
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSustained delivery of growth factors with high loading efficiency in a layer by layer assembly†
Febriyani F. R.
Damanik
ab,
Marzia
Brunelli
ac,
Laura
Pastorino
c,
Carmelina
Ruggiero
c,
Clemens
van Blitterswijk
ab,
Joris
Rotmans
d and
Lorenzo
Moroni
 *ab
*ab
aUniversity of Twente, Drienerlolaan 5, Zuidhorst 145, 7522 NB Enschede, The Netherlands. E-mail: l.moroni@maastrichtuniversity.nl
bMaastricht University, MERLN Institute for Technology-Inspired Regenerative Medicine, Universiteitsingel 40, 6229 ER Maastricht, The Netherlands
cLaboratory of Nanobioscience and Medical Informatics, Dept. Informatics, Bioengineering, Robotics and Systems Engineering (DIBRIS), University of Genoa, Genoa, Italy
dDepartment of Nephrology, Leiden University Medical Center, Albinusdreef 2, 2333ZA Leiden, The Netherlands
First published on 17th October 2019
Abstract
Layer by layer (LBL) assembly has garnered considerable interest due to its ability to generate multifunctional films with high tunability and versatility in terms of substrates and polyelectrolytes, allowing the option to use complex devices and drugs. Polyelectrolytes, such as growth factors (GFs), are essential, but costly, delicate, biological molecules that have been used in various tissue regeneration applications. For this reason, the controlled drug delivery of efficiently loaded GFs via LBL assembly (GF-LBL) can contribute to the establishment of cost-effective biologically triggered biomedical applications. We have developed an LBL method to load GFs (specifically, transforming growth factor beta 1, platelet-derived growth factor ββ, and insulin growth factor 1), with up to 90% efficiency approximately, by gas plasma surface activation and tuning the pH to increase the ionic strength of polyelectrolytes. Poly(styrenesulfonate) (PSS) and poly(ethyleneimine) (PEI) have been used to provide the initial necessary charge for multilayer build-up. Heparin and dextran sulphate have been investigated as counter polyelectrolytes to enhance the activity of GFs by protecting their ligands, where heparin resulted in the highest achievable loading efficiency for all GFs. Oxygen gas plasma and acidic pH levels also resulted in a significant increase in GF loading efficiency. The three GFs were released by diffusion and erosion in a controlled manner over lengthy time scales and the bioactivity was maintained for up to 14 days. When tested as implants in vitro, GF-LBL constructs increased fibroblast proliferation, influenced cell morphology and migration, and enhanced myofibroblast differentiation, indicating that the biological functionalities of the GFs were preserved. In conclusion, this developed LBL assembly method can provide a simple drug delivery system, which may yield more effective applications for tissue regeneration as well as biomedical sciences at large.
Introduction
Over the past few decades, layer by layer (LBL) assembly methods have evolved to become a versatile, gentle and simple, bottom-up nanofabrication technique to generate ultra-thin films.1,2 In the assembly process, multilayers of films are deposited onto the surface of the substrate via alternative adsorption of the interacting materials.3 An LBL assembly is generally fabricated on top of a charged substrate. Unlike spin4 and spray5 deposition, or more complex assemblies like electromagnetic6 and fluid assembly7 that can only be done on planar or particulate surfaces, classic immersion LBL deposition provides an extensive choice of materials, shapes and sizes.2 Hence, charged substrates can range from flat, smooth silicon wafers8 to rough, irregularly shaped ceramics and polymer scaffolds9 and rods, and from large-scale surface coatings10 to nanoscale electrospun fibres11 and nanoparticles.12 This allows us to use LBL methods on our implants and many other devices with complex shapes and materials.As not all LBL substrates have a sufficient amount of initial charge, different techniques have been used to activate their surfaces. Persulfonation through piranha solution or photocatalytic oxidation activates the substrate's surface, but limits the vast options of the substrate material, due to its aggressive oxidation technique.13 Materials that are sensitive to such priming methods, for instance many polymeric thin layered substrates, can be activated via a more controlled surface method, such as gas plasma treatment.14 As the fabrication of these multilayer LBL assembly films is carried out by exposing the charged substrate alternatively with positively and negativity charged solutions, called polyelectrolytes,1 almost any variety of charged species, such as nanoparticles,15 dendrimers,16 polysaccharides,17 nucleic acids18 and viruses19 can be successfully used as polyelectrolytes to generate LBL films. Direct polymeric absorption of strong polyelectrolytes, for example poly(styrenesulfonate) (PSS, basal isoelectric point 1) and poly(ethyleneimine) (PEI, basal isoelectric point 11) used for our implants, provide negatively and positively charged substrates, respectively, and have been deployed to provide the initial necessary charge for multilayer build-up.20,21
Alternated depositions of oppositely charged polyelectrolytes allow great freedom in the layer sequence.22 A simple way to increase loaded solution or thickness of the LBL film is by increasing the number of layers used. Additionally, factors such as temperature and ionic strength through the modulation of pH are known to be strong influencers, while deposition time and polyelectrolyte concentration are known to be weak influencers that can modulate the loading capacity of the polyelectrolytes.23 Tuning these factors increases the loading capacity of the chosen polyelectrolyte until a plateau is achieved, indicating the maximum capacity of up to 90% loading approximately that we have achieved in this study, with a controlled release that has never been reported in a simple modelled LBL study before. The balance between adhesion strength and loading capacity is necessary to provide mild conditions and to preserve sensitive polyelectrolyte functions.
Due to its versatility to be applied to almost any type of charged substance and substrate with high tunability, LBL coating is used in many different fields, particularly in biomedical applications, to fabricate thin films.24 In the field of tissue regeneration, LBL has been used for wound healing dressings, cardiovascular devices, bone and ligament grafts, and cartilage scaffolds among others.25 Growth factors (GFs) are important mediators of tissue regeneration and have been incorporated into various drug delivery systems. For example, transforming growth factor beta 1 (TGF-β1), platelet-derived growth factor ββ (PDGF-ββ) and insulin growth factor 1 (IGF-1) are involved in tissue morphogenesis and can influence the foreign body response. Nevertheless, current drug delivery systems for GFs, specifically LBL, suffer from several disadvantages. First, many do not offer high loading capacities,26 which would be beneficial given the high costs of GFs. Second, few offer tuneable controlled release,24 a critical parameter given the short half-life of GFs and consequences of GF overdosing, which includes uncontrolled cell growth or apoptosis. Hence, due to its capacity to sequester the high concentration of biological components under mild aqueous conditions, preserving fragile protein activity,27 the LBL assembly technique is highly suitable for the incorporation of GFs. Moreover, anionic sulphated polysaccharides such as heparin (HEP, basal isoelectric point 3.5–4) and dextran sulphate (DS, basal isoelectric point 4–5) can be used as counter polyelectrolytes to enhance the activity of GFs by protecting their ligands.28 For these reasons, developing a growth factor delivery system using a highly optimized LBL assembly process would provide highly promising loading capacity and tunable controlled release beneficial for various biomedical applications. Here we report the development, optimisation, and evaluation of an LBL assembly technique to incorporate a high concentration of GFs, specifically TGF-β1, PDGF-ββ, and IGF-1, into surface-activated polymeric implants. PEI and PSS were used to provide the initial charge on the polymeric devices, on top of which the GF multilayer construct was built. HEP and DS were considered as GF counter polyelectrolytes thanks to their capacity to protect and preserve the activity of GFs. Loading capacity and release rate were analysed to identify the essential parameters needed to provide an ideal delivery system for foreign body response modulation. Various factors such as surface activation, counter polyelectrolyte and polyelectrolyte deposition, time, pH, temperature and sequence were systematically tuned to achieve the ideal balance of parameters (Scheme 1). We hypothesized that the fine-tuning of strong polyelectrolyte influencers, such as available surface area and charge, and ionic strength, would result in enhanced GF loading efficiency while preserving their bioactivity. Moreover, in vitro studies using fibroblasts, measuring metabolic activity, proliferation, morphology and differentiation to myofibroblasts confirmed the bioactivity of the released GFs. Together, these studies demonstrate that our novel LBL assembly method would be an effective and affordable delivery strategy for GFs and other critical biomolecules, and has important applications in tissue engineering and other biomedical sciences.
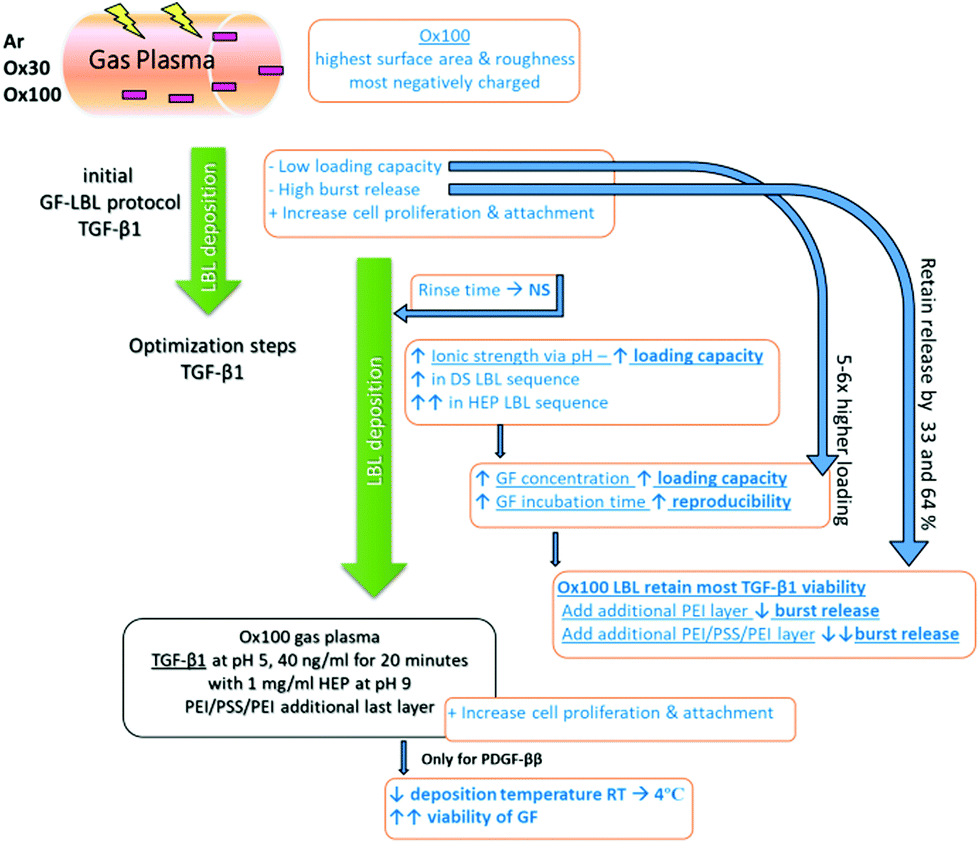 | ||
| Scheme 1 Schematic overview of the GF-LBL optimization procedure. Fabricated polymeric implant rod was firstly plasma treated to create a negatively charged surface for polyelectrolyte deposition. Evaluation showed Ox100 to have the highest surface and roughness, as well as most negatively charged. Then the initial LBL assembly procedure was performed and despite the low loading capacity and high burst release, TGF-β1 bioactivity was intact, as shown in the increased cell proliferation and attachment. A series of optimization steps (pH, concentration, deposition sequence) showed an increase of 5–6× in loading capacity and a 64% retained release. This hence resulted in an improved cell proliferation and attachment. An additional temperature optimization for PDGF-ββ was needed to maintain GF bioactivity. | ||
Experimental
Fabrication of polymeric implants
Polymeric implant rods were manufactured with a bioplotter (Envisiontec GmbH), an XYZ plotter device as previously described by Moroni et al.29 Briefly, poly(ethylene oxide terephthalate)/poly(butylene terephthalate) (PEOT/PBT) resins were loaded into a steel syringe and heated at 180–200 °C. With the computer-aided manufacturing software (CAM, PrimCAM) governing the bioplotter, fibres were extruded at 4–5 bars to fabricate cylindrical implants of 1.75 mm in diameter.Additionally, polymeric implant sheets of 500 μm thickness were prepared using a hot-embossed compression moulding technique as previously described.30 Briefly, resins of PEOT/PBT were dispersed inside circular punched moulds of thin stainless steel and sandwiched between two functionalized silicon wafers (FDTS, Sigma-Aldrich). The wafer–mould–wafer stack was positioned in the aperture of a temperature hydraulic press (Fortune Holland) at 10 bar and 180 °C. After 5 min, the press was cooled to 60 °C and the pressure was released. The mould and wafer were separated manually to provide smooth PEOT/PBT implant sheets. Both rods and sheets were used in the loading capacity, release profile and cell culture analysis, and are mentioned as “samples” to indicate that both types were used.
Surface activation and charge staining
Surface activation on PEOT/PBT implants was conducted to provide higher surface area charge and initial charge for alternate polyelectrolyte absorption. PEOT/PBT surface activation was performed by gas plasma treatment using inert argon plasma treatment (PDC-002; Harrick Plasma, Ithaca, USA) at 100 mTorr, 30 W for 45 min (Ar) or reactive oxygen plasma etching (Etch RIE Tetske, Nanolab, University of Twente) for 5 min at 100 mTorr, with a power of 30 W or 100 W (respectively, Ox30 and Ox100). After activation, charge staining was done to identify the surface charge: 0.5% of crystal violet and 0.5% safranin solution were used as the cationic and anionic dye, respectively. Activated implants were exposed to either crystal violet or safranin dye for 1 min and rinsed twice to remove the excess colour. Images were obtained using a light stereomicroscope at 1× magnification.Scanning electron microscopy (SEM) and atomic force microscopy (AFM)
The samples were gold sputtered at 40 mA and 100 mTorr for 30 s. SEM images were obtained using a Philips XL30 ESEM-FEG SEM at 10 kV and a working distance of 10 mm. Surface roughness and area analyses were performed through AFM using tapping mode (PicoScan Controller 2500, Molecular Imaging, USA) with a super sharp TESP cantilever: 42 N m−1, 320 kHz, 2–5 nm ROC, No Coatings (Bruker AFM probes). The roughness measurements (Ra, Rq, and Rmax) as well as available surface area were determined using the Scanning Probe Image Processor (SPIPTM, version 4.2.2.0) software. Measurements were performed in areas of 1 and 25 μm2. High-quality images in three dimensions (3D) of the polymer's surface before and after polyelectrolyte deposition were recorded and this was repeated three times at randomly different surface locations to verify the reproducibility of the observed characteristics.LBL assembly
Polyelectrolyte solutions of PEI (pH 3) and PSS (pH 10) were diluted in MilliQ water at a concentration of 2 mg ml−1. DS was dissolved in buffer solution (0.15 M NaCl) at pH 7 to a concentration of 1 or 2 mg ml−1. HEP was diluted in buffer solution to 1 mg ml−1 at pH 7 and later at pH 9. Powdered collagen (COL, basal isoelectric point 7.8) from calf skin was dissolved in 1 M acetic acid overnight and diluted with MilliQ water to a concentration of 0.2 mg ml−1, at pH 3 or pH 7. All the above solutions were filtered through a 0.02 μm filter before use. Sterile 0.1% bovine serum albumin (BSA) in 1× phosphate buffer solution (PBS, Invitrogen Life Technologies) was used to dilute the TGF-β1 (basal isoelectric point 8.83) concentration to 10 ng ml−1 for initial optimization studies at pH 7 and later to 40 ng ml−1 at pH 5, PDGF-ββ (basal isoelectric point 9.8) to 40 ng ml−1 at pH 5, and IGF-1 (basal isoelectric point 9.78) to 100 ng ml−1 at pH 5. GF volumes of 1 ml were used for 10 ng ml−1 concentration and 250 μl for 40 and 100 ng ml−1 to keep the amount of GF available to be loaded the same. All reagents were purchased from Sigma Aldrich, if not mentioned otherwise. All GFs were purchased from R&D Systems.All depositions were done at room temperature (RT), with the exception of PDGF-ββ, which was performed at either RT or 4 °C. PEI, PSS, HEP and DS were deposited over 10 min and rinsed with MilliQ water for 30 s. For GF deposition, Corning Costar® Ultra-Low attachment multiwell plates (Sigma Aldrich) and Eppendorf Protein LoBind Microcentrifuge Tubes (Fischer Scientific), pre-treated with positively charged PEI, were used. GFs were deposited for 10 or 20 min and then rinsed with 1× PBS solution for 5 min. Final assemblies were (COL/DS)4, (TGF-β1/DS)4, (TGF-β1/HEP)4, (PDGF-ββ/HEP)4 and (IGF-1/HEP)4 with or without the addition of PEI or PEI/PSS/PEI deposition at the end. For simplification of all final assemblies with either COL or GF, (PEI/PSS)2 coating was done beforehand. For (COL/DS)4 deposition on a flat surface, a gold plate was cleaned with H2SO4 at 150 °C for 20 min followed by washing in MilliQ water. Polyelectrolyte solutions were alternatively introduced into the measurement chamber and left in contact with the gold plate. After each adsorption step, pure water was poured into the chamber and left in contact with the plate for 1 min in order to remove the unabsorbed polyelectrolytes.
Quartz crystal microbalance (QCM) and Brunauer–Emmett–Teller (BET) assays
The (COL/DS)4 multilayer assembly process was monitored in situ and in real time by the QCM-D technique using a dissipative QCM (QCM-Z500, KSV Instruments, Finland). QSX 301 Au quartz crystals (Q-sense), having a diameter of 1.5 cm and a fundamental frequency of 5 MHz, were cleaned with H2SO4 at 150 °C for 20 min followed by washing in MilliQ water, prior to assembling in the measurement chamber. A Teflon liquid chamber with a volume of 2 ml was used in the experiments. Polyelectrolyte solutions were alternatively introduced into the measurement chamber and left in contact with the quartz sensor. After each adsorption step, pure water was poured into the chamber and left in contact with the plate for 1 minute in order to remove the unabsorbed polyelectrolytes. QCM data analysis was performed using the QCM impedance analysis software (KSV Instruments, version 3.11) in order to calculate the areal mass density for each deposited layer. The surface area of (PEI/PSS)2 deposited alone, and with addition of (TGF-β1/DS)4 and (COL/DS)4 in sequence (n = 3) was measured through BET analysis using an N2 adsorption–desorption isotherm TriStar 3000 automated gas adsorption analyser (Tristar 3000 V6.03 A).Sircol assay
The loading capacity of COL on surface-treated rods (n = 3) was measured by collagen extraction using 0.1 mg ml−1 pepsin (0.5 M acetic acid, overnight at 4 °C). The acidic extract was then neutralized with an acid neutralising reagent (Sircol, Biocolor Ltd) and incubated overnight at 4 °C in an Isolation & Concentration Reagent (Sircol, Biocolor Ltd). Quantifications were performed according to the picrosirius red-based colorimetric Sircol collagen dye binding assay kit (Biocolor Ltd) and measured at 540 nm.Enzyme-linked immunosorbent assay (ELISA)
ELISAs (n = 3) were conducted to quantify GF loading capacity in each layer and the release rate profile. Loading capacity was measured in solution (PBS or culture medium) obtained before and after GF deposition, and subtracted to determine the amount that was incorporated. For the release rate profile, implants were incubated in 1× PBS solution for 4 h, and ELISA assays were conducted at 1, 2, 3, 4, 6, 7, 9, 10, 12, 14, and 16 d according to the manufacturer's instructions (DuoSet ELISA development kit, R&D Systems Europe Ltd). The initial release profile was measured by incubating TGF-β1 implants (n = 3) in 1 ml of 1× PBS, and 100 μl was collected per time point without refreshing. After optimisation, the incubated solution was removed and refreshed for every time point. GF release profiles were fitted using the Korsmeyer–Peppas model,31 following the equation:| Q0 = Kntn |
Cell culture and cell seeding
The TK173 human renal fibroblast cell line (from human kidney and previously characterized32) and neonatal rat dermal fibroblasts (nRDFs, R106-05n, Tebu-bio Cell Application, Inc.) were cultured in a basic culture medium comprising α-MEM (Gibco), fetal bovine serum (10%, Lonza), L-glutamine (2 mM, Gibco), penicillin (100 U ml−1) and streptomycin (100 mg ml−1, Gibco). Both fibroblast cell types were expanded at an initial seeding density of 3000 cells per cm2. The culture was refreshed every 2–3 days. The cells were harvested at 80–90% confluency before trypsinisation for cell seeding.The TK173 cell line was used for the bioactivity analysis of the initial protocol with (TGF-β1/DS)4 and (TGF-β1/HEP)4. Rod implants (n = 3) of 2 cm in length were sterilized with 70% ethanol. Agarose molds (3% wt/v) were placed below the rods to prevent cell attachment to the bottom of a 12 well-plate and for optimum static cell seeding. The cell seeding density was 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells in a 250 μl volume. The samples were rinsed with 1× PBS and collected for DNA assay and SEM at 4 h for attachment and on day 4 for proliferation.
000 cells in a 250 μl volume. The samples were rinsed with 1× PBS and collected for DNA assay and SEM at 4 h for attachment and on day 4 for proliferation.
nRDFs were used to analyse the optimised protocol with (TGF-β1/HEP)4, using two seeding models to separate the effect of the implant substrate on GF release. To study the direct cell contact to the PEOT/PBT surface, as well as the release of GFs, 50000 nRDFs were seeded directly on polymeric sheets. To examine individually the effect on GFs released in medium without the presence of cell attachment, 50000 nRDFs were plated on a 24-well tissue culture plate. After 24 h, the rods were inserted into each well to provide the release of GFs. For nRDFs on both sheets and rods, DNA assays were performed on days 1 and 4, and the resulting data were used to determine the proliferation rate. For imaging, polymeric sheets were seeded with 10000 cells and examined on days 1, 4, and 7. All cell experiments were performed under a 5% CO2 humid atmosphere at 37 °C.
DNA assay
The proliferation rate from day 1 and 4 was measured using a CyQuant Cell Proliferation Assay kit (Molecular Probes). For each in vitro study, the samples (n = 3) were washed twice with 1× PBS, collected into a 500 μl Eppendorf tube and frozen at −80 °C. After undergoing three freeze-thaw cycles, 1× lysis buffer was added to the samples at RT over 1 h. The samples were incubated for an additional 1 h with lysis buffer RNase. Subsequently, cell lysate and CyQuant GR dye (1×) were mixed 1:1 in a 96-well plate and incubated in the dark for 15 min. Fluorescence was measured at excitation and emission wavelengths of 480 and 520 nm, respectively, using a spectrophotometer (the VICTOR3 Multilabel Plate Reader Perkin Elmer Corporation).
Live/dead assay, immunostaining and SEM sample preparation
:200) conjugated with goat anti-mouse Alexa Fluor® 488 (Invitrogen, 1:1000) as the secondary antibody, Phalloidin-Texas Red (Molecular Probes, 1:100) and 4′,6-diamidino-2-phenylindole (Dapi) (1:100) with multiple rinses in between and after to remove non-specific staining. Images were obtained by using a fluorescence microscope (Nikon Eclipse E600).
Statistical analysis
Statistical analysis was performed using Graphpad and expressed as mean ± standard deviation. Biochemical assays were performed in triplicate, if not stated otherwise. Statistical analysis was done by a two-way Analysis of Variance (ANOVA) with Bonferroni's multiple comparison test (p < 0.05), unless otherwise indicated in the figure legends. For all figures, the following applies: * p < 0.05, ** p < 0.01, and *** p < 0.001.Results
Substrate selection, surface activation and polyelectrolyte deposition
For LBL assembly, we selected a polymeric implant composed of poly(ethylene oxide terephthalate)/poly(butylene terephthalate) (PEOT/PBT), which has shown successful applications in several tissue regeneration applications. Effective multilayer LBL build-up requires sufficient surface area and charge. We accomplished this by surface activation using argon (Ar) and oxygen (Ox30 and Ox100) plasma treatment. We observed a rougher surface with random rough fragments with Ar, and more ordered shorter and higher peaks with Ox30 and Ox100, respectively (Fig. 1A). Ox100 provided the highest surface roughness and area increase compared to unmodified (X) PEOT/PBT controls (Table 1). For surface charge, Ox100 implants showed the highest negative surface charge, with lesser charge in Ox30 and much lesser in Ar implants, determined by crystal violet staining (Fig. 1B). Safranin staining for positively charged surfaces was not observed for any of the three activation conditions, showing that all surface activated implants have negatively charged surfaces (Fig. 1B). Hence, all surface activated implants showed potential in providing a larger capacity for LBL absorption and stronger interaction to the counter charged polyelectrolyte. | ||
| Fig. 1 Surface activation and polyelectrolyte deposition. (A) SEM images of the PEOT/PBT surface before (X) and after gas plasma treatment of argon at 30 W (Ar) and oxygen at 30 W and 100 W (Ox30 and Ox100, respectively). The non-activated X implant showed a smooth surface, while Ar, Ox30 and Ox100 treatment provided a rougher surface. Scale bars: 10 μm. (B) Stereomicroscopy image of Ox100 sheets (left, top row) and rods (left, bottom); Ox30 sheets (right, top); and Ar (right, bottom). Materials were stained with crystal violet (left sheet for each pair, and top rod in Ox100) and safranin (right sheet for each pair, bottom rod in Ox100) to detect their charge. Crystal violet staining was highest in Ox100 implants whereas no safranin staining was observed in any implants. Scale bar: 1 cm. (C) SEM image of Ar surface-activated rod deposited partially with (PEI/PSS)2 as depicted by the white arrow, where the LBL occurred. The surface treated samples provided the same depiction (not shown). Scale bar: 2 mm. (D) 3D images of AFM analysis on surface topography of Ar and Ox30-treated surfaces before (top row) and after (PEI/PSS)2 deposition (bottom row), indicating a decrease in surface roughness after LBL deposition. Image surface area: 1 μm2. | ||
| X | Ar | Ox30 | Ox100 | |
|---|---|---|---|---|
| Before deposition | ||||
| Surface area difference (%) | 2.3 ± 0.7 | 13.4 ± 3.5 | 15.1 ± 1.9 | 19.2 ± 4.7 |
| R q (nm) | 5.5 ± 0.6 | 13.1 ± 2.2 | 15.5 ± 4.5 | 54.9 ± 5.3 |
| R a (nm) | 4.7 ± 0.5 | 10.8 ± 1.7 | 13.3 ± 3.8 | 40.6 ± 3.6 |
| After (PEI/PSS) 2 | ||||
| Surface area difference (%) | 0.3 ± 0.1 | 1.8 ± 0.4 | 1.5 ± 0.5 | 2.7 ± 1.2 |
| R q (nm) | 5.2 ± 0.5 | 4.9 ± 1.1 | 10.8 ± 2.3 | 29.8 ± 3.5 |
| R a (nm) | 3.8 ± 0.4 | 3.9 ± 0.7 | 8.7 ± 1.2 | 23.4 ± 2.4 |
Next, we evaluated whether the surfaces were capable of building up polyelectrolytes, an essential parameter for LBL assembly. For these studies, we first evaluated the deposition of PEI and PSS followed by collagen (COL) and DS by QCM. The LBL deposition consists of two layers of alternating polycation PEI and polyanion PSS, referred to as (PEI/PSS)2, followed by four layers of alternating positively charged COL and negatively charged DS referred to as (COL/DS)4. The deposition sequence showed differences based on the pH of COL: we observed linear growth at pH 7 and exponential growth at pH 3 (ESI Fig. 1†). Absorbed (PEI/PSS)2 indicating LBL deposition on rods was observed by SEM (Fig. 1C and ESI Fig. 2†). Polyelectrolyte deposition can also be determined by surface area changes measured by AFM and BET. AFM analysis showed different surface topographies after (PEI/PSS)2 deposition (Fig. 1D), and the corresponding surface area and roughness measurements (Table 1) were decreased. Surface area differences before and after deposition were 116, 136 and 165 nm for every 1 μm2 surface area of Ar, Ox30 and Ox100, respectively, compared to 20 nm for controls. Surface roughness (expressed as Rq and Ra measurements) was also reduced by 2.7-, 1.5- and 1.8-fold after (PEI/PSS)2 deposition for Ar, Ox30 and Ox100, respectively. BET analysis showed a decrease in surface area, indicating the build-up of layers after polyelectrolyte deposition (ESI Fig. 3†), while collagen quantification assay displayed the amount of loaded collagen (ESI Fig. 4†).
Loading GFs on LBL and nomenclature description
Having established that (PEI/PSS)2 can be deposited in multilayers on surface-activated PEOT-PBT implants, we aimed at loading different GFs on these assemblies. We selected three cationic GFs (TGF-β1, PDGF-ββ, and IGF-1) that are chemotactically able to recruit fibroblasts to the implantation site33 and involved in the generation of tissue engineered blood vessels (TEBVs) for myofibroblast and smooth muscle cell differentiation. From the results, here on, all implants will be coated with (PEI/PSS)2 and hence for simplification, the nomenclature of (PEI/PSS)2 will be dropped. The nomenclature of, for example, Ox30 (TGF-β1/DS)4 refers to surface implants activated by Ox30, coated with (PEI/PSS)2 and further deposited with four layers of alternating growth TGF-β1 and counter polyelectrolyte DS. Furthermore, loaded layers were evaluated by deducting the initial amount by remaining amount of GF used for deposition, GF release in solution, and bioactivity of the released GF.Selecting the optimum surface activation and parameters for growth factor drug delivery substrate
To find the optimum surface activation for GF delivery, we assessed the different plasma activations and its ability to support TGF-β1 loading, control release and bioactivity. Initially, we evaluated the LBL coating methodology with two counter polyelectrolytes DS and HEP (1 mg ml−1), TGF-β1 (10 ng ml−1 at pH 7) and compared it with X, Ar, Ox30 and Ox100 treated substrates. The results showed a low average loading capacity of 8.22% ± 1.38% and 8.63% ± 1.05% for TGF-β1 with DS and HEP as the counter polyelectrolyte, respectively (Fig. 2A and B). High TGF-β1 burst release was seen in both PBS and culture medium of around 36.65% and 30.15% in implants with DS and HEP, respectively, and no significant viable release was seen further after day 1 (ESI Fig. 5†). Nevertheless, cell culture studies to analyse the effect of TGF-β1 LBL rod implants on the attachment and proliferation of TK173 cells showed positive results (ESI Fig. 6†). On day 1, (TGF-β1/DS)4 implants displayed a significantly greater number of attached cells compared to the corresponding surface-treated rods without (TGF-β1/DS)4. On day 4, cell proliferation was significantly higher with TGF-β1 implants compared to the same rods without TGF-β1, showing the impact of the loaded TGF-β1. Among the TGF-β1-coated rods, the rods surface-treated with Ox100 showed the highest cell proliferation rate. Similar results were observed with Ar-treated rods with and without (TGF-β1/HEP)4 (ESI Fig. 6B†). Furthermore, SEM analysis showed higher cell attachment on TGF-β1-coated rods compared to non-coated rods (ESI Fig. 7†).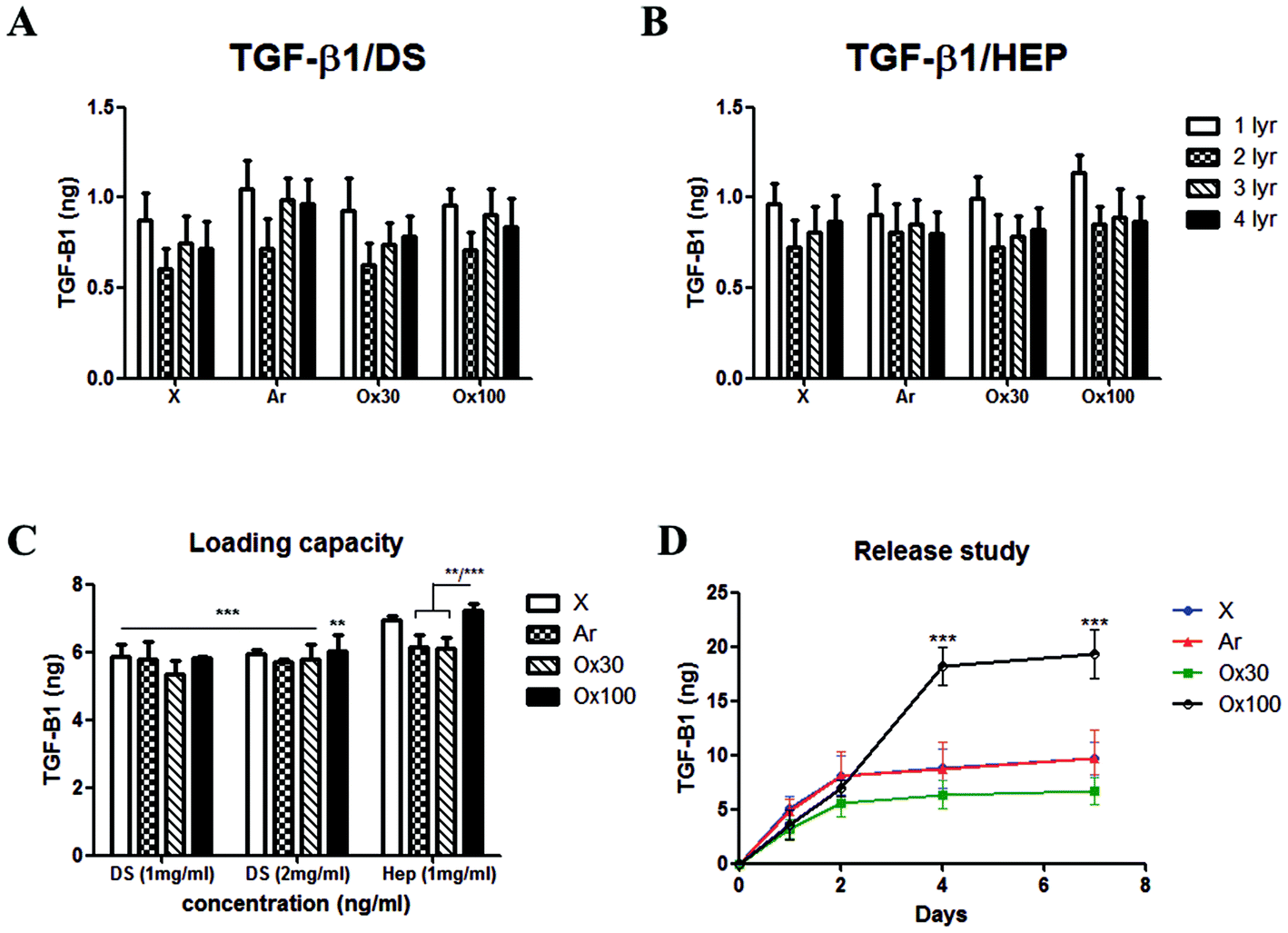 | ||
| Fig. 2 Loading capacity and release profile of different surface activation techniques (Ar, Ox30, Ox100), pH (7 or 5), and counter polyelectrolyte (DS or HEP) deposition. Loading capacity of TGF-β1 (10 ng ml−1, pH 7) in (A) (TGF-β1/DS)4 with 1 mg ml−1 DS and (B) (TGF-β1/HEP)4 implants surface-activated with Ar, Ox30, Ox100. Non-activated surfaces with (PEI/PSS)2 coating (X) were used as a control. The first layer (1 lyr) provided highest loading for all 8 conditions. (C) Loading capacity of TGF-β1 (40 ng ml−1, pH 5) on different surface activation and counter polyelectrolytes. Loading capacity was highest in Ox100-treated (TGF-β1-HEP)4 implants compared to all other samples tested. (D) Release rate profile of loaded TGF-β1 (40 ng ml−1, pH 5) from (TGF-β1/HEP)4PEI/PSS/PEI on days 1, 2, 4, and 7 showed a retained release with a significantly higher release of Ox100-treated LBL implants compared to others (*** P < 0.001). | ||
Despite the significant increase in proliferation, the above-mentioned LBL assembly still required extensive tuning. Various parameters, including the washing procedure, pH and concentration, were tuned to optimise the loading and release of TGF-β1. Firstly, changing the rinsing duration of the procedure provided no significant differences. However, increasing the ionic strength of DS and TGF-β1 by changing their pH to 9 and 5 provided a statistically significant increase in TGF-β1 loading compared to their loading efficiency at pH 7 (ESI Fig. 8A and B†). HEP counter polyelectrolyte implants had superior loading capacity, where the change in pH tripled TGF-β1 loading efficiency. Hence, for next optimization, we only used the HEP counter polyelectrolyte and coupled it with TGF-β1 solution loaded at different durations and concentrations (10 and 20 minutes; 10, 20 and 40 ng ml−1) (ESI Fig. 8C†). The volume of TGF-β1 solution used for loading is adjusted to create a fixed 10 ng of TGF-β1 for every loading layer. Increasing TGF-β1 loading solution to 40 ng ml−1 increased l TGF-β1 loading capacity, while increasing the duration of the loading to 20 minutes gave better loading reproducibility, resulting in loading capacities of 41.72, 46.41, 42.91 and 50.18% for X, Ar, Ox30 and Ox100, respectively, 5–6× higher than initial loading. Changing the pH of TGF-β1 to 3 showed no viable signal (data not shown). Comparing the loading parameters of LBL assembly with DS as the counter polyelectrolyte at different concentrations (1 and 2 mg ml−1) to HEP Ox100-treated LBL implants provided the highest loading efficiency of 72.26% (Fig. 2C).
Using these optimised conditions, we performed a cumulative release study in which (TGF-β1-HEP)4 implants were incubated in PBS, and solutions were collected, refreshed, and analysed on days 1, 2, 4, and 7 (ESI Fig. 9A†). Half of all TGF-β1 loaded was released after one day of incubation, with Ox100-treated LBL implants having the highest total release of TGF-β1. After day 7, X, Ar, and Ox30-treated LBL implants showed no significant release and hence analysis was stopped on day 7. The release profile results indicated that Ox100-treated LBL implants were able to retain the viability of TGF-β1, while the other LBL implants were not. To improve the release profile by inhibiting burst release, we added an additional layer of PEI or PEI/PSS/PEI (Fig. 2D and ESI Fig. 9B†). The additional PEI and PEI/PSS/PEI layer was able to retain TGF-β1 release by 32.61% and 64.39%, a significant decrease from no additional layer. From these collective studies, we determined the (PEI/PSS)2 LBL assembly conditions for loading and release of TGF-β1: Ox100 surface treatment, 40 ng ml−1 of TGF-β1 at pH 5 deposited for 20 min, with 1 mg ml−1 HEP at pH 9 as the counter polyelectrolyte, and an additional layer of PEI/PSS/PEI deposited.
Efficient loading and release of TGF-β1, PDGF-ββ and IGF-1 and their effect on cell proliferation
After achieving a highly efficient TGF-β1 loading, the maintenance of loaded and released TGF-β1 bioactivity was confirmed by analysing its effect on cell proliferation. An in vitro study was done to analyse separately the effect of TGF-β1 release in medium (Fig. 3A, right schematics) and cell–material interactions in combination with TGF-β1 release (Fig. 3B, left schematics) on the proliferation rate of nRDFs. All seeded wells containing TGF-β1 LBL rod implants provided statistically significantly higher proliferation than non-treated rod implants. Ox100 TGF-β1 LBL rod implants provided the highest cell proliferation compared to non-coated unmodified rods. The sheets were used instead of rods to secure maximum cell and material interactions to assess the TGF-β1 effect on adhered cells to the implant (Fig. 3B). When the cells were directly seeded onto the sheet implant, all surface treated TGF-β1 LBL sheet implants displayed a statistically significantly higher cell proliferation rate compared to non-coated sheet implants, and Ox100 provided the highest proliferation rate of cells.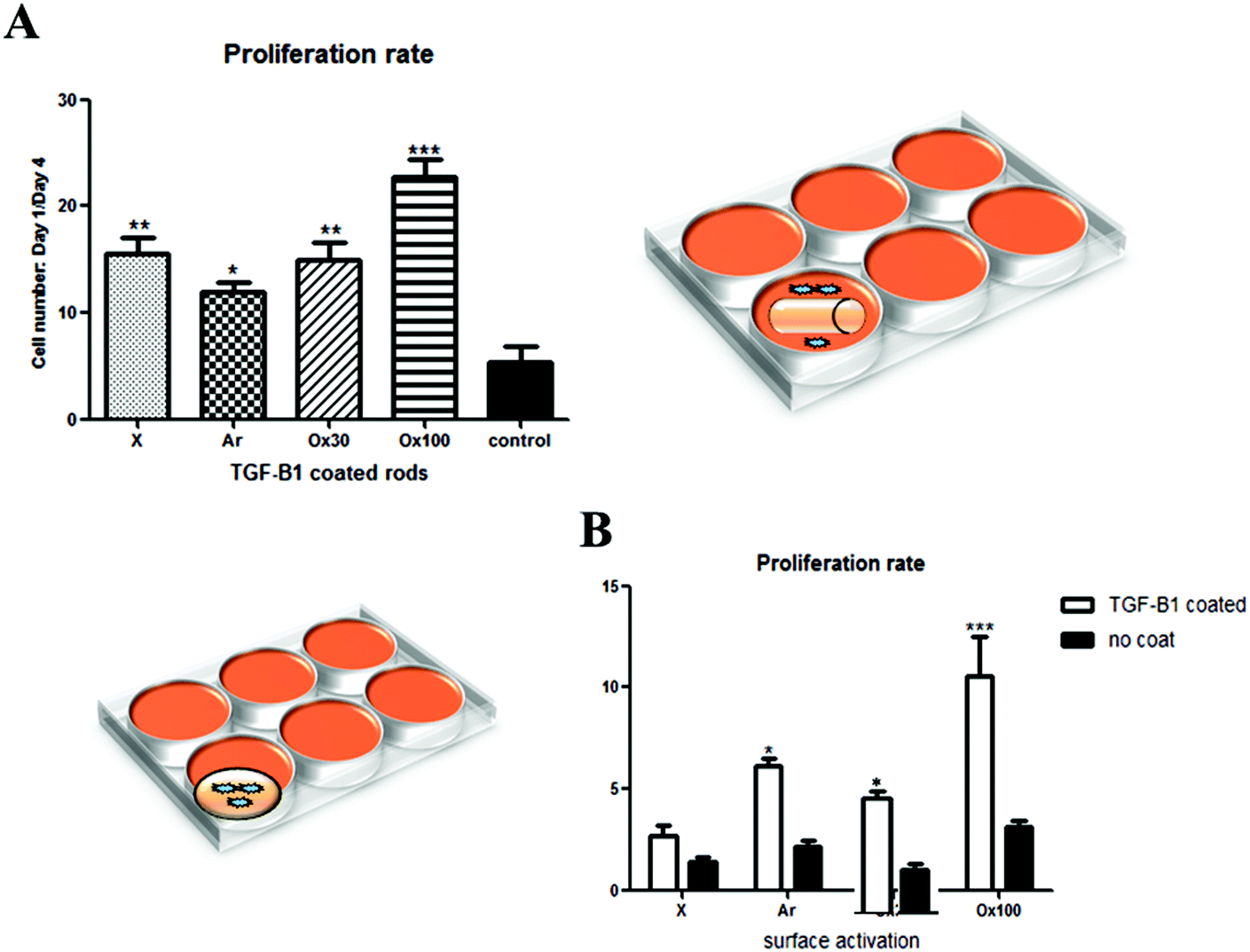 | ||
| Fig. 3 Cell proliferation study on different surface activated LBL TGF-β1 implants. (A) Proliferation rate study of nRDFs seeded on a 24 well plate with exposure to TGF-β1 LBL and non-coated rods after 24 hours of seeding to analyse the effect on cell proliferation through release of TGF-β1 in the medium. Control refers to wells with unmodified rods. Wells with TGF-β1 LBL rod implants had statistically higher proliferation compared to control. Ox100 TGF-β1 LBL rod implants provided the highest proliferation. (B) Proliferation rate analysis of nRDFs seeded directly to TGF-β1 LBL sheets to show the effect of cell–material interaction as well as TGF-β1 released in medium. Stars (*P < 0.05, ** P < 0.01, *** P < 0.001) indicate statistically significant values compared to control or non-coated implants. | ||
Due to the increase in the ionic strength of GF adhesion from the change of pH, a final optimisation step was carried out to avoid unspecific GF deposition. All further GF deposition took place in a low binding reservoir pre-coated with positively charged PEI to prevent undesirable binding of positively charged TGF-β1 onto the loading reservoir. This modification resulted in an increase in the TGF-β1 loading capacity of the implants. Fig. 4a shows a significant increase of loading capacity of 82 ± 5.8% (average of all 4 layers, with the highest loading to be 92%) on Ox100 LBL implants. Additionally, we assessed PDGF-ββ and IGF-1 loading capabilities. Fitting with the Korsmeyer–Peppas model revealed the release to be governed by a combination between diffusion and erosion.34 Indeed, the n exponent of the power equation amounts to 0.71, 0.56 and 0.74 for TGF-β1 (R2 = 0.999), PDGF-ββ (R2 = 0.988) and IGF-1 (R2 = 0.999) respectively, suggesting a non-Fickian diffusion mechanism.35 IGF-1 LBL Ox100 implants provided a high average loading capacity of 75.42 ± 9.16% and a sustainable controlled release. For PDGF-ββ, despite the obtained high loading (ESI Fig. 10†), its release was scarcely detected for both DS and HEP sequences (PDGF-ββ (RT)). A lower temperature solution of 4 °C was used to further optimize the LBL deposition of PDGF-ββ. The tuned method provided sufficient loading of PDGF-ββ of 71.4 ± 3.94% and a viable controlled release. Similar to the proliferation studies of TGF-β1 LBL implants, PDGF-ββ and IGF-1 LBL Ox100 implants were examined to investigate their effect on cells (Fig. 4c). Rods were used to examine the effect of released GF in the medium, while sheets were chosen for the maximum effect on cell and material interactions with exposure to released GF. The release of GFs alone from PDGF-ββ (4 °C) and IGF-1 LBL Ox100 rod implants displayed a statistically significant increase in proliferation. Furthermore, with the combination of cell and material interactions on PDGF-ββ (4 °C) and IGF-1 Ox100 LBL sheet implants, a statistically significant increase of cell proliferation was also observed. PDGF (RT) LBL Ox100 implants showed only a slight increase compared to control in both cell culture studies.
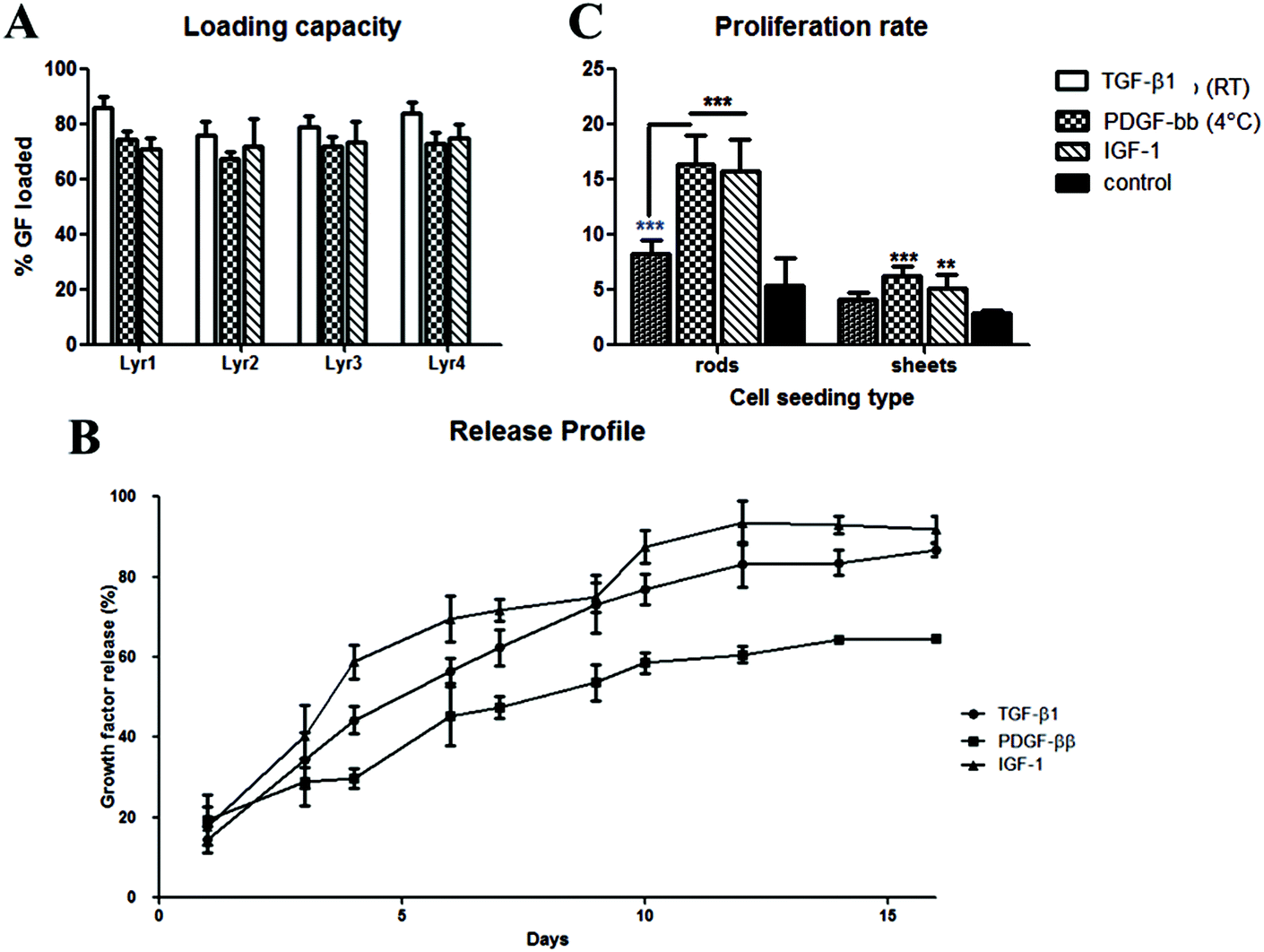 | ||
| Fig. 4 Loading capacity and release profile of TGF-β1, PDGF-ββ and IGF-1, and their effect on cell proliferation. (A–B) Optimized and tuned LBL deposition displayed high loading capacity of GFs (up to 92%, 82%, and 85% for TGF-β1, PDGF-ββ, and IGF-1 respectively) and its sustainable controlled release up to 16 days. (C) Proliferation rate of nRDF on wells exposed to the released GF from LBL rod implants and direct seeding to the LBL sheet implant. Cell proliferation on PDGF-ββ (4 °C) and IGF-1 Ox100 LBL implants displayed statistically significant higher values compared to control, with only a slight higher proliferate rate in implants with PDGF-ββ deposited in RT. Cells in both PDGF-ββ (4 °C) and IGF-1 Ox100 LBL rod implants showed higher proliferation than PDGF-ββ (RT). Control in rods refers to wells with non-coated Ox100 LBL rods, while control in sheets refers to nRDFs seeded on non-coated Ox100 sheets. Stars (*P < 0.05, ** P < 0.01, *** P < 0.001) indicate statistically significant values compared to control, while blue stars refer to statistically significant values between the GF-LBL implants. | ||
Bioactivity of TGF-β1, PDGF-ββ and IGF-1 released from GF-LBL Ox100 implants
Further bioactivity of TGF-β1, PDGF-ββ and IGF-1 released from LBL implant sheets can be analysed by cell staining to examine cell behaviour (Fig. 5). Cell spreading of nRDFs improved with enhanced actin filaments when exposed to GF-LBL Ox100 implants, compared to non-coated implants. To be able to show a possible effect on cell migration, Ox100 sheets were only coated with PDGF-ββ at the edge of the circumference. GF-LBL Ox100 implants induced fibroblast migration, as seen in PDGF-ββ LBL Ox100 implants through elongated filopodia and nRDF attachment protruding and migrating from the center of the implants, where the cells were seeded, towards the source of released PDGF-ββ at the edge of the sheet. GF-LBL Ox100 implants displayed viable cell attachment on days 4 and 7. Cell staining supported DNA assay in which LBL implants provided a high proliferation rate compared to non-coated implants. Finally, LBL Ox100 implants loaded with GFs such as TGF-β1 showed a higher population of α-SMA positive cells compared to LBL implants without GF. Hence, effectively loaded GF-LBL implants provided a sustainable controlled release of GF with intact bio-functionalities (Scheme 2).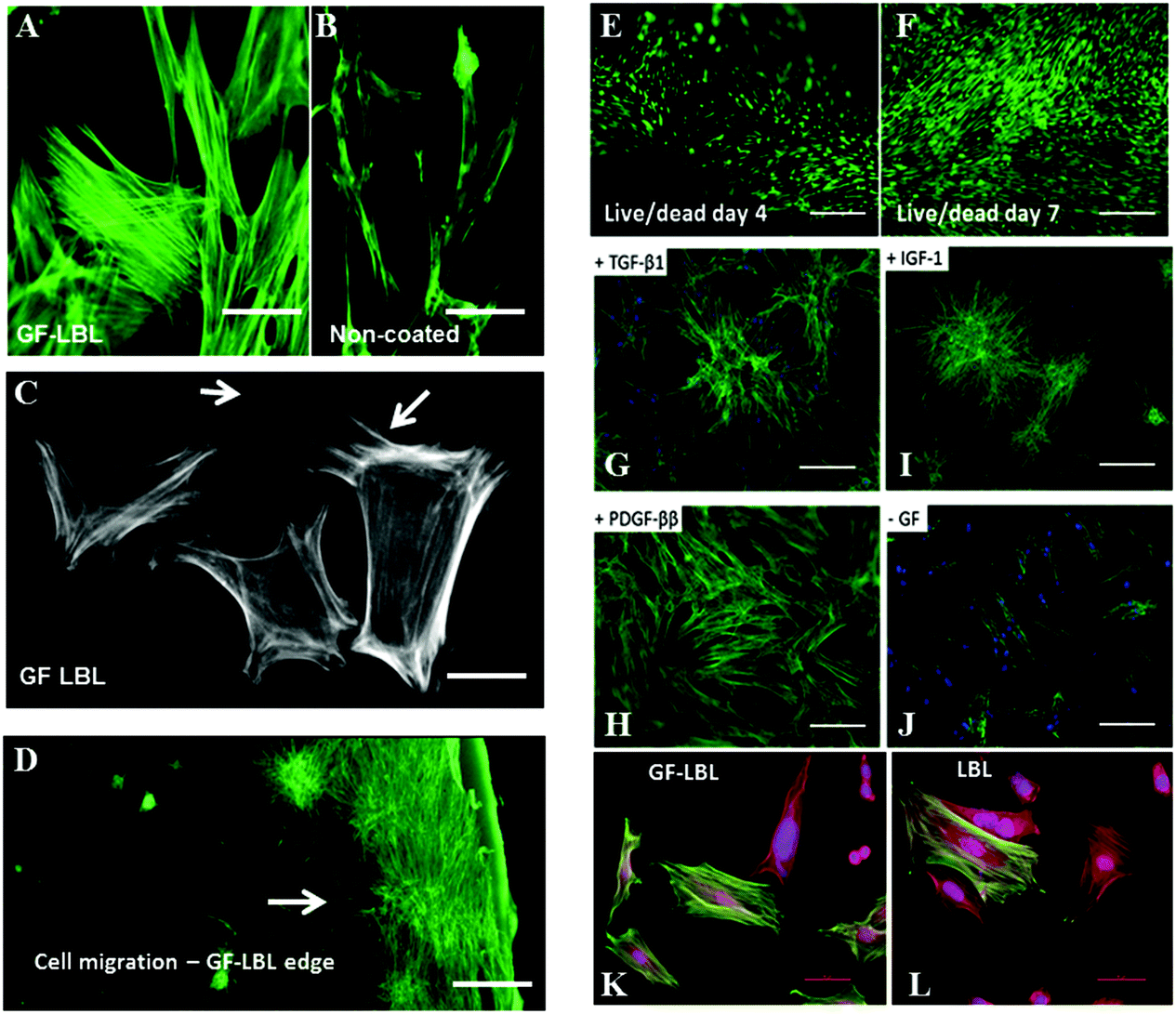 | ||
| Fig. 5 Cell staining images of nRDFs seeded on GF-LBL and non-coated sheet implants. (a) LBL implants showed enhanced actin filaments compared to (b) non-coated implants. (c) Cell morphology staining of actin filaments on PDGF-ββ LBL implants showed elongated filopodia. (d) Cell staining showed nRDFs migration and attachment on the coated area (edge) of the GF-LBL implant. (e and f) Live dead assay displayed live cells (green), and no attached dead cells (red) on GF-LBL implants on days 4 and 7. (g–j) Cell staining on day 4 showed high cell attachment in TGF-β1, PDGF-ββ and IGF-1 implants compared to non-coated implants. Star-like groups of cells were seen on TGF-β1 and IGF-β1 implants. (k and l) α-SMA staining on GF-LBL displays a higher amount of cells positive with α-SMA compared to LBL implants without TGF-β1. Scale bar: (a and b) 20 μm, (c) 10 μm, (d) 200 μm, (e and f) 10 μm, (g–j) 50 μm, (k and l) 20 μm. | ||
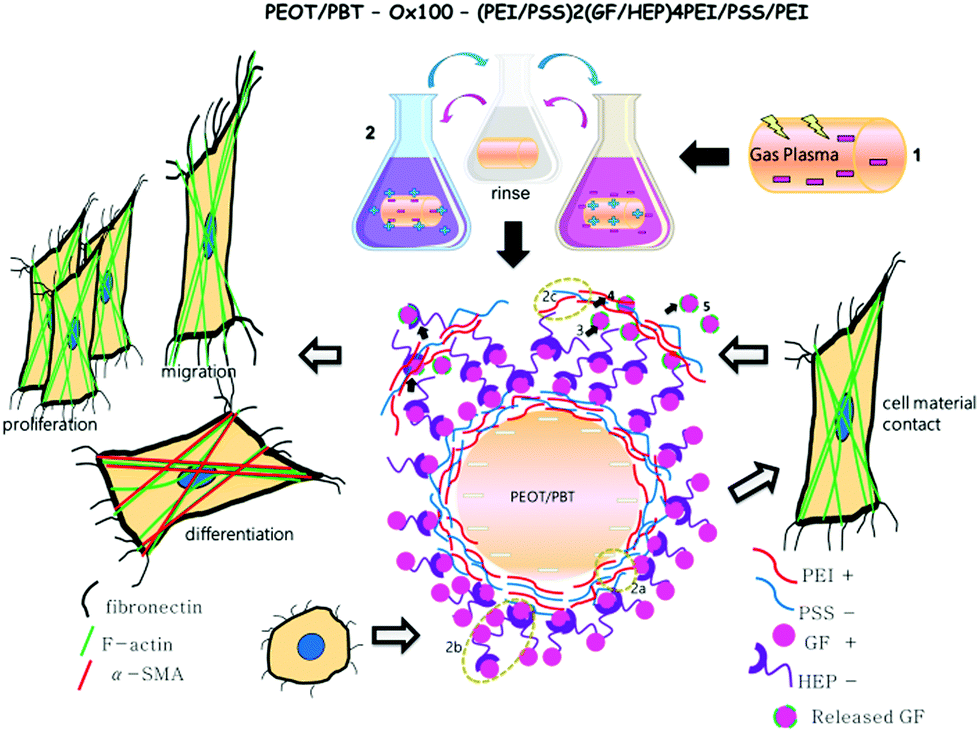 | ||
| Scheme 2 Schematic overview of the GF-LBL procedure, mechanism and cellular response upon seeding HDF on LBL implants. (1) The fabricated polymeric implant rod was first gas plasma treated to create a negatively charged surface for polyelectrolyte deposition. (2) Polyelectrolyte deposition step comprises incubating the implant to positively and negatively charged solutions of PEI and PSS (2a, first 4 layers), GF and HEP (2b, next 8 layers) and again PEI and PSS (2c, for the last 3 layers). (3–5) In culture medium, GF starts to escape from the LBL assembly, passing through the top PEI-PSS-PEI layer by diffusion. Upon seeding HDF on the LBL implant, the response to the initial cell–material contact and additionally sensing GFs release to the culture medium influence cell migration, proliferation and differentiation. | ||
Discussion
LBL assembly has been the substantial focus in many research fields for its ability to have nanometre control over film thickness, which corresponds to the quantity of the loaded targeted drug.2 Different methods to analyse layer deposition were used depending on the substrate and polyelectrolyte type. QCM has been the standard technique to monitor LBL deposition and provide the ideal parameters for layer deposition. However, QCM requires a flat conducting substrate.36 Hence, quartz crystals with gold electrodes were used to examine the (COL/DS)4 sequence, as the basis template for the optimization of the LBL assembly technique specialized for GF delivery, in which COL is then substituted with a GF of interest. When transferring this sequence to polymeric rods or sheets other techniques need to be explored. As we have investigated here, SEM analysis can provide insight into changes in topography and charge qualitatively,37 while other techniques such as AFM and BET can provide quantitative analysis on the surface area, which corresponds to the quantity of layer deposition. AFM measures actual surface area, roughness as well as indentation experiments to investigate the adhesion of the polyelectrolytes,38,39 but is limited to microscale areas, while BET provides a larger overview of surface analysis but measurements of surface area are superficial and time consuming for small areas.40 With regard to GF loading, a straightforward technique would be to obtain the difference of deposition solution before and after each layer coating, and measure it with ELISA. Release and cell culture studies provided added value for the indication of viable GF release.The initial setup of depositing a neutral pH polyelectrolyte with a standard incubation time and volume/concentration setup onto our GF-LBL implants provided a similar loading efficiency of many LBL assemblies41 and a statistically significant increase in cell proliferation. Nevertheless, optimisation by tuning of the LBL parameters shown in this study has provided a much higher loading capacity than the so-called “high density drug loading” often ranging between 10–40%.41 Deposition parameters such as time, temperature, pH and concentration have been tuned to provide the ideal conditions for the fabrication of a specific functional LBL assembly. Low influencers,23 such as GF concentration, provided a significant increase of loading capacity only on specific surface treated implants, namely Ar and Ox100, while the deposition time provided no significant differences in loading capacity, though beneficially reduced the loading deviation of samples. Another tuneable parameter is temperature, which is known to be a strong influencer. PDGF-ββ is a highly sensitive growth factor with a life-time of seconds to a couple of minutes.42 For this reason, PDGF-ββ deposition using a colder temperature solution can preserve the lifetime of PDGF-ββ up until binding with HEP, allowing it to shrink the existing deposited layers. However, as temperature increased back to RT, the deposited layers swell, which allows an increased absorption of PDGF-ββ within the existing deposited layers.43 Ionic strength, a strong influencer as well, provided a statistically significant increase in loading capacity when tuning the pH of GFs and the other polyelectrolytes, as seen also in COL-LBL deposition. When loading sensitive proteins such as GFs, however, care must be taken to avoid the risk of degradation.44 Despite the increased loading capacity of collagen, TGF-β1 was denatured at pH 3, but not at pH 5. This is similarly seen with FGF, in which the GF seemed to be unstable at pH below 5.45 Aggregation could have also developed and most likely reduced loading capacity. Additionally, the pH change could provoke the swelling of the deposited structure resulting in increased wall permeability and diffusion,46 which enhance the time of release if desired. Besides parameter tuning, attention must be given to unwanted absorbance on deposition reservoirs due to high GF ionic strength. This could be avoided by using low binding positively charged loading reservoirs, as observed in this study. Moreover, anionic sulphated polysaccharides such as HEP and DS have specific biological sites to immobilise GF,47 thus providing a secondary interaction for GF stability while being exposed to physiological or even harsh conditions.
GF delivery systems with sustainable controlled release is crucial, due to GF's short half-life and harmful side effects in undesirable concentrations.24 LBL assemblies have been known for their initial burst release due to diffusion being the main mechanism of release.48 Additional deposition of PEI and PSS minimised initial burst release. This concept is similarly observed by Tan et al.,49 who described the correlation between the polyelectrolyte layers and release time of the targeted drug. Additionally, our optimised technique explored the benefits of “mixed” interactions of strong and weak polyelectrolytes. PEI and PSS are strong polyelectrolytes while GFs, COL, HEP and DS are less strong. As mentioned by Glinel et al., the exponential growth of multilayers could be due to weakly charged polyelectrolytes, while strong interactions offer a linear growth.46 When combining this observation with the design of Garza et al.50 of alternating exponentially growing polyelectrolyte multilayers acting as reservoirs and linearly growing polyelectrolyte layer as a barrier, the LBL sequence of (PEI/PSS)2/(GF/HEP)4 provided the ideal template as a protein delivery system. In addition to increase loading by adding HEP into the LBL sequence, HEP has been known to potentiate TGF-β1 biological activity. Models have been described in which HEP could bind at two distinct sites of the TGF-β dimer.51 This interaction tripled the half-life by protecting TGF-β1 from proteolytic degradation and doubled the amount of cell-associated TGF-β1 in vitro.52 Moreover, HEP provided a better bond in protecting PDGF-ββ than DS, as shown in the release profile. HEP has also been shown to enhance PDGF-ββ intracellular signalling and chemotaxis of fibroblasts.53 Furthermore, HEP plays a role in regulating IGF-1 and IGF-1 binding protein complexes. The fusion of HEP binding domains and IGF-1 can trigger proteoglycan synthesis in cartilage54 and HEP can inhibit the IGF-1 and IGF binding protein complex, thus making IGF-1 available to bind its receptor. By the combination of strong and compact additional layers’ ionic and biological interactions, our optimised GF-LBL implants can sustain a controlled release for up to 14 days. Although we haven't performed further assays, we expect the release to be continued for longer times as the loaded quantity of GFs was not exhausted in the tested time window.
Another factor that seemed to have affected the GF release profile is the topography and the initial base charge of the implant due to surface activation. Gas plasma treatment can selectively modify surface properties for enhanced deposition without effecting the bulk characteristics of the implant.55 A prerequisite for achieving a stable LBL implant resides in introducing reactive groups on the substrate's surface through gas plasma treatment.1 Despite having similar roughness and surface area availability for polyelectrolyte absorption, active oxygen plasma treatment (Ox30) provided a more charged surface than inert Ar. This explains the greater decrease of surface area difference before and after deposition on Ox30 implants. High power oxygen plasma treated implants (Ox100) provided a higher surface area and charge, and triumphed over all the other surface plasma treated surfaces shown by its increase in loading and release rate profile. This is in accordance with the fact that electrostatic interaction as well as polyelectrolyte bridging phenomena are the main driving force in the formation of multi-layered structures of oppositely charged electrolytes on a substrate, leading – as shown in this study – to increased loading efficiency for a more charged surface. Cell studies on GF-LBL Ox100 implants provided higher cell proliferation, emphasising the importance of initial ionic strength for effective drug delivery. However, despite the need for initial ionic strength for the stability of an LBL build-up, the significance of the initial ionic strength could be overwhelmed by a stronger influencer of the initial strong polyelectrolyte absorption, being here PEI and PSS.20 This was proven from the no significant difference seen between TGF-β1 in X and Ox100 implants with regard to GF loading. Nevertheless, the surface topography and roughness of the Ox100 substrate may have provided a way of securely loading and retaining the GFs to provide controlled released and intact bio-functionalities. Future studies investigating the mechanism of how surface topography coupled with surface roughness and charge could affect the release of drug loading LBL assembly would be beneficial for providing additional tuning parameters for controlled LBL delivery systems.
Released GFs can instruct cell behaviour through binding of transmembrane cell receptors. Hence, the low GF release from GF-LBL Ox100 implants (ESI Fig. 5†) that were uncounted for, possibly due to a short half-life,24 could however be immediately taken up by the seeded fibroblasts avoiding unnecessary loss. This could explain the higher cell proliferation on PDGF-ββ (RT) LBL Ox100 implants compared to control, despite their low measured release profile. The bioactivity of released GFs could also be analysed through cell imaging studies. GF-LBL Ox100 implants provided an ideal and biocompatible template for fibroblasts to grow as shown by cell viability and morphology. The last layer of PEI could be the reason for increased cell anchoring due to its cationic nature and interaction with the negatively charged outer surface of cells.56 Nevertheless, further studies to measure the amount of PEI possibly being released from the LBL system would be beneficial as it has been known to be toxic in high concentrations.56 The chemotactic function of released GFs was investigated and confirmed through elongated filopodia.57 Further confirmation was displayed by the migration of fibroblasts to the release source of PDGF-ββ, similarly shown by Grinnell et al.58 Besides higher proliferation, TFG-β1 and IGF-1 LBL Ox100 implants displayed adhered fibroblast forming colonies that could possibly suggest active collagen secretion.59,60 While PDGF-ββ showed the distribution of active proliferation, GFs have been known to be capable of inducing cell growth. High expression of collagen has been shown to allow significant build-up of the extracellular matrix (ECM) promoting fibroblast proliferation.61 Nonetheless, additional collagen measurements would be useful to understand which mechanism drives proliferation.
Extracellular matrix (ECM) is crucial to provide cell adhesion, proliferation, migration and differentiation,62 leading to successful tissue regeneration. Differentiated myofibroblasts are characterized by the increased production of ECM proteins, such as collagen and by the development of α-SMA-positive stress fibres that are connected with the ECM.63 During wound healing, myofibroblasts are pivotal as primary extracellular matrix (ECM)-secreting cells are involved in wound strengthening and contractility.64 α-SMA, a myofibroblast marker, was observed more in GF-LBL implants compared to LBL implants without GF incorporation. TGF-β1, in the presence of mechanical stress such as rigid polymeric implants, leads to the further differentiation of myofibroblasts by inducing de novo expression of α-SMA.65
Implants composed of PEOT/PBT block copolymers provide easy tunability by varying the PEOT/PBT ratio and length of the PEO segment, and for its in vitro and in vivo biocompatibility.66,67 We also have previously investigated PEOT/PBT rods as an in vivo bioreactor device and showed their ability to modulate the foreign body response,30 in which various GFs play an important role. By combining these features, our GF-LBL implant can be a promising application for induced and controlled tissue regeneration.
Conclusion
We have optimised an LBL assembly technique to effectively incorporate GFs on polymeric implants and provide a controlled sustainable release. We investigated in a systematic manner different tuneable parameters, such as the time and concentration of weak influencers, and the ionic strength and temperature of strong influencers, to enhance the loading capacity of GFs. Taking advantage of the “mixed” interaction of strong and weak polyelectrolytes, secondary interaction through biological site binding, and substrate surface properties, the optimal conditions to provide a sustainable controlled release of different GFs were discovered. In vitro studies displayed viable, highly proliferating, well anchored, α-SMA positive cells on GF-LBL Ox100 implants, confirming the bioactivity of the released GFs. Hence, the developed GF-LBL technique represents an excellent method for preparing the next generation of highly functional materials for tissue regeneration applications.Conflicts of interest
Authors declare no conflicts of interest.Acknowledgements
This research forms part of the Project P3.03 DialysisXS of the research program of the BioMedical Materials institute, co-funded by the Dutch Ministry of Economic Affairs, Agriculture and Innovation. The financial contribution of the Nierstichting Nederland is gratefully acknowledged. This research project was also possible, thanks to the Dutch Province of Limburg.References
- G. Decher and J.-D. Hong, Buildup of ultrathin multilayer films by a self-assembly process, 1 consecutive adsorption of anionic and cationic bipolar amphiphiles on charged surfaces, Makromol. Chem., Macromol. Symp., 1991, 46(1), 321–327 CrossRef CAS.
- J. J. Richardson, M. Bjornmalm and F. Caruso, Multilayer assembly. Technology-driven layer-by-layer assembly of nanofilms, Science, 2015, 348(6233), aaa2491 CrossRef PubMed.
- S. Park, U. Han, D. Choi and J. Hong, Layer-by-layer assembled polymeric thin films as prospective drug delivery carriers: design and applications, Biomater. Res., 2018, 22, 29 CrossRef PubMed.
- P. A. Chiarelli, M. S. Johal, J. L. Casson, J. B. Roberts, J. M. Robinson and H. L. Wang, Controlled Fabrication of Polyelectrolyte Multilayer Thin Films Using Spin-Assembly, Adv. Mater., 2001, 13(15), 1167–1171 CrossRef CAS.
- J. B. Schlenoff, S. T. Dubas and T. Farhat, Sprayed Polyelectrolyte Multilayers, Langmuir, 2000, 16(26), 9968–9969 CrossRef CAS.
- P. R. Van Tassel, Polyelectrolyte adsorption and layer-by-layer assembly: Electrochemical control, Curr. Opin. Colloid Interface Sci., 2012, 17(2), 106–113 CrossRef CAS.
- H. J. Kim, K. Lee, S. Kumar and J. Kim, Dynamic sequential layer-by-layer deposition method for fast and region-selective multilayer thin film fabrication, Langmuir, 2005, 21(18), 8532–8538 CrossRef CAS PubMed.
- H. Lee, Y. Lee, A. R. Statz, J. Rho, T. G. Park and P. B. Messersmith, Substrate-Independent Layer-by-Layer Assembly by Using Mussel-Adhesive-Inspired Polymers, Adv. Mater., 2008, 20(9), 1619–1623 CrossRef CAS PubMed.
- T. Paulraj, N. Feoktistova, N. Velk, K. Uhlig, C. Duschl and D. Volodkin, Microporous polymeric 3D scaffolds templated by the layer-by-layer self-assembly, Macromol. Rapid Commun., 2014, 35(16), 1408–1413 CrossRef CAS.
- A. J. Mateos, A. A. Cain and J. C. Grunlan, Large-Scale Continuous Immersion System for Layer-by-Layer Deposition of Flame Retardant and Conductive Nanocoatings on Fabric, Ind. Eng. Chem. Res., 2014, 53(15), 6409–6416 CrossRef CAS.
- Y. B. Truong, V. Glattauer, K. L. Briggs, S. Zappe and J. A. Ramshaw, Collagen-based layer-by-layer coating on electrospun polymer scaffolds, Biomaterials, 2012, 33(36), 9198–9204 CrossRef CAS PubMed.
- Z. J. Deng, S. W. Morton, E. Ben-Akiva, E. C. Dreaden, K. E. Shopsowitz and P. T. Hammond, Layer-by-layer nanoparticles for systemic codelivery of an anticancer drug and siRNA for potential triple-negative breast cancer treatment, ACS Nano, 2013, 7(11), 9571–9584 CrossRef CAS.
- H. Zhao, P. Yang, J. Deng, L. Liu, J. Zhu, Y. Sui, J. Lu and W. Yang, Fabrication of a molecular-level multilayer film on organic polymer surfaces via chemical bonding assembly, Langmuir, 2007, 23(4), 1810–1814 CrossRef CAS.
- X. Liu, S. Tang, H.-K. Choi and H.-S. Choi, Effect of plasma-treated polymer substrates on fabricating surface microsystems through LbL coating, Macromol. Res., 2010, 18(5), 413–420 CrossRef CAS.
- N. A. Kotov, I. Dekany and J. H. Fendler, Layer-by-Layer Self-Assembly of Polyelectrolyte-Semiconductor Nanoparticle Composite Films, J. Phys. Chem., 1995, 99(35), 13065–13069 CrossRef CAS.
- J.-A. He, R. Valluzzi, K. Yang, T. Dolukhanyan, C. Sung, J. Kumar, S. K. Tripathy, L. Samuelson, L. Balogh and D. A. Tomalia, Electrostatic Multilayer Deposition of a Gold–Dendrimer Nanocomposite, Chem. Mater., 1999, 11(11), 3268–3274 CrossRef CAS.
- S. Ye, C. Wang, X. Liu and Z. Tong, Multilayer nanocapsules of polysaccharide chitosan and alginate through layer-by-layer assembly directly on PS nanoparticles for release, J. Biomater. Sci., Polym. Ed., 2005, 16(7), 909–923 CrossRef CAS PubMed.
- Y. Lvov, G. Decher and G. Sukhorukov, Assembly of thin films by means of successive deposition of alternate layers of DNA and poly(allylamine), Macromolecules, 1993, 26(20), 5396–5399 CrossRef CAS.
- P. J. Yoo, K. T. Nam, J. Qi, S. K. Lee, J. Park, A. M. Belcher and P. T. Hammond, Spontaneous assembly of viruses on multilayered polymer surfaces, Nat. Mater., 2006, 5(3), 234–240 CrossRef CAS.
- A. Delcorte, P. Bertrand, E. Wischerhoff and A. Laschewsky, Adsorption of Polyelectrolyte Multilayers on Polymer Surfaces, Langmuir, 1997, 13(19), 5125–5136 CrossRef CAS.
- A. M. Aslandaş, Y. Onganer and K. Meral, Polyelectrolytes-assisted layer-by-layer assemblies of graphene oxide and dye on glass substrate, RSC Adv., 2015, 5(23), 18051–18056 RSC.
- 5 – Self-assembling in natural, synthetic, and hybrid materials with applications in controlled drug delivery, in Controlled Drug Delivery, ed. M. A. Mateescu, P. Ispas-Szabo and E. Assaad, Woodhead Publishing, 2015, pp. 163–223 Search PubMed.
- S. T. Dubas and J. B. Schlenoff, Factors Controlling the Growth of Polyelectrolyte Multilayers, Macromolecules, 1999, 32(24), 8153–8160 CrossRef CAS.
- K. Lee, E. A. Silva and D. J. Mooney, Growth factor delivery-based tissue engineering: general approaches and a review of recent developments, J. R. Soc., Interface, 2011, 8(55), 153–170 CrossRef CAS.
- P. Gentile, I. Carmagnola, T. Nardo and V. Chiono, Layer-by-layer assembly for biomedical applications in the last decade, Nanotechnology, 2015, 26(42), 422001 CrossRef CAS.
- H. S. Bernstein, Tissue Engineering in Regenerative Medicine, Humana Press, 2011 Search PubMed.
- M. L. Macdonald, R. E. Samuel, N. J. Shah, R. F. Padera, Y. M. Beben and P. T. Hammond, Tissue integration of growth factor-eluting layer-by-layer polyelectrolyte multilayer coated implants, Biomaterials, 2011, 32(5), 1446–1453 CrossRef CAS.
- T. Katagiri, S. Tsukamoto, K. Osawa and S. Kokabu, Ligand–Receptor Interactions and Their Implications in Delivering Certain Signaling for Bone Regeneration, 2015, pp. 1–15 Search PubMed.
- L. Moroni and L. P. Lee, Micropatterned hot-embossed polymeric surfaces influence cell proliferation and alignment, J. Biomed. Mater. Res., Part A, 2009, 88(3), 644–653 CrossRef.
- F. F. Damanik, T. C. Rothuizen, C. van Blitterswijk, J. I. Rotmans and L. Moroni, Towards an in vitro model mimicking the foreign body response: tailoring the surface properties of biomaterials to modulate extracellular matrix, Sci. Rep., 2014, 4, 6325 CrossRef CAS.
- G. Singhvi and R. Singh, In vitro drug release characterization models, Int. J. Pharm. Stud. Res., 2011, II(1), 77–84 Search PubMed.
- G. A. Muller, Human renal fibroblast cell lines (tFKIF and tNKF) are new tools to investigate pathophysiologic mechanisms of renal interstitial fibrosis, Exp. Nephrol., 1995, 3, 127–133 CAS.
- P. Teller and T. K. White, The physiology of wound healing: injury through maturation, Surg. Clin. North Am., 2009, 89(3), 599–610 CrossRef.
- T. Nandi, A. Rahman, N. Jahan, R. Islam and O. Pavel, Determination of in vitro release kinetics of metformin hydrochloride from six brands of metformin hydrochloride tablets available in Bangladesh using water media: A UV spectroscopic analysis, Spec. J. Med. Res. Health Sci., 2017, 2(2), 8–16 CrossRef.
- J. H. Petropoulos and P. P. Roussis, Study of ‘non-Fickian’ diffusion anomalies through time lags. I. Some time-dependent anomalies, J. Chem. Phys., 1967, 47(4), 1491–1496 CrossRef CAS.
- K. A. Marx, Quartz crystal microbalance: a useful tool for studying thin polymer films and complex biomolecular systems at the solution-surface interface, Biomacromolecules, 2003, 4(5), 1099–1120 CrossRef CAS.
- S. Sunny, N. Vogel, C. Howell, T. L. Vu and J. Aizenberg, Lubricant-Infused Nanoparticulate Coatings Assembled by Layer-by-Layer Deposition, Adv. Funct. Mater., 2014, 24(42), 6658–6667 CrossRef CAS.
- A. Díez-Pascual and P. Shuttleworth, Layer-by-Layer Assembly of Biopolyelectrolytes onto Thermo/pH-Responsive Micro/Nano-Gels, Materials, 2014, 7(11), 7472–7512 CrossRef.
- O. Mermut, J. Lefebvre, D. G. Gray and C. J. Barrett, Structural and Mechanical Properties of Polyelectrolyte Multilayer Films Studied by AFM, Macromolecules, 2003, 36(23), 8819–8824 CrossRef CAS.
- S. M. Ubnoske, Q. Peng, E. R. Meshot, C. B. Parker and J. T. Glass, Protocol for High-Sensitivity Surface Area Measurements of Nanostructured Films Enabled by Atomic Layer Deposition of TiO2, J. Phys. Chem. C, 2015, 119(46), 26119–26127 CrossRef CAS.
- P. T. Hammond, Building biomedical materials layer-by-layer, Mater. Today, 2012, 15(5), 196–206 CrossRef CAS.
- H. Seppa, Platelet-derived growth factor in chemotactic for fibroblasts, J. Cell Biol., 1982, 92(2), 584–588 CrossRef CAS.
- H. L. Tan, M. J. McMurdo, G. Pan and P. G. Van Patten, Temperature Dependence of Polyelectrolyte Multilayer Assembly, Langmuir, 2003, 19(22), 9311–9314 CrossRef CAS.
- M. Vazquez-Rey and D. A. Lang, Aggregates in monoclonal antibody manufacturing processes, Biotechnol. Bioeng., 2011, 108(7), 1494–1508 CrossRef CAS.
- H. Fan, S. N. Vitharana, T. Chen, D. O'Keefe and C. R. Middaugh, Effects of pH and polyanions on the thermal stability of fibroblast growth factor 20, Mol. Pharm., 2007, 4(2), 232–240 CrossRef CAS.
- K. Glinel, C. Déjugnat, M. Prevot, B. Schöler, M. Schönhoff and R. V. Klitzing, Responsive polyelectrolyte multilayers, Colloids Surf., A, 2007, 303(1–2), 3–13 CrossRef CAS.
- X. Liu, Y. Won and P. X. Ma, Surface modification of interconnected porous scaffolds, J. Biomed. Mater. Res., Part A, 2005, 74(1), 84–91 CrossRef.
- R. C. Smith, M. Riollano, A. Leung and P. T. Hammond, Layer-by-layer platform technology for small-molecule delivery, Angew. Chem., Int. Ed., 2009, 48(47), 8974–8977 CrossRef CAS.
- J. P. Tan, Q. Wang and K. C. Tam, Control of burst release from nanogels via layer by layer assembly, J. Controlled Release, 2008, 128(3), 248–254 CrossRef CAS.
- J. M. Garza, N. Jessel, G. Ladam, V. Dupray, S. Muller, J. F. Stoltz, P. Schaaf, J. C. Voegel and P. Lavalle, Polyelectrolyte multilayers and degradable polymer layers as multicompartment films, Langmuir, 2005, 21(26), 12372–12377 CrossRef CAS PubMed.
- M. Lyon, G. Rushton and J. T. Gallagher, The Interaction of the Transforming Growth Factor- s with Heparin/Heparan Sulfate Is Isoform-specific, J. Biol. Chem., 1997, 272(29), 18000–18006 CrossRef CAS.
- T. A. McCaffrey, D. J. Falcone, D. Vicente, B. Du, S. Consigli and W. Borth, Protection of transforming growth factor-beta 1 activity by heparin and fucoidan, J. Cell Physiol., 1994, 159(1), 51–59 CrossRef CAS.
- C. Rolny, D. Spillmann, U. Lindahl and L. Claesson-Welsh, Heparin amplifies platelet-derived growth factor (PDGF)- BB-induced PDGF alpha -receptor but not PDGF beta -receptor tyrosine phosphorylation in heparan sulfate-deficient cells. Effects on signal transduction and biological responses, J. Biol. Chem., 2002, 277(22), 19315–19321 CrossRef CAS.
- R. E. Miller, A. J. Grodzinsky, K. Cummings, A. H. Plaas, A. A. Cole, R. T. Lee and P. Patwari, Intraarticular injection of heparin-binding insulin-like growth factor 1 sustains delivery of insulin-like growth factor 1 to cartilage through binding to chondroitin sulfate, Arthritis Rheum., 2010, 62(12), 3686–3694 CrossRef CAS.
- D. Hegemann, H. Brunner and C. Oehr, Plasma treatment of polymers for surface and adhesion improvement, Nucl. Instrum. Methods Phys. Res., Sect. B, 2003, 208, 281–286 CrossRef CAS.
- A. R. Vancha, S. Govindaraju, K. V. Parsa, M. Jasti, M. Gonzalez-Garcia and R. P. Ballestero, Use of polyethyleneimine polymer in cell culture as attachment factor and lipofection enhancer, BMC Biotechnol., 2004, 4, 23 CrossRef.
- P. K. Mattila and P. Lappalainen, Filopodia: molecular architecture and cellular functions, Nat. Rev. Mol. Cell Biol., 2008, 9(6), 446–454 CrossRef CAS.
- F. Grinnell, L. B. Rocha, C. Iucu, S. Rhee and H. Jiang, Nested collagen matrices: a new model to study migration of human fibroblast populations in three dimensions, Exp. Cell Res., 2006, 312(1), 86–94 CAS.
- X. Pan, Z. Chen, R. Huang, Y. Yao and G. Ma, Transforming growth factor beta1 induces the expression of collagen type I by DNA methylation in cardiac fibroblasts, PLoS One, 2013, 8(4), e60335 CrossRef CAS.
- C. D. Blackstock, Y. Higashi, S. Sukhanov, S. Y. Shai, B. Stefanovic, A. M. Tabony, T. Yoshida and P. Delafontaine, Insulin-like growth factor-1 increases synthesis of collagen type I via induction of the mRNA-binding protein LARP6 expression and binding to the 5′ stem-loop of COL1a1 and COL1a2 mRNA, J. Biol. Chem., 2014, 289(11), 7264–7274 CrossRef CAS.
- V. J. Thannickal, D. Y. Lee, E. S. White, Z. Cui, J. M. Larios, R. Chacon, J. C. Horowitz, R. M. Day and P. E. Thomas, Myofibroblast differentiation by transforming growth factor-beta1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase, J. Biol. Chem., 2003, 278(14), 12384–12389 CrossRef CAS.
- C. Frantz, K. M. Stewart and V. M. Weaver, The extracellular matrix at a glance, J. Cell Sci., 2010, 123(Pt 24), 4195–4200 CrossRef CAS.
- V. V. Petrov, R. H. Fagard and P. J. Lijnen, Stimulation of Collagen Production by Transforming Growth Factor- 1 During Differentiation of Cardiac Fibroblasts to Myofibroblasts, Hypertension, 2002, 39(2), 258–263 CrossRef CAS.
- F. Klingberg, B. Hinz and E. S. White, The myofibroblast matrix: implications for tissue repair and fibrosis, J. Pathol., 2013, 229(2), 298–309 CrossRef CAS.
- B. Hinz, P. Pittet, J. Smith-Clerc, C. Chaponnier and J. J. Meister, Myofibroblast development is characterized by specific cell-cell adherens junctions, Mol. Biol. Cell, 2004, 15(9), 4310–4320 CrossRef CAS PubMed.
- D. Bakker, C. A. van Blitterswijk, S. C. Hesseling, J. J. Grote and W. T. Daems, Effect of implantation site on phagocyte/polymer interaction and fibrous capsule formation, Biomaterials, 1988, 9(1), 14–23 CrossRef CAS.
- R. van Dijkhuizen-Radersma, F. L. A. M. A. Péters, N. A. Stienstra, D. W. Grijpma, J. Feijen, K. de Groot and J. M. Bezemer, Control of vitamin B12 release from poly(ethylene glycol)/poly(butylene terephthalate) multiblock copolymers, Biomaterials, 2002, 23(6), 1527–1536 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9bm00979e |
| This journal is © The Royal Society of Chemistry 2020 |