
Organic chemistry at anodes and photoanodes
Lacey M.
Reid†
 a,
Tengfei
Li†
a,
Yang
Cao
a and
Curtis P.
Berlinguette
*abc
a,
Tengfei
Li†
a,
Yang
Cao
a and
Curtis P.
Berlinguette
*abc
aDepartment of Chemistry, The University of British Columbia, 2036 Main Mall, Vancouver, BC V6T 1Z1, Canada. E-mail: cberling@chem.ubc.ca
bDepartment of Chemical & Biological Engineering, The University of British Columbia, 2360 East Mall, Vancouver, BC V6T 1Z3, Canada
cStewart Blusson Quantum Matter Institute, The University of British Columbia, 2355 East Mall, Vancouver, BC V6T 1Z4, Canada
First published on 18th June 2018
Abstract
Solar-driven electrolytic water splitting is a promising means of storing renewable electricity, but the kinetic limitations of the anodic oxygen evolution reaction (OER) have impeded the deployment of electrolyzers that produce hydrogen fuels derived from water. In this review, we summarize alternative anodic chemistries being considered as a means of lowering the amount of electricity required to produce hydrogen at the cathode, or simply driving chemistry that forms products more valuable than oxygen at the anode. The potential for an organic oxidation reaction to instead occur at the anode presents a new opportunity for the production of value-added chemical products from cheap, readily available and, in some cases, renewable feedstocks.
1. Introduction
The high gravimetric energy density of molecular hydrogen makes H2 ideal as a carrier of stored energy which can later be used to drive a fuel cell to recover the stored energy as electricity.1–5 Chemists have been splitting water into molecular hydrogen and oxygen using electricity since the invention of the voltaic cell,6 yet nearly all commercial hydrogen is produced by the steam-reforming of methane. The amount of electricity required to drive the concurrent electrochemical water-oxidation reaction at the anode—237 kJ of electrical energy per mole of oxidized water—is typically more costly than the value of hydrogen produced at the cathode.7 The economically viable conversion and storage of solar energy as hydrogen fuels will require the engineering of innovative electrochemical or photoelectrochemical devices with highly efficient catalysts for the anodic oxidation reaction.A solar photovoltaic (PV) cell can operate in tandem with a non-photoactive, or “dark”, electrochemical (EC) water-splitting cell to power the cathodic hydrogen evolution reaction (HER) and anodic oxygen evolution reaction (OER) with solar-derived electricity.3,8–10 Alternatively, sunlight can be converted directly into hydrogen fuels via photoelectrochemical (PEC) water splitting, where energetic electrons and/or holes generated upon the absorption of light drive HER or OER directly at a semiconductor–liquid interface.2,11–14
Water electrolysis is a well-established commercial technology; however, the economical production of hydrogen is curbed by the large overpotential required to overcome the sluggish kinetics of the anodic OER,1,3,8 and moreover the anodic production of oxygen gas which holds little economic value.15–17 One solution may be to produce a more valuable product at the anode to satisfy the industrial demand for chemicals of global importance. Both of these challenges can be addressed by choosing a suitable electrochemical reaction that occurs at a lower redox potential. To this end, the oxidation of organic substrates has recently emerged as an alternative to the production of oxygen gas at the anode. Organic chemistry at the anode has been demonstrated for the sustainable and cost-effective synthesis of fine chemicals, pharmaceuticals and biomass valorization, combined with production of clean hydrogen at the cathode.15,18–22 In this review, we will describe the fundamental principles of anodic water oxidation and the limitations of EC and PEC water-splitting for the production of clean hydrogen. Several examples of organic oxidation reactions are described which potentially offer high-value alternatives to OER. We outline three common anodic organic electrooxidation reactions paired with cathodic HER: alcohol oxidation; C–H functionalization; and alkene functionalization. We also highlight the emerging development of PEC devices for generating value-added products. The discussion of these systems will focus on how these electrochemical reaction systems can be further optimized, including how the choice of electrode materials, feedstock chemicals and redox mediators affect the anodic organic oxidation process and overall efficiency of the system.
While many more organic oxidation reactions can be feasibly carried out at the anode of an EC cell, we have targeted cases that have been demonstrated in tandem with HER generation, and that have the potential to operate on a larger scale. A more comprehensive account of organic transformations via electrochemistry can be found elsewhere.20,21 Further information on the operation and design of electrode materials for PEC and EC cells for water-splitting can be found in other reviews.4,23,24
1.1. Fundamental physics of electrochemical and photoelectrochemical water splitting
In a typical electrochemical water-splitting cell, protons from the electrolyte are reduced at the cathode to produce dihydrogen while water oxidation at the anode generates protons to complete the circuit and balance proton consumption at the cathode (Fig. 1a). Water splitting requires an applied voltage (Vapp) of at least 1.23 V (the difference between the standard electrochemical potentials of OER (EH2O/O2) and HER (EH2/H2O)), but a voltage closer to 2 V is required in practice to overcome any voltage inefficiencies inherent to the cell. Although this overpotential can be associated with both anode and cathode reactions, it is the sluggish kinetics of the four-electron transfer process of OER and the large energy barrier of dioxygen formation that limits the overall reaction rate of water electrolysis.1,4,8,9 Significant research efforts have aimed to develop more efficient OER catalysts to reduce the high overpotential required for water splitting.23–29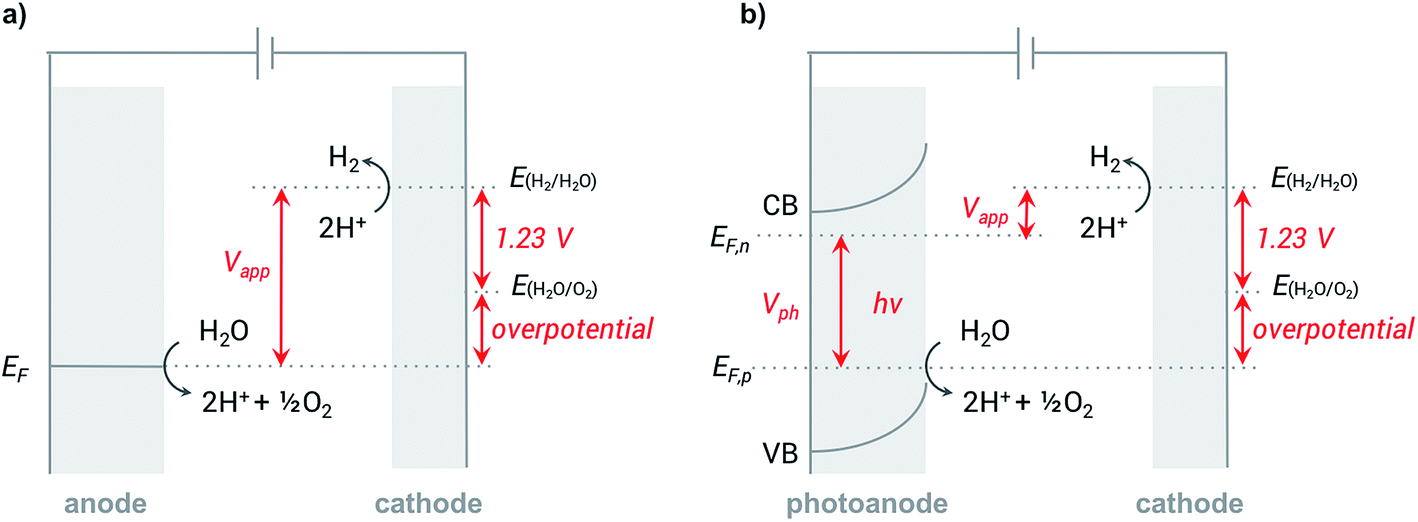 | ||
| Fig. 1 Schematics of (a) electrochemical (EC) and (b) photoelectrochemical (PEC) water oxidation processes at the (photo)anode and corresponding proton reduction processes at the cathode. A voltage must be applied to overcome the electrochemical potential (EF) of water oxidation. Solar energy capture supplies part of this voltage to the photoanode in a photoelectrochemical cell. Electrons and holes are created in the conduction band (CB) and valence band (VB) of the semiconductor, respectively. The quasi Fermi levels of electrons (EF,n) and holes (EF,p) define the photovoltage (Vph) that can be extracted from the photoanode. | ||
The direct conversion of sunlight into chemical fuels in a PEC water splitting cell is enabled by semiconducting photoelectrodes at the semiconductor–liquid junction (Fig. 1b). Non-equilibrium charge populations are created in the conduction band (CB) and valence band (VB) of the semiconductor as a result of the separation of electrons and holes that occurs when photons of appropriate energy are absorbed by the photoanode.2,11,12 The overall photovoltage (Vph) that can be extracted from the photoanode is determined by the potential difference between the quasi Fermi levels of electrons (EF,n) and holes (EF,p).2,30–32 Water is oxidized at the photoanode by holes and reduced on cathode by the photo-generated electrons that travel from the photoanode to the cathode under the applied external bias. An external voltage (Vapp) is still required in a PEC cell; however, this potential is much smaller than that required in an electrochemical cell due to the photovoltage supplied by captured solar energy.
1.2. Challenges of OER and opportunities for anodic organic chemistry
A key challenge in designing an OER catalyst is the long-term stability of the anode in aqueous media. The stability of the electrode material during water electrolysis is a more serious issue for photoelectrocatalytic materials. Very few non-precious metal OER electrocatalysts have been reported which can retain high catalytic activity over 1000 hours.27,33 Even fewer photoanode materials are able to retain high OER performance (photocurrent density > 10 mA cm−2) over 100 h of operation.34,35 The mechanism of photoanode degradation is not yet fully understood; however, it has been widely observed that the surface of the photoanode begins to dissolve in aqueous solution within hours of PEC electrolysis and generally leads to a loss of PEC activity.34,36–38The large overpotential required for OER remains a key challenge, thus providing the impetus to prepare (photo)electrocatalysts that can facilitate OER at lower overpotentials. Different strategies have been applied to mediate OER more effectively; e.g., the use of amorphous mixed-metal electrocatalysts to reduce the overpotential of OER.24,39 Catalyst stability and activity in neutral pH conditions has been promoted via formation of the active electrocatalyst in situ.23 Recent reports have described IrOx/SrIrO3,27 and Fe(PO3)2/Ni2P foam33 electrocatalysts for OER that exhibit a current density of 10 mA cm−2 with an applied voltage of less than 1.5 V.
Improvements in PEC OER activity have focused on the design of composite photoanode-electrocatalyst assemblies35,40,41 as well as the application of high-surface-area, nanostructured photoanode materials which may suppress charge recombination.42,43 Photoanodes comprising layers of BiVO4/FeOOH/NiOOH or NiOx/CoOx/n-Si have recently achieved photocurrent densities of 3 mA cm−2 (0.6 V applied voltage)42 and 30 mA cm−2 (1.3 V voltage under illumination),35 respectively. Additionally, Grätzel and colleagues reported a perovskite PV-electrolyzer tandem apparatus in 2014 that achieved a solar-to-hydrogen efficiency of 12.3% using a NiFe electrode.10 However, the persistence of the higher overpotential of water oxidation compared to the hydrogen evolution potential raises the question of whether there is a higher-value alternative to driving anode reaction.
In most water splitting architectures, the anodic product, oxygen gas, is vented rather than captured.15–17 The fundamental purpose of OER as the anodic water-electrolysis reaction is simply to complete the electrical circuit and to provide protons to offset the proton consumption by HER at the cathode. Many chemical oxidation reactions can be achieved electrochemically and are typically accompanied by a deprotonation process.19,21 Replacement of anodic OER with an alternative organic oxidation reaction has the potential to generate products with higher economic value than oxygen gas (e.g., fine chemicals, pharmaceutical precursors) while the desired product of water splitting (H2) continues to be produced at the cathode. Furthermore, using the anode to drive organic transformations can also provide an opportunity to alleviate the degradation of photoelectrodes that occurs during water oxidation (see Section 2.2). In the following sections, the anodic oxidation of organic substrates will be discussed as a promising strategy to address the limitations of the traditional anodic oxidation of water.
2. Driving organic transformations at the anode
2.1. Principles of (photo)electrochemical organic oxidation
Organic redox reactions can be readily achieved using chemical oxidizing reagents; however, these oxidants are often expensive or toxic (e.g., CrO3, Pb(OAc)4, KMnO4, OsO4), and can produce harmful waste. Electrochemical oxidation reactions afford the use of catalytic rather than stoichiometric amounts of oxidizing reagents via electrochemical regeneration, offering an environmentally friendly alternative to traditional chemical oxidation methods.44 Furthermore, an electricity-driven redox process affords the ability to tune the oxidative strength of the reaction to the redox potential of a substrate so that catalytic redox reagents are not necessarily required.45 Electricity drives the electron transfer processes instead of chemical oxidants, shuttling electrons between the surface of anode and the organic substrates. Selectivity between different functional groups is controlled by the electric potential applied to the working electrode rather than the oxidative strength of a selected chemical oxidant.45 The relatively mild reaction conditions required for electrochemistry—along with the ability to target desired functional groups for oxidation—are features of anodic oxidation which can impart high selectivity alongside operational simplicity.Electron transfer between electrode and substrate is an interfacial heterogeneous process that may be expected to be kinetically slower than a homogeneous chemical reaction. The surface area of the electrode can be increased, or a redox shuttle or “mediator” can be employed, to more efficiently transfer electrons between anode and substrate (Fig. 2a) (analogous to the role of redox photosensitizers in photocatalytic oxidation reactions, Fig. 2c). A more detailed discussion of the redox mediator will be provided in Section 2.3. Photoelectrochemical oxidation of an organic substrate (Fig. 2b) follows a similar electron transfer mechanism to the EC system. A lower potential is required to drive electron transfer between the photoanode and substrate, owing to the photovoltage generated by the semiconductor. Both photocatalytic and electrocatalytic oxidations are promising for environmentally friendly, sustainable and cost-effective syntheses of organic commodity chemicals; however, this review will focus solely on using EC and PEC systems to drive organic transformations.
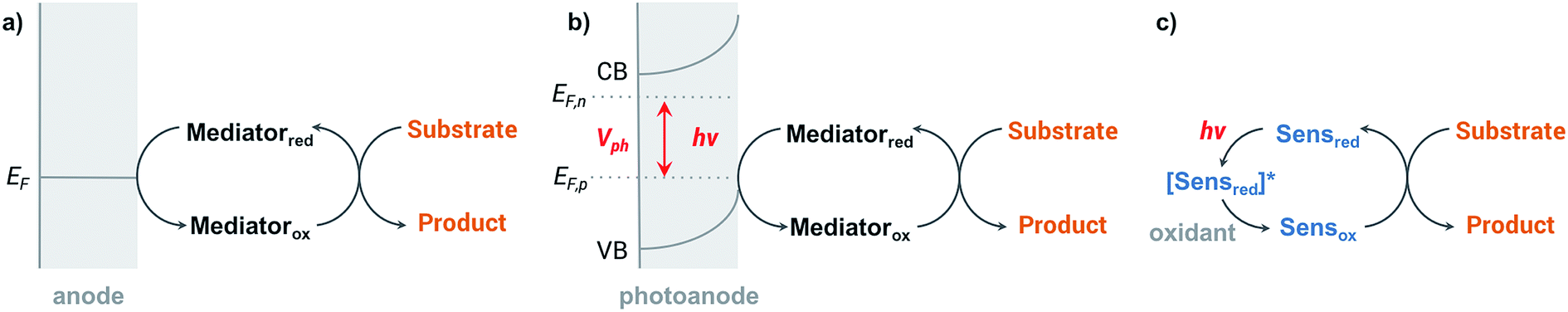 | ||
| Fig. 2 General principles of (a) electrocatalytic, (b) photoelectrocatalytic, and (c) photocatalytic oxidations of an organic substrate. In (a) and (b) the potential supplied to the anode results in the oxidation of a redox mediator (mediatorred → mediatorox) that subsequently oxidizes the substrate at a comparable chemical potential. In photocatalytic cells the process is mediated by a sensitizer molecule (Sens) through an analogous mechanism. | ||
2.2. Solvents: aqueous vs. organic media
Common solvents for electrochemical experiments include water, organic liquids, and low-melting-point salts known as ionic liquids. Ionic liquids have received significant research interest due to their high conductivity, wide potential window and non-flammability;46 however, this paper will primarily focus on the discussion of aqueous and organic solvents in this section.Polarity is one of the first considerations when choosing a solvent for anodic oxidation because many organic substrates are insoluble in a highly polar solvent (e.g., water), and a supporting electrolyte, which is required to provide conductivity during electrolysis, is insoluble in non-polar solvents (e.g., pentane, toluene). Solvents with intermediate polarity (e.g., acetone, nitromethane, acetonitrile) are ideal to balance the solubilities of the organic substrate and supporting electrolyte, while nonpolar solvents such as hexane and benzene should be avoided in general. Secondly, the potential range beyond which a solvent starts to degrade determines whether that solvent can be used for the oxidation of a certain substrate. The anodic potential window of several common solvents and the oxidation potentials of several common types of organic substrates are shown in Fig. 3. Oxidation of organic substrates with low redox potentials (e.g., alcohols) can be achieved directly on the surface of the anode, whereas direct electrolysis of organic substrates with high redox potentials (e.g., olefins, unactivated hydrocarbons) usually lead to oxidative degradation of the solvent before the substrate can be oxidized. Redox mediators are therefore required in most anodic organic oxidation systems to facilitate the oxidation of the substrate at lower potentials, avoiding undesirable solvent oxidation.
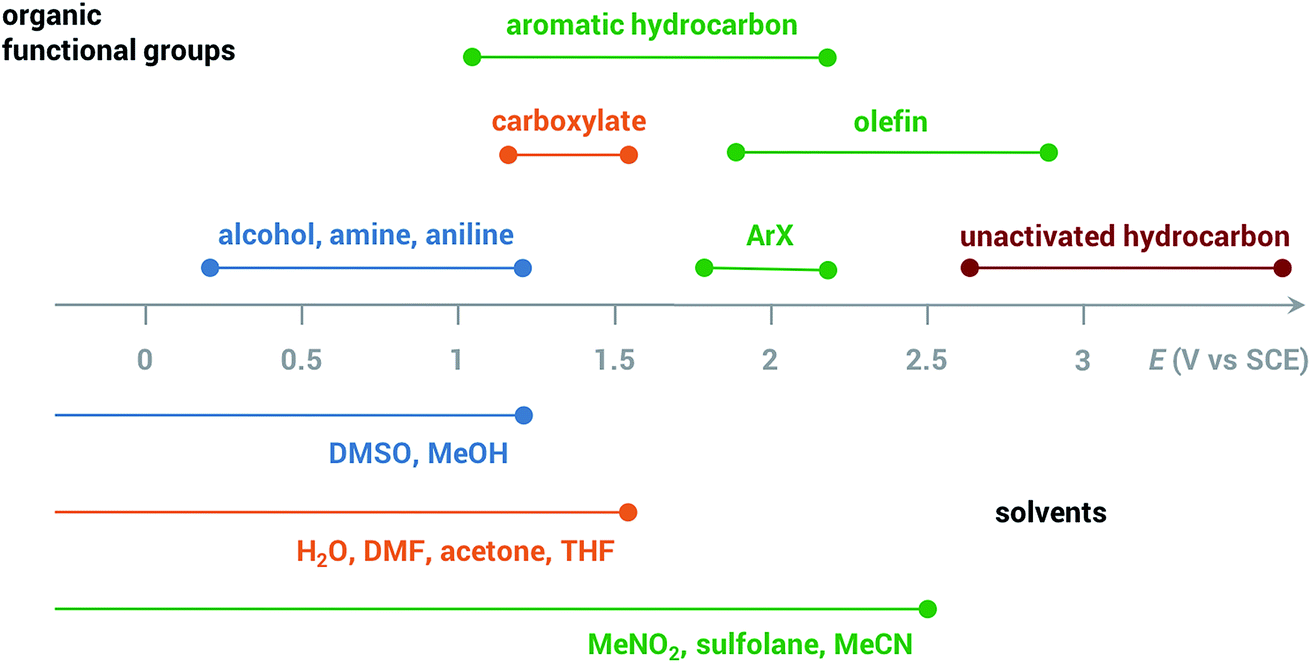 | ||
| Fig. 3 Anodic potential range of dimethyl sulfoxide (DMSO), methanol (MeOH), H2O, dimethylformamide (DMF), acetone, tetrahydrofuran (THF), nitromethane (MeNO2), sulfolane, acetonitrile (MeCN) and approximate redox potentials of several common organic functional groups. All potentials are referenced to the saturated calomel electrode (SCE). Adapted from ref. 47. | ||
Lastly, the stability of the electrode plays an important role in choosing a solvent, especially in photoelectrochemical oxidation systems. Although common electrode materials (e.g., platinum or carbon electrode) used in electrocatalytic organic oxidation are stable in most solvents, one of the key challenges for PEC is the photocorrosion of the semiconductor electrode over time.36,38 While photocorrosion in water can be suppressed by a protective (electrocatalytic) metal oxide layer (e.g., CoOx, NiOx, FeOx),36,37,48 these layers tend to suppress the oxidation of organic compounds in favor of OER which occurs at similar applied potentials.15,49 We demonstrated that photoelectrochemistry carried out in MeCN rather than water suppressed photocorrosion of a BiVO4 photoanode (Fig. 4).17 Photoanodes studied after 96 h of exposure to PEC conditions in H2O showed a much more significant reduction in light absorption and photocurrent than those operating in MeCN (Fig. 4a and b). The accelerated photocorrosion of BiVO4 in water was confirmed by scanning electron microscope images of photoanodes before and after 96 h of PEC operation. Photoanodes subjected to 96 h of PEC electrolysis in MeCN showed full coverage of FTO with spherical grains of BiVO4 while those photoanodes operating in water showed visible areas of FTO afterward (Fig. 4c). These data collectively indicate that the use of organic media in place of water affords markedly higher semiconductor photostability, ultimately enabling sustained photoelectrochemical conversion over four days.
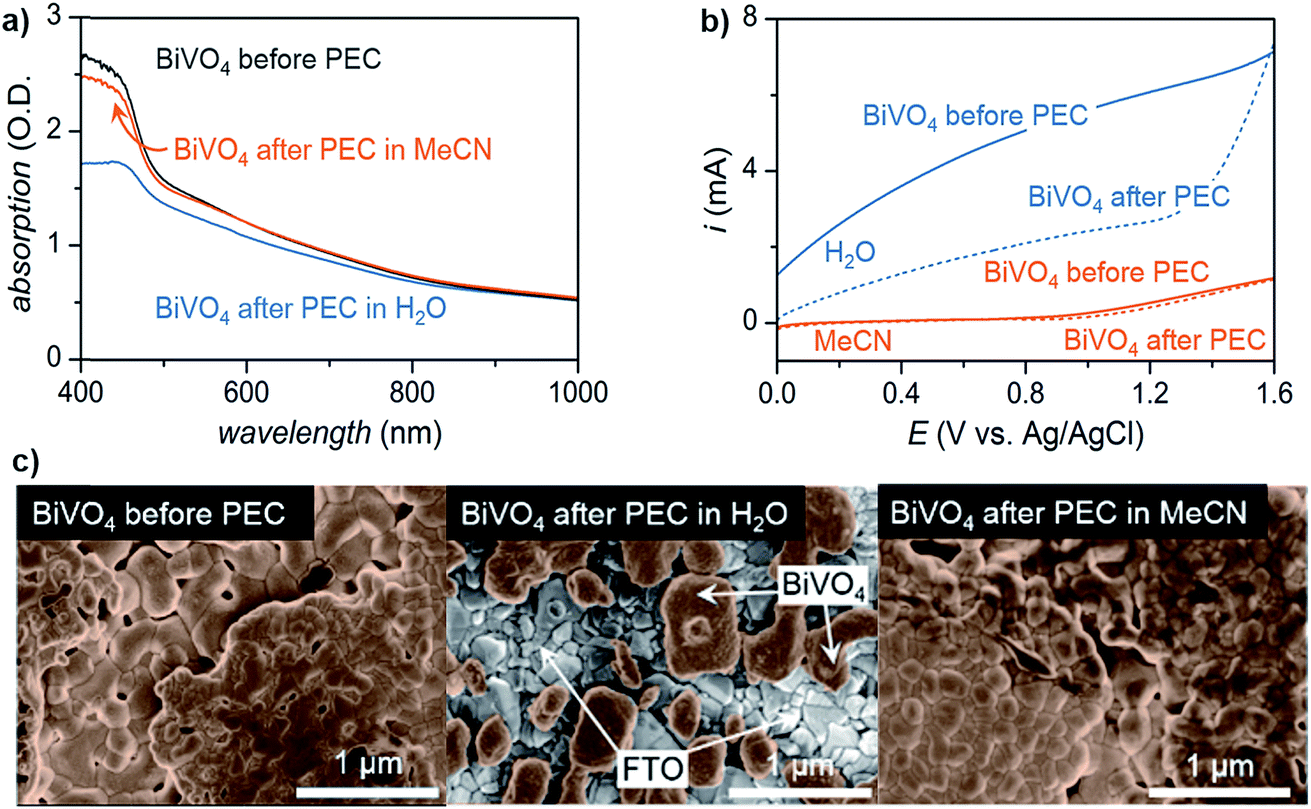 | ||
| Fig. 4 Comparison of BiVO4 photoanodes before and after sustained PEC electrolysis for 96 h in either water or acetonitrile at a constant potential of 0.1 V or 1.6 V (vs. Ag/AgCl), respectively, as illustrated by: (a) UV-vis absorption spectra; (b) linear sweep voltammogram curves of photocurrents; and, (c) scanning electron microscopy (SEM) images. PEC electrolysis in water degraded the BiVO4 photoanode to the extent that the underlying FTO was visible in the SEM image (c; center), consistent with the decreased optical absorption and generated photocurrent. Copyright 2017, Nature Publishing Group.17 | ||
2.3. Redox mediators
The redox mediator facilitates the transfer of electrons between the anode and organic substrate in an indirect EC oxidation process (Fig. 5), a combination of heterogeneous electron transfer and homogeneous redox catalysis. An activated form of the mediator (Medox) is generated when the applied potential reaches the redox potential of the mediator (EMediator). A charge-transfer process then occurs between Medox and substrate to generate a highly reactive radical or cation.50 The equilibrium electron transfer should favour the production of substrate over the product if the applied potential is less positive than the redox potential of substrate (ESubstrate); however, the equilibrium can be shifted towards product when the reactive intermediate subsequently undergoes several rapid and irreversible chemical reactions. These follow-up reactions consume the product of the thermodynamically unfavorable electron-transfer step, removing this intermediate from equilibrium.19,50 The oxidation of an organic substrate can be carried out at a potential equal to EMediator and lower than ESubstrate by this process since the overall reaction rate is dictated by the equilibrium constant.19,50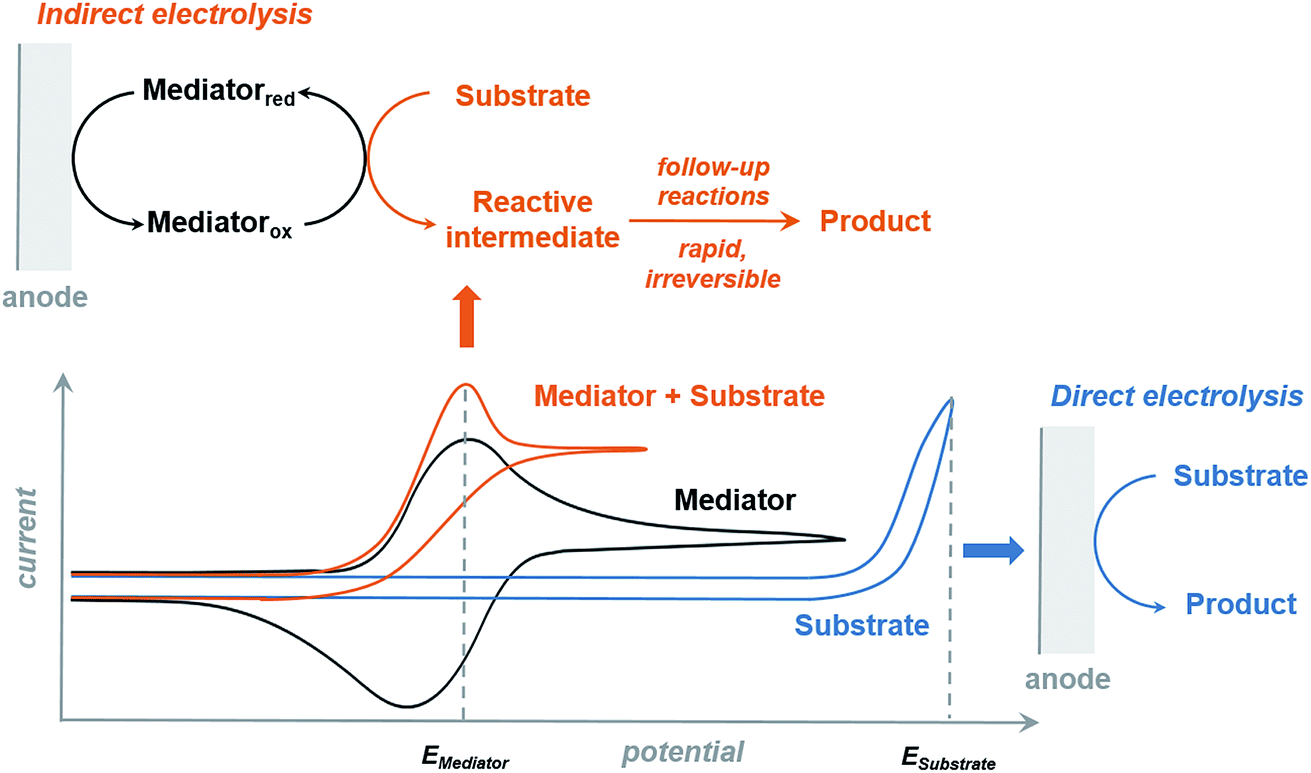 | ||
| Fig. 5 Cyclic voltammogram plots of a redox mediator, substrate (direct electrolysis) and mediator + substrate combination (indirect electrolysis), and the corresponding charge-transfer process. Indirect electrolysis carried out at the redox potential of the mediator (EMediator) converts the substrate into a reactive intermediate, followed by rapid, irreversible chemical reactions to yield the final product. Direct electrolysis (without a redox mediator) requires a much higher potential equal to ESubstrate. | ||
Less electrical energy is consumed in an indirect electrolysis process carried out at the redox potential of the mediator rather than that of the substrate. The reaction can be performed under mild conditions (e.g., low overpotential, room temperature) to protect functional groups that could otherwise be sensitive to oxidation. If the potential difference between EMediator and ESubstrate is too large then the reaction rate could be too slow to yield appreciable amounts of product; therefore, a mediator having a redox potential 0.5–1.5 V lower than that of the target substrate is normally selected. Redox potentials of several common mediators and functional groups are shown in Fig. 6.
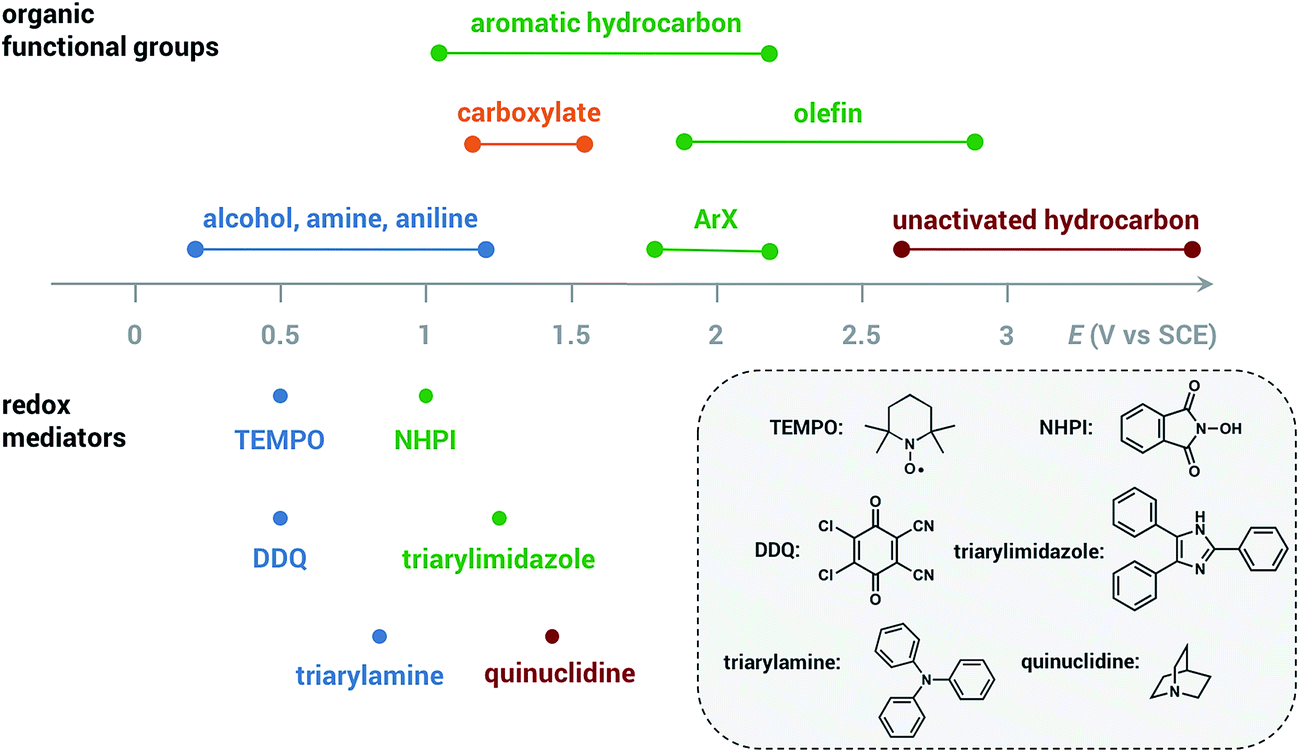 | ||
| Fig. 6 Approximate redox potentials of several common organic functional groups and redox mediators. The redox potentials of the mediator can be shifted by structural or functional-group modification. All potentials are referenced to the saturated calomel electrode (SCE). Data from ref. 19 and 47. | ||
Higher substrate chemoselectivity can be achieved by redox-mediated EC compared to direct oxidation of organic substrates. The advantage is two-fold: (i) a relatively lower applied voltage avoids the oxidation of functional groups with lower standard potentials; and (ii) transient interactions or chemical bonding occurring between the substrate and mediator can show selectivity for particular functional groups.19 For example, 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) is known to selectively oxidize primary alcohols over secondary alcohols in basic conditions, whereas secondary alcohols are favoured over primary alcohols in acidic conditions.51 The selectivity in basic conditions is usually attributed to the large steric effect of the secondary alcohol when a chemical bond is formed between the nitrogen atom of TEMPO and the oxygen atom of the alcohol (Fig. 7a). The mechanism of TEMPO-mediated alcohol oxidation under acidic conditions is believed to involve hydride transfer between TEMPO and the alcohol, favouring electron-rich secondary alcohols (Fig. 7b).51
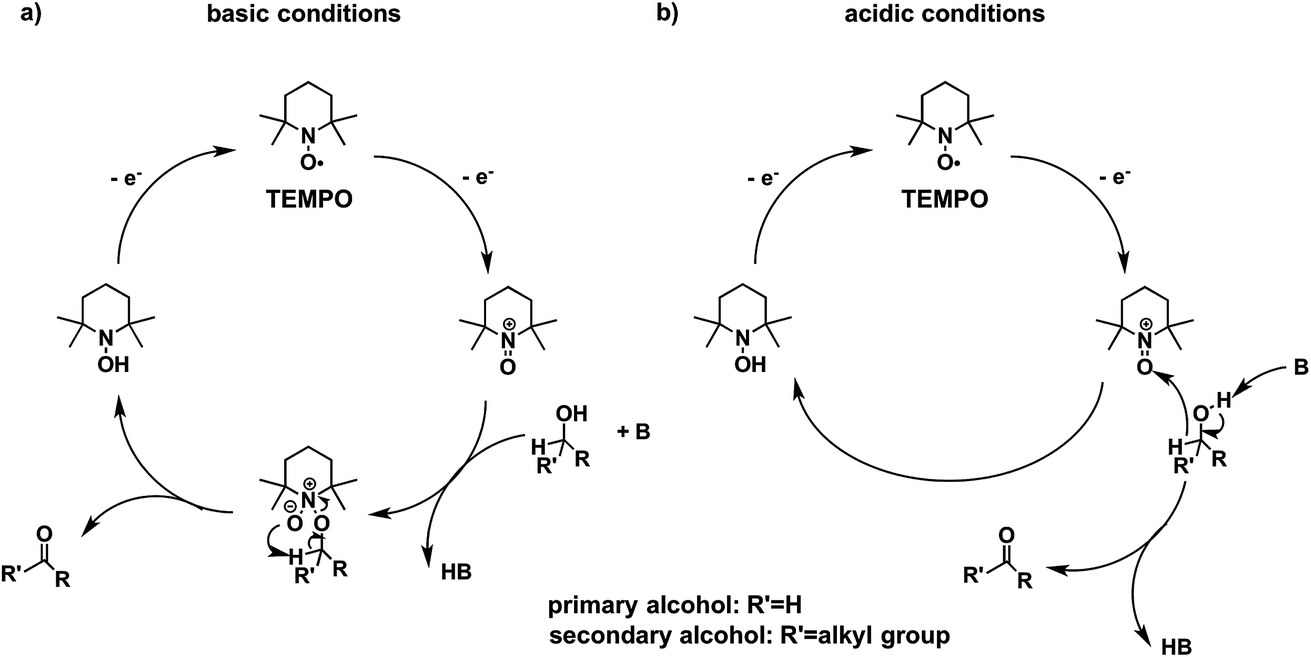 | ||
| Fig. 7 The mechanism of TEMPO-mediated alcohol oxidation proceeds through an alkoxide adduct under basic conditions, or through a bimolecular hydride transfer under acidic conditions, to impart substrate chemoselectivity. Adapted from ref. 51. Copyright 2015, American Chemical Society. | ||
3. Electrochemical organic oxidations
The use of redox mediators has remained a useful strategy for lowering the overpotential in an electrochemical cell so that less electrical energy is required; however, the fundamental thermodynamic limitations of electrochemical water splitting still apply to systems where the anode current primarily drives OER. Direct replacement of OER with the oxidation of a readily-oxidized species would establish a new benchmark for hydrogen production at overpotentials that are inherently lower than in a typical water-splitting cell. This strategy has been successfully applied for substrates such as ammonia,52 methanol,53 ethanol,54 and other small chemical feedstock molecules having oxidation products which are more commercially valuable than oxygen.55,56 The integration of HER at the cathode with the synthesis of value-added oxidation products at the anode is an emerging field of “electrochemical reforming” of chemical feedstocks.It is important to consider a few key criteria in selecting an oxidation reaction to replace OER. Namely, the organic substrate must be soluble and have a lower (less positive) anodic oxidation potential than the aqueous medium to prevent OER from competing with the organic oxidation. Furthermore, the feedstock chemical should be chosen to maximize the value of the oxidation product so that hydrogen is generated in a more cost-effective process. The reaction should be selective to avoid costly separation steps where multiple oxidation products are possible. A repertoire of organic reactions exist which fulfill some or all of these criteria and can readily be applied to an electrochemical reforming process. Baran and colleagues have exhaustively reviewed a large scope of electrochemical organic transformations that have been developed since 2000.21
This review will focus on a few model reactions that have been developed for electrochemical reforming purposes and fulfill most or all of the criteria for effectively replacing OER, including: alcohol oxidation; C–H functionalization; and alkene functionalization. The following sections will discuss strategies for enhancing faradaic efficiencies of these reactions, while avoiding over-oxidation by enhancing selectivity.
3.1. Electrochemical alcohol oxidation
Alcohol oxidation reactions are important chemical transformations for synthetic chemistry. Primary alcohols are typically oxidized at an anodic potential between 0.5–1.3 V affording aldehyde and carboxylic acid products that can be used as building blocks for a multitude of commodity chemicals. When designing a catalyst for electrochemical alcohol oxidation there are many parameters that can be optimized to dial into a system with the highest catalytic activity at the lowest overpotential. The choice of electrode material and type of mediator are often the first parameter adjusted in order to optimize activity and we can use the growing body of literature to guide research into better catalyst design.With the goal of designing low-cost electrodes from first-row transition metals, Sun and colleagues reported a porous Ni3S2/Ni foam anode (Fig. 8a) for the oxidation of several alcohols under alkaline conditions.16 Nickel water-splitting catalysts typically display poor stability and low efficiency in alkaline electrolyzers. Phosphorus, chalcogens or transition metal elements are often incorporated within the nickel to prepare composite catalysts stable to the alkaline environment.57 This particular nickel sulfide catalyst on nickel foam reported by Sun in 2016 (ref. 16) was previously shown to be highly active and stable for water splitting at pH 14, catalyzing both HER and OER as both the cathode and anode material.58 The 2016 report described the oxidation of ethanol, benzyl alcohol, furfuryl alcohol, furfural, and 5-hydroxymethylfurfural (HMF) employing the Ni3S2/Ni foam anode and carbon rod counter electrode at an applied potential of 1.423 V (vs. RHE), 160–200 mV lower than that required for overall water splitting with this catalyst.16 Moderate current densities (100 mA cm−2) were observed and the low redox potential of the nickel sulfide catalyst enabled the direct oxidation of substrates in the absence of an organic mediator. At this overpotential (1.423 V), all aldehyde species were further oxidized; all five feedstocks, including the primary ethanol, benzyl and furfuryl alcohols, were converted to the corresponding carboxylate products with high faradaic efficiencies (nearly unity). The Ni3S2/Ni foam was then employed as a bifunctional catalyst for both the anodic (oxidation) and cathodic (HER) reactions. HMF oxidation to 2,5-furandicarboxylic acid (FDCA) was demonstrated at cell voltages of 1.46, 1.52, 1.58, and 1.64 V (vs. RHE) to achieve benchmark current densities of 10, 20, 50 and 100 mA cm−2, respectively. FDCA was produced in ∼98% faradaic efficiency at 1.50 V (vs. RHE) after 58 C of charge was passed. The same group later reported the oxidation of HMF and other primary alcohols using a bifunctional hierarchically-porous nickel (hp-Ni) catalyst prepared by a benign one-step electrodeposition process,59 achieving a similar performance to the Ni3S2/Ni foam.
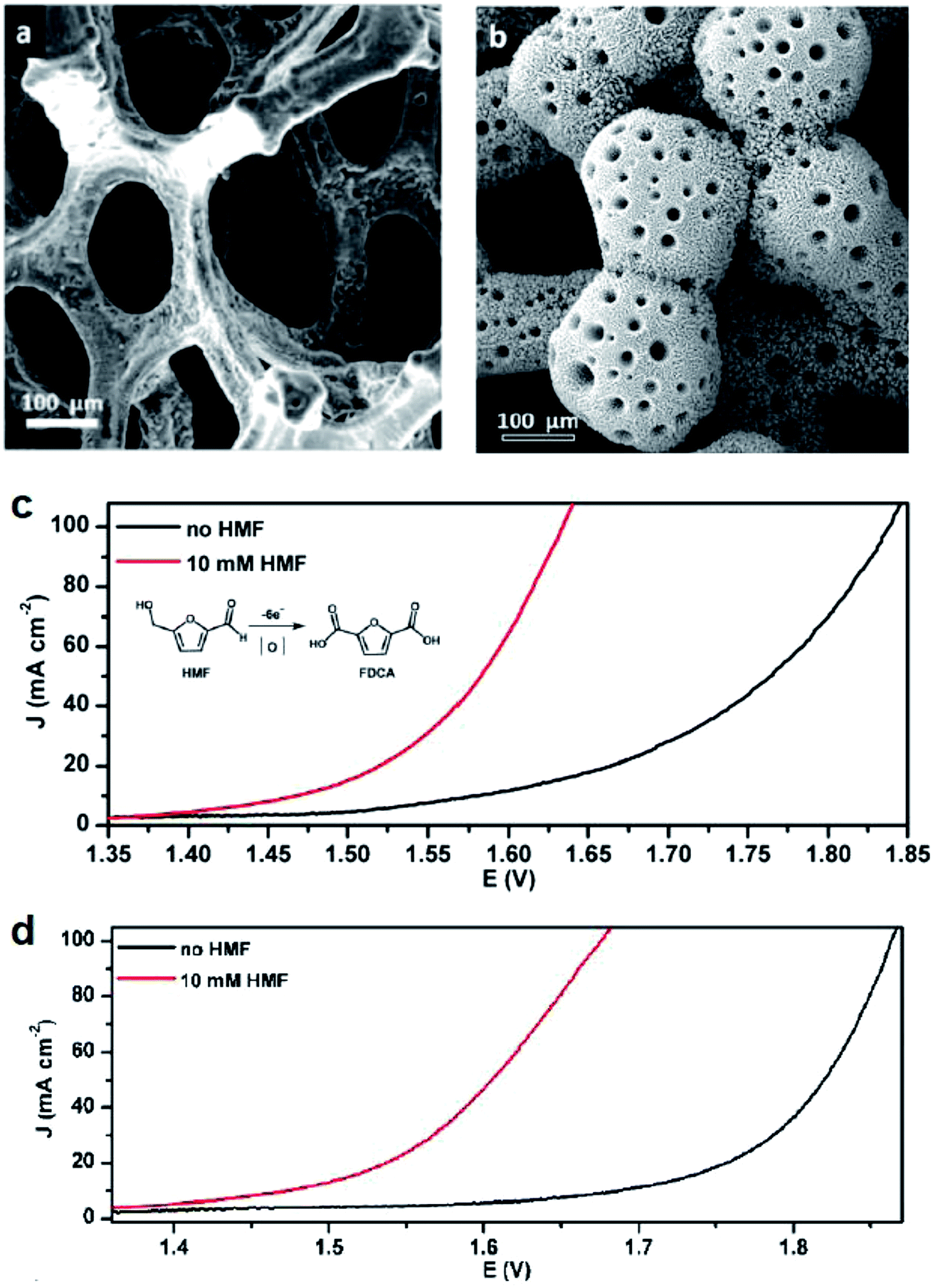 | ||
| Fig. 8 (a and b) SEM images of electrodes employed for the electrochemical oxidation of 5-hydroxymethylfurfural (HMF) to the value-added 2,5-furandicarboxylic acid (FDCA) in tandem with HER: (a) Ni3S2/Ni-foam (anode); (b) 3D hierarchically porous Ni (hp-Ni) (bifunctional cathode and anode); corresponding linear sweep voltammograms of (c) Ni3S2/Ni-foam and (d) 3D hp-Ni in 1.0 M KOH, in the presence (red) or absence (black) of the HMF substrate. LSV scan rate: 2 mV s−1. Copyright 2016, American Chemical Society.16,59 | ||
Vizza and coworkers reported the oxidation of ethanol in basic electrolyte using a PEM-type electrolyzer54 employing a Pd/TiO2 nanomaterial catalyst similar to that used in a previous report for direct methanol fuel cell catalysis.60 The anode material described in this work consisted of a web of TiO2 nanotube arrays on Pt/C embedded with palladium nanoparticles with Pd and Pt loadings of 1.7 and 0.3 mg cm−2 respectively. Oxidation of ethanol was kinetically slow at room temperature, yet high current densities of 1.5–2.00 A cm−2 (0.92 V) could be obtained at a cell temperature of 80 °C. No oxygen evolution was detected under the cell working potential of <1.0 V and the sole product of ethanol oxidation (sodium acetate) was confirmed by HPLC and spectral analysis of the anode exhaust. The high electrochemical surface area provided by the nanoparticles within the porous anode showed high activity not only for the oxidation of ethanol but also for biomass-derived alcohols glycerol, ethylene glycol and 1,2-propanediol, although a distribution of oxidation products was observed (Fig. 9).
 | ||
| Fig. 9 A high-surface-area anode featuring palladium nanoparticles on a web of TiO2 nanotube arrays (a) was used for the electrocatalytic oxidation of different biomass-derived alcohols in a PEM-type electrolyzer. The oxidation of ethanol was selective for acetate while ethylene glycol, glycerol and 1,2-propanediol oxidation each gave a mixture of products (b). Adapted from ref. 54. Copyright 2014, Nature Publishing Group. | ||
It is well known that in basic conditions TEMPO is chemoselective for the oxidation of primary over secondary alcohols due to the steric bulk delivered by the pendant methyl groups on the nitroxyl mediator. For the electro-oxidation of sterically hindered secondary alcohols, Onomura and coworkers have reported the use of azabicyclo-N-oxyl mediators under basic conditions at constant current (50 mA).62 Where TEMPO afforded low to moderate yields (23–61%) for the oxidation of sterically encumbered alcohols, the azabicyclo-N-oxyl mediators in most cases yielded the ketones in >75% (Table 1).
| Substrate alcohol | Redox mediator | |||||
|---|---|---|---|---|---|---|
|
|
|
|
|
|
|
|

|
76 | 86 | 99 | 82 | 92 | 23 |
|
82 | 65 | 87 | 86 | 90 | 61 |
|
99 | 74 | 85 | 99 | 94 | 41 |
|
74 | 75 | 72 | 99 | 97 | 56 |
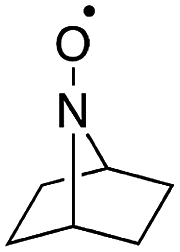
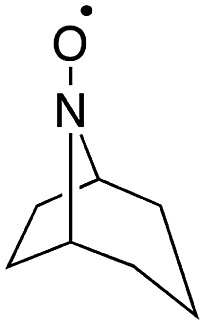
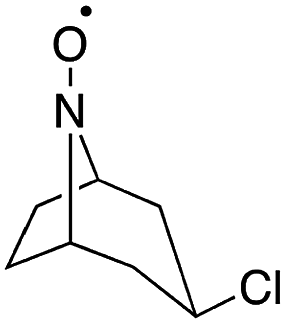
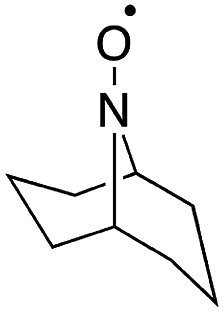
Stahl and colleagues conducted mechanistic studies of series of cyclic nitroxyl compounds (Fig. 10) for the electrocatalytic oxidation of benzylic and aliphatic primary and secondary alcohols under alkaline conditions where steric effects typically dictate catalytic activity toward bulkier substrates.51 Despite the steric bulk of 4-acetamido-TEMPO (ACT), this mediator largely outperformed the derivatized nitroxyl compounds and the classic TEMPO mediator in this study. At high pH values, the ACT mediator was highly active, showing TOFs of 1000 and 6000 h−1 for the room-temperature oxidation of 2-butanol and 1-butanol, respectively, at 0.7 V applied potential. The authors have provided a case for anode chemistry where the operating potential can be adjusted to the redox potential of the nitroxyl mediator to maximize activity.
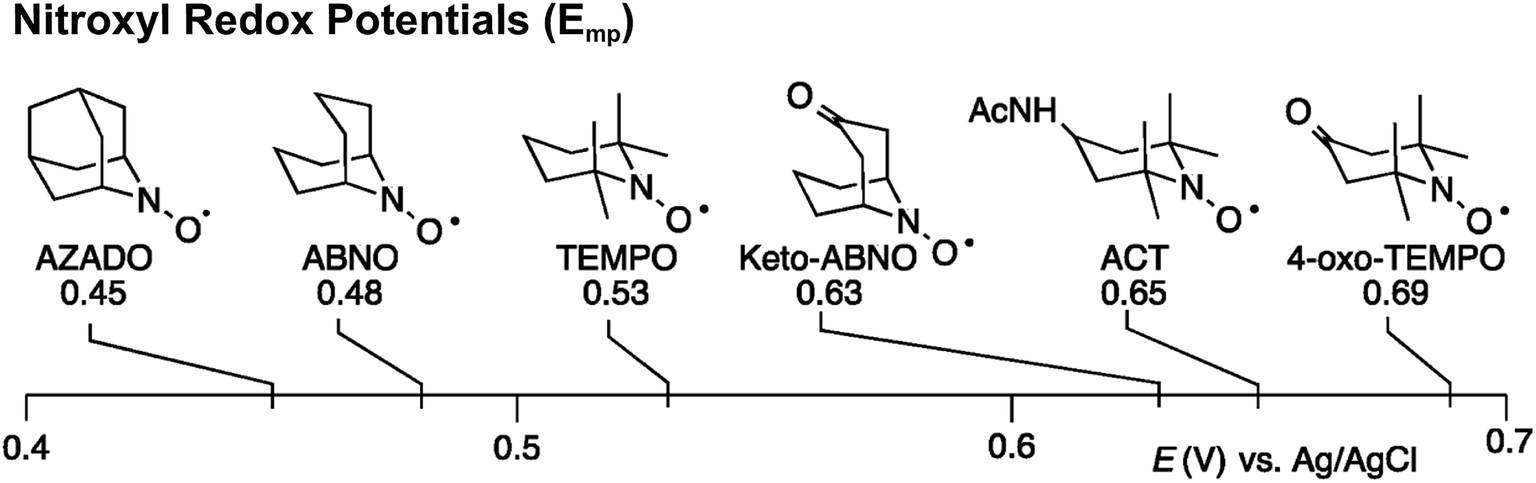 | ||
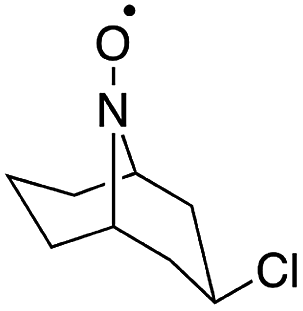
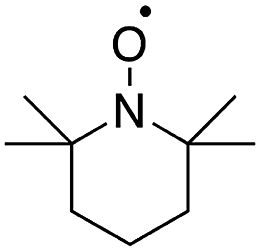
| Fig. 10 Structures of cyclic nitroxyl mediators and midpoint potentials (Emp) of the corresponding one-electron nitroxyl/oxoammonium redox couple. Adapted from ref. 51. Copyright 2015, American Chemical Society. | ||
In follow up work, the same group demonstrated that a proton-coupled redox process using TEMPO and a Cu(II) bipyridine complex catalyzed the same alcohol oxidation reaction at the redox potential of the (bpy)Cu(II)/(bpy)Cu(I) complex, effectively lowering the operating electrode potential by 500 mV.63 The cooperative 2 × 1 electron process generated the TEMPO˙ radical at −0.14 V (vs. Cp2Fe+/0) instead of the TEMPO+ species which requires a higher potential (Fig. 11a). The reactive oxyammonium radical could then serve as a one-electron, one-proton acceptor with Cu(II) acting as a one-electron oxidant. By comparison, the oxidation of benzyl alcohol by the TEMPO/Brønsted base-catalyzed system proceeded at an applied potential of 0.36 V. The kinetic studies in this work provide an account of the two differing mechanisms for (bpy)Cu/TEMPO and TEMPO/base catalysis, which is a useful guide for mediator design for particular substrates. The (bpy)Cu/TEMPO system was thought to proceed through a Cu(II)–alkoxide complex in the rate limiting step (Fig. 11a) and therefore this reaction was accelerated with more highly acidic alcohol substrates (Fig. 11b).
 | ||
| Fig. 11 (a) Catalytic cycle proposed for the (bpy)Cu/TEMPO redox-mediated electrochemical oxidation of alcohols. (b) Comparison of the relative catalytic activity of (bpy)Cu/TEMPO, (bpy)Cu/ABNO, and NMI/TEMPO (NMI = N-methylimidazole) for the oxidation of several small alcohols. Copyright 2016, Nature Publishing Group.63 | ||
Triarylimidazole and triarylamine redox mediators have been reported as an alternative to nitroxyl-based mediators for alcohol oxidation.65,66 The groups of Zeng and Little have together reported a library of triarylimidazoles in order to correlate electronic properties to the first oxidation potential.65 The triarylimidazoles typically displayed three oxidation peaks by CV, with the first oxidation peak (E1ox) corresponding to a quasi-reversible redox couple occurring between 0.7–1.45 V (vs. Ag/AgCl) depending on the substituents on the pendent aryl groups. Electron withdrawing groups placed on the aryl groups caused E1ox to increase, whereas electron donating groups afforded oxidation at lower potentials. The authors proposed a guideline for selecting an appropriate mediator for a catalytic system: a mediator should be suitable for the oxidation of a substrate when the oxidation potential of the substrate does not exceed E1ox of the mediator by >500 mV. Use of this guideline was demonstrated for a mono-brominated triarylimidazole mediator (E1ox = 1.28) employed for the oxidation of 4-methoxybenzyl alcohol (Esubstrate = 1.52 V) at an applied potential of 1.28 V (vs. Ag/AgCl). The ketone product was afforded in 65% yield.
Catalyst discovery and optimization for electrochemical oxidations has been largely empirical to date. The examples described in this section have pursued small gains in catalytic activity by achieving small diminutions in the mediator redox potential. The Minteer and Sigman research groups have together developed a more nuanced mechanistic understanding of the empirical reports of TEMPO-based mediators and have applied computational modelling as a tool to predict trends in catalytic activity.64 In this report, the pH dependence of the electrochemical characteristics and catalytic activity of TEMPO for glycerol oxidation was investigated to inform on the nature of the rate limiting step, which was the one-electron oxidation of TEMPO-H to TEMPO˙ radical described by the second oxidation potential (E2a). When TEMPO was modified with functional groups that stabilized the TEMPO-H intermediate, for example by preventing protonation to TEMPOH-H+, the catalytic activity of the redox mediator increased for glycerol oxidation. Fundamentally this increased reactivity was a result of the lowering of E2a with respect to E1a (pH independent). For a library of TEMPO compounds, E2a − E1a or ΔEa can be estimated computationally to predict the mediators that have the highest catalytic activities. In addition to predictions on the redox states, density functional theory (DFT) was used to derive structure–function relationships for catalytic activity based on how functional groups introduced to TEMPO might stabilize or destabilize the catalytic intermediates TEMPO-H and TEMPO˙+ radical cation, thereby affecting the magnitude of E2a and E1a, respectively. The resulting plot of normalized catalytic activity versus redox potential was used to visually sort functionalized TEMPO mediators based on suitable applications (Fig. 12). For example, mediators falling within quadrant I may be useful for anodic catalysts in fuel cell applications where low Ea and high catalytic activity are ultimate design goals, whereas mediators falling in quadrant II have higher redox potentials that may be beneficial to synthetic applications where the oxidation potential is tuned to the desired substrate to enhance selectivity.
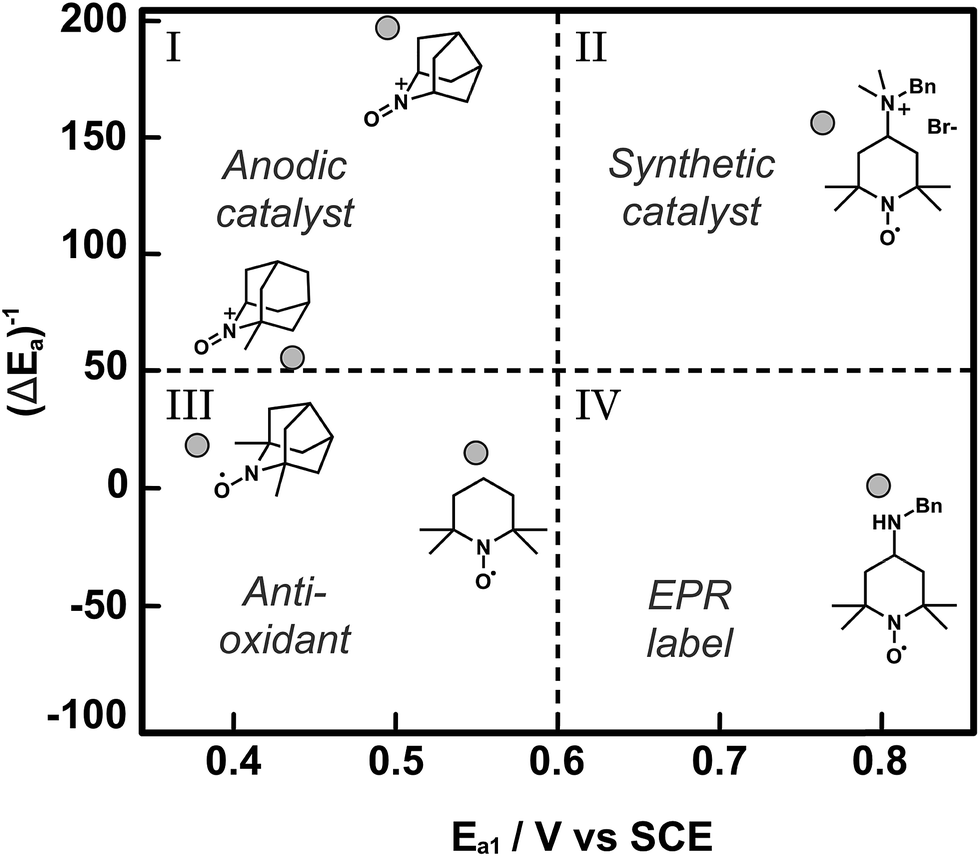 | ||
| Fig. 12 A plot of normalized catalytic activity of representative examples (grey circles) of modified TEMPO-based mediators for the electrochemical oxidation of glycerol. The statistical mean of catalytic activity (maximum current density) and redox potential (E1a) divide the space into quadrants to provide a visual guide to determine the suitability of the mediators for different applications. Adapted from ref. 64. Copyright 2015, American Chemical Society. | ||
Toward the goal of enhancing the performance of organic redox-mediated electrocatalytic cells, several groups have described the immobilization of redox mediators onto the electrode surface.67–71 Fixing the mediator to the electrode has the added benefit of catalyst reuse; however, the problem of slow diffusional kinetics through the immobilization medium must be addressed since this can lead to a significant drop in current response.69 The covalent attachment of TEMPO to a polymer through the Lewis basic sites of poly(ethylenimine) yielded a highly active immobilized TEMPO catalyst for the electrochemical oxidation of saccharides and bioavailable alcohols.67 Moderate current densities of 2–8 mA cm−2 (0.8 V vs. SCE) were achieved for the oxidation of methanol, ethanol, isopropanol and glycerol in neutral pH at room temperature, an order of magnitude higher than the values obtained using a homogeneous methoxy-TEMPO catalyst. The authors proposed that the polymer-bound TEMPO may self-react with immobilized tertiary amine sites to enhance the catalytic activity as well as being assisted by the localized buffer regions offered by the basic sites of the polymer backbone.67 Other groups have reported the immobilization of TEMPO onto thin films of mesoporous silica,72 or multi-walled carbon nanotubes (MWCNTs)71 on glassy carbon electrodes for the electrocatalytic oxidation of a wide scope of benzyl alcohols. The TEMPO-functionalized MCM-41 coated electrodes showed the highest activity for primary benzyl alcohol substrates bearing electron donating groups, with a maximum turnover frequency of 3070 h−1. The system was also tolerant of pyridyl, furfuryl, allylic and secondary benzyl alcohols. Electrochemical preparatory-scale (20 mmol) synthesis of these substrates was achieved after 4 h at an operating potential of 1.0 V.72
Preparative-scale electrolysis was also achieved with a pyrene-tethered TEMPO mediator immobilized on a MWCNT-decorated carbon electrode by noncovalent π–π interactions (Fig. 13a).71 Electrochemical oxidation of several para- and ortho-substituted benzyl alcohols was achieved in >90% yield within 80 min in most cases, at an applied potential of 0.7 V (vs. Ag/AgCl). Substrates with electron donating groups showed the highest activity with a turnover frequency of ∼4000 h−1 observed for the oxidation of 4-methoxybenzyl alcohol; however, modest yields were obtained even with electron-poor and nitro-bearing alcohols (Fig. 13b). The authors also reported the oxidation of hydroxymethylpyrimidine in 91% yield, demonstrating the application of chemically modified electrodes toward the preparative-scale electroorganic synthesis of important pharmaceuticals and natural products.71
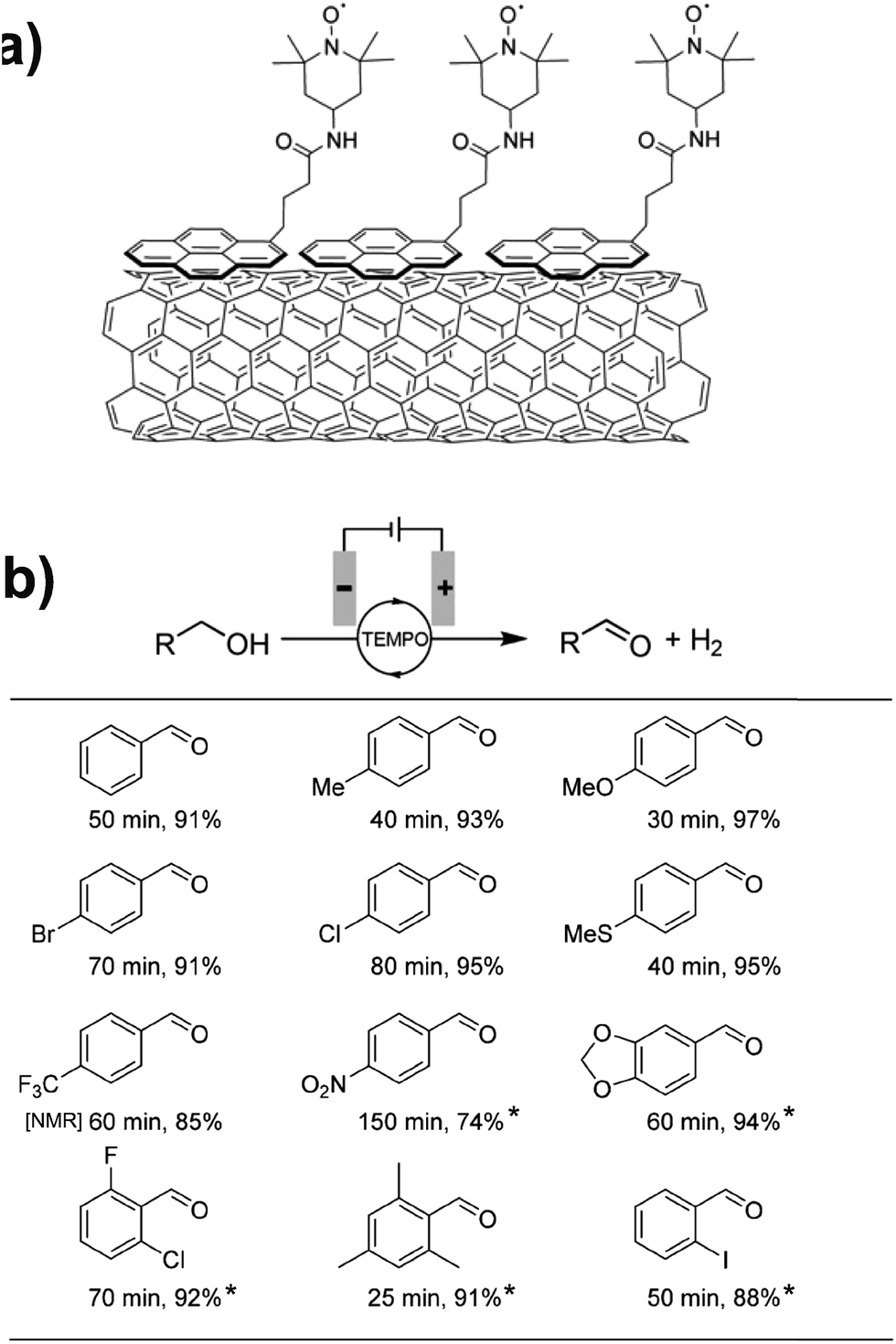 | ||
Fig. 13 (a) A pyrene-tethered TEMPO mediator immobilized on a carbon nanotube-decorated electrode was employed for (b) preparative scale electrochemical oxidation of a range of benzyl alcohols at 0.7 V (vs. Ag/AgCl) with reaction times (min) and isolated yields (%) indicated. Mediator loading: 0.05 mol% pyrene-TEMPO; solvent: 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 or (*) 4:1 water:acetonitrile with 0.2 M NaHCO3/Na2CO3 (1:1). [NMR] indicates NMR yield. Adapted from ref. 71. Copyright 2017, Wiley-VCH. :1 or (*) 4:1 water:acetonitrile with 0.2 M NaHCO3/Na2CO3 (1:1). [NMR] indicates NMR yield. Adapted from ref. 71. Copyright 2017, Wiley-VCH. | ||
Although a large focus has remained on TEMPO-decorated anodes, we can envision other classes of organic redox mediators realizing enhanced stability when immobilized on the electrode surface. A glassy-carbon supported phenanthoimidazole mediator was reported to yield turnover numbers of 15000 and 14000 for the electrochemical oxidation of 4-methoxybenzyl alcohol and the related benzyloxy methyl ether, respectively, after 5 h electrolysis at 1.37 V (vs. SCE) in the presence of 2,6-lutidine base (Fig. 14).73 The p-anisyl aldehyde and benzyl ester products were obtained with 78% and 70% faradaic efficiency, respectively, without the generation of overoxidized side products. The molecular arylimidazole was relatively unstable in the homogeneous system by comparison, yielding lower turnover numbers by three orders of magnitude. The authors proposed that immobilization of this mediator shut down the decomposition pathway that would normally be initiated by the radical ionic character of the oxidized form of the mediator.73
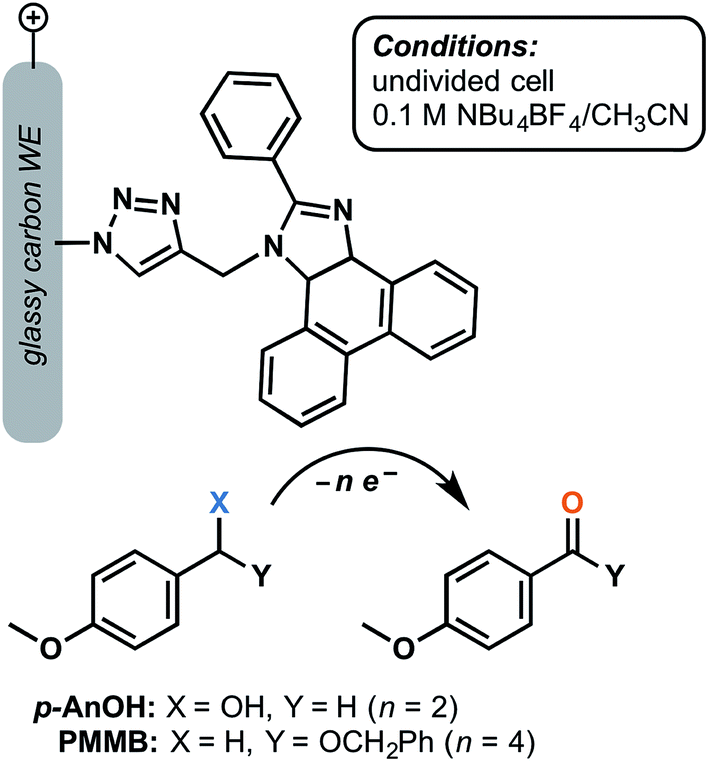 | ||
| Fig. 14 Electrochemical oxidation of p-anisyl alcohol (p-AnOH) and 1-((benzyloxy)methyl)-4-methoxybenzene (BMMB) to the respective p-anisyl aldehyde and (benzyloxy)methyl p-anisyl-carboxylate was achieved using a phenanthroimidazole redox mediator tethered to a glassy carbon working electrode. Adapted from ref. 73. Copyright 2017, Royal Society of Chemistry. | ||
HMF is a particularly useful substrate to study as a product obtained from cellulose biomass via the dehydration of fructose and sucrose sugars.74 HMF can be oxidized into FDCA, a substrate which has been identified as a potential alternative to terephthalic acid. This industrially-important precursor to polyamides, polyesters and polyurethanes makes FDCA a value-added product of biomass alcohol reforming.74 Chemical oxidation of HMF into FDCA has traditionally been performed with stoichiometric oxidants, toxic (e.g., Pb) or expensive (e.g., Au, Pd, Pt) catalysts.75,76 This transformation has been achieved electrochemically using supported Au and Pd bimetallic nanoparticles in an alkaline anion-exchange membrane (AEM) flow-cell reactor; however the selectivity for FDCA over other oxidation products (2-formyl-5-furancarboxylic acid; 5-hydroxymethyl-2-furancarboxylic acid) was generally low.77 HMF oxidation to FDCA was achieved with a molar yield of 83% (FDCA selectivity × HMF conversion) after 1 h using a Pd1Au2/C electrocatalyst in the flow-cell reactor (0.9 V vs. RHE; 35 °C; flow = 20 mL min−1).
Cha and Choi reported in 2015 the electrochemical oxidation of HMF coupled with HER using a gold electrode and TEMPO redox mediator.78 HMF oxidation was achieved in 99% yield at potential of 1.54 V (vs. RHE) passing 40 C, corresponding to a faradaic efficiency of 94%.78 Sun and colleagues reported in 2016 the oxidation of HMF and other biomass-derived alcohols in basic conditions using a porous Ni3S2/Ni foam anode, as discussed in Section 3.1.1.16 Electrochemical oxidation of HMF to FDCA was achieved in >99% yield using the Ni3S2/Ni foam catalyst with a faradaic efficiency of 98% at 1.50 V (vs. RHE) after passing 58 C of charge.
We have recently reported the electrochemical reduction of CO2 to CO paired with anodic oxidation of alcohols (Fig. 15).17 Primary and secondary aliphatic and benzyl alcohols were electrochemically oxidized using a copper–indium cathode and TEMPO redox mediator with sustained electrolytic conversion of CO2 into CO over 3 h as a proof of concept experiment. The carbonyl products were afforded in >78% yield at an applied potential of −0.70 V (vs. RHE) corresponding to a faradaic efficiency of >70%.
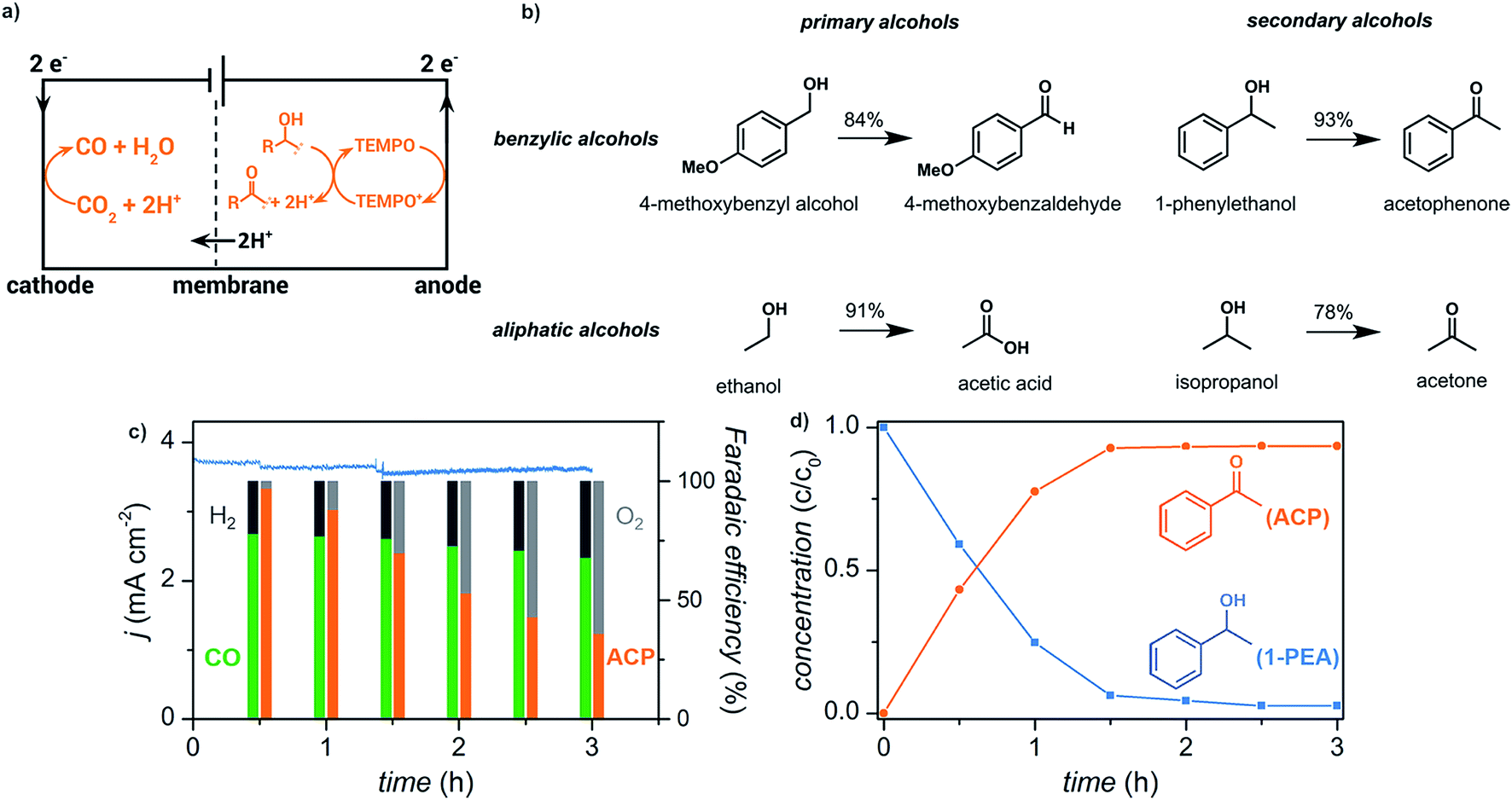 | ||
| Fig. 15 (a) Schematic depicting paired electrolysis of cathodic CO2 reduction and anodic TEMPO-mediated alcohol oxidation. (b) Reaction scope for the oxidation of primary and secondary benzylic alcohols, and primary and secondary aliphatic alcohols. (c) Current density (blue trace) and faradaic efficiencies (columns) over 3 h of paired electrolysis at an external bias of −0.70 V (vs. RHE) that converts CO2 into CO and 1-phenylethanol (1-PEA) into acetophenone (ACP). The faradaic efficiencies for the cathodic products CO (green) and H2 (black) and anodic products acetophenone (orange) and O2 (gray) are indicated. (d) Relative concentrations of 1-phenylethanol (blue) and acetophenone (orange) over the course of the 3 h experiment as quantified by 1H NMR spectroscopy. Adapted from ref. 81. Copyright 2017, American Chemical Society. | ||
3.2. Anodic transformation of olefins
Olefins such as ethylene, propylene, and butenes are major feedstocks for the industrial production of plastics/fibers, alcohols, aldehydes and other important products, and are readily available from natural gas and petroleum sources.82–84 Electrochemical oxidation and functionalization of these inexpensive unsaturated hydrocarbons by means of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond oxidation or addition of various functional groups can be carried out with or without redox-mediators (Scheme 1). As a result, anodic oxidation of simple olefins can give rise to value-added products such as ketones, epoxides, 1,2-diols, aldehydes and carboxylic acids.85–88 This section of the review will focus on promising anodic transformations of olefin feedstocks having industrial significance. Examples of electrochemical synthesis involving multi-step anodic transformations on complex and/or expensive olefin substrates have been described elsewhere.21,89
C bond oxidation or addition of various functional groups can be carried out with or without redox-mediators (Scheme 1). As a result, anodic oxidation of simple olefins can give rise to value-added products such as ketones, epoxides, 1,2-diols, aldehydes and carboxylic acids.85–88 This section of the review will focus on promising anodic transformations of olefin feedstocks having industrial significance. Examples of electrochemical synthesis involving multi-step anodic transformations on complex and/or expensive olefin substrates have been described elsewhere.21,89
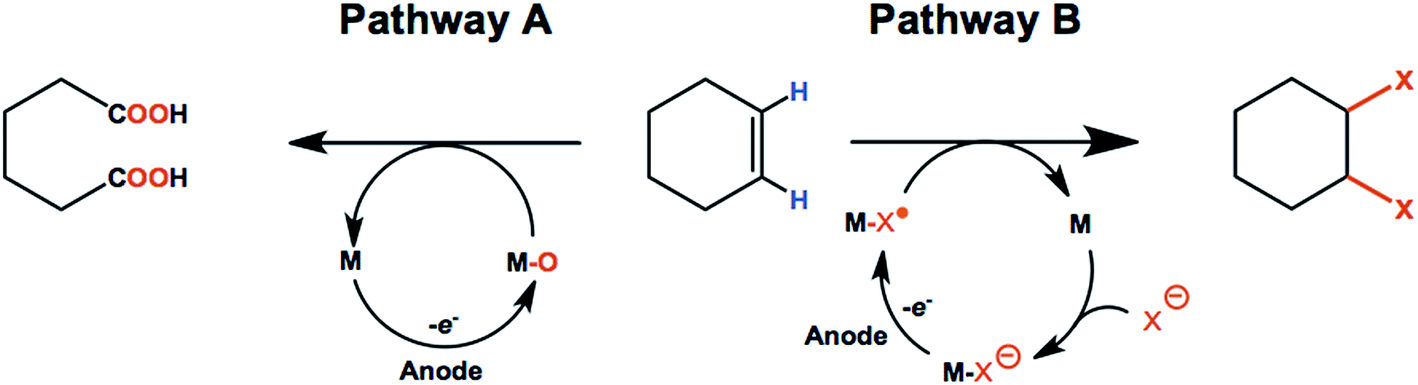 | ||
| Scheme 1 General mechanism for the anodic oxygenation or functionalization of olefins via redox-mediated (M = mediator) processes. In pathway A, a redox mediator is activated anodically to form a highly oxidizing species (M–O) that facilitates the oxygenation of the alkene into a dicarboxylic acid. In pathway B, the electroactive species is a mediator-halide or mediator–chalcogenide radical (M–X˙) that delivers functional groups to the olefin. | ||
C bonds. Substrates that are electron-rich (e.g., enols and enolates) will readily undergo direct anodic oxidation to form reactive radical cations; however, the electrochemical oxidation of unactivated alkenes must proceed via an indirect mechanism due to the higher standard potentials of isolated CC bonds. Such transformations often require powerful chemical species (e.g., halogen radicals/cations, high-oxidation-state metal complexes) to be generated at the anode which may also yield unwanted byproducts. Careful selection of oxidative intermediates and clever control over their reactivity are required to achieve high chemical yields and faradaic efficiencies for meaningful electrolyzer applications.46
Another important aspect of olefin oxidation or functionalization is the choice of solvent. Most industry-relevant olefins are insulating and sparingly soluble in the aqueous media ideal for cathodic HER. Biphasic or partially aqueous solvent mixtures such as CH2Cl2/H2O and CH3CN/H2O systems have been traditionally employed; however, these mixtures require vigorous mechanical stirring and/or additional biphasic extraction and evaporation steps to recover products.86,90 Ionic liquids have been successfully employed in chemical epoxidation of alkenes therefore can potentially be used as the anolyte, even though they may be less cost-effective than water/organic mixtures due to higher costs.91
C bond cleavage and addition of oxygen atoms to avoid the use of stoichiometric oxidants. Improved reaction performance can be expected with the help of redox mediators in that non-mediated oxidation reactions are often less efficient and more difficult to control. For example, an ozone-generating lead dioxide anode was reported for direct oxidation of olefins into carboxylic acids.86 The carboxylic acid products were obtained in up to 90% yield; however, the resulting faradaic efficiencies were lower than 5%.86 A Ru(III)/IO4-mediated reaction using the same type of anode material showed significantly higher performance by comparison (Scheme 2, left), generating the carboxylic acid products in up to 91% yield, with faradaic efficiencies up to 78%.90
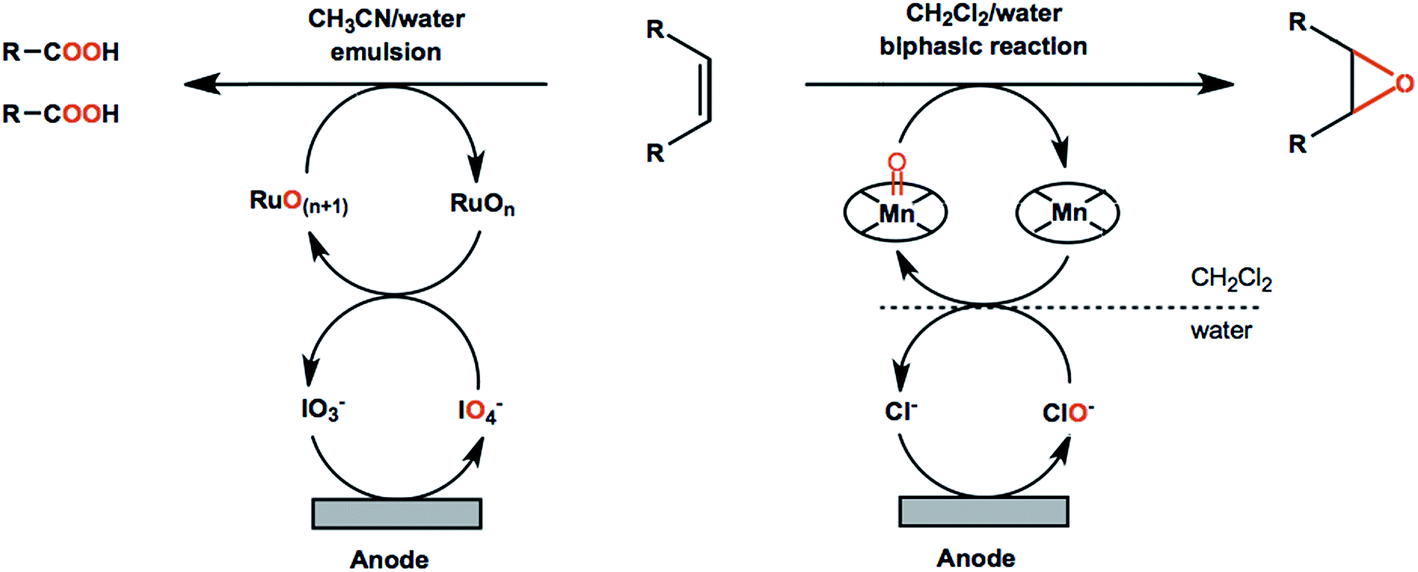 | ||
| Scheme 2 Metal-oxo catalysts are the most commonly used mediators in electrochemical olefin oxidations. Examples of (Ru/IO4−)-mediated olefin oxidation (left) and olefin epoxidation mediated by a Mn–salen complex (right) are shown. | ||
Metal-oxo catalyst systems employing ruthenium, iron, manganese, chromium and osmium metals are the most commonly used mediators in electrochemical oxygenation of olefins.92 The chemistry of metal-oxo oxidants has been extensively studied as these species capable of initializing several types of transformations including oxygen atom transfer and the hydrogen abstraction of strong C–H, N–H, and O–H bonds.93 In EC systems the metal-oxo species are generated from low-valent metal–aquo complexes in solution and are subsequently reactive toward the oxygenation of alkene functional groups. Electrochemical olefin epoxidation mediated by a manganese–salen redox couple was reported in the early 2000s.94 The reactions were carried out in a CH2Cl2/H2O–NaCl biphasic system in an undivided cell (Scheme 2, right) at a constant current density of 6.7 mA cm−2 with an estimated faradaic efficiency of 33% based on the data provided.94
Iron–porphyrin complexes give rise to some of the best homogeneous olefin oxygenation electrocatalysts in terms of chemical yield and faradaic efficiency.95 For instance, iron–porphyrin electrocatalysts (TMP)FeIIIOH (TMP = meso-tetramesitylporphyrin) have been reported by Groves and co-workers in the 1980s showing a calculated faradaic efficiency up to 74% in dichloromethane. The reaction yield was improved to >90% in a later report by Hickman et al. which used an analogous Fe–TPP catalyst (TPP = meso-tetraphenylporphyrin).96 Water-soluble Fe–porphyrin catalysts reported by Su and co-workers were found to promote allylic hydrogen abstraction in aqueous media at room temperature while epoxidation only occurred at potentials above which the catalyst was no longer stable.97,98 Chemoselective catalytic systems with high faradaic efficiencies and long-term stabilities are yet to be discovered for the future commercial application of electrochemical oxygenation of olefins. Halogenated Fe–porphyrin catalysts used in the chemical oxidation of olefins are worth mentioning,99,100 and the factors controlling their activity and selectivity are relatively well-understood.101–103 For instance, in catalyzed epoxidation of cyclooctene by H2O2, the formation of inactive μ-oxo iron porphyrin dimers can be avoided when certain positions on the catalyst molecule were fluorinated.102 Similar strategies may also be useful in the development of olefin electrocatalysts.
C bond. Such a mechanism involves two separate steps, the first of which must be carried out in an anhydrous organic solvent under cryogenic conditions;87 therefore, the cation pool strategy can be synthetically useful (but is not suitable for large-scale electrolysis).
Radical intermediates are typically water insensitive therefore radical olefin functionalization can be carried out in aqueous or partially-aqueous media. A common disadvantage of radical reactions is that the intermediates are highly reactive and the reaction outcome is often difficult to control. A promising approach has been reported by Lin et al. in which redox-active metal-containing mediators were employed to stabilize the anodically-generated radical intermediates.104–106
An example of anodic radical functionalization of alkenes was recently reported by Lin et al.104 Electrochemical diazidation of alkenes was achieved via a radical mechanism whereby the azidyl radical (N3˙) was generated at the anode from sodium azide (NaN3) followed by the addition of N3˙ to the CC bond to form a carbon radical adduct. The carbon radical intermediate was trapped by TEMPO, resulting in the formation of a TEMPO-N3 adduct in high yield. Less than 5% of the desired 1,2-diazide was obtained in the absence of TEMPO, suggesting that the formation of the second C–N bond was unfavourable. A catalytic amount (5 mol%) of Mn2+ was added to facilitate successive additions of N3˙ to the CC bond to generate product in greater than 80% yield (Fig. 16b). The electrolysis was carried out at a relatively low anode potential due to the low redox potential of N3˙/N3− (∼0.7 V vs. Fc/Fc+). Furthermore, this diazidation strategy exhibited a broad substrate scope with compatibility across a variety of functional groups. A Mn-assisted mechanism was proposed to explain the role of Mn in diazidation. MnII and N3− first formed a MnII–N3 complex and subsequently generated MnIII–N3 by anodic oxidation, followed by azide transfer to the CC bond (Fig. 16b). The MnIII–N3 complex was believed to play a central role in the excellent reactivity and chemoselectivity; the introduction of Mn-bound complex was proposed to diminish the high propensity of N3˙ to undergo dimerization, C–H or electron abstraction, and other side reactions.
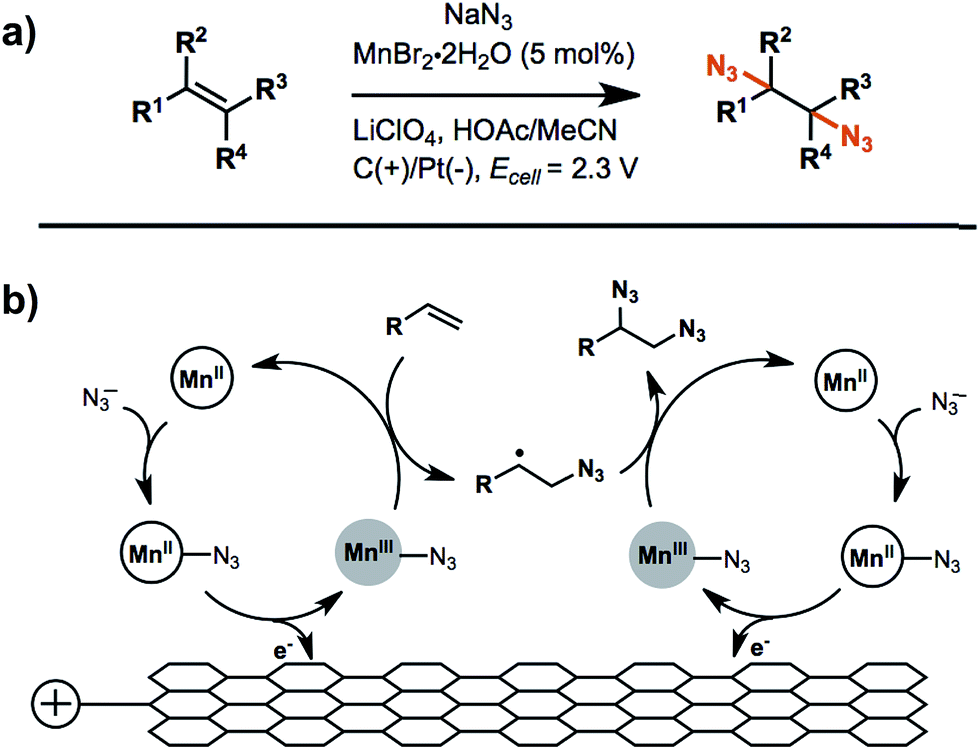 | ||
| Fig. 16 (a) Electrochemical diazidation of substituted olefins mediated by a Mn–salen complex in an undivided cell using a reticulated vitreous carbon anode and platinum cathode. (b) Proposed catalytic cycle for the anodic reaction. Adapted from ref. 104. Copyright 2017, American Association for the Advancement of Science. | ||
3.3. Electrochemical C–H functionalization
The activation and subsequent functionalization of C–H bonds is one of the key challenges in organic synthesis and is typically carried out under extreme conditions (>100 °C) using strong oxidants or noble-metal catalysts.107 The nonpolar nature and high activation energy of aliphatic and allylic C–H bonds offers a formidable challenge in transforming these groups in the presence of other functional groups. Difficult C–H bond transformations can be achieved electrochemically, however, with the proper selection of electrode materials and redox mediators to overcome the high C–H oxidation potential.21,89 The high-value commodity chemicals afforded by electrochemical C–H functionalization makes this class of reaction ideal for pairing with cathodic hydrogen generation, provided that the challenges in reaction chemoselectivity can be addressed. This section reviews the reports of electrochemical C–H activation to install simple functional groups such as halogens, amines, carbonyls and trifluoromethyl groups.The use of redox mediators offers the ability to lower the working potential and access transformations of more challenging substrates. Additionally, greater reaction chemoselectivity is afforded for substrates bearing functional groups having different standard potentials. A mediator may also function as both an electron carrier and a proton shuttle. For example, N-hydroxyphthalimide (NHPI) was shown to experience a drop in standard potential from 1.44 V to 0.85 V (vs. SCE) in the presence of added pyridine base as a result of the proton-coupled oxidation of NHPI to phthalimido-N-oxyl (PINO) that occurs at the anode.108 NHPI was reported to mediate the hydrogen-atom transfer of activated C–H at a lower potential than that required for direct anodic oxidation of the benzylic substrate (Fig. 17).109 The careful selection of mediators, co-oxidants and reaction conditions offers a pathway to tune the oxidative strength of the reaction for the activation of specific types of C–H bonds.
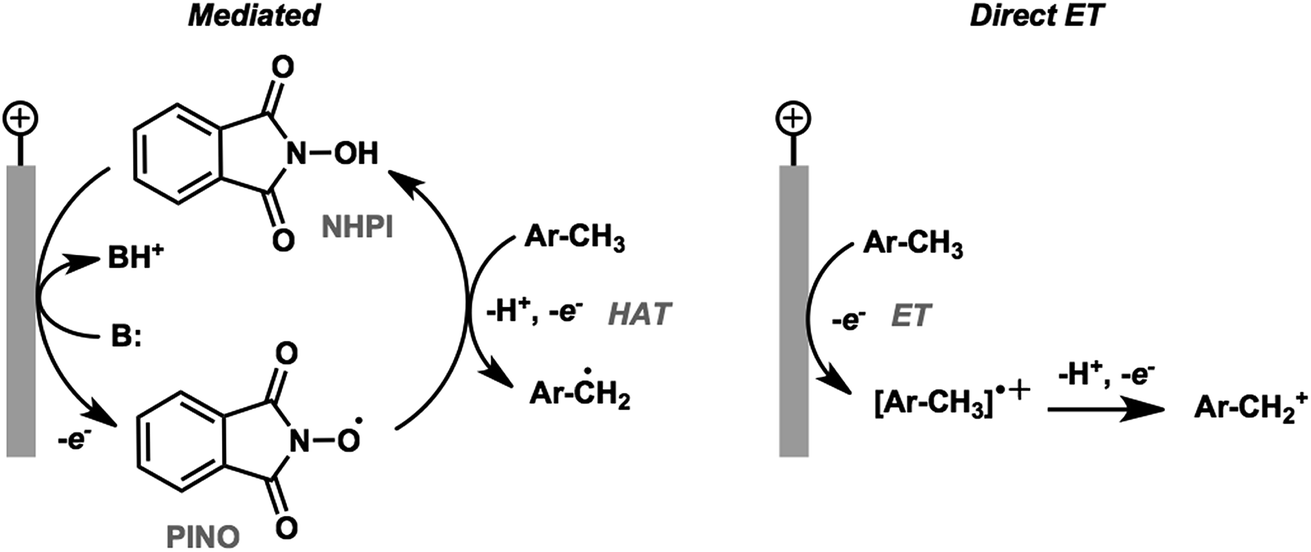 | ||
| Fig. 17 Comparison of NHPI/PINO-mediated and direct anodic initiation of benzylic C–H activation. The mediated process involves a hydrogen-atom transfer (HAT) from the activated benzylic C–H to the PINO mediator to generate an active benzyl radical. Direct anodic electron transfer at a higher overpotential generates a reactive radical cation. Adapted from ref. 109. Copyright 2017, American Chemical Society. | ||
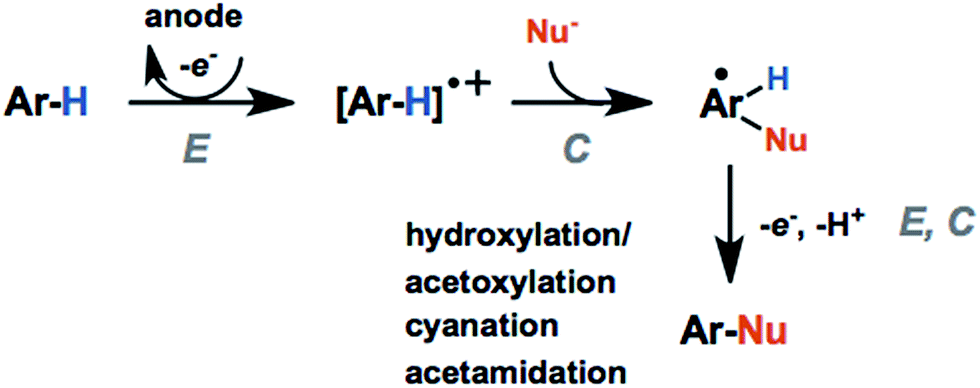 | ||
| Scheme 3 Direct anodic oxidation of an arene generates the radical cation intermediate via an electrochemical (E) step followed by a chemical nucleophilic reaction (C). The subsequent electron transfer and deprotonation steps complete the ECEC process to yield a variety of products depending on the nucleophile (Nu−) present. | ||
Electron donating groups (e.g., hydroxy, methyl ether) activate the arene toward electrochemical generation of the radical cation. These groups direct the regioselectivity of the arene functionalization to the positions para- or ortho- to the electron-donating substituents, as with typical electrophilic aromatic substitution reactions. Functionalization at the meta-position using meta-directing groups is more challenging due to the higher oxidation potentials of electron-deficient arenes.21 Alkyl substituents can activate the arene and direct the regioselectivity toward certain products; however, chemoselectivity can also be a problem. C–H activation of benzylic C–H groups can compete with the C(sp2)–H activation resulting in benzylic functionalization or arene–arene coupling.110,111 For example, direct anodic oxidation of mesityl in LiClO4/MeCN at 1.6 V (vs. SCE) gave the following products: 3,5-dimethylbenzaldehyde; bimesityl; termesityl; 2,2′,4,4′,6,6′-hexamethyldiphenylmethane; 2,4,6,3′,5′-pentamethyldiphenylmethane; and N-3,5-dimethylbenzylacetamide, formed via anodic coupling of the benzyl radical cation with the MeCN solvent.111
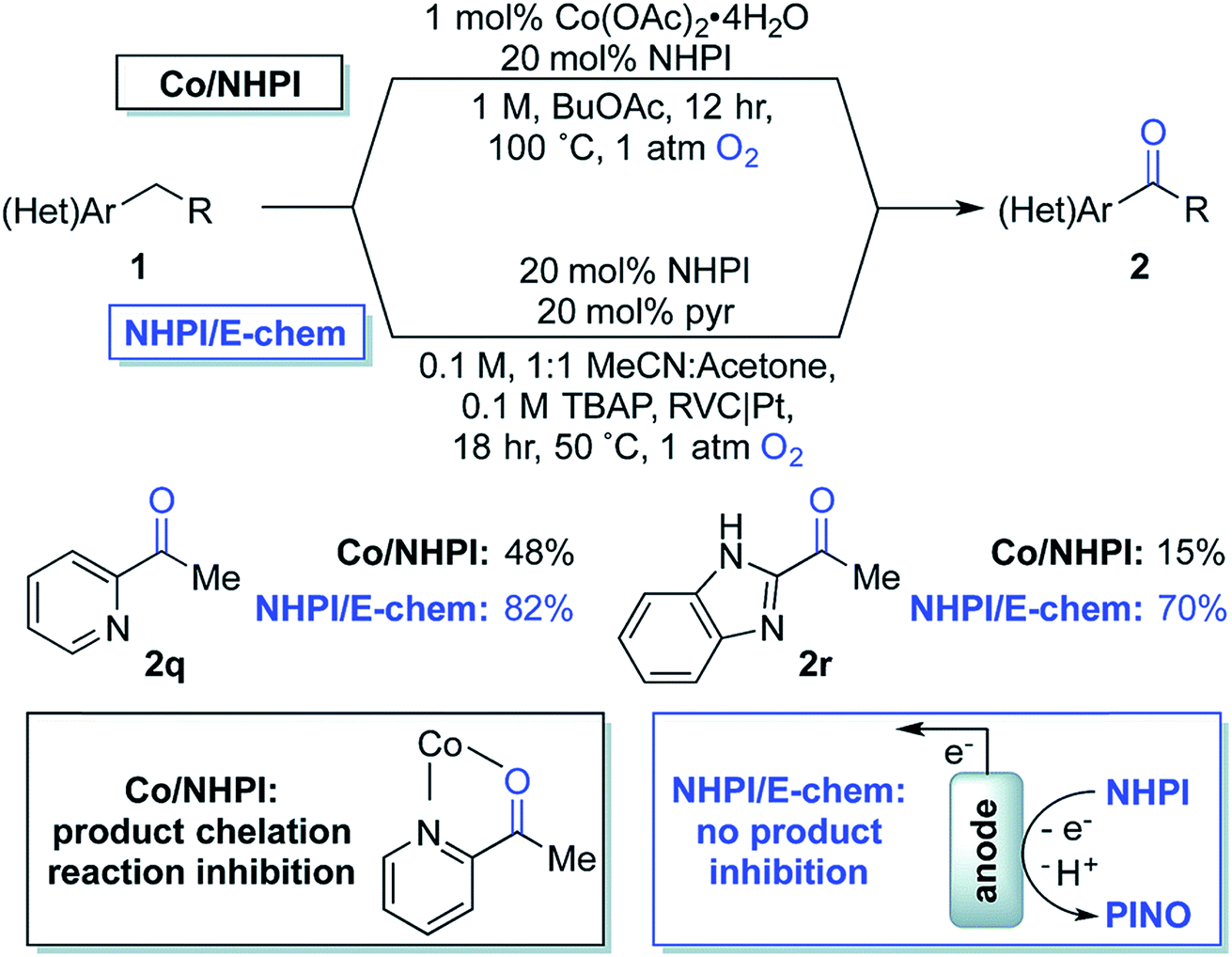 | ||
| Fig. 18 Chemical oxidation (with Co2+/NHPI; NHPI = N-hydroxyphthalimide) and electrochemical oxidation (with NHPI only) of benzylic C–H to yield benzyl ketones. NHPI is anodically oxidized to the phthalimido-N-oxyl (PINO) radical which mediates the selective abstraction of benzylic C–H bonds to generate an organic radical, affording the oxygenated products on quenching with dioxygen. Copyright 2017, Royal Society of Chemistry.115 | ||
In 2017, Stahl and colleagues identified a cobalt(II)/NHPI catalyst system for the aerobic oxidation of benzylic C–H bonds.115 These reactions were typically run for 12 h in organic solvent at 90–100 °C under 1 atm O2. This method was tolerant of heterocycles and highly selective for benzylic methylene groups; however, the reaction was low yielding for some benzyl pyridines and imidazoles due to chelation of the cobalt co-catalyst (Fig. 18). The authors developed an electrochemical oxidation system for the oxygenation of these substrates which did not require the cobalt co-catalyst nor the high operating temperature (reduced to 50 °C) of the chemical system.115 Electrochemical oxidation was instead achieved using a NHPI redox mediator and RVC anode (Pt cathode) in an undivided 3-electrode cell at 0.65–0.75 V (vs. Ag/AgCl) under 1 atm O2. EC oxidation of the benzylic C–H bonds in absence of Co2+ gave substantially higher product yields than the aerobic oxidation system that was prone to catalyst chelation.115
Chemical functionalization of allylic C–H bonds is widely known and well developed; however, most reaction conditions require toxic reagents (e.g., CrO3, SeO2) and/or expensive metal catalysts (e.g., Rh, Pd) precluding their development for environmentally sustainable, large-scale syntheses.116,117 Electrochemical functionalization of allylic C–H bonds is more difficult to achieve than aromatic C–H functionalization due to the higher redox potential of allylic C–H bonds (Fig. 3). Direct EC allylic C–H oxidation was first reported in 1968 on α-pinene,118 followed by the first report of indirect allylic oxidation mediated by NHPI in 1985.119 Unfortunately, electrochemical strategies toward allylic C–H activation have been limited by relatively low product yields. This challenge could potentially be overcome by developing an efficient mediator having the required redox potential to activate allylic C–H bonds.
In 2016, Baran and coworkers reported a study which evaluated a series of redox mediators for the allylic C–H oxidation of the sesquiterpene valencene into the corresponding α,β-unsaturated ketone nootkatone (Fig. 19a).18 This investigation was an extension of the 1985 report on the indirect (NHPI-mediated) allylic oxidation119 which sought to improve upon the original conditions to deliver a proof-of-concept experiment for a scalable electrochemical synthesis. The authors identified two other modifications to improve the original EC allylic oxidation in addition to the selection of a new mediator: (1) addition of tert-butyl hydroperoxide (tBuOOH) as a co-oxidant; and (2) the design of an optimized EC cell using inexpensive carbon electrodes.18 Among the different mediators tested, tetrachloro-N-hydroxyphthalimide (Cl4NHPI) showed the best performance with a 20% increase in yield for the nootkatone product compared to simple NHPI. The presence of the electron-withdrawing Cl groups on Cl4NHPI afforded a +0.1 V gain in redox potential compared to NHPI, resulting in a more reactive Cl4NHPI radical. A scope of 40 allylic C–H substrates were electrochemically oxidized to enones on 0.5 mmol scale with moderate to high yields (40–90%) at a constant current of 10 mA per mmol of substrate. Three of these substrates were converted at 100 gram scale in a beaker open to the air (Fig. 19b), demonstrating a scalable and operationally-simple electrochemical oxidation of allylic C–H bonds without using toxic or expensive chemical reagents.
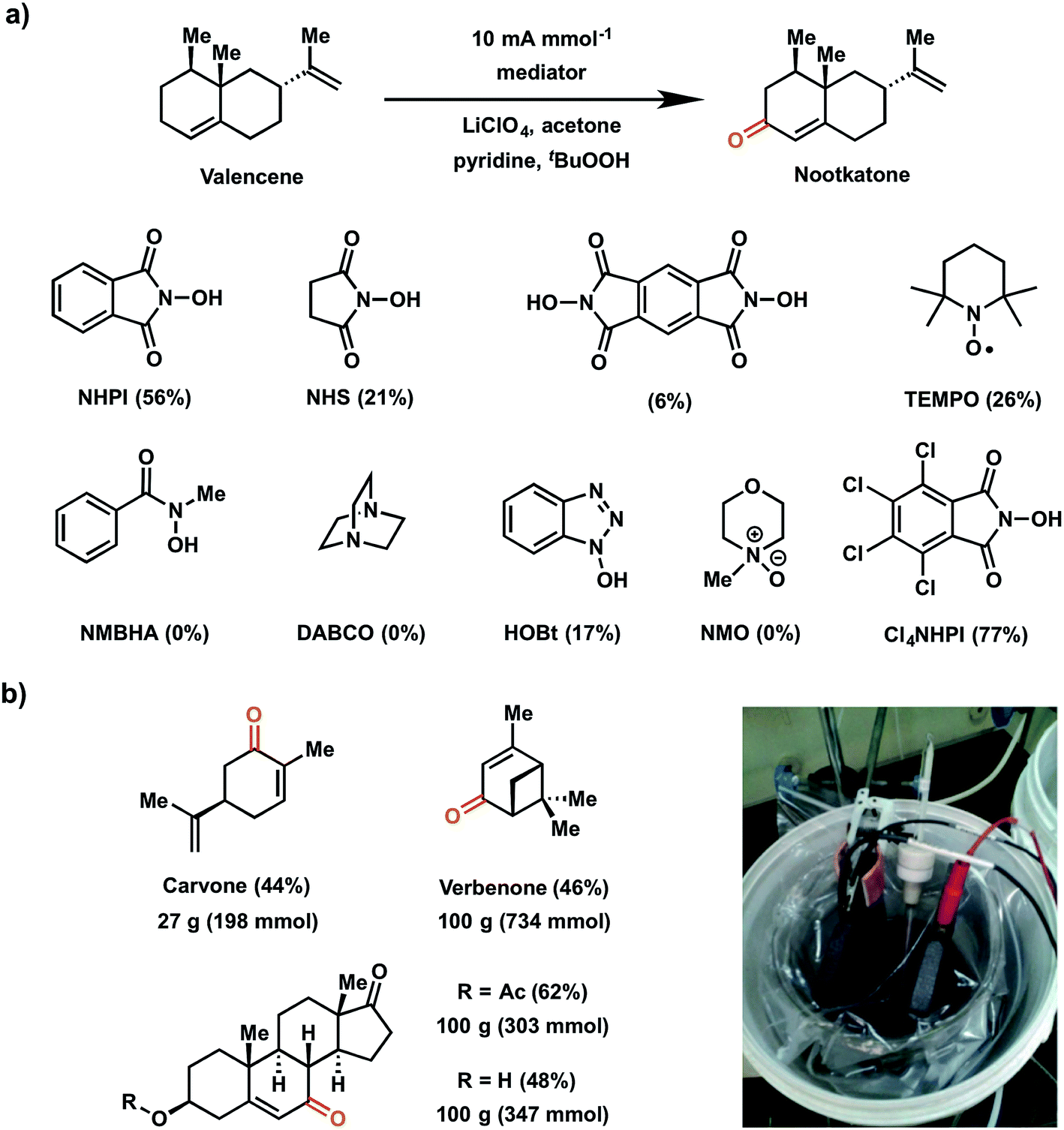 | ||
| Fig. 19 (a) Scope of redox mediators explored for electrochemical allylic C–H oxidation of valencene into nootkatone. Conditions: Cl4NHPI redox mediator (0.2 equiv.), tBuOOH co-oxidant (1.5 equiv.), pyridine base (2 equiv.), LiClO4 electrolyte (0.6 equiv.), acetone solvent (0.16 M in substrate), RVC electrodes. (b) Large-scale allylic C–H oxidation of selected substrates in a beaker open to air (pictured in inset). Data from ref. 18. Copyright 2016, Nature Publishing Group. | ||
Stahl and coworkers applied a similar strategy toward benzylic/allylic C–H functionalization to yield a larger scope of products in addition to ketones.109 Radicals generated by the NHPI-mediated HAT process could be captured by iodine (I2) rather than O2 or tBuOOH to yield benzyl iodides from the methylarene substrates (Fig. 20a). The benzyl iodides could be sequentially reacted with a choice of nucleophiles to yield a variety of C–N, C–O or C–C products (Fig. 20b). The NHPI-mediated HAT process was similar to Baran's prior report18 in that the anodic oxidation of NHPI into phthalimido-N-oxyl (PINO) mediated the generation of the benzylic radical.109 The use of NHPI as a mediator allowed C–H functionalization to proceed at potentials ∼1 V lower than that of direct electrolysis, demonstrating another example of how indirect electrolysis with a redox mediator is able to lower the required working voltage.
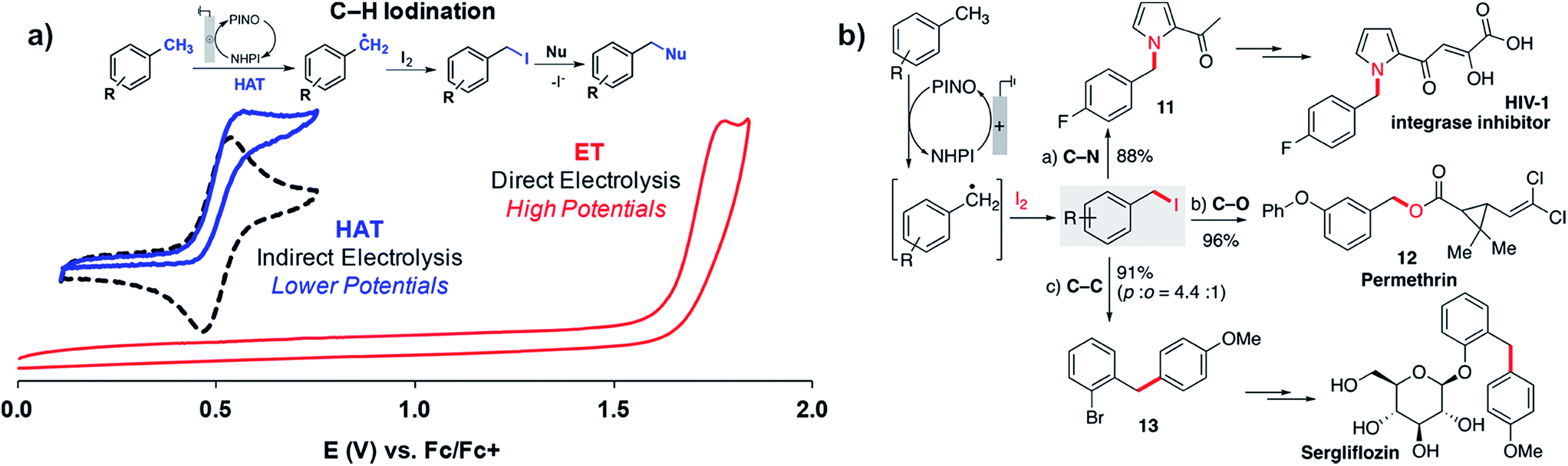 | ||
| Fig. 20 (a) NHPI/PINO-mediated C–H iodination at an anode with I2 trapping of the benzylic radical generated by the hydrogen-atom-transfer (HAT) process (NHPI = N-hydroxyphthalimide; PINO = phthalimido-N-oxyl). (b) Sequential reaction of a benzyl iodide with different nucleophiles provides access to pharmaceutical intermediates and compounds. Copyright 2018, American Chemical Society.109 | ||
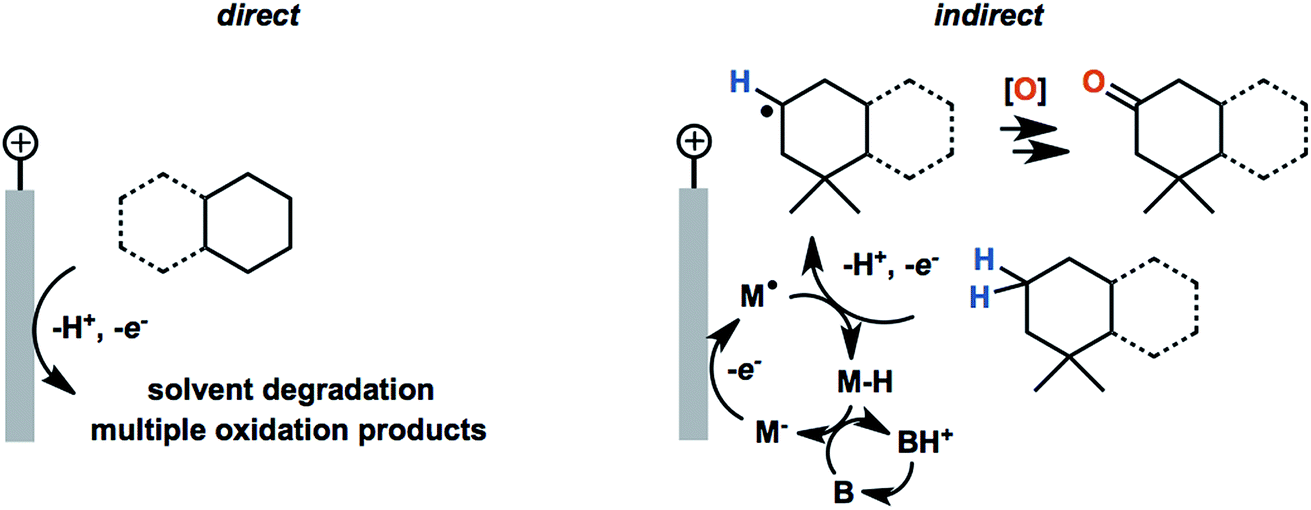 | ||
| Fig. 21 Comparison of the direct anodic oxidation of aliphatic C–H substrates and an indirect redox-mediated process (M = mediator; B = base). The generation of the radical intermediate proceeds at a lower electrode potential in the mediated process, suppressing the oxidation of solvent. | ||
In 2017, Baran and coworkers reported a protocol to drive the anodic C–H oxidation of the aliphatic backbone of a sesquiterpene lactone, sclareolide, using a simple redox mediator.47 Quinuclidine-mediated oxidation of the unactivated C–H groups was achieved over 12 h under a constant current of 5 mA. Optimization of the EC system also avoided the use of expensive electrode materials, rather employing RVC (anode) and nickel (cathode). EC oxidation using quinuclidine gave the highest yield (51%) and chemoselectivity towards oxidation of C2 methylene compared to the other redox mediators tested (Fig. 22). Both methanediyl and methanetriyl C–H bonds were oxidized into ketone and alcohol products, respectively, due to the redox potential (1.2 V) of quinuclidine. The anodic chemistry was conducted in open flask and scaled up to 50 g, demonstrating a scalable, chemoselective route for the oxidation of unactivated C–H bonds in complex organic substrates.47 The authors noted that the total cost of all reagents, additives, solvents, and electricity for the 50 gram reaction cost ∼$ 2500 whereas the cost for solely the methyl(trifluoromethyl)dioxirane catalyst for the analogous chemical oxidation would have surpassed $ 9000.
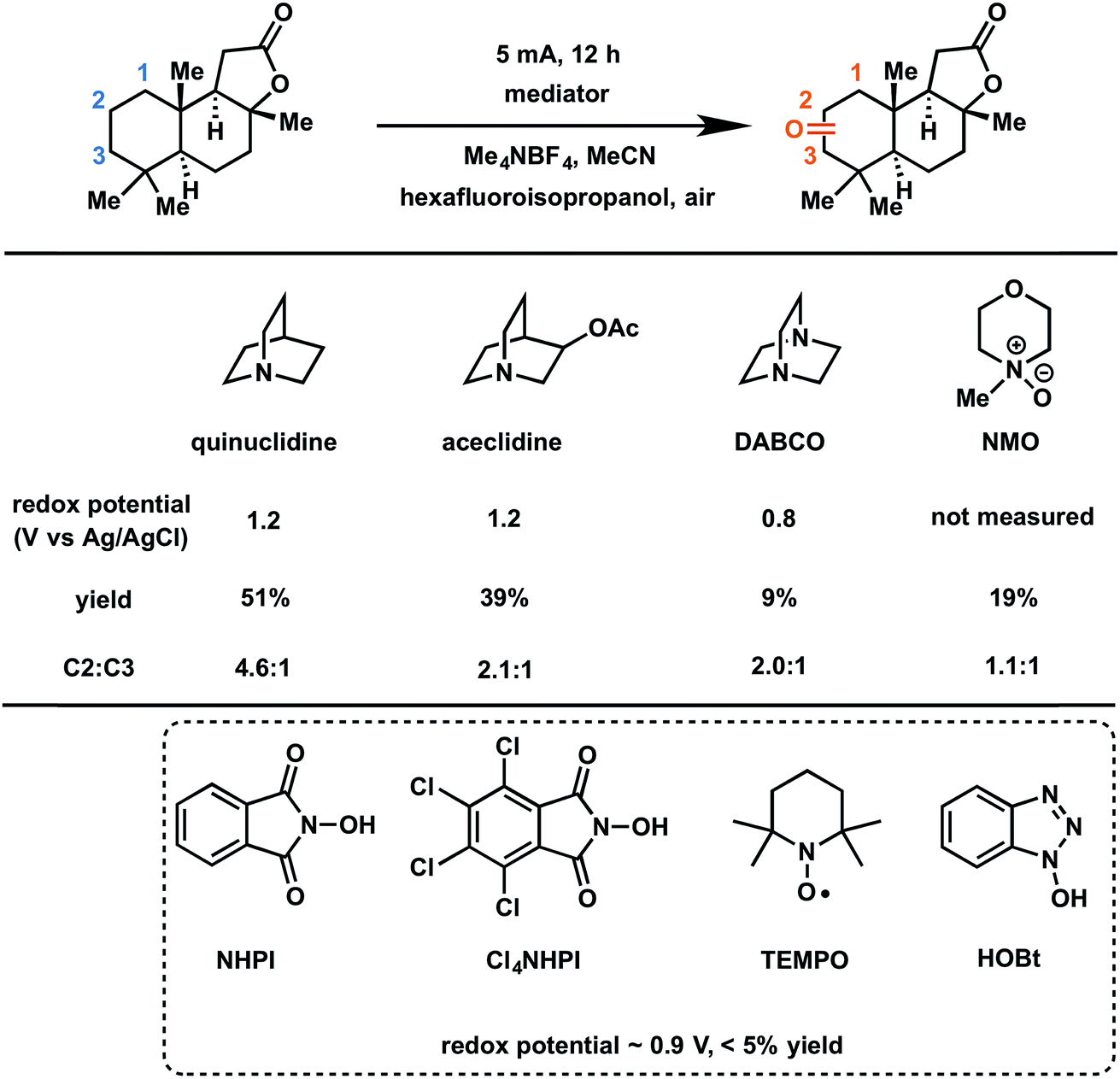 | ||
| Fig. 22 Optimization of redox mediators for the EC C–H oxidation of a sesquiterpene lactone. Typical conditions: lactone substrate (0.2 mmol), mediator (0.2 mmol), hexafluoroisopropanol additive (2 mmol), Me4NBF4 electrolyte (0.2 mmol), MeCN solvent, RVC anode, nickel cathode. Data from ref. 47. Copyright 2017, American Chemical Society. | ||
The reports on C–H functionalization summarized herein have described transformations of a wide scope of C–H substrates under mild conditions. The sensitivity of these reactions to the electrode potential and to reaction conditions has dictated that reaction optimization has been typically realized on a case-by-case basis. The scalability of these reactions, however, offers an opportunity for development toward paired chemical synthesis with electrolyzer technology. Further study must be undertaken to investigate the faradaic efficiencies of these reactions toward HER and C–H functionalization, as well as the stability of the electrode and mediator systems over time.
4. Photoelectrochemical organic oxidations
Photoelectrochemical (PEC) oxidation of organic substrates has emerged as a new route to convert sunlight into valuable chemical products. PEC oxidative degradation of organic wastes is widely known,124–127 and the concept of using biomass-derived feedstocks as sacrificial agents (whose oxidative degradation occurs at a lower overpotential than water oxidation) has also been reported.128,129 The first report of PEC valorization of organic substrates into commodity chemicals, however, was only recently reported.15 In this study, the PEC oxidation of 5-hydroxymethylfurfural into 2,5-furandicarboxylic acid (FDCA, a key monomer to produce polymer materials) was achieved using a BiVO4 photoanode and TEMPO redox mediator (Fig. 23a). The onset potential for TEMPO-mediated oxidation of HMF was negatively shifted by ∼0.7 V compared to water oxidation (Fig. 23b), indicating that the organic oxidation chemistry was more favourable than the oxidation of water. HMF oxidation was achieved with nearly 100% faradaic efficiency whereas the competing water oxidation reaction was completely suppressed. | ||
| Fig. 23 (a) Schematic depiction for the PEC oxidation of 5-hydroxymethylfurfural (HMF) into 2,5-furandicarboxylic acid (FDCA) on a BiVO4 photoanode mediated by TEMPO. (b) Linear sweep voltammograms obtained in aqueous buffer solution (black), buffer solution containing TEMPO (blue), and buffer solution containing TEMPO and HMF (red). Copyright 2015, Nature Publishing Group.15 | ||
The PEC oxidation of HMF previously reported by Cha and Choi78 demonstrated a proof-of-concept design for producing value-added products at the photoanode; however, the use of aqueous media continues to limit the scope of possible organic chemical reactions and can lead to extensive photoanode degradation as discussed in Section 2.2. Our group has recently demonstrated the PEC oxidation of organic substrates in organic media,17 where MeCN was used as solvent to broaden substrate scope (e.g., alcohol, hydrocarbons) and suppress corrosion of the BiVO4 photoanode. The PEC oxidation of benzyl alcohol into benzaldehyde mediated by N-hydroxysuccinimide (NHS) was performed as a proof-of-concept experiment to test photoelectrochemistry in organic media. Linear sweep voltammetry (LSV) curves were measured for blank MeCN solution and after successive addition of a series of chemicals (Fig. 24) and a 41% yield of benzaldehyde was achieved after 8 h of PEC electrolysis at 0.8 V (vs. Ag/AgCl). Similar photocurrent profiles were obtained for the more challenging C–H oxidation (Fig. 24b and c) and the organic PEC experiments afforded cyclohexanone from cyclohexene (38% yield after 8 h) and 1-tetralone from tetralin (75% yield after 24 h). This photoelectrochemistry in organic media highlights the potential to generate a variety of higher-value products compared to the O2 generate by traditional PEC cells.
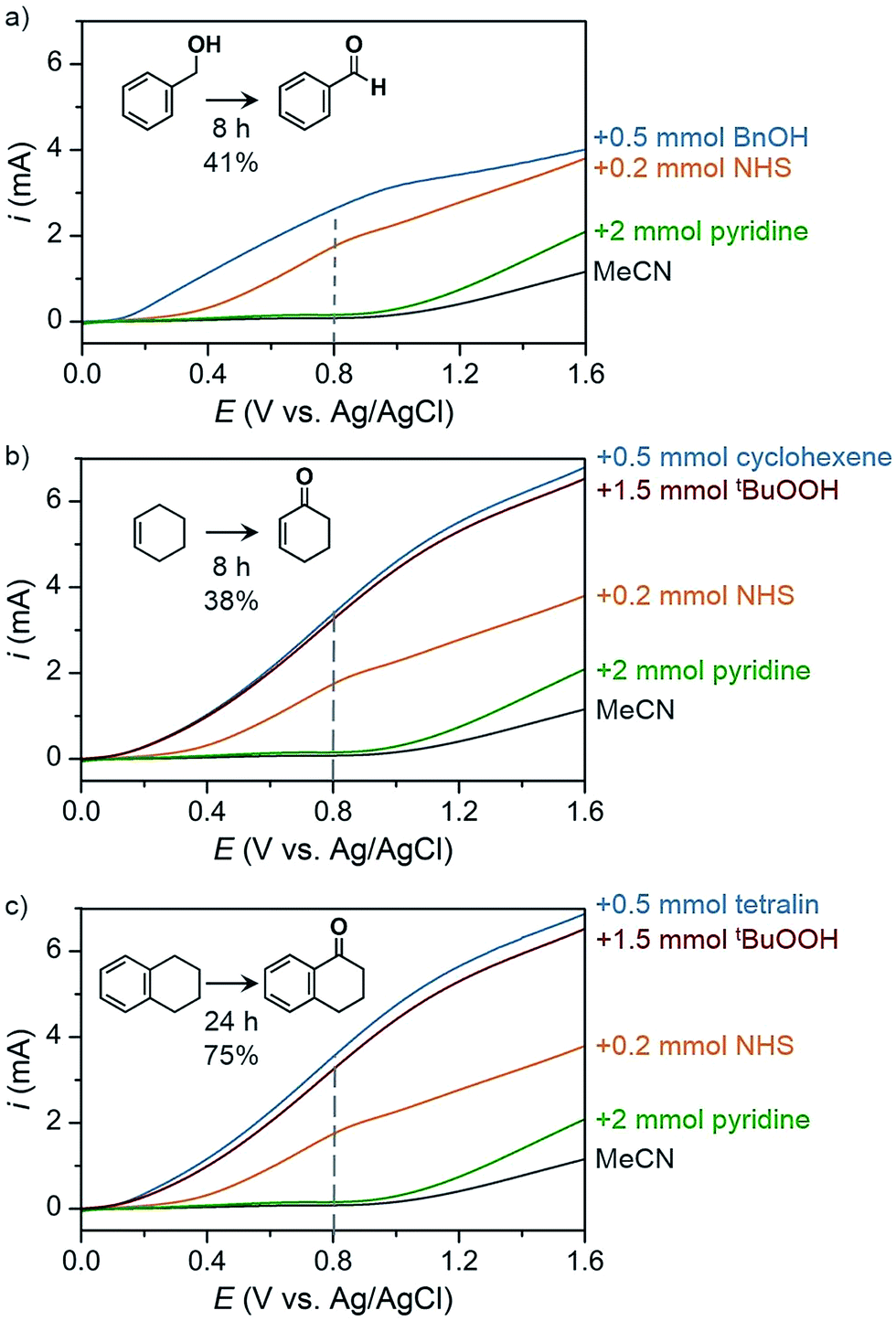 | ||
| Fig. 24 Linear sweep voltammogram curves of PEC organic oxidation profiles corresponding to oxidations of (a) benzyl alcohol to benzaldehyde, (b) cyclohexene to cyclohexanone, and (c) tetralin to 1-tetralone using a BiVO4 photoanode immersed in 25 mL MeCN containing 0.1 M LiClO4. Photocurrent profiles correspond to the solvent and electrolyte solution (black) after the successive addition of 2 mmol pyridine (green), 0.2 mmol NHS (orange), 1.5 mmol of tBuOOH (red) and 0.5 mmol of the respective substrate (blue). The dashed line indicates the potential that was used as the applied voltage for PEC electrolysis experiments. Copyright 2017, Nature Publishing Group.17 | ||
5. Summary
Organic oxidation reactions in tandem with electrolytic HER offer a pathway to clean and affordable hydrogen fuel. This review has focused on examples of non-photoactive electrochemical systems that can operate in tandem with solar PV electricity. PEC organic oxidation provides a direct route for the conversion of solar energy into commodity chemicals and clean hydrogen, but currently remains at a research and development stage. Improvements in PEC organic chemistry are required in order to address the current performance challenges of these cells, including the development of photoanodes that can achieve higher photocurrent density, and redox mediators that can catalyze more types of organic transformations with higher yield and selectivity.Our analyses have focused on three potentially high-value classes of reactions for electrochemical anodic oxidation in tandem with H2 generation, based on availability of feedstock chemicals, oxidation potential of the starting materials and value of oxidized products, among other criteria. Electrochemical oxidation of alcohols, olefins and hydrocarbon substrates has been demonstrated in tandem with HER, albeit on different scales as a result of the different device challenges that each system faces. Organic transformations at the electrochemical anode are highly substrate dependent and therefore no single solution can be applied as a guideline for the development of improved EC and PEC devices.
Electrochemical alcohol oxidation is the most researched for electrolyzer applications among these three categories of anodic reactions, with several recent reports demonstrating the utility of tandem HER with oxidation of biomass-derived alcohols and pushing the limits of energy efficiency via electrode materials development. When the lifecycle analysis of the feedstock is factored into the calculation of the energy consumption of the electrolyzer, anodic oxidation of biomass-derived alcohols offers a pathway to clean hydrogen generation that is economically competitive with water-splitting;54 however, this technology is still in a very early stage and currently far from being economically competitive with petroleum-derived hydrogen. Major breakthroughs in olefin oxidation within the past several years have changed the way we think about controlling reactivity in these types of systems by trapping cation and radical intermediates. With further development of optimal reaction conditions to tackle the problem of product selectivity, olefin oxidation reactions have the potential to be employed in electrolysers for the supply of high-value substituted alkene chemicals. Lastly, electrochemical C–H activation of hydrocarbon feedstocks has been recently demonstrated for the functionalization of chemical substrates into a variety of oxygenated products. Activated C–H groups can be readily oxidized at an anode with careful selection of an appropriate redox mediator, whereas unactivated C–H groups require a large overpotential and yield and selectivity remains poor. Both activated and unactivated C–H oxidations are still in the stage of “benchtop-scale” laboratory research and require further proof-of-concept demonstrations and explicit study of anodic oxidation and cathodic HER faradaic efficiencies.
Access to affordable electrolytic hydrogen is dependent, among several other factors, on the materials development and device engineering in order to produce more efficient electrode materials and appropriate substrate/mediator pairs. This can be achieved by incorporating modeling and predictive tools to guide the exploration of the large parameter space defined by the electronics of redox mediators and the material properties of EC and PEC electrodes.
Conflicts of interest
There are no conflicts to declare.References
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef PubMed.
- N. S. Lewis, Science, 2016, 351(aad1920), 1–9 Search PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, 1–12 CrossRef PubMed.
- J. H. Montoya, L. C. Seitz, P. Chakthranont, A. Vojvodic, T. F. Jaramillo and J. K. Nørskov, Nat. Mater., 2016, 16, 70–81 CrossRef PubMed.
- J. M. Andújar and F. Segura, Renewable Sustainable Energy Rev., 2009, 13, 2309–2322 CrossRef.
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901–4934 CrossRef.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef PubMed.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1(003), 1–13 Search PubMed.
- J. Luo, J.-H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, N.-G. Park, S. D. Tilley, H. J. Fan and M. Grätzel, Science, 2014, 345, 1593–1596 CrossRef PubMed.
- M. Grätzel, Nature, 2001, 414, 338–344 CrossRef PubMed.
- K. Sivula, J. Phys. Chem. Lett., 2013, 4, 1624–1633 CrossRef PubMed.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- W. A. Smith, I. D. Sharp, N. C. Strandwitz and J. Bisquert, Energy Environ. Sci., 2015, 8, 2851–2862 RSC.
- H. G. Cha and K.-S. Choi, Nat. Chem., 2015, 7, 328–333 CrossRef PubMed.
- B. You, X. Liu, N. Jiang and Y. Sun, J. Am. Chem. Soc., 2016, 138, 13639–13646 CrossRef PubMed.
- T. Li, T. Kasahara, J. He, K. E. Dettelbach, G. M. Sammis and C. P. Berlinguette, Nat. Commun., 2017, 8, 1–5 CrossRef PubMed.
- E. J. Horn, B. R. Rosen, Y. Chen, J. Tang, K. Chen, M. D. Eastgate and P. S. Baran, Nature, 2016, 533, 77–81 CrossRef PubMed.
- R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC.
- E. J. Horn, B. R. Rosen and P. S. Baran, ACS Cent. Sci., 2016, 2, 302–308 CrossRef PubMed.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef PubMed.
- C. A. Mesa, A. Kafizas, L. Francàs, S. R. Pendlebury, E. Pastor, Y. Ma, F. Le Formal, M. T. Mayer, M. Grätzel and J. R. Durrant, J. Am. Chem. Soc., 2017, 139, 11537–11543 CrossRef PubMed.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef PubMed.
- R. D. L. Smith, M. S. Prévot, R. D. Fagan, Z. Zhang, P. A. Sedach, M. K. J. Siu, S. Trudel and C. P. Berlinguette, Science, 2013, 340, 60–63 CrossRef PubMed.
- M. W. Louie and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 12329–12337 CrossRef PubMed.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef PubMed.
- L. C. Seitz, C. F. Dickens, K. Nishio, Y. Hikita, J. Montoya, A. Doyle, C. Kirk, A. Vojvodic, H. Y. Hwang, J. K. Norskov and T. F. Jaramillo, Science, 2016, 353, 1011–1014 CrossRef PubMed.
- D. A. Salvatore, K. E. Dettelbach, J. R. Hudkins and C. P. Berlinguette, Sci. Adv., 2015, 1, 1–7 Search PubMed.
- J. W. D. Ng, M. García-Melchor, M. Bajdich, P. Chakthranont, C. Kirk, A. Vojvodic and T. F. Jaramillo, Nat. Energy, 2016, 1, 1–8 Search PubMed.
- C. Du, X. Yang, M. T. Mayer, H. Hoyt and J. Xie, Angew. Chem., Int. Ed. Engl., 2013, 52, 12692–12695 CrossRef PubMed.
- T. Li, J. He, B. Peña and C. P. Berlinguette, Angew. Chem., Int. Ed. Engl., 2016, 55, 1769–1772 CrossRef PubMed.
- B. J. Trześniewski, I. A. Digdaya, T. Nagaki, S. Ravishankar, I. Herraiz-Cardona, D. A. Vermaas, A. Longo, S. Gimenez and W. A. Smith, Energy Environ. Sci., 2017, 10, 1517–1529 RSC.
- H. Zhou, F. Yu, J. Sun, R. He, S. Chen, C. W. Chu and Z. Ren, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5607–5611 CrossRef PubMed.
- M. R. Shaner, S. Hu, K. Sun and N. S. Lewis, Energy Environ. Sci., 2014, 8, 203–207 RSC.
- X. Zhou, R. Liu, K. Sun, D. Friedrich, M. T. McDowell, F. Yang, S. T. Omelchenko, F. H. Saadi, A. C. Nielander, S. Yalamanchili, K. M. Papadantonakis, B. S. Brunschwig and N. S. Lewis, Energy, 2015, 8, 2644–2649 Search PubMed.
- J. A. Seabold and K.-S. Choi, Chem. Mater., 2011, 23, 1105–1112 CrossRef.
- T. W. Kim and K.-S. Choi, J. Phys. Chem. Lett., 2016, 7, 447–451 CrossRef PubMed.
- J. W. Ager, M. R. Shaner, K. A. Walczak, I. D. Sharp and S. Ardo, Energy Environ. Sci., 2015, 8, 2811–2824 RSC.
- R. D. L. Smith, M. S. Prévot, R. D. Fagan, S. Trudel and C. P. Berlinguette, J. Am. Chem. Soc., 2013, 135, 11580–11586 CrossRef PubMed.
- F. A. L. Laskowski, M. R. Nellist, R. Venkatkarthick and S. W. Boettcher, Energy Environ. Sci., 2017, 10, 570–579 RSC.
- D. K. Zhong, S. Choi and D. R. Gamelin, J. Am. Chem. Soc., 2011, 133, 18370–18377 CrossRef PubMed.
- T. W. Kim and K.-S. Choi, Science, 2014, 343, 990–994 CrossRef PubMed.
- T. Li, J. He, B. Peña and C. P. Berlinguette, ACS Appl. Mater. Interfaces, 2016, 8, 25010–25013 CrossRef PubMed.
- E. Steckhan, T. Arns, G. Heineman WRHilt, D. Hoormann, J. Jörissen, L. Kröner, B. Lewall and H. Pütter, Chemosphere, 2001, 43, 63–73 CrossRef PubMed.
- B. A. Frontana-Uribe, R. Daniel Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Green Chem., 2010, 12, 2099–2119 RSC.
- J.-I. Yoshida, K. Kataoka, R. Horcajada and A. Nagaki, Chem. Rev., 2008, 108, 2265–2299 CrossRef PubMed.
- Y. Kawamata, M. Yan, Z. Liu, D.-H. Bao, J. Chen, J. T. Starr and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 7448–7451 CrossRef PubMed.
- Q. Jia, K. Iwashina and A. Kudo, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11564–11569 CrossRef PubMed.
- K. R. Vuyyuru and P. Strasser, Catal. Today, 2012, 195, 144–154 CrossRef.
- E. Steckhan, Angew. Chem., Int. Ed., 1986, 25, 683–701 CrossRef.
- M. Rafiee, K. C. Miles and S. S. Stahl, J. Am. Chem. Soc., 2015, 137, 14751–14757 CrossRef PubMed.
- F. Vitse, M. Cooper and G. G. Botte, J. Power Sources, 2005, 142, 18–26 CrossRef.
- T. Take, K. Tsurutani and M. Umeda, J. Power Sources, 2007, 164, 9–16 CrossRef.
- Y. X. Chen, A. Lavacchi, H. A. Miller, M. Bevilacqua, J. Filippi, M. Innocenti, A. Marchionni, W. Oberhauser, L. Wang and F. Vizza, Nat. Commun., 2014, 5, 1–6 Search PubMed.
- W. Yan, D. Wang and G. G. Botte, Appl. Catal., B, 2012, 127, 221–226 CrossRef.
- A. T. Marshall and R. G. Haverkamp, Int. J. Hydrogen Energy, 2008, 33, 4649–4654 CrossRef.
- Y. Yan, B. Y. Xia, B. Zhao and X. Wang, J. Mater. Chem. A, 2016, 4, 17587–17603 RSC.
- L.-L. Feng, G. Yu, Y. Wu, G.-D. Li, H. Li, Y. Sun, T. Asefa, W. Chen and X. Zou, J. Am. Chem. Soc., 2015, 137, 14023–14026 CrossRef PubMed.
- B. You, X. Liu, X. Liu and Y. Sun, ACS Catal., 2017, 7, 4564–4570 CrossRef.
- M. Wang, D. Guo and H. Li, J. Solid State Chem., 2005, 178, 1996–2000 CrossRef.
- M. F. Semmelhack, C. R. Schmid and D. A. Cortés, Tetrahedron Lett., 1986, 27, 1119–1122 CrossRef.
- Y. Demizu, H. Shiigi, T. Oda, Y. Matsumura and O. Onomura, Tetrahedron Lett., 2008, 49, 48–52 CrossRef.
- A. Badalyan and S. S. Stahl, Nature, 2016, 535, 406–410 CrossRef PubMed.
- D. P. Hickey, D. A. Schiedler, I. Matanovic, P. V. Doan, P. Atanassov, S. D. Minteer and M. S. Sigman, J. Am. Chem. Soc., 2015, 137, 16179–16186 CrossRef PubMed.
- N.-T. Zhang, C.-C. Zeng, C. M. Lam, R. K. Gbur and R. D. Little, J. Org. Chem., 2013, 78, 2104–2110 CrossRef PubMed.
- N.-N. Lu, S. J. Yoo, L.-J. Li, C.-C. Zeng and R. D. Little, Electrochim. Acta, 2014, 142, 254–260 CrossRef.
- D. P. Hickey, R. D. Milton, D. Chen, M. S. Sigman and S. D. Minteer, ACS Catal., 2015, 5, 5519–5524 CrossRef.
- R. Ciriminna, G. Palmisano and M. Pagliaro, ChemCatChem, 2015, 7, 552–558 CrossRef.
- A. Kolodziej, S. D. Ahn, M. Carta, R. Malpass-Evans, N. B. McKeown, R. S. L. Chapman, S. D. Bull and F. Marken, Electrochim. Acta, 2015, 160, 195–201 CrossRef.
- E. Megiel, Adv. Colloid Interface Sci., 2017, 250, 158–184 CrossRef PubMed.
- A. Das and S. S. Stahl, Angew. Chem., Int. Ed. Engl., 2017, 56, 8892–8897 CrossRef PubMed.
- B. Karimi, M. Rafiee, S. Alizadeh and H. Vali, Green Chem., 2015, 17, 991–1000 RSC.
- B. M. Johnson, R. Francke, R. Daniel Little and L. A. Berben, Chem. Sci., 2017, 8, 6493–6498 RSC.
- R.-J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef PubMed.
- O. Casanova, S. Iborra and A. Corma, ChemSusChem, 2009, 2, 1138–1144 CrossRef PubMed.
- S. E. Davis, L. R. Houk, E. C. Tamargo, A. K. Datye and R. J. Davis, Catal. Today, 2011, 160, 55–60 CrossRef.
- D. J. Chadderdon, L. Xin, J. Qi, Y. Qiu, P. Krishna, K. L. More and W. Li, Green Chem., 2014, 16, 3778–3786 RSC.
- H. G. Cha and K.-S. Choi, Nat. Chem., 2015, 7, 328–333 CrossRef PubMed.
- C. Costentin, M. Robert and J.-M. Savéant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC.
- H.-R. Jhong, S. Ma and P. J. A. Kenis, Curr. Opin. Chem. Eng., 2013, 2, 191–199 CrossRef.
- T. Li, Y. Cao, J. He and C. P. Berlinguette, ACS Cent. Sci., 2017, 3, 778–783 CrossRef PubMed.
- B. S. Greensfelder, H. H. Voge and G. M. Good, Ind. Eng. Chem., 1949, 41, 2573–2584 CrossRef.
- J. A. Curiale and E. B. Frolov, Org. Geochem., 1998, 29, 397–408 CrossRef.
- Products & Technology Olefins, https://www.americanchemistry.com/ProductsTechnology/Olefins/, (accessed 20 March 2018).
- Y. Ashikari, A. Shimizu, T. Nokami and J.-I. Yoshida, J. Am. Chem. Soc., 2013, 135, 16070–16073 CrossRef PubMed.
- U.-S. Bäumer and H. J. Schäfer, J. Appl. Electrochem., 2005, 35, 1283–1292 CrossRef.
- A. Shimizu, R. Hayashi, Y. Ashikari, T. Nokami and J.-I. Yoshida, Beilstein J. Org. Chem., 2015, 11, 242–248 CrossRef PubMed.
- Y. Ashikari, T. Nokami and J.-I. Yoshida, Org. Lett., 2012, 14, 938–941 CrossRef PubMed.
- A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef PubMed.
- U.-S. Bäumer and H. J. Schäfer, Electrochim. Acta, 2003, 48, 489–495 CrossRef.
- C. Dai, J. Zhang, C. Huang and Z. Lei, Chem. Rev., 2017, 117, 6929–6983 CrossRef PubMed.
- K.-C. Cheung, W.-L. Wong, D.-L. Ma, T.-S. Lai and K.-Y. Wong, Coord. Chem. Rev., 2007, 251, 2367–2385 CrossRef.
- W. P. Griffith, Coord. Chem. Rev., 1970, 5, 459–517 CrossRef.
- H. Tanaka, M. Kuroboshi, H. Takeda, H. Kanda and S. Torii, J. Electroanal. Chem., 2001, 507, 75–81 CrossRef.
- N. Phougat, P. Vasudevan, N. K. Jha and D. K. Bandhopadhyay, Transition Met. Chem., 2003, 28, 838–847 CrossRef.
- D. L. Hickman, A. Nanthakumar and H. M. Goff, J. Am. Chem. Soc., 1988, 110, 6384–6390 CrossRef.
- M.-H. Liu and Y. O. Su, J. Electroanal. Chem., 1998, 452, 113–125 CrossRef.
- T.-S. Lee and Y. O. Su, J. Electroanal. Chem., 1996, 414, 69–73 CrossRef.
- M. W. Grinstaff, M. G. Hill, J. A. Labinger and H. B. Gray, Science, 1994, 264, 1311–1313 CrossRef PubMed.
- E. R. Birnbaum, M. W. Grinstaff, J. A. Labinger, J. E. Bercaw and H. B. Gray, J. Mol. Catal. A: Chem., 1995, 104, L119–L122 CrossRef.
- N. A. Stephenson and A. T. Bell, J. Mol. Catal. A: Chem., 2007, 275, 54–62 CrossRef.
- N. A. Stephenson and A. T. Bell, J. Mol. Catal. A: Chem., 2007, 272, 108–117 CrossRef.
- N. A. Stephenson and A. T. Bell, J. Am. Chem. Soc., 2005, 127, 8635–8643 CrossRef PubMed.
- N. Fu, G. S. Sauer, A. Saha, A. Loo and S. Lin, Science, 2017, 357, 575–579 CrossRef PubMed.
- N. Fu, G. S. Sauer and S. Lin, J. Am. Chem. Soc., 2017, 139, 15548–15553 CrossRef PubMed.
- K.-Y. Ye, G. Pombar, N. Fu, G. S. Sauer, I. Keresztes and S. Lin, J. Am. Chem. Soc., 2018, 140, 2438–2441 CrossRef PubMed.
- J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740–4761 RSC.
- M. Masui, T. Ueshima and S. Ozaki, J. Chem. Soc., Chem. Commun., 1983, 479–480 RSC.
- M. Rafiee, F. Wang, D. P. Hruszkewycz and S. S. Stahl, J. Am. Chem. Soc., 2018, 140, 22–25 CrossRef PubMed.
- L. Eberson and K. Nyberg, Acc. Chem. Res., 1973, 6, 106–112 CrossRef.
- K. Nyberg, Acta Chem. Scand., 1971, 25, 534–542 CrossRef.
- W. R. Gutekunst and P. S. Baran, Chem. Soc. Rev., 2011, 40, 1976–1991 RSC.
- R. A. F. Tomás, J. C. M. Bordado and J. F. P. Gomes, Chem. Rev., 2013, 113, 7421–7469 CrossRef PubMed.
- W. Partenheimer, Catal. Today, 1995, 23, 69–158 CrossRef.
- D. P. Hruszkewycz, K. C. Miles, O. R. Thiel and S. S. Stahl, Chem. Sci., 2017, 8, 1282–1287 RSC.
- V. Weidmann and W. Maison, Synthesis, 2013, 45, 2201–2221 CrossRef.
- S. M. Weinreb and B. M. Trost, Comprehensive Organic Synthesis, Elsevier Ltd, 1991 Search PubMed.
- T. Shono, Tetrahedron Lett., 1968, 9, 6207–6208 CrossRef.
- M. Masui, K. Hosomi, K. Tsuchida and S. Ozaki, Chem. Pharm. Bull., 1985, 33, 4798–4802 CrossRef.
- R. Curci, L. D'Accolti and C. Fusco, Acc. Chem. Res., 2006, 39, 1–9 CrossRef PubMed.
- T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362–3374 CrossRef PubMed.
- A. E. Shilov and G. B. Shul'pin, Chem. Rev., 1997, 97, 2879–2932 CrossRef PubMed.
- M. S. Chen and M. C. White, Science, 2010, 327, 566–571 CrossRef PubMed.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef.
- H. Park, H.-I. Kim, G.-H. Moon and W. Choi, Energy Environ. Sci., 2016, 9, 411–433 RSC.
- M. S. Koo, K. Cho, J. Yoon and W. Choi, Environ. Sci. Technol., 2017, 51, 6590–6598 CrossRef PubMed.
- D. Raptis, V. Dracopoulos and P. Lianos, J. Hazard. Mater., 2017, 333, 259–264 CrossRef PubMed.
- D. V. Esposito, R. V. Forest, Y. Chang, N. Gaillard, B. E. McCandless, S. Hou, K. H. Lee, R. W. Birkmire and J. G. Chen, Energy Environ. Sci., 2012, 5, 9091–9099 RSC.
- E. Kalamaras, V. Dracopoulos, L. Sygellou and P. Lianos, Chem. Eng. J., 2016, 295, 288–294 CrossRef.
Footnote |
| † L. M. R. and T. L. contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |