CrossRef; (b) R. Hu, N. L. C. Leung and B. Z. Tang, Chem. Soc. Rev., 2014, 43, 4494 RSC.
- Z. Zhao, H. Nie, C. Ge, Y. Cai, Y. Xiong, J. Qi, W. Wu, R. T. K. Kwok, X. Gao, A. Qin, J. W. Y. Lam and B. Z. Tang, Adv. Sci., 2017, 4, 1700005 CrossRef PubMed.
- (a) Y. Chen, W. Zhang, Z. Zhao, Y. Cai, J. Gong, R. T. K. Kwok, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams and B. Z. Tang, Angew. Chem., Int. Ed., 2018, 57, 5011 CrossRef CAS PubMed; (b) D. Wang, H. Su, R. T. K. Kwok, X. Hu, H. Zou, Q. Luo, M. M. S. Lee, W. Xu, J. W. Y. Lam and B. Z. Tang, Chem. Sci., 2018, 9, 3685 RSC.
- X. Gu, E. Zhao, T. Zhao, M. Kang, C. Gui, J. W. Y. Lam, S. Du, M. M. T. Loy and B. Z. Tang, Adv. Mater., 2016, 28, 5064 CrossRef CAS PubMed.
- G. Feng and B. Liu, Acc. Chem. Res., 2018, 51, 1404 CrossRef CAS PubMed.
- X. Shi, C. Y. Y. Yu, H. Su, R. T. K. Kwok, M. Jiang, Z. He, J. W. Y. Lam and B. Z. Tang, Chem. Sci., 2017, 8, 7014 RSC.
- M. Kang, X. Gu, R. T. K. Kwok, C. W. T. Leung, J. W. Y. Lam, F. Li and B. Z. Tang, Chem. Commun., 2016, 52, 5957 RSC.
- (a) Q. Qi, Y. Li, X. Yan, F. Zhang, S. Jiang, J. Su, B. Xu, X. Fu, L. Sun and W. Tian, Polym. Chem., 2016, 7, 5273 RSC; (b) C. Gui, E. Zhao, R. T. K. Kwok, A. C. S. Leung, J. W. Y. Lam, M. Jiang, H. Deng, Y. Cai, W. Zhang, H. Su and B. Z. Tang, Chem. Sci., 2017, 8, 1822 RSC.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC.
- (a) K. Li, W. Qin, D. Ding, N. Tomczak, J. Geng, R. Liu, J. Liu, X. Zhang, H. Liu, B. Liu and B. Z. Tang, Sci. Rep., 2013, 3, 1150 CrossRef PubMed; (b) W. Qin, D. Ding, J. Liu, W. Z. Yuan, Y. Hu, B. Liu and B. Z. Tang, Adv. Funct. Mater., 2012, 22, 771 CrossRef CAS.
- Y. Chen, J. W. Y. Lam, R. T. K. Kwok, B. Liu and B. Z. Tang, Mater. Horiz., 2019, 6, 428 RSC.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718 CrossRef CAS PubMed.
- H. Qian, M. E. Cousins, E. H. Horak, A. Wakefield, M. D. Liptak and I. Aprahamian, Nat. Chem., 2016, 9, 83 CrossRef PubMed.
- C. J. Bhongale, C.-W. Chang, C.-S. Lee, E. W.-G. Diau and C.-S. Hsu, J. Phys. Chem. B, 2005, 109, 13472 CrossRef CAS PubMed.
- (a) S. Grimme, Angew. Chem., Int. Ed., 2008, 47, 3430 CrossRef CAS PubMed; (b) H. Saigusa and E. C. Lim, J. Phys. Chem., 1995, 99, 15738 CrossRef CAS.
- (a) Y.-X. Li, X.-F. Yang, J.-L. Miao and G.-X. Sun, J. Phys. Chem. C, 2016, 120, 21722 CrossRef CAS; (b) S. Samanta, U. Manna and G. Das, New J. Chem., 2017, 41, 1064 RSC.
- N. Meher and P. K. Iyer, Angew. Chem., Int. Ed., 2018, 57, 8488 CrossRef CAS PubMed.
- J. N. Zhang, H. Kang, N. Li, S. M. Zhou, H. M. Sun, S. W. Yin, N. Zhao and B. Z. Tang, Chem. Sci., 2017, 8, 577 RSC.
- L. Shi, K. Li, P.-C. Cui, L.-L. Li, S.-L. Pan, M.-Y. Li and X.-Q. Yu, J. Mater. Chem. B, 2018, 6, 4413 RSC.
- M. Jiang, X. Gu, R. T. K. Kwok, Y. Li, H. H. Y. Sung, X. Zheng, Y. Zhang, J. W. Y. Lam, I. D. Williams, X. Huang, K. S. Wong and B. Z. Tang, Adv. Funct. Mater., 2018, 28, 1704589 CrossRef.
- J. Han, J. You, X. Li, P. Duan and M. Liu, Adv. Mater., 2017, 29, 1606503 CrossRef PubMed.
- (a) M. Jiang, X. Gu, J. W. Y. Lam, Y. Zhang, R. T. K. Kwok, K. S. Wong and B. Z. Tang, Chem. Sci., 2017, 8, 5440 RSC; (b) H. Lu, Y. Zheng, X. Zhao, L. Wang, S. Ma, X. Han, B. Xu, W. Tian and H. Gao, Angew. Chem., Int. Ed., 2016, 55, 155 CrossRef CAS PubMed.
- D. Wang, H. Su, R. T. K. Kwok, G. Shan, A. C. S. Leung, M. M. S. Lee, H. H. Y. Sung, I. D. Williams, J. W. Y. Lam and B. Z. Tang, Adv. Funct. Mater., 2017, 27, 1704039 CrossRef.
- (a) Z. Song, W. Zhang, M. Jiang, H. H. Y. Sung, R. T. K. Kwok, H. Nie, I. D. Williams, B. Liu and B. Z. Tang, Adv. Funct. Mater., 2016, 26, 824 CrossRef CAS; (b) A. C. Shaikh, D. S. Ranade, S. Thorat, A. Maity, P. P. Kulkarni, R. G. Gonnade, P. Munshi and N. T. Patil, Chem. Commun., 2015, 51, 16115 RSC.
- (a) B.-K. An, J. Gierschner and S. Y. Park, Acc. Chem. Res., 2012, 45, 544 CrossRef CAS PubMed; (b) K. A. N. Upamali, L. A. Estrada, P. K. De, X. Cai, J. A. Krause and D. C. Neckers, Langmuir, 2011, 27, 1573 CrossRef CAS PubMed; (c) L. Shi, K. Li, L.-L. Li, S.-Y. Chen, M.-Y. Li, Q. Zhou, N. Wang and X.-Q. Yu, Chem. Sci., 2018, 9, 8969 RSC.
- Y. Li, F. Li, H. Zhang, Z. Xie, W. Xie, H. Xu, B. Li, F. Shen, L. Ye, M. Hanif, D. Ma and Y. Ma, Chem. Commun., 2007, 231 RSC.
- M. Gao, H. Su, Y. Lin, X. Ling, S. Li, A. Qin and B. Z. Tang, Chem. Sci., 2017, 8, 1763 RSC.
- S. Xu, T. Liu, Y. Mu, Y.-F. Wang, Z. Chi, C.-C. Lo, S. Liu, Y. Zhang, A. Lien and J. Xu, Angew. Chem., Int. Ed., 2015, 54, 874 CrossRef CAS PubMed.
- N. Zhao, Z. Yang, J. W. Y. Lam, H. H. Y. Sung, N. Xie, S. Chen, H. Su, M. Gao, I. D. Williams, K. S. Wong and B. Z. Tang, Chem. Commun., 2012, 48, 8637 RSC.
- G. Haberhauer, R. Gleiter and C. Burkhart, Chem. – Eur. J., 2016, 22, 971 CrossRef CAS PubMed.
- L. Stojanović and R. Crespo-Otero, ChemPhotoChem, 2019, 3, 907 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and methods: synthesis and cell culture; structural characterization: single crystal, NMR and ESI-MS data; photophysical supplementary data and bioimaging supplementary data, etc. CCDC 1907963, 1907968 and 1907970. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ma00022a |
| This journal is © The Royal Society of Chemistry 2020 |