
Photoredox systems with biocatalysts for CO2 utilization
Y.
Amao
 *ab
*ab
aAdvanced Research Institute for Natural Science and Technology, Osaka City University, Sugimoto 3-3-138, Sumiyoshi-ku, Osaka 558-8585, Japan. E-mail: amao@ocarina.osaka-cu.ac.jp
bResearch Center for Artificial Photosynthesis, Osaka City University, Sugimoto 3-3-138, Sumiyoshi-ku, Osaka 558-8585, Japan
First published on 14th June 2018
Abstract
Visible-light driven redox systems have been attracting significant research attention due to their ability to produce hydrogen and reduce and utilize CO2 for the production of solar fuels and chemicals. Generally, visible-light driven redox systems consist of an electron donor, a photocatalytic dye, an electron mediator and a catalyst. One of the important components in visible-light driven redox systems is an effective catalyst for hydrogen production and reduction and utilization of CO2. The catalysts used in visible-light driven redox systems are classified into metal nanoparticles, molecular catalysts and biocatalysts. Among them, biocatalysts are promising due to their excellent reaction and substrate selectivity, especially for the reduction and utilization of CO2, which is remarkably higher than that of other catalysts. Among the biocatalysts for CO2 reduction and utilization, NAD(P)+-dependent dehydrogenases, which are commercially available, are widely used in visible-light driven redox systems. Formate dehydrogenase (FDH) from Candida boidinii is a typical NAD(P)+-dependent dehydrogenase for the visible-light driven redox reduction of CO2 to formate. Furthermore, the addition of commercially available aldehyde (aldDH), formaldehyde (FldDH) and alcohol (ADH) dehydrogenase to this system results in the reduction of CO2 to methanol via formate and formaldehyde as intermediates in visible-light driven redox systems. Furthermore, NAD(P)+-dependent dehydrogenases for decarboxylation are also commercially available and widely used for the visible-light driven formation of carbon–carbon bonds from CO2 and organic molecules. Malic enzyme (ME) from chicken liver is a typical NAD(P)+-dependent dehydrogenase with decarboxylating ability for the visible-light driven production of malate, which is based on the formation of carbon–carbon bonds from CO2 and pyruvate. In this review, visible-light driven CO2 reduction and utilization systems involving the photoreduction of NAD(P)+ and biocatalysts are introduced. Furthermore, visible-light driven CO2 reduction and utilization systems involving the photoreduction of bipyridinium salt (viologen)-based electron mediators and biocatalysts also are introduced. In particular, the simplification of visible-light driven CO2 reduction and utilization systems by utilizing viologen-based electron mediators and the improvement in their efficiency without changing the structure of the biocatalyst are also discussed.
 Y. Amao | Prof. Yutaka Amao received his PhD in 1997 from the Tokyo Institute of Technology. He worked as a researcher at the Kanagawa Academy of Science and Technology (1997–1998), and then at the National Aerospace Laboratory (1998–2001). He became an Associate Professor in Oita University. He also started work as a Precursory Research for Embryonic Science and Technology researcher at the Japan Science and Technology Agency (2011–2015). In 2013, he was appointed a full Professor of Osaka City University. He also was appointed the Director of the Research Center for Artificial Photosynthesis (2015-present). In 2018, he was awarded the Fellow of Royal Chemistry Society (FRSC). |
Introduction
Greenhouse gases are chemical compounds that induce the greenhouse effect. The rapid increase in the atmospheric concentrations of the three main human-made greenhouse gases, CO2, methane, and nitrous oxide, in the Earth is clear from the data sets for these gases over the last 400![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 years. Among the greenhouse gasses, CO2 is the most important greenhouse gas produced by human activity, which is primarily through the combustion of fossil fuels. Its concentration in the Earth's atmosphere has increased by more than 30% since the Industrial Revolution. Thus, the development of technology for a drastic reduction in CO2 gas is important for the future.1 The protocol concerning the reduction of CO2 in the atmosphere was deliberated at the Kyoto Conference on Climate Change (COP3).2 Furthermore, at the 2009 U.N. Summit on Climate Change held at New York, the Prime Minister of Japan declared that the Japanese administration would aim to cut greenhouse gas emissions by more than 25% by 2020 from the 1990 levels. Recently, the Paris Climate Accord (COP21) was implemented, which is an agreement dealing with greenhouse gas emission mitigation, adaptation, and finance starting in the year 2020. The COP21 was negotiated in Paris and adopted by consensus on the 12th December 2015, which states that CO2 needs to be drastically reduced (more than 30%) worldwide from 2030 to 2050.3 Thus, the production of low-carbon fuels, hydrogen, CO2-based alcohol, etc. using renewable energy such as solar energy is important to mitigate global warming. Consequently, visible-light driven redox systems are attracting significant attention for the production of solar fuels and chemicals due to their ability to produce hydrogen and reduce and utilize CO2 using solar energy, which consist of a photocatalytic dye and catalyst. Generally, visible-light driven redox systems consist of an electron donor (D), a photocatalytic dye (P), an electron mediator (C) and a catalyst, as shown in Fig. 1.4–10
000 years. Among the greenhouse gasses, CO2 is the most important greenhouse gas produced by human activity, which is primarily through the combustion of fossil fuels. Its concentration in the Earth's atmosphere has increased by more than 30% since the Industrial Revolution. Thus, the development of technology for a drastic reduction in CO2 gas is important for the future.1 The protocol concerning the reduction of CO2 in the atmosphere was deliberated at the Kyoto Conference on Climate Change (COP3).2 Furthermore, at the 2009 U.N. Summit on Climate Change held at New York, the Prime Minister of Japan declared that the Japanese administration would aim to cut greenhouse gas emissions by more than 25% by 2020 from the 1990 levels. Recently, the Paris Climate Accord (COP21) was implemented, which is an agreement dealing with greenhouse gas emission mitigation, adaptation, and finance starting in the year 2020. The COP21 was negotiated in Paris and adopted by consensus on the 12th December 2015, which states that CO2 needs to be drastically reduced (more than 30%) worldwide from 2030 to 2050.3 Thus, the production of low-carbon fuels, hydrogen, CO2-based alcohol, etc. using renewable energy such as solar energy is important to mitigate global warming. Consequently, visible-light driven redox systems are attracting significant attention for the production of solar fuels and chemicals due to their ability to produce hydrogen and reduce and utilize CO2 using solar energy, which consist of a photocatalytic dye and catalyst. Generally, visible-light driven redox systems consist of an electron donor (D), a photocatalytic dye (P), an electron mediator (C) and a catalyst, as shown in Fig. 1.4–10
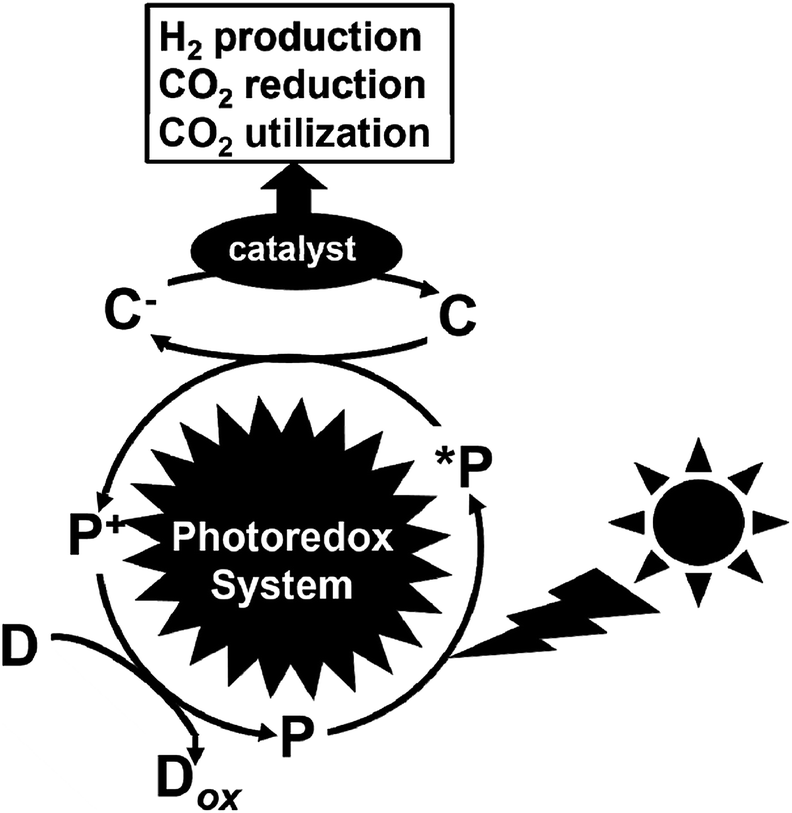 | ||
| Fig. 1 Schematic illustration of a visible-light driven redox system consisting of an electron donor (D), photocatalytic dye (P), electron mediator (C) and catalyst. | ||
One of the important components in these systems is an effective catalyst for hydrogen production, and reduction and utilization of CO2, which is classified into metal nanoparticles, molecular catalysts and biocatalysts. For example, platinum nanoparticles,11–21 and cobalt,23–26 platinum,27–33 and iron-based molecular catalysts34–44 are used for visible-light driven hydrogen production with a photocatalytic dye. The biocatalyst hydrogenase (H2ase)45–53 is also used as a catalyst for visible-light driven hydrogen production. Studies using H2ase as a catalyst in visible-light driven hydrogen production systems with various photocatalytic dyes, ruthenium polypyridyl coordinate complexes, metalloporphyrins, semiconductor-based metal oxides, etc. have been conducted since the 1970s.54–66
In contrast, copper-based catalysts,67–76 silver-loaded metal oxides,77–82 and rhenium- and iron-based molecular catalysts83–90 are used for the visible-light driven reduction of CO2 to CO, formate, etc. with a photocatalytic dye. However, the problems associated with using these catalysts for the visible-light driven reduction of CO2 include low product selectivity, and simultaneous hydrogen production. When copper-based catalysts are used for the reduction and utilization of CO2 in electrochemical reactions, CO, formate, methane, ethylene, ethanol, n-propanol and hydrogen are observed as products. As an example, a copper-based catalyst with a gallium nitride-based semiconductor was applied for the light-driven reduction of CO2.91 In this system, the solar energy conversion yield was estimated to be 0.2%. However, the products obtained were CO, formate, methane, ethylene, ethanol, etc. This is because its CO2 reduction selectivity for products is low, thus, it was difficult to obtain the desired products based on the reduction of CO2 in this system.
Visible-light driven CO2 reduction systems have been studied using cobalt polypyridyl, macrocyclic complexes and rhenium bipyridyl complexes. Visible-light driven CO2 reduction to CO has also been accomplished using a rhenium bipyridyl complex system, in which simultaneous hydrogen production was observed. A system consisting of triethylamine (TEA) as the electron donor, tris(2-phenylpyridinato)iridium(III) (Ir(ppy)3) as the photocatalytic dye, and an Fe(III)-porphyrin-based catalyst (chloro iron(III) 5,10,15,20-tetra(40-N,N,N-trimethylanilinium) porphyrin; Fe-p-TMA, chloro iron(III) 5,10,15,20-tetrakis(2′,6′-dihydroxyphenyl) porphyrin; and Fe-o-OH or chloro iron(III) tetraphenylporphyrin; FeTPP) for the visible-light driven reduction of CO2 to methane with high selectivity was also reported.92 In this system, CO, methane and hydrogen were produced upon visible-light irradiation (>420 nm). By using Fe-p-TMA as the catalyst for CO2 reduction, CO was the main product from the direct photoreduction of CO2. Using a two-pot procedure that first reduces CO2 and then reduces CO to generate methane with a selectivity of up to 82% and quantum yield of 0.18%, the proposed mechanism for the visible-light driven reduction of CO2 to methane with the Fe(III) porphyrin-based catalyst is as follows. Initially, the Fe(III) porphyrin-based catalyst is reduced with 3-electrons to catalytically produce Fe(0) active species. Then, the Fe(0) species reduces CO2, and Fe(I) is regenerated through electron transfer from the photoexcited Ir(ppy)3. The CO produced binds to Fe(II) and is further reduced with a total of 6-electrons, and then transferred from the photoexcited Ir(ppy)3 and 6-protons to generate methane via Fe(I)-formyl as the proposed intermediate. Also, some studies on the light driven reduction of CO2 with silver nanoparticle-loaded gallium oxide have been reported. In this system, CO2 reduction to CO and simultaneous hydrogen production are observed. A system consisting of a semiconductor photocatalyst, bismuth vanadium oxide (BiVO4) and a cobalt(II) chlorin (Co(II) Chl)-based molecular catalyst for the visible-light driven reduction of CO2 to CO with high selectivity has also been reported. This system employed a BiVO4 fluorine-doped tin oxide (FTO) photoanode surface-modified with iron(III) oxide(hydroxide) (FeO(OH)) (FeO(OH)/BiVO4/FTO) and Co(II) Chl as the cathode active material adsorbed on multiwalled carbon nanotubes (Co(II) Chl-modified cathode). Photoelectrochemical CO2 reduction occurred using this system to produce CO with 83% Faradaic efficiency at an applied bias voltage of −1.3 V at the Co(II) Chl-modified cathode vs. the FeO(OH)/BiVO4/FTO photoanode under visible light irradiation in a CO2-saturated aqueous solution (pH 4.6). The difference in the oxidation potential of the FeO(OH)/BiVO4/FTO electrode in the dark and under visible-light irradiation was estimated to be ca. 1.5 V, which is smaller than the band gap of BiVO4 (band gap energy: 2.4 eV). This indicates that the FeO(OH)/BiVO4/FTO photoanode lowers the total bias, which enables the simultaneous oxidation of water and reduction of CO2 to CO.93 From these results, it is important to improve the CO2 photoreduction product selectivity using metal-based, molecular and photo-catalysts.
Among the catalysts for the reduction and utilization of CO2, biocatalysts are promising due to their excellent reaction and substrate selectivity in homogenous aqueous media. Organic molecule syntheses based on biocatalytic methods have been given much attention due to their regio- and stereo-selectivity, and mild reaction conditions. Biocatalytic syntheses compete with conventional methods based on chemical synthesis, especially in organic molecules syntheses that cannot be successfully conducted using chemical catalysts. Examples of beneficial biocatalytic reactions include the oxidation of alkanes, alkenes, and aromatics.94 Since biocatalytic reactions proceed in aqueous solution as the reaction medium, biocatalytic methods are attractive for green processes in the reduction and utilization of CO2.
For example, formate dehydrogenase (FDH) catalyzes the oxidation and reduction of formate and CO2 with the redox couple NAD+/NADH as the co-enzyme. Thus, visible-light driven CO2 reduction to formate can occur in photoredox systems, as shown in Fig. 1, which consist of an electron donor, photocatalytic dye, and electron mediator in the presence of FDH as a catalyst.
Biocatalysts for the reduction and utilization of CO2 are classified into two categories: (1) CO2 reduction to CO or formate and (2) formation of carbon–carbon bonds from CO2 and organic molecules to produce carboxylic acid. Carbon monoxide dehydrogenase (CODH) and FDH are biocatalysts for the reduction of CO2 to CO and formate, respectively. Systems consisting of FDH, aldehyde dehydrogenase (AldDH) and alcohol dehydrogenase (ADH) are used for the reduction of CO2 to methanol via formate and formaldehyde as intermediates.
Malic enzyme (ME) and isocitrate dehydrogenase (IDH) are biocatalysts for the formation of carbon–carbon bonds from CO2 and pyruvate and 2-oxoglutarate to produce malate and isocitrate, respectively.
In the visible-light driven reduction and utilization of CO2, photocatalytic dyes with high visible-light sensitization activity are necessary to act as models for natural photosynthetic dyes such as chlorophyll. The model compounds used as photosynthetic dyes are classified into four main categories.95 The first category comprises porphyrin compounds. The second category is ruthenium polypyridyl complexes. The third category is natural photosynthesis dyes, such as chlorophyll and its derivatives, and the fourth category is photocatalytic materials such as CdS and TiO2.
Electron mediators for the visible-light driven reduction and utilization of CO2 with biocatalysts are easily reduced with a photocatalytic dye and need to function as co-enzymes for the biocatalysts. NAD(P)+, bipyridinium salts (so-called viologen) and rhodium complexes are widely used as electron mediators for the visible-light driven reduction and utilization of CO2 with biocatalysts. The photocatalytic dyes (P), electron mediators (C) and biocatalyst utilized in the visible-light driven reduction and utilization of CO2 are summarized in Fig. 2.
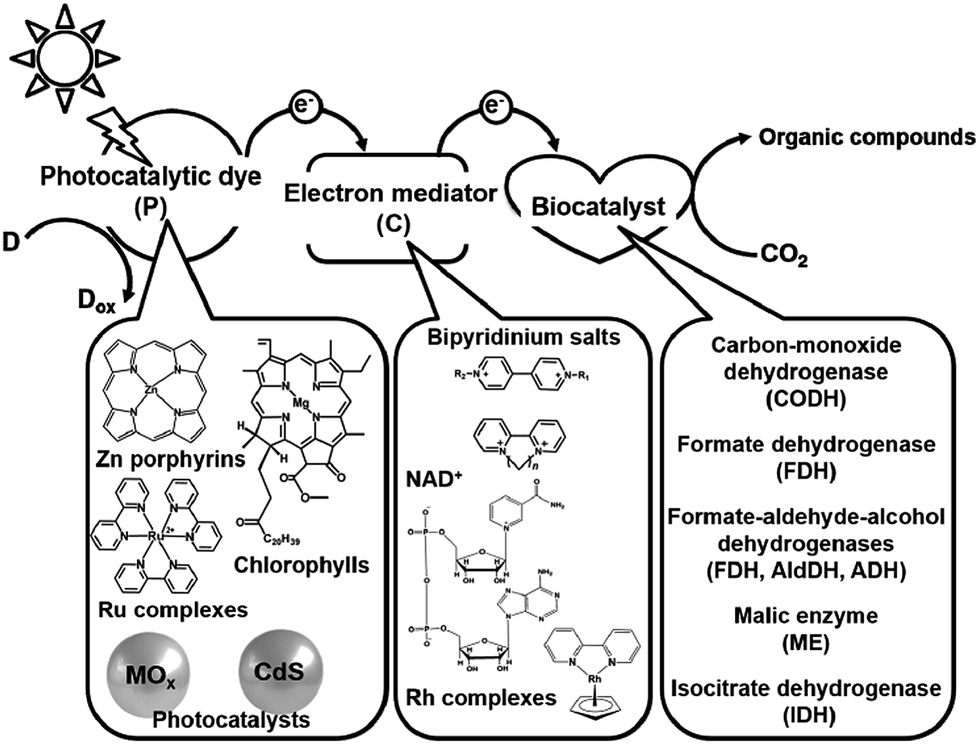 | ||
| Fig. 2 Summary of the photocatalytic dyes (P), electron mediators (C) and biocatalysts utilized in the visible-light driven reduction and utilization of CO2. | ||
In this review, the properties of the commercially available biocatalysts for the reduction and utilization of CO2 are introduced. Also, visible-light driven CO2 reduction and utilization systems involving the photoreduction of NAD(P)+ and biocatalysts are introduced. Furthermore, the visible-light driven CO2 reduction and utilization systems involving the photoreduction of viologen-based electron mediators and biocatalysts are also introduced. In particular, by utilizing viologen-based electron mediators,96 the simplification of the visible-light driven CO2 reduction and utilization systems and improvement in their efficiency without changing the structure of the biocatalyst are also mentioned.
Biocatalysts for the reduction and utilization of CO2
Biocatalysts for CO2 reduction
Biocatalysts with the ability to reduce CO2 in light driven redox systems are introduced in this section. Biocatalysts for the light driven reduction of CO2 are a family of enzymes that catalyze the oxidation of C1 materials such as formate and CO to CO2 by donating electrons to a second substrate, NAD(P)+, and the reverse reaction of CO2 reduction, donating electrons to a second substrate NAD(P)H. These biocatalysts are so-called NAD(P)+-dependent dehydrogenases. The typical NAD(P)+-dependent dehydrogenase reaction the for reduction of CO2 are shown in Fig. 3.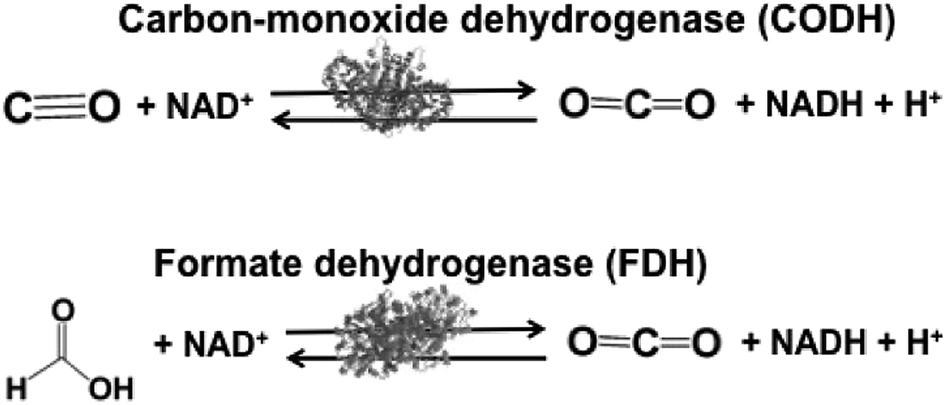 | ||
| Fig. 3 Reaction schemes of the biocatalysts carbon-monoxide (CODH) and formate (FDH) dehydrogenases. | ||
Biocatalysts for the reduction of CO2 to CO and formic acid are carbon-monoxide dehydrogenase (CODH)97–101 and formate dehydrogenase (FDH),102–105 respectively. Especially, FDH obtained from Candida boidinii (EC 1.2.1.2), which is a commercially available biocatalyst, is widely used in various systems for the reduction of CO2 to formic acid using light driven redox, photoelectrochemical and thermal catalytic reactions.
The reduction of CO2 to methanol has been paid attention for the production of low-carbon alcohol fuel by combining FDH, commercially available aldehyde (AldDH)106–108 from Yeast (EC 1.2.1.5) and alcohol dehydrogenase (ADH)109,110 from Yeast (EC 1.1.1.1). AldDH catalyzes the oxidation of formaldehyde to formic acid in the presence of NAD(P)+ and the reverse reaction of formic acid to formaldehyde in the presence of NAD(P)H. ADH catalyzes the oxidation of methanol to formaldehyde in the presence of NAD(P)+ and the reverse reaction of formaldehyde to methanol in the presence of NAD(P)H (Fig. 4).
 | ||
| Fig. 4 Reaction schemes of the biocatalysts aldehyde (AldDH) and alcohol (ADH) dehydrogenases. | ||
By using the same co-enzyme NAD(P)H for FDH, FldDH and ADH, the reduction of CO2 to methanol via formate and formaldehyde as intermediates was developed, as shown in Fig. 5.
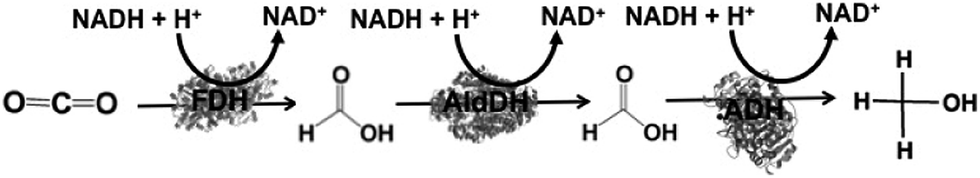 | ||
| Fig. 5 Reaction scheme of CO2 reduction to methanol using FDH, FldDH, and ADH in the presence of NADH. | ||
For example, the reduction CO2 to methanol in FDH, FldDH, and ADH immobilized silica sol–gel matrices has been accomplished in the presence of NADH. The yield of NADH to methanol was estimated to be 21.0% in the aqueous system, in contrast to 91.2% using the silica sol–gel system.111 The reduction of CO2 to methanol using FDH, FldDH, and ADH immobilized on polystyrene particles has also been developed with glutamate dehydrogenase (GluDH) for the utilization of the NAD+/NADH redox couple. In solution, the yield of CO2 reduction to methanol was estimated to be 12%, whereas it was estimated to be 80% using biocatalysts co-immobilized the polystyrene particle system.112
Formaldehyde dehydrogenase (FldDH) from Pseudomonas sp. (EC 1.2.1.46) specialized for formaldehyde oxidation and formate reduction with the NAD+/NADH redox couple is also commercially available.
Furthermore, the visible-light driven reduction of CO2 can be developed by combining the photoreduction of NAD+ to NADH and a biocatalyst such as FDH in the complex system of FDH, FldDH and ADH.
Biocatalysts for CO2 utilization
Biocatalysts with the function of CO2 utilization based on the formation of carbon–carbon bonds in light-driven redox systems are introduced in this section. Biocatalysts for CO2 utilization in light-driven redox systems are a set of enzymes that catalyze decarboxylation or carboxylation using the NADP+/NADPH redox couple. Malic enzyme (ME)113–118 and isocitrate dehydrogenase (IDH)119–122 are typical biocatalysts for CO2 utilization, as shown in Fig. 6.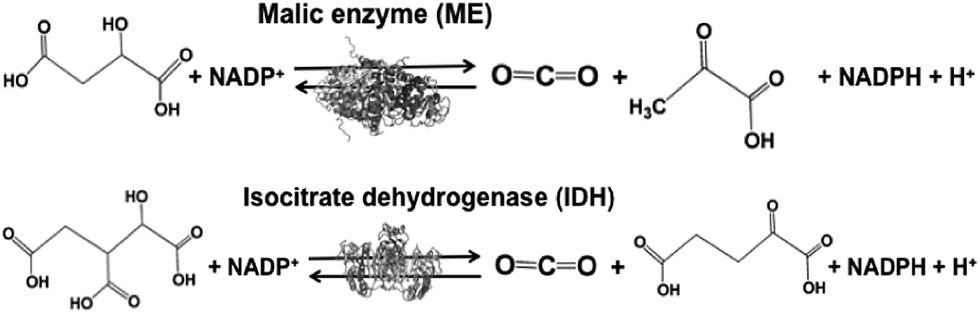 | ||
| Fig. 6 Reaction schemes of the biocatalysts malic enzyme (ME) and isocitrate dehydrogenase (IDH). | ||
ME from Chicken liver (EC 1.1.1.40) and IDH from Yeast (EC 1.1.1.41) are commercially available biocatalysts. ME catalyzes the reaction of malate conversion to pyruvate and CO2, and the reverse reaction of pyruvate and CO2 conversion to malate using the NADP+/NADPH redox couple. IDH catalyzes the reaction of isocitrate conversion to 2-oxoglutarate and CO2, and the reverse reaction of 2-oxoglutarate and CO2 conversion to isocitrate using the NADP+/NADPH redox couple.
For example, the conversion of pyruvate and CO2 to malate using ME has been developed121,122 with glucose-6-phosphate dehydrogenase for the utilization of the NADP+/NADPH redox couple. Under optimal conditions using this system, the ratio of CO2 and pyruvate to malate was estimated to be about 38% after 24 h of incubation.121
The visible-light driven CO2 utilization based on the formation of carbon–carbon bonds can be developed by combining the photoreduction of NADP+ to NADPH and a biocatalyst such as ME and IDH.
Photoredox systems for the light-driven reduction of CO2
Photoredox systems for the light-driven reduction of CO2 with a biocatalyst via the NAD(P)+/NAD(P)H redox couple
As a simple idea, the use of FDH and complex system of FDH, AldDH and ADH as CO2 reduction catalysts can applied in the visible-light driven redox system of NAD+/NADH. For example, visible-light driven CO2 reduction to formate can be developed with a redox system consisting of an electron donor (D), photocatalytic dye (P), and NAD+ in the presence of FDH, as shown in Fig. 7.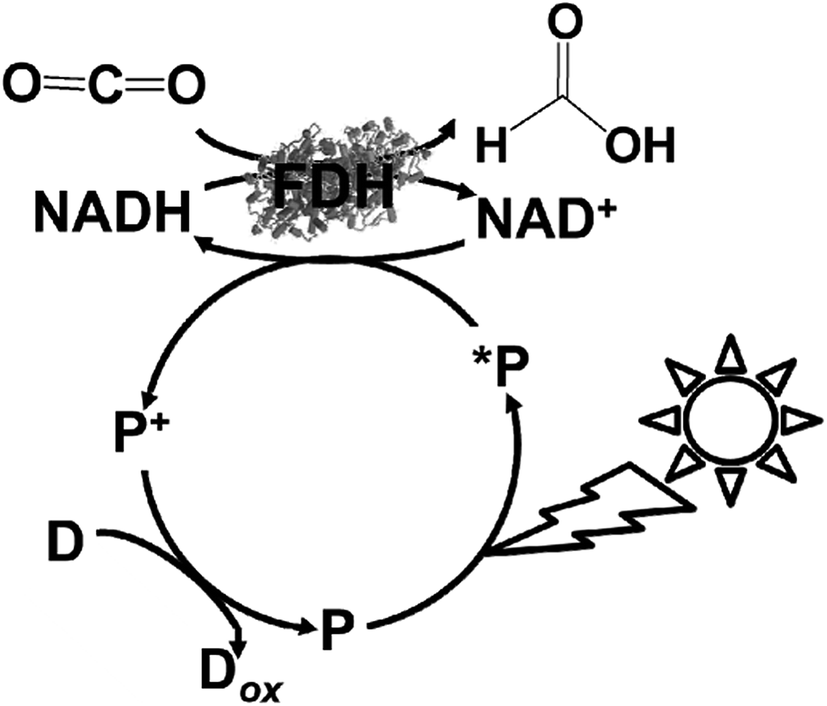 | ||
| Fig. 7 Scheme of the visible-light driven redox system for CO2 reduction to formate consisting of an electron donor (D), photocatalytic dye (P), NAD+ and FDH. | ||
However, the NAD dimer, (NAD)2, forms during the reaction of the single-electron reduced NAD+ with a photocatalytic dye, such as tris(bipyridine)ruthenium(II) (Ru(bpy)32+), as shown in Fig. 8. Moreover, the reaction between NAD+ and (NAD)2 is irreversible.123–127 Since (NAD)2 is an inactive co-enzyme for NAD+-dependent dehydrogenases such as FDH, AldDH, and ADH, it is difficult to achieve a photoredox system based on the combination of NAD+ photoreduction and FDH for the reduction of CO2 to formate.
 | ||
| Fig. 8 Dimerization process of the single-electron reduced NAD+ (NAD˙). | ||
In contrast, some studies on the visible-light driven reduction of NAD+ to NADH by a photocatalytic dye via a second catalyst, such as rhodium complexes and ferredoxin-NADP+ reductase (FNR) (EC 1.18.12) have been reported. In this system, for example, NAD+ dimerization is suppressed via the iridium or rhodium complex.
The NAD+ reduction to NADH with the iridium complex Ir(III) (Cp*)(4-(1H-pyrazol-1-yl-κN2)benzoate-κC3)(H2O)+ (Cp*: 1,2,3,4, 5-pentamethylcyclopenta-dienyl) is introduced as an example,128 and the redox process of the iridium complex Ir(III) (Cp*)(4-(1H-pyrazol-1-yl-κN2)benzoate-κC3)(H2O)+ is shown in Fig. 9.
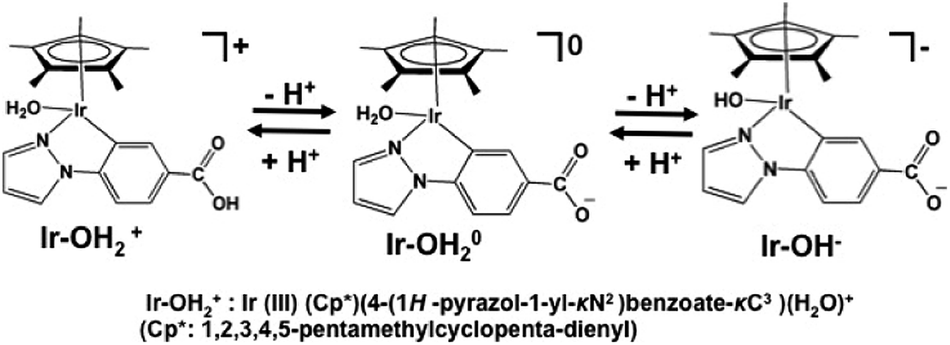 | ||
| Fig. 9 Redox reaction of iridium complex, Ir (Cp*)(4-(1H-pyrazol-1-yl-κN2)benzoate-κC3)(H2O)+ (Cp*: 1,2,3,4,5-pentamethylcyclopentadienyl). | ||
The hydrogen gas induced NAD+ reduction to NADH proceeds with the redox coupling of the iridium complex, as shown in Fig. 10. In this process, the iridium hydride complex (Ir–OH−) produced under an atmospheric of hydrogen gas undergoes 1,4-selective hydrogenation of NAD+ to form NADH.
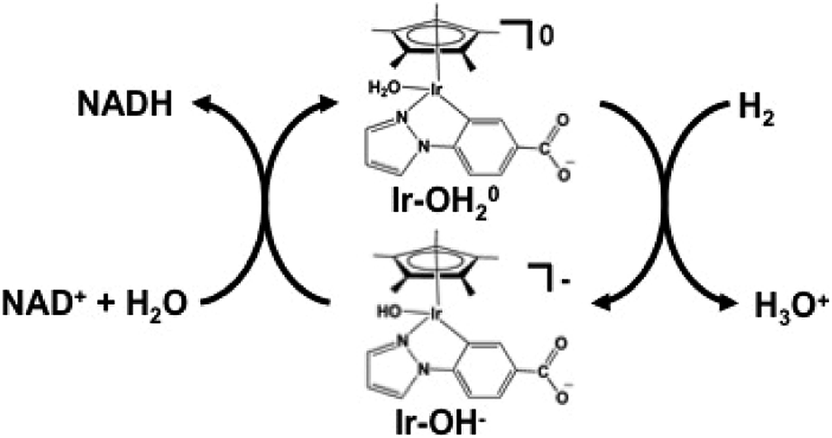 | ||
| Fig. 10 NAD+ reduction to NADH with an iridium complex and hydrogen gas. | ||
The visible-light driven NAD+ reduction to NADH a system comprised of a photocatalytic dye and rhodium complex, Cp*Rh(bpy)(OH2)2+, as an electron mediator has been developed, as shown in Fig. 11.129–132
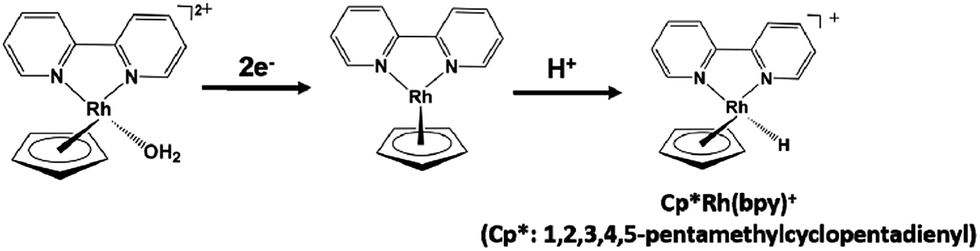 | ||
| Fig. 11 Redox process of the rhodium complex Cp*Rh(bpy)(OH2)2+. | ||
Carbon-doped TiO2, boron-doped TiO2, and multianthraquinone-substituted porphyrin (MAQSP) with chemically converted graphene (CCG) (MAQSP/CCG) are used as photocatalytic dyes in the Cp*Rh(bpy) (OH2)2+ electron-mediated reduction of NAD+ to NADH. The conversion yields of NAD+ to NADH for the carbon-doped TiO2, boron-doped TiO2, MAQSP/CCG, MAQSP, and W2Fe4Ta2O17 in this system are estimated to be 94.2%, 94.0%, 44.5%, 23.8% and 14.5% (2 h irradiation), respectively.131 The conversion yields of NAD+ to NADH using trisubstituted isatin on the porphyrin ring provided 1,1′,1′′-((20-(2-((7-amino-9,10-dioxo-9,10-dihydroanthracen-2-yl)amino)quinolin-3-yl) porphyrin-5,10,15-triyl) tris (quinoline-3,2-diyl)) tris(indoline-2,3-dione) (IP) with CCG (IP/CCG) and IP as the photocatalytic dye in this system were estimated to be 38.9% and 7.03% (90 min irradiation), respectively.132
Table 1 shows a summary of the conversion yields of NAD+ to NADH in the light driven redox system comprised of a photocatalytic dye and Cp*Rh(bpy) (OH2)2+.
By using Cp*Rh(bpy) (OH2)2+ as an electron mediator, a light-driven system for the reduction of NAD+ to NADH has been developed with a photocatalytic dye. By using MAQSP/CCG as the photocatalytic dye in this system, the visible-light driven reduction of CO2 to formate proceeds with FDH as the catalyst, as shown in Fig. 12. In this system, W2Fe4Ta2O17 and MAQSP are also are used as the photocatalytic dye.132
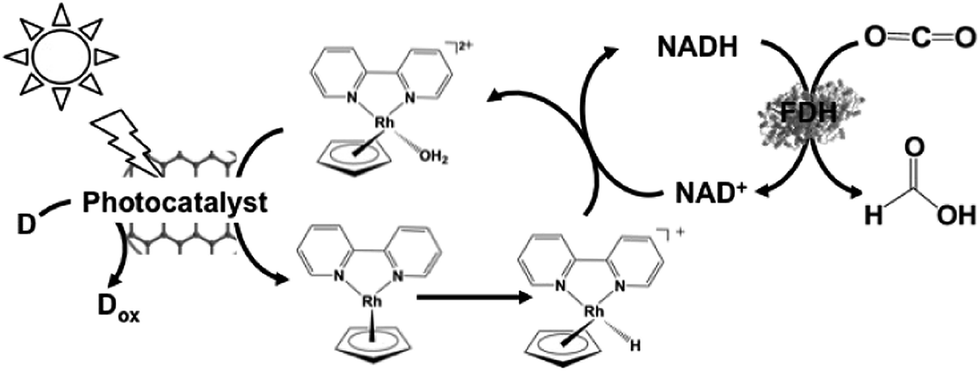 | ||
| Fig. 12 Scheme of the visible-light driven redox system for CO2 reduction to formate consisting of an electron donor (D), photocatalyst, Cp*Rh(bpy) (OH2)2+, NAD+ and FDH. | ||
The typical reaction conditions are photocatalytic dye (0.5 mg), NAD+ (1.24 μmol), rhodium complex (0.62 μmol), and FDH (3 units) in 3.1 mL of sodium phosphate buffer (100 mM, pH 7.0) with TEOA (1.24 mmol) in the presence of CO2. The efficiency of formate production in the system of MAQSP/CCG, Cp*Rh(bpy), NAD+ and FDH was 110.55 μmol after 2 h irradiation, while that for the systems using W2Fe4Ta2O17 and MAQSP were 14.25 and 46.53 μmol, respectively.132
Table 2 shows a summary of formate production after 2 h irradiation in the light-driven CO2 reduction system of a photocatalytic dye and Cp*Rh(bpy) (OH2)2+via the NAD+/NADH redox couple with FDH.
Another visible-light driven redox system for the reduction of CO2 to formate consists of an electron donor (D), a photocatalyst, Cp*Rh(bpy) (OH2)2+, NAD+, FDH, and chemically converted graphene (CCG) covalently bonded to the light harvesting BODIPY molecule (1-picolylamine-2-aminophenyl-3-oxyphenyl-4,40-difluoro-1,3,5,7-tetr-amethyl-2,6-diethyl-4-bora-3a,4adiaza-s-indacene-triazine) (CCG-BODIP) as the photocatalytic dye.134
The photocatalyst-biocatalyst coupled system developed using CCG–BODIPY as the photocatalyst led to NADH regeneration of about 54%, followed by its consumption to produce about 144 μmol of formate from CO2.
Since the visible-light driven redox system for CO2 reduction to formate consists of an electron donor (D), a photocatalyst, Cp*Rh(bpy) (OH2)2+, NAD+ and FDH, methanol production was realized by adding FldDH and ADH to this system, as shown in Fig. 13. In this system, IP/CCG, MAQSP/CCG and IP were used as the photocatalytic dyes.132
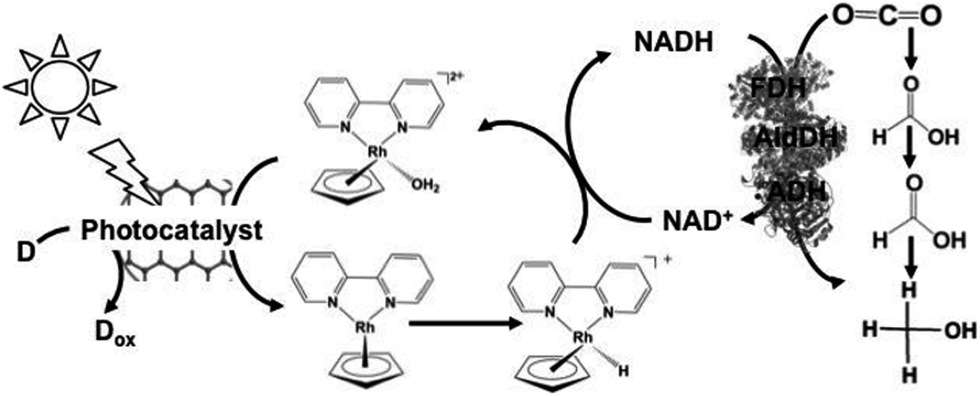 | ||
| Fig. 13 Scheme of the visible-light driven redox system for CO2 reduction to methanol consisting of an electron donor (D), a photocatalyst, Cp*Rh(bpy) (OH2)2+, NAD+, FDH, FldDH and ADH. | ||
The typical reaction conditions include photocatalytic dye (0.5 mg), NAD+ (1.24 μmol), rhodium complex (0.62 μmol), FDH (9 units), FldDH (9 units) and ADH (9 units) in 3.1 mL of sodium phosphate buffer (100 mM, pH 7.0) with TEOA (1.24 mmol) in the presence of CO2.
The methanol concentration in the system using IP/CCG as the photocatalytic dye was estimated to be 11.21 μM after 1 h visible-light irradiation. On the other hand, only 5.62 μM of methanol was obtained with CCGCMAQSP as the photocatalytic dye. Moreover, no methanol production was observed in the system using IP as the photocatalytic dye. From these results, the CO2 reduction yield using the system composed of a photocatalytic dye, Cp*Rh(bpy) (OH2)2+, NAD+ and biocatalyst depends on the efficiency of NAD+ reduction to NADH.
Table 3 shows a summary of the methanol production after 1 h irradiation in the light-driven CO2 reduction system composed of a photocatalytic dye and Cp*Rh(bpy) (OH2)2+via the NAD+/NADH redox couple with FDH, FldDH and ADH.
A semi-biological light driven CO2 reduction to formate system combining the reduction of NADP+ with photosynthetic protein and FDH has also been constructed. For example, the light-driven reduction of CO2 to formate by the photosynthetic protein cyanobacterial photosystem I (PSI) and NADPH-dependent FDH via the NADP+/NADPH redox couple has been reported, as shown in Fig. 12. This system involves NADP+ reduction to NADPH with FNR via the redox protein ferredoxin (Fd), as show in Fig. 14.135
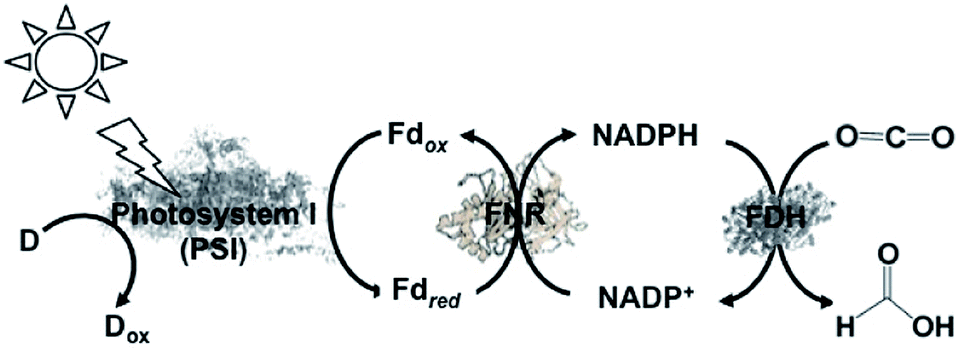 | ||
| Fig. 14 Scheme of the visible-light driven redox system for CO2 reduction to formate consisting of an electron donor (D), PSI as the photocatalytic material, Fd, FNR, NADP+ and FDH. | ||
In this system, FDH from the Pseudomonas sp. 101 (EC 1.2.1.2) mutant (PsFDH[QN]) is used as the biocatalyst for CO2 reduction. The typical reaction for the light-driven reduction of CO2 to formate is performed in 3 mL phosphate buffer using PS I at a concentration of 100 μg chlorophyll/mL, 10 mM ascorbate, 10 μM plastocyanin (PC), 2 μM Fd, 0.5 μM FNR, 1 mM NADP+, 4 mM NADPH, 56 μM PsFDH[QN], 0.05% Triton X-100, and 10% sucrose. In this system, PC acts as the electron donor. The formate concentration linearly increased with irradiation time after about a 30 min lag time. The maximum rate for the production of formate was estimated to be 26 μM h−1. In the reaction under a 10% O2 and 90% CO2 gas saturated atmosphere, no formate was detected, which indicates the O2-sensitivity of this system. In addition, this study introduced the NADPH-dependent FDH mutant into heterocysts of the cyanobacterium Anabaena sp. PCC 7120 and demonstrated an increase in formate concentration in all the cells. These results provide a new possibility for the visible-light driven reduction of CO2.
With a new concept for NADP+ reduction, an amino group-modified dendrimer (Den-NH2) was used as an electron mediator for NADP+ reduction to NADPH in a visible-light driven redox system. For example, the visible-light driven reduction of CO2 to methanol using the combination of FDH, FldDH and ADH, and reduction of NADP+ to NADHP with the system of a photocatalytic dye and Den-NH2 as the electron mediator was also developed, as shown in Fig. 15.136
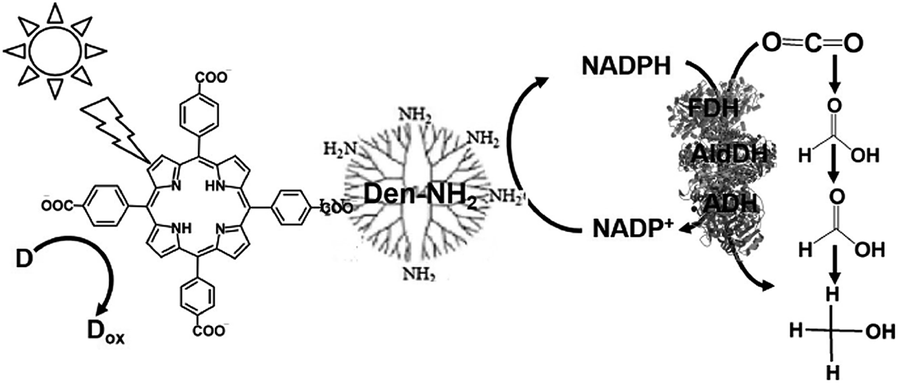 | ||
| Fig. 15 Scheme of the visible-light driven redox system for CO2 reduction to methanol consisting of an electron donor (D), tetrakis(4-carboxyphenyl)porphyrin (TCCP) as the photocatalytic dye, Den-NH2, NADP+, FDH, FldDH and ADH. | ||
Attempts were made to improve the reaction efficiency of this system by adsorbing CO2, NADP+ or NADPH, FDH, FldDH and ADH on Den-NH2. For CO2 reduction to methanol, TCPP, Den-NH2, FDH, FldDH, ADH and NADPH were immobilized on organoclay-incorporated 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO)-oxidized cellulose nanofiber (TOCNF) films (Den-TCPP-NADPH (FDH/FldDH/ADH)-TOCNF). By using Den-TCPP-NADPH (FDH/FldDH/ADH)-TOCNF, methanol production was observed upon laser irradiation of 488 nm. The proposed mechanism of NADP+ reduction in this system is described in Fig. 16.136
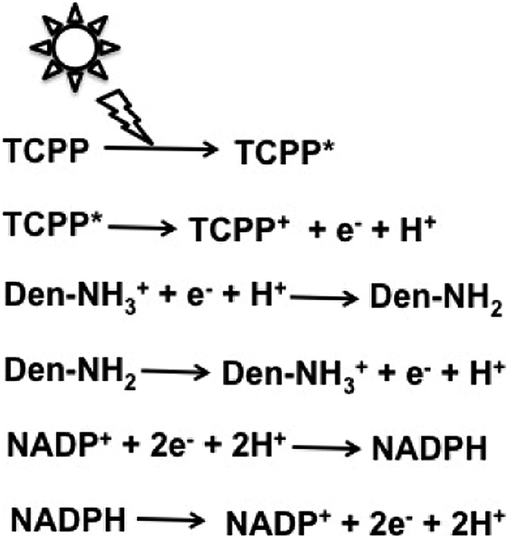 | ||
| Fig. 16 Proposed mechanism of NADP+ reduction to NADPH in the visible-light driven redox system consisting of an electron donor (D), tetrakis(4-carboxyphenyl)porphyrin (TCCP), Den-NH2 and NADP+. | ||
From the proposed mechanism shown in Fig. 16, NADP+ reduction to NADPH proceeds with the redox couple Den-NH2/Den-NH3+ and then CO2 reduction to methanol with FDH, FldDH and ADH occurs using the redox couple NADP+/NADPH.
The authors of that paper claimed that the four-electron oxidation of water to O2 proceeds with the photosensitization of TCPP, or in other words, water molecules work as an electron donor to the oxidized TCPP+. However, O2 evolution in this system has not been measured yet. Since water does not act as an electron donor in the photoredox system with TCPP, in general, NADPH works as an effective electron donor in this system. In this system, since NADPH is loaded on Den-TCPP-TOCNF, the substantial photoreduction efficiency of NADP+ to NADPH is not clear. Thus, in the future, discussion including the photoreduction efficiency of NADP+ to NADPH in addition to CO2 reduction to methanol with Den-TCPP-NADPH (FDH/FldDH/ADH)-TOCNF is necessary.
Photoredox systems for the light-driven reduction of CO2 with biocatalyst via the bipyridinium salt redox couple
The relationship between the NAD(P)+-dependent dehydrogenases and natural co-enzyme NAD(P)+ or NAD(P)H is considered again. For example, the kinetic parameters for the Michaelis constants (Km) of NAD+ and NADH for FDH from Candida boidinii in the oxidation of formate to CO2 and the reverse reaction have been determined. The Km values for NAD+ in the oxidation of formate and NADH in the CO2 reduction to FDH were estimated to be 50 and 2087 mM, respectively. Thus, FDH can be activated by a lower concentration of NAD+ than NADH (1/400), and the affinity of NAD+ for FDH is higher than that of NADH. Then, what are the Km values of NAD+ and NADH for other commercially available NAD(P)+-depend dehydrogenases, such as FldDH, FldDH and ADH? Table 4 shows a summary of the previously reported Km values of NAD+ and NADH for the commercially available ADH,137 AldDH,138,139 FldDH,140 and FDH.141,142| Dehydrogenase | K m (μM) | Reference | |
|---|---|---|---|
| NAD+ | NADH | ||
| FDH | 15 | 9980 | 141 and 142 |
| AldDH | 1100 | — | 138 and 139 |
| FldDH | 350 | — | 140 |
| ADH | 5.9 | 122 | 136 |
From Table 4, the affinity of NAD+ for all the dehydrogenases is overwhelmingly higher than that of NADH. The Km values of NADH for AldDH from Yeast and FldDH from Pseudomonas sp. have not been reported thus far to the best of our knowledge. Also, no formate reduction to formaldehyde with FldDH from Pseudomonas sp. in the presence of NADH has been observed. Once the NAD(P)+/NADH redox couple is used, the affinity between NAD(P)H and commercially available NAD(P)+-dependent dehydrogenases does not change. Therefore, the catalytic activities of dehydrogenases cannot be controlled in visible-light driven redox systems using the NAD(P)+/NAD(P)H redox couple. Moreover, since the produced NAD(P)H acts as an electron donor and is consumed, the NAD(P)+/NAD(P)H redox couple is not suitable for use in visible-light driven redox systems consisting of a photocatalytic dye and commercially available NAD(P)+-dependent dehydrogenases. Moreover, NAD(P)+ is a very expensive biological reagent.
As mentioned above, NAD(P)+/NAD(P)H redox couple-free visible-light driven redox systems with commercially available NAD(P)+-dependent dehydrogenases are desirable. Thus, it is necessary to design and synthesize electron mediator molecules with simple chemical structures, which can be easily reduced via the visible-light sensitization of a photocatalytic dye and act as a co-enzyme for commercially available NAD(P)+-depend dehydrogenases. Among the various electron mediators, methylviologen (1,1′-dimethyl-4,4′-bipyridinium salt; MV: chemical structure is shown in Fig. 17) is widely used as an electron mediator in visible-light driven redox systems with NAD(P)+-depend dehydrogenases for hydrogen production and CO2 reduction.143
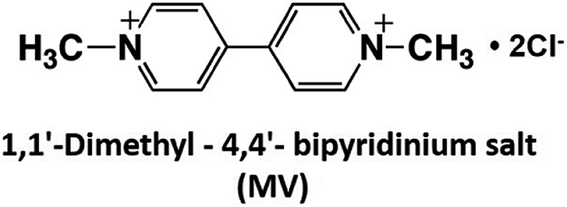 | ||
| Fig. 17 Chemical structure of 1,1′-dimethyl-4,4′-bipyridinium salt (MV). | ||
However, there is a serious problem with the use of MV in visible-light driven redox systems consisting of a photocatalytic dye and biocatalyst. The single-electron reduced MV is easily oxidized to MV by oxygen, thus it is used under a saturated atmosphere of inert gas such as nitrogen or argon in visible-light driven redox systems using a photocatalytic dye and biocatalyst for hydrogen production as an example. Since dissolved oxygen in the sample solution can be removed with CO2 gas, MV can be effectively used as an electron mediator in visible-light driven CO2 reduction and utilization systems using a photocatalytic dye and biocatalyst. Since 4,4′- or 2,2′-bipyridinium salt (BPs) is easily chemically modified, various BPs have been synthesized as effective electron mediators for visible-light driven redox systems with NAD(P)+-dependent dehydrogenases. The chemical structures of various BPs are shown in Fig. 18.143
 | ||
| Fig. 18 Chemical structures of various 4,4′- and 2,2′-bipyridinium salts (BPs). | ||
Visible-light driven CO2 reduction systems coupling the reduction of various BPs with Ru(bpy)32+, water soluble zinc porphyrin or chlorophyll-a and commercially available NAD(P)+-dependent dehydrogenases have been reported. In these systems, the single-electron reduced BPs act as a co-enzyme for NAD(P)+-dependent dehydrogenases instead of NAD(P)H.
The visible-light driven reduction of CO2 to formate involving various BPs, Ru(bpy)32+ as a photocatalytic dye, alkanethiol (RSH) as an electron donor and FDH has been reported, as shown in Fig. 19 for the first time.144–147 The redox processes of 4,4′- and 2,2′-BPs are also shown in Fig. 19.
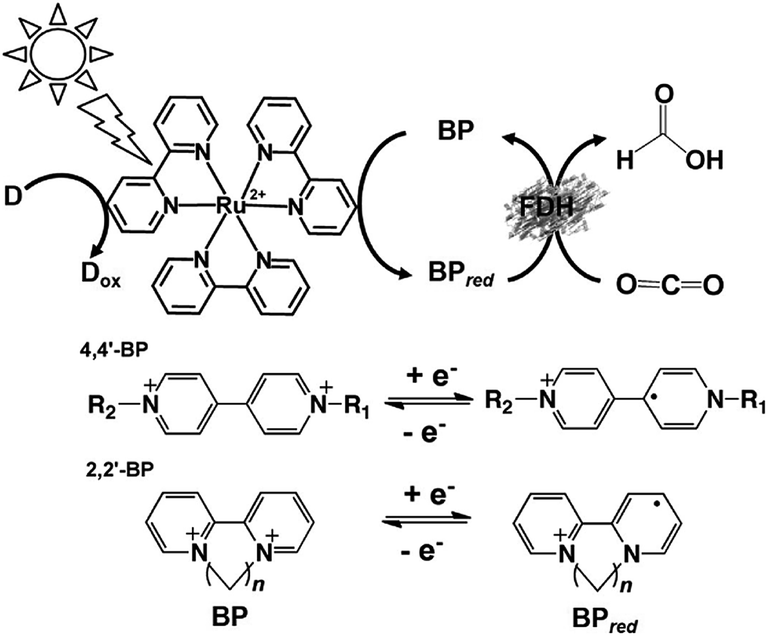 | ||
| Fig. 19 Scheme of the visible-light driven redox system for the reduction of CO2 to formate consisting of an electron donor (D), Ru(bpy)32+ as the photocatalytic dye, BP and FDH. The redox processes of 4,4′- and 2,2′-BPs are also indicated. | ||
In that report, DM, TB, QB and MV were used as an electron mediator and the single-electron reduced BPs acted as the co-enzyme for FDH. The typical reaction sample consisted of RSH (10 mM), Ru(bpy)32+ (30 μM), various BPs (1.0 mM) and FDH (0.89 units) in CO2-saturated buffer solution. The rate of formate production using DM, TB, QB and MV as the electron mediator were estimated to be 0.21, 0.60, 0.30 and 0.75 mM h−1, respectively. By using TB or MV as an electron mediator, an effective visible-light driven system for the reduction of CO2 to formate with FDH was developed.
Furthermore, the effects of the chemical structure and redox potentials of various 2,2′-BPs on the visible-light driven reduction of CO2 to formate with Ru(bpy)32+ and FDH have been reported.148 The typical reaction sample consisted of triethanolamine TEOA (0.5 M), Ru(bpy)32+ (0.5 mM), various 2,2′-BPs (3.0 mM) and FDH (8 mg) in CO2-saturated buffer solution. The concentrations of formate produced after 7 h irradiation using DB, TB, QB and MV as the electron mediator were estimated to be 0.50, 0.95, 0.40 and 0.50 mM, respectively. This study showed that compared to systems using other 2,2′-BPs, the most effective visible-light driven CO2 reduction to formate with Ru(bpy)32+ and FDH is achieved by using TB.
The visible-light driven reduction of CO2 to formate using the water-soluble zinc porphyrin zinc tetraphenylporphyrin tetrasulfonate (ZnTPPS) in place of Ru(bpy)32+ as the photocatalytic dye has also been developed, as shown in Fig. 20.149
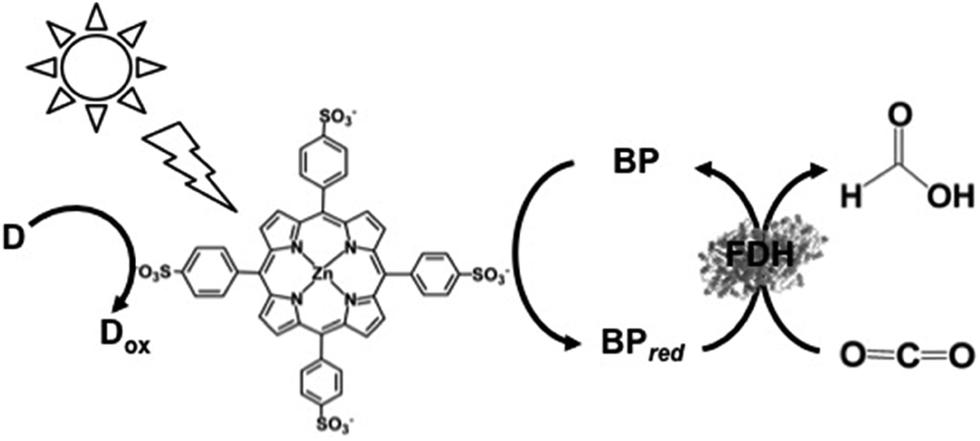 | ||
| Fig. 20 Scheme of the visible-light driven redox system for the reduction of CO2 to formate consisting of an electron donor (D), ZnTPPS as the photocatalytic dye, BP and FDH. | ||
The typical reaction sample consisted of TEOA (0.3 M), ZnTPPS (10 μM), 2,2′-BPs (DM, DB, TB, QB and MV) (0.1 mM) and FDH (9.3 μM) in CO2-saturated buffer solution. The rate of formate production using DM, DB, TB, QB and MV as the electron mediator were estimated to be 18.0, 35.0, 23.5, 13.5 and 22.0 μM h−1, respectively. By using DB as an electron mediator, the effective visible-light driven reduction of CO2 to formate was developed using ZnTPPS.
Table 5 shows a summary of the rate of formate production in the visible-light driven redox system composed of an electron donor, a photocatalytic dye, 2,2′-BPs and FDH.
Here, the effects of the chemical structure based on the dihedral angles and redox potentials of 2,2′-BPs on the visible-light driven reduction of CO2 to formate with photocatalytic dye and FDH are discussed. The redox potentials (E1/2) for DB, TB, QB and DM were estimated to be −0.55, −0.72, −0.82 and −0.89 V (vs. Ag/AgCl), respectively. The dihedral angles of the single-electron reduced DB, TB, QB and DM calculated by the molecular mechanics method were estimated to be about 21, 37, 45, and 55°, respectively. From the data, 2,2′-BPs with a higher redox potential and small dihedral angle between pyridine-rings are suitable electron mediators for the visible-light driven reduction of CO2 to formate with a photocatalytic dye and FDH.
In the visible-light driven reduction of CO2 to formate with the photocatalytic dye, 2,2′-BPs and FDH, the single-electron reduced 2,2′-BPs act as a co-enzyme for FDH instead of NAD(P)H. As mentioned above, the total yield of CO2 reduction to formate in the system consisting of an electron donor, a photocatalytic dye, 2,2′-BPs, and FDH depends on the relative magnitude of the redox potentials of the 2,2′-BPs. Since the reduction efficiency of each 2,2′-BP using the visible-light sensitization of a photocatalytic dye is not the same, the direct interaction between the single-electron reduced 2,2′-BPs and FDH is unclear. The kinetic parameters of the single-electron reduced 2,2′-BPs for the reduction of CO2 to formate with FDH using an enzymatic kinetic analysis were recently reported for the first time.150,151 For the enzymatic kinetic analysis, sodium dithionate was used to chemically prepare the single-electron reduced 2,2′-BPs. It is confirmed that no CO2 reduction proceeds with only sodium dithionate (in the absence of FDH or 2,2′-BPs). Considering the affinity of the single-electron reduced 2,2′-BPs for FDH, the Km values of the single-electron reduced DB, TB, QB and DM in the CO2 reduction to formate with FDH were estimated to be 78.4, 142, 165 and 220 μM, respectively. The Km values of the single-electron reduced 2,2′-BPs for FDH also depend on the dihedral angle of the pyridine rings of the reduced form 2,2′-BPs. Since the dihedral angle of the pyridine rings in the reduced 2,2′-BPs become smaller, the Km values become smaller. The single-electron reduced 2,2′-BP with a lower Km value, DB or TB, tends to interact with FDH at a lower concentration than that of QB and DM. The Km values for FDH, the dihedral angle and redox potential of the reduced form of the 2,2′-BPs are listed in Table 6.
| 2,2′-BP | K m (μM) | Dihedral angle (°) | Redox potential (V) | Reference |
|---|---|---|---|---|
| DB | 78.4 | 21 | −0.55 | 150 and 151 |
| TB | 142 | 37 | −0.72 | 151 |
| QB | 165 | 45 | −0.82 | 151 |
| DM | 220 | 55 | −0.89 | 150 and 151 |
Comparing the Km value and the formate production rate in the visible-light driven reduction of CO2 with a photocatalytic dye and FDH, efficient CO2 reduction is achieved by using a 2,2′-BPs with a lower Km value. Since the single-electron reduced DB with a small dihedral angle easily interacts with the binding site of FDH due to small steric hindrance, effective CO2 reduction with FDH is accomplished using other single-electron reduced 2,2′-BPs.
Various 4,4′-BPs are widely used as electron mediators as well as 2,2′-BPs in the visible-light driven reduction of CO2 to formate with a photocatalytic dye and FDH. As mentioned above, the visible-light driven reduction of CO2 to formate with Ru(bpy)32+ and FDH using MV as an electron mediator was reported in the 1980s. Subsequently, the visible-light driven reduction of CO2 to formate with Ru(bpy)32+ and FDH using various 4,4′-BPs (PV, BV and BisEV) as an electron mediator was reported. The typical reaction sample consisted of triethanolamine TEOA (0.5 M), Ru(bpy)32+ (0.5 mM), various 4,4′-BPs (3.0 mM) and FDH (8 mg) in CO2-saturated buffer solution. The concentrations of formate produced after 7 h irradiation using PV, BV, and BisEV as the electron mediator were estimated to be 0.20, 0.40 and 0.40 mM, respectively.
The first example using a water-soluble zinc porphyrin as a photocatalytic dye as well as Ru(bpy)32+, the visible-light driven reduction of CO2 to formate involving zinc tetrakis(4-methylpyridiyl)porphyrin (ZnTMPyP) MV, TEOA and FDH was also recently reported, as shown in Fig. 21.152–154
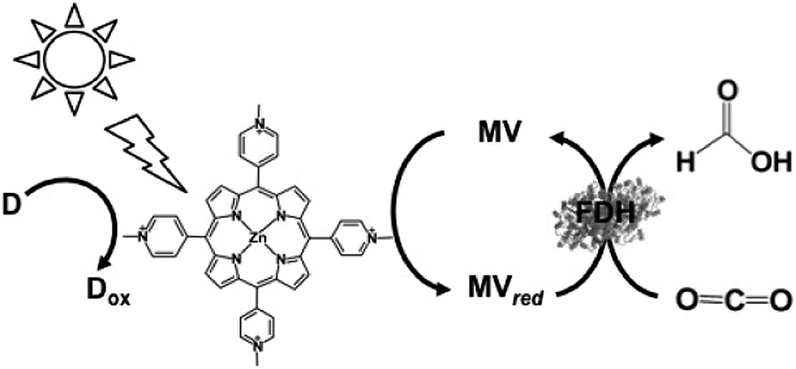 | ||
| Fig. 21 Scheme of the visible-light driven redox system for the reduction of CO2 to formate consisting of an electron donor (D), ZnTMPyP, MV and FDH. | ||
The typical reaction sample consisted of TEOA (0.3 M), ZnTMPyP (9.0 μM), MV (15 mM), and FDH in CO2 saturated buffer solution. For the effective reduction of CO2 to formate, units of FDH in solution were varied between 2.5 and 30 units in the report. It was observed that formate production increased with FDH activity up to 20 units and then decreased. The concentration of formate produced under the optimum conditions was estimated to be 60 μM after 3 h irradiation. Furthermore, the turnover number of MV in this system was estimated to be 0.03 min−1.
ZnTMPyP is an excellent photocatalytic dye, but its stability is poor for continuous visible-light irradiation. As mentioned above, in contrast, ZnTPPS is an excellent photocatalytic dye with photostability and is widely used in visible-light driven redox systems, as shown in Fig. 21. The visible-light driven reduction of CO2 to formate was performed with ZnTPPS and FDH using various 4,4′-BPs as electron mediators (Fig. 19).
The effect of the ionic-group of 4,4′-BPs as the electron mediator on the visible-light driven reduction of CO2 to formate with the system consisting of ZnTPPS and FDH in the presence of TEOA was studied.152,153 1,1′-Diaminoethyl- (DAV), 1-aminoethyl-1′-methyl- (AMV), 1-carboxymethyl-1′-methyl- (CMV) and 1,1′-dicarboxymethyl-4,4′-bipyridinium salt (DCV) were synthesized as 4,4′-BPs with an ionic-group. The typical reaction sample consisted of TEOA (0.3 M), ZnTPPS (10 μM), 4,4′-BPs (DAV, AMV, CMV and DCV) (0.1 mM) and FDH (9.3 μM) in CO2-saturated buffer solution. The rate of formate produced using DAV, AMV, CMV and DCV as the electron mediator was estimated to be 120, 100, 50.0 and 40.0 μM h−1, respectively. By using 4,4′-BPs with a cationic amino-group, DAV or AMV, as the electron mediator, the effective visible-light driven reduction of CO2 to formate was observed compared with the 4,4′-BPs with an anionic carboxy-group, CMV and DCV. The formate production rate with DAV is about 3.2 times larger than that of the system with DCV. The single-electron reduced DAV and MAV were used as effective electron mediators for FDH in the visible-light driven reduction of CO2 to formate.
Since the carbamoyl group of NAD+ is captured at the active site of FDH via a hydrogen bond, the single-electron reduced 4,4′-BPs with a carbamoyl group are an effective co-enzyme of FDH in the reduction of CO2 to formate.157 1,1′-Dicarbamoylmethyl-4,4′-bipyridinium salt (DCarV) and 1-carbamoylmethyl-1′-methyl-4,4′-bipyridinium salt (MCarV) as 4,4′-BPs with a carbamoyl group were synthesized to improve the affinity for FDH. The typical reaction sample consisted of TEOA (0.3 M), ZnTPPS (10 μM), 4,4′-BPs with a carbamoyl-group (DCarV and MCarV) (0.1 mM) and FDH (9.3 μM) in CO2-saturated buffer solution. The rate of formate produced using DCarV and MCarV as the electron mediator was estimated to be 95.0 and 120 μM h−1, respectively. By using MCarV, the efficiency of the visible-light driven reduction of CO2 to formate with the system consisting of ZnTPPS and FDH in the presence of TEOA was improved by about 60% compared to that using MV. From these results, the carbamoyl group in 4,4′-BP affects the electron transfer, leading an improvement in CO2 reduction to formic acid with FDH.
Table 7 shows a summary of the rate of formate production in the visible-light driven redox system consisting of an electron donor, a photocatalytic dye, 4,4′-BPs and FDH.
| Photocatalytic dye | Electron donor | 4,4′-BPs | Rate of formate production (μM h−1) | Reference |
|---|---|---|---|---|
| Ru(bpy)32+ | TEOA | MV | 71.4 | 148 |
| PV | 28.6 | 148 | ||
| BV | 57.1 | 148 | ||
| BisEV | 57.1 | 148 | ||
| ZnTMPyP | TEOA | MV | 20.0 | 152–154 |
| ZnTPPS | TEOA | MV | 60.5 | 155 and 156 |
| DAV | 120 | 155 and 156 | ||
| MAV | 100 | 156 | ||
| MCV | 50.0 | 156 | ||
| DCV | 40.0 | 156 | ||
| DCarV | 95.0 | 157 | ||
| MCarV | 120 | 157 |
The direct interaction between the single-electron reduced 4,4′-BPs and FDH is unclear. The kinetic parameters of the single-electron reduced 4,4′-BPs for the reduction of CO2 to formate with FDH by using an enzymatic kinetic analysis were also reported recently for the first time.158,159 Considering the affinity of the single-electron reduced 4,4′-BPs for FDH, the Km values of the single-electron reduced DAV, MAV, MV, MCV and DCV in the reduction of CO2 to formate with FDH were estimated to be 17.0, 118, 212, 292 and 370 μM, respectively. The Km values of the single-electron reduced 4,4′-BPs for FDH also depend on the number of positive charges in the reduced form 4,4′-BPs. The number of electron charges in the single-electron reduced 4,4′-BPs are DAV (+3), MAV (+2), MV (+1), MCV (0) and DCV (−1).
The formate production is proportional to the number of electron charges in the single-electron reduced 4,4′-BPs. These results indicate that the formate production in the reaction depends on the ionic-substituted groups in the single-electron reduced 4,4′-BPs. The Km values for FDH, and number of electron charges of the reduced form 4,4′-BPs are listed in Table 8.
| 4,4′-BPs | K m (μM) | Number of electron charges of reduced form 4,4′-BPs | Reference |
|---|---|---|---|
| DAV | 17.0 | 3 | 158 |
| MAV | 118 | 2 | 158 and 159 |
| MV | 212 | 1 | 158 and 159 |
| MCV | 292 | 0 | 159 |
| DCV | 370 | −1 | 159 |
Comparing the Km value and the formate production rate in the visible-light driven CO2 reduction with ZnTPPS and FDH, efficient CO2 reduction is achieved by using a DAV or MAV with a lower Km value.
More notably, oxidized BPs do not act as a co-enzyme for FDH in the oxidation of formate to CO2. That is, only single-electron reduced BPs act as a co-enzyme for FDH in the reduction of CO2 to formate. This property of BPs is not found in the natural co-enzymes NAD+ and NADH.
As described above, by using various BPs, the rate of CO2 reduction to formate can be controlled without changing the structure of FDH.
In addition, the visible-light driven reduction of CO2 to formate with the system consisting of chlorophyll-a (MgChl-a) as the photocatalytic dye, MV and FDH in the presence of an electron donor is shown in Fig. 22.160
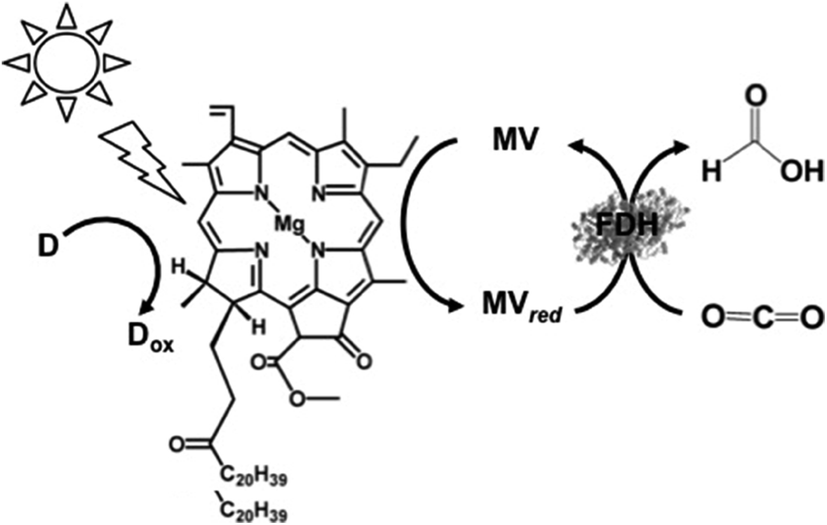 | ||
| Fig. 22 Scheme of the visible-light driven redox system for CO2 reduction to formate consisting of an electron donor (D), chlorophyll-a (MgChl-a), MV and FDH. | ||
Since the visible-light driven redox system for the reduction of CO2 to formate consists of an electron donor (D), a photocatalytic dye, BP and FDH, methanol production can be realized by adding AldDH and ADH in this system, as shown in Fig. 23. By using this system, ZnTPPS or metal-free TPPS (H2TPPS) can be used as the photocatalytic dye. Also, the visible-light driven reduction of CO2 to methanol with the system of TEOA, ZnTPPS, MV, FDH, AldDH, and ADH was reported recently for the first time.161–163
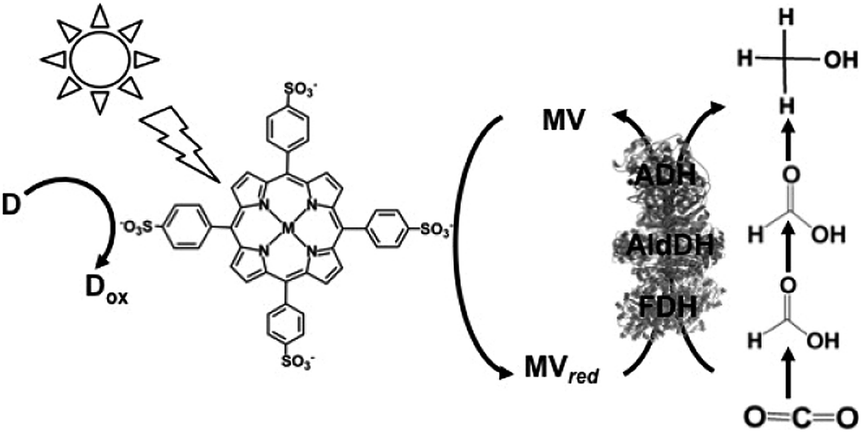 | ||
| Fig. 23 Scheme of the visible-light driven redox system for CO2 reduction to methanol consisting of an electron donor (D), MTPPS (M: Zn or H2), MV, FDH, AldDH and ADH. | ||
The typical sample solution consisted of TEOA (0.3 M), ZnTPPS (0.1 μM), MV (0.1 mM), FDH (12.5 units), AldDH (12.5 units), ADH (12.5 units) and sodium bicarbonate (NaHCO3) instead of CO2 in the buffer solution. When the sample solution containing ZnTPPS, MV, TEOA, FDH, AldDH, ADH and NaHCO3 was irradiated, the concentration of methanol produced was detected to be 4.5 μM after 3 h irradiation. The conversion yield of HCO3− to methanol was estimated to be 4.5% after 3 h irradiation (initial concentration of HCO3− 100 μM). In this system, the single-electron reduced MV acts as a shared co-enzyme for the three dehydrogenases FDH, AldDH and ADH.
However, the yield of CO2 reduction to methanol with this system is low. Thus, to improve the efficiency of CO2 reduction to methanol using this system, it is necessary to clarify the interaction between FDH, AldDH or ADH and the single-electron reduced MV. The kinetic parameters for the reduction of formaldehyde to methanol with ADH and the single-electron reduced MV were determined using enzymatic kinetic analysis. The Km value of the single-electron reduced MV for ADH in the reduction of formaldehyde to methanol was estimated to be 312 μM. The Km value of the single-electron reduced MV for ADH is almost the same value for FDH, which indicates that the affinity of the single-electron reduced MV for ADH is equal to that for FDH.163
Based on this result, an improvement in the reduction of CO2 to methanol under visible-light irradiation was attempted with a system consisting of TEOA, H2TPPS, MV, FDH, AldDH and ADH. The three dehydrogenases were adjusted to the same concentration (not same units). When the sample solution consisting of H2TPPS (100 μM), MV (2.0 mM), TEOA (0.3 M), FDH (2.0 μM), AldDH (2.0 μM) and ADH (2.0 μM) in CO2 saturated 50 mM sodium pyrophosphate buffer was irradiated, the production of methanol was observed with an increase in irradiation time and the methanol concentration produced was estimated to be 6.8 μM after 100 min irradiation. Further improvement in the efficiency of the visible-light driven reduction of CO2 to methanol is desirable in the system consisting of TEOA, H2TPPS, MV, FDH, AldDH and ADH.
In addition, the light driven reduction of CO2 to methanol using methanol dehydrogenase (MDH) from Bacillus (EC 1.1.1.244), ZnS as the photocatalytic dye and pyrroloquinoline quinone (PQQ)164 as the electron mediator has also been reported, as shown in Fig. 24. The redox process of PQQ165–169 also is indicated in Fig. 24.
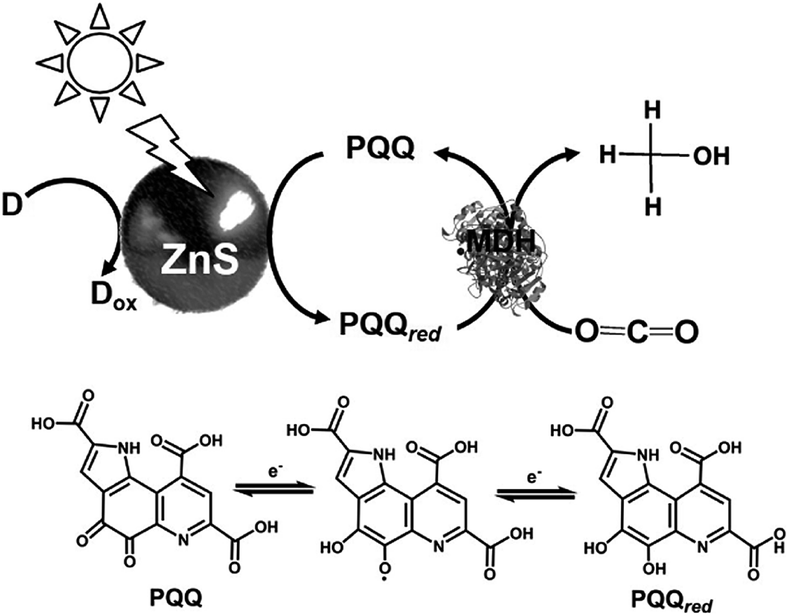 | ||
| Fig. 24 Scheme of the light-driven redox system for the reduction of CO2 to methanol consisting of an electron donor (D), ZnS, PQQ and MDH. | ||
In this system, 2-propanol is used as the electron donor. The typical sample solution consisted of CO2-saturated ZnS microcrystalline colloid containing PQQ (5 mM) and MDH (2 units). When the sample containing ZnS, PQQ and MDH in CO2 saturated solution was irradiated with UV-light, formate and methanol production was observed. A saturation tendency of the methanol production appeared when the amount of methanol accumulated to ca. 0.25 mM after 1 h irradiation. On the other hand, the concentration of formate increased with an increase in irradiation time. After 3 h irradiation, 0.50 mM of formate was produced with this system.
The proposed mechanism of the light-driven reduction of CO2 to methanol with the system consisting of an electron donor, ZnS, PQQ and MDH is shown in Fig. 25. In this system, there are two photoredox processes: (1) reduction of CO2 to formate with the photocatalytic reaction of ZnS and (2) reduction of formate to methanol with ZnS via the PQQ/PQQred redox couple. Thus, rate of CO2 reduction to formate with the photocatalytic reaction of ZnS is higher than that of formate reduction to methanol.
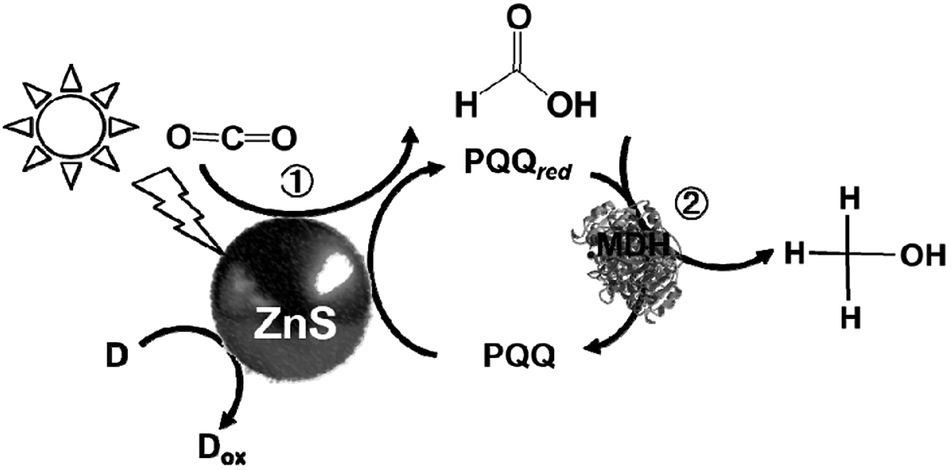 | ||
| Fig. 25 Proposed mechanism for the light-driven reduction of CO2 to methanol involving of an electron donor (D), ZnS, PQQ and MDH. | ||
Next, the light-driven reduction of CO2 to CO with systems consisting of a photocatalytic dye and carbon monoxide dehydrogenase (CODH) from Carboxydothermus hydrogenoformans (EC1.2.7.4) are introduced. By using CODH in the light-driven reduction of CO2 to CO with a photocatalytic dye, an electron mediator such as MV is unnecessary.
The typical light-driven reduction of CO2 to CO with the system consisting of a photocatalytic dye and CODH is shown in Fig. 26.170–173
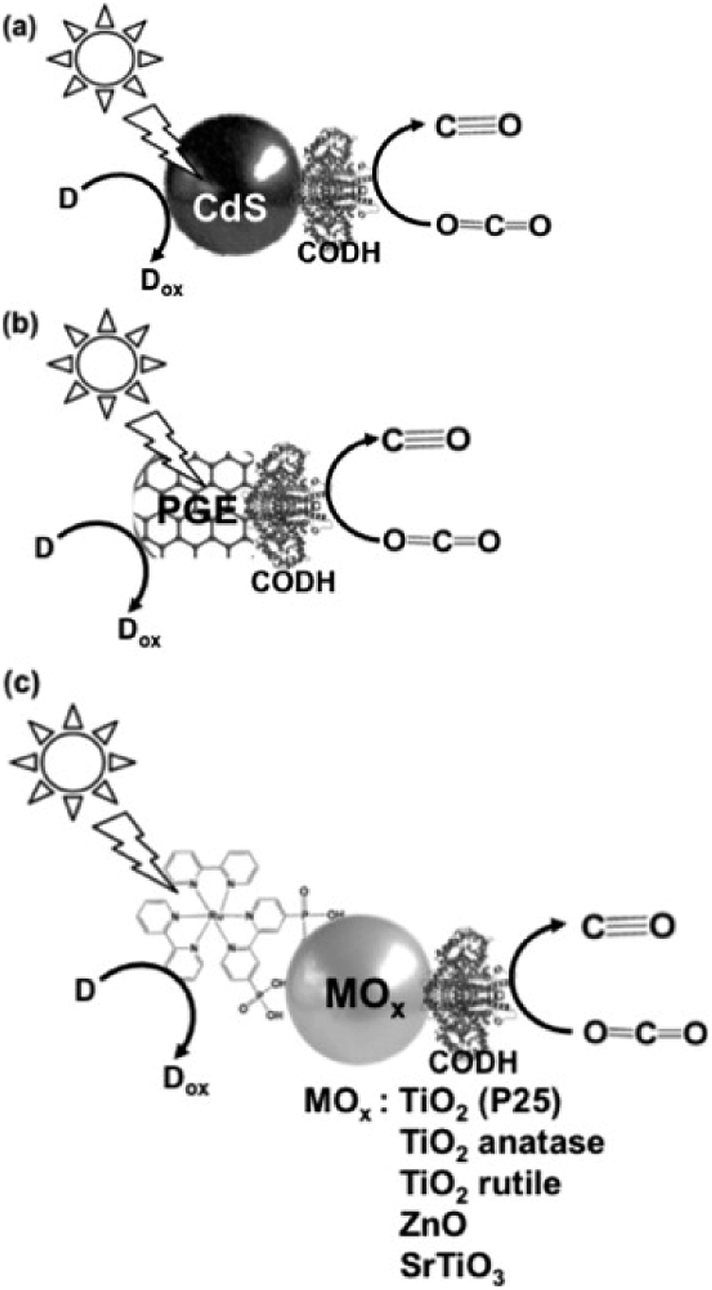 | ||
| Fig. 26 Scheme of the light-driven redox system for the reduction of CO2 to CO consisting of an electron donor (D), a photocatalytic dye (a) CdS, (b) pyrolytic graphite “edge” (PGE), and (c) dye-modified metal oxide and CODH. | ||
The light-driven reduction of CO2 to CO with systems consisting of a photocatalytic dye and CODH are classified into three categories based on the photocatalytic dye: (a) CdS,172 (b) pyrolytic graphite “edge” (PGE)173 and (c) dye-modified metal oxide (TiO2, ZnO or SrTiO3).170,171
The effect of an electron donor on the light-driven reduction of CO2 to CO with the system consisting of CdS and CODH has been investigated. The typical reaction sample consists of CODH immobilized CdS based quantum dots (CODH-QDs) and various electron donors (ascorbate, EDTA and TEOA: 0.2 M and KI: 0.3 M) in CO2 saturated 0.35 M 2-(N-morpholino)ethanesulfonic acid buffer. By using EDTA as the electron donor, 3.0 μmol of CO was evolved in the light-driven redox system with CODH-QDs after 5 h irradiation. In contrast, by using TEOA, 0.5 μmol of CO was evolved after 5 h irradiation. Moreover, no CO evolution was observed in the system using ascorbate or KI.
By using CODH and [Ru(bpy)2(4,4′-(PO3H2)2-bpy)]Br2 (RuP) modified TiO2 (P25) as the photocatalytic dye (RuP-TiO2–CODH) at pH 6 and 20 °C, the visible-light driven reduction of CO2 to CO proceeded and 5 μmol of CO was produced after 4 h irradiation. At the reaction temperature of 50 °C, 12 μmol of CO was produced in this system after 4 h irradiation. By using CODH and [Ru(bpy)2(4,4′-(PO3H2)2-bpy)]Br2 (RuP) modified TiO2 (anatase), TiO2 (rutile), ZnO and SrTiO3, 4.3, 0, 0.55 and 0.5 μmol of CO was produced after 4 h irradiation, respectively.
Also, using CODH immobilized PGE as the photocatalytic dye, only the photoelectrochemical properties of CO2 reduction to CO were investigated.173
Photoredox systems for light-driven CO2 utilization
In this section, we focus on light-driven redox systems with biocatalysts for the formation of carbon–carbon bonds from CO2 as the carbon feedstock. As mentioned above, ME and IDH are attractive biocatalysts for this target, and CO2 is bonded to organic molecules as a carboxy group.174
A visible-light driven redox system with ME that exploits the reduction of NADP+ to NADPH as the electron was described recently for the first time. In this case, the use of Ru(bpy)32+, MV, FNR and 2-mercaptoethanol led to malate production from HCO3− and pyruvate, as shown in Fig. 27.145,175
 | ||
| Fig. 27 Scheme of the visible-light driven redox system for malate production from pyruvate and CO2 consisting of Ru(bpy)32+, MV, FNR, NADP+, ME and 2-mercaptoethanol. | ||
In this system, the single-electron reduced MV does not act as a co-enzyme for ME in the reaction of malate production from CO2 and pyruvate. Thus, the redox system of NADP+/NADPH with FNR is needed in the system.
FNR catalyzes the reduction of NADP+ to NADPH by coupling with ferredoxin, as shown in Fig. 12. Moreover, FNR also catalyzes the reduction of NADP+ to NADPH by coupling with MV and the single-electron reduced MV (MVred), as shown in Fig. 28.180
 | ||
| Fig. 28 Reaction scheme of FNR for NADP+ reduction to NADPH in the presence of the single-electron reduced MV (MVred). | ||
This system was composed of Tris buffer (4.2 mL; 0.2 M; pH 8.0) containing pyruvic acid (470 mM), MV (0.19 mM), NADP+ (0.18 mM), Ru(bpy)32+ (21 μM), MgCl2, (9.5 μM), NaHCO3 (0.2 M) and 2-mercaptoethanol (19 mM). Then FNR (0.2 units) and ME (1.33 units) were added to the above solution. After 2 h irradiation to this system, 3.0 mM of malate was produced and the yield of malate produced from HCO3− and pyruvate was estimated to be 3.6% after 2 h irradiation.
IDH biocatalyst coupled with a visible-light driven redox system consisting of NADP+, Ru(bpy)32+, MV, FNR and DL-dithiothreitol (DTT) as an electron donor for isocitrate production from HCO3− and α-oxoglutarate was also reported, as shown in Fig. 29.145,180
 | ||
| Fig. 29 Scheme of the visible-light driven redox system for isocitrate production from α-oxoglutarate and CO2 consisting of Ru(bpy)32+, MV, FNR, NADP+, IDH and DTT. | ||
This system was composed of Tris buffer (4.2 mL; 0.2 M; pH 7.2) containing Ru(bpy)32+ (14 μM), MV (0.17 mM), NADP+ (0.17 mM), MnCl2, (1.7 mM), NaHCO3 (0.17 M), α-oxoglutaric acid (42 mM) and DTT (8.3 mM). Then the biocatalysts FNR (0.2 units) and IDH (0.47 units) immobilized on poly(acrylamide-co-N-acryloxysuccinimide) were added to the above solution. After 2 h irradiation to this system, 0.6 mM of isocitrate was produced and yield for isocitrate production from HCO3− and α-oxoglutarate was estimated to be 1.9% after 2 h irradiation.
A different approach involving ME and the reduction of NADP+ with a system containing TiO2 semiconductor or CdS photocatalyst, MV, FNR and lactate or 2-mercaptoethanol as the electron donor for malate production from CO2 and pyruvate acid was reported, as shown in Fig. 30.181
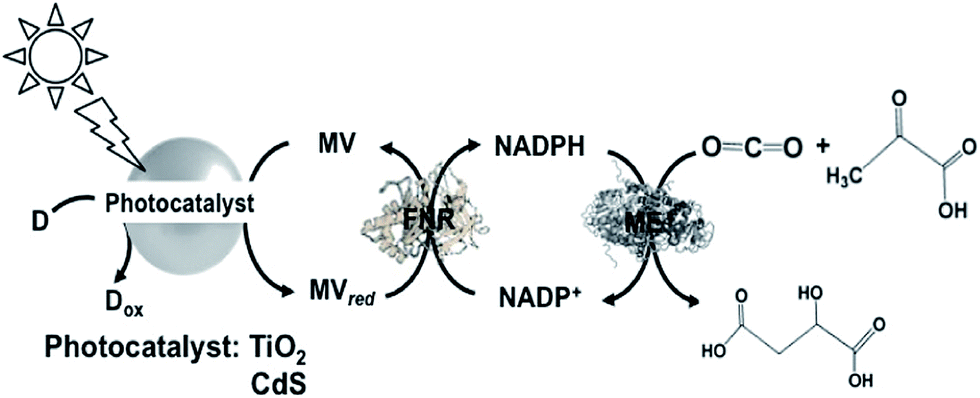 | ||
| Fig. 30 Scheme of the light-driven redox system for malate production from pyruvate and CO2 consisting of a photocatalyst (TiO2 or CdS), MV, FNR, NADP+, ME and lactate or 2-mercaptoethanol. | ||
When TiO2 was used as the photocatalyst, the reaction solution consisted of 10 mL of CO2-saturated Tris buffer containing MV (1.0 mM), NADP+ (0.1 mM), FNR (0.2 unit), ME (1.0 unit), pyruvic acid (2.0 mM), 2-mercaptoethanol (27 mM) and TiO2. When this solution was irradiated with UV light, 0.3 mM of malate was produced after 5 h irradiation. In contrast, when CdS was used as the photocatalyst, 1.0 mM of malate was produced after 5 h irradiation under the same conditions. It is thought that under the optimum conditions, ca. 50% of pyruvate is converted to malate using CdS and ca. 15% using TiO2 semiconductor.
Since the oxidation of lactate to pyruvate proceeds with the photocatalytic function of CdS, the light-driven production of malate from CO2 and lactate via pyruvate with the system of CdS and ME was realized. The sample solution contained ME (5.0 units) in 10 cm3 of 10 mL of CO2-saturated Tris buffer containing MV (1.0 mM), NADP+ (0.1 mM), FNR (0.2 unit), NADP+ (0.1 mM), sodium lactate (100 mM) and CdS (1.25 mM). After 6 h irradiation, the concentrations of malate and pyruvate were determined to be 0.2 and 0.7 mM, respectively. This system achieved the production of malate from CO2 and pyruvate as an intermediate produced from lactate (electron donor) oxidation with CdS.
A visible-light driven catalytic system consisting of NADH as the electron donor, ZnChl-e6 (Chlorin-e6, formed by the hydrolysis of chlorophyll-a), MV, FNR, NADP+ and ME was reported, as shown in Fig. 31.182,183
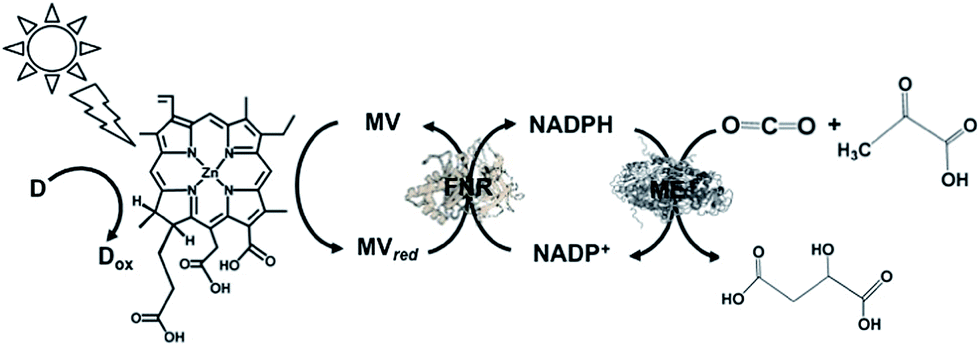 | ||
| Fig. 31 Scheme of the visible-light driven redox system for malate production from pyruvate and CO2 consisting of ZnChl-e6, MV, FNR, NADP+, ME and NADH. | ||
The sample solution was composed of NADH (30 mM), Zn Chl-e6 (50 μM), MV (1.0 mM), FNR (4.0 units), NADP+ (10 mM), pyruvic acid (10 mM), NaHCO3 (10 mM) and ME (4.5 units) in Bis–Tris buffer (pH 8.0). After 3 h irradiation, the concentration of malate produced was 0.65 mM and the ratio of pyruvate to malate was estimated to be about 6.5%.
These results show that malate production proceeds via the combination of NADP+ reduction to NADPH using the photocatalytic dye with FNR via the MV/MVred redox couple and biocatalytic conversion of pyruvate and CO2 to malate. To simplify this system, NADP+ reduction to NADPH with the system of chemically-modified Chl-a and FNR without the MV/MVred redox couple was attempted.
A visible-light driven redox system with ME and reduction of NADP+ with a system containing ascorbate as the electron donor, polyethylene glycol modified chlorophyll-a (PEG-Chl-a) and FNR, as shown in Fig. 32, was exploited for the production of malate from CO2 and pyruvate.184 Since NADP+ is directly reduced to NADPH with FNR and PEG-Chl-a in this reaction, it is not necessary to add MV as an electron mediator.
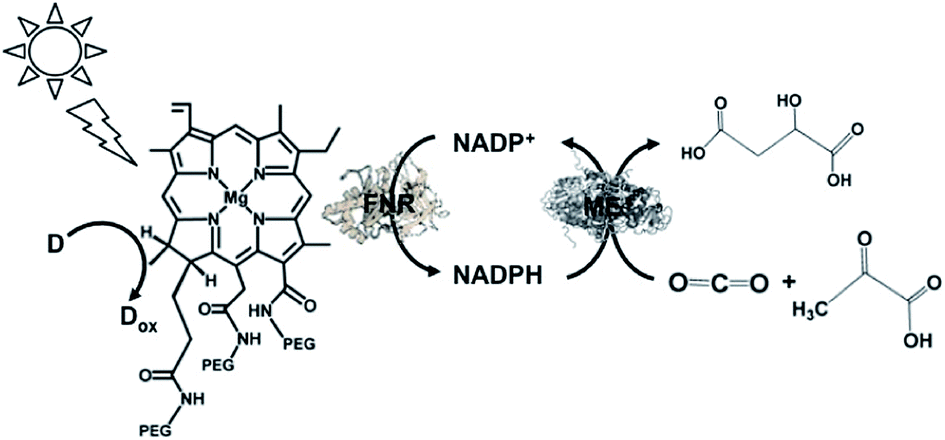 | ||
| Fig. 32 Scheme of the visible-light driven redox system for malate production from pyruvate and CO2 consisting of PEG-Chl-a, FNR, NADP+, ME and ascorbate. | ||
The sample solution was composed of the PEG-chlorophyllin conjugate (15 μM), ascorbate (6.0 mM), NaHCO3 (180 mM), MgCl2 (15 mM), pyruvate (0.8 mM), NADP+ (3.2 mM), FNR (2.5 units) and ME (5.0 units) in 10 mL of 50 mM phosphate buffer at pH 7.4. The concentration of malate produced was estimated to be 75 μM after 3 h visible light irradiation.
The yield of malate from pyruvate and HCO3− was estimated to be 0.041%.
Photoredox systems for light-driven CO2 utilization with biocatalysts via the bipyridinium salt redox couple
The light-driven redox system for the formation of carbon–carbon bonds from CO2 and organic molecules with a photocatalytic dye and biocatalyst using the NADP+/NADPH redox couple is very complicated; however, it is necessary to simplify it with an electron mediator instead of NADP+ reduction with FNR, as shown in Fig. 33.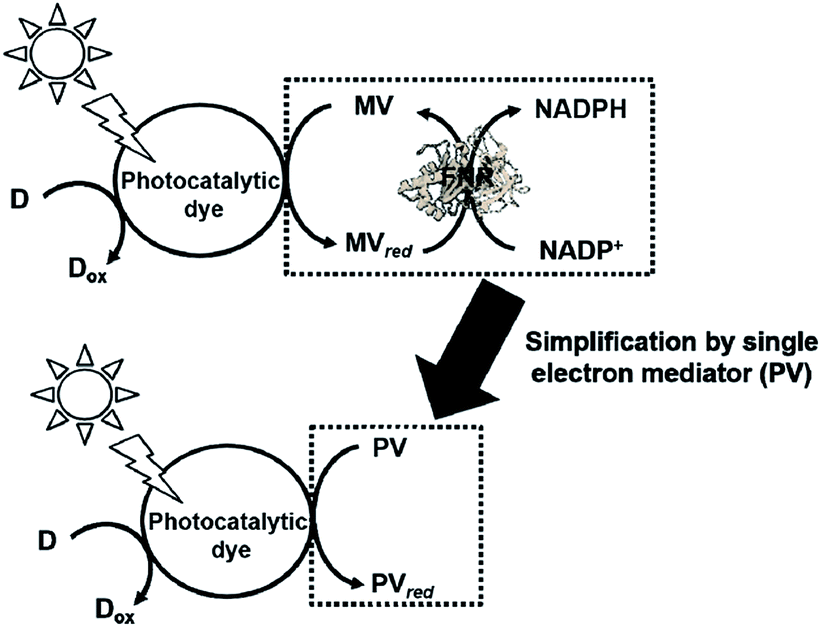 | ||
| Fig. 33 Scheme of the light driven redox system for NADP+ reduction to NADPH with the system of an electron donor, a photocatalytic dye, MV, and FNR (upper) and simplified system using a single electron mediator (lower). | ||
To develop a simplified visible-light driven redox system for the formation of carbon–carbon bonds from CO2 and organic molecules using the system of a photocatalytic dye and biocatalyst without the NADP+/NADPH redox couple, an electron mediator that shows the same behavior as NADP+ or NADPH is necessary. 1,1′-Diphenyl-4,4′-bipyridinium salt (phenylviologen) derivatives (PVs) have received much attention as electron mediators with the same behavior as NADP+ or NADPH because PVs have lower first and second reduction potentials compared with 2,2′- or 4,4′-BPs.185 The chemical structures of 1,1′-diphenyl-4,4′-bipyridinium salt derivatives (PVs) are shown in Fig. 34.
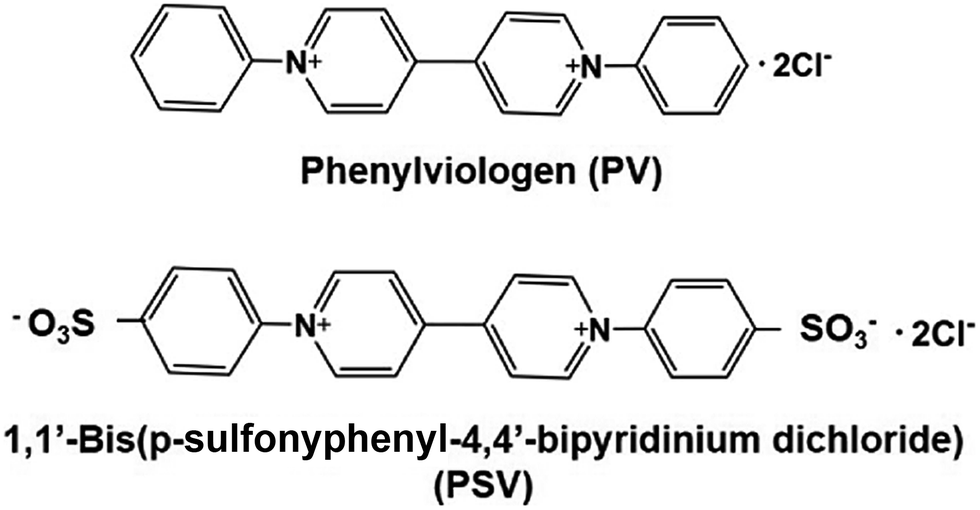 | ||
| Fig. 34 Chemical structures of 1,1′-diphenyl-4,4′-bipyridinium salt (PV) and 1,1′-bis (p-sulfonyphenyl)-4,4′-bipyridinium dichloride (PSV). | ||
A visible-light driven redox system with ME that exploits the reduction of PV as the electron mediator was recently described for the first time. In this case, the use of MTPPS (M: Zn and H2), PV and TEOA led to malate production from CO2 and pyruvate, as shown in Fig. 35.186–188 The redox process of PV is also indicated in Fig. 35.
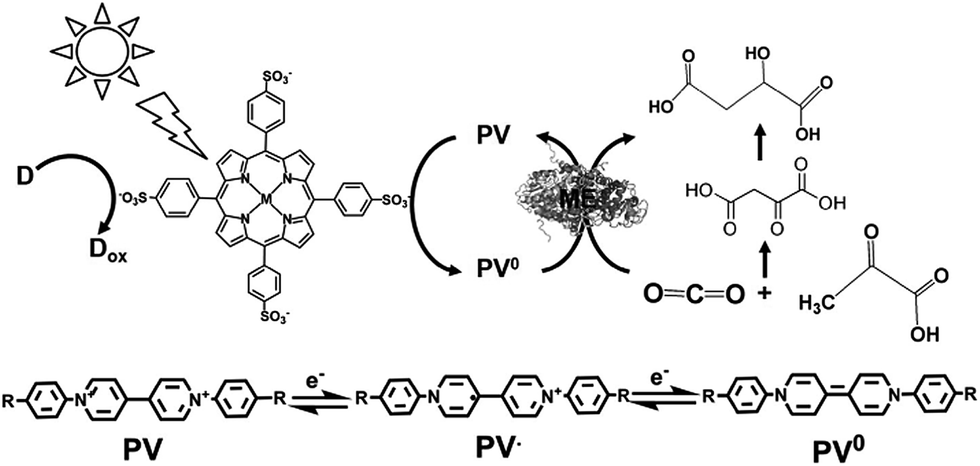 | ||
| Fig. 35 Scheme of the visible-light driven redox system for malate production from pyruvate and CO2 consisting of an electron donor, MTPPS, PV and ME. The redox process of PV is also indicated. The single- and double- electron reduced PV are abbreviated as PV˙ and PV0, respectively. | ||
The reduction potentials of PSV and PV are summarized in Table 9. The reduction potentials of MV are also listed as a reference in the table. The single- (Ered1) and double- (Ered2) electron reduction potentials of PVs were determined by cyclic voltammetry using an Ag/AgCl electrode as the reference electrode.
| BPs | Single-electron reduction potential (Ered1) (V) | Double-electron reduction potential (Ered2) (V) | Reference |
|---|---|---|---|
| PSV | −0.38 | −0.72 | 187 and 188 |
| PV | −0.39 | −0.74 | 186 |
| MV | −0.67 | −1.05 | 186–188 |
The single- and double-electron reduction potentials of PV were estimated to be −0.39 and −0.74 V, respectively. On the other hand, the single- and double-electron reduction potentials of PSV were estimated to be −0.38 and −0.72 V, respectively. The reduction potentials of PV and PSV are very close, thus it is considered that the sulfo group has little effect on the reduction potential. The single and double electron reduction potentials of PVs are lower than that of MV (−0.67 and −1.05 V, respectively). Thus, the single and double electron reduced PVs could be easily produced with a photocatalytic dye.
By using PV as an electron mediator, the visible-light driven malate production from pyruvate and CO2 with the system of TEOA, H2TPPS, PV and ME was developed. For PV photoreduction, the sample solution consisted of TEOA (0.3 M), ZnTPPS (10 mM) and PV (0.1 mM) in 5.0 mL of 10 mM bis–Tris buffer (pH 7.4). When this sample solution was irradiated, the absorption band due to the double-electron reduced PV increased with an increase in irradiation time. The redox potentials of the exited triplet state of H2TPPS in aqueous buffer, E(H2TPPS+/3H2TPPS*) and E(3H2TPPS*/H2TPPS), were estimated to be −0.70 and 0.40 V using electrochemical and photochemical measurements, respectively, thus allowing for the production of single- or double-electron reduced PV.
For the visible-light driven production of malate from CO2 and pyruvate with H2TPPS, PV and ME in the presence of co-factor MgCl2, the sample solution was composed of TEOA (0.2 M), H2TPPS (40 μM), PV (0.4 mM), pyruvate (12 mM), and MgCl (10 mM). When this sample solution was irradiated, the concentration of malate and oxaloacetate increased with the irradiation time. The concentration of malate produced was estimated to be 565 μM after 3 h irradiation. On the other hand, the concentration of oxaloacetate was estimated to be 1.0 mM after 3 h irradiation. The yield of pyruvate to malate was calculated to be 4.7%.
Since both the single- and the double-electron reduction states of PV are insoluble in an aqueous solution, PV is not suitable for homogenous visible-light driven redox systems. Thus, the water-soluble PV derivative, PSV is used as an electron mediator in visible-light driven redox systems.
For PSV photoreduction, the sample solution consisted of TEOA (0.3 M), ZnTPPS (10 μM) and PSV (0.1 mM) in 5.0 mL of 10 mM bis–Tris buffer (pH 7.4). When this sample solution was irradiated, the absorption band due to the double-electron reduced PSV also increased with an increase in irradiation time.
For the visible-light driven production of malate from CO2 and pyruvate with TEOA, H2TPPS, PV and ME in the presence of co-factor MgCl2, the sample solution was composed of TEOA (0.2 M), H2TPPS (40 μM), PSV (0.4 mM), pyruvate (12 mM), and MgCl (10 mM). When this sample solution was irradiated, the concentration of malate and oxaloacetate increased with the irradiation time. In the system with PSV, upon visible-light irradiation, the concentration of oxaloacetate increased within 1 h, and then decreased. On the other hand, the concentration of malate increased with a decrease in the concentration of oxaloacetate. Thus, oxaloacetate was formed as an intermediate and then reduced to malate. The malate concentration after 3 h irradiation was estimated to be 604 μM. The yield of pyruvate to malate was calculated to be 5.0%.
Table 10 shows a summary of the visible-light driven production of malate from CO2 and pyruvate with TEOA, H2TPPS, PVs and ME.
| PVs | Malate produced after 1 h irradiation (μM) | Yield of pyruvate to malate | Reference |
|---|---|---|---|
| PV | 565 | 4.7 | 186 |
| PSV | 604 | 5.0 | 187 and 188 |
| MV | 0 | 0 | 186–188 |
From these results, PVs0 is used for the formation of C–C bonds from CO2 and pyruvate to produce oxaloacetate and PSV˙ is used for the reduction of the carbonyl group of oxaloacetate to malate. The possible mechanism in the visible-light driven redox system for malate production based on the formation of C–C bonds from CO2 and pyruvate is shown in Fig. 36.
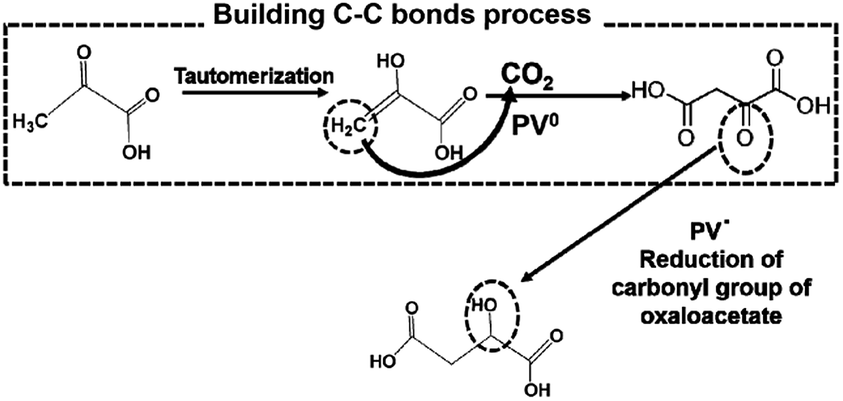 | ||
| Fig. 36 Possible mechanism for the production of malate based on the formation of C–C bonds from CO2 and pyruvate using PV as an electron mediator in the visible-light driven redox system. | ||
Initially, the hydrogen in pyruvate is abstracted by PV0 and then CO2 is bonded to the pyruvate derivative as a carboxy group based on the formation of C–C bonds from CO2, resulting in the production of oxaloacetate. Finally, oxaloacetate is reduced to malate by PV˙.
Therefore, the visible light-driven production of oxaloacetate and malate based on the ME-catalyzed formation of carbon–carbon bonds from pyruvate and CO2via the photoreduction of PV as an electron mediator with water-soluble porphyrin in the presence of TEOA was developed for the first time.
The detailed underlying mechanisms for the production of oxaloacetate from CO2 and pyruvate are based on the formation of carbon–carbon bonds using the double-electron reduced PV produced in the photoredox system.
The light driven production of isocitrate from CO2 and α-oxoglutarate with a photocatalytic dye, CdS and IDH using the MV redox couple without FNR, as shown in Fig. 37, has been reported.189
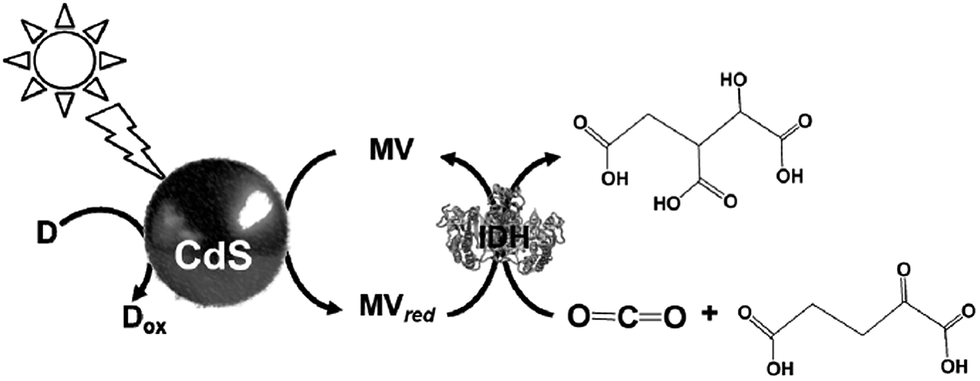 | ||
| Fig. 37 Scheme of the light-driven redox system for the production of isocitrate from CO2 and α-oxoglutarate with a photocatalytic dye, CdS and IDH using the MV redox couple. | ||
The sample solution was composed of 0.2 M Tris buffer containing TEOA (2.0 M), MV (1.0 mM), IDH (0.8 unit), CdS (0.016 mol), and α-oxoglutaric acid (10 mM). The production of isocitrate proceeds linearly with the irradiation time for 8 h and then beyond that decreases gradually, resulting in complete suppression after 20 h. The amount of isocitrate was estimated to be 8.0 μmol after 20 h.
Other photoredox systems for light-driven molecular conversion
Visible-light driven redox systems using photocatalytic dyes and biocatalysts have not only been developed for the reduction and utilization of CO2 but also for molecular conversion. In this section, some studies on visible-light driven redox systems for molecular conversion with a photocatalytic dye and biocatalyst are introduced.The visible-light driven reduction of acetate to ethanol with an electron donor (in this system NADPH), ZnChl-e6, MV, AldDH and ADH was developed, as shown in Fig. 38.190 In this system, acetate is reduced to ethanol via acetaldehyde with AldDH and ADH using the single-electron reduced MV by the visible-light sensitization of ZnChl-e6.
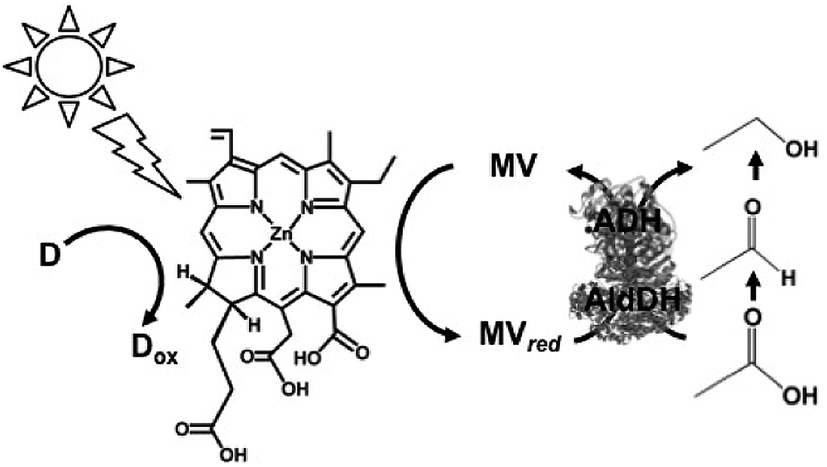 | ||
| Fig. 38 Scheme of the visible-light driven redox system for the reduction of acetate to ethanol with an electron donor, ZnChl-e6, MV, AldDH and ADH. | ||
The sample solution consisted of ZnChl-e6 (100 μM), MV (12 mM), NADPH (3.3 mM), AldDH (0.22 μM), ADH (6.7 nM) and sodium acetate (30 mM) in 50 mM sodium pyrophosphate buffer (pH 7.4). When this sample solution was irradiated, 1.4 mM of ethanol was produced after 150 min irradiation. In this system, the single-electron reduced MV acts as a co-enzyme for AldDH and ADH in the reduction of acetate and acetaldehyde.
Lactate-based polymers (PLAs) were introduced by Watson and have been received much attention as biodegradable polymers and environmentally friendly materials.191 To develop functional PLAs, optically pure lactate is desirable. The biocatalytic reduction of pyruvate to lactate with commercially available lactate dehydrogenase (LDH) from Pig heart (EC 1.1.1.27) with high optical purity and is suitable to obtain the starting material for PLAs. LDH catalyzes the reaction of lactate oxidation to pyruvate in the presence of NAD+ and the reversed reaction of pyruvate reduction to lactate in the presence of NADH, as shown in Fig. 39.182,193
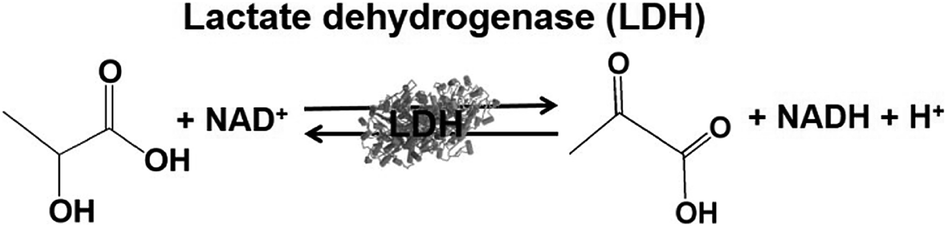 | ||
| Fig. 39 Reaction scheme of lactate dehydrogenase (LDH). | ||
By using LDH as a biocatalyst, the visible-light driven reduction of pyruvate to lactate with an electron donor (in this system TEOA), ZnTMPyP MV and LDH was developed, as shown in Fig. 40.194
 | ||
| Fig. 40 Scheme of the visible-light driven redox system for the reduction of pyruvate to lactate with an electron donor (in this system TEOA), ZnTMPyP MV and LDH. | ||
For the visible-light driven production of lactate, the sample solution consisted of TEOA (0.3 M), ZnTMPyP (9.0 μM), MV (60 μM), pyruvic acid (0.1 mM) and LDH (20 units) in 3.0 mL of 10 mM phosphate buffer (pH 7.0). When this sample solution was irradiated, 0.17 mM of lactate was produced after 4 h irradiation. In this system, the single-electron reduced MV acts as a co-enzyme for LDH in the reduction of pyruvate to lactate.
Biocatalysts for the visible-light driven utilization of ammonia have also been paid attention. Among them, NAD(P)+-dependent dehydrogenases can be used for visible-light driven redox systems with a photocatalytic dye. The commercially available glutamate dehydrogenase (GluDH) from Yeast (EC 1.4.1.4) catalyzes the production of 2-oxoglutarate and NH4+ from L-glutamate in the presence of NADP+ and the reversed reaction of L-glutamate production from 2-oxoglutarate and NH4+ in the presence of NADPH, as shown in Fig. 41.195–197
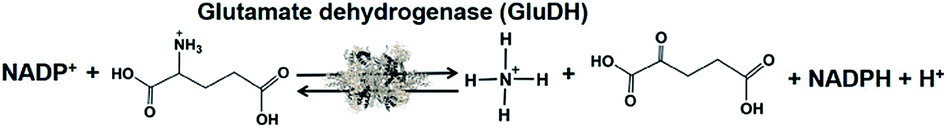 | ||
| Fig. 41 Reaction scheme of glutamate dehydrogenase (GluDH). | ||
By using GluDH as a catalyst for the production of L-glutamate from 2-oxoglutarate and NH4+ in the presence of NADPH, the formation of nitrogen-carbon bonds has been developed. Thus, the visible-light driven formation of nitrogen–carbon bonds from NH3 as a feedstock bonded to organic molecules with GluDH and the NADP+/NADPH redox couple using photocatalytic dye is accomplished.
The single-electron reduced MV does not act as a co-enzyme for GluDH in the production of L-glutamate from 2-oxoglutarate and NH4+. To apply GluDH in visible-light driven redox systems with a photocatalytic dye, the redox system of NADP+/NADPH with FNR has to be integrated into the system.
The visible-light driven redox system with GluDH and reduction of NADP+ with a system containing an ascorbate as the electron donor, polyethylene glycol-modified chlorophyll-a (PEG-Chl-a) and FNR, as shown in Fig. 42, was exploited for the production of L-glutamate from 2-oxoglutarate and NH4+ for the first time.198 As mentioned above, NADP+ is directly reduced to NADPH with FNR and PEG-Chl-a in this reaction, thus it is not necessary to add MV as an electron mediator.
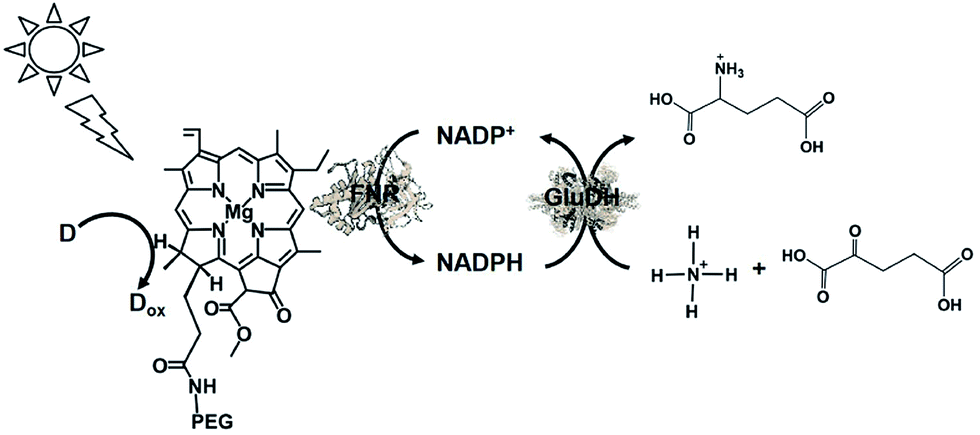 | ||
| Fig. 42 Scheme of the visible-light driven redox system for the production of L-glutamate from α-oxoglutarate and NH4+ and CO2 consisting of PEG-Chl-a, FNR, NADP+, ascorbate and GluDH. | ||
The sample solution consisted of PEG-Chl-a (22.2 μM), ascorbate (8 mM), NADP+ (3.2 mM), α-oxoglutaric acid (8 mM), NH4Cl (8.0 mM), GluDH (40 units), and FNR (2.5 units) in 10 mL of 100 mM phosphate buffer (pH 7.8). The amount of glutamate increased with the visible light irradiation time. During irradiation for 44 h, the amount of glutamate produced from α-oxoglutarate and NH4Cl was determined to be 0.98 mM, of which approximately 12% of α-oxoglutarate (8.0 mM) was converted to glutamate.
Unfortunately, this visible-light driven system with the formation of nitrogen-carbon bonds from NH3 is the only example of this type of reaction system, and simple electron mediators such as PVs have not been applied yet.
Conclusions and outlook
In this review, we focused on biocatalysts for the visible-light driven reduction and utilization of CO2 in systems consisting of a photocatalytic dye and an electron mediator, and the following points were outlined.(1) Representative examples of biocatalysts for the reduction and utilization of CO2 were introduced.
(2) The visible-light driven reduction and utilization of CO2 with a photocatalytic dye and biocatalyst via the NAD(P)+/NADPH or BP/BPred redox couple were introduced.
Especially, the visible-light driven reduction and utilization of CO2 with a photocatalytic dye and biocatalyst via the NAD(P)+/NADPH or BP/BPred redox couple were described in detail. There is no doubt that it is effective to use the NAD(P)+/NAD(P)H redox couple for the visible-light driven reduction and utilization of CO2 with biocatalysts. These systems can be applied for utilization in vivo since they are capable of regenerating NA(P)+/NADPH. The affinity between the natural co-enzyme, NAD(P)+ or NAD(P)H and biocatalyst does not change; however, the catalytic activity of the biocatalyst cannot be controlled with the NAD(P)+/NAD(P)H redox couple. This is because the produced NAD(P)+/NADPH works as an electron donor, in other words, as a sacrificial reagent, and is consumed. Moreover, the NAD(P)+/NADPH redox couple is not suitable for the visible-light driven reduction and utilization of CO2 with a photocatalytic dye and biocatalyst. Even if effective NAD(P)+ reduction to NAD(P)H with visible-light driven redox systems can be achieved, NAD(P)+ is a very expensive biological reagent. Furthermore, since NAD(P)+ cannot be reduced directly to NAD(P)H with a photocatalytic dye, a second electron mediator such as a rhodium complex or second catalyst such as FNR is indispensable for the development of visible-light driven CO2 reduction and utilization systems. If researchers refer to the contribution to the substantial CO2 reduction by visible-light driven redox systems consisting of a photocatalytic dye and biocatalyst using use the NAD(P)+/NAD(P)H redox couple in vitro, the complexity of the system cannot be denied. Unfortunately, it is obvious that visible-light driven redox systems consisting of a photocatalytic dye and biocatalyst using use the NAD(P)+/NAD(P)H redox couple in vitro have limitations unless using a commercially available biocatalyst and the corresponding co-enzyme. Thus, it is necessary to design and prepare simple molecules that are easily reduced with a photocatalytic dye and act as a co-enzyme for biocatalysts. 4,4′- or 2,2′-BPs, which have been widely used as electron mediator molecules in visible-light driven redox systems, have been paid significant attention because BPs can be easily chemically modified. By using chemically modified BPs, the catalytic activity of biocatalysts for the reduction and utilization of CO2 can be controlled. There are reports that it is better to use natural co-enzymes than the NAD(P)+/NAD(P)H redox couple because BP especially MV is a toxic material.133 However, this opinion is an irrelevant idea because the catalytic activity of commercially available biocatalyst cannot be controlled with the NAD(P)+/NAD(P)H redox couple. Thus, controlling the catalytic activity of biocatalysts with cheap molecules is an important point for the practical application of visible-light driven redox systems with a commercially available biocatalyst in vitro. By using a single-electron mediator based on simple molecules for the visible-light driven reduction and utilization of CO2 with a photocatalytic dye and biocatalyst, the reaction system is simpler than that of systems using the NAD(P)+/NAD(P)H redox couple.
For example, simple device for the visible-light driven CO2 reduction to formate consisting of a Chl-e6, a BP with long alkyl chain (CH3V(CH2)9COOH) and FDH has been developed as shown in Fig. 43.199
 | ||
| Fig. 43 Device for the visible-light driven reduction of CO2 to formate consisting of Chl-e6, BP with long alkyl chain (CH3V(CH2)9COOH) and FDH. | ||
By using this device, formate production based on the reduction of CO2 with visible-light irradiation in the presence of a suitable electron donor has been developed. After 3 h visible light irradiation, 2.0 mM formate was produced using this device.
Of course, BPs are not versatile as electron mediators for biocatalysts in light-driven redox systems with a photocatalytic dye. BPs are used under a saturated atmosphere of inert gas such as nitrogen or argon in visible-light driven redox systems due to the deactivation of the single-electron reduced BPs by oxygen. Since dissolved oxygen in the sample solution can be removed with CO2 as an inert gas, MV is used as an electron mediator in visible-light driven CO2 reduction and utilization systems using a photocatalytic dye and biocatalyst.
Moreover, most of the systems introduced in this review require an electron donor, in other words, a sacrificial reagent, and the use of electron sources such as water is an important factor in the future. There is also a proposal to replace the process involving the sacrificial reagent in visible-light driven CO2 reduction systems consisting of photocatalytic dye and biocatalyst with a hydrolysis system of saccharide or only saccharide. For example, the visible-light driven reduction of CO2 to formate with FDH coupling the glucose oxidation with glucose dehydrogenase (GDH) from Thermoplasma acidophilum (EC 1.1.1.47) and the reduction of MV by the photosensitization of ZnTPPS via the NAD+/NADH redox cycle has been reported, as shown in Fig. 44.200
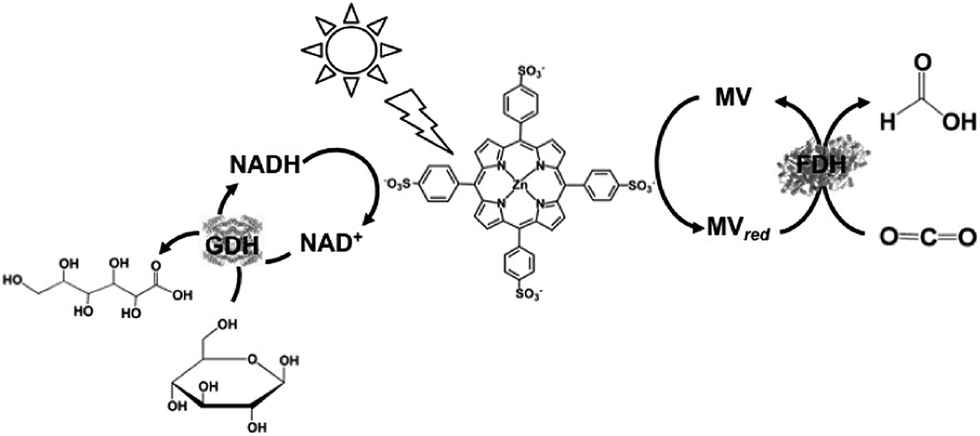 | ||
| Fig. 44 Scheme of the visible-light driven reduction of CO2 to formate with FDH coupling the glucose oxidation with GDH and the reduction of MV by the photosensitization of ZnTPPS via the NAD+/NADH redox cycle. | ||
As mentioned above, NADH acts as a sacrificial reagent and NAD+ is consumed in visible-light driven redox systems consisting of ZnTPPS, MV and FDH. In the system shown in Fig. 43, as NADH is regenerated with GDH, the visible visible-light driven redox system is accomplished without NAD+ consumption.
Oxygen produced due to water oxidation in visible-light driven redox systems deactivates the biocatalyst or electron mediator, thus it is necessary to separate the oxygen production system from the CO2 reduction system. The visible-light driven reduction of CO2 with a photocatalytic dye and a biocatalyst using water as an electron donor also has been reported.201 A visible-light driven electrochemical biofuel-based cell consisting of the thylakoid membrane of microalgae Spirulina platensis immobilized on a TiO2 layer electrode as a photoanode, FDH/CH3V(CH2)9COOH co-immobilized electrode as a cathode, and CO2-saturated buffer solution as the redox electrolyte was developed, as shown in Fig. 45. By using this cell, formate and oxygen are produced stoichiometrically with visible light irradiation. Unfortunately, the efficiency of the CO2 reduction is still low in this system, but efficient CO2 reduction systems using water as an electron donor will be achieved using a photoanode composed of effective photocatalytic materials such as BiVO4.
 | ||
| Fig. 45 Schematic of the device consisting of a visible-light driven cell with a thylakoid membrane on a TiO2 layer electrode as the anode and FDH/CH3V(CH2)9COOH co-immobilized electrode as the cathode. | ||
In the future, it is expected that new technologies with biocatalysts will be developed for the effective and practical visible-light driven reduction and utilization of CO2 with the system of a photocatalytic dye, an effective simple and cheap electron mediator and biocatalyst.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Our work introduced in this review is partially supported by Precursory Research for Embryonic Science and Technology (10104062) (PRESTO, Japan Science and Technology Agency JST), Grant-in-Aid for Challenging Exploratory Research (Japan Society for the Promotion of Science) (15K14239), and Grant-in-Aid for Scientific Research on Innovative Areas “Artificial Photosynthesis (2406)” and “Innovations for Light-Energy Conversion (4906, 18H05174)”.Notes and references
- E. S. Sanz-Pérez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840 CrossRef PubMed.
- K. Takeuchi, 6th International Conference on Greenhouse Gas Control Technologies, Elsevier, Kyoto 2002, p. 849 Search PubMed.
- https://unfccc.int/resource/docs/2015/cop21/eng/l09r01.pdf(Adoption Of The Paris Agreement).
- J. R. Darwent, P. Douglas, A. Harriman, G. Porter and M. C. Richoux, Coord. Chem. Rev., 1982, 44, 93 CrossRef.
- P. A. Brugger, P. Cuendet and M. Grätzel, J. Am. Chem. Soc., 1981, 103, 2923 CrossRef.
- I. Okura, Coord. Chem. Rev., 1985, 68, 53 CrossRef.
- I. Okura, Biochimie, 1986, 68, 189 CrossRef PubMed.
- Y. Amao, Curr. Nanosci., 2008, 4, 45 CrossRef.
- Y. Amao and I. Okura, Sensitization by Metal Complexes Towards Future Artificial Photosynthesis, in Photocatalysis, Kodansha Springer, 2002 Search PubMed.
- A. Harriman, G. Porter and M.-C. Richoux, J. Chem. Soc., Faraday Trans. 2, 1981, 77, 833 RSC.
- I. Okura and N. Kim-Thuuan, J. Mol. Catal., 1979, 6, 227 CrossRef.
- I. Okura, M. Takeuchi and N. Kim-Thuuan, Photochem. Photobiol., 1981, 33, 413 CrossRef.
- I. Okura, M. Takeuchi, S. Kusunoki and S. Aono, Inorg. Chim. Acta, 1982, 63, 157 CrossRef.
- I. Okura, N. Kaji, S. Aono, T. Kita and A. Ymada, Inorg. Chem., 1985, 24, 451 CrossRef.
- Y. Amao, Y. Tomonou, Y. Ishikawa and I. Okura, Int. J. Hydrogen Energy, 2002, 27, 621 CrossRef.
- Y. Tomonou and Y. Amao, BioMetals, 2002, 15, 391 CrossRef PubMed.
- Y. Tomonou and Y. Amao, BioMetals, 2003, 16, 419 CrossRef PubMed.
- N. Sugiyama, M. Toyoda and Y. Amao, Colloids Surf., A, 2006, 284–285, 384 CrossRef.
- Y. Fuchino and Y. Amao, Biophysics, 2006, 2, 57 CrossRef PubMed.
- Y. Amao, Y. Maki and Y. Fuchino, J. Phys. Chem. C, 2009, 113, 16811 CrossRef.
- S. Ishigure, A. Okuda, K. Fujii, Y. Maki, M. Nango and Y. Amao, Bull. Chem. Soc. Jpn., 2009, 82, 93 CrossRef.
- C. V. Krishnan, B. S. Brunschwig, C. Creutz and N. Sutin, J. Am. Chem. Soc., 1985, 107, 2005 CrossRef.
- C. Baffert, V. Artero and M. Fontecave, Inorg. Chem., 2007, 46, 1817 CrossRef PubMed.
- X. L. Hu, B. S. Brunschwig and J. C. Peters, J. Am. Chem. Soc., 2007, 129, 8988 CrossRef PubMed.
- O. Pantani, E. Anxolabehere-Mallart, A. Aukauloo and P. Millet, Electrochem. Commun., 2007, 9, 54 CrossRef.
- P. Du, K. Knowles and R. Eisenberg, J. Am. Chem. Soc., 2008, 130, 12576 CrossRef PubMed.
- K. Sakai and H. Ozawa, Coord. Chem. Rev., 2007, 251, 2753 CrossRef.
- M. Wang, Y. Na, M. Görlov and L. Sun, Dalton Trans., 2009, 6458 RSC.
- K. Sakai, Y. Kizaki, T. Tsubomura and K. Matsumoto, J. Mol. Catal., 1993, 79, 141 CrossRef.
- H. Ozawa, Y. Yokoyama, M. Haga and K. Sakai, Dalton Trans., 2007, 36, 1197 RSC.
- H. Ozawa, M. Haga and K. Sakai, J. Am. Chem. Soc., 2006, 128, 4926 CrossRef PubMed.
- H. Ozawa, M. Kobayashi, B. Balan, S. Masaoka and K. Sakai, Chem.–Asian J., 2010, 5, 1860 CrossRef PubMed.
- S. Masaoka, Y. Mukawa and K. Sakai, Dalton Trans., 2010, 39, 5868 RSC.
- J. W. Peters, W. N. Lanzilotta, B. J. Lemon and L. C. Seefeldt, Science, 1998, 282, 1853 CrossRef PubMed.
- Y. Nicolet, C. Piras, P. Legrand, C. E. Hatchikian and J. C. Fontecilla-Camps, Structure, 1999, 7, 13 CrossRef PubMed.
- Y. Na, M. Wang, J. Pan, P. Zhang, B. Åkermark and L. Sun, Inorg. Chem., 2008, 47, 2805 CrossRef PubMed.
- L.-C. Song, M.-Y. Tang, F.-H. Su and Q.-M. Hu, Angew. Chem., Int. Ed., 2006, 45, 1130 CrossRef PubMed.
- L.-C. Song, M.-Y. Tang, S.-Z. Mei, J.-H. Huang and Q.-M. Hu, Organometallics, 2007, 26, 1575 CrossRef.
- R. Lomoth and S. Ott, Dalton Trans., 2009, 38, 9952 RSC.
- D. Streich, Y. Astuti, M. Orlandi, L. Schwartz, R. Lomoth, L. Hammarstrcm and S. Ott, Chem.–Eur. J., 2010, 16, 60 CrossRef PubMed.
- S. Ott, M. Kritikos, B. Åkermark and L. Sun, Angew. Chem., Int. Ed., 2003, 42, 3285 CrossRef PubMed.
- S. Salyi, M. Kritikos, B. Åkermark and L. Sun, Chem.–Eur. J., 2003, 9, 557 CrossRef PubMed.
- S. Ott, M. Borgström, M. Kritikos, R. Lomoth, J. Bergquist, B. Åkermark, L. Hammarström and L. Sun, Inorg. Chem., 2004, 43, 4683 CrossRef PubMed.
- J. Ekström, M. Abrahamsson, C. Olson, J. Bergquist, F. B. Kaynak, L. Eriksson, L. Sun, H.-C. Becker, B. Åkermark, L. Hammarström and S. Ott, Dalton Trans., 2006, 35, 4599 RSC.
- C. Wombwell, C. A. Caputo and E. Reisner, Acc. Chem. Res., 2015, 48, 2858 CrossRef PubMed.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081 CrossRef PubMed.
- K. A. Vincent, A. Parkin and F. A. Armstrong, Chem. Rev., 2007, 107, 4366 CrossRef PubMed.
- S. Yoshizawa and A. Böck, Biochim. Biophys. Acta, 2009, 1790, 1404 CrossRef PubMed.
- C. S. A. Baltazar, M. C. Marques, C. M. Soares, A. M. DeLacey, I. A. C. Pereira and P. M. Matias, Eur. J. Inorg. Chem., 2011, 948 CrossRef.
- E. Garcin, X. Vernede, E. C. Hatchikian, A. Volbeda, M. Frey and J. C. Fontecilla-Camps, Structure, 1999, 7, 557 CrossRef PubMed.
- F. M. A. Valente, A. S. F. Oliveira, N. Gnadt, I. Pacheco, A. V. Coelho, A. V. Xavier, M. Teixeira, C. M. Soares and I. A. Pereira, J. Biol. Inorg Chem., 2005, 10, 667 CrossRef PubMed.
- A. Parkin, G. Goldet, C. Cavazza, J. C. Fontecilla-Camps and F. A. Armstrong, J. Am. Chem. Soc., 2008, 130, 13410 CrossRef PubMed.
- L. Sun, B. Akermark and S. Ott, Coord. Chem. Rev., 2005, 249, 1653 CrossRef.
- A. A. Krasnovski, G. P. Brin and U. V. Nikandrov, Dokl. Acad. Nauk. SSSR., 1975, 228, 1214 Search PubMed.
- J. A. M. Smith and D. Mauzerall, Photochem. Photobiol., 1981, 34, 407 CrossRef.
- G. McLendon and D. S. Miller, J. Chem. Soc., Chem. Commun., 1980, 533 RSC.
- I. Okura, M. Takeuchi, S. Kusunoki and S. Aono, Inorg. Chim. Acta, 1982, 63, 157 CrossRef.
- C. A. Caputo, M. A. Gross, V. W. Lau, C. Cavazza, B. Lotsch and E. Reisner, Angew. Chem., Int. Ed., 2014, 53, 11538 CrossRef PubMed.
- T. Sakai, D. Mersch and E. Reisner, Angew. Chem., Int. Ed., 2013, 52, 12313 CrossRef PubMed.
- E. Reisner, J. C. Fontecilla-Camps and F. A. Armstrong, Chem. Commun., 2009, 550–552 RSC.
- E. Reisner, Eur. J. Inorg. Chem., 2011, 1005 CrossRef.
- C. A. Caputo, L. Wang, R. Beranek and E. Reisner, Chem. Sci., 2015, 6, 5690 RSC.
- Y. Amao and I. Okura, J. Mol. Catal. A: Chem., 1996, 105, 125 CrossRef.
- Y. Amao and I. Okura, J. Mol. Catal. A: Chem., 1995, 103, 69 CrossRef.
- Y. Amao, Y. Tomonou and I. Okura, Sol. Energy Mater. Sol. Cells, 2003, 79, 103 CrossRef.
- T. Itoh, A. Ishii, Y. Kodera, A. Matsushima, M. Hiroto, H. Nishimura, T. Tsuzuki, T. Kamachi, I. Okura and Y. Inada, Bioconjugate Chem., 1998, 9, 409 CrossRef PubMed.
- Y. Hori, K. Kikuchi, A. Murata and S. Suzuki, Chem. Lett., 1986, 15, 897 CrossRef.
- Y. Hori, A. Murata and R. Takahashi, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 2309 RSC.
- J. Yuan, L. Liu, R. R. Guo, S. Zeng, H. Wang and J. X. Lu, Catalysts, 2017, 7, 220 CrossRef.
- Q. Li, J. J. Fu, W. L. Zhu, Z. Z. Chen, B. Shen, L. H. Wu, Z. Xi, T. Y. Wang, G. Lu and J. J. Zhu, J. Am. Chem. Soc., 2017, 139, 4290 CrossRef PubMed.
- C. H. Lee and M. W. Kanan, ACS Catal., 2015, 5, 465 CrossRef.
- X. F. Feng, K. L. Jiang, S. S. Fan and M. W. Kanan, J. Am. Chem. Soc., 2015, 137, 4606 CrossRef PubMed.
- C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 7231 CrossRef PubMed.
- M. Gattrell and N. Gupta, J. Electroanal. Chem., 2006, 594, 1 CrossRef.
- R. Reske, H. Mistry, F. Behafarid, B. R. Cuenya and P. Strasser, J. Am. Chem. Soc., 2014, 136, 6978 CrossRef PubMed.
- K. P. Kuhl, E. R. Cave, D. N. Abramc and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050 RSC.
- Z. Wang, K. Teramura, Z. Huang, S. Hosokawa, Y. Sakata and T. Tanaka, Catal. Sci. Technol., 2016, 6, 1025 RSC.
- K. Teramura, H. Tatsumi, W. Zheng, S. Hosokawa and T. Tanaka, Bull. Chem. Soc. Jpn., 2015, 88, 431 CrossRef.
- Z. Wang, K. Teramura, S. Hosokawa and T. Tanaka, Appl. Catal., B, 2015, 163, 241 CrossRef.
- M. Yamamoto, T. Yoshida, N. Yamamoto, T. Nomoto, Y. Yamamoto, S. Yagi and H. Yoshida, J. Mater. Chem. A, 2015, 3, 16810 RSC.
- T. Yoshida, N. Yamamoto, T. Mizutani, M. Yamamoto, S. Ogawa, S. Yagi, H. Nameki and H. Yoshida, Catal. Today, 2018, 303, 320 CrossRef.
- M. Yamamoto, S. Yagi and T. Yoshida, Catal. Today, 2018, 303, 334 CrossRef.
- H. Takeda, K. Koike, H. Inoue and O. Ishitani, J. Am. Chem. Soc., 2008, 130, 2023 CrossRef PubMed.
- B. Gholamkhass, H. Mametuska, K. Koike, T. Tanabe, M. Furue and O. Ishitani, Inorg. Chem., 2005, 44, 2326 CrossRef PubMed.
- K. Kiyosawa, N. Shiraishi, T. Shimada, D. Masui, H. Tachibana, S. Takagi, O. Ishitani, D. A. Tryk and H. Inoue, J. Phys. Chem. C, 2009, 113, 11667 CrossRef.
- H. Kumagai, G. Sahara, K. Maeda, M. Higashi, R. Abe and O. Ishitani, Chem. Sci., 2017, 8, 4242 RSC.
- Y. Tamaki and O. Ishitani, ACS Catal., 2017, 7, 3394 CrossRef.
- H. Takeda, C. Cometto, O. Ishitani and M. Robert, ACS Catal., 2017, 7, 70 CrossRef.
- G. Sahara, H. Kumagai, K. Maeda, N. Kaeffer, V. Artero, M. Higashi, R. Abe and O. Ishitani, J. Am. Chem. Soc., 2016, 138, 14152 CrossRef PubMed.
- J. Rohacova and O. Ishitani, Chem. Sci., 2016, 7, 6728 RSC.
- M. Deguchi, S. Yotsuhashi, Y. Yamada and K. Ohkawa, Adv. Condens. Matter Phys., 2015, 537860 Search PubMed.
- H. Rao, L. C. Schmidt, J. Bonin and M. Robert, Nature, 2017, 548, 74 CrossRef PubMed.
- S. Aoi, K. Mase, K. Ohkubo, T. Suenobu and S. Fukuzumi, ACS Energy Lett., 2017, 2, 532 CrossRef.
- K. Faber, Biotransformations in Organic Chemistry, Springer, Berlin, 3rd edn, vol. 201, 1997 Search PubMed.
- Y. Amao, ChemCatChem, 2011, 3, 458 CrossRef.
- C. L. Bird and A. T. Kuhn, Chem. Soc. Rev., 1981, 10, 49 RSC.
- O. Meyer and H. G. Schlegel, J. Bacteriol., 1980, 141, 74 Search PubMed.
- S. W. Ragsdale, J. E. Clark, L. G. Ljungdahl, L. L. Lundie and H. L. Drake, J. Biol. Chem., 1983, 258, 2364 Search PubMed.
- T. I. Doukov, T. M. Iverson, J. Seravalli, S. W. Ragsdale and C. L. Drennan, Science, 2002, 298, 567 CrossRef PubMed.
- C. L. Drennan, J. Heo, M. D. Sintchak, E. Schreiter and P. W. Ludden, Proc. Natl. Acad. Sci., 2001, 98, 11973 CrossRef PubMed.
- H. Dobbek, V. Svetlitchnyi, L. Gremer, R. Huber and O. Meyer, Science, 2001, 293, 1281 CrossRef PubMed.
- D. C. Davison, Biochem. J., 1950, 49, 520 CrossRef.
- J. R. Quayle, Methods Enzymol., 1966, 9, 360 Search PubMed.
- D. R. Jollie and J. D. Lipscomb, J. Biol. Chem., 1991, 266, 21853 Search PubMed.
- E. Racker, J. Biol. Chem., 1950, 184, 313 Search PubMed.
- B. A. Manjasetty, J. Powlowski and A. Vrielink, Proc. Natl. Acad. Sci., 2003, 100, 6992 CrossRef PubMed.
- S. Harada, J. Anthropol. Sci., 1991, 99, 123 Search PubMed.
- Y. Lei, P. D. Pawelek and J. Powlowski, Biochemistry, 2008, 47, 6870 CrossRef PubMed.
- E. G. Brandt, M. Hellgren, T. Brinck, T. Bergman and O. Edholm, Phys. Chem. Chem. Phys., 2009, 11, 975 RSC.
- P. Zucca, M. Littarru, A. Rescigno and E. Sanjust, Biosci., Biotechnol., Biochem., 2009, 73, 1224 CrossRef PubMed.
- R. Obert and B. C. Dave, J. Am. Chem. Soc., 1999, 121, 12192 CrossRef.
- B. El-Zahab, D. Donnelly and P. Wang, Biotechnol. Bioeng., 2007, 99, 508 CrossRef PubMed.
- M. Ando, T. Yoshimoto, S. Ogushi, K. Rikitake, S. Shibata and D. Tsuru, J. Biochem., 1979, 85, 1165 Search PubMed.
- W. Hohnloser, B. Osswald and F. Lingens, Z. Physiol. Chem., 1980, 361, 1763 CrossRef.
- I. Harary, S. R. Korey and S. Ochoa, J. Biol. Chem., 1953, 203, 595 Search PubMed.
- S. Ochoa, A. H. Mehler and A. Kornberg, J. Biol. Chem., 1948, 74, 979 Search PubMed.
- W. J. Rutter and H. A. Lardy, J. Biol. Chem., 1958, 233, 374 Search PubMed.
- D. B. Cherbavaz, M. E. Lee, R. M. Stroud and D. E. Koshland, J. Mol. Biol., 2000, 295, 377 CrossRef PubMed.
- H. Tarhonskaya, A. M. Rydzik, I. K. H. Leung, N. D. Loik, M. C. Chan, A. Kawamura, J. S. McCullagh, T. D. W. Claridge, E. Flashman and C. J. Schofield, Nat. Commun., 2014, 5, 3423 CrossRef PubMed.
- F. J. Corpas, J. B. Barroso, L. M. Sandalio, J. M. Palma, J. A. Lupiáñez and L. A. del Río, Plant Physiol., 1999, 121, 921 CrossRef PubMed.
- Y. Ohno, T. Nakamori, H. Zheng and S. Suye, Biosci., Biotechnol., Biochem., 2008, 72, 1278 CrossRef PubMed.
- H. Zheng, T. Nakamori and S. Suye, J. Biosci. Bioeng., 2009, 107, 16 CrossRef PubMed.
- K. Hironaka, S. Fukuzumi and T. Tanaka, J. Chem. Soc., Perkin Trans. 2, 1984, 1705 RSC.
- H. Wu, C. Tian, X. Song, C. Liu, D. Yang and Z. Jiang, Green Chem., 2013, 15, 1773 RSC.
- F. Hollmann, I. W. C. E. Arends and K. Buehler, ChemCatChem, 2010, 2, 762 CrossRef.
- F. Hollmann, B. Witholt and A. Schmid, J. Mol. Catal. B: Enzym., 2002, 19–20, 167 CrossRef.
- D. E. Torres Pazmino, M. Winkler, A. Glieder and M. W. Fraaije, J. Biotechnol., 2010, 146, 9 CrossRef PubMed.
- Y. Maenaka, T. Suenobu and S. Fukuzumi, J. Am. Chem. Soc., 2012, 134, 367 CrossRef PubMed.
- Z. Y. Jiang, C. Q. Lü and H. Wu, Ind. Eng. Chem. Res., 2005, 44, 4165 CrossRef.
- Q. Shi, D. Yang, Z. Y. Jiang and J. Li, J. Mol. Catal. B: Enzym., 2006, 43, 44 CrossRef.
- C. B. Park, S. H. Lee, E. Subramanian, B. B. Kale, S. M. Lee and J. O. Baeg, Chem. Commun., 2008, 5423 RSC.
- R. K. Yadav, J.-O. Baeg, G. H. Oh, N.-J. Park, K.-J. Kong, J. Kim, D. W. Hwang and S. K. Biswas, J. Am. Chem. Soc., 2012, 134, 11455 CrossRef PubMed.
- R. K. Yadav, G. H. Oh, N.-J. Park, A. Kumar, K.-J. Kong and J.-O. Baeg, J. Am. Chem. Soc., 2014, 136, 16728 CrossRef PubMed.
- R. K. Yadav, J.-O. Baeg, A. Kumar, K.-J. Kong, G. H. Oh and N.-J. Park, J. Mater. Chem. A, 2014, 2, 5068 RSC.
- M. Ihara, Y. Kawano, M. Urabe and A. Okabe, PLoS One, 2013, 8, e71581 CrossRef PubMed.
- K. J. Shah and T. Imae, J. Mater. Chem. A, 2017, 5, 9691 RSC.
- C. Drewke and M. Ciriacy, Biochim. Biophys. Acta, 1988, 950, 54 CrossRef.
- H. Wang, D. Xiao, C. Zhou, L. Wang, L. Wu, Y. Lu, Q. Xiang, K. Zhao, X. Li and M. Ma, Appl. Microbiol. Biotechnol., 2017, 101, 4507 CrossRef PubMed.
- X. Wang, C. J. Mann, Y. Bai, L. Ni and H. Weiner, J. Bacteriol., 1998, 180, 822 Search PubMed.
- S. Ogushi, M. Ando and D. Tsuru, Agric. Biol. Chem., 1984, 48, 597 Search PubMed.
- A. Andreadeli, D. Platis, V. Tishkov, V. Popov and N. E. Labrou, FEBS J., 2008, 275, 3859 CrossRef PubMed.
- T. Schmidt, C. Michalik, M. Zavrel, A. Spiess, W. Marquardt and M. B. Ansorge-Schumacher, Biotechnol. Prog., 2010, 26, 73 Search PubMed.
- Y. Amao, Chem. Lett., 2017, 46, 780 CrossRef.
- D. Mandler and I. Willner, J. Chem. Soc., Perkin Trans. 2, 1988, 997 RSC.
- I. Willner and D. Mandler, J. Am. Chem. Soc., 1989, 111, 1330 CrossRef.
- I. Willner, N. Lapidot, A. Riklin, R. Kasher, E. Zahavy and E. Katz, J. Am. Chem. Soc., 1994, 116, 1428 CrossRef.
- I. Willner and N. Lapidot, J. Am. Chem. Soc., 1990, 112, 6438 CrossRef.
- M. Kodaka and Y. Kubota, J. Chem. Soc., Perkin Trans. 2, 1999, 891 RSC.
- Y. Amao, R. Abe and S. Shiotani, J. Photochem. Photobiol., A, 2015, 313, 149 CrossRef.
- S. Ikeyama, R. Abe, S. Shiotani and Y. Amao, Chem. Lett., 2016, 45, 979 CrossRef.
- S. Ikeyama, R. Abe, S. Shiotani and Y. Amao, Bull. Chem. Soc. Jpn. DOI:10.1246/bcsj.20180013 , accepted.
- R. Miyatani and Y. Amao, Biotechnol. Lett., 2002, 24, 1931 CrossRef.
- R. Miyatani and Y. Amao, J. Mol. Catal. B: Enzym., 2004, 27, 121 CrossRef.
- R. Miyatani and Y. Amao, J. Jpn. Pet. Inst., 2004, 47, 27 CrossRef.
- S. Ikeyama and Y. Amao, Sustainable Energy Fuels, 2017, 1, 1730 RSC.
- S. Ikeyama and Y. Amao, Photochem. Photobiol. Sci., 2018, 17, 60 RSC.
- S. Ikeyama, T. Katagiri and Y. Amao, J. Photochem. Photobiol., A, 2018, 358, 362 CrossRef.
- S. Ikeyama and Y. Amao, Chem. Lett., 2016, 45, 1259 CrossRef.
- S. Ikeyama and Y. Amao, ChemCatChem, 2017, 9, 833 CrossRef.
- I. Tsujisho, M. Toyoda and Y. Amao, Catal. Commun., 2006, 7, 173 CrossRef.
- Y. Amao and T. Watanabe, Chem. Lett., 2004, 33, 1544 CrossRef.
- Y. Amao and T. Watanabe, Appl. Catal., B, 2009, 86, 109 CrossRef.
- Y. Amao and R. Kataoka, Catal. Today, 2018, 307, 243 CrossRef.
- S. Kuwabata, K. Nishida, R. Tsuda, H. Inoue and H. Yoneyama, J. Electrochem. Soc., 1994, 141, 1488 Search PubMed.
- M. G. van Kleef, J. Jongejan, and J. Duine, in PQQ and Quinoproteins, ed. J. A. Jongejan and J. A. Duine, Springer Netherlands, p. 217, 1989 Search PubMed.
- S. Itoh, M. Kinugawa, N. Mita and Y. Ohshiro, J. Chem. Soc., Chem. Commun., 1989, 11, 694 RSC.
- E. Katz, D. D. Schlereth and H.-L. Schmidt, J. Electroanal. Chem., 1994, 367, 59 CrossRef.
- E. Katz, D. D. Schlereth, H.-L. Schmidt and A. J. J. Olsthoorn, J. Electroanal. Chem., 1994, 368, 165 CrossRef.
- I. Emahi, M. Mitchell and D. A. Baum, J. Electrochem. Soc., 2017, 164, 3097 CrossRef.
- T. W. Woolerton, S. Sheard, E. Reisner, E. Pierce, S. W. Ragsdale and F. A. Armstrong, J. Am. Chem. Soc., 2010, 132, 2132 CrossRef PubMed.
- T. W. Woolerton, S. Sheard, E. Pierce, S. W. Ragsdale and F. A. Armstrong, Energy Environ. Sci., 2011, 4, 2393 RSC.
- Y. S. Chaudhary, T. W. Woolerton, C. S. Allen, J. H. Warner, E. Pierce, S. W. Ragsdale and F. A. Armstrong, Chem. Commun., 2012, 48, 58 RSC.
- A. Bachmeier, V. C. C. Wang, T. W. Woolerton, S. Bell, J. C. Fontecilla-Camps, M. Can, S. W. Ragsdale, Y. S. Chaudhary and F. A. Armstrong, J. Am. Chem. Soc., 2013, 135, 15026 CrossRef PubMed.
- Y. Amao, SPR Photochemistry, 2018, 45, 163 Search PubMed.
- L. Willner, D. Mandler and A. Riklin, J. Chem. Soc., Chem. Commun., 1986, 1022 RSC.
- T. Omura, E. Sanders and R. W. Estabrook, Arch. Biochem. Biophys., 1966, 117, 660 CrossRef.
- M. Shin, K. Tagawa and D. I. Arno, Biochem. Z., 1963, 338, 84 Search PubMed.
- J. M. Berg, J. L. Tymoczko and L. Stryer, Biochemistry, W. H. Freeman, New York, 6th edn, 2007 Search PubMed.
- A. Aliverti, V. Pandini, A. Pennati, M. de Rosa and G. Zanetti, Arch. Biochem. Biophys., 2008, 474, 283 CrossRef PubMed.
- J. P. Benz, M. Lintala, J. Soll, P. Mulo and B. Bölter, Trends Plant Sci., 2010, 15, 608 CrossRef PubMed.
- H. Inoue, M. Yamachika and H. Yoneyama, J. Chem. Soc., Faraday Trans., 1992, 88, 2215 RSC.
- Y. Amao and M. Ishikawa, J. Jpn. Pet. Inst., 2007, 50, 272 CrossRef.
- Y. Amao and M. Ishikawa, Catal. Commun., 2007, 8, 423 CrossRef.
- T. Itoh, H. Asada, K. Tobioka, Y. Kodera, A. Matsushima, M. Hiroto, H. Nishimura, T. Kamachi, I. Okura and Y. Inada, Bioconjugate Chem., 2000, 11, 8 CrossRef PubMed.
- H. Kamogawa and S. Sato, Bull. Chem. Soc. Jpn., 1991, 64, 321 CrossRef.
- T. Katagiri, S. Ikeyama and Y. Amao, J. Photochem. Photobiol., A, 2018, 358, 368 CrossRef.
- Y. Amao, S. Ikeyama, T. Katagiri and K. Fujita, Faraday Discuss., 2017, 198, 73 RSC.
- T. Katagiri, K. Fujita, S. Ikeyama and Y. Amao, Pure Appl. Chem. Search PubMed , in submitted.
- H. Inoue, Y. Kubo and H. Yoneyama, J. Chem. Soc., Faraday Trans., 1991, 87, 553 RSC.
- Y. Amao, N. Shuto and H. Iwakuni, Appl. Catal., B, 2016, 180, 403 CrossRef.
- P. D. Watson, Ind. Eng. Chem., 1948, 40, 1393 CrossRef.
- C. J. Valvona, H. L. Fillmore, P. B. Nunn and G. J. Pilkington, Brain Pathol., 2016, 26, 3 CrossRef PubMed.
- J. R. Pineda, R. Callender and S. D. Schwartz, Biophys. J., 2007, 93, 1474 CrossRef PubMed.
- R. Miyatani and Y. Amao, Photochem. Photobiol. Sci., 2004, 3, 681 RSC.
- J. W. Coulton and M. Kapoor, Can. J. Microbiol., 1973, 19, 427 CrossRef PubMed.
- S. Grisolia, C. L. Quijada and M. Fernandez, Biochim. Biophys. Acta, 1964, 81, 61 Search PubMed.
- I. Shiio and H. Ozaki, J. Biochem., 1970, 68, 633 CrossRef PubMed.
- H. Asada, T. Itoh, Y. Kodera, A. Matsushima, M. Hiroto, H. Nishimura and Y. Inada, Biotechnol. Bioeng., 2001, 76, 86 CrossRef PubMed.
- Y. Amao, N. Shuto, K. Furuno, A. Obata, Y. Fuchino, K. Uemura, T. Kajino, T. Sekito, S. Iwai, Y. Miyamoto and M. Matsuda, Faraday Discuss., 2012, 155, 289 RSC.
- Y. Amao, S. Takahara and Y. Sakai, Int. J. Hydrogen Energy, 2014, 39, 20771 CrossRef.
- Y. Amao, M. Fujimura, M. Miyazaki, M. Nakamura, A. Tadokoro and N. Shuto, New J. Chem., 2018, 42, 9269 RSC.
| This journal is © The Royal Society of Chemistry 2018 |