
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel synthesis of platinum complexes and their intracellular delivery to tumor cells by means of magnetic nanoparticles†
Alessandra
Quarta
*a,
Manuel
Amorín
b,
María José
Aldegunde
b,
Laura
Blasi
c,
Andrea
Ragusa
ad,
Simone
Nitti
e,
Giammarino
Pugliese
e,
Giuseppe
Gigli
af,
Juan R.
Granja
 *b and
Teresa
Pellegrino
*e
*b and
Teresa
Pellegrino
*e
aCNR NANOTEC – Institute of Nanotechnology, c/o Campus Ecotekne, via Monteroni, 73100, Lecce, Italy. E-mail: alessandra.quarta@nanotec.cnr.it
bCentro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CIQUS) and Departamento de Química Orgánica, Universidade de Santiago de Compostela, 15782 Santiago de Compostela, Spain. E-mail: juanr.granja@usc.es
cCNR, Institute for Microelectronics and Microsystems, Via Monteroni, Lecce, 73100, Italy
dDipartimento di Scienze e Tecnologie Biologiche e Ambientali, Università del Salento, Campus Ecotekne, via Monteroni, 73100, Lecce, Italy
eIstituto Italiano di Tecnologia, via Morego 30, 16163, Genova, Italy. E-mail: teresa.pellegrino@iit.it
fDipartimento di Matematica e Fisica E. De Giorgi, Università del Salento, Campus Ecotekne, via Monteroni, 73100, Lecce, Italy
First published on 6th December 2019
Abstract
Platinum-based drugs are popular in clinics as chemotherapeutic agents to treat solid tumors. However, severe side effects such as nephro- and neurotoxicity impose strict dosage limitations that can lead to the development of drug resistance and tumor relapse. To overcome these issues Pt(IV) prodrugs and platinum delivery systems might represent the next generation of platinum-based drugs. In this study four novel Pt(II) complexes (namely, PEG-Glu-Pt-EDA, PEG-Glu-Pt-DACH, PEG-Mal-Pt-EDA and PEG-Mal-Pt-DACH) were synthesized and a general strategy to covalently bind them to iron oxide nanoparticles was developed. The intracellular uptake and cell distribution studies of Pt-tethered magnetic nanoparticles on breast and ovarian cancer cell line models indicate that binding of the Pt complexes to the nanoparticles facilitates, for all the complexes, cellular internalization. Moreover, the magnetic nanoparticles (MNPs), as shown in a magnetofection experiment, enhance the uptake of MNP-Pt conjugates if a magnet is placed beneath the culture dish of tumor cells. As shown by a Pt release experiment, intranuclear platinum quantification and TEM analysis on cell sections, the presence of a pH-sensitive dicarboxylic group coordinating the Pt complex, triggers platinum dissociation from the NP surface. In addition, the triazole moiety facilitates endosomal swelling and the leakage of platinum from the endosomes with intranuclear localization of platinum release by the NPs. Finally, as assessed by MTT, caspase, calcein/ethidium bromide live/dead assays, among the four NP-Pt conjugates, the NP-Glu-Pt-EDA complex having a glutamate ring and ethylenediamine as a chelating amine group of the platinum showed higher cytotoxicity than the other three MNP–platinum conjugates.
Introduction
Pt-Based drugs cover almost 50% of chemotherapeutic agents, and they are used to treat malignant solid tumors (i.e. pancreatic, testicular, cervical, ovarian cancers, etc.).1,2 Despite cisplatin, only other two platinum based molecules, namely carboplatin and oxaliplatin, have been approved for use on humans.3,4 These drugs are all square-planar platinum(II) complexes with a Pt atom that is coordinated to two leaving (the carboxylic groups) and two non-leaving amine groups.5 The nature of these two types of chemical groups affects the solubility, stability, efficacy and toxicity of the drugs.6Pt compounds however are hampered by dose-limiting toxic side effects, including nephrotoxicity, neurotoxicity, drug resistance and tumor relapse.7 The latter two effects generally arise from poor accumulation levels of platinum in tumor tissues, low cellular uptake,8 drug inactivation by intracellular glutathione,9 and the DNA repairing mechanisms developed by tumor cells.10,11 Nanocarriers could mitigate these side effects at different levels: (i) they improve the solubility of the drug while prolonging its circulation time; (ii) they penetrate deep the tumor; (iii) they enable a triggered release of the Pt complexes, which consequently reduce the drug resistance and allow lower drug doses to be used.12,13
Stimuli responsive nano- and mesoscale carriers in which a chemotherapeutic agent can be simply encapsulated or covalently attached to the surface and be released under specific stimuli have shown promising results.14–17 Some of these systems have also been successfully tested in animal models, showing a remarkable reduction of unwanted side effects and decreased drug resistance.13,18–21 The composition of the carrier together with the structure of the drug dictates the loading capability and the release profile of the drug itself.22 In addition, its ability to tune size and surface chemistry of nanoparticles also allows the control of their circulation time, biodistribution, immune sequestration and clearance.23 The optimization of these features over the last few years has led to the first FDA approved nanoformulations for drug delivery in chemotherapy (i.e. Doxil, Abraxane, and Onivyde).24 In the case of platinum, a liposomal formulation of cisplatin, namely Lipoplatin, is currently under clinical validation for the treatment of ovarian and pancreatic cancer.25
In addition to lipid-based vesicles,26 other targeted nanoparticles with triggered release are under development, such as those made of an inorganic core.14,22
For example, gold nanoparticles have been engineered through linker molecules that anchor cisplatin27 or to bind oxaliplatin,28,29 or with a specific oligonucleotide sequence to link Pt(IV).30 In another study, cyclodextrin-capped gold particles were conjugated to an adamantine–platinum complex.31
Gold-based heterostructures such as gold-coated iron oxide nanoparticles32 and dumbbell-like gold–magnetite nanocrystals have also been exploited for platinum delivery onto cancer cell lines.32–35 Despite the successful targeting delivery and intracellular toxicity of these nanosystems, the clinical application of gold-based nanoparticles is limited by their non-degradable composition. An in vivo study on gold–iron oxide heterostructures showed that after one year from the injection, the iron oxide domain had been metabolized while the gold one accumulated in several organs.36 More studies have confirmed the slow intracellular degradation and clearance of differently coated magnetic iron oxide nanoparticles.15,33,37 The response of magnetic nanoparticles to magnetic stimulation makes them eligible candidates not only as vectors for anticancer drugs12 but also as contrast agents in Magnetic Resonance Imaging (MRI)38 and heat agents for magnetic hyperthermia.39–42
Platinum-based pharmaceuticals have been encapsulated into host matrices at the surface of individual and/or clustered MNPs.43–47 Different matrices such as gelatin,43 hyaluronic acid,47 poly(ethyl-2-cyanoacrylate),48 and copolymers of poly(methacrylic acid)-grafted-poly(ethyleneglycol methacrylate) have been exploited,45 all with the purpose to protect the drug molecules from inactivation. In two distinct studies, magnetic micelles bearing a cisplatin prodrug were prepared by assembling MNPs and phospholipids44 or MNPs and thermo-responsive polymers.49 Sun's group used porous hollow magnetite nanoparticles as a hosting structure for cisplatin.43 More recently, iron oxide nanoparticles co-loaded with both a Pt(IV) prodrug were tested in tumor-bearing mice.50 It is worth noting that in most of the aforementioned cases the drug was not covalently attached to the surface of the magnetic nanoparticles. Directly linking the drug to the nanoparticles may affect its internalization, biodistribution, and toxicity.
In this study, we have investigated the chemotherapeutic potential of four novel Pt(II) complexes, synthesized by us, and covalently conjugated to iron oxide nanoparticles. They are formed by a cis-platinum complex containing two different diamine groups and two different dicarboxylic moieties with a long PEG side chain for binding to the nanoparticle. The platinum complexes are specifically designed to change their stability at endosomal pH to release Pt(II) ions and at the same time reincorporate protons into the endosomes. The internalization and the resulting cytotoxic effect of the nanoconjugates were proven in two cancer cell lines and compared to those of the free drugs and commercially available cisplatin. The cellular data show that the internalization of the Pt complexes conjugated to the nanoparticles is always enhanced compared to that of free Pt complexes and it is even higher when the uptake is performed in a magnetofection experiment with a magnet placed beneath the cell dish. Among the four MNP-platinum complexes developed, Pt-MNP nanoconjugates having Pt linked through a glutamate ligand as the chelating dicarboxylate and having EDA as an unexchangeable bidentate ligand in their structure were the most effective anti-tumoral compounds.
Results and discussion
Synthesis of Pt complexes
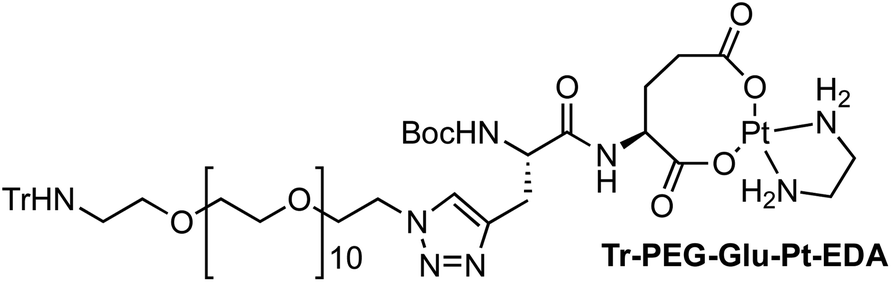
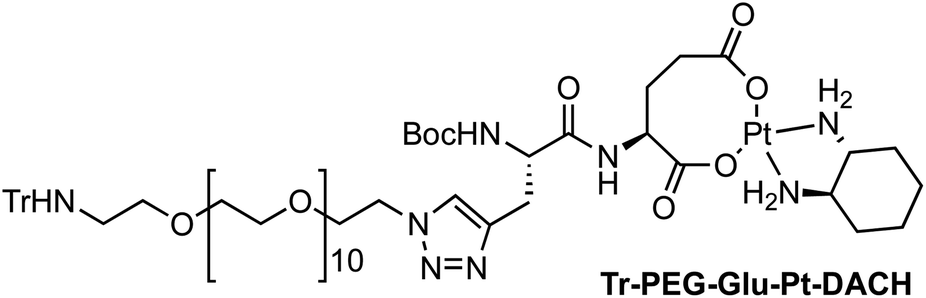
Four Pt complexes with different chelating dicarboxylates, either malonate or glutamate, and diamino groups, either ethylenediamine or a chiral diaminocyclohexane, were synthesized (Fig. 1).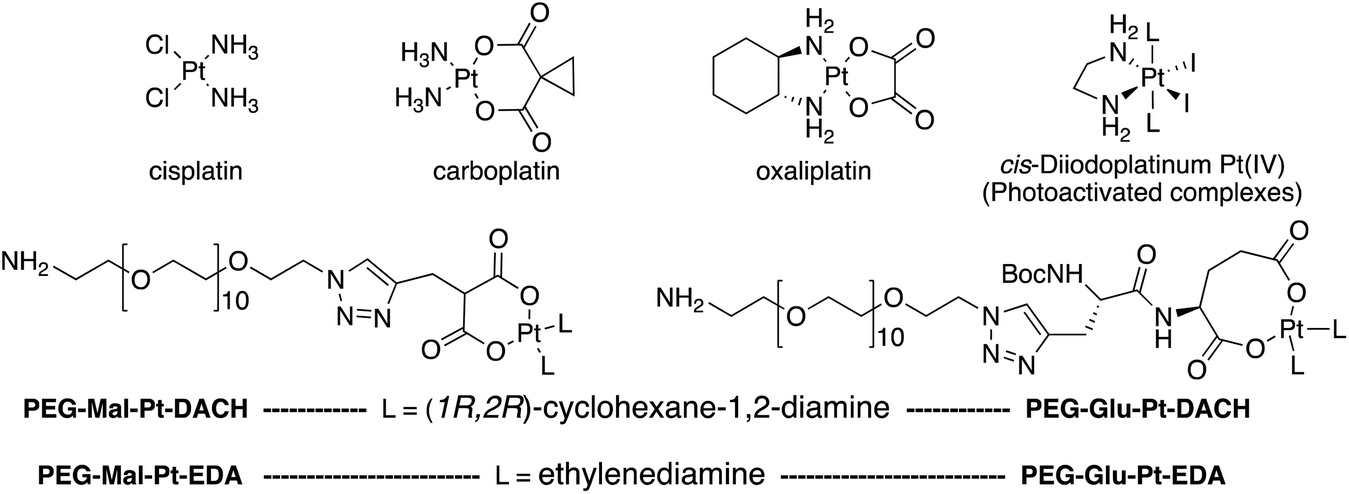 | ||
| Fig. 1 Top: The structure of commercially available cisplatin derivatives and photoactivated diodoplatinum(VI) complexes. Bottom: The platinum complexes prepared in this work. | ||
On the one hand, the malonate derivatives (PEG-Mal-Pt-DACH and PEG-Mal-Pt-EDA) seemed particularly attractive because their ring structure is similar to that of carboplatin, although its spiro-cyclopropyl moiety was replaced with a spacer to connect the drug to the nanoparticle. On the other hand, the glutamic acid derivatives (PEG-Glu-Pt-DACH and PEG-Glu-Pt-EDA) have an eight-membered ring chelating dicarboxylate that, in principle, should facilitate platinum release. Similar to the malonate derivatives, a spacer was bound to the α-amino group for anchoring the metal complex to the nanoparticle. In both cases, the copper catalysed 3 + 2 cycloaddition reaction (click chemistry) between a terminal alkyne on the dicarboxylate side chain and an azido modified poly(ethylene glycol) (PEG) spacer provided a simple strategy for the incorporation of an amino moiety at the end of the chain (Scheme 1). This group easily allowed the binding of the drug derivatives to the nanoparticle surface through simple amide bond formation. In addition to the two carboxylates, two different bidentate amine ligands were selected for this study. The first one, namely (1R,2R)-1,2-diaminocyclohexane (DACH), is identical to the one used in the commercial oxaliplatin. The second one, ethylenediamine (EDA), was chosen due to its structural simplicity and because it has already been shown to give promising results with regard to its use in photoactive Pt(IV) complexes.51,52
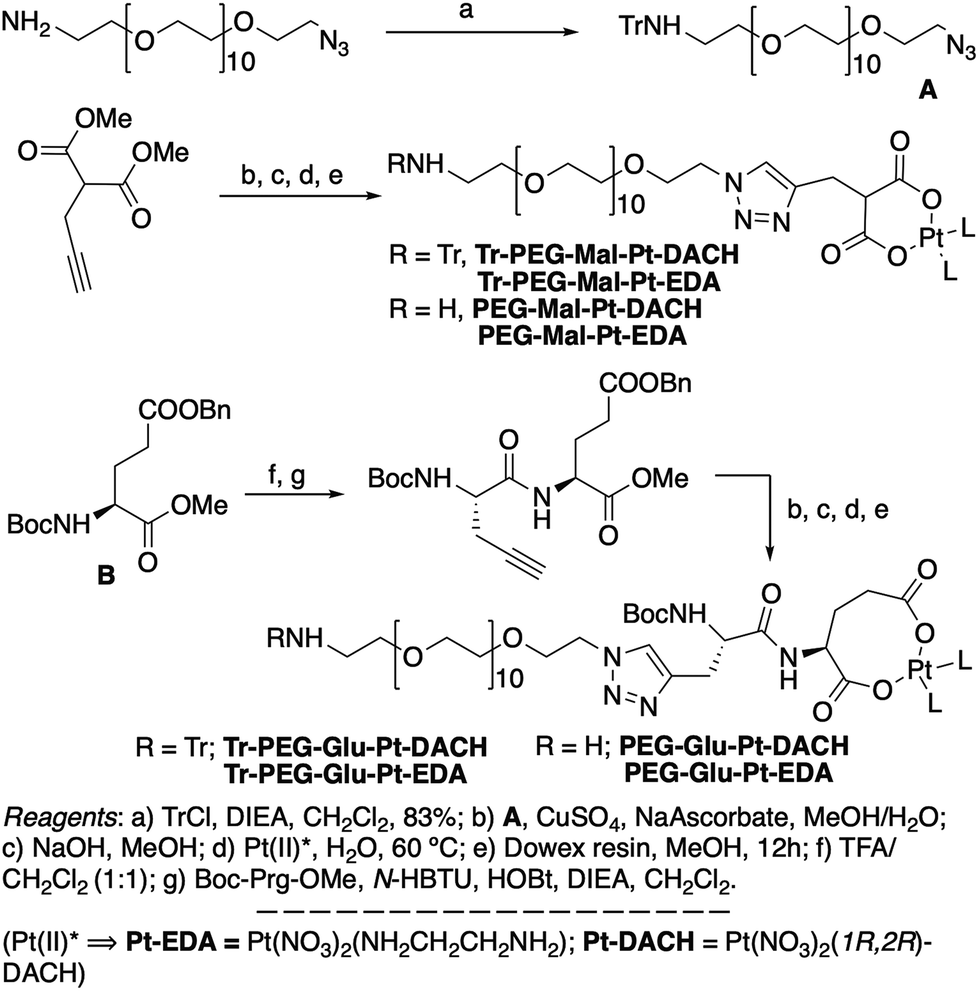 | ||
| Scheme 1 Synthetic strategy used for the preparation of PEG-Glu-Pt-DACH, PEG-Glu-Pt-EDA, PEG-Mal-Pt-DACH, and PEG-Glu-Pt-EDA platinum complexes. | ||
The synthesis of the platinum complexes is illustrated in Scheme 1. The first step was the protection of the amino group on a commercially available PEG derivative that was treated with trityl chloride in the presence of diisopropylethylamine to provide azide A with a yield of 83% (as determined by NMR, see the ESI† for the NMR spectra of the compounds involved in the synthesis). In parallel, N-Boc-Glu(Bn)-OMe was deprotected with trifluoroacetic acid (TFA) and then coupled to Boc-protected propargylglycine to obtain dipeptide B with a yield of 93%. The click reaction between the azido derivative A and either dimethyl propargylmalonate or dipeptide B resulted in the corresponding triazole derivatives with a yield of 71% and 72%, respectively. The platinum complexes were prepared by reacting dinitroplatinum compounds, either Pt(NO3)2-DAE or Pt(NO3)2-DACH, with the dicarboxylate salts of the triazole derivatives previously obtained by the basic hydrolysis of methyl esters. Finally, the trityl protecting groups were removed after treatment with the acidic Dowex resin thus yielding the four Pt-derivatives (PEG-Glu-Pt-DACH, PEG-Glu-Pt-EDA, PEG-Mal-Pt-DACH, and PEG-Glu-Pt-EDA) ready for conjugation to the magnetic nanoparticles (MNPs) through simple amide bonds (Fig. 2).
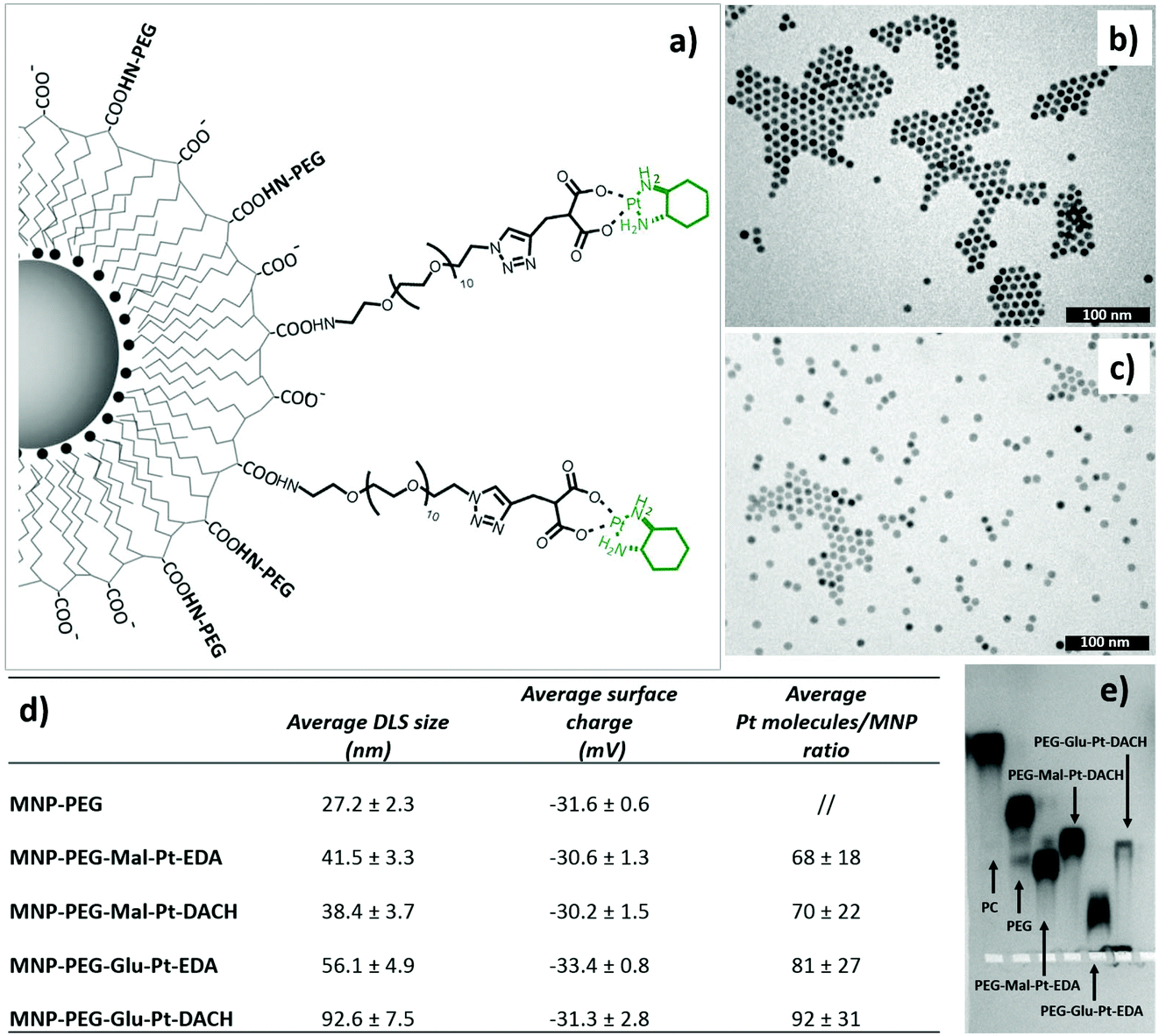 | ||
| Fig. 2 (a) Representative drawing of the MNP-PEG-Glu-Pt-DACH conjugate. TEM images of the nanoparticles after water solubilization (b) and (c) surface functionalization with PEG-Glu-Pt-EDA. (d) The table reports the average hydrodynamic size and surface charge values for the nanoparticles conjugated to the different platinum complexes, as determined by DLS, as well as the average number of platinum complexes linked per type of nanoparticle, as assessed by elemental analysis using the Pt/Fe ratio. (e) Agarose gel electrophoresis characterization showing the mobility of the nanoparticles after each step of surface functionalization. | ||
Preparation and characterization of the nanoparticles conjugated to the Pt-complexes
Magnetic iron oxide nanoparticles (with an average size of 10 nm) were prepared according to a thermal decomposition method,53 and then transferred into water by means of a polymer encapsulation procedure with polymaleic anhydride octadecene.54 After solubilization, the amphiphilic polymer exposes to the outer environment carboxylic groups, which were used to link the platinum derivatives. Before attaching the drug, it was necessary to pre-functionalize the surface of the nanoparticles with some PEG molecules to ensure their stability in DMSO, the solvent used during the coupling reaction to dissolve the Pt complexes. The final nanoparticle–platinum complexes were transferred into PBS buffer and kept at 4 °C prior to use. It is worth mentioning that the choice of this polymer coated and PEG functionalized MNPs was mainly dictated by their safety and biocompatibility even at an injection dose as high as 0.8–1.0 mg of iron, as previously demonstrated by us in an in vivo biodistribution study on a murine model.55The four MNP-Pt conjugates were characterized by means of dynamic light scattering (DLS), gel electrophoresis, elemental analysis (Fig. 2), transmission electron microscopy (TEM) (Fig. 1SI†), and Fourier Transform Infrared (FTIR) spectroscopy (Fig. 3).
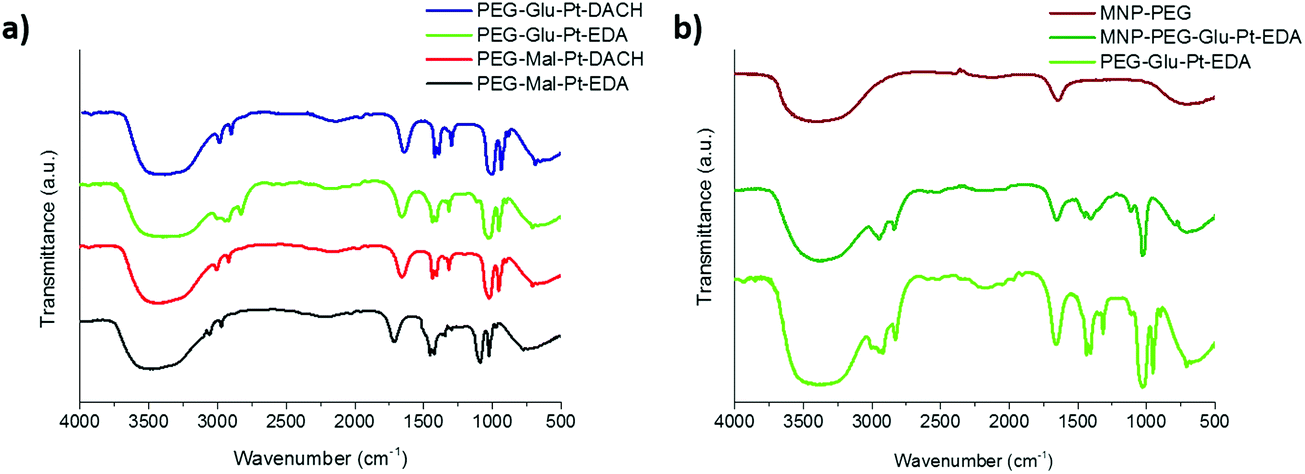 | ||
| Fig. 3 FTIR spectra of (a) the synthesized Pt complexes and (b) the MNP before and after conjugation to PEG-Glu-Pt-EDA. In panel (b) the spectrum of PEG-Glu-Pt-EDA is also reported for comparison. | ||
For their conjugation to the MNPs, the number of Pt molecules to be added per nanoparticle needed to be optimized in order to achieve the right balance between linking a sufficiently toxic amount of Pt complexes to each nanoparticle and to be able to preserve the stability and solubility of the nanoparticles under physiological conditions.
The hydrodynamic diameters verified that the size of the MNP-Pt conjugates shifted towards larger values than that of the sample coated with only PEG molecules, but the overall size was smaller than 100 nm (Fig. 2, panel d). The MNPs bearing the PEG-Glu-Pt-DACH complex showed the largest diameter, likely due to the tendency to form clusters and interparticle interactions, as also observed by gel electrophoresis (Fig. 2, panel e). The surface charge was quite negative for all the samples, with an average value close to −30 mV. The average amount of Pt complexes linked to each nanoparticle was estimated by means of elemental analysis, ranging from about 70 to 100 metal complexes attached per nanoparticle depending on the preparation conditions and on the type of Pt complex (last column of the table of Fig. 2). The pattern of gel migration clearly showed the different delays of the nanoparticle bands at each step of functionalization especially when comparing the polymer coated NPs with the PEG coated up to those conjugated to the Pt-complexes. As an example, in the agarose gel electrophoresis shown in Fig. 2e, the MNP-Pt loaded for the electrophoretic run carried an average amount of 50 Pt molecules per nanoparticle. The gel electrophoresis pattern of the four MNP-Pt conjugates showed that, under the same conjugation conditions, the bands corresponding to the complexes that contain glutamate are more delayed than those containing malonate. It should be noted that the MNP-Glu-Pt-DACH sample was partially stuck in the well, highlighting the tendency of the conjugate to aggregate.
Additionally, representative TEM images of the nanoparticles at each step of preparation, showing well dispersed nanoparticles, with the exception of the MNP-PEG-Glu-Pt-DACH sample that evidenced a tendency to aggregate, confirmed the reduced stability of this nanoconjugate compared to the others (Fig. 1b and c and Fig. 1SI†).
The FTIR spectra of the Pt complexes and those of the MNP at different steps of the preparation were recorded to verify the change in surface functionalization (Fig. 3). On the one hand, for the four Pt complexes (Fig. 3a) the peaks of the C–O stretching, C–H and O–H bending in the region from about 950 to 1700 cm−1 can be recognized, together with the C–H stretching in the region from 2850 and 3000 cm−1 were observed. On the other hand, the spectrum of the MNP-PEG showed the typical signals of the free carboxylic acids exposed on the outer surface, i.e. the asymmetric carbonyl stretching of the carboxylate group (1645 cm−1, quite lower than typical wavenumbers probably because of being involved in hydrogen bonding) and the corresponding hydroxyl stretching (broad peak at about 3400 cm−1) (Fig. 3b, brown curve). Despite some PEG molecules being linked to the MNP, only very small bumps could be observed for their typical C–O and C–H symmetric and asymmetric stretching (1073, 2922 and 2850 cm−1, respectively). Successful binding of the MNP to PEG-Glu-Pt-EDA was confirmed by the spectrum of the resulting MNP-Pt conjugates (Fig. 3b, dark green curve) which clearly presented the peaks belonging to both Pt complexes and PEG molecules. The FTIR spectra of the other nanoconjugates presented similar profiles, also indicating successful reactions (data not shown).
To study the pH-dependent release of the conjugated platinum, the MNP-Pt nanoconjugates were kept at 37 °C for either 24 or 48 h in solutions at a well-defined pH in the range of 3 to 7. The amount of Pt that separated from the nanoparticles was recovered by filtration and quantified by elemental analysis, and the data obtained were plotted as a percentage of the initial platinum amount (Fig. 4). Having proven the presence of platinum on the NPs, we further studied the Pt release for all the Pt-conjugates by keeping the MNP-Pt conjugates in solution while gradually reducing the pH towards acidic values and evaluating the free platinum after separation from the NP fraction. At a pH of 3, almost 40% of Pt was released into the solution after 24 h, and this value increased up to 80% and 85% after 48 h in the case of MNP-Glu-Pt-EDA and MNP-Mal-Pt-EDA, respectively. At a pH of 4.0–4.5, which corresponds to the lysosomal pH, approximately 20% of Pt was detected after 24 h, whereas the average free platinum for all the nanoconjugates ranged between 50 and 70% after 48 h. On the other hand, at the physiological pH 7 the amount of Pt released was as low as about 10% and 20% after 24 h and 48 h, respectively, suggesting a potential good stability of the drugs before cellular internalization.
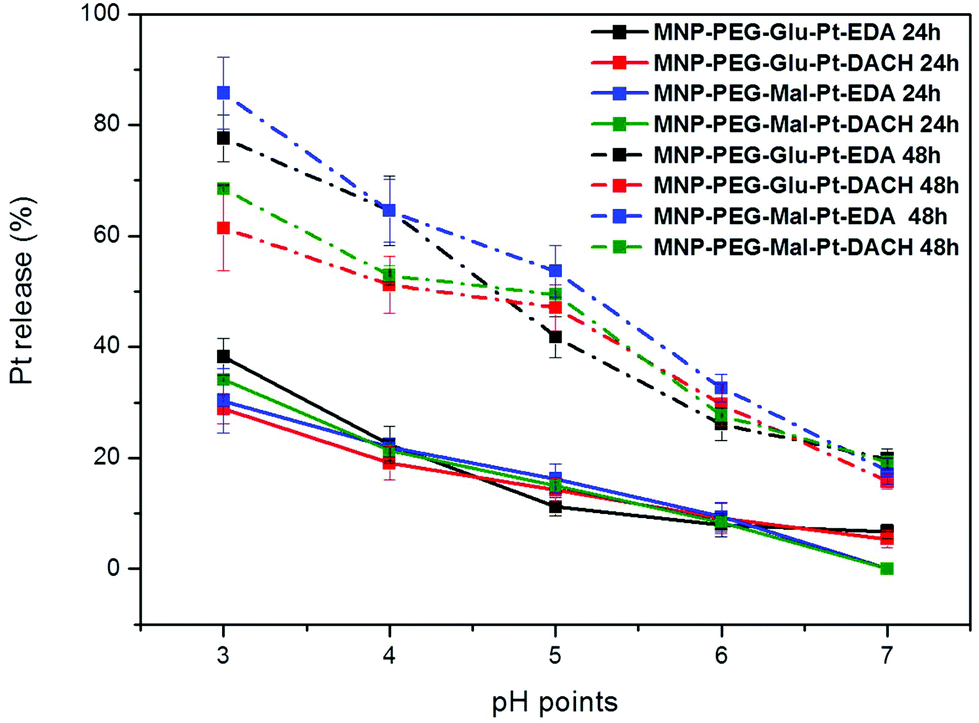 | ||
| Fig. 4 Release profile of Pt from MNP-Pt conjugates after 24 h and 48 h at various pH values. The lines are traced only for guiding the eyes; they do not represent any mathematical fit. For each data point, an average of at least 3 measurements has been recorded. | ||
It is widely known that in an endocytic pathway, the pH drops from 6.5 in early endosomes to 4 in mature lysosomes and might facilitate the dissociation of platinum from the nanoparticle surface.56,57
Cellular studies
Based on these Pt loading and release findings, cellular studies were performed to assess the cytotoxic activity of the free Pt complexes and of the MNP-Pt conjugates in two tumour cell lines that display different sensitivities to platinum-based chemotherapy. In detail, ovarian cancer cells (IGROV-1) and breast cancer cells (MDA-MB-231) were chosen as in vitro models, the latter being generally recognized as cells quite resistant to platinum-based drug treatments.58–60Viability and apoptotic cellular assays (namely MTT and caspase) were performed to investigate the efficacy of both free and nanoparticle-bound platinum complexes on both cell lines.
First, a MTT proliferation assay was carried out to determine the inhibitory concentration (IC50) values of Pt complexes (either free or conjugated to the MNP) after 24 h of incubation with IGROV-1 and MDA-MB-231 cells. Cisplatin was also tested as an additional reference for being routinely used in the clinical treatment of several cancers. Remarkably, all the free Pt complexes showed greater toxicity (IC50 between 6 and 8 μM) as compared to cisplatin (IC50 around 98 μM and 80 μM for the MDA-MB-231 and IGRO-1 cells, respectively). This means that the bare Pt complexes are on average 10 times more toxic than standard cisplatin. In addition, the IC50 values further decreased when the Pt drugs were bound to the nanoparticles (Table 1). As expected, the IGROV-1 cells were more sensitive to the toxic activity of the Pt derivatives than the MDA-MB-231 ones. Indeed, in the case of IGROV-1 that had been administered with MNP-Glu-Pt-EDA and MNP-Glu-Pt-DACH, the IC50 dropped down to 1.6 and 1.2 μM, respectively, while MDA-MB-231 cells that had been incubated with the same nanoparticles showed an IC50 equal to 5.3 and 5.2 μM, respectively. This corresponds to a further 3–4 fold decrease of IC50 as compared to the corresponding free drug. The same trend was also detected when the two cell lines were incubated with MNP-Mal-Pt-EDA and MNP-Mal-Pt-DACH, but the difference in the IC50 was less significant (the values for the IGROV-1 were 4.2 and 4 μM, and the values for the MDA-MB-231 cells were 6.2 and 5.8 μM, respectively).
| MDA-MB-231 | IGROV-1 | |
|---|---|---|
| Tested sample | IC50 (μM) | IC50 (μM) |
| PEG-Glu-Pt-EDA | 7.1 ± 0.2 | 5.8 ± 0.3 |
| PEG-Glu-Pt-DACH | 8.3 ± 0.1 | 7.9 ± 0.1 |
| PEG-Mal-Pt-EDA | 6.2 ± 0.1 | 6.8 ± 0.1 |
| PEG-Mal-Pt-DACH | 7.4 ± 0.1 | 7.5 ± 0.1 |
| MNP-PEG-Glu-Pt-EDA | 5.3 ± 0.2 | 1.6 ± 0.4 |
| MNP-PEG-Glu-Pt-DACH | 5.2 ± 0.4 | 1.2 ± 0.4 |
| MNP-PEG-Mal-Pt-EDA | 6.2 ± 0.1 | 4.2 ± 0.1 |
| MNP-PEG-Mal-Pt-DACH | 5.8 ± 0.1 | 4.0 ± 0.4 |
| Cisplatin | 97.6 ± 5.1 | 79.8 ± 0.3 |
These results suggest that the Pt complexes and MNPs-Pt conjugates affect cell survival significantly as compared to cisplatin and there are correlations between the structures of the nanoconjugates, platinum complexes and cell line types. Indeed, as a first observation, the Pt-based drugs seem to induce a more cytotoxic effect when bound to the nanoparticles, with respect to that of the free Pt complexes at the same dose of administration. These results are in line with the literature, as it has already been reported that the use of drug carriers could facilitate drug uptake and accumulation by overcoming several limitations of traditional drugs.3,61
The cell response also depends on the genetic and epigenetic characteristics of the cell lineage and on the acquisition of drug resistance upon exposure.62 Several drug resistance mechanisms have already been reported, from the overexpression of efflux pump Pgp to the activation of DNA repair mechanisms.63 Thus, it might be assumed that under the same experimental conditions, the MDA-MB-231 cells exhibited some resistance to the Pt-based drugs (or at least a reduced sensitivity to drug action), since this behaviour has been partially identified over the last few years.64
In an attempt to define the interactions between MDA-MB-231 or IGROV-1 cells and Pt compounds, a caspase-3 apoptotic assay was performed. The two cell lines were incubated with either the free Pt complexes or the MNP derivatives at a Pt concentration equal to 2 μM – a value that is close to the IC50 of the most effective nanoconjugates.
As reported in Fig. 5, the activation of caspase 3, which is a common killing factor in the apoptotic process, was detected by using a fluorogenic substrate which is cleaved upon stimulation. It is evident that, while MNP-PEG induced an activation close to 20% in both cell lines, the free Pt compounds and their nanoconjugates triggered a significant activation of caspase 3. This process seemed to be more prominent in IGROV-1 cells than in MDA-MB-231 ones, and it was boosted in cells that had been administered with MNP bearing the Pt complexes with respect to those that had been incubated with free Pt molecules. These data were in accordance with those obtained by the MTT assay, and they confirmed that IGROV-1 cells are more sensitive than MDA-MB-231 cells to the Pt drugs.
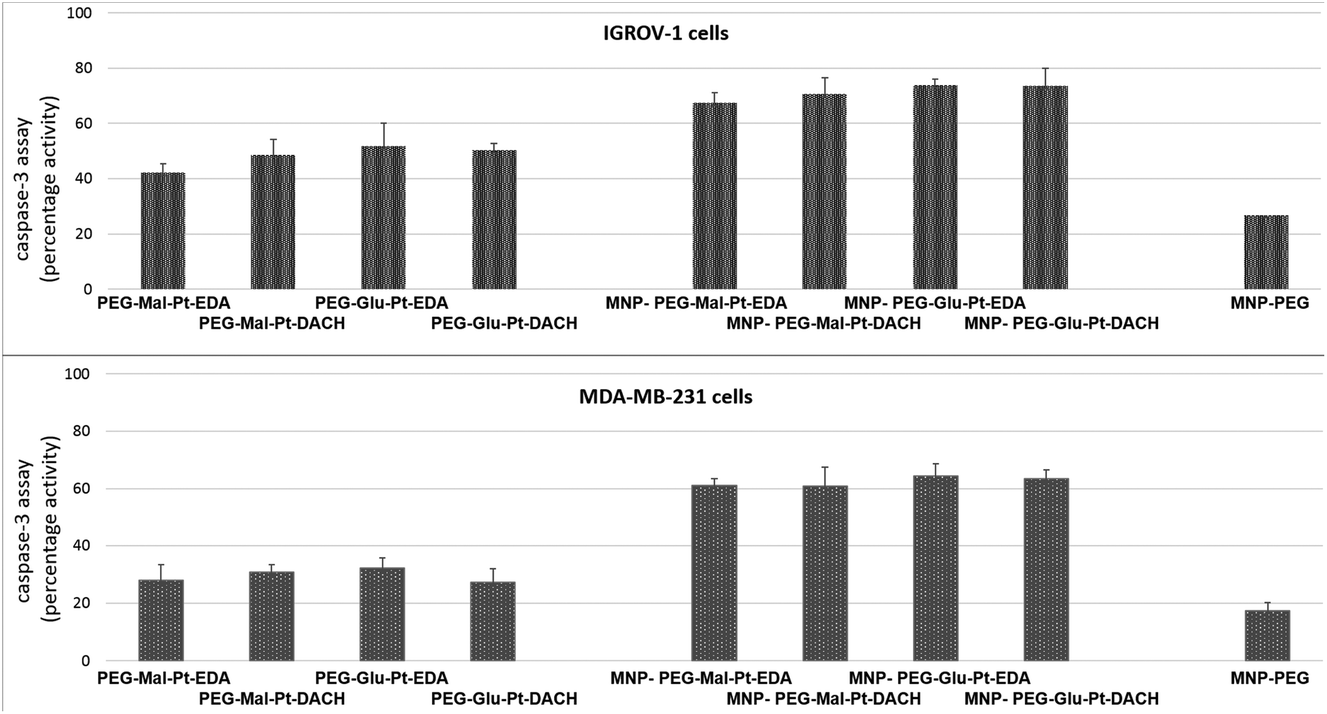 | ||
| Fig. 5 Caspase-3 assay performed with IGROV-1 and MDA-MB-231 cells incubated for 24 h with the free Pt complexes, the four MNP-Pt conjugates, and MNP-PEG. The results are reported as a percentage of the fluorescence intensity of each sample with respect to the untreated control cells. The values and the SD were obtained from 3 independent experiments. The t-test carried out on the two cellular data sets revealed a significant difference (p < 0.01) among the cells incubated with the free Pt complexes and those administered with the Pt-MNP conjugates. | ||
Activation of the apoptotic process seemed to be relatively constant over time, which is likely due to the progressive uptake of either the Pt compounds or the MNPs, subsequently prolonging their effects on cell homeostasis.
Furthermore, unlike in the case of the MTT assay, there were no considerable differences within each group of samples for each cell line. Indeed, the fluorescence intensity detected in the cells after incubation with the Pt complexes at both time points was rather similar (the values ranged from 42% to 51% in the case of IGROV-1, and from 26% to 36% in the case of MDA-MB-231). Similar results were found for the four MNP-Pt conjugates (the values ranged from 67% to 74% in the case of IGROV-1, and from 54% to 64% in the case of MDA-MB-231).
It might be expected that the activation of the caspase cascade would be more consistent in the case of Pt-tethered nanoparticles as compared to free Pt complexes, but it should be noted that, as described in the next sections, the uptake mechanism and the intracellular trafficking of the Pt conjugates are different from those of the free complexes, thus likely leading to cytotoxic effects that involve multiple molecular events.
Since the best overall results from the characterization performed so far were obtained with the PEG-Glu-Pt-EDA drug and the corresponding nanoconjugate, we decided to focus our attention on those compounds.
A live/dead assay was also performed on MDA-MB-231 cells, which were incubated with either PEG-Glu-Pt-EDA or MNP-Glu-Pt-EDA for 24 h. The cells treated with MNP-PEG-Glu-Pt-EDA exhibited a very poor fluorescence in the green channel and a strong fluorescence in the ethidium bromide channel, thus indicating a loss of membrane integrity and cellular damage with consequent necrosis (Fig. 2SI†). In the case of the cells that were incubated with the free platinum complex, PEG-Glu-Pt-EDA, the merged channel provided an image with orange cells due to the overlap between the calcein and ethidium bromide channel signals. Thus, it is likely that these cells did not suffer as much as those that were incubated with MNP-PEG-Glu-Pt-EDA. In contrast, control cells showed green fluorescence in the calcein channel, which is a sign of vital status.
Quantitative analyses of the fluorescent pixels in both channels (Fig. 3SI†) confirmed that the signal associated with ethidium bromide tremendously increased in the Pt-treated cells, while the signal associated with calcein diminished.
The results obtained by the two viability assays and the live/dead assay (the latter in the case of MDA-MB-231 cells only) suggest that, upon exposure to Pt compounds, the amplitude of the cellular damage and the activation of multiple subcellular processes could lead to the activation of either apoptosis or necrosis, depending on the cell status and the ability to repair DNA damage. However, regardless of the type of Pt complex tested, it seems that the toxic effect of the drug is more pronounced when it is conjugated to the nanoparticle, which is likely due to an enhanced and faster internalization.
Effect of a magnet on the cellular uptake
The magnetic response of iron oxide nanoparticles is a valuable tool to drag any cargo associated with the magnetic nanoparticles.65 Here, in a magnetofection experiment a NdFeB magnet was placed under the cell culture well while the cells were challenged with either PEG-Glu-Pt-EDA or MNP-PEG-Glu-Pt-EDA. The effect of magnetic delivery on cell viability is shown in Fig. 6.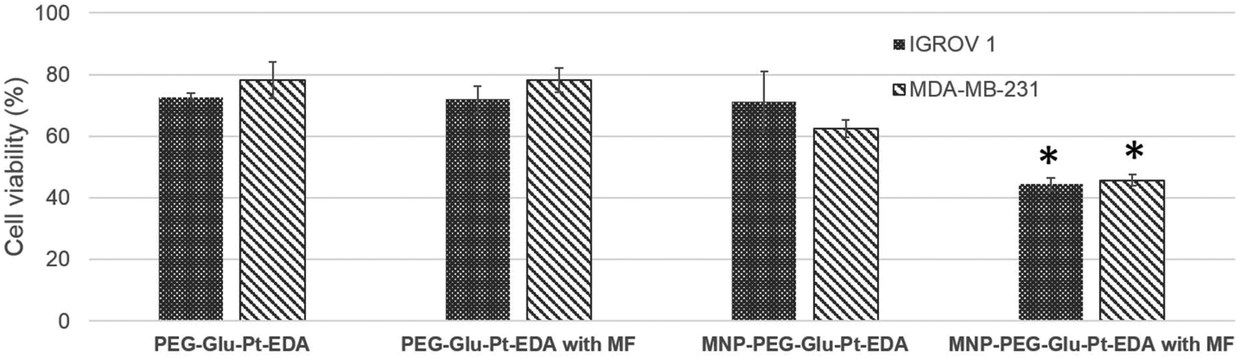 | ||
| Fig. 6 Effect of the application of a magnetic field (MF) on the viability of IGROV-1 and MDA-MB-231 cells incubated for 24 h with either PEG-Glu-Pt-EDA or MNP-PEG-Glu-Pt-EDA (* indicates p < 0.05 when compared with MNP-PEG-Glu-Pt-EDA. Statistical analysis was performed using the t-test). | ||
Cells were incubated with a concentration of Pt (either as a Pt complex or a MNP-Pt conjugate) equal to 750 nM (an amount below the IC50), corresponding to 5.5 nM iron oxide nanoparticles for MNP-PEG-Glu-Pt-EDA. Application of the magnet for 24 h under the cell culture plate boosted the Pt delivery with an evident increase of the cellular cytotoxicity associated with the enhanced platinum delivery. Indeed, when the magnet was applied, the viability of the cells incubated with MNP-PEG-Glu-Pt-EDA dropped from 70 and 62% in the case of IGROV-1 and MDA-MB-231 (in the absence of the magnet) to 44 and 45% (in the presence of a magnet). As expected, no differences were observed with or without the magnetic field when the cells were incubated with the free Pt complex.
Nanoparticle internalization and localization
To elucidate the contribution of the nanoparticle-mediated delivery in combination with the magnetic guidance, a quantitative estimation of Pt internalized by the cells was performed by elemental analysis (Fig. 7). Both IGROV-1 and MDA-MB-231 cells were treated for 24 h with either PEG-Glu-Pt-EDA or MNP-PEG-Glu-Pt-EDA with or without the application of a magnetic field. Then the cells were processed to detect Pt internalization for the Pt-MNP conjugates, the free Pt complexes and the free cisplatin. After 24 h, the highest intracellular content of Pt was detected when the Pt complex was linked to the nanoparticle (reaching 1.2 × 10−11 g per cell in IGROV-1 and 2.0 × 10−11 g per cell in MDA-MB-231, respectively) and it was more than double when a magnetic field was applied under the well (2.9 × 10−11 g per cell in IGROV-1 and 5.0 × 10−11 g per cell in MDA-MB-231, respectively). However, the intracellular amount of Pt was around one order of magnitude smaller when the cells were administered with the free PEG-Glu-Pt-EDA complex (1.8 × 10−12 g per cell in IGROV-1 and 2.9 × 10−12 g per cell in MDA-MB-231, respectively). Remarkably, in the case of cisplatin the intracellular uptake was even less, since the Pt amount detected was 1.5 × 10−13 g per cell in IGROV-1 and 2.1 × 10−13 g per cell in MDA-MB-231, respectively.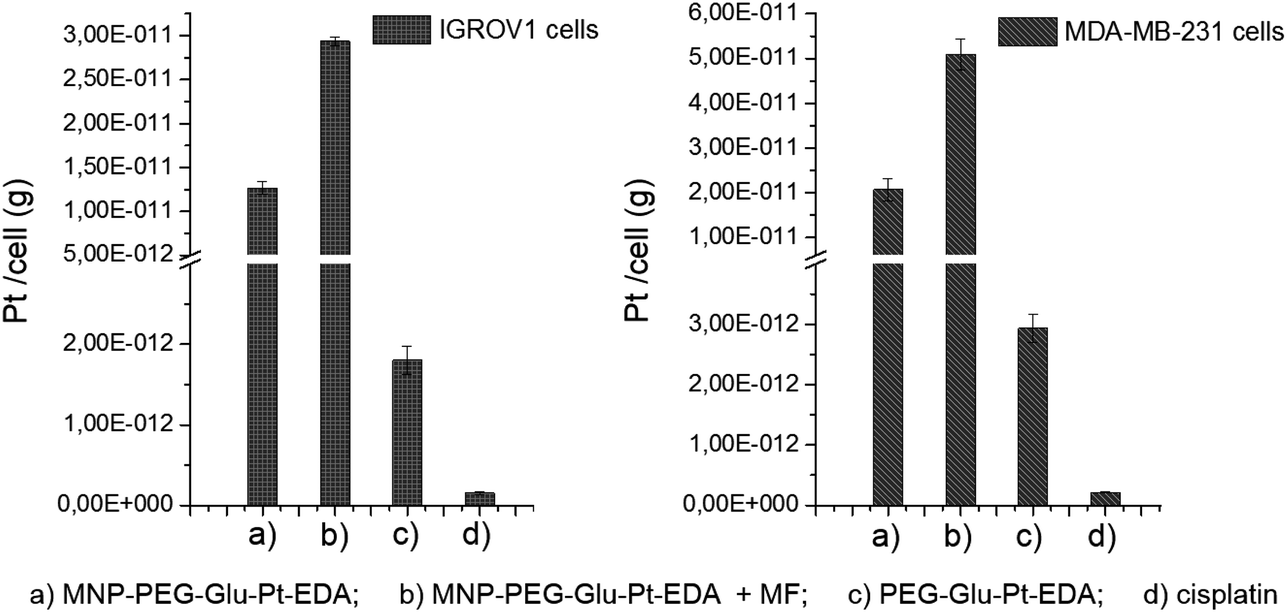 | ||
| Fig. 7 Pt determination via elemental analysis on IGROV-1 cells (left panel) and MDA-MB-231 cells (right panel) incubated with either MNP-PEG-Glu-Pt-EDA (a) without and (b) with the application of a magnetic field (MF), or (c) the free PEG-Glu-Pt-EDA complex or (d) cisplatin for 24 h. | ||
These results may be explained on the basis of the poor solubility of the free cisplatin under physiological conditions, which would explain the low cytotoxicity measured by the MTT assay.
In contrast, the magnetic nanoparticles accelerated the Pt internalization, facilitating its uptake even without applying a magnetic field, probably through endocytosis, as already reported in the literature.12 According to this process, the nanoparticles, once engulfed within vesicular structures, are usually confined to the perinuclear region where they remain localized unless a physicochemical process (mediated by the chemical composition or stimuli responsiveness of the nanoparticles, such the triazole moiety of the platinum complexes) is activated. The triazole moieties by reincorporating protons and counter ions from the cytoplasm into the endosomes may cause rupture or destabilization of the endolysosome membranes, and consequently the release of their content inside the cytosol.66
With the aim of following the intracellular pathway of the MNP-Pt complex, structural analysis of the cells incubated with either MNP-PEG-Glu-Pt-EDA or MNP-PEG at sub-lethal doses (Pt concentration equal to 0.25 μM) was performed by electron microscopy (Fig. 8). After 1 h of incubation, the nanoparticles were clearly present in the endosomal vesicles, and the morphology of the endosomes containing either MNP-PEG (Fig. 8a) or MNP-PEG-Glu-Pt-EDA (Fig. 8b) was similar.
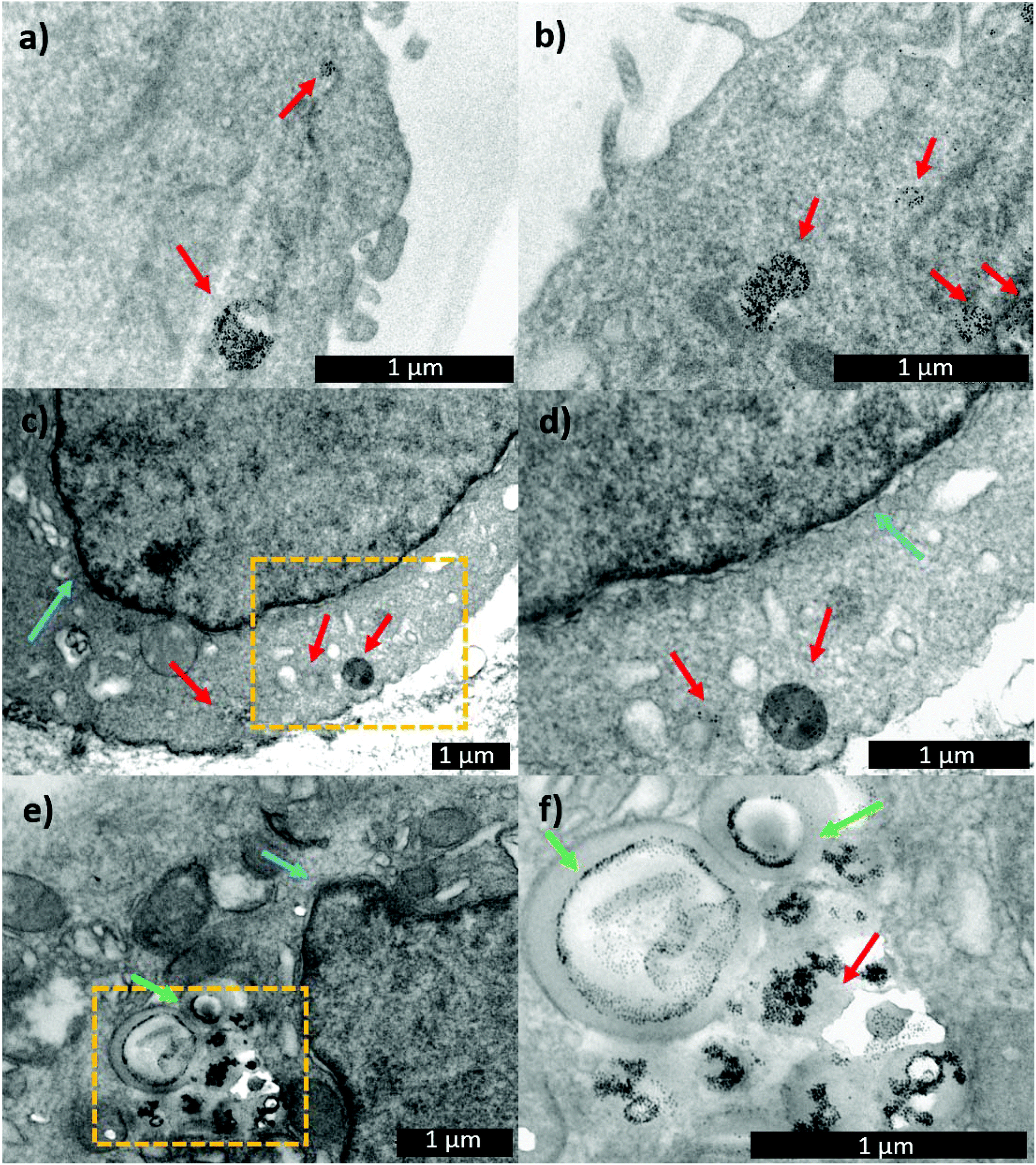 | ||
| Fig. 8 TEM images of IGROV-1 cells incubated for 1 h with (a) MNP-PEG and (b) MNP-PEG-Glu-Pt-EDA. TEM images of IGROV-1 cells incubated for 24 h with (c and d) MNP-PEG and (e and f) MNP-PEG-Glu-Pt-EDA. The red arrows indicate the endolysosomes containing the nanoparticles. The green arrows indicate the swelling of the vesicular structures, observed only for MNP-PEG-Glu-Pt-EDA, in which the nanoparticles appear to be arranged in circular geometries. The blue arrows point to the nuclear membrane. (d) and (f) are the zoomed areas of (c) and (e), respectively. | ||
In another experiment, after the cells were incubated for 1 h with MNP-PEG-Glu-Pt-EDA or MNP-PEG, the cell medium was replaced with a fresh one that did not contain nanoparticles or the Pt complex, and the cultures were kept for an additional 24 h at 37 °C. The TEM images of the intracellular uptake pathway showed that the endosome structure was dramatically altered, they appeared swollen and exhibited the nanoparticles in a corona-like distribution (Fig. 8e and f). Indeed, it seems that the MNP-PEG-Glu-Pt-EDA conjugate promoted an osmotic imbalance, accompanied by water diffusion and a consequent swelling of the endosomes. This effect was not detected in the case of MNP-PEG that was incubated under the same conditions (Fig. 8c and d; additional TEM images in Fig. 4 and 5 of the ESI†). By analyzing and comparing the size distribution of the endosomal vesicles (Fig. 6SI†), it became apparent that they have a broad distribution in the cells incubated with the MNP-Pt conjugate, ranging from 100 to 800 nm, with two intense peaks at 250 and 500 nm, the latter population being the most abundant. In the case of MNP-PEG, the analysis showed a much more uniform distribution, between 200 and 400 nm, with a median endosomal size of 400 nm.
As suggested by our data, this peculiar behaviour can be attributed to the structure of the Pt complexes. Indeed, the presence of a triazole group has been often used as a pH-sensitive group to trigger endosomal protonation and drug escape in others types of cargo systems (i.e. cation lipids and modified antibody structures).67,68
The triazole group in the Pt complex may facilitate the entrance of protons, anions and water, favouring the high osmotic pressure inside the endosome. The consequent swelling of the endosomal vesicles and the acidic hydrolysis of the platinum(II) complex enable the metal release and therefore the endosomal escape, as shown in Fig. 8SI.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 69
69
Interestingly, the TEM images suggest that the swelling process did not lead to the rupture of the endosomal vesicles but only to the increase of their volume, which likely allowed for the Pt escape, while the bulkier MNPs remained confined to the endosomes (at least at sub-lethal doses), as already reported by some of us.70
After 96 h, the enlarged vesicles were still visible in the cytosol, although they looked less swelled, likely as the consequence of the cellular attempt to restore the physiological endosomal sizes (Fig. 7SI†).
To verify if the platinum complex reaches the nucleus and binds itself to the deoxyguanosine residues, subsequently hindering the DNA replication process, as already reported,71 we have quantitatively determined the amount of intra-nuclear platinum. To this aim, the DNA of the cells incubated with either MNP-PEG-Glu-Pt-EDA or PEG-Glu-Pt-EDA (under the same conditions used for the estimation of the intracellular Pt) was extracted prior to the detection of Pt via elemental analysis. The results show that the absolute amount of intranuclear Pt is higher when the cells were incubated with the MNP bearing the Pt complex (1.5 and 0.78 pg in the case of IGROV-1 and MDA-MB-231, respectively) than with the free complex (0.21 and 0.16 pg in the case of IGROV-1 and MDA-MB-231, respectively) (Table 2).
| Sample name: | Pt (g per nucleus) | |
|---|---|---|
| IGROV-1 | MNP-PEG-Glu-Pt-EDA | 1.5 × 10−12 ± 1.2 × 10−12 |
| PEG-Glu-Pt-EDA | 2.1 × 10−13 ± 2.6 × 10−14 | |
| MDA-MB-231 | MNP-PEG-Glu-Pt-EDA | 7.8 × 10−13 ± 1.7 × 10−14 |
| PEG-Glu-Pt-EDA | 1.6 × 10−13 ± 7.6 × 10−15 |
Conclusions
In this study, four novel platinum complexes with amino terminated PEG chain linkers were synthesized and then attached to the surface of iron oxide nanoparticles. The platinum-based drugs were specifically designed to have a chelating dicarboxylic acid moiety, as in the case of oxaliplatin, either malonate or glutamate, and a diamino group, either EDA or DACH. The amino-PEG on the side chain of the platinum complex allowed its binding to iron oxide nanoparticles. The cytotoxicity assays showed that the MNP-Pt complexes were generally more cytotoxic than the corresponding free Pt-compounds and cisplatin used as a gold standard. This was directly related to the enhanced drug uptake when it is linked to the MNP rather than the free complex. In the case of the MNP-derivatives, internalization was further accelerated when an external magnetic field was applied.Finally, a possible explanation of the mechanism through which the Pt complex detaches from nanoparticles and escapes from the endosomal compartment has been provided. As a first step, the pH-hydrolysis of the platinum–ligand complex on the platinum–MNP conjugates, also verified in a test tube, enables the uncoupling of the Pt ions from the nanoparticle surface, while the presence of a triazole moiety in the platinum complex promotes the swelling of the cellular endosomes, the latter observed under TEM analysis, allowing the endosomal escape of the platinum.
Indeed, the endocytic pathway often limits the intracellular transport of drugs/nanoparticles since most of the carriers and cargos become trapped within the nano-meso scale, generally entering the cell via an endocytosis mechanism and remaining confined within the vesicles without reaching their biological target.72
Finally, the platinum intranuclear localization, as measured by platinum associated with the DNA, enables the execution of the anti-proliferation action. The highest cytotoxic effect was observed for the Pt-nanoconjugates containing the glutamate ligand as the chelating dicarboxylate and EDA as the diamino group, yielding a nanodrug that proved to be more than order of magnitude more efficient than standard cisplatin. This nanoconjugate also reached lower IC50 concentrations when compared to previous literature studies on Pt-based drugs bound to iron oxide nanoparticles, such as porous hollow Fe3O4 nanoparticles43 and dumbbell-like Au–Fe3O4 nanoparticles73 for cisplatin delivery, or other magnetic nanoparticles designed to deliver different platinum(IV) prodrugs,46,50 thus being a promising candidate for future in vivo studies. As a next step, efforts should be made to develop a single magnetic Pt-nanoconjugate that can merge the physical magnetic accumulation and cytotoxicity effects of the platinum compound with biomolecular targeting that can recognize receptors overexpressing on the specific tumor under treatment (such as AFRA antibody fragments that can link folate receptors overexpressing on ovarian cancer cells55). This would make the anti-tumoral treatment very specific sparing healthy cells that do not express the receptors, and at the same time, it would require fewer doses of anti-tumoral agents thus minimizing side effects.
Materials and methods
Chemicals
1-[Bis(dimethylamino)methylene]-1H-benzotriazolium hexafluorophosphate 3-oxide (N-HBTU), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), alpha-amino acids, amines, etc. were purchased from Novabiochem, Applied Biosystems, Global Sales Manager, GL Biochem (Shanghai) Ltd, China, Sigma-Aldrich or Across. The azido-PEG-amine [N3–CH2–CH2–(O–CH2–CH2)10–O–CH2–CH2–NH2] was purchased from Polypure AS (Oslo, Norway), and the Pt compounds [PtCl2-EDA and K2PtCl4] were purchased from Alfa Aesar. QuadraSil® AP was purchased from Aldrich. All reagents and solvents were used as received unless otherwise stated. Solvent mixtures for chromatography are reported as v/v ratios. Column chromatography was performed on EM Science silica gel 60 (230–400 mesh). CH2Cl2 and DIEA were distilled from CaH2 over argon immediately prior to use.General techniques
1H NMR spectra were recorded on a Bruker AMX 500 MHz spectrometer or a Varian Mercury 300 MHz spectrometer. Chemical shifts (δ) were reported in parts per million (ppm, δ) relative to tetramethylsilane (δ = 0.00 ppm) or by the deuterium solvent. 1H NMR splitting patterns are designated as singlet (s), doublet (d), triplet (t), or quartet (q). Splitting patterns that could not be easily interpreted are designated as multiplet (m) or broad (br). 13C NMR spectra were recorded on a Bruker AMX 500 MHz spectrometer or a Varian Mercury 300 MHz spectrometer. Carbon resonances were assigned by using DEPT spectra obtained with phase angles of 135°. Electrospray ionization (ESI) mass spectra were recorded on a Bruker BIOTOF II mass spectrometer.Synthesis of Pt complexes
A solution of Boc-L-Glu(OBn)-OMe (108 mg, 0.31 mmol) in a mixture of TFA/CH2Cl2 (1
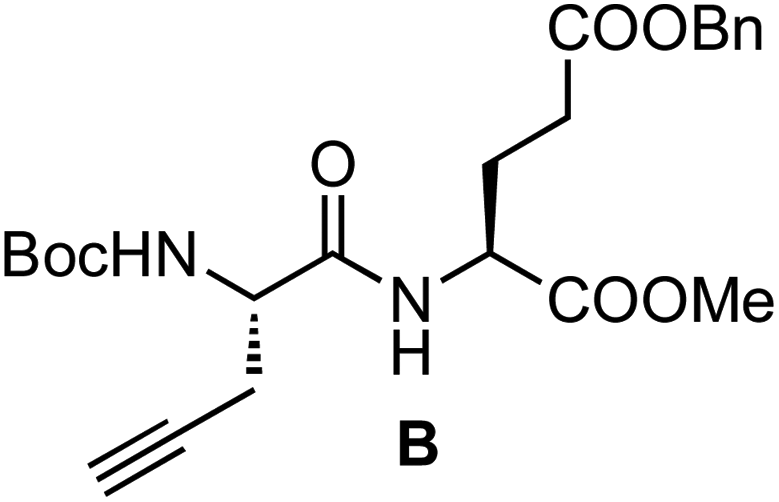 :1, 4 mL) was stirred at room temperature. After 15 min, the solvent was removed under reduced pressure and the resulting solid was dried under high vacuum for 3 h. The resulting TFA salt was dissolved in CH2Cl2 (3 mL), and then Boc-L-Pra-OH (66 mg, 0.31 mmol), N-HBTU (129 mg, 0.34 mmol) and DIEA (0.22 μL, 1.20 mmol) were successively added. The mixture was stirred at room temperature for 2 h and the solution was poured into a separation funnel and washed with an aqueous solution of HCl (10%, 3 × 3 mL) and NaHCO3 (sat. sol., 3 × 3 mL). The organic layer was dried with anhydrous Na2SO4, and then it was filtered and concentrated under reduced pressure. The resulting yellow oil was purified by flash chromatography (50% EtOAc in hexanes) to give 129 mg of the dipeptide B as a white foam [93%, Rf = 0.55 (50% EtOAc in hexanes)]. 1H NMR (CDCl3, 500 MHz, δ): 7.29 (m, 5H), 7.12 (d, J = 7.8 Hz, 1H), 5.42 (d, J = 8.0 Hz, 1H), 5.06 (s, 2H), 4.61 (td, J = 7.9 and 5.1 Hz, 1H), 4.29 (br, 1H), 3.67 (s, 3H), 2.72 (ddd, J = 17.0, 5.8 and 2.7 Hz, 1H), 2.57 (ddd, J = 17.0, 5.8 and 2.3 Hz, 1H), 2.51–1.91 (m, 4H), 2.01 (t, J = 2.7 Hz, 1H), 1.43 (s, 9H) ppm. 13C NMR (CDCl3, 75 MHz, δ): 172.4 (CO), 171.8 (CO), 170.2 (CO), 155.3 (CO), 135.7 (C), 128.5 (CH), 128.4 (CH), 128.2 (CH), 80.4 (C), 79.3 (C), 71.7 (CH), 66.4 (CH2), 52.9 (CH), 52.5 (CH), 51.6 (CH3), 30.1 (CH2), 28.2 (CH3), 27.3 (CH2), 22.3 (CH2) ppm. MS (ESI-TOF+) [m/z (%)]: 469.2 ([MNa]+, 100), 413.1 ([MNa − tBu]+, 7), 369.1 ([MNa − Boc]+, 8). HRMS (ESI) calculated for C23H30N2NaO7: 469.1945, found: 469.1945.
:1, 4 mL) was stirred at room temperature. After 15 min, the solvent was removed under reduced pressure and the resulting solid was dried under high vacuum for 3 h. The resulting TFA salt was dissolved in CH2Cl2 (3 mL), and then Boc-L-Pra-OH (66 mg, 0.31 mmol), N-HBTU (129 mg, 0.34 mmol) and DIEA (0.22 μL, 1.20 mmol) were successively added. The mixture was stirred at room temperature for 2 h and the solution was poured into a separation funnel and washed with an aqueous solution of HCl (10%, 3 × 3 mL) and NaHCO3 (sat. sol., 3 × 3 mL). The organic layer was dried with anhydrous Na2SO4, and then it was filtered and concentrated under reduced pressure. The resulting yellow oil was purified by flash chromatography (50% EtOAc in hexanes) to give 129 mg of the dipeptide B as a white foam [93%, Rf = 0.55 (50% EtOAc in hexanes)]. 1H NMR (CDCl3, 500 MHz, δ): 7.29 (m, 5H), 7.12 (d, J = 7.8 Hz, 1H), 5.42 (d, J = 8.0 Hz, 1H), 5.06 (s, 2H), 4.61 (td, J = 7.9 and 5.1 Hz, 1H), 4.29 (br, 1H), 3.67 (s, 3H), 2.72 (ddd, J = 17.0, 5.8 and 2.7 Hz, 1H), 2.57 (ddd, J = 17.0, 5.8 and 2.3 Hz, 1H), 2.51–1.91 (m, 4H), 2.01 (t, J = 2.7 Hz, 1H), 1.43 (s, 9H) ppm. 13C NMR (CDCl3, 75 MHz, δ): 172.4 (CO), 171.8 (CO), 170.2 (CO), 155.3 (CO), 135.7 (C), 128.5 (CH), 128.4 (CH), 128.2 (CH), 80.4 (C), 79.3 (C), 71.7 (CH), 66.4 (CH2), 52.9 (CH), 52.5 (CH), 51.6 (CH3), 30.1 (CH2), 28.2 (CH3), 27.3 (CH2), 22.3 (CH2) ppm. MS (ESI-TOF+) [m/z (%)]: 469.2 ([MNa]+, 100), 413.1 ([MNa − tBu]+, 7), 369.1 ([MNa − Boc]+, 8). HRMS (ESI) calculated for C23H30N2NaO7: 469.1945, found: 469.1945.
A solution of Boc-L-Pra-L-Glu(OBn)-OMe (116 mg, 0.26 mmol) in a mixture of H2O/MeOH (1
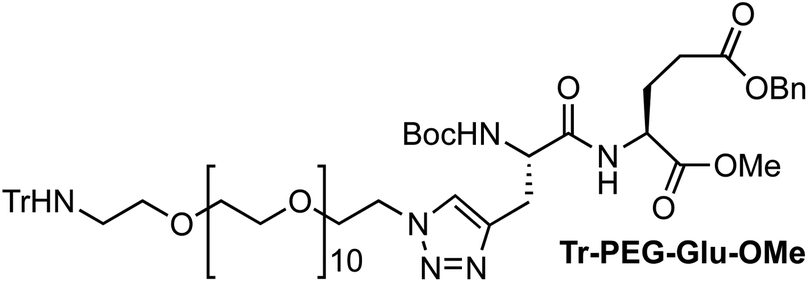 :1, 40 mL) was treated with CuSO4 (104 mg, 0.65 mmol) and sodium L-ascorbate (620 mg, 3.1 mmol). The mixture was stirred at room temperature for 15 min, and then a solution of azido-PEG-amine-Trt (A) (211 mg, 0.26 mmol) in a mixture of H2O/MeOH (1:1, 5 mL) was added. The mixture was stirred at room temperature for 24 h, and then the solution was treated with QuadraSil® AP resin to remove any excess Cu. After stirring for 20 min, the resin was filtered and washed with MeOH, and then the solvent was removed under reduced pressure. The resulting residue was purified by flash chromatography (1–3% MeOH in CH2Cl2) to give 236 mg of Tr-PEG-Glu-OMe as a colorless oil [72%, Rf = 0.60 (10% MeOH/CH2Cl2)]. 1H NMR (CDCl3, 300 MHz, δ): 7.54 (s, 1H), 7.49–7.46 (m, 6H), 7.35 (m, 4H), 7.29–7.14 (m, 12H), 6.09 (br, 1H), 5.10 (s, 2H), 4.59–4.48 (br, 2H), 4.38 (q, J = 5.1 Hz, 2H), 3.77 (t, J = 5.2 Hz, 2H), 3.69 (s, 3H), 3.64–3.51 (m, 42H), 3.30 (dd, J = 14.9 and 5.2 Hz, 1H), 3.07 (dd, J = 15.0 and 5.4 Hz, 1H), 2.35 (t, J = 5.4 Hz, 2H), 2.24–2.08 (m, 4H), 1.45 (s, 9H) ppm. 13C NMR (CDCl3, 75 MHz, δ): 172.3 (CO), 171.7 (CO), 171.2 (CO), 146.2 (CO), 143.3 (C), 135.8 (C), 128.7 (CH), 128.6 (CH), 128.4 (CH), 128.3 (CH), 127.8 (CH), 126.2 (CH), 123.6 (CH), 80.3 (C), 77.6 (C), 71.3 (CH2), 70.7 (CH2), 70.6 (CH2), 70.5 (CH2), 70.1 (CH2), 69.4 (CH2), 66.5 (CH2), 54.0 (CH), 52.5 (CH), 51.5 (CH3), 50.1 (CH2), 42.1 (CH2), 29.9 (CH2), 28.3 (CH3), 27.6 (CH2), 27.4 (CH2) ppm. MS (ESI-TOF+) [m/z (%)]: 1281.7 ([MNa]+, 12), 1259.7 ([MH]+, 6), 1039.5 ([MNa − Trt]+, 20), 1017.6 ([MH − Trt]+, 100). HRMS (ESI) calculated for C66H94N6O18: 1259.6697, found: 1259.6718.
:1, 40 mL) was treated with CuSO4 (104 mg, 0.65 mmol) and sodium L-ascorbate (620 mg, 3.1 mmol). The mixture was stirred at room temperature for 15 min, and then a solution of azido-PEG-amine-Trt (A) (211 mg, 0.26 mmol) in a mixture of H2O/MeOH (1:1, 5 mL) was added. The mixture was stirred at room temperature for 24 h, and then the solution was treated with QuadraSil® AP resin to remove any excess Cu. After stirring for 20 min, the resin was filtered and washed with MeOH, and then the solvent was removed under reduced pressure. The resulting residue was purified by flash chromatography (1–3% MeOH in CH2Cl2) to give 236 mg of Tr-PEG-Glu-OMe as a colorless oil [72%, Rf = 0.60 (10% MeOH/CH2Cl2)]. 1H NMR (CDCl3, 300 MHz, δ): 7.54 (s, 1H), 7.49–7.46 (m, 6H), 7.35 (m, 4H), 7.29–7.14 (m, 12H), 6.09 (br, 1H), 5.10 (s, 2H), 4.59–4.48 (br, 2H), 4.38 (q, J = 5.1 Hz, 2H), 3.77 (t, J = 5.2 Hz, 2H), 3.69 (s, 3H), 3.64–3.51 (m, 42H), 3.30 (dd, J = 14.9 and 5.2 Hz, 1H), 3.07 (dd, J = 15.0 and 5.4 Hz, 1H), 2.35 (t, J = 5.4 Hz, 2H), 2.24–2.08 (m, 4H), 1.45 (s, 9H) ppm. 13C NMR (CDCl3, 75 MHz, δ): 172.3 (CO), 171.7 (CO), 171.2 (CO), 146.2 (CO), 143.3 (C), 135.8 (C), 128.7 (CH), 128.6 (CH), 128.4 (CH), 128.3 (CH), 127.8 (CH), 126.2 (CH), 123.6 (CH), 80.3 (C), 77.6 (C), 71.3 (CH2), 70.7 (CH2), 70.6 (CH2), 70.5 (CH2), 70.1 (CH2), 69.4 (CH2), 66.5 (CH2), 54.0 (CH), 52.5 (CH), 51.5 (CH3), 50.1 (CH2), 42.1 (CH2), 29.9 (CH2), 28.3 (CH3), 27.6 (CH2), 27.4 (CH2) ppm. MS (ESI-TOF+) [m/z (%)]: 1281.7 ([MNa]+, 12), 1259.7 ([MH]+, 6), 1039.5 ([MNa − Trt]+, 20), 1017.6 ([MH − Trt]+, 100). HRMS (ESI) calculated for C66H94N6O18: 1259.6697, found: 1259.6718.
A solution of dimethyl 2-(prop-2-yn-1-yl)malonate (48 mg, 0.28 mmol) in a mixture of H2O/MeOH (1
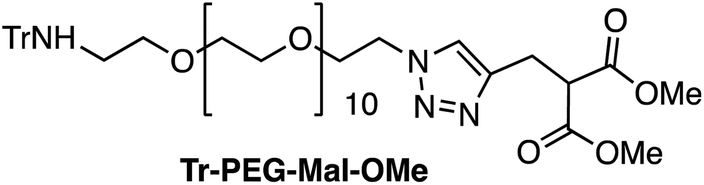 :1; 46 mL) was treated with CuSO4 (204 mg, 1.27 mmol) and sodium L-ascorbate (1.26 g, 6.37 mmol). The mixture was stirred at room temperature for 15 min, and then a solution of azido-PEG-amine-Trt (A) (207 mg, 0.25 mmol) in a mixture of H2O/MeOH (1:1, 4 mL) was added. The mixture was stirred at room temperature for 24 h, and then the solution was treated with QuadraSil® AP resin to remove any excess Cu. After stirring for 20 min, the resin was filtered and washed with MeOH, and then the solvent was removed under reduced pressure. The resulting residue was purified by flash chromatography (1–10% MeOH in CH2Cl2) to give 178 mg of Tr-PEG-Mal-OMe as a light-yellow oil [71%, Rf = 0.54 (10% MeOH/CH2Cl2)]. 1H NMR (CDCl3, 300 MHz, δ): 7.62 (s, 1H), 7.51–7.47 (m, 6H), 7.30–7.16 (m, 10H), 4.51 (t, J = 5.0 Hz, 2H), 3.93 (br, 1H), 3.86 (t, J = 5.0 Hz, 2H), 3.74 (s, 6H), 3.66–3.51 (m, 42H), 3.35 (br, 2 H), 2.36 (t, J = 5.4 Hz, 2H) ppm. 13C NMR (CDCl3, 75 MHz, δ): 168.9 (CO), 145.9 (C), 128.5 (CH), 127.6 (CH), 126.0 (CH), 71.0 (CH2), 70.4 (CH2), 70.2 (CH2), 69.9 (CH2), 69.2 (CH2), 52.5 (CH3), 51.1 (CH), 50.2 (CH2), 42.8 (CH2), 24.8 (CH2) ppm. MS (ESI-TOF+) [m/z (%)]: 1005.5 ([MNa]+, 9), 983.5 ([MH]+, 8), 763.4 ([MNa − Trt]+, 11), 741.4 ([MH − Trt]+, 100). HRMS (ESI) calculated for C51H75N4O15: 983.5223, found: 983.5257.
:1; 46 mL) was treated with CuSO4 (204 mg, 1.27 mmol) and sodium L-ascorbate (1.26 g, 6.37 mmol). The mixture was stirred at room temperature for 15 min, and then a solution of azido-PEG-amine-Trt (A) (207 mg, 0.25 mmol) in a mixture of H2O/MeOH (1:1, 4 mL) was added. The mixture was stirred at room temperature for 24 h, and then the solution was treated with QuadraSil® AP resin to remove any excess Cu. After stirring for 20 min, the resin was filtered and washed with MeOH, and then the solvent was removed under reduced pressure. The resulting residue was purified by flash chromatography (1–10% MeOH in CH2Cl2) to give 178 mg of Tr-PEG-Mal-OMe as a light-yellow oil [71%, Rf = 0.54 (10% MeOH/CH2Cl2)]. 1H NMR (CDCl3, 300 MHz, δ): 7.62 (s, 1H), 7.51–7.47 (m, 6H), 7.30–7.16 (m, 10H), 4.51 (t, J = 5.0 Hz, 2H), 3.93 (br, 1H), 3.86 (t, J = 5.0 Hz, 2H), 3.74 (s, 6H), 3.66–3.51 (m, 42H), 3.35 (br, 2 H), 2.36 (t, J = 5.4 Hz, 2H) ppm. 13C NMR (CDCl3, 75 MHz, δ): 168.9 (CO), 145.9 (C), 128.5 (CH), 127.6 (CH), 126.0 (CH), 71.0 (CH2), 70.4 (CH2), 70.2 (CH2), 69.9 (CH2), 69.2 (CH2), 52.5 (CH3), 51.1 (CH), 50.2 (CH2), 42.8 (CH2), 24.8 (CH2) ppm. MS (ESI-TOF+) [m/z (%)]: 1005.5 ([MNa]+, 9), 983.5 ([MH]+, 8), 763.4 ([MNa − Trt]+, 11), 741.4 ([MH − Trt]+, 100). HRMS (ESI) calculated for C51H75N4O15: 983.5223, found: 983.5257.
A solution of dichloro(ethylenediamine)platinum(II) (31 mg, 0.10 mmol) in H2O (2 mL) was treated with AgNO3 (32 mg, 0.19 mmol). The mixture was protected from light using aluminium foil, and it was stirred under Ar at 60 °C for 20 h. After this time, the mixture was cooled down to room temperature, filtered through a Celite pad and washed with H2O (3 × 2 mL). The solvent was concentrated under reduced pressure until approximately 1 mL.60
Compound Tr-PEG-Glu-OMe (40 mg, 0.032 mmol) was dissolved in MeOH (1 mL) before being treated with an aqueous solution of NaOH (0.5 M, 320 μL, 0.16 mmol), and then the mixture was stirred at room temperature for 12 h. After HPLC-MS analysis had confirmed the disappearance of the starting material, the solvents were concentrated under reduced pressure and the solid was dried under high vacuum.
The resulting residue was treated with the [Pt(NO3)2EDA] aqueous solution, which had previously been prepared, and the solution was stirred at 60 °C for 24 h. Then the reaction mixture was cooled down to room temperature and concentrated under reduced pressure. The resulting residue was dissolved in H2O, and the solution was cooled in an ice/water bath. This formed a precipitate, which was then removed by centrifugation and decantation. The solution was lyophilized, giving 45 mg of Tr-PEG-Glu-Pt-EDA as a yellow solid (quantitative). 1H NMR (D2O, 300 MHz, δ): 7.96 (s, 1H), 7.44–7.18 (m, 6H), 7.15–6.79 (m, 9H), 4.62 (br, 2H), 4.23 (m, 1H), 3.95 (br, 2H), 3.80–3.22 (m, 43H), 3.00–2.55 (m, 6H), 2.52–1.87 (m, 6H), 1.38 (s, 9H) ppm. MS (ESI-TOF+) [m/z (%)]: 1190.5 ([MNa − Trt]+, 3), 1168.5 ([MH − Trt]+, 100), 935.5 ([MNa − Trt − Pt- EDA]+, 6), 913.5 ([MH − Trt − Pt-EDA]+, 584.2 ([MH − Trt]2+, 1). HRMS (ESI) calculated for C41H80N8O18Pt: 1167.5236, found: 1167.5256.
This compound was prepared following the same procedure described above for Tr-PEG-Glu-Pt-EDA, but [Pt(Cl2)(1R,2R)-DACH] (36 mg, 0.10 mmol) and compound Tr-PEG-Glu-OMe (30 mg, 0.024 mmol) were used to give 35 mg of Tr-PEG-Glu-Pt-DACH as a yellow solid (quantitative). 1H NMR (D2O, 300 MHz, δ): 7.97 (s, 1H), 7.40–7.18 (m, 6H), 7.11–6.81 (m, 9H), 4.65 (m, 2H), 4.24 (m, 1H), 3.98 (br, 2H), 3.79–3.12 (m, 43H), 2.60–1.85 (m, 10H) 1.83–1.06 (m, 17H) ppm. MS (ESI-TOF+) [m/z (%)]: 1484.6 ([MNa]+, 1), 1462.7 ([MH]+, 1), 1242.5 ([MNa − Trt]+, 7), 1220.6 ([MH − Trt]+, 100), 1177.6 (2), 935.5 ([MNa − Trt − Pt- DACH]+, 8), 913.5 ([MH − Trt − Pt-DACH]+, 13). HRMS (ESI) calculated for C64H99N8O18Pt: 1462.6724, found: 1462.6692.
This compound was prepared following the same procedure described above for Tr-PEG-Glu-Pt-EDA, but dichloro(ethylenediamine)platinum(II) (29 mg, 0.09 mmol) and compound Tr-PEG-Mal-Me (30 mg, 0.03 mmol) were used to give 36 mg of Tr-PEG-Mal-Pt-EDA as a dark-yellow solid (quantitative). 1H NMR (D2O, 300 MHz, δ): 8.06 (s, 1H), 7.45–7.15 (m, 6H), 7.08–6.76 (m, 9H), 4.66 (br, 2H), 3.99 (d, 2H, J = 16.3 Hz), 3.80–3.13 (m, 45H), 3.01–2.44 (m, 6H), 2.22 (m, 2H) ppm. MS (ESI-TOF+) [m/z (%)]: 967.4 ([MH − Trt]+, 5), 922.4 ([MH − Trt − CO2]+, 100), 713.4 ([MH − Trt − Pt-EDA]+, 18), 691.4 ([MNa − Trt − Pt-EDA − CO2]+, 23), 669.4 ([MH − Trt − Pt-EDA − CO2]+, 62). HRMS (ESI) calculated for C32H64N6O15Pt: 967.4077, found: 967.4063.

This compound was prepared using the same procedure described above for Tr-PEG-Glu-Pt-EDA, but [Pt(Cl2)(1R,2R)-DACH] (34 mg, 0.09 mmol) and compound Tr-PEG-Mal-Me (30 mg, 0.030 mmol) were used to give 35 mg of the mentioned compound as a dark yellow solid (quantitative). 1H NMR (D2O, 300 MHz, δ): 8.03 (s, 1H), 7.61–7.16 (m, 6H), 7.12–6.72 (m, 9H), 4.67 (br, 2H), 4.01 (br, 2H), 3.83–3.15 (m, 43H), 2.84–1.92 (m, 6H), 1.80–1.06 (m, 8H) ppm. MS (ESI-TOF+) [m/z (%)]: 1284.5 ([MNa]+, 4), 1262.6 ([MH]+, 37), 1042.4 ([MNa − Trt]+, 18), 1020.5 ([MH − Trt]+, 79), 976.5 ([MH − Trt − CO2]+, 100), 713.4 ([MH − Trt − Pt-DACH]+, 15), 669.4 ([MH − Trt − Pt-DACH − CO2]+, 21). HRMS (ESI) calculated for C55H83N6O15Pt: 1262.5563, found: 1262.5569.
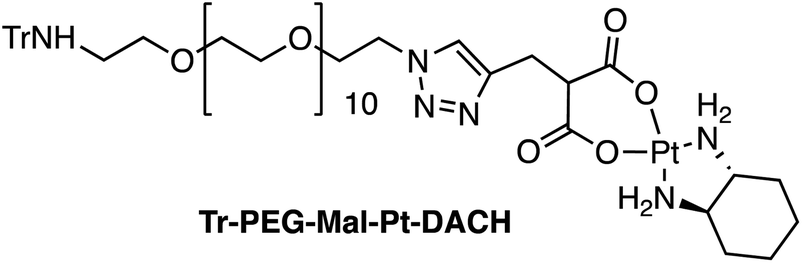
Synthesis of MNP-Pt conjugates
MNPs with an average size of 10 nm were prepared by means of a thermal decomposition approach that has already been reported. Oleic acid capped MNPs were transferred into water by a polymer coating procedure that was developed by us. Then, the surface of the nanoparticles was functionalized with monoamino-PEG by linking the amino moieties of PEG to the outstretched carboxy groups of the polymer via EDC chemistry. This step was introduced to enable the nanoparticles to dissolve in DMSO. In detail, 500 molecules of PEG per nanoparticle and 30000 molecules of EDC per nanoparticle were used, respectively, starting from a nanoparticle concentration equal to 1 μM. The reaction occurred in pH 7.4 PBS and, after 3 h stirring at rt, the PEG-functionalized NPs were washed on centrifuge filters at least 5 times.
Prior to conjugation to the nanoparticles, the trityl-protecting group was removed from the Pt complexes through the use of a strongly acidic cation exchange resin. In details, the Pt complexes were dissolved in methanol (1 mg mL−1), and then the mixture was kept for 18 h in the resin. The resulting mixture was passed through a column, and the collected solution was left to dry completely. Finally, the complex was dissolved in DMSO immediately before the coupling reaction. A recent study demonstrated that the use of DMSO may result in ligand displacement in and changes to the structure of the complexes, thus inhibiting their anticancer ability.74 Thus, DMSO was used only for the conjugation step.
The Pt complexes were bound to the nanoparticle surface as per the following procedure: the nanoparticle solutions in a PBS/DMSO mixture (1:1, 0.5 μM, 200 μL) were treated with an aqueous solution of EDC (25 mM, 100 μL) for 30 minutes. Then, the Pt-complex in DMSO (1 mg mL−1, 50 μL) was added, and the solution was left under stirring for 2 h. Finally, the MNP-Pt conjugates were thoroughly washed on centrifuge filters before being dissolved in PBS. The resulting nanoparticles were stored in a fridge. In order to modulate the number of Pt molecules that were linked to each nanoparticle, the amount of EDC and Pt-complexes was respectively adjusted.
Structural and morphological characterization of MNP-Pt conjugates
Low-magnification TEM images were recorded on a JEOL Jem1011 microscope operating at an accelerating voltage of 100 kV. Electrophoretic characterization was carried out by running the nanoparticles through 2% agarose gel immersed in TBE buffer (pH 8.0) for 1 h at 100 V. DLS and zeta potential measurements were performed on a Zetasizer Nano ZS90 (Malvern, USA) equipped with a 4.0 mW He–Ne laser operating at 633 nm and with an avalanche photodiode detector. DLS measurements were performed in PBS (pH 7.4, with 137 mM NaCl, 10 mM phosphate, 2.7 mM KCl). FT-IR spectra were recorded in transmittance mode on a Jasco 6300 spectrometer (Jasco Corp., Tokyo, Japan) between 4000 and 400 cm−1 with 40 scans and a resolution of 4 cm−1 and analyzed with the Spectra Manager software (Jasco). Samples were dissolved in methanol and 50 μL of each solution was poured on the ATR crystal surface. The spectrum of each sample was acquired against a background obtained with the crystal without any sample. All analyses were carried out at room temperature.The amount of Pt molecules that were linked to each nanoparticle was estimated via elemental analysis using an Inductively Coupled Plasma Atomic Emission Spectrometer (ICP-AES) Varian 720-ES. The calibration curves for both Fe and Pt were prepared, and the relative amount of metal per sample was estimated upon acidic digestion in HNO3/HCl (1/3) solution.
Cellular studies
Two human adenocarcinoma cell lineages were used in this study: ovarian (IGROV-1, a gift from Dr Silvana Canevari, Fondazione IRCCS Istituto Nazionale dei Tumori, Milano, Italy) and breast (MDA-MB-231, American Type Culture Collection) cancer cell lines. The cells were grown at 37 °C and 5% CO2 in RPMI-1640 medium supplemented with L-glutamine, penicillin, streptomycin, and 10% heat-inactivated fetal bovine serum.MTT viability assay, caspase-3 assay and live/dead assay
To assess the cytotoxic effect of the free Pt complexes and the MNP-Pt conjugates, three different types of assays were performed: MTT assays, caspase-3 assays, and live/dead assays. The MTT and caspase-3 assays are both enzymatic tests that detect the formation of a colorimetric product (MTT) or a fluorescent product (caspase-3) upon the enzymatic cleavage of specific substrates.For the MTT assay, the cells (5 × 104 cells) suspended in the medium (1 mL) were seeded in each well of a 12 well-plate. After 24 h incubation at 37 °C, a fresh medium containing either MNP-Pt conjugates, MNP-PEG or free Pt complexes at Pt concentrations from 0 to 20 μM was added. The cells were kept under incubation with either free or bound drugs for 24 h. Next, the medium was removed, and the cells were washed twice with a phosphate buffer (pH 7.4). 1 mL of serum-free medium containing 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (1 mg mL−1) was then added into each well. After 3 h of incubation at 37 °C, the dark insoluble formazan obtained was dissolved in DMSO (2 mL). The absorbance signal at 570 nm was correlated to the percentage of vital cells by comparing data from treated cells to those of the control cells. Finally, the IC50 values were determined by plotting the viability percentage against the concentration of the administered drugs.
In the case of the caspase-3 assay, the cells (5 × 104 in 1 mL of medium) were seeded in each well of a 12 well-plate. After 24 h incubation at 37 °C, a fresh medium containing either MNP-Pt conjugates, MNP-PEG or free Pt complexes at Pt concentrations equal to 2 μM was added. In the case of MNP-PEG, the concentration of nanoparticles that corresponded to the highest amount of MNP-Pt conjugates was used (0.24 μM). Two time points (24 and 48 h) were set. Then the fluorometric caspase-3 Apoptosis Detection Kit from Santa Cruz was used, and the procedure provided by the company was followed. In detail, after the lysis and preparation of the cellular samples, 10 μL of the fluorogenic substrate, DEVD-AFC, were added to the cell lysate and left under incubation for 1 h at 37 °C. Upon caspase-3 activity, the substrate was cleaved, releasing the fluorescent AFC (7-amino-4-trifluoromethyl coumarin). The molecule was detected by spectrofluorimetry (Cary Eclipse Fluorimeter) at an excitation wavelength of 400 nm, and the emission peaked at 505 nm. The caspase-3 activity and thus the apoptotic status of the treated cells were compared with that of the control cells.
The live/dead assay (purchased from Invitrogen) was performed on 6 well-plates in which glass coverslips were placed and then 1 × 105 MDA-MB-231 cells were seeded. After 24 h of incubation at 37 °C, a fresh medium containing MNP or Pt complexes (at the same concentration as that reported for the caspase assay) was added to each well. After 24 h, the cells were washed with PBS prior to proceeding with the assay protocol. In detail, calcein AM (2 μM) and EthD-1 (4 μM) were added directly to the cells and after 40 minutes of incubation at room temperature, the solution was removed and fresh PBS was added. Soon after, the cells were analysed under a confocal microscope (TCS-SP5, Leica, Mannheim, Germany) using excitation wavelengths of 488 and 543 nm for calcein AM and EthD-1, respectively. The fluorescence windows were collected separately, at 530 nm for calcein and at 645 nm for EthD-1. A quantitative analysis of the pixel intensities for each fluorescent channel was carried out using Leica Application Suite AF 2.6.3 software (Leica, Mannheim, Germany). The fluorescence intensity within a defined region of interest (ROI) was normalized with respect to the background signal of the scanned area.
Quantification of the cellular and nuclear uptake of Pt
The cells (2 × 105 cells) were seeded in each well of a 6 well-plate (in 2 mL of culture medium). After 24 h, the medium was replaced with 2 mL of a fresh medium containing either MNP-PEG-Glu-Pt-EDA or PEG-Glu-Pt-EDA or cisplatin at a sub-lethal Pt concentration equal to 0.25 μM, corresponding approximately to a nanoparticle concentration of 5 nM. After 24 h, they were processed for elemental analysis. Next, the cells were washed three times with PBS and trypsinized. The cell suspension was then centrifuged, the supernatant was removed, and a concentrated solution of HCl/HNO3 (3/1, 1 mL) was added to digest the cells. The intracellular Pt concentration was measured by means of elemental analysis using an Inductively Coupled Plasma Atomic Emission Spectrometer (ICP-AES).To estimate the nuclear uptake of Pt, once the cells were incubated with either MNP-PEG-Glu-Pt-EDA or PEG-Glu-Pt-EDA, they were trypsinized, resuspended in TE buffer (10 mM Tris base, pH 7.6, 0.5 mM EDTA, pH 8.0) with 1% SDS and then incubated with proteinase K 20 mg mL−1 overnight at 55 °C. Then, the standard protocol for DNA extraction was used. Finally, a concentrated solution of HCl/HNO3 (3/1, 1 mL) was added to the DNA prior to run the ICP-AES measurements.
Ultrastructural analysis of cells and tissues
The cells (1 × 106 cells) suspended in 5 mL of medium were seeded in a culture dish. After 24 h of incubation at 37 °C, the medium was replaced with a fresh medium containing either MNP-PEG-Glu-Pt-EDA or MNP-PEG at a sub-lethal concentration of Pt equal to 0.25 μM, corresponding approximately to a nanoparticle concentration of 30 nM. The cells were then incubated at 37 °C for 1 h or 24 h. Then, they were washed with PBS and fixed with glutaraldehyde (2.5%) in cacodylate buffer (0.1 M) at 4 °C for 30 min. The fixed specimens were washed three times with the same buffer, and 1% osmium tetroxide in a cacodylate buffer was added for 1 h. Next, the cells were washed again and dehydrated with 25%, 50%, 75% and 100% acetone. Two steps of infiltration in a mixture of resin/acetone (1/1 and 2/1 ratios) were performed, and then the specimens were embedded in 100% resin at 60 °C for 48 h. Ultrathin sections (100 nm thick) were cut on an Ultramicrotome, then stained with lead citrate and observed under an electron microscope.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was funded by the European Research Council (starting grant ICARO, Contract No. 678109) and partially supported by the European project Magnifyco (Contract No. NMP4-SL-2009-228622) and by the Project FISR—C.N.R. “Tecnopolo di Nanotecnologia e Fotonica per la Medicina di Precisione”— (CUPB83B17000010001). This work was also supported by the Spanish Agencia Estatal de Investigación (AEI) and the ERDF (CTQ2016-78423-R), and by the Xunta de Galicia and the ERDF (EM2014/011) and Centro singular de investigación de Galicia accreditation 2016–2019, (ED431G/09).References
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS PubMed.
- B. W. Harper, A. M. Krause-Heuer, M. P. Grant, M. Manohar, K. B. Garbutcheon-Singh and J. R. Aldrich-Wright, Chemistry, 2010, 16, 7064–7077 CrossRef CAS PubMed.
- N. J. Wheate, S. Walker, G. E. Craig and R. Oun, Dalton Trans., 2010, 39, 8113–8127 RSC.
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 CrossRef CAS PubMed.
- T. C. Johnstone, J. J. Wilson and S. J. Lippard, Inorg. Chem., 2013, 52, 12234–12249 CrossRef CAS PubMed.
- J. T. Hartmann and H. P. Lipp, Expert Opin. Pharmacother., 2003, 4, 889–901 CrossRef CAS PubMed.
- R. Oun, Y. E. Moussa and N. J. Wheate, Dalton Trans., 2018, 47, 6645–6653 RSC.
- H. Burger, W. J. Loos, K. Eechoute, J. Verweij, R. H. Mathijssen and E. A. Wiemer, Drug Resist. Updates, 2011, 14, 22–34 CrossRef CAS PubMed.
- M. Pasello, F. Michelacci, I. Scionti, C. M. Hattinger, M. Zuntini, A. M. Caccuri, K. Scotlandi, P. Picci and M. Serra, Cancer Res., 2008, 68, 6661–6668 CrossRef CAS PubMed.
- L. R. Kelland, Drugs, 2000, 59(Suppl 4), 1–8 CrossRef CAS PubMed.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed.
- P. a. Ma, H. Xiao, C. Li, Y. Dai, Z. Cheng, Z. Hou and J. Lin, Mater.Today, 2015, 18, 554–564 CrossRef CAS.
- H. S. Oberoi, N. V. Nukolova, A. V. Kabanov and T. K. Bronich, Adv. Drug Delivery Rev., 2013, 65, 1667–1685 CrossRef CAS PubMed.
- R. J. Browning, P. J. T. Reardon, M. Parhizkar, R. B. Pedley, M. Edirisinghe, J. C. Knowles and E. Stride, ACS Nano, 2017, 11, 8560–8578 CrossRef CAS PubMed.
- B. T. Mai, P. B. Balakrishnan, M. J. Barthel, F. Piccardi, D. Niculaes, F. Marinaro, S. Fernandes, A. Curcio, H. Kakwere, G. Autret, R. Cingolani, F. Gazeau and T. Pellegrino, ACS Appl. Mater. Interfaces, 2019, 11, 5727–5739 CrossRef PubMed.
- M. A. C. Stuart, W. T. Huck, J. Genzer, M. Mueller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk and M. Urban, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- X. Xu, W. Ho, X. Zhang, N. Bertrand and O. Farokhzad, Trends Mol. Med., 2015, 21, 223–232 CrossRef CAS PubMed.
- R. Du, H. Xiao, G. Guo, B. Jiang, X. Yan, W. Li, X. Yang, Y. Zhang, Y. Li and X. Jing, Colloids Surf., B, 2014, 123, 734–741 CrossRef CAS PubMed.
- X. Ling, X. Chen, I. A. Riddell, W. Tao, J. Q. Wang, G. Hollett, S. J. Lippard, O. C. Farokhzad, J. J. Shi and J. Wu, Nano Lett., 2018, 18, 4618–4625 CrossRef CAS PubMed.
- X. Y. Wang, Z. Chang, X. Nie, Y. Y. Li, Z. P. Hu, J. L. Ma, W. Wang, T. Song, P. Zhou, H. Q. Wang and Z. Yuan, Nanomedicine, 2019, 15, 153–163 CrossRef CAS PubMed.
- B. T. Mai, S. Fernandes, P. B. Balakrishnan and T. Pellegrino, Acc. Chem. Res., 2018, 51, 999–1013 CrossRef CAS PubMed.
- S. Tran, P.-J. DeGiovanni, B. Piel and P. Rai, Clin. Transl. Med., 2017, 6, 44 CrossRef PubMed.
- D. Bobo, K. J. Robinson, J. Islam, K. J. Thurecht and S. R. Corrie, Pharm. Res., 2016, 33, 2373–2387 CrossRef CAS PubMed.
- G. P. Stathopoulos and T. Boulikas, J. Drug Delivery, 2012, 2012, 10 Search PubMed.
- D. Liu, C. He, A. Z. Wang and W. Lin, Int. J. Nanomed., 2013, 8, 3309–3319 Search PubMed.
- J. Comenge, C. Sotelo, F. Romero, O. Gallego, A. Barnadas, T. G. Parada, F. Dominguez and V. F. Puntes, PLoS One, 2012, 7, e47562 CrossRef CAS PubMed.
- S. D. Brown, P. Nativo, J.-A. Smith, D. Stirling, P. R. Edwards, B. Venugopal, D. J. Flint, J. A. Plumb, D. Graham and N. J. Wheate, J. Am. Chem. Soc., 2010, 132, 4678–4684 CrossRef CAS PubMed.
- G. E. Craig, S. D. Brown, D. A. Lamprou, D. Graham and N. J. Wheate, Inorg. Chem., 2012, 51, 3490–3497 CrossRef CAS PubMed.
- S. Dhar, W. L. Daniel, D. A. Giljohann, C. A. Mirkin and S. J. Lippard, J. Am. Chem. Soc., 2009, 131, 14652–14653 CrossRef CAS PubMed.
- Y. Shi, J. Goodisman and J. C. Dabrowiak, Inorg. Chem., 2013, 52, 9418–9426 CrossRef CAS PubMed.
- A. J. Wagstaff, S. D. Brown, M. R. Holden, G. E. Craig, J. A. Plumb, R. E. Brown, N. Schreiter, W. Chrzanowski and N. J. Wheate, Inorg. Chim. Acta, 2012, 393, 328–333 CrossRef CAS.
- F. Mazuel, A. Espinosa, N. Luciani, M. Reffay, R. Le Borgne, L. Motte, K. Desboeufs, A. Michel, T. Pellegrino, Y. Lalatonne and C. Wilhelm, ACS Nano, 2016, 10, 7627–7638 CrossRef CAS PubMed.
- C. George, D. Dorfs, G. Bertoni, A. Falqui, A. Genovese, T. Pellegrino, A. Roig, A. Quarta, R. Comparelli, M. L. Curri, R. Cingolani and L. Manna, J. Am. Chem. Soc., 2011, 133, 2205–2217 CrossRef CAS PubMed.
- H. Kakwere, M. E. Materia, A. Curcio, M. Prato, A. Sathya, S. Nitti and T. Pellegrino, Nanoscale, 2018, 10, 3930–3944 RSC.
- J. Kolosnjaj-Tabi, Y. Javed, L. Lartigue, J. Volatron, D. Elgrabli, I. Marangon, G. Pugliese, B. Caron, A. Figuerola, N. Luciani, T. Pellegrino, D. Alloyeau and F. Gazeau, ACS Nano, 2015, 9, 7925–7939 CrossRef CAS PubMed.
- A. Laskar, M. Ghosh, S. I. Khattak, W. Li and X. M. Yuan, Nanomedicine, 2012, 7, 705–717 CrossRef CAS PubMed.
- J. Estelrich, M. J. Sánchez-Martín and M. A. Busquets, Int. J. Nanomed., 2015, 10, 1727–1741 CAS.
- W. Wu, C. Z. Jiang and V. A. L. Roy, Nanoscale, 2016, 8, 19421–19474 RSC.
- V. F. Cardoso, A. Francesko, C. Ribeiro, M. Banobre-Lopez, P. Martins and S. Lanceros-Mendez, Adv. Healthcare Mater., 2018, 7, 1–35 Search PubMed.
- F. M. Kievit and M. Zhang, Acc. Chem. Res., 2011, 44, 853–862 CrossRef CAS PubMed.
- D. K. Chatterjee, P. Diagaradjane and S. Krishnan, Ther. Delivery, 2011, 2, 1001–1014 CrossRef CAS PubMed.
- K. Cheng, S. Peng, C. Xu and S. Sun, J. Am. Chem. Soc., 2009, 131, 10637–10644 CrossRef CAS PubMed.
- J. Hernandez-Gil, M. Cobaleda-Siles, A. Zabaleta, L. Salassa, J. Calvo and J. C. Mareque-Rivas, Adv. Healthcare Mater., 2015, 4, 1034–1042 CrossRef CAS PubMed.
- E. Voulgari, A. Bakandritsos, S. Galtsidis, V. Zoumpourlis, B. P. Burke, G. S. Clemente, C. Cawthorne, S. J. Archibald, J. Tucek, R. Zboril, V. Kantarelou, A. G. Karydas and K. Avgoustakis, J. Controlled Release, 2016, 243, 342–356 CrossRef CAS PubMed.
- Z. Cheng, Y. Dai, X. Kang, C. Li, S. Huang, H. Lian, Z. Hou, P. Ma and J. Lin, Biomaterials, 2014, 35, 6359–6368 CrossRef CAS PubMed.
- H. Unterweger, R. Tietze, C. Janko, J. Zaloga, S. Lyer, S. Durr, N. Taccardi, O. M. Goudouri, A. Hoppe, D. Eberbeck, D. W. Schubert, A. R. Boccaccini and C. Alexiou, Int. J. Nanomed., 2014, 9, 3659–3676 CrossRef CAS PubMed.
- J. Yang, H. Lee, W. Hyung, S. B. Park and S. Haam, J. Microencapsulation, 2006, 23, 203–212 CrossRef CAS PubMed.
- E. A. Vitol, E. A. Rozhkova, V. Rose, B. D. Stripe, N. R. Young, E. E. W. Cohen, L. Leoni and V. Novosad, Adv. Mater. Interfaces, 2014, 1, 1400182 CrossRef.
- C. Yu, B. Ding, X. Zhang, X. Deng, K. Deng, Z. Cheng, B. Xing, D. Jin, P. a. Ma and J. Lin, Biomaterials, 2018, 155, 112–123 CrossRef CAS PubMed.
- P. J. Bednarski, R. Grunert, M. Zielzki, A. Wellner, F. S. Mackay and P. J. Sadler, Chem. Biol., 2006, 13, 61–67 CrossRef CAS PubMed.
- P. J. Bednarski, F. S. Mackay and P. J. Sadler, Adv. Anticancer Agents Med. Chem., 2007, 7, 75–93 CrossRef CAS PubMed.
- T. Hyeon, S. S. Lee, J. Park, Y. Chung and H. B. Na, J. Am. Chem. Soc., 2001, 123, 12798–12801 CrossRef CAS PubMed.
- R. Di Corato, A. Quarta, P. Piacenza, A. Ragusa, A. Figuerola, R. Buonsanti, R. Cingolani, L. Manna and T. Pellegrino, J. Mater. Chem., 2008, 18, 1991–1996 RSC.
- A. Quarta, D. Bernareggi, F. Benigni, E. Luison, G. Nano, S. Nitti, M. C. Cesta, L. Di Ciccio, S. Canevari, T. Pellegrino and M. Figini, Nanoscale, 2015, 7, 2336–2351 RSC.
- J. Mindell, Annu. Rev. Physiol., 2012, 74, 69–86 CrossRef CAS PubMed.
- R. Duncan and S. C. W. Richardson, Mol. Pharm., 2012, 9, 2380–2402 CrossRef CAS PubMed.
- N. Eckstein, J. Exp. Clin. Cancer Res., 2011, 30, 91–91 CrossRef CAS PubMed.
- B. Stordal, M. Hamon, V. McEneaney, S. Roche, J.-P. Gillet, J. J. O'Leary, M. Gottesman and M. Clynes, PLoS One, 2012, 7, e40717–e40717 CrossRef CAS PubMed.
- M. K. Kim, T. Osada, W. T. Barry, X. Y. Yang, J. A. Freedman, K. A. Tsamis, M. Datto, B. M. Clary, T. Clay, M. A. Morse, P. G. Febbo, H. K. Lyerly and D. S. Hsu, Mol. Cancer Ther., 2012, 11, 1500–1509 CrossRef CAS PubMed.
- M. G. Apps, E. H. Choi and N. J. Wheate, Endocr.-Relat. Cancer, 2015, 22, R219–R233 CAS.
- D.-W. Shen, L. M. Pouliot, M. D. Hall and M. M. Gottesman, Pharmacol. Rev., 2012, 64, 706–721 CrossRef CAS PubMed.
- C. A. Rabik and M. E. Dolan, Cancer Treat. Rev., 2007, 33, 9–23 CrossRef CAS PubMed.
- D. J. Stewart, Crit. Rev. Oncol. Hematol., 2007, 63, 12–31 CrossRef PubMed.
- K. Li, H. Nejadnik and H. E. Daldrup-Link, Drug Discovery Today, 2017, 22, 1421–1429 CrossRef CAS PubMed.
- A. Akinc and G. Battaglia, Cold Spring Harbor Perspect. Biol., 2013, 5, 1–24 Search PubMed.
- V. Muripiti, B. Lohchania, S. K. Marepally and S. V. Patri, MedChemComm, 2018, 9, 264–274 RSC.
- R. S. Singh, C. Goncalves, P. Sandrin, C. Pichon, P. Midoux and A. Chaudhuri, Chem. Biol., 2004, 11, 713–723 CrossRef CAS PubMed.
- A. El-Sayed, S. Futaki and H. Harashima, AAPS J., 2009, 11, 13–22 CrossRef CAS PubMed.
- A. Quarta, M. Rodio, M. Cassani, G. Gigli, T. Pellegrino and L. L. Del Mercato, ACS Appl. Mater. Interfaces, 2017, 9, 35095–35104 CrossRef CAS PubMed.
- S. Dasari and P. B. Tchounwou, Eur. J. Pharmacol., 2014, 364–378 CrossRef CAS PubMed.
- H. K. Shete, R. H. Prabhu and V. B. Patravale, J. Nanosci. Nanotechnol., 2014, 14, 460–474 CrossRef CAS PubMed.
- C. Xu, B. Wang and S. Sun, J. Am. Chem. Soc., 2009, 131, 4216–4217 CrossRef CAS PubMed.
- M. D. Hall, K. A. Telma, K. E. Chang, T. D. Lee, J. P. Madigan, J. R. Lloyd, I. S. Goldlust, J. D. Hoeschele and M. M. Gottesman, Cancer Res., 2014, 74, 3913–3922 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nr07015j |
| This journal is © The Royal Society of Chemistry 2019 |