
A triple chain polycationic peptide-mimicking amphiphile – efficient DNA-transfer without co-lipids†
Shashank Reddy
Pinnapireddy‡
 a,
Julia
Giselbrecht‡
b,
Boris
Strehlow
a,
Christopher
Janich
a,
Catharina
Husteden
b,
Annette
Meister
c,
Harald
Loppnow
d,
Daniel
Sedding
d,
Frank
Erdmann
e,
Gerd
Hause
f,
Gerald
Brezesinski
g,
Thomas
Groth
h,
Andreas
Langner
b,
Udo
Bakowsky
*a and
Christian
Wölk
*b
a,
Julia
Giselbrecht‡
b,
Boris
Strehlow
a,
Christopher
Janich
a,
Catharina
Husteden
b,
Annette
Meister
c,
Harald
Loppnow
d,
Daniel
Sedding
d,
Frank
Erdmann
e,
Gerd
Hause
f,
Gerald
Brezesinski
g,
Thomas
Groth
h,
Andreas
Langner
b,
Udo
Bakowsky
*a and
Christian
Wölk
*b
aDepartment of Pharmaceutics and Biopharmaceutics, University of Marburg, Robert-Koch-Str. 4, 35037, Marburg, Germany. E-mail: UBakowsky@aol.com
bMartin Luther University Halle-Wittenberg, Institute of Pharmacy, Department of Medicinal Chemistry, Wolfgang-Langenbeck-Str. 4, 06120 Halle (Saale), Germany. E-mail: christian.woelk@pharmazie.uni-halle.de
cMartin Luther University Halle-Wittenberg, Institute of Biochemistry and Biotechnology, Kurt-Mothes-Str. 3a, 06120 Halle, Germany
dMedical Faculty of Martin Luther University Halle-Wittenberg, Internal Medicine III, Ernst-Grube-Str. 40, 06120 Halle (Saale), Germany
eMartin Luther University Halle-Wittenberg, Institute of Pharmacy, Department of Pharmacology, Wolfgang-Langenbeck-Str. 4, 06120 Halle (Saale), Germany
fMartin Luther University Halle-Wittenberg, Biocenter, Weinbergweg 22, 06120 Halle, Germany
gMax Planck Institute of Colloids and Interfaces, Science Park Potsdam-Golm, Am Mühlenberg 1, 14476 Potsdam, Germany
hMartin Luther University Halle-Wittenberg, Institute of Pharmacy, Department Biomedical Materials, Heinrich-Damerow-Str. 4, 06120 Halle (Saale), Germany
First published on 23rd October 2019
Abstract
Non-viral gene delivery in its current form is largely dependent upon the ability of a delivery vehicle to protect its cargo in the extracellular environment and release it efficiently inside the target cell. Also a simple delivery system is required to simplify a GMP conform production if a marketing authorization is striven for. This work addresses these problems. We have developed a synthetic polycationic peptide-mimicking amphiphile, namely DiTT4, which shows efficient transfection rates and good biocompatibility without the use of a co-lipid in the formulation. The lipid–nucleic acid complex (lipoplex) was characterized at the structural (electron microscopy), physical (laser Doppler velocimetry and atomic force microscopy) and molecular levels (X-ray scattering). Stability studies of the lipoplexes in the presence of serum and heparin indicated a stable formation capable of protecting the cargo against the extracellular milieu. Hemocompatibility studies (hemolysis, complement activation and erythrocyte aggregation) demonstrated the biocompatibility of the formulation for systemic administration. The transfection efficiency was assessed in vitro using the GFP assay and confocal laser scanning microscopy studies. With the chorioallantoic membrane model, an animal replacement model according to the 3R strategy (replacement, refinement, and reduction), initial in vivo experiments were performed which demonstrate fast and efficient transfection in complex tissues and excellent biocompatibility.
Introduction
The therapeutic efficacy of non-viral gene delivery vehicles has remained largely unchanged over the past decade. Several delivery vehicles have been structurally and chemically modified in an attempt to mimic and reach the efficiency and speed with which viral delivery vehicles deliver and integrate their genetic material into the host cell. The main drawback of the viral vectors is their excessive cost of production, safety concerns and immunogenic responses towards them, which have restricted their clinical use to only a few cases.1 Non-viral vectors offer several benefits with respect to their ease of production, low manufacturing costs and the ability to easily optimize them depending upon individual needs and the area of application.2 In order to achieve the desired therapeutic effect, non-viral delivery vehicles should retain their integrity within the extracellular environment, enter the target cells efficiently and subsequently release the cargo at the place of action (nucleus or cytosol). Among others, liposomes, nanoparticles, polymers or a combination of these are one of the most frequently used delivery vehicles for gene delivery.3–5 Liposomes are the most preferred, and are routinely used as drug delivery vehicles due to their compatibility with biological membranes and their ability to deliver a wide range of biomolecules.6 A variety of lipids could be incorporated into the formulations in order to optimize the liposomes to suit the target. With advancements in the human genome project, the number of therapeutically relevant targets has been steadily increasing thereby providing ample opportunity for gene therapy which has witnessed an ever-growing interest in the recent past.7The main limitations of non-viral gene delivery vehicles compared to viral ones are the degradation of the cargo and premature release prior to entering the cells,8,9 and a lack of specific mechanisms to overcome cellular barriers, such as those present in viral vectors,10,11 and, therewith, lower gene expression rates. Nevertheless, non-specific physical mechanisms exist, which allow non-viral vectors to overcome cellular barriers which are tunable to increase the efficiency.12–14
The quest for a lipid based delivery vehicle which consists of only one lipid has led us to the development of the peptide-mimicking amphiphile DiTT4. DNA-loaded nanoparticles (lipoplexes) solely prepared from this lipid (without a typically used co-lipid) were extensively characterized in terms of their size, structure and morphology (CryoTEM and atomic force microscopy), surface charge/zeta potential (laser Doppler velocimetry), thermotropic phase behavior (differential scanning calorimetry) and supramolecular structure (small angle X-ray scattering). The stability of the formulation was determined in the presence of serum and a competing polyanion with high charge density, such as heparin, to demonstrate an appropriate stability for systemic administration. Hemolysis, erythrocyte aggregation, and complement system activation tests were performed to evaluate the effect of the formulation on blood. To determine the efficiency of transgene expression, transfection studies were carried out in tumor cell lines. DiTT4 lipoplexes were fluorescently labeled and their internalization was tracked inside fluorescently labeled cells at regular time intervals using confocal microscopy. The chorioallantoic membrane of the chicken embryo, an animal replacement model according to the 3R strategy (replacement, refinement, and reduction),15 was used as an in vivo model to demonstrate the high transfection efficiency and speed as well as the absence of acute toxic effects. DiTT4 lipoplexes have exhibited excellent transfection efficiency in a complex tissue together with a biocompatible profile thereby making it a prospective vehicle for gene delivery.
Materials and methods
Materials
If not stated otherwise, chemicals were purchased from Sigma-Aldrich. Green fluorescence protein expressing plasmid pCMV-GFP and luciferase expressing plasmid pCMV-luc were purchased from Plasmid Factory (Bielefeld, Germany). Enhanced green fluorescent protein expressing plasmid peGFP-C2 obtained from Clontech (Saint-Germain-en-Laye, France) was transformed in competent cells (E. coli DH5α, Invitrogen, Darmstadt, Germany). The plasmid DNA was isolated using an EndoFree Plasmid Mega Kit (Qiagen, Hilden, Germany). Pathway inhibitors dynasore and filipin III were purchased from VWR Chemicals (Darmstadt, Germany). The solvents used in this study were all of analytical grade. Filter sterilized (0.22 μm syringe filter) MilliQ water (Millipore, Billerica, USA) was used for all the experiments. The synthesis of DiTT4 was described previously.16 ATTORho6G-N-hydroxysuccinimidester was purchased from Atto-TEC (Siegen, Germany). YoYo®-1 Iodide (1 mM in DMSO) and wheat germ agglutinin (WGA)–Alexa Fluor® 633 conjugate were obtained from Thermo Fisher Scientific. DOPE and Rhod-DOPE (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (ammonium salt; λmaxabs 560 nm/λmaxem 583 nm) were purchased from Avanti Polar Lipids.Labelling of DiTT4 with ATTORho6G
DiTT4 (1.51 μmol) was dissolved in dry dichloromethane (500 μL) and the fluorescence label ATTORho6G-N-hydroxysuccinimidester (1.51 μmol, dissolved in 200 μL CH2Cl2) and triethylamine (2 μmol) were added to it. The mixture was stirred for 12 h at room temperature under an argon atmosphere. Afterwards, the solvent was evaporated under reduced pressure and the labeled lipid DiTT4-AttoRho6G was purified by preparative layer chromatography using glass plates (20 × 20 cm) with silica gel 60, 0.5 mm thickness (Merck, Darmstadt, Germany) and chloroform/methanol/ammonia (65/35/5, v/v/v), as the eluent. By dissolving and centrifugation of the silica gel, the purified lipid was obtained in several steps.Lipid formulation and lipoplex preparation
Lipid formulations (micelles or liposomes) were prepared by the film hydration procedure. DiTT4, DiTT4-AttoRho6G and Rhod-DOPE were separately dissolved in chloroform/methanol (8/2, v/v). In general, DiTT4 was used alone or mixed to an appropriate molar ratio with a fluorescence labeled lipid. The solvent was evaporated for 1 h at 200 mbar and a further 3 h at 10 mbar. After the formation of dry lipid films, a sterile filtered 10 mM HEPES buffer solution (pH 7.3) was added to give the final concentration. Afterwards, the lipid dispersions were incubated at 50 °C while shaking (1400 rpm) for 30 min (Eppendorf Thermomixer 5436) followed by sonication at 37 kHz for 3 min at 25 °C.For the lipoplex preparation plasmid DNA was complexed at different N/P ratios (ratio of the primary amines in the cationic lipid to the phosphate groups of the DNA) together with a DiTT4 lipid formulation in HEPES buffer (pH 7.3) by gentle pipetting and subsequent incubation for 15 min at 25 °C (1 step mixing procedure by adding DNA to the lipid).
Zeta-potential measurements
The zeta potential measurements were performed by laser Doppler velocimetry (lDv) using a Zetasizer Nano ZS ZEN3600 (Malvern Instruments, Worcestershire, UK) and a clear disposable folded capillary cell (DTS1060, Malvern Instruments). Three independent measurements involving 30 runs with a voltage of 60 V were performed at 25 °C. For the calculations, the viscosity (η = 0.8872 mPa s), dielectric constant (ε = 78.5 F m−1) and refractive index (n = 1.33) of water were assumed. The mobility μ of the diffusing aggregates was converted into the ζ potential using the Smoluchowski relationship ζ = μ η/ε (Zetasizer Software 6.34).Differential scanning calorimetry (DSC)
DiTT4 dispersed in buffer (1 mg mL−1) or lipoplexes with a fixed amount of DNA at 1 mg mL−1 (amount of lipid results from the different N/P ratios) was used. The samples were then degassed for 20 min by slow stirring under vacuum. The DSC measurements were performed on a MicroCal VP-DSC calorimeter (MicroCal Inc., Northampton, MA). The scanned temperature range was between 2 and 95 °C, the heating rate was 60 K h−1, and each heating and cooling scan was repeated to confirm reproducibility. The first scan was discarded for experiments without DNA. For experiments with DNA the first scan was evaluated because here the information of DNA melting appears. The reference cell was filled with pure buffer. The buffer–buffer baseline was subtracted from the thermograms of the samples, and the DSC scans were evaluated using MicroCal Origin 8.0 software.Small angle X-ray scattering (SAXS)
Lipid dispersions were prepared as described above. Lipid concentrations of 20 wt% (200 mg mL−1) in buffer were used. Samples were transferred into open glass capillaries (inner diameter: 1.5 mm, WJM-GLAS, Berlin, Germany), which were then sealed on both sides. After three days storage at 4 °C SAXS was performed using a NanoStar spectrometer (high-flux configuration) with a sealed tube, crossed Göbel mirrors and a HiStar multiwire proportional counter (Bruker). The incoming beam had a wavelength of 0.154 nm, and the exposure time was 2 h. The temperature was fixed to the appropriate temperature during the measurements. Dry rat tail collagen was used for calibration.Transmission electron microscopy (TEM)
For TEM, lipoplexes with a concentration of 0.05 mg mL−1 were used. The negatively stained samples were prepared by spreading the dispersion (5 μL) onto a Cu-grid coated with a Formvar film (Plano, Wetzlar, Germany). After 1 min, the excess liquid was removed by blotting with filter paper and 1% aq. uranyl acetate (5 μL) was placed onto the grid and drained off after 1 min. The dried specimens were examined using an EM 900 transmission electron microscope (Carl Zeiss Microscopy GmbH, Oberkochen, Germany). Micrographs were acquired using a SSCCD SM-1k-120 camera (TRS, Moorenweis, Germany).Cryo-transmission electron microscopy (CryoTEM)
Lipoplex dispersions of 2 mg mL−1 were used for CryoTEM investigations. Vitrified specimens for CryoTEM were prepared using a blotting procedure, performed in a chamber with controlled temperature and humidity using an EM GP grid plunger (Leica, Wetzlar, Germany). A drop of the sample solution was placed onto an EM-grid coated with a holey carbon film (C-flat, Protochips Inc., Raleigh, NC). Excess solution was then removed by blotting with filter paper to leave a thin film of the solution spanning the holes of the carbon film on the EM-grid. Vitrification of the thin film was achieved by rapid plunging of the grid into liquid ethane held just above its freezing point. The vitrified specimen was kept below 108 K during storage, transfer to the microscope and investigation. The specimens were examined under a Libra 120 Plus transmission electron microscope (Carl Zeiss Microscopy GmbH), operating at 120 kV. The microscope was equipped with a Gatan 626 cryotransfer system. Images were acquired using a BM-2k-120 dual speed on-axis SSCCD camera (TRS).Atomic force microscopy (AFM)
AFM was used to determine the surface morphology of the lipoplexes and to complement the particle size analysis performed by PCS. 10 μL of the sample was pipetted onto silica wafers which were glued onto glass slides. The samples were allowed to settle onto the wafers before being gently washed with water followed by nitrogen gas. AFM was performed under ambient conditions using a Nanowizard® 3 Nanoscience AFM (JPK Instruments, Berlin, Germany) in intermittent contact mode with an aluminum coated silicon nitride probe (HQ:NSC14/Al BS; Mikromasch, Tallinn, Estonia) at scan rates between 0.5 and 1 Hz as described previously.17Gel retardation assay
For the gel retardation assay 20 μL of a lipoplex dispersion (containing 0.4 μg DNA) were used and briefly mixed with 4 μL of blue/orange 6× loading dye (G190A). All samples were electrophoresed in a 1% agarose gel containing 0.308 μg mL−1 EtBr in 1% TAE buffer (pH 8) for 1 h at 90 V. Additionally, a 1 kb DNA ladder (G571A) (Promega, Madison, WI, USA) was used. The fluorescent pDNA bands were visualized and photographed with a UVP UVsolotouch (Jena Analytics, Germany).Heparin destabilization assay
A heparin ammonium salt from porcine intestinal mucosa was used (termed heparin in the text). 40 μL of lipoplex solutions (2.83 μg pDNA, N/P 4) were prepared as described above. 5 μL of lipoplexes were added to different amounts of heparin which was dissolved in HEPES buffer (10 mM, pH 7.3). The volume was adjusted to 10 μL with HEPES buffer (10 mM, pH 7.3). The samples were incubated for 1 h at room temperature (25 °C). The mixtures were analyzed with the gel retardation assay.Cell culture
A549 (human alveolar carcinoma) and HeLa (human cervical cancer) cells procured from the ATCC were cultured in DMEM (Capricorn Scientific) supplemented with 4.5 mg mL−1 glucose and 10% FBS. HUVEC cells purchased from Promega were cultured in Endothelial Cell Growth Medium (Promega, Madison, WI, USA) complemented with the Growth Medium Supplement Mix (Promega, Madison, WI, USA). The culture conditions were 37 °C and 5% CO2. The cells were grown as monolayers and were passaged upon reaching 80–90% confluency. For experiments, cells within passage numbers 10–30 (A549, HeLa) and 3–8 (HUVEC) were used.eGFP assay
Cells were seeded onto 96-well plates at a density of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–11000 cells per well 24 h before transfection. The cells were washed once with PBS buffer (pH 7.3) containing Ca2+ and Mg2+. Lipoplexes were added to the cells (containing 0.1 μg plasmid DNA per well) and made up with DMEM containing 10% FBS to a final volume of 100 μL per well. After 24 h the cells were washed twice with PBS before further analysis. Afterwards, eGFP fluorescence was measured using a BMG 10 filter (λabs = 485 nm, λem = 520 nm) with a fixed detector gain. The transfection efficiency was background-fluorescence-corrected and presented as relative fluorescence units (RFU), normalized to the cell count.
000–11000 cells per well 24 h before transfection. The cells were washed once with PBS buffer (pH 7.3) containing Ca2+ and Mg2+. Lipoplexes were added to the cells (containing 0.1 μg plasmid DNA per well) and made up with DMEM containing 10% FBS to a final volume of 100 μL per well. After 24 h the cells were washed twice with PBS before further analysis. Afterwards, eGFP fluorescence was measured using a BMG 10 filter (λabs = 485 nm, λem = 520 nm) with a fixed detector gain. The transfection efficiency was background-fluorescence-corrected and presented as relative fluorescence units (RFU), normalized to the cell count.
Luciferase and BCA assays
Cells were seeded onto 96-well plates at a density of 10000–11000 cells per well 24 h before transfection. The cells were washed once with PBS buffer (pH 7.3) containing Ca2+ and Mg2+. Lipoplexes were added to the cells (containing 0.1 μg pCMV-luc per well) and made up with DMEM containing 10% FBS to a final volume of 100 μL per well. After 24 h the cells were washed twice with PBS and lysed using cell culture lysis buffer (Promega, Mannheim, Germany) and the lysates were transferred onto an opaque white bottomed plate and a transparent plate for the luciferase and BCA assay, respectively. The luciferase activity was measured immediately upon injecting the luciferase assay mixture using a luminescence plate reader (FLUOStar Optima, BMG Labtech, Offenburg, Germany). The protein amount was also quantified using the same device at an absorbance wavelength of 540 nm. The results were expressed as relative luciferase units (RLU) per mg protein.
Flow cytometry
For the transfection experiments the HUVEC cells were seeded in a 6-well plate with a density of 1.5 × 105 cells per mL and 24 h after seeding the lipoplexes were added after complete complex formation with an amount of 1.6 μg pDNA per well. After an incubation of 24 h at 37 °C the GFP-expression was measured by FACS analysis. Viability was determined via propidium iodide (PI) staining with a concentration of 50 μg mL−1. The relative fluorescence units were measured with a BD Accuri™ C6 Plus flow cytometer (BD Bioscience, USA). Trypsinate cells were centrifuged (220g, 5 min, RT) and resuspended in PBS. Per measurement, 10000 cells were analyzed in each sample (GFP: λex: 488 nm, λem: 510 nm; PI: λex: 488 nm, λem: 617 nm). Single cells were gated by size (FSC-H) and granularity (SSC) with the device associated analysis software. The calculated single cell population was gated to detect GFP and PI expressing cells to calculate the relative number of transfected cells and dead cells.
Confocal laser scanning microscopy
The experiments were performed with a confocal laser scanning microscope (Zeiss LSM 710, Carl Zeiss, Jena) using an inverse microscope (AxioObserver.Z1) with a fast z-scanning stage and 40× and 63× Plan APOCHROMAT objectives. Zeiss Software ZEN2008 was used for generation of images and controlling the microscope. Zeiss Software ZEN2011 was used for data processing. A549 cells were seeded at 4 × 105 cells per well in a 4-well CELLview™ cell culture dish 24 h prior to transfection to 70–80% confluence. Afterwards the cells were transfected with tagged lipoplexes. The transfection was performed in a manner similar to the GFP assay with the exception that a 5-fold amount of all substances was used. For nucleus staining the cells were permeabilized using 0.1% (v/v) Triton X-100 (Sigma-Aldrich) for 10 min before the nucleus is stained with YoYo-1® followed by washing with PBS (2 × 0.5 mL). For visualizing the cell membrane, the cells were treated with Wheat Germ Agglutinin Alexa Fluor® 633 (λmaxex = 632 nm, λmaxem = 647 nm) at a concentration of 2.5 μg mL−1 for 3 min followed by washing with PBS (2 × 0.5 mL). The cells were fixed with 0.5 mL of 2% formaldehyde solution and incubated at room temperature in the dark for 15 min. The cells were washed twice again with 0.5 mL PBS (pH 7.3). A drop of fixation solution (Aquatex®, Merck, Schwalbach, Germany) was placed on the cells. Lipid-labeled lipoplexes where prepared from liposomes consisting of DiTT4/DiTT4-AttoRho6G (λmaxabs 533 nm/λmaxem 577 nm) 1/0.002 (n/n).Cell viability assay
The cytotoxicity assay was performed parallel to the transfection experiments using the same procedure. 100 μL of a 2% AlamarBlue®/PBS solution (Invitrogen) was added to each well. The plates were incubated for 1 h at 37 °C. The fluorescence signal was measured using a BMG 10 filter (λex = 544 nm, λem = 590 nm) with a fixed detector gain.Inhibition of endocytotic pathways
The experiments were performed similar to transfection experiments with the difference that before addition of the lipoplexes, the cells were incubated for 30 min with either Dynasore (80 μM in DMEM prepared with a 7.5 mM stock solution in DMSO) or a filipin complex from Streptomyces filipensis (5 μg mL−1 in DMEM prepared with a 0.4 mg mL−1 stock solution in DMSO). The control experiment was performed with an additional incubation period of 30 min with DMEM. Before incubation with the pathway inhibitors, the cells were carefully washed with PBS (pH 7.3) to remove serum which can have inactivating effects on the inhibitors. The control was incubated with medium during the 30 min incubation period.Chorioallantoic membrane model
Certified pathogen free fertilized chicken eggs were obtained from VALO BioMedia (Osterholz-Scharmbeck, Germany) and incubated at 37 °C under a humidified atmosphere (>60% RH) inside a hatching incubator (Ehret KMB 6, Dipl. Ing. W. Ehret, Emmendingen, Germany). Chorioallantoic membrane model (CAM) experiments were performed as previously described.18 On egg development day (EDD) 3, a ∅ 3 mm hole was drilled into the basal part of the shell and approximately 3 mL of albumin was drawn out of the egg. The apical part of the shell was cut open (∅ 30 mm) to expose the CAM. The bottom end was sealed using cellophane tape and the apical part using a paraffin film to prevent contamination. The eggs were further incubated. On EDD 10, 100 μL of lipoplexes (with 0.5 μg eGFP plasmid, either DiTT4 formulation or Lipofectamine 2000) were injected either topically or into the mesoderm and the eggs were returned to the incubator. The CAM was cut out after 4 h and 24 h intervals using a scalpel, washed with isotonic NaCl and analyzed under a CLSM (LSM 510; Carl Zeiss) regarding GFP (λmaxabs 488 nm/λmaxem 510 nm) and lissamine rhodamine B (λmaxabs 560 nm/λmaxem 583 nm) fluorescence. Similar detector gain was used throughout all the acquisitions. Every transfection experiment was repeated once. For the determination of the transfected area the Fiji software was used (for details see the ESI†).19 For screening of acute toxicity (embryo death) and vascular damage (hemorrhagic or thrombotic effects) on EDD 10, 100 μL of lipoplexes (containing 1 μg eGFP plasmid) were injected intravenously into 6 eggs and the eggs were returned to the incubator. At regular intervals the eggs were screened for hemorrhagic, thrombotic or necrotic events at the blood vessels and effects on the development of embryo until EDD 14.Hemolysis and erythrocyte aggregation
Blood compatibility tests of the lipoplexes were performed according to the guidelines of ISO 10993-4. For this purpose, human erythrocytes were isolated from “buffy coats” of healthy donors, obtained from the “Transfusion Medicine” of the Medical Faculty of the Martin Luther University Halle-Wittenberg. The voluntary donors gave written informed consent. For the isolation, Biocoll solution (25 mL) was overlaid with a buffy coat, mixed with heparin-containing PBS (30 + 30 mL) and centrifuged at 400g for 30 min. The supernatant was removed and the erythrocyte pellet was washed thrice with PBS (30 mL; 2660g; 7 min). The erythrocytes were then diluted with PBS to obtain a concentration of 8 × 109 cells per mL. The lipoplex samples (0.5 mL) were diluted with the erythrocyte solution (0.5 mL) and incubated for 1 h at 37 °C. Subsequently, the samples were centrifuged at 2300g for 5 min and 100 μL of each sample or control replicate were transferred to a well of a 96-well plate. The absorbance of the samples (Asample) was then measured at 540 nm with an ELISA reader (Spectrafluor, Tecan, Crailsheim, Germany). Various concentrations of lipoplexes were tested. As controls PBS and 1% Triton X-100® were used. The absorbance values of Triton X-100® were considered as 100% hemolysis (Apositive control). Untreated erythrocytes were used as a negative control (Anegative control). The samples and controls were tested in triplicate and the experiment repeated once. The percentage of hemoglobin release was calculated according to the following equation: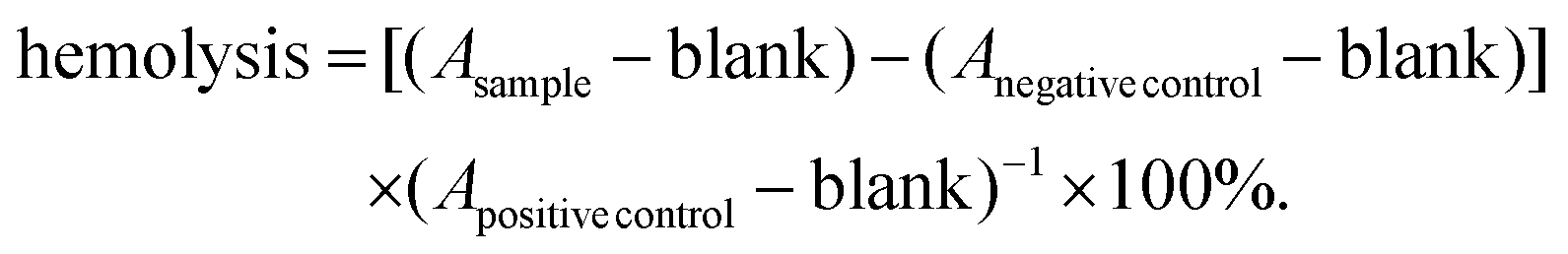
The erythrocyte aggregation was evaluated by microscopy. For this purpose, different amounts of lipoplexes were diluted in PBS (150 μL) and incubated with 150 μL of an erythrocyte suspension (9.5 × 106 erythrocytes per mL) in a 24-well plate for 2 h at 37 °C. The erythrocyte aggregation was analyzed by microscopy. The samples were tested in duplicate and the experiment repeated once.
Complement activation assay
The complement hemolytic activity was tested using the AP50 and CH50 test kits which were purchased from Haemoscan (Groningen, Netherland) using the manufacturer's protocol. According to the manufacturer, both kits are suited to evaluate the hemocompatibility of biomaterials and medical devices according to the international standard ISO 10993-4 after blood, plasma, or serum contact. The alternative pathway method (AP50) is based on the lysis of sensibilized rabbit erythrocytes in the presence of Mg2+ if the alternative pathway is activated and the classical pathway method (CH50) is based on the lysis of sensibilized sheep erythrocytes in the presence of Ca2+ and Mg2+ if the classical complement pathway is activated. To prepare the AP50 or CH50 erythrocyte suspension, 100 μL of the erythrocyte suspension (purchased with the AP50 or CH50 test kits) was added slowly to 0.5 mL of dilution buffer. The dispersion was centrifuged for 10 min at 400g. The cell pellet was collected by aspirating the supernatant. Multiple washing steps were required to obtain a colorless supernatant. The pellet was suspended in an appropriate amount of dilution buffer (adjusted to the desired absorption values for 100% hemolysis: AP50 A415 0.4–0.5; CH50 A415 0.8–0.9). The dilution buffer was used as a negative control (Anegative) while the lysis fluid served as a positive control (Apositive). The samples were prepared by adding DiTT4 lipoplexes (N/P 4) in HEPES buffer (containing 2 μg DNA and 8.2 μg DiTT4) to human serum (1:9 v:v; Capricorn Scientific, Germany). The samples were incubated for 10 min at 37 °C on an orbital shaker (300 rpm). After centrifugation at 30000g for 10 min to sediment the lipoplexes, the supernatant was used to prepare different serum dilutions with dilution buffer (Asample). Two references provided by the manufacturer were used: reference 1 was plasma with a normal complement and reference 2 was with a low complement. References were also used in a dilution range from 4 to 64 times. To the 50 μL serum dilutions and reference dilutions as well as negative and positive controls 50 μL of the prepared AP50 or CH50 erythrocyte suspension was added. After incubation for 30 min at 37 °C, 100 μL of stop solution (Life Technologies, NY, USA) was added. All samples were centrifuged at 400g for 10 min. Supernatants (100 μL) were collected and transferred to 96-well plates (Grainer Bio One, Frickenhausen, Germany) and the absorption of the samples was determined at 415 nm with a Polarstar Omega (BMG Labtech, Ortenberg, Germany). AP50 and CH50 values were obtained by plotting the % lysis values against the dilution factor while % lysis was calculated using the formula: %Lysis = (Asample − Anegative)/(Apositive − Anegative).
Results and discussion
Structural investigations and DNA complexation
DiTT4 is a peptide-mimicking amphiphile developed for transfection. The synthesis, pH dependent self-association behavior in the absence of DNA, and preliminary investigation regarding DNA complexation and in vitro transfection have already been published.16 It was demonstrated that DiTT4 exhibits a pH dependent change of the self-assembling behavior: at pH 7.3 it forms liposomes, while at pH 5, the value reached in endosomes, rod-like micelles are the preferred lipid associates (Fig. 1). To which extent these structures are relevant in the presence of DNA is still an open question and part of this section.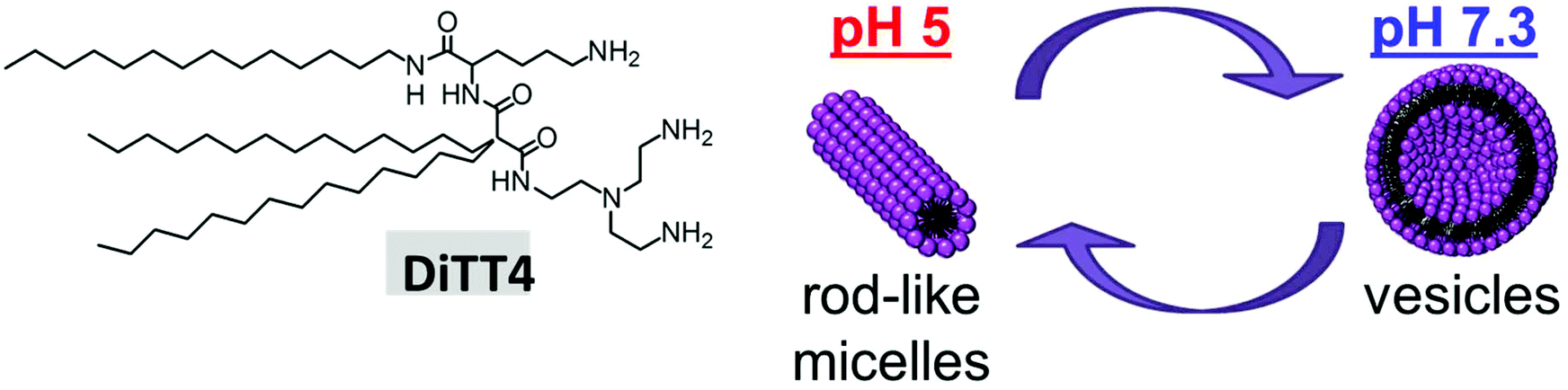 | ||
| Fig. 1 Structure of DiTT4. Scheme demonstrating the pH dependent self-assembling of the lipid in the absence of DNA. | ||
DiTT4 efficiently forms complexes with plasmid DNA and total DNA encapsulation occurs at an N/P ratio above 2.5.16 In Fig. 2A the SAXS patterns of the lipoplex formulation with N/P 4 at pH 7.3 and pH 5 are shown. Both show diffraction peaks which can be assigned to a lamellar mesophase. Two Bragg reflexes in the q ratio of 1:2 appear. The first reflex results in a lamellar space distance of d = 2π/q = 58.7 Å for both pH values. This means that the pH induces no changes in the lamellar repeat distance. The d value is 5 Å higher than the values reported for the DNA free systems,16 but DNA strands have a diameter of around 20 Å.20 This suggests entrapment of DNA between the lipid bilayers, though additional evidence is required to support this theory. The TEM image with negative staining showed the typical structure of a lamellar lipoplex (Fig. 2B, white arrow),21–23 which was first described by Rädler et al.24 These lipoplexes, also termed Lcα liquid crystalline structures, are supra-molecular structures where DNA is entrapped between lipid bilayers in a sandwich-like manner (Fig. 2E). A small increase of only 5 Å compared to the DNA free formulation determined by SAXS means that the DNA is compacted in these lipoplexes (DNA is tightly bound and penetrates partially in the head group part of the lipid bilayer) which was also confirmed with the ethidium bromide exclusion method.16
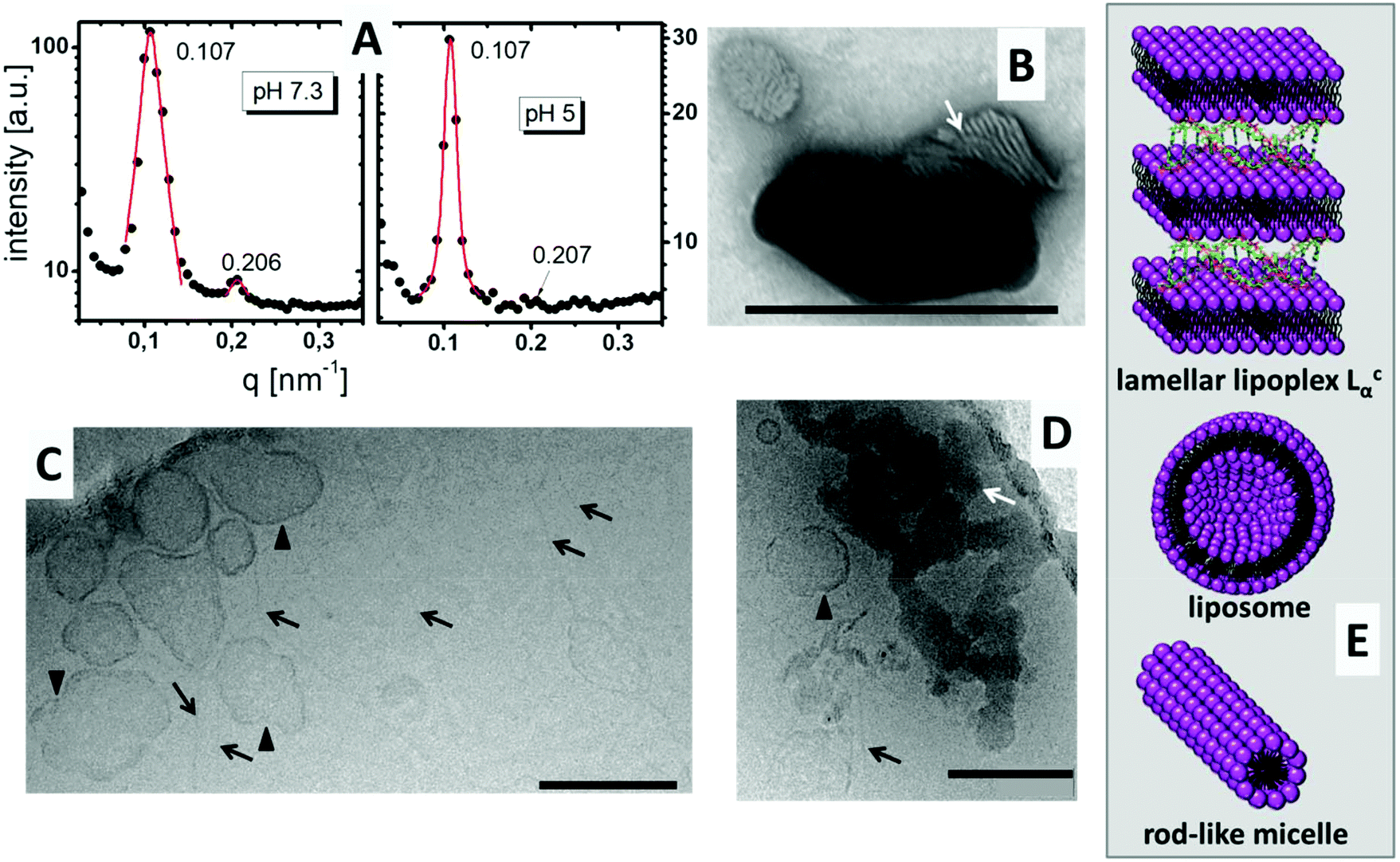 | ||
| Fig. 2 (A) Small Angle X-ray scattering (SAXS) patterns of DiTT4 lipoplexes (N/P 4) at pH 7.3 and pH 5 with a 10% (wt) dispersion in HEPES buffer (0.1 M, pH 7.3) and acetate buffer (0.1 M, pH 5), respectively. (B) TEM image of negatively stained DiTT4 lipoplexes (N/P 4) in HEPES buffer (10 mM, pH 7.3). White arrow indicates the lamellar structure. The scale bar indicates 200 nm. (C/D) CryoTEM images of DiTT4 lipoplexes at N/P 4 in a 2 mg mL−1 dispersion in acetate buffer at pH 5 (10 mM). The black arrows indicate fibre-like structures, the black triangles indicate vesicular structures and the white arrows indicate lipoplexes with a lamellar substructure. The scale bars indicate 200 nm. (E) Schematic illustrations of the structures which appear in a DiTT4 lipoplex formulation (N/P 4) at pH 5. | ||
Fig. 2C and D show CryoTEM images of DiTT4 lipoplex formulations (N/P 4) at pH 5. Comparable to the DNA free sample (see Wölk et al.)16 fibers were present in the sample (Fig. 2C and D, black arrows). We assume that these rod-like micelles contain no DNA. Also liposomes are present, which is not typical of the lipid formulation without DNA.16 The irregular shape of these unilamellar liposomes (Fig. 2C and D, black triangles) could indicate a possible coating with DNA which stabilizes the liposomes and prevents the formation of the rod-like micelles; nevertheless this is only a thesis which is hard to prove. For instance, gold labeling of DNA results in fast sample melting and is not suitable for the used CryoTEM setup while the liposomes and the fibers cannot be observed in the TEM image with negative staining, obviously due to destruction during the preparation procedure (data not shown). Additionally, the CryoTEM micrographs show dense structures with high contrast at pH 5 (Fig. 2D, white arrow). These structures were assigned to the lamellar lipoplexes, while the lamellar structure cannot be visualized as in Fig. 2B due to the absence of a contrast reagent (uranyl acetate preferentially binds to the negatively charged DNA due to the positive charge of the uranyl ion). A CryoTEM image of the formulation at pH 7.3 was not possible due to fast sample melting induced by the dense lipoplex structures. The structural information concerning the lipoplex formulation at pH 5 obtained by TEM are summarized in Fig. 2E, while we assumed that DNA is mostly encapsulated in Lcα lipoplexes, rarely attached to liposomes and not associated with rod-like micelles.
The lipoplex structures found in the CryoTEM experiment (Fig. 2D, white arrow) could indicate a possible aggregation of the particles in the DiTT4 (N/P 4) lipoplex formulation. The PCS results show a z-average radius of 287 nm with a polydispersity index of 0.249.16 Additional AFM experiments were performed (Fig. 3A) which support these data. AFM analysis revealed that the particle size of supra-molecular structures found in the DiTT4 (N/P 4) lipoplex formulation were significant below 1 μm and therefore suitable for systemic administration from the size point of view. The micrograph shows a relatively homogeneous distribution of the nano-scaled lipoplexes with a spherical morphology.
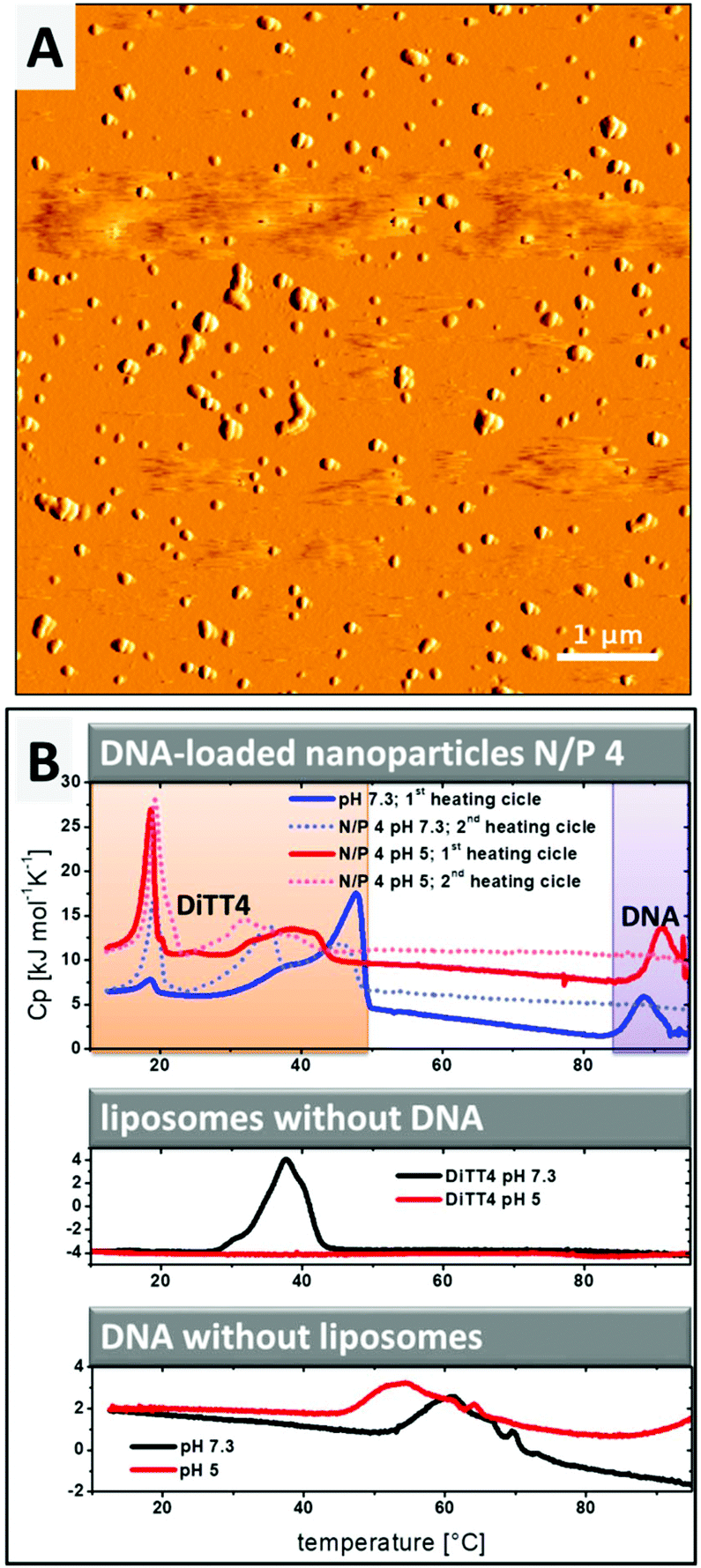 | ||
| Fig. 3 (A) AFM images showing the morphology of DiTT4 lipoplexes (N/P 4) in HEPES buffer (10 mM, pH 7.3). The image is presented in the amplitude mode. (B) DSC heating curves of DiTT4 lipoplexes (N/P 4) in comparison with DNA and DiTT4 dispersion. HEPES buffer (10 mM, pH 7.3) and acetate buffer (10 mM, pH 5) were used as the solvent. The concentration of the lipid in samples without DNA was 1 mg mL−1. For the DNA control and lipoplexes a DNA concentration of 1 mg mL−1 was used. For the experiments with lipoplexes the first and second heating scans are shown. For experiments with the lipid dispersion only the second scan is shown. | ||
Investigation of the stability of DNA complexation
Information about the strength of interaction between DiTT4 and DNA and the encapsulation efficiency of the nucleic acids in the lipid nanoparticles was obtained by DSC experiments. The rod-like micelles at pH 5 show no phase transition in the investigated temperature range, while in the case of liposomes at pH 7.3 a transition with the maximum at Tm = 39.8 °C was observed (Fig. 3B, diagram in the middle; compare red line and black line). Uncomplexed DNA (without a lipid in the sample) at pH 7.3 shows a broad exothermic peak between 50 °C and 70 °C which results from the melting, implying the denaturation of the double strand to single strands (Fig. 3B, lower diagram, black line). This melting process depends on the guanine to adenine ratio and the size of the DNA,25,26 as well as the ionic strength and the pH.27–29 At pH 5 (Fig. 3B, lower diagram, red line) the melting point shifts to lower temperatures compared to pH 7.3. In the second scan no melting peak is observed because the DNA single strands did not anneal in the timescale of the DSC experiment (2nd scan of DNA samples without DNA is not shown in Fig. 3B). If the complexation of DNA with polycationic synthetic delivery vehicles results in a compaction of the DNA, what we assume from the SAXS and ethidium bromide experiments, the melting point is shifted to higher temperatures.30–32 This effect is observed for DiTT4 lipoplexes (N/P 4) at pH 5 and pH 7.3 (Fig. 3B, upper diagram). A peak at around 90 °C occurs which does not appear in the second scan. Because the melting peak of uncomplexed DNA vanishes, this peak can be assigned to the melting of compacted DNA (Fig. 3B, upper diagram, peaks labeled with a purple box). The lipoplex formulations show additional peaks between 10 °C and 50 °C (Fig. 3B, upper diagram, peaks labeled with the orange box). Considering the fact that at N/P 4 an excess of DiTT4 occurs, these peaks can be assigned to phase transitions of DiTT4 in different self-assembled associates, for instance, liposomes, lipoplexes, or DNA coated liposomes. This observation fits perfectly with the various structures found in the CryoTEM images (Fig. 2C and D). Furthermore, the data showed a lipid exchange between those structures, due to the change in peak intensities between the first and second scans. Additionally, the curves of the DSC experiment with DiTT4 lipoplexes (N/P 4) at pH 5 support the presence of lipoplexes and liposomes in this formulation, while DiTT4 alone only forms rod-like micelles which exhibit no phase transition in the investigated temperature range.Summarizing, DiTT4 forms lamellar lipoplexes with comparable structural parameters at pH 5 and 7.3 (SAXS experiments) and a high degree of compaction (DSC experiments) which is also not affected by the pH shift. Nevertheless, at pH 5 multiple self-assembled structures are present (fibers, liposomes, and lipoplexes).
The encapsulation of DNA by DiTT4 lipid nanoparticles depends on the ionic strength. If the complex formation is performed in DMEM, which is a typical cell culture medium containing different salts, amino acids and glucose, the complex formation between lipid and DNA is very inefficient. The zeta potential measurements and the gel-retention assay showed that even at an N/P ratio of 6 a high amount of DNA is still uncomplexed, which is indicated by a high negative zeta potential and electrophoretic signals assigned to free DNA (Fig. 4A). In 10 mM HEPES buffer, pH 7.3, a medium also used for the structural investigation described above, the complex formation was more effective. At N/P ratios between 2 and 2.5, total complexation and compaction of DNA occurred due to the positive shift of the zeta potential caused by the excess of the lipid. Hence, no signal of free DNA appeared in the gel retention assay (Fig. 4A). At N/P 2.5, even the fluorescence in the starting lane disappeared because of the exclusion of ethidium bromide from the lipoplexes due to the high degree of compaction of encapsulated DNA inside this lipid nanoparticles, an observation which fits with previous studies.16 Based on these results, HEPES (pH 7.3) was chosen as a buffer of choice for the biological experiments using DiTT4 lipoplexes.
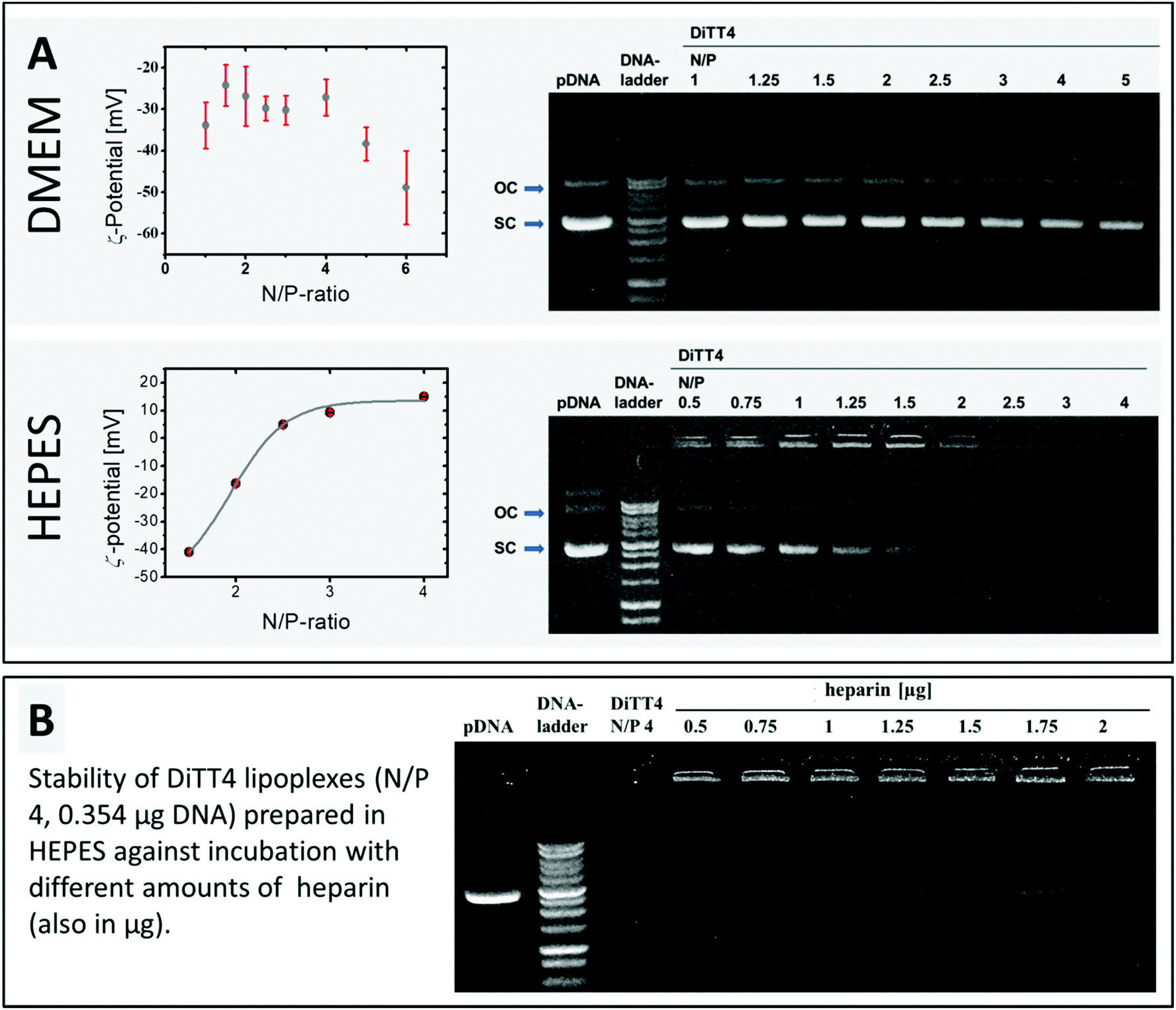 | ||
| Fig. 4 (A) Results of the zeta potential measurement and the gel retardation assay for the lipoplex formation of DiTT4 with pDNA at different N/P ratios. The complex formation was achieved in DMEM or HEPES buffer (10 mM, pH 7.3). The zeta potential curve obtained in HEPES was additionally fitted with a Boltzmann fit. (B) Effect of heparin on the DiTT4 lipoplex formulation (N/P 4). The lipoplexes were treated with different amounts of heparin and investigated by the gel retardation assay. Each sample and the pDNA control contain 0.354 μg peGFP-C2 allowing a comparison of the intensity on each gel. | ||
DiTT4 lipoplexes prepared in HEPES buffer at an N/P ratio of 4 (finished DNA encapsulation according to zeta potential measurements and gel retardation assay, Fig. 4A) are stable even after the treatment with heparin, a negatively charged polymer with a high charge density (higher than DNA). Heparin can be used as a model for glycosaminoglycanes (GAGs) of the extracellular matrix and the glycocalix of cells. Therefore, this assay provides information about a possible GAG-triggered premature DNA release before entering a cell,33 which should be avoided. Even at high amounts of heparin (more than the fivefold amount of heparin referred to DNA) only a weak signal of free plasmid DNA was found (Fig. 4B). This indicates the high compacting effect of the cationic lipid formulation on the DNA,34 which is concordant with the ethidium bromide exclusion (lipid formulation in HEPES at N/P 2.5 and higher, Fig. 4A). At this point cell culture experiments are necessary to prove DNA transfer activity despite the high degree of compaction. An efficient and stable DNA encapsulation is a benefit for retaining the nanoparticle integrity of DiTT4 lipoplexes, but an efficient intracellular release should be ensured for an effective system.
Cell culture experiments
In vitro cell culture experiments serve as a valuable system for the optimization and testing of the formulations in terms of efficiency, internalization mechanism and cytotoxicity. Pre-screening cell-culture experiments have shown that the DiTT4 lipoplex formulation at N/P 4 is the most efficient one.16 The transfection efficiency of DiTT4 N/P 4 was determined using the GFP assay in human alveolar epithelial carcinoma cells (A549) and human cervical cancer cells (HeLa), two different epithelial cell types used as initial screening cell lines. In comparison with the commercially available Lipofectamine 2000®, the gold standard for in vitro transfection, the lipoplex formulation DiTT4 N/P 4 shows a significantly lower GFP expression level after 24 h; nevertheless the value was high above zero which allows us to term the formulation as efficient (Fig. 5A). Additional toxicity studies were performed to determine the in vitro cell viability (Fig. 5A). Regarding the viability test the lipoplex formulation DiTT4 N/P 4 is significantly better than Lipofectamine 2000®. Nevertheless, the viability of A549 cells after treatment with DiTT4 lipoplexes is around 70% while in HeLa cells values around 30% are reached. This could be either due to the lipid formulation or an overexpression of the eGFP. The common reporter genes GFP, eGFP and β-galactosidase can have toxic effects if they are overexpressed.35–37 Therefore we screened the liposome formulation in the absence of DNA for toxic effects (Fig. 5B). The viability values range between 70% and 80% in A549 and are comparable to the values with DiTT4 lipoplexes (Fig. 5A). In this cell line the measured toxicity of the lipoplexes seems to be a result of the lipid formulation. The situation in HeLa cells is quite different. The treatment with DiTT4 liposomes results in viability values around 100% (Fig. 5B). Therefore, we assume that the viability of 30% observed after treatment of HeLa cells with lipoplexes (Fig. 5A) is strongly caused by the overexpression of eGFP. To prove such a vector dependent cytotoxicity we exchanged the eGFP (peGFP-C2) encoding plasmid DNA with a luciferase encoding plasmid DNA (pCMV-luc) for the experiments in HeLa cells (Fig. 5C). The viability of DiTT4 and Lipofectamine 2000 treated HeLa cells using the pCMV-luc plasmid is above 90%, and therewith much better than the values determined with the peGFP-C2 reporter gene vector (compare Fig. 5A and C). This result supports the thesis that the expression product using the peGFP-C2 reporter gene causes the toxic effect observed in HeLa cells. An additional positive outcome of the reporter gene vector exchange is that the transfection efficiency of DiTT4 is comparable to Lipofectamine 2000® using the luciferase encoding plasmid in HeLa cells (Fig. 5C). Different expression behavior of the two plasmids can explain this difference between peGFP-C2 (Lipofectamine 2000 was more efficient than DiTT4) and pCMV-luc (Lipofectamine 2000 was comparable to DiTT4) in HeLa cells. Summarizing, DiTT4 can efficiently transfer the reporter gene vector in two different tumor endothelial cell lines. We further tested the efficiency in HUVEC cells as a primary endothelial cell line, which belong to the hard to transfect cell lines for plasmid DNA.38 DiTT4 lipoplexes show a transfection rate around 20% with a high deviation in four independent experiments while the viability is above 90% (Fig. 5D). Lipofectamine 2000® has a comparable efficiency (Fig. 5D, compare also Fig. 5G) with H. One transfection experiment with DiTT4 lipoplexes on HUVEC cells at passage 8 yields a transfection efficiency around 50% (Fig. 5E). The comparison of DiTT4 lipoplex treated HUVEC cells with liposome treated HUVEC cells shows that the measured green fluorescence results from the pDNA expression and not from a lipid induced increase of the autofluorescence which can appear for example by the lipid triggered induction of stress fibers (compare Fig. 5E with F).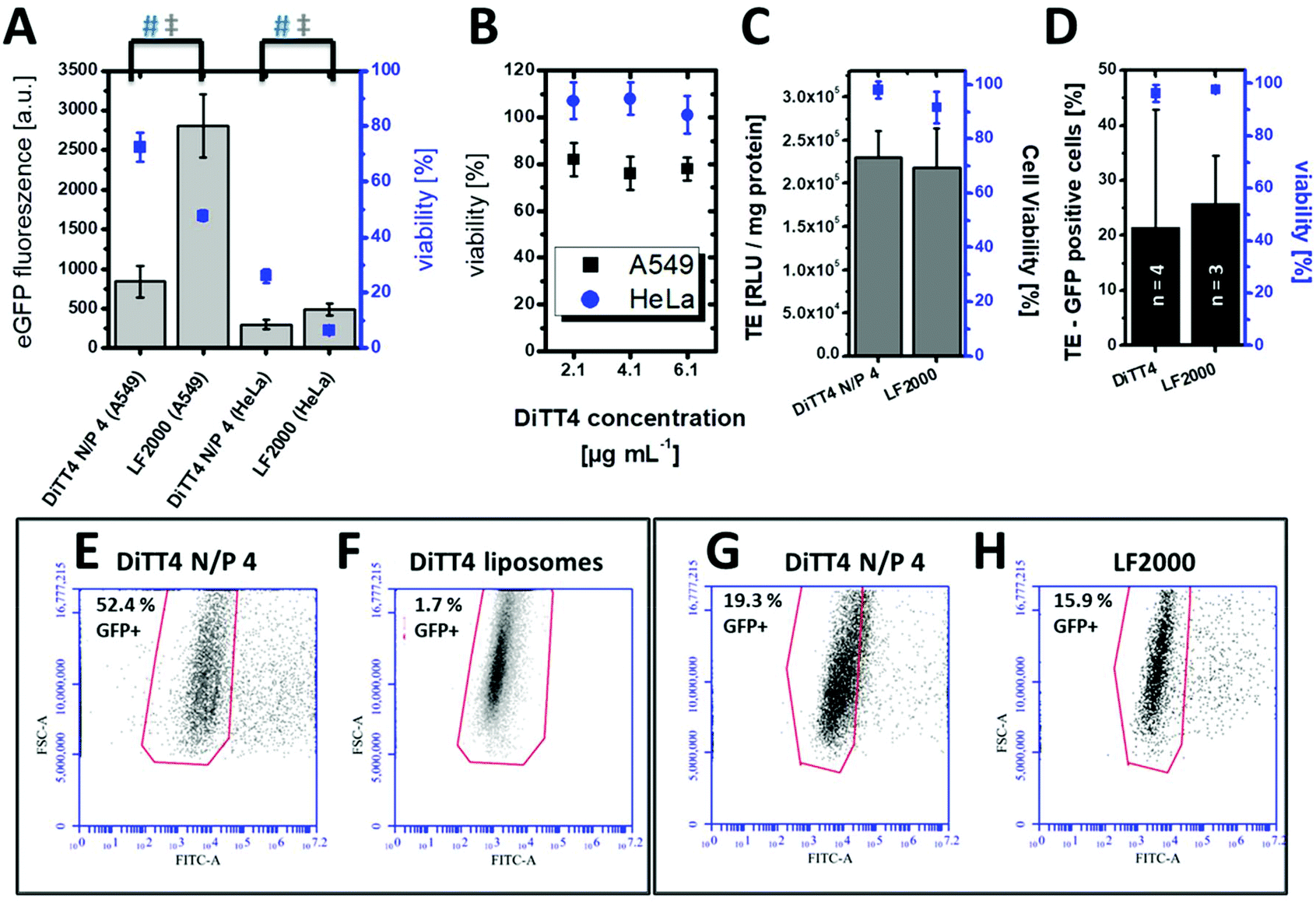 | ||
| Fig. 5 (A) Transfection studies in A549 and HeLa cells. Bars (Y-axis on the left side for scaling) show eGFP fluorescence 24 h after incubation with DiTT4/peGFP-C2 lipoplexes N/P 4 (pDNA = 0.1 μg per well in a 96 well plate) compared to the standard Lipofectamine 2000® (LF2000). The corresponding viability (AlamarBlue assay) is given as squares (Y-axis on the right side for scaling). Statistically significant differences are indicated by # (viability) and ‡ (eGFP fluorescence) using the one way ANOVA (n = 12, α = 0.05). (B) Cell viability (AlamarBlue assay) of A549 and HeLa cells after treatment with different concentrations of cationic liposomes. The concentration 4.1 μg mL−1 corresponds to the lipid concentration of lipoplexes which are used in the transfection experiments in A. (C) Transfection studies with DiTT4/pCMV-luc lipoplexes N/P 4 (pDNA = 0.1 μg per well in a 96 well plate, encoding for luciferase) in HeLa cells after 24 h in comparison with Lipofectamine 2000®. (D) Transfection efficiency (TE, relative value of GFP positive cells, Y-axis on the left side for scaling) and viability (determined by propidium iodide fluorescence, Y-axis on the right side for scaling) of DiTT4/pCMV-GFP lipoplexes N/P 4 (pDNA = 1.6 μg per well in a 6 well plate) in HUVEC cells determined by FACS for 4 independent measurements compared with Lipofectamine 2000® (3 independent measurements). (E–H) Transfection efficiency as GFP fluorescence in HUVEC cells treated with (E and G) DiTT4/pCMV-GFP lipoplexes N/P 4 (pDNA = 1.6 μg per well in a 6 well plate, two independent measurements which are also included in the calculation for figure D). (F) DNA free DiTT4 liposomes with the same lipid concentration, and (H) Lipofectamine 2000®/pCMV-GFP lipoplexes (pDNA = 1.6 μg per well in a 6 well plate). | ||
To throw light into the uptake mechanism behind the internalization of DNA loaded DiTT4 nanoparticles, pathway inhibitors were used in experiments with the cell line A549. The dynamin-dependent endocytosis inhibitor dynasore39,40 and the inhibitor of clathrin-independent endocytosis filipin III39,41 were employed for this purpose. The results show no significant change in the transfection efficiency in the presence of filipin III (Fig. 6A). In contrast, a substantial decrease in the eGFP expression could be noticed in the presence of dynasore (Fig. 6A) suggesting an uptake via dynamin-dependent endocytosis. Dynamin is essential for clathrin-dependent endocytosis and some pathways of the clathrin-independent endocytosis (caveolin-mediated endocytosis is dynamin-dependent and flotillin-mediated endocytosis is either dynamin dependent or independent).13 Toxicity studies were performed to determine whether the observed decrease in GFP fluorescence resulted from a real inhibition of an endocytotic pathway, or only from the toxic effects of the inhibitor (Fig. 6A). All measured viabilities in the presence of an inhibitor were significantly lower than the value determined for the control, but for the filipin treatment experiments the viability was more or less on the level of the inhibitor free experiment. Differentially, for the dynasore treated samples a substantial decrease of the viability was observed. Consequently, it cannot be determined to which extent the decrease of eGFP fluorescence is a result of dynamin inhibition and what is the effect of reduced fluorescence due to cell death. Finally we can conclude that dynamin-dependent and clathrin-independent endocytotic pathways are partially involved in the lipoplex uptake, but dynamin-independent and clathrin-independent uptake mechanisms are also possible. For example an uptake via direct membrane fusion can be discussed.
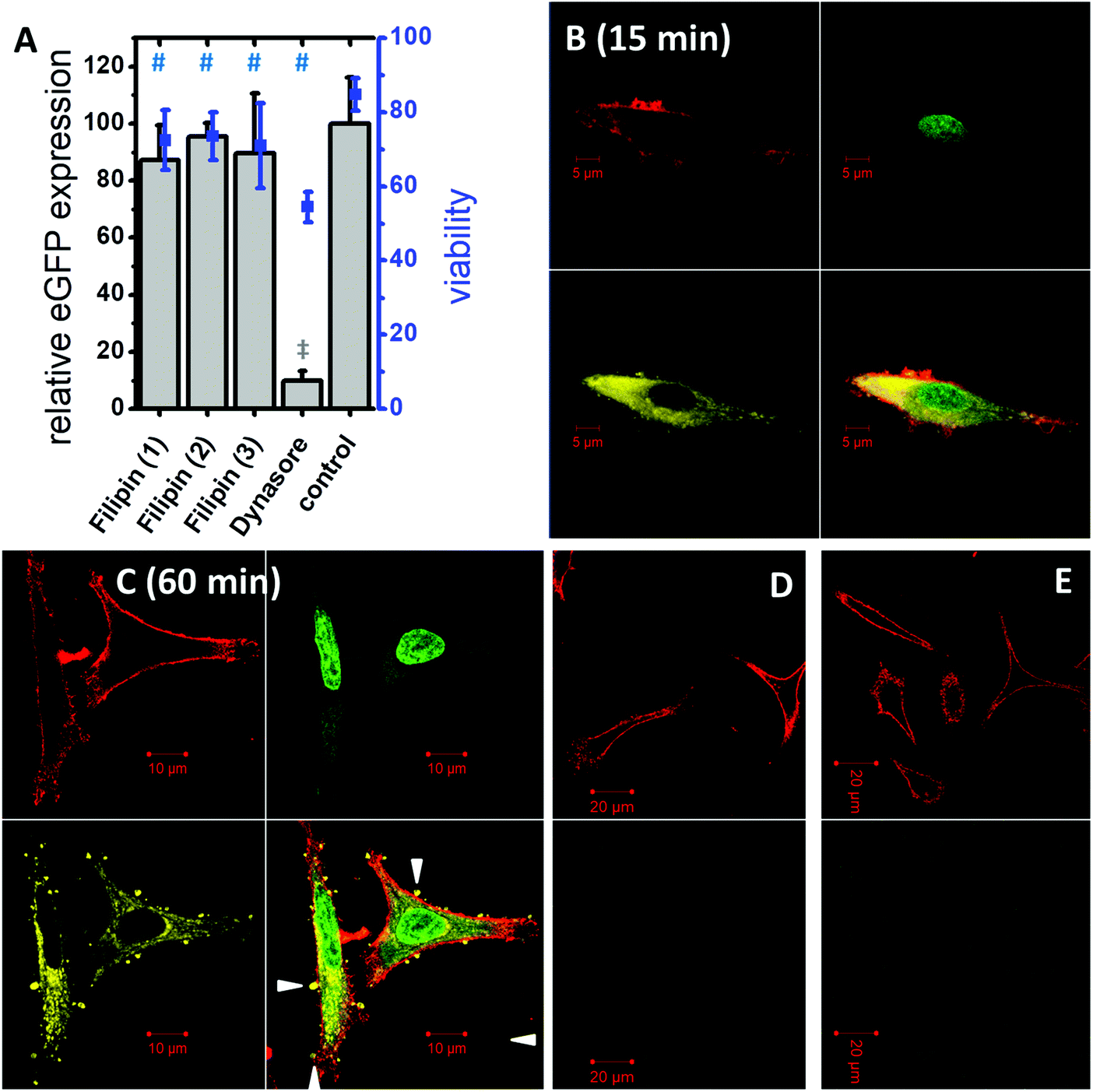 | ||
| Fig. 6 Pathway inhibition experiments showing relative eGFP expression (bars) compared with the inhibitor free experiment (control) and the corresponding cytotoxic effects (squares) of DiTT4/peGFP-C2 lipoplexes (pDNA 0.1 μg per well) at N/P 4 after 24 h in the absence and presence of dynasore (80 μM) or filipin [(1) 3 μg mL−1, (2) 3.75 μg mL−1, and (3) 5 μg mL−1] during a 30 min incubation period. Statistically significant differences are indicated by # (viability) and ‡ (eGFP fluorescence) using the one way ANOVA (n = 12). (B and C) Confocal images of A549 cells at 15 min (B) and 60 min (C) after transfection with DiTT4/peGFP-C2 lipoplexes at a N/P ratio of 4. Following colors are chosen: DiTT4-AttoRho6G (yellow), DNA label YoYo®-1 iodide for nucleus labeling (green), and membrane label Alexa Fluor® 633 for cell membrane labeling (red). In each image the upper panels and the lower panel left show the single channel images and the lower panel right shows the merged image. The white triangles point to DiTT4-containing structures on the cell surface. (D and E) Control of the autofluorescence in the yellow channel (D, upper panel: Alexa Fluor® 633 and lower panel: DiTT4-AttoRho6G channel), and in the green channel (E, upper panel: Alexa Fluor® 633 and lower panel: YoYo®-1 iodide channel) with the same system setup. | ||
The uptake of DiTT4 lipoplexes was investigated by confocal laser scanning microscopy using fluorescently tagged lipoplexes. As a label for the lipid formulation a rhodamine-tagged DiTT4 (DiTT4-AttoRho6G) was used. Fig. 6B demonstrates that the lipoplexes were internalized within 15 min by A549 cells. The lipid label (yellow) is localized in the cytoplasm. A comparable situation was seen after 60 min (Fig. 6C). The lipid label is only located in the cytoplasm and not in the nucleus. Furthermore, after 60 min lipoplex incubation, restricted structures with DiTT4-AttoRho6G fluorescence were found at the cell surface (Fig. 6C, white triangles). Whether these lipid structures are exogenous particles attached to the cell surface or result from exocytotic clearance processes of the cell cannot be answered with the present experiments. Nevertheless, both scenarios are possible. In recent research evidence for exocytotic clearance of internalized lipoplexes has also been described.42 Finally we demonstrated that the lipoplex formulation is efficiently internalized into the cytoplasm and the lipid itself does not appear in the nucleus within 60 min. Nevertheless, the measured eGFP fluorescence in the transfection experiments (Fig. 5A) demonstrates that at a certain time value the pDNA must have entered the nucleus. How this happens is not a topic of this research.
Interaction with blood components
A crucial requirement of lipoplexes for systemic administration is the stability in blood. Therefore, the stability of the DiTT4 lipoplexes after 6 h incubation with 10% foetal bovine serum (FBS) and human serum (HS), respectively, was investigated at 3 different N/P ratios. This assay provides information whether serum components can trigger a premature release of DNA and/or the degradation. The results are presented in Fig. 7A and B. For the lipoplexes at N/P 0.5, 2 and 4 no free plasmid DNA was observed after treatment with FBS or HS. This can be due to the high stability of the complex (complexed DNA does not migrate and also excludes ethidium bromide intercalation),43 or due to the digestion of the released DNA by DNase. Both kinds of serum show DNase activity; therefore the controls are named pDNA + FBS and pDNA + HS. The latter also demonstrates that serum components can form aggregates with plasmid DNA (a new signal near the start lane). The treatment of serum incubated lipoplexes with SDS results in complex disassembling and releases the plasmid DNA from the serum treated lipoplexes. The released DNA now can migrate in the gel (Fig. 7A and B). It can be seen that all three lipoplex formulations show bands of open circular and supercoiled plasmid DNA. The transformation from supercoiled DNA to open circular DNA results from single cut events.43 At N/P 4, the protective effect is more pronounced, because the intensity of the pDNA bands is higher compared to those of the other lipoplex formulations and the supercoiled pDNA is dominant. Overall, in DiTT4 lipoplexes (N/P 4), the DNA is sufficiently protected against DNase digestion and disintegration by serum components.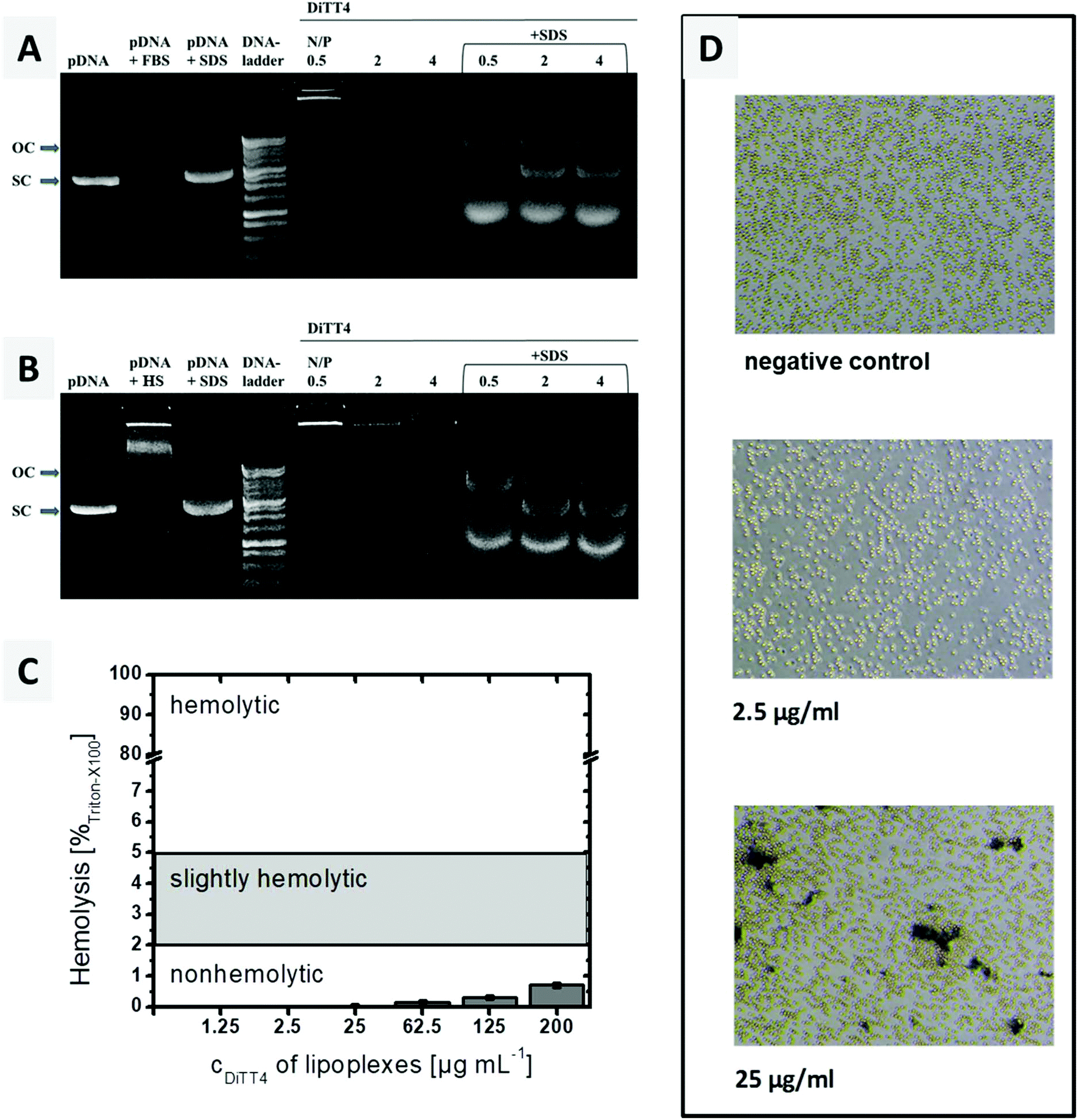 | ||
| Fig. 7 (A and B) Gel retardation assay after incubation of DiTT4 lipoplexes at three different N/P ratios with 10% FBS (A) or 10% HS (B) at 37 °C for 6 h. To facilitate DNA release from the lipoplexes after serum incubation and allow electrophoretic mobility of incorporated DNA, 1.8 μL SDS solution (0.35 M) was added. The DNA concentration was the same in all samples. OC = open circular pDNA, SC = supercoiled pDNA. (C) Hemolysis assay with the increasing concentration of lipoplexes (DiTT4 N/P 4). The limit values are according to the ASTM International Designation F756-17. (D) Results of the erythrocyte aggregation assay for two different concentrations of DiTT4 lipoplexes (N/P 4) and the negative control. | ||
Hemocompatibility studies (Fig. 7C and D) are a prerequisite for any formulation before any systemic application in animals and humans may be performed. The lysis of erythrocytes is followed by the release of hemoglobin, which can be measured spectrophotometrically at 540 nm. Up to a concentration of 200 μg mL−1, a value higher than the concentration used in the in vitro and CAM experiments (see below), DiTT4 lipoplexes are non-hemolytic according to the ASTM International Designation F756-17D guidelines.
Erythrocyte aggregation, which can result in emboli, was also evaluated. Schlenk et al. classified erythrocyte aggregation in 3 stages: stage 1 = no aggregation, stage 2 = moderate aggregation with rouleaux formation, but majority of erythrocytes staying discrete, and stage 3 = aggregation of almost all erythrocytes in clusters.44 At a lipoplex concentration of 2.5 μg mL−1 the erythrocyte suspension looks comparable to the negative control which means a classification in stage 1 of erythrocyte aggregation i.e. no aggregation (Fig. 7D). At a 10-fold higher concentration (25 μg mL−1), an aggregation of stage 2 occurred. This concentration is two times higher than the maximum tested by Schlenk et al. (12.5 μg mL−1). Therefore a low tendency towards erythrocyte aggregation can be assumed. Nevertheless, the CAM model will be analysed below, in order to evaluate the hemocompatibility of the lipoplexes in a physiological blood circle.
A further part of hemocompatibility testing was the investigation of the complement activating potential of the DiTT4 lipoplex formulation (N/P 4). The complement cascade is part of the innate immune system and is composed of protein cascades consisting of proteins present in the blood. It can be activated by three pathways: the classical pathway, the alternative pathway, and the mannose-binding lectin pathway.45 The activation results in direct cell lysis or recruitment of leucocytes, and therewith induces inflammatory processes. Foreign surfaces, even nanoparticles, can activate the complement cascade after blood contact via the classical or the alternative route.46–49 The activation potential of DiTT4 lipoplexes at N/P 4 on the complement cascade was determined using established assays to prescreen possible complement activation:50 (i) the AP50 test which is based on the lysis of sensitized rabbit erythrocytes after activation of the alternative pathway of the complement in serum or plasma in the presence of Mg2+ and (ii) and the CH50 test which is based on the lysis of sensitized sheep erythrocytes in the presence of Ca2+ and Mg2+. The results are presented in Fig. 8. AP50 and CH50 values are the serum dilution factor that represents 50% lysis of complement-sensitized red blood cells which results from complement system components in the blood after incubation with DiTT4 lipoplexes (N/P 4). According to the manufacturer's protocol a complement activation results in the consumption of complement proteins, which reduces the CH50 or AP50 level. First we want to examine the AP50 test. The value of the DiTT4 formulation (AP50DiTT4 = 6) was lower than those of the two standardized plasma references (AP50reference 1 = 7.5 and AP50reference 2 = 12), indicating that the DiTT4 lipoplexes activate the alternative pathway. Whether or not this degree of activation is clinically relevant, for instance causing complement activation-related pseudo-allergy CARPA,48 has to be investigated by specific tests in ongoing research, which is not part of this study. In contrast, the classical pathway did not seem to be activated by the lipoplex formulation, since it has a CH50DiTT4 value of 24, much higher than those of the two references (CH50reference 1 = 7; CH50reference 2 = 11).
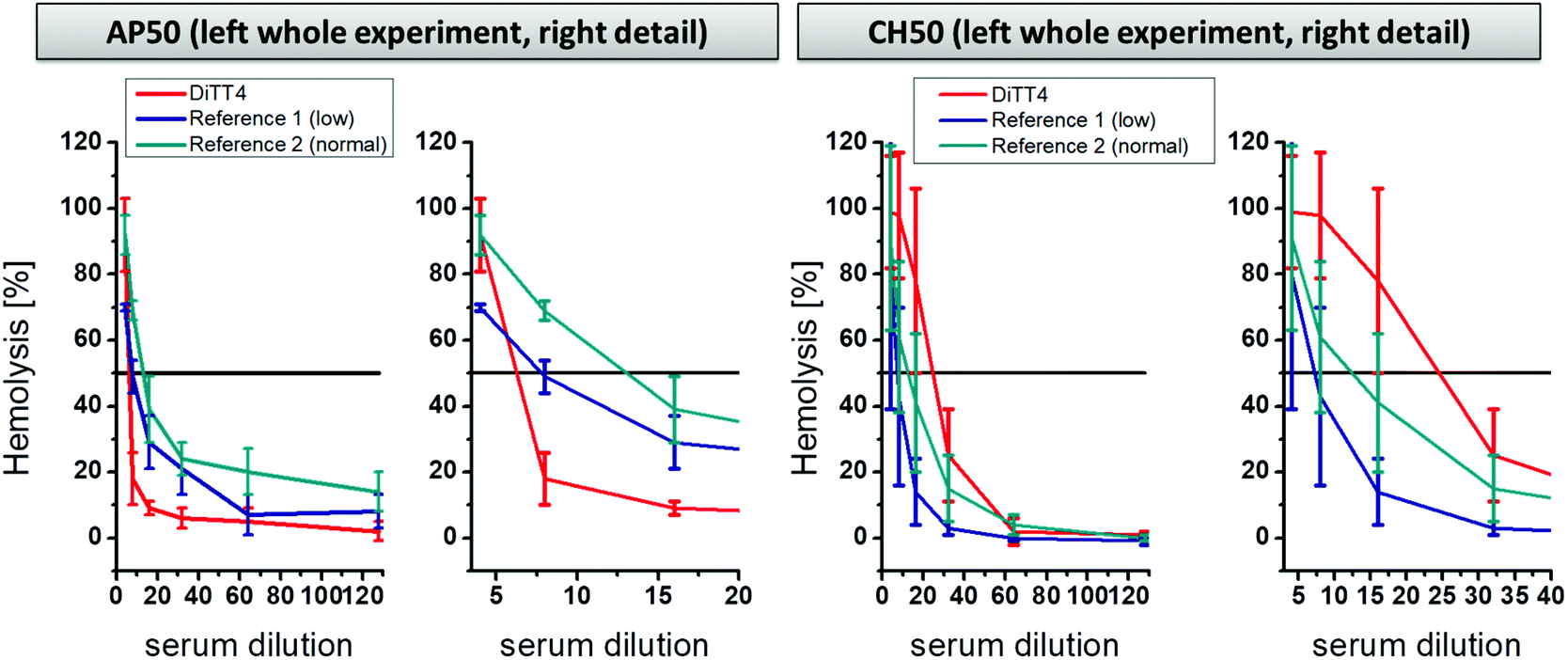 | ||
| Fig. 8 Activation of complement pathways determined by the AP50 test (alternative pathway method) and the CH50 test (classical pathway method) of DiTT4 lipoplexes at NP 4 compared with two reference plasma samples (provided by the manufacturer of the test kits). Reference 1 is standard low complement plasma and reference 2 is standard normal complement plasma. | ||
CAM experiments
The CAM model was utilized to determine the transfection efficiency and biocompatibility of the DiTT4 lipoplexes in vivo as an intermediate step between in vitro experiments and experiments in mammalian animal models. Compared to mammalian models, the CAM model offers a cost effective, 3R (replacement, refinement, and reduction) conforming, and an uncomplicated method to study the effect of drug delivery vehicles in vivo in a vertebrate model,51–54 and was also utilized as an in vivo model for non-viral gene transfection.5,55–57 Consequently, the CAM model enables the testing of gene delivery vehicles in a complex tissue of primary cells. Furthermore we used the model system to test for toxic effects, because it is known that there can be huge gaps between in vitro and in vivo results for nanoparticles.58 The biocompatibility testing in hen's egg CAM models with the main focus on hemocompatibility is an accepted in vivo model allowing screening for hemocompatibility and acute toxicity regarding the chicken embryo.44 Initially, to determine the biocompatibility (hemocompatibility and acute toxicity), 100 μL of DiTT4 lipoplexes (N/P 4; containing 1 μg pDNA) were injected intravenously into the hen's egg CAM on EDD 10. There was no influence on the development of the egg even upon reaching EDD 14. No signs of hemorrhage, stasis, or destruction of the microvasculature of the CAM or toxicity could be found (Fig. 9B). Also the embryo was viable until the experiment had to be abandoned on EDD 14 (data not shown). Nevertheless, this model is not suitable for testing the inflammatory response because the chicken embryos become immune competent at EDD 18.54 For the transfection experiments we injected Rhod-DOPE labeled DiTT4 lipoplexes (N/P 4; containing 0.5 μg pCMV-GFP) topically on the chorionic epithelium (consisting of epithelial cells; we also used epithelial cells in the in vitro experiments) or into the mesoderm (consisting of blood vessels and different cell types), according to Fig. 9A. After mesodermal or topical injection of DiTT4 lipoplexes, approximately 1 cm2 of the CAM was cut out and investigated for GFP expression using confocal microscopy after 4 h and 24 h time intervals. The expression could be seen in both topically and mesodermally injected eggs soon after 4 h (Fig. 9C and E), concluding that the efficient transgene expression was not restricted to the in vitro scenario described above. For the topical administration we have to take into account that the epithelial cell layer undergoes a striking proliferation,59 which hypothetically can result in easier transfection properties (cell cycle dependence of gene transfer).60 The mesoderm is a more complex tissue and its maximum of proliferation is finished at EDD 8, while full differentiation is reached at EDD 13.61 In the case of the mesodermal injection almost 22% of the analyzed area (1.25 × 1.25 mm around the point of injection, Fig. 9C, ESI S1†) showed GFP-positive cells after 4 h, a remarkably fast and effective lipofection. For comparison, Lipofectamine 2000® shows <1% transfected mesodermal cells after 4 h (see ESI S1†). After 24 h, around 75% of the analyzed area showed GFP expressing cells (Fig. 9D, ESI S1†), while for Lipofectamine 2000® below 5% of the evaluated area is transfected (Fig. 9F). Consequently, DiTT4 lipoplexes show remarkable efficiency in a complex tissue like the mesoderm, which is built of an extracellular matrix composed of fibronectin, laminin, collagen type IV, and specific aminoglycanes,62 in which the mesodermal blood vessels, lymphatic vessels and mesenchymal cells are embedded.52,61,63 It is necessary to mention that the bright green fluorescent cell population 4 h after mesodermal injection is a different one compared to that at 24 h. After 4 h the bright fluorescent cells have an irregular shape with a lot of cell protuberances (Fig. 9C and ESI S2†) while after 24 h the main green fluorescent cell population has a more or less circular shape (Fig. 9D). Considering the fact that fibroblasts are the major cell type of the mesoderm,64 we postulate that these are different developmental stages of fibroblasts.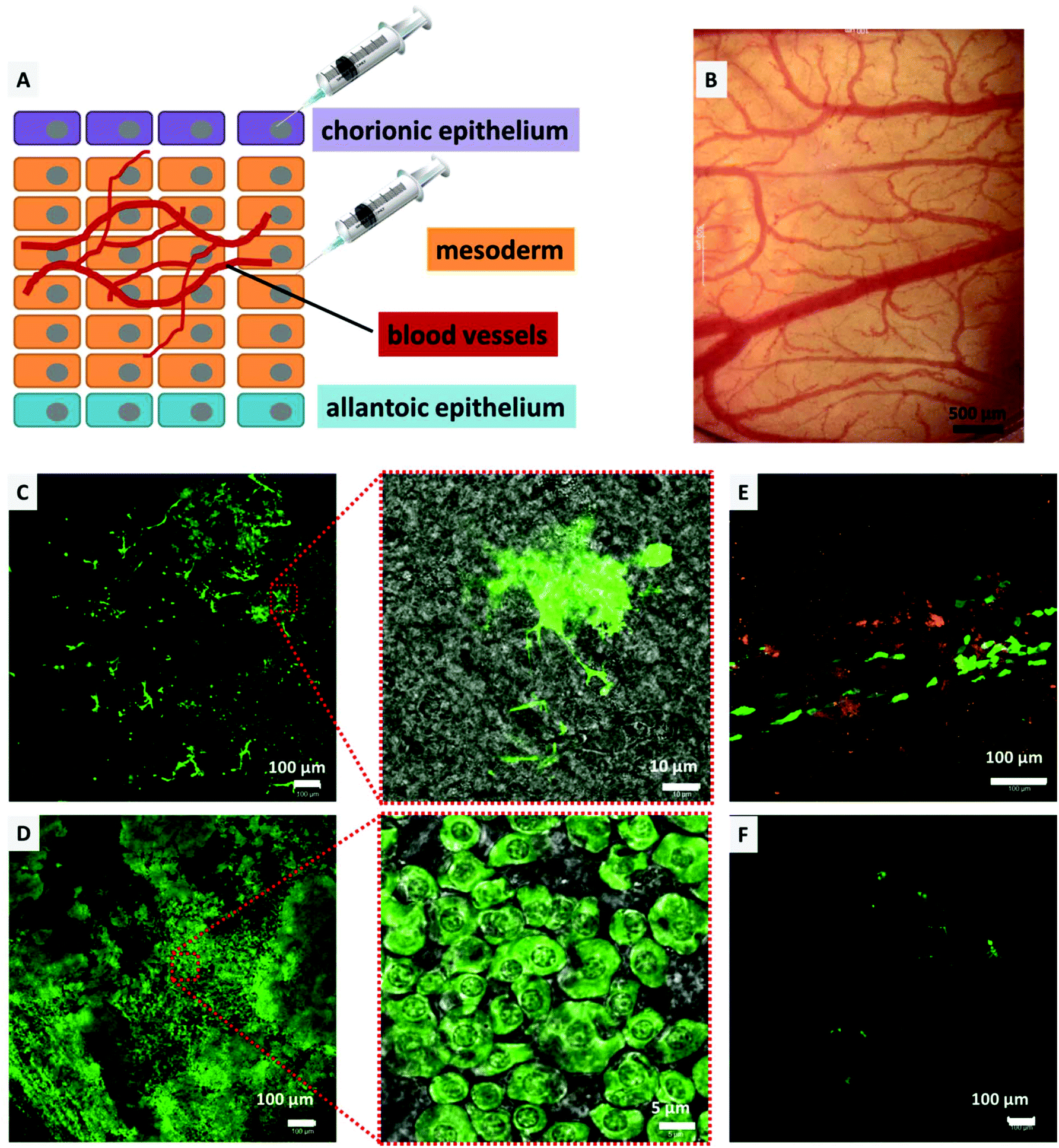 | ||
| Fig. 9 (A) Schematic illustration of the HET-CAM transfection experiment. The lipoplexes were injected on the chorionic epithelium and into the mesoderm. (B) Visualization of the chorioallantoic membrane 24 h after injecting DiTT4 lipoplexes (N/P 4 containing 1 μg DNA). (C/D) CLSM micrographs showing GFP expression (green) in the mesoderm 4 h (C) and 24 h (D) after injection of DiTT4 pCMV-GFP lipoplexes (N/P 4, 0.5 μg DNA). Insets are the magnified micrographs of the images along with a transmitted light channel to give an impression of the cellular morphology. (E) CLSM micrograph showing GFP expression (green) in the chorionic epithelium 4 h after injection of DiTT4 pCMV-GFP lipoplexes (N/P 4, 0.5 μg DNA) labelled with 1% rhodamine-DOPE (orange). (F) CLSM micrographs showing GFP expression (green) in the mesoderm 24 h after injection of Lipofectamine 2000® pCMV-GFP lipoplexes. | ||
Summarizing, the absence of acute toxicity and damage of microvasculature together with the speed and efficiency of transfection confirm the biocompatible and efficient nature of the DNA-loaded DiTT4 lipoplexes in this vertebrate model. The experiments also demonstrate that DiTT4 is much more efficient in a complex tissue than Lipofectamine 2000®.
Despite the high lipofection efficacy in primary cells of the CAM, the rapid gene expression in vivo is an outcome of this study which needs additional attention. A rapid gene expression 4 h post-administration is possible by the use of microinjection or electroporation, both of which are not feasible for in vivo gene delivery.65,66 With this kinetics DiTT4 is extremely fast compared to viral systems. In an early report, Subramanian et al. investigated the kinetics of GFP expression in Chinese hamster ovary cells after transfection of a GFP-encoding plasmid using Lipofectamine®.67 They demonstrated with experimental and simulation methods that the gene expression maximum was reached 24 h after transfection. Nevertheless, GFP fluorescence was also detected earlier. Similar behavior was shown for rat mammary carcinoma cells (MAT B III) transfected with a GFP encoding plasmid using Lipofectamine 2000®.68 Pampinella et al. transfected C2C12 myoblasts and primary myoblasts with luciferase encoding plasmid using multicomponent lipopolyplexes loaded with luciferase encoding plasmid. They demonstrate first relevant gene expression after 3–5 h. It is relevant to note that they transfected primary cells really fast. Nevertheless, a missing “gold standard” makes it hard to estimate if the transfection efficiency is high. Also Brunner et al. demonstrate a clearly detectable luciferase expression after 8 h in HeLa with PEI derivatives and Lipofectamine® in different stages of the cell cycle, while the latter reagent was not effective after 8 h for cells in the pure G1 phase.69 The authors assume that the fast and cell cycle independent acting transfection reagent PEI22 (a linear PEI derivative) efficiently overcomes the nuclear barrier. We also postulate this for DiTT4, to explain the fast and efficient plasmid transfer in primary cells such as the cells in the CAM tissue. The possibility to form micellar structures under weakly acidic conditions (pH 5, comparable to endosomal pH values),70 even in the lipoplex formulations (Fig. 1 and 2) raises the hypothesis that fusion processes between the lipid nanoparticles and the membranes of the cells play a major role, because micelles as highly curved lipid assemblies can effectively interact with membranes. The detailed evaluation of this transfer mechanism of DiTT4 is the focus of the on-going investigation. Whether the fast transfection which was proved in the CAM model can be used for therapeutic issues is part of ongoing research.
As a summary of this chapter we note that DiTT4 has shown fast and efficient plasmid transfer compared to the “gold standard” of lipofection, Lipofectamine 2000®, in a complex tissue composed of primary cells embedded in a complex extracellular matrix.
Conclusion
Structural investigations demonstrate efficient nucleic acid complexation by DiTT4 lipoplexes under formation of small lipoplexes in the nanometer scale. The in vitro studies revealed the efficient transfection potential of the DiTT4 lipoplexes and also demonstrate a save hemocompatibility profile. The latter includes serum stability (resistance against serum induced disintegration of lipoplexes and DNA-digestion), low hemolysis of red blood cells, and low tendency towards erythrocyte aggregation in high concentrations. Some evidence of a slight complement activation via the alternative pathway could be seen. More detailed investigations are necessary to understand the relevance of this outcome for clinical use. The hemocompatibility was also confirmed in the in vivo experiments by intravenous injection in the CAM model where no hemorrhagic or thrombotic effects at the CAM membrane and no acute toxic effects on the embryo were observed. The subsequent in vivo experiments in the chorioallantoic membrane model complemented the in vitro studies and showed that the delivery potential of the DiTT4 lipoplexes was not limited to an in vitro environment, but rather showed results highly superior to the standard Lipofectamine 2000®.Summarizing, this study demonstrates the efficiency and biocompatibility of DNA loaded DiTT4 nanoparticles also in complex living tissues and paves the way towards potential therapeutic applications which have to be investigated in mammalian models, while systemic and local administration routes can be considered. Systemic administration needs more detailed investigation of the complement activating effect. The CAM experiments reflect the possibility of using DiTT4 in therapeutic strategies, where fibroblasts have to be transfected. Possible therapeutic applications are in cardiac diseases,71,72 tissue engineering,73,74 and neurological diseases.75
Abbreviations
| AFM | Atomic force microscopy |
| CAM | Chorioallantoic membrane model |
| CryoTEM | Cryo-transmission electron microscopy |
| DiTT4 | N-[6-Amino-1-oxo-1-(N-tetradecylamino)hexan-(2S)-2-yl]-N′-2,2-ditetradecylpropandiamide |
| DSC | Differential scanning calorimetry |
| lDv | Laser Doppler velocimetry |
| PCS | Photon correlation spectrometry |
| N/P ratio | Ratio of the primary amines in the cationic lipid to the phosphate groups of the DNA |
| SAXS | Small angle X-ray scattering |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors express gratitude towards PD Dr Simon Drescher (Martin Luther University Halle-Wittenberg) for the preparation of the negatively stained TEM sample. Furthermore, the authors thank Prof. Dariush Hinderberger and Dr Andreas Kerth for providing the opportunity to use DSC. The project is funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – project grant number 396823779 (C. W.). We further thank S. Koch and C. Pilowski for technical assistance.References
- M. A. Kotterman, T. W. Chalberg and D. V. Schaffer, Annu. Rev. Biomed. Eng., 2015, 17, 63–89 CrossRef CAS PubMed
.
- M. Ramamoorth and A. Narvekar, J. Clin. Diagn. Res., 2015, 9, GE01–GE06 Search PubMed
- C. Wölk, C. Janich, A. Meister, S. Drescher, A. Langner, G. Brezesinski and U. Bakowsky, Bioconjugate Chem., 2015, 26(12), 2461–2473 CrossRef
- S. R. Pinnapireddy, L. Duse, B. Strehlow, J. Schäfer and U. Bakowsky, Colloids Surf., B, 2017, 158, 93–101 CrossRef CAS PubMed
- E. Baghdan, S. R. Pinnapireddy, B. Strehlow, K. H. Engelhardt, J. Schäfer and U. Bakowsky, Int. J. Pharm., 2018, 535, 473–479 CrossRef CAS
-
D. D. Lasic and F. J. Martin, Stealth Liposomes, CRC Press, 1995 Search PubMed
- M. Foldvari, D. W. Chen, N. Nafissi, D. Calderon, L. Narsineni and A. Rafiee, J. Controlled Release, 2016, 240, 165–190 CrossRef CAS
- M. Rezaee, R. K. Oskuee, H. Nassirli and B. Malaekeh-Nikouei, J. Controlled Release, 2016, 236, 1–14 CrossRef CAS
- C. H. Jones, C.-K. Chen, A. Ravikrishnan, S. Rane and B. A. Pfeifer, Mol. Pharm., 2013, 10, 4082–4098 CrossRef CAS
- J. L. Anderson and T. J. Hope, Gene Ther., 2005, 12, 1667 CrossRef CAS
- M. Nonnenmacher and T. Weber, Gene Ther., 2012, 19, 649 CrossRef CAS
- T. Bus, A. Traeger and U. S. Schubert, J. Mater. Chem. B, 2018, 6, 6904–6918 RSC
- D. Vercauteren, J. Rejman, T. F. Martens, J. Demeester, S. C. De Smedt and K. Braeckmans, J. Controlled Release, 2012, 161, 566–581 CrossRef CAS PubMed
- Z. U. Rehman, I. S. Zuhorn and D. Hoekstra, J. Controlled Release, 2013, 166, 46–56 CrossRef
- C. S. Kue, K. Y. Tan, M. L. Lam and H. B. Lee, Exp. Anim., 2015, 64, 129–138 CrossRef CAS PubMed
- C. Wölk, C. Janich, D. Pawlowska, S. Drescher, A. Meister, G. Hause, B. Dobner, A. Langner and G. Brezesinski, ChemPhysChem, 2015, 16, 2115–2126 CrossRef PubMed
- J. Sitterberg, A. Ozcetin, C. Ehrhardt and U. Bakowsky, Eur. J. Pharm. Biopharm., 2010, 74, 2–13 CrossRef CAS PubMed
- A. Ozcetin, A. Aigner and U. Bakowsky, Eur. J. Pharm. Biopharm., 2013, 85, 711–715 CrossRef
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J.-Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676 CrossRef CAS
- R. Podgornik, D. C. Rau and V. A. Parsegian, Macromolecules, 1989, 22, 1780–1786 CrossRef CAS
- C. Wölk, C. Janich, U. Bakowsky, A. Langner and G. Brezesinski, Adv. Colloid Interface Sci., 2017, 248, 20–34 CrossRef
- V. Alfredsson, Curr. Opin. Colloid Interface Sci., 2005, 10, 269–273 CrossRef CAS
- M. Schmutz, D. Durand, A. Debin, Y. Palvadeau, A. Etienne and A. R. Thierry, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 12293–12298 CrossRef CAS
- J. O. Rädler, I. Koltover, T. Salditt and C. R. Safinya, Science, 1997, 275, 810–814 CrossRef
-
M. Mandel and J. Marmur, in Methods in Enzymology, Academic Press, 1968, vol. 12, pp. 195–206 Search PubMed
- D. M. Crothers, N. R. Kallenbach and B. H. Zimm, J. Mol. Biol., 1965, 11, 802–820 CrossRef CAS
- P. L. Privalov, O. B. Ptitsyn and T. M. Birshtein, Biopolymers, 1969, 8, 559–571 CrossRef CAS
- A. Tempestini, V. Cassina, D. Brogioli, R. Ziano, S. Erba, R. Giovannoni, M. G. Cerrito, D. Salerno and F. Mantegazza, Nucleic Acids Res., 2013, 41, 2009–2019 CrossRef CAS
- D. Y. Lando, S. G. Haroutiunian, A. M. Kul'ba, E. B. Dalian, P. Orioli, S. Mangani and A. A. Akhrem, J. Biomol. Struct. Dyn., 1994, 12, 355–366 CrossRef CAS PubMed
- C. Wölk, D. Pawlowska, S. Drescher, A. Auerswald, A. Meister, G. Hause, A. Blume, A. Langner, G. Brezesinski and B. Dobner, Langmuir, 2014, 30, 4905–4915 CrossRef
- J. Rumschöttel, S. Kosmella, C. Prietzel, D. Appelhans and J. Koetz, Colloids Surf., B, 2017, 154, 74–81 CrossRef
- D. Dey, C. Maiti, S. Maiti and D. Dhara, Phys. Chem. Chem. Phys., 2015, 17, 2366–2377 RSC
- C. Janich, C. Wölk, F. Erdmann, T. Groth, G. Brezesinski, B. Dobner and A. Langner, J. Controlled Release, 2015, 220, 295–307 CrossRef CAS
- J. P. N. Silva, A. C. N. Oliveira, M. P. P. A. Casal, A. C. Gomes, P. J. G. Coutinho, O. P. Coutinho and M. E. C. D. Real Oliveira, Biochim. Biophys. Acta, Biomembr., 2011, 1808, 2440–2449 CrossRef CAS
- A. M. Ansari, A. K. Ahmed, A. E. Matsangos, F. Lay, L. J. Born, G. Marti, J. W. Harmon and Z. Sun, Stem Cell Rev. Rep., 2016, 12, 553–559 CrossRef CAS
- D. Ganini, F. Leinisch, A. Kumar, J. Jiang, E. J. Tokar, C. C. Malone, R. M. Petrovich and R. P. Mason, Redox Biol., 2017, 12, 462–468 CrossRef CAS
- E. R. Detrait, W. J. Bowers, M. W. Halterman, R. E. Giuliano, L. Bennice, H. J. Federoff and E. K. Richfield, Mol. Ther., 2002, 5, 723–730 CrossRef CAS
-
O. Gresch and L. Altrogge, in Protein Expression in Mammalian Cells: Methods and Protocols, ed. J. L. Hartley, Humana Press, Totowa, NJ, 2012, pp. 65–74, DOI:10.1007/978-1-61779-352-3_5
- D. Dutta and J. G. Donaldson, Cell. Logist., 2012, 2, 203–208 CrossRef PubMed
- E. Macia, M. Ehrlich, R. Massol, E. Boucrot, C. Brunner and T. Kirchhausen, Dev. Cell, 2006, 10, 839–850 CrossRef CAS
- J. E. Schnitzer, P. Oh, E. Pinney and J. Allard, J. Cell Biol., 1994, 127, 1217–1232 CrossRef CAS
- C. Janich, S. R. Pinnapireddy, F. Erdmann, T. Groth, A. Langner, U. Bakowsky and C. Wolk, Eur. J. Pharm. Biopharm., 2017, 118, 38–47 CrossRef CAS
- J. Giselbrecht, C. Janich, S. R. Pinnapireddy, G. Hause, U. Bakowsky, C. Wölk and A. Langner, Int. J. Pharm., 2018, 541, 81–92 CrossRef CAS PubMed
- F. Schlenk, S. Werner, M. Rabel, F. Jacobs, C. Bergemann, J. H. Clement and D. Fischer, Arch. Toxicol., 2017, 91, 3271–3286 CrossRef CAS PubMed
- M. J. Walport, N. Engl. J. Med., 2001, 344, 1058–1066 CrossRef CAS
- K. N. Ekdahl, J. D. Lambris, H. Elwing, D. Ricklin, P. H. Nilsson, Y. Teramura, I. A. Nicholls and B. Nilsson, Adv. Drug Delivery Rev., 2011, 63, 1042–1050 CrossRef CAS
- G. L. Szeto and E. B. Lavik, J. Mater. Chem. B, 2016, 4, 1610–1618 RSC
- O. M. Merkel, R. Urbanics, P. Bedőcs, Z. Rozsnyay, L. Rosivall, M. Toth, T. Kissel and J. Szebeni, Biomaterials, 2011, 32, 4936–4942 CrossRef CAS
- A. Chonn, P. R. Cullis and D. V. Devine, J. Immunol., 1991, 146, 4234–4241 CAS
- U. R. Nilsson and B. Nilsson, J. Immunol. Methods, 1984, 72, 49–59 CrossRef CAS
- N. A. Lokman, A. S. F. Elder, C. Ricciardelli and M. K. Oehler, Int. J. Mol. Sci., 2012, 13, 9959–9970 CrossRef CAS
- D. Ribatti, Mech. Dev., 2016, 141, 70–77 CrossRef CAS
- T. I. Valdes, D. Kreutzer and F. Moussy, J. Biomed. Mater. Res., 2002, 62, 273–282 CrossRef CAS
- A. Vargas, M. Zeisser-Labouèbe, N. Lange, R. Gurny and F. Delie, Adv. Drug Delivery Rev., 2007, 59, 1162–1176 CrossRef CAS
- Y. Lei, M. Rahim, Q. Ng and T. Segura, J. Controlled Release, 2011, 153, 255–261 CrossRef CAS
- G. Chouinard-Pelletier, M. Leduc, D. Guay, S. Coulombe, R. L. Leask and E. A. Jones, Biomed. Eng. Online, 2012, 11, 67 CrossRef
- I. Tariq, S. R. Pinnapireddy, L. Duse, M. Y. Ali, S. Ali, M. U. Amin, N. Goergen, J. Jedelská, J. Schäfer and U. Bakowsky, Eur. J. Pharm. Biopharm., 2019, 135, 72–82 CrossRef CAS PubMed
- F. Joris, B. B. Manshian, K. Peynshaert, S. C. De Smedt, K. Braeckmans and S. J. Soenen, Chem. Soc. Rev., 2013, 42, 8339–8359 RSC
- M. G. Gabrielli and D. Accili, J. Biomed. Biotechnol., 2010, 2010, 12 Search PubMed
- S. Brunner, T. Sauer, S. Carotta, M. Cotten, M. Saltik and E. Wagner, Gene Ther., 2000, 7, 401 CrossRef CAS
- P. Nowak-Sliwinska, T. Segura and M. L. Iruela-Arispe, Angiogenesis, 2014, 17, 779–804 CrossRef CAS
- D. Ribatti, B. Nico, A. Vacca, L. Roncali, P. H. Burri and V. Djonov, Anat. Rec., 2001, 264, 317–324 CrossRef CAS
- A. N. Makanya, I. Dimova, T. Koller, B. Styp-Rekowska and V. Djonov, PLoS One, 2016, 11, e0152821 CrossRef
- G. Melkonian, C. Le, W. Zheng, P. Talbot and M. Martins-Green, Toxicol. Appl. Pharmacol., 2000, 163, 26–37 CrossRef CAS
- J. A. Gershan, B. D. Johnson, J. Weber, D. W. Schauer, N. Natalia, S. Behnke, K. Burns, K. W. Maloney, A. B. Warwick and R. J. Orentas, Genet. Vaccines Ther., 2005, 3, 4 CrossRef PubMed
- H. Pollard, J.-S. Remy, G. Loussouarn, S. Demolombe, J.-P. Behr and D. Escande, J. Biol. Chem., 1998, 273, 7507–7511 CrossRef CAS PubMed
- S. Subramanian and F. Srienc, J. Biotechnol., 1996, 49, 137–151 CrossRef CAS
- S. Mehier-Humbert, T. Bettinger, F. Yan and R. H. Guy, J. Controlled Release, 2005, 104, 203–211 CrossRef CAS
- S. Brunner, E. Fürtbauer, T. Sauer, M. Kursa and E. Wagner, Mol. Ther., 2002, 5, 80–86 CrossRef CAS
- S. Padilla-Parra, P. M. Matos, N. Kondo, M. Marin, N. C. Santos and G. B. Melikyan, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 17627–17632 CrossRef CAS PubMed
- R. Kumarswamy, I. Volkmann, V. Jazbutyte, S. Dangwal, D.-H. Park and T. Thum, Arterioscler., Thromb., Vasc. Biol., 2012, 32, 361–369 CrossRef CAS
- M. Jayawardena Tilanthi, B. Egemnazarov, A. Finch Elizabeth, L. Zhang, J. A. Payne, K. Pandya, Z. Zhang, P. Rosenberg, M. Mirotsou and J. Dzau Victor, Circ. Res., 2012, 110, 1465–1473 CrossRef CAS
- F. Bastami, P. Nazeman, H. Moslemi, M. Rezai Rad, K. Sharifi and A. Khojasteh, Cell Proliferation, 2017, 50, e12321 CrossRef
- D. F. E. Ker, R. Sharma, E. T. H. Wang and Y. P. Yang, PLoS One, 2015, 10, e0139054 CrossRef
- M. H. Tuszynski, L. Thal, M. Pay, D. P. Salmon, H. U. Sang, R. Bakay, P. Patel, A. Blesch, H. L. Vahlsing, G. Ho, G. Tong, S. G. Potkin, J. Fallon, L. Hansen, E. J. Mufson, J. H. Kordower, C. Gall and J. Conner, Nat. Med., 2005, 11, 551 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9bm01093a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |