
Electrodeposited Cu–Pd bimetallic catalysts for the selective electroreduction of CO2 to ethylene†
Ruting
Feng
a,
Qinggong
Zhu
b,
Mengen
Chu
a,
Shuaiqiang
Jia
a,
Jianxin
Zhai
a,
Haihong
Wu
 *a,
Peng
Wu
*a and
Buxing
Han
*ab
*a,
Peng
Wu
*a and
Buxing
Han
*ab
aShanghai Key Laboratory of Green Chemistry and Chemical Processes, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, P. R. China. E-mail: hhwu@chem.ecnu.edu.cn; pwu@chem.ecnu.edu.cn; hanbx@iccas.ac.cn
bBeijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Colloid and Interface and Thermodynamics, CAS Research/Education Center for Excellence in Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Zhongguancun North First Street 2, Beijing, 100190, P. R. China
First published on 29th September 2020
Abstract
Cu–Pd bimetallic catalysts were fabricated on carbon paper (CP) by the electrodeposition method via a dynamic hydrogen bubble template approach. At a potential of −1.2 V vs. RHE, the Faradaic efficiency (FE) of C2H4 could reach 45.2% with a current density of 17.4 mA cm−2 in an H-type electrolytic cell. Detailed studies suggest that the enhanced performance of Cu–Pd/CP was attributed mainly to the synergistic effect, low interfacial charge transfer resistance, and the 3D architecture of the catalysts.
Carbon dioxide (CO2) reduction has garnered intense interest with the global energy and environmental crises.1,2 An ideal route for the chemical utilization of CO2 is to develop catalytic processes for the selective conversion of CO2 into value-added chemicals and fuels.3–5 Among all the developed techniques, electrocatalytic reduction is a promising route for the utilization of CO2 as a carbon feedstock to value-added chemicals.6–11 To date, extensive efforts have been devoted to the development of electrocatalysts for the CO2 reduction reaction (CO2RR) to higher value products.12–14
Ethylene (C2H4) is a popular chemical commodity used in industry.15,16 Compared with C1 products, C2H4 possesses an impressive energy density and higher economic value.13,17,18 Currently, copper (Cu) is the promising candidate as a catalyst that could produce C2 products, but often a wide range of products are formed with low product selectivity.19,20 To overcome the poor selectivity of pure Cu toward C2 products, various strategies have been proposed, including alloying,21 doping,22 changing the composition and morphology, and creating special electrolytic devices.23 For instance, Cu-doped carbon catalysts have been reported to enhance the electroreduction of CO2 to C2H4.24 Plasma-activated oxide derived-Cu (OD-Cu) catalysts with high stability of Cu+ species have been reported to promote the C2 product formation.25 While significant progress has been achieved in the CO2RR to C2H4,26,27 the exploration of electrochemical systems for electrocatalytic C–C coupling in aqueous electrolytes is still an open challenge due to low selectivity, activity, and large overpotential.28–30 The design of the catalyst holds the key to address these challenges.31,32
Recently, Cu-based bimetallic catalysts have shown great potential in enhancing the catalytic ability toward C2 products by increasing strains and the synergistic effects of atoms in the catalyst.20,33,34 Enhancement in the selectivity for C2H4 has been observed on various bimetallic catalysts. For example, Au–Cu bimetallic catalysts have been investigated to increase the C2H4 selectivity to 46.7%.35 Ag–Cu nanoparticles showed a selectivity of the CO2RR toward C2H4 with the FE of 41.3%.36 Besides, the Cu–Ru bimetallic alloy could catalyze CO2 conversion to C2H4 with the FE of 19%.37 The nanoporous Cu–Ag bimetallic catalyst was also reported to reduce CO2 to C2H4 with the FE of 60%.38 However, even after such extensive research efforts, achieving high selectivity for C2H4 on facilely-prepared bimetallic catalysts is still highly desired in this field.39,40
Self-supporting bimetallic catalysts by electrodeposition is a very useful technique that can form a three-dimensional (3D) porous structure and an increased surface area by a one-step strategy of potentiostatic co-electrodeposition utilizing hydrogen bubble dynamic templates.41,42 Compared with other synthesized methods, the corresponding technology is often known as electroplating, which can simplify the experimental process, further reduce the cost and increase the electrode efficiency due to their extraordinary chemical properties in the electrolyte during the electrodeposition.43 Electrocatalysts also provide some obvious advantages in the CO2RR, such as fast electron transfer rate, a large specific area and high electrochemical performance.12
In this work, we focus on enhancing the selectivity of the CO2RR to C2H4 on Cu–Pd bimetallic catalysts, which were prepared by galvanostatic electrodeposition using the self-supporting technique. The resulting Cu–Pd bimetallic catalysts had a high selectivity toward C2H4 (45.2%) at an applied potential of −1.2 V vs. RHE and a current density of 17.4 mA cm−2 in aqueous electrolyte. The high catalytic activity of bimetallic Cu–Pd catalysts was attributed to the synergistic effect between Cu and Pd, fast electron transfer rate, and the 3D architecture of the catalyst.
The Cu–Pd bimetallic catalysts were synthesized by a facile electrodeposition method, which is schematically described in Fig. 1A. In the deposition process, Cu and Cu–Pd samples were electrodeposited in an acid solution containing Cu2+ and Pd2+ at pH = 1. During the deposition, the dynamic hydrogen bubble was used as a template to prepare functional porous materials. Cu–Pd was electrodeposited galvanostatically at a constant current density. The catalysts were characterized by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). Fig. 1B–D provide the SEM images of CP, Cu/CP and Pd/CP. It is shown that a rough surface of Cu deposit and a half-baked dendritic 3D structure for Pd were formed on CP. Unlike these catalysts, Cu–Pd bimetallic catalysts show observable structural changes (Fig. 1E), and a tetrahedron structure was formed with nanostructured features on the scale of 90–200 nm (Fig. 1F, TEM). We also examined Cu–Pd/CP catalysts using elemental distribution mappings (EDS, Fig. 1G–I), which show that Cu (green) and Pd (red) atoms are homogeneously distributed, forming a bimetallic structure.
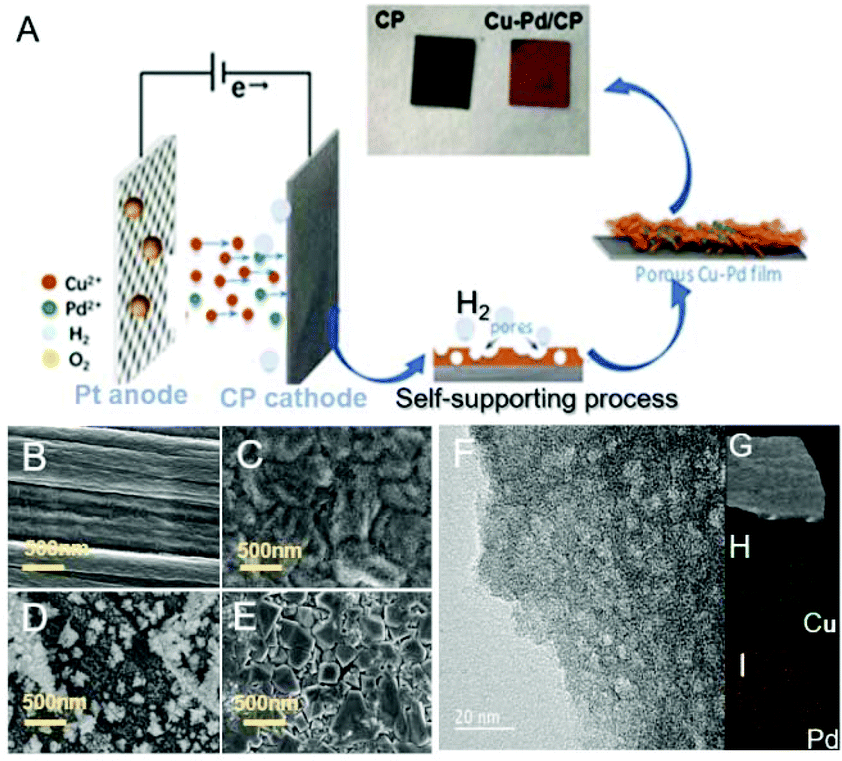 | ||
| Fig. 1 (A) Schematic illustration of the procedure for preparing Cu–Pd bimetallic catalysts; SEM images: (B) CP; (C) Cu/CP catalysts; (D) Pd/CP catalysts; (E) Cu–Pd/CP catalysts; (F) TEM image of Cu–Pd/CP catalysts; (G) the elemental mappings of Cu–Pd/CP catalysts. The images in green and red represent (H) Cu and (I) Pd, respectively. | ||
According to X-ray diffraction (XRD, Fig. 3A), the diffraction peak located at 24.8° was assigned to the (002) plane of the CP. Cu diffraction peaks of the (111), (200), and (220) facets are present for the bimetallic samples. The diffraction peaks of these samples had an appreciable shift in comparison with that of Pd. No Pd-related peaks were found, indicative of the low amount of Pd in the bimetallic catalysts. The Cu–Pd bimetallic samples also exhibited Cu2O diffraction peaks of (110), (111), and (220), which are associated with the presence of the Cu2O phase in the bimetallic catalysts.
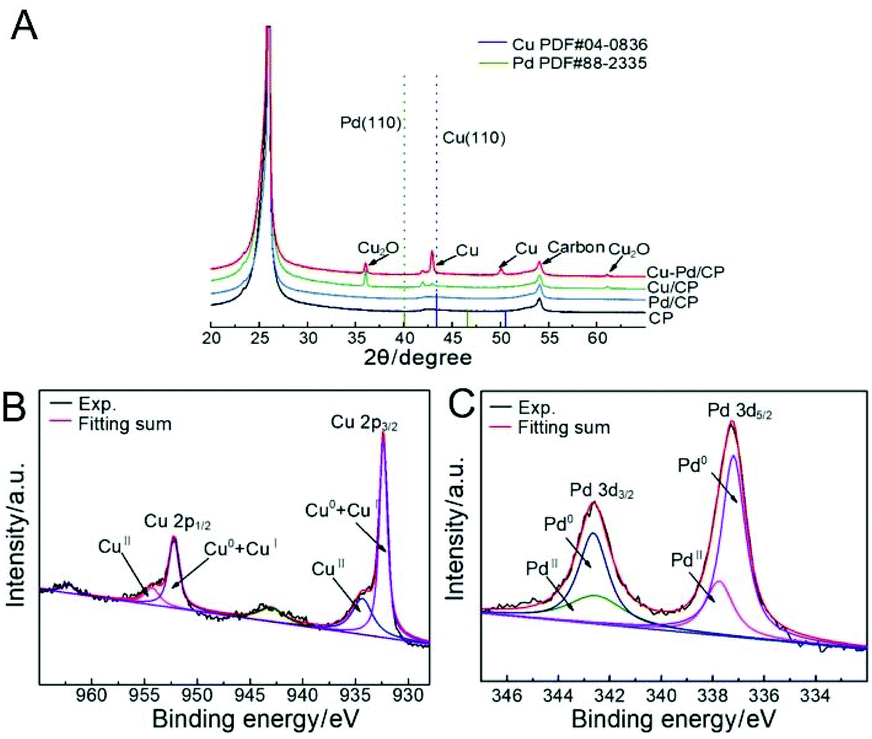 | ||
| Fig. 2 (A) XRD patterns of Cu–Pd/CP, Cu/CP, Pd/CP, and CP; XPS spectra of (B) Cu 2p and (C) Pd 3d levels of catalysts. | ||
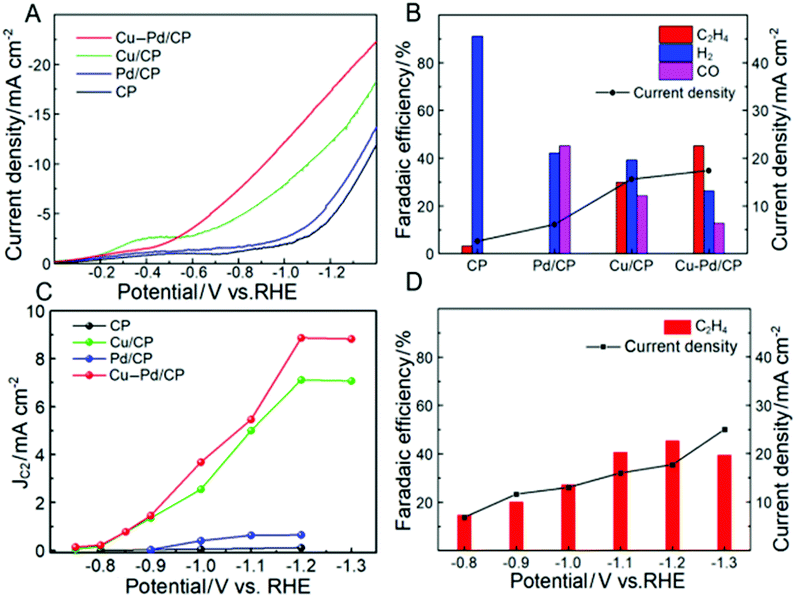 | ||
| Fig. 3 (A) LSV curves of various electrodes with a scan rate of 50 mV−1; (B) the FE of C2H4 at −1.2 V (vs. RHE) over various electrodes; (C) partial current density of C2H4 over various electrodes at different applied potentials; (D) the FE and current density of C2H4 over Cu–Pd/CP at different applied potentials in CO2-saturated 0.1 M KCl solution. | ||
X-ray photoelectron spectroscopy (XPS) further identified the elemental contents and valence states of Cu and Pd in the catalysts (Fig. 2B and C,). The results of XPS investigations on Cu/CP and Pd/CP are consistent with the results of XRD. For Cu–Pd/CP, it can be seen that the Cu/Pd ratio is 6.83, which indicates the formation of bimetallic catalysts (Table S1†). The binding energies of Cu 2p spectra were fitted with two components, which are CuI + Cu0 (2p3/2, 932.4 eV; 2p1/2, 952.0 eV) and CuII (2p3/2, 934.2 eV; 2p1/2, 954.0 eV), respectively. The intensities of Pd 3d5/2 peaks of Cu–Pd catalysts at 337.2 eV and 337.8 eV correspond to the metallic Pd0 and PdII, and the Pd 3d3/2 peaks at 342.5 eV and 342.8 eV correspond to Pd0 and PdII. This indicates that O species exist on the catalyst surface. The presence of O species may cause an enhancement in the C2H4 production efficiency.38
The electrocatalytic performances of Cu–Pd/CP were investigated in CO2-saturated KCl aqueous solution in a typical three-electrode electrochemical system. Linear sweep voltammetry (LSV) was conducted to investigate the performance of Cu–Pd in 0.1 M KCl aqueous solution, and the LSV curves were swept in the potential range from 0 to −1.4 V (vs. RHE) at a sweep rate of 50 mV s−1. As shown in Fig. 3A, Cu–Pd/CP exhibited a higher current density than other electrodes (CP, Pd/CP, and Cu/CP), suggesting that Cu–Pd bimetallic catalysts were favorable to the binding of CO2. In addition, the much higher current density of the CO2-saturated catholyte than the N2-saturated catholyte implied the reduction of CO2 (Fig. S1†).
Controlled potential electrolysis was performed using a typical H-type electrolysis cell in the same system. After electrolysis, the gas and liquid products were analyzed by gas chromatography (GC) and nuclear magnetic resonance (1H-NMR) analysis, respectively. The main gaseous products were C2H4, H2 and a trace amount of CO; no liquid product was detected by NMR. Fig. 3B shows the FE of the main products over various electrodes; it indicates that CP and Pd/CP have little catalytic effect on the electrocatalytic reduction of CO2 to C2H4. The main product on Pd/CP was syngas, which contains H2 and CO. On Cu/CP, the FE of C2H4 was 29.97% with a CO FE of 24.3%. Among these samples, Cu–Pd/CP was the best catalyst with a C2H4 FE of 45.23% at −1.2 V (vs. RHE). Compared with the reported results using different bimetallic electrocatalysts (Table S2†), the FE and current density were high in the aqueous electrolyte using an H-type cell.
To gain kinetic insights into the CO2RR to C2H4 on Cu–Pd catalysts, the current density for C2H4 at various overpotentials was measured. As shown in Fig. 3C, the onset potential over Cu–Pd/CP was −0.65 V, which was more positive than those of Cu/CP (−0.72 V) and Pd/CP (−0.88 V), indicating that the initial electron transfer to CO2 to form an adsorbed CO2− intermediate was much easier over Cu–Pd/CP.44 We also studied the change of C2H4 over Cu–Pd/CP at different applied potentials. As the electrolytic potential increased, the FE of C2H4 increased first and then decreased. The FE of C2H4 reached 45.2% with a current density of 17.4 mA cm−2 at −1.2 V vs. RHE (Fig. 3D). The decrease in the FE of C2H4 at a high applied potential resulted from the enhancement in the production rate of H2.
The electrochemical activity of Cu–Pd/CP was also characterized according to the Randle–Sevcik equation. The reduction current density plotted against the root of the scan rate is illustrated in Fig. S2.† According to the result, the Cu–Pd/CP has the largest slope among the electrodes, indicating the highest reduction rate for the CO2RR. This result is consistent with the electrochemical impedance spectroscopy (EIS) result (Fig. S3†–4), which provides the charge transfer rate at the electrode/electrolyte interface. The results indicated that the charge transfer resistance (Rct) of the Cu–Pd/CP was lower than that of the Cu/CP. The above result shows that Pd or Cu could not promote the generation of the C2H4 product efficiently, and the excellent performance of Cu–Pd/CP can be attributed to the synergistic effect between Cu and Pd in the catalysts. On one hand, Cu-based bimetallic catalysts have shown great potential in enhancing the catalytic ability toward C2 products by increasing strains and the synergistic effects of atoms in the catalyst. Oxygen-bearing Cu catalysts could enable the C–C coupling in the electrocatalytic CO2RR systems. The mixed valence form of copper (Cu0 and CuI) may be the dominant species during the CO2RR, which could be the most active species for the C2 product formation. The Cu+/Cu0 interfaces formed through stabilized Cu+ facilitate the *CO–CO dimerization, promoting the formation of C2+ products and suppressing the conversion to C1 products. On the other hand, Pd is known to be an excellent catalyst for the CO2RR to CO.21,40 A trace amount of Pd could promote the adsorption of the *CO intermediate and facilitate their dimerization to form C2 products.38,45
We also studied the electrolyte effect over the Cu–Pd/CP electrode. In addition to 0.1 M KCl electrolyte, other electrolytes that contain different cations and anions were used in this work (Fig. S5†). For example, in the NaCl electrolyte, H2 was the main product. CO2RR exhibited better performance for C2H4 with the FE of 35% in 0.1 M KHCO3 electrolyte. In 0.1 M KH2PO4 and K2HPO4 electrolyte, the FE of C2H4 was very low with H2 as the main product. In the KCl electrolyte, even if the electrolyte concentration is increased, the FE of C2H4 did not increase significantly, which indicates that 0.1 M KCl was the best electrolyte. We think that the electrolyte affects CO2 reduction mainly in two ways. First, the alkali metal cation (such as Na+, K+) in the electrolyte is known to influence the electrocatalytic ability of CO2 reduction. Density functional theory (DFT) calculations have shown that the alkali metal cations influence the distribution of products formed as a consequence of the electrostatic interactions between the solvated cations present at the outer Helmholtz plane and the adsorbed species having large dipole moments. Therefore, an increased alkali metal size leads to higher selectivity to C2 products.46,47 Second, the local buffering capability of cations could increase the rate of the proton-transfer reaction, which affects the activity and selectively for the CO2RR. When the cation concentration was too low or too high, it will inhibit the proton-transfer reaction. Therefore, a moderate concentration facilitates proton transfer and improves the catalytic activity and selectivity.48,49 These arguments are consistent with our experimental results.
We assume that the excellent activity of Cu–Pd bimetallic catalysts toward C2H4 resulted from the synergistic effect of Cu and Pd in Cu–Pd catalysts. Therefore, we prepared the Cu–Pd catalysts using different electrodeposition methods (CV and amperometric mode), which may contain different atomic ratios of the elements (Table S1†). The corresponding electrodes were then named Cu–Pd/CP-CV and Cu–Pd/CP-IT (see the Experimental section in the ESI†). The SEM image of Cu–Pd/CP-CV and Cu–Pd/CP-IT electrodes showed a symmetrical and a homogeneous morphology, respectively (Fig. S6†). After electrolysis, we found that the FE of C2H4 was approximately 25% over both electrodes with high hydrogen evolution reaction (HER) than the Cu–Pd/CP electrode (Fig. 4A). Therefore, we characterized the Cu–Pd catalysts prepared using different methods by XRD (Fig. S7†) and XPS (Fig. S8†) to get more information about the surface elemental states and catalytic performance. Among the three methods, Cu–Pd/CP showed the lowest (Cu0 + CuI)/CuII and Pd0/PdII ratio, which suggests the existence of oxygen vacancies in the catalysts (Table S1†). Therefore, it is beneficial for the adsorption of the intermediates and facilitates the formation of C2H4. The EIS results (Fig. S9, Table S3†) also showed that Cu–Pd/CP had a lower Rct (Nyquist plot) and a larger phase angle in the low-frequency region (Bode plot) than that of Cu–Pd/CP-CV and Cu–Pd/CP-IT, resulting in the increase of electron transfer and the diffusion ability of Cu–Pd/CP.
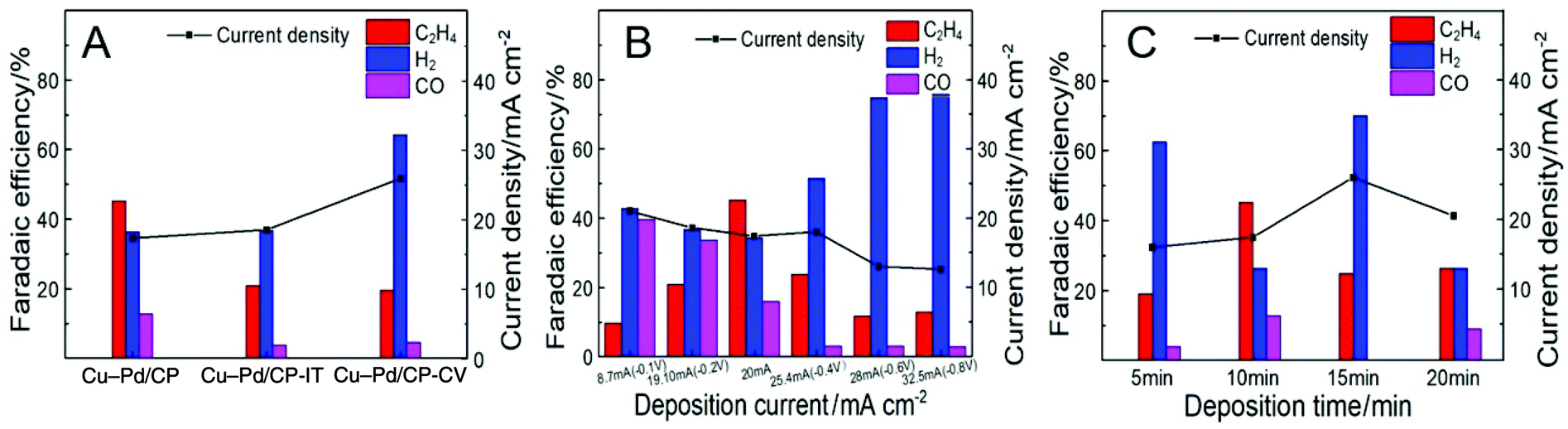 | ||
| Fig. 4 The FE of C2H4 over Cu–Pd bimetallic catalysts (A) using different electrodeposition methods; (B) under different current densities; (C) obtained at various deposition times. | ||
The effect of deposition current density on the properties of the catalysts was also studied. The deposition of Cu–Pd bimetallic catalysts under different current densities not only affected the co-deposition process, but also changed the morphology of the catalyst. Therefore, we fixed the total amount of electrons to 12 Coulomb and obtained Cu–Pd catalysts with different morphologies under different current densities (Fig. S10†). Fig. 4B shows that the deposition current density of Cu–Pd catalysts had a great influence on the FE of C2H4. It may be due to the different atomic amounts in the catalysts. As shown in Fig. S11,† the bimetallic solution has an intermediate potential between the other two monometallic solutions. It indicates that at a high deposition potential and high current density, the deposition tended to deposit Cu. At a low deposition potential and low current density, the content of Pd in the catalysts increased. According to this tendency, the FE of C2H4 was first increased and then decreased with increasing deposition current density, which indicates that an appropriate Cu–Pd ratio was beneficial for the reaction. This can also be known from the fact that the FE of C2H4 over electrodeposited Cu/CP or Pd/CP was much lower (Fig. 3B).
The excellent activity of Cu–Pd catalysts may also result partially from the 3D architecture of the film. Therefore, we performed the CO2 reduction using Cu–Pd catalysts obtained at various deposition times. Fig. S12† shows that the amount of the catalyst increased with increasing deposition time. The CO2RR current density increased continuously with deposition time, but the FE of C2H4 first increased and then decreased (Fig. 4C). This can be explained by the fact that the 3D structure gradually formed with increasing deposition time. This structure may provide more opportunities for the C–C coupling for the intermediates. When the film of the 3D structure was thick enough (>10 min), excessive catalyst agglomeration was not conducive to the reaction.
Conclusion
In summary, we developed a facile electrodeposition method to fabricate Cu–Pd catalysts onto conductive substrates through the dynamic hydrogen bubble template approach. The synthesized Cu–Pd/CP is a highly effective catalyst for CO2 reduction to C2H4. The FE and current density of C2H4 could reach 45.23% and 17.4 mA cm−2 in 0.1 M KCl aqueous electrolyte, respectively. A detailed study indicates that the synergistic effect between Cu and Pd in the Cu–Pd catalysts, low interfacial charge transfer resistance, the 3D architecture of the catalysts, and a suitable electrolyte are all favourable for the formation of C2H4. We believe that the strategy to prepare bimetallic catalysts by co-electrodeposition coupled with an effective electrolyte can be used to design more efficient catalytic systems for CO2 reduction to C2 products.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the National Natural Science Foundation of China (21533011, 21733011), the National Key Research and Development Program of China (2017YFA0403102), and the Chinese Academy of Sciences (QYZDY-SSW-SLH013).Notes and references
- M. He, Y. Sun and B. Han, Angew. Chem., Int. Ed., 2013, 52, 9620–9633 CrossRef CAS.
- Z. Sun, T. Ma, H. Tao, Q. Fan and B. Han, Chem, 2017, 3, 560–587 CAS.
- W. Lu, B. Jia, B. Cui, Y. Zhang, K. Yao, Y. Zhao and J. Wang, Angew. Chem., Int. Ed., 2017, 56, 11851–11854 CrossRef CAS.
- S. A. Abbas, S.-H. Kim, H. Saleem, S.-H. Ahn and K.-D. Jung, Catalysts, 2019, 9, 367 CrossRef CAS.
- L. Fan, C. Xia, F. Yang, J. Wang, H. Wang and Y. Lu, Sci. Adv., 2020, 6, eaay3111 CrossRef.
- Q. Zhu, J. Ma, X. Kang, X. Sun, J. Hu, G. Yang and B. Han, Sci. China: Chem., 2016, 59, 551–556 CrossRef CAS.
- H. Yang, L. Wu, H. Wang and J. Lu, Chin. J. Catal., 2016, 37, 994–998 CrossRef CAS.
- A. Loiudice, P. Lobaccaro, E. A. Kamali, T. Thao, B. H. Huang, J. W. Ager and R. Buonsanti, Angew. Chem., Int. Ed., 2016, 55, 5789–5792 CrossRef CAS.
- X. Kang, Q. Zhu, X. Sun, J. Hu, J. Zhang, Z. Liu and B. Han, Chem. Sci., 2016, 7, 266–273 RSC.
- Y. Chen and T. Mu, Green Chem., 2019, 21, 2544–2574 RSC.
- X. Sun, Q. Zhu, X. Kang, H. Liu, Q. Qian, Z. Zhang and B. Han, Angew. Chem., Int. Ed., 2016, 55, 6771–6775 CrossRef CAS.
- P. Moreno-García, N. Schlegel, A. Zanetti, A. Cedeño López, M. d. J. Galvez-Vazquez, A. Dutta, M. Rahaman and P. Broekmann, ACS. Appl. Mater. Interfaces, 2018, 10, 31355–31365 CrossRef.
- A. Eilert, F. S. Roberts, D. Friebel and A. Nilsson, J. Phys. Chem. Lett., 2016, 7, 1466–1470 CrossRef CAS.
- Z. Q. Liang, T. T. Zhuang, A. Seifitokaldani, J. Li, C. W. Huang, C. S. Tan, Y. Li, P. De Luna, C. T. Dinh, Y. Hu, Q. Xiao, P. L. Hsieh, Y. Wang, F. Li, R. Quintero-Bermudez, Y. Zhou, P. Chen, Y. Pang, S. C. Lo, L. J. Chen, H. Tan, Z. Xu, S. Zhao, D. Sinton and E. H. Sargent, Nat. Commun., 2018, 9, 3828 CrossRef.
- F. S. Roberts, K. P. Kuhl and A. Nilsson, Angew. Chem., Int. Ed., 2015, 54, 5179–5182 CrossRef CAS.
- S. Sen, D. Liu and G. T. R. Palmore, ACS. Catal., 2014, 4, 3091–3095 CrossRef CAS.
- C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 7231–7234 CrossRef CAS.
- Z. Tan, T. Peng, X. Tan, W. Wang, X. Wang, Z. Yang, H. Ning, Q. Zhao and M. Wu, ChemElectroChem, 2020, 7, 2020–2025 CrossRef CAS.
- Y. Zheng, A. Vasileff, X. Zhou, Y. Jiao, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2019, 141, 7646–7659 CrossRef CAS.
- C. Chen, X. Sun, L. Lu, D. Yang, J. Ma, Q. Zhu, Q. Qian and B. Han, Green Chem., 2018, 20, 4579–4583 RSC.
- S. Ma, M. Sadakiyo, M. Heima, R. Luo, R. T. Haasch, J. I. Gold, M. Yamauchi and P. J. Kenis, J. Am. Chem. Soc., 2017, 139, 47–50 CrossRef CAS.
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. De Luna, H. Yuan, J. Li, Z. Wang, H. Xie, H. Li, P. Chen, E. Bladt, R. Quintero-Bermudez, T. K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2018, 10, 974–980 CrossRef CAS.
- J. Du, S. Li, S. Liu, Y. Xin, B. Chen, H. Liu and B. Han, Chem. Sci., 2020, 11, 5098–5104 RSC.
- H. Ning, X. Wang, W. Wang, Q. Mao, Z. Yang, Q. Zhao, Y. Song and M. Wu, Carbon, 2019, 146, 218–223 CrossRef CAS.
- H. Mistry, A. S. Varela, C. S. Bonifacio, I. Zegkinoglou, I. Sinev, Y. W. Choi, K. Kisslinger, E. A. Stach, J. C. Yang, P. Strasser and B. R. Cuenya, Nat. Commun., 2016, 7, 12123 CrossRef.
- D. Ren, Y. Deng, A. D. Handoko, C. S. Chen, S. Malkhandi and B. S. Yeo, ACS. Catal., 2015, 5, 2814–2821 CrossRef CAS.
- J.-C. Lee, J.-Y. Kim, W.-H. Joo, D. Hong, S.-H. Oh, B. Kim, G.-D. Lee, M. Kim, J. Oh and Y.-C. Joo, J. Mater. Chem. A, 2020, 8, 11632–11641 RSC.
- D. Raciti, K. J. Livi and C. Wang, Nano Lett., 2015, 15, 6829–6835 CrossRef CAS.
- L. Zhang, Z. J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2017, 56, 11326–11353 CrossRef CAS.
- K. Yao, Y. Xia, J. Li, N. Wang, J. Han, C. Gao, M. Han, G. Shen, Y. Liu, A. Seifitokaldani, X. Sun and H. Liang, J. Mater. Chem. A, 2020, 8, 11117–11123 RSC.
- F. Li, L. Chen, G. P. Knowles, D. R. MacFarlane and J. Zhang, Angew. Chem., Int. Ed., 2017, 56, 505–509 CrossRef CAS.
- X. Wang, Z. Chen, X. Zhao, T. Yao, W. Chen, R. You, C. Zhao, G. Wu, J. Wang, W. Huang, J. Yang, X. Hong, S. Wei, Y. Wu and Y. Li, Angew. Chem., Int. Ed., 2018, 57, 1944–1948 CrossRef CAS.
- M. J. Mao, M. D. Zhang, D. L. Meng, J. X. Chen, C. He, Y. B. Huang and R. Cao, ChemCatChem, 2020, 12, 3530–3536 CrossRef CAS.
- S. Jia, Q. Zhu, H. Wu, M. e. Chu, S. Han, R. Feng, J. Tu, J. Zhai and B. Han, Chin. J. Catal., 2020, 41, 1091–1098 CrossRef CAS.
- Y. Chen, Z. Fan, J. Wang, C. Ling, W. Niu, Z. Huang, G. Liu, B. Chen, Z. Lai, X. Liu, B. Li, Y. Zong, L. Gu, J. Wang, X. Wang and H. Zhang, J. Am. Chem. Soc., 2020, 142, 12760–12766 CrossRef CAS.
- J. Huang, M. Mensi, E. Oveisi, V. Mantella and R. Buonsanti, J. Am. Chem. Soc., 2019, 141, 2490–2499 CrossRef CAS.
- J. T. Billy and A. C. Co, Appl. Catal., B, 2018, 237, 911–918 CrossRef CAS.
- T. T. H. Hoang, S. Verma, S. Ma, T. T. Fister, J. Timoshenko, A. I. Frenkel, P. J. A. Kenis and A. A. Gewirth, J. Am. Chem. Soc., 2018, 140, 5791–5797 CrossRef CAS.
- L. Lu, X. Sun, J. Ma, D. Yang, H. Wu, B. Zhang, J. Zhang and B. Han, Angew. Chem., Int. Ed., 2018, 57, 14149–14153 CrossRef CAS.
- D. Chen, Q. Yao, P. Cui, H. Liu, J. Xie and J. Yang, ACS Appl. Energy Mater., 2018, 1, 883–890 CrossRef CAS.
- B. J. Plowman, L. A. Jones and S. K. Bhargava, Chem. Commun., 2015, 51, 4331–4346 RSC.
- X. Lu and C. Zhao, Nat. Commun., 2015, 6, 6616 CrossRef CAS.
- P. Ganesan, A. Sivanantham and S. Shanmugam, ACS. Appl. Mater. Interfaces., 2017, 9, 12416–12426 CrossRef CAS.
- Q. Zhu, X. Sun, D. Yang, J. Ma, X. Kang, L. Zheng, J. Zhang, Z. Wu and B. Han, Nat. Commun., 2019, 10, 3851 CrossRef.
- T. Takashima, T. Suzuki and H. Irie, Electrochim. Acta, 2017, 229, 415–421 CrossRef CAS.
- J. Resasco, L. D. Chen, E. Clark, C. Tsai, C. Hahn, T. F. Jaramillo, K. Chan and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 11277–11287 CrossRef CAS.
- A. S. Varela, W. Ju, T. Reier and P. Strasser, ACS. Catal., 2016, 6, 2136–2144 CrossRef CAS.
- M. R. Singh, Y. Kwon, Y. Lum, J. W. Ager 3rd and A. T. Bell, J. Am. Chem. Soc., 2016, 138, 13006–13012 CrossRef CAS.
- J. Fu, W. Zhu, Y. Chen, Z. Yin, Y. Li, J. Liu, H. Zhang, J. J. Zhu and S. Sun, Angew. Chem., Int. Ed., 2019, 58, 14100–14103 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc03051a |
| This journal is © The Royal Society of Chemistry 2020 |