
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceArtificial simulation of salivary and gastrointestinal digestion, and fermentation by human fecal microbiota, of polysaccharides from Dendrobium aphyllum
Huifan Liu a,
Fan Gongb,
Fashan Weib and
Hui Wu*a
a,
Fan Gongb,
Fashan Weib and
Hui Wu*a
aDepartment of Food Quality and Safety, College of Food Science and Engineering, South China University of Technology, Wushan Road 381, Guangzhou, Guangdong 510640, China. E-mail: fehwu@scut.edu.cn; Fax: (+86) 20-87112532; Tel: (+86) 20-87112853
bHenan Institute of Product Quality Inspection and Supervision, Zhengzhou, Henan 450000, China
First published on 16th April 2018
Abstract
In vitro salivary and gastrointestinal (GI) digestion and fermentation of polysaccharides extracted from Dendrobium aphyllum were investigated in this study. Salivary amylase showed no influence on D. aphyllum polysaccharides (DAP). The molecular weight of DAP decreased dramatically during the first 0.5 h of gastric digestion, and then reduced steadily during the subsequent GI tract consumption. The content of reducing sugars increased steadily during GI digestion. Only released free mannose of DAP was detectable by gas chromatography-mass spectrometry analysis during the first 12 h of fermentation, which was contributed by fecal microbiota metabolism. In terms of the fermentation pattern, the pH dropped significantly due to the formation of six types of short-chain fatty acids (SCFAs). This study demonstrates that polysaccharides extracted from D. aphyllum can be digested by the GI tract and are physiologically active in the human large bowel by lowering the pH of the large intestinal environment and promoting the production of SCFAs.
1. Introduction
Bioactive carbohydrates have shown satisfactory benefits to human health through their antioxidant, immune-boosting, antidiabetic, antilipidemic, and antineoplastic effects.1,2 However, as functional foods, the actual effects of macromolecular carbohydrates on the human body can only be determined after digestion, glycolysis, and absorption.3,4 The human consumption process may directly or indirectly alter carbohydrate structure and content due to the presence of strong acids, increased bile levels, and complicated intestinal microbiota conditions, all of which may also alter their bioactive effects.5 Based on human physiological gastric and intestinal pH conditions, in vivo digestion models have been developed because they are simple, cheap, and reproducible tools, and do not require ethical considerations.6Carbohydrates are a type of prebiotics that can selectively contribute to the diversity and quantity of intestinal colonized microbiota, such as Lactobacillus and Bifidobacterium species, and can decrease the risk of various chronic diseases in humans.7,8 Fermentation is one of the pathways utilized to generate energy through the breakdown of carbohydrates to yield short-chain fatty acids (SCFAs); this is the primary activity of the large-intestinal microflora.9 In addition to providing the body with energy, SCFAs can stimulate salt and water absorption, and affect epithelial cell growth and cell proliferation in the jejunum and ileum.10 Moreover, SCFAs can inhibit the breeding of harmful pathogens and colorectal cancer by decreasing the pH of the internal environment.6 In vitro fermentation using the TIM-2 system is a satisfactory method for evaluating the fermentation process and metabolic characteristics of large-intestinal microflora on carbohydrates.5 After collecting intestinal microbiota from human feces, a simulated human intestinal fermentation environment is implemented using a vacuum and temperature control.11 The advantages of this method are the absence of ethical constraints, lack of influence of other digested compounds, and easy monitoring of the degradation conditions; however, the fecal inoculum is not a good representation of the entire colon.12
Dendrobium is the second largest genus in Orchidaceae, and Dendrobium species have been used as traditional medicine and edible food sources for decades in Asia.13 Among 78 Dendrobium species, Dendrobium aphyllum is used as a functional food and is widely planted in tropical and subtropical regions of Asia, Australia, and the Pacific islands for its cultivation advantages.14 Polysaccharides extracted from D. aphyllum are a source of dietary fiber from carbohydrates digested in the gastrointestinal (GI) tract and is utilized by human large-intestinal microflora. Polysaccharides can decrease the pH in parts of the colon, regulate the intestinal immune system, and mediate species and quantity of large-intestinal microflora in order to decrease the risk of various chronic diseases.9,15 As large biological molecules, different polysaccharides have different digestive characteristics and fermentation capabilities due to their complex structures,16 so it is valuable to evaluate D. aphyllum polysaccharides that have not previously been analyzed.
In our previous study, D. aphyllum polysaccharides (DAP; molecular weight [Mw], 471.586 kDa), was composed of mannose (71.3%) and glucose (28.7%), and its backbone was composed of β-D-mannopyranose and β-D-glucopyranose residues with mainly 1,4-linked glycosidic bonds.17 This work was designed for the development of an in vitro digestion model for subsequent in vitro fermentation with human fecal cultures to provide human consumption information. Changes in pH, molecular weight, SCFA production, and total and reducing sugar contents were analyzed. In addition, the relationship between carbohydrate consumption and SCFA production was evaluated. The study may provide some useful information for the study of artificial stimulated GI digestion and colon fermentation on carbohydrates, especially on D. aphyllum species.
2. Materials and methods
2.1 Materials
D. aphyllum samples were collected from the GAP farm located in Xishuangbanna, Yunnan province, China. These samples were authenticated by experts from the Department of Life Science, South China Agricultural University. Standard monosaccharides (L-rhamnose, D-ribose, L-(+)-arabinose, D-(+)-xylose, D-mannose, and D-(+)-glucose) were purchased from Sigma (St. Louis, MO, USA). Acetic acid (100% purity) and i-valeric acid (99.9% purity) were purchased from Merck (Darmstadt, Germany). Propionic acid (100% purity) was purchased from Janssen Chimica (Belgium). Butyric acid (99.9% purity), i-butyric acid (100% purity), and valeric acid (100% purity) were purchased from Sigma (St. Louis, MO, USA). All other chemicals and reagents used were of analytical grade and commercially available.2.2 DAP sample preparation
DAP was collected as described in our previous study.17 Briefly, after decolorization and removal of water-soluble impurities, the collected sample powder (10 g) was extracted in a material to solvent ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :85 (w/v), temperature of 80 °C, and time of 4.6 h. After using the Sevag method for deproteinization, the protein-free crude polysaccharide (10 mL, 10 μg mL−1) was loaded onto a DEAE-Sepharose Fast Flow chromatography column (1.6 × 35 cm) at a flow rate of 1 mL min−1, and then eluted with ultrapure water. Eluents were analyzed by the phenol–sulfuric acid method, and the polysaccharide fraction was then concentrated, dialyzed, and lyophilized. A polysaccharide fraction sample of 1.95 g was dissolved in 1.3 L ultrapure water, and 1 mL of this sample was loaded onto a Sephadex G-200 chromatography column (1.6 × 60 cm). The polysaccharide fraction was washed with 300 mL ultrapure water at a flow rate of 1 mL min−1 for a total of 300 min. Following elution and lyophilization, a DAP fraction of 1.376 g was obtained.
:85 (w/v), temperature of 80 °C, and time of 4.6 h. After using the Sevag method for deproteinization, the protein-free crude polysaccharide (10 mL, 10 μg mL−1) was loaded onto a DEAE-Sepharose Fast Flow chromatography column (1.6 × 35 cm) at a flow rate of 1 mL min−1, and then eluted with ultrapure water. Eluents were analyzed by the phenol–sulfuric acid method, and the polysaccharide fraction was then concentrated, dialyzed, and lyophilized. A polysaccharide fraction sample of 1.95 g was dissolved in 1.3 L ultrapure water, and 1 mL of this sample was loaded onto a Sephadex G-200 chromatography column (1.6 × 60 cm). The polysaccharide fraction was washed with 300 mL ultrapure water at a flow rate of 1 mL min−1 for a total of 300 min. Following elution and lyophilization, a DAP fraction of 1.376 g was obtained.
2.3 Simulated saliva digestion
2.4 Simulated artificial gastric and intestinal digestion in vitro
2.5 In vitro fermentation of polysaccharides
Twenty anaerobically sealed tubes were prepared, 5 of each containing 20% DAP-I (100 mg in total), mannose (71.26 mg in total), glucose (28.74 mg in total), or water, respectively. The mass of mannose and glucose was calculated according to the monosaccharide composition assay of DAP.17 To each anaerobically sealed tube, 40% human fecal preculture (as described in 2.5.2) and 40% growth medium were added for in vitro fermentation with the precultured human fecal microbiota. Under conditions of 10% H2, 10% CO2, and 80% N2 in a Forma Anaerobic System (Thermo Electron Crop., Marietta, Ohio, USA), the anaerobic sealed tubes were incubated in a TC-2212B Thermostat shaker (37 °C, 160 rpm). Fermentation of the samples (DAP-F, mannose, and glucose) and the controls (Control-F) were analyzed before or after 0.5, 6, 12, and 24 h culture by removing the corresponding tubes and plunging them into iced water for 20 min to inactive the fermentation.
2.6 Determination of pH for in vitro fermentation of polysaccharide
The pH of the fermentation cultures from 2.5.3 were measured with a micro-pH meter (Tianjing Analytic Corp., Tianjing, China). In order to better analyze fermentation patterns, the anaerobically sealed tubes containing DAP-F, mannose, glucose, or control removed after 24 h were continuously cultured in a TC-2212B Thermostat shaker (37 °C, 160 rpm) for a further 24 h, and the pH was measured again by micro-pH meter.2.7 Determination of molecular weight changes
High-performance gel permeation chromatography (HPGPC) was conducted to determine the weight average Mw of DAP, DAP-Sa, DAP-S, DAP-I, and DAP-F. A Waters HPLC system (Waters, Milford, MA, USA), serially linked with two gel columns (TSK-G5000PWXL column, 7.8 × 300 mm inner diameter, 10 ìm; and TSK G-3000PWXL column, 7.8 × 300 mm inner diameter, 6 μm) was used, and samples were eluted with 0.02 M KH2PO4 and detected with a Waters 2414 differential refractive index detector with a flow rate of 0.6 mL min−1 and column temperature of 35 °C. A total of 10 μL sample solution was injected, and chromatography was performed for 45 min. Dextran standards with different molecular weights (Mw 5.2 × 103, 1.16 × 105, 2.38 × 104, 4.86 × 104, 1.48 × 105, 2.73 × 105, 4.1 × 105, and 6.68 × 105 Da) were used for data analysis.2.8 Determination of reducing sugar content
The reducing sugar content in each sample after artificial digestion and fermentation was assessed using an original sugar content kit (Suzhou Comin Biotechnology Co., Ltd., Suzhou, China).2.9 Determination of released free monosaccharides
Six monosaccharides (L-rhamnose, D-ribose, L-(+)-arabinose, D-(+)-xylose, D-mannose, and D-(+)-glucose) were weighed (2 mg), prepared as concentration gradients, and then 1 mg myoinositol, 10 mg hydroxylamine hydrochloride, and 1 mL pyridine were added for derivatization. After mixing thoroughly, the samples were placed in a water bath for 30 min at 90 °C, 1 mL of acetic anhydride was added, and samples were incubated for another 30 min in the water bath at 90 °C. Sample solutions (1 mL) were passed through 0.22 μm organic phase filter membranes. Sample (DAP, DAP-Sa, DAP-S, DAP-I, and DAP-F) and control (Control-S, Control-I) solutions (5 mL) were analyzed after derivatization as above. After preparation, 1 μL of each sample was analyzed by gas chromatography (GC) (7890B, Agilent, Santa Clara, CA, USA) with a DB-170 chromatographic column (30 mm × 0.53 mm × 0.25 μm) from 100 °C to 230 °C at 10°C min−1, then from 230 °C to 250 °C at 50°C min−1, and 1 min retention. A flame ionization detector was used with parameters of 230 °C at 300 mL min−1 for the air rate, 30 mL min−1 for the hydrogen rate, and 5 mL min−1 for the nitrogen rate.2.10 Determination of SCFAs by GC-mass spectroscopy (GC-MS)
The DAP-F solutions were centrifuged (3900 × g, 15 min) and the supernatants were collected for the determination of SCFAs. SCFA standards, including concentration gradients of acetic acid, propionic acid, i-butyric acid, butyric acid, i-valeric acid, and valeric acid, were analyzed. Chromatographic analysis was carried out using the Agilent 7890 N GC and Agilent 5975C QMS system. A GC column (HP-INNOWAX, 190901N-213, J &W Scientific, Agilent Technologies Inc., USA) of 30 m × 250 μm in support-coated with 0.25 μm film thickness was used. Nitrogen was supplied as the carrier gas at a flow rate of 1.2 mL min−1 with a split ratio of 1:50. The initial column temperature was held at 40 °C for 1 min, then programmed at a rate of 5°C min−1 to 250 °C for 10 min. The temperature of the FID and injection port was 250 °C. The injected standard SCFA and sample volume for GC analysis was 0.5 μL, and the running time for each analysis was 50.5 min.
2.11 Carbohydrate contribution for SCFA production
The production of SCFAs in the fermentation cultures with added glucose or mannose were determined by GC-MS as described in 2.10. The contribution of glucose or mannose in the DAP for SCFA production (mainly acetic, propionic, valeric, and butyric acids) at different fermentation time points was calculated as follows:24| Y (%) = Ca/Cb × 100, |
2.12 Statistical analysis
Data are expressed as the mean ± standard deviation (SD) of three replicates. Significant differences between the means of parameters were calculated using Duncan's multiple-range test with SPSS 17.0 software (SPSS, Inc., Chicago, IL, USA). P < 0.05 was considered to indicate statistical significance.3. Results and discussion
3.1 Molecular changes of DAP after digestion and fermentation
We hypothesized that in most cases, the gastric emptying process can last 2 h, and may be less than 2 h for these polysaccharides. However, because artificial stimulation was in vitro in this study, 6 h was used for the stomach and intestinal digestion model. This allowed for a sufficient simulation of GI digestive conditions of DAP for a more reliable and logical characterization, as well as to maintain consistency with a previously reported study.18 The molecular changes during specific digestion and fermentation processes are shown in Table 1. In general, the molecular weights dropped steadily from 471.586 ± 67.2 to 0.37 ± 0.1 kDa, and were not detected after the fermentation process in which macromolecular DAP were digested to low molecular weight polysaccharides and subsequently utilized by human microbiota to oligosaccharides and monosaccharides.| Samples | MW (kDa) | CR (mM) | Released free monosaccharide (ng μl) | |
|---|---|---|---|---|
| Glucose | Mannose | |||
| a Data are shown as mean ± standard (n = 3).b Mean values in the same column with different letters were significantly different (Tukey test, p < 0.05).c Not detected or lower than limit of quantification. | ||||
| Before digestion | 471.586 ± 67.2ab | 0.1189 ± 0.00382a | NDc | ND |
| Saliva (4 h) | 471.231 ± 71.1a | 0.1126 ± 0.01218a | ND | ND |
| Stomach (0.5 h) | 19.83 ± 6.3b | 0.2847 ± 0.00322b | ND | ND |
| Stomach (1 h) | 17.1 ± 2.3b | 0.2849 ± 0.00684b | ND | ND |
| Stomach (2 h) | 15.9 ± 1.2b | 0.2887 ± 0.00150b | ND | ND |
| Stomach (3 h) | 13.2 ± 1.9c | 0.2899 ± 0.00578b | ND | ND |
| Stomach (4 h) | 11.4 ± 1.6c | 0.2900 ± 0.00188b | ND | ND |
| Stomach (5 h) | 9.2 ± 0.3d | 0.3010 ± 0.00344c | ND | ND |
| Stomach (6 h) | 8.8 ± 0.5d | 0.3042 ± 0.00146c | ND | ND |
| Intestine (0.5 h) | 8.2 ± 0.1d | 0.3121 ± 0.00818c | ND | ND |
| Intestine (1 h) | 6.9 ± 0.2e | 0.31894 ± 0.01480c | ND | ND |
| Intestine (2 h) | 6.3 ± 0.8e | 0.3191 ± 0.00280c | ND | ND |
| Intestine (3 h) | 6.4 ± 0.1e | 0.3519 ± 0.00993d | ND | ND |
| Intestine (4 h) | 4.8 ± 0.1f | 0.3570 ± 0.00049d | ND | ND |
| Intestine (5 h) | 3.9 ± 0.1f | 0.3727 ± 0.00038e | ND | ND |
| Intestine (6 h) | 2.1 ± 0.2h | 0.3820 ± 0.00179e | ND | ND |
| Fermentation (0.5) | 1.92 ± 0.5h | 0.466 ± 0.00311f | ND | 428.6 |
| Fermentation (6) | 0.87 ± 0.2h | 0.3859 ± 0.00104e | ND | 787.496 |
| Fermentation (12) | 0.37 ± 0.1I | 0.3797 ± 0.00038e | ND | 987.183 |
| Fermentation (24) | ND | 0.2956 ± 0.00098b | ND | ND |
In terms of saliva digestion, the constituents and their amounts vary in different human saliva samples, but salivary amylase content is similar, with a certain range of amylase activity and approximate pH of 7.2. Salivary amylase is the major factor during saliva digestion for breaking down polysaccharides and has been reported to have significant effects on amylose, amylopectin, and glycogen.25 However, there were no significant differences before and after salivary digestion; the DAP molecular weight changed from 471.586 ± 67.2 (before digestion) to 471.231 ± 71.1 kDa (DAP-Sa). Because salivary amylase activity was assessed as 157 ± 5 D units mL−1, agreeing with the value (18–208 D units mL−1) reported previously by Ruth and Roozen (2000)25, the value of salivary amylase was appropriate. However, DAP molecules were not liberated, and this may be because salivary amylase has been shown to affect molecules containing α-limit dextrin,26,27 but the glycosidic bonds in dendrobium polysaccharides are mainly β-(1,4)-linkages.28
During artificially simulated gastric digestion, the molecular weight of DAP-S dropped rapidly for 0.5 h at the beginning and subsequently decreased steadily over the following 5.5 h. Because the gastric medium has a strong acidic pH of 3, and carbohydrates have been shown to be very sensitive to acidic pH,18 the molecular breakdown of DAP-S was more efficient in the acid medium. During the 6 h simulated intestinal digestion, molecular weights of DAP-I also decreased steadily, almost generating oligosaccharides (the Mw of oligosaccharides were 300–2000 Da). There are two main pathways for the molecular loss of DAP; one is the breakdown of glycosidic bonds.29 Meanwhile, the active forces between glycosidic bonds or monosaccharide groups induce polysaccharides to form aggregates in aqueous systems, whereas strong acids, high concentrations of bile salts, and agglomerates contained in the GI medium destroy the spatial structure of DAP.30 Moreover, the molecular weights of DAP decrease significantly in the stimulated stomach environment but minimally in the intestinal condition. The pH and salts were able to depolymerize and break of macromolecular substance.31,32 When processing the artificially stimulated GI digestion, Hu et al. (2013) found that the pH was the major cause for the significant decrease in molecular weight of Plantago asiatica L., and the variations of molecular weight were in line with our results.18 Therefore, we concluded that the very low pH in the gastric environment resulted in the depolymerization and breakage of DAP, which is consistent with specific kinds of macromolecular carbohydrates reported previously.33
A continuous molecular weight reduction occurred during the culture with human feces, and after this fermentation process, the molecular weight was no longer detectable. The steady loss of DAP-F molecules was attributed to microbial metabolism, including hydrogen carbon dioxide and SCFA generation.34
3.2 Content change of reducing sugars in DAP after digestion and fermentation
As discussed above, the molecular loss of DAP may due to glycosidic bond breakdown, or composition or spatial structure disruption, but the content variation of reducing sugars can be used determine the exact reason.35 As shown in Table 1, the reducing sugar content before the digestion was 0.1189 ± 0.00382 mM, and after the simulated gastric digestion, this increased significantly to 0.3042 ± 0.00146 mM. It has been reported that quantitative reducing ends would increase when the loss of molecules was caused by the breakdown of glycosidic bonds.36 Moreover, the acidic pH would cause generate reducing end through the breakdown of glycosidic bonds.18 After intestinal digestion, the reducing sugar content increased slightly to 0.3820 ± 0.00179 mM, but the molecular weight of DAP-I was significantly decreased; therefore, the reason for the intestinal molecular lost may be due to the disruption of aggregates by substances contained in the intestinal medium, such as the high concentration of bile salt or other agglomerates. When focusing on the fermentation pattern, the reducing sugar content increased during the first 0.5 h to 0.466 ± 0.00311 mM and subsequently decreased from 0.3859 ± 0.00104 mM to 0.2956 ± 0.00098 mM. The reason for the increase in reducing sugars may be due to the degradation by microbiota of polysaccharides to generate reducing ends.18 It was discovered that polysaccharides, as an energy source, can be utilized by gut bacteria to produce hydrogen and carbon dioxide during respiration and flatus production,10 which leads to a decrease in reducing sugars. In fact, microbial metabolism is a complex process in which various types and amounts of bacteria modulate individually or interdependently during fermentation;16 therefore, the variation in reducing sugar content may be attributed to the combined effects of microbial degradation and utilization.3.3 Determination of released free DAP monosaccharides after digestion and fermentation
As shown in Fig. 1A, six standard monosaccharides, L-rhamnose, D-ribose, L-(+)-arabinose, D-(+)-xylose, D-mannose, and D-(+)-glucose were satisfactorily separated within 13 min on GC-MS analysis. Based on the retention time and corresponding peaks in Fig. 1B–E, there were no monosaccharide-related peaks before or after gastrointestinal digestion when compared with the retention time of standard monosaccharides as shown in Fig. 1A, which infers that non-detectable released of free monosaccharides were generated during gastrointestinal digestion. We should note that the lower peaks shown on Fig. 1B–E were other substances in the gastrointestinal digestion medium captured by GC-MS. The absence of free monosaccharide production during digestion proves that acidic pH and substances contained within the gastrointestinal medium, especially bile salts, can only break down the glycosidic bonds or disrupt the structure sufficiently to lower the molecular weight and generate reducing ends. However, when focusing on the fermentation process, although DAP contains mannose and glucose, as shown in our previous study,17 only mannose was detected in steadily increasing quantities from the beginning to 12 h (Fig. 2A–C) and was non-detectable after a 24 h fermentation (Fig. 2D). The concentrations of released free monosaccharides is shown in Table 1 as 428.6 ng μL−1, 787.496 ng μL−1, and 987.183 ng μL−1 for DAP-F fermentation at 0.5, 6, and 12 h, respectively. The combined effects of microbial degradation and utilization by human fecal microbiota leads to the change in free monosaccharides, which indicates that consumed polysaccharides in the human GI tract were escaping digestion and small intestinal absorption but reached the large intestine and were altered by its multiple microbiota. Meanwhile, the inability to detect glucose during fermentation may not be because free glucose was not released, but because glucose, as the major carbohydrate source, was very rapidly absorbed by microbiota when released.16 At the same time, based on our previous study, the molar ratio of glucose and mannose was 1:2.48 (ref. 17); the high quantity of mannose leads to easier detection by GC-MS. However, mannose was not detectable after 24 h of fermentation, and one of the reasons may be that as culture time increased, the carbohydrate source contained in the fermentation medium was exhausted and only mannose could be used for microbial metabolism.37
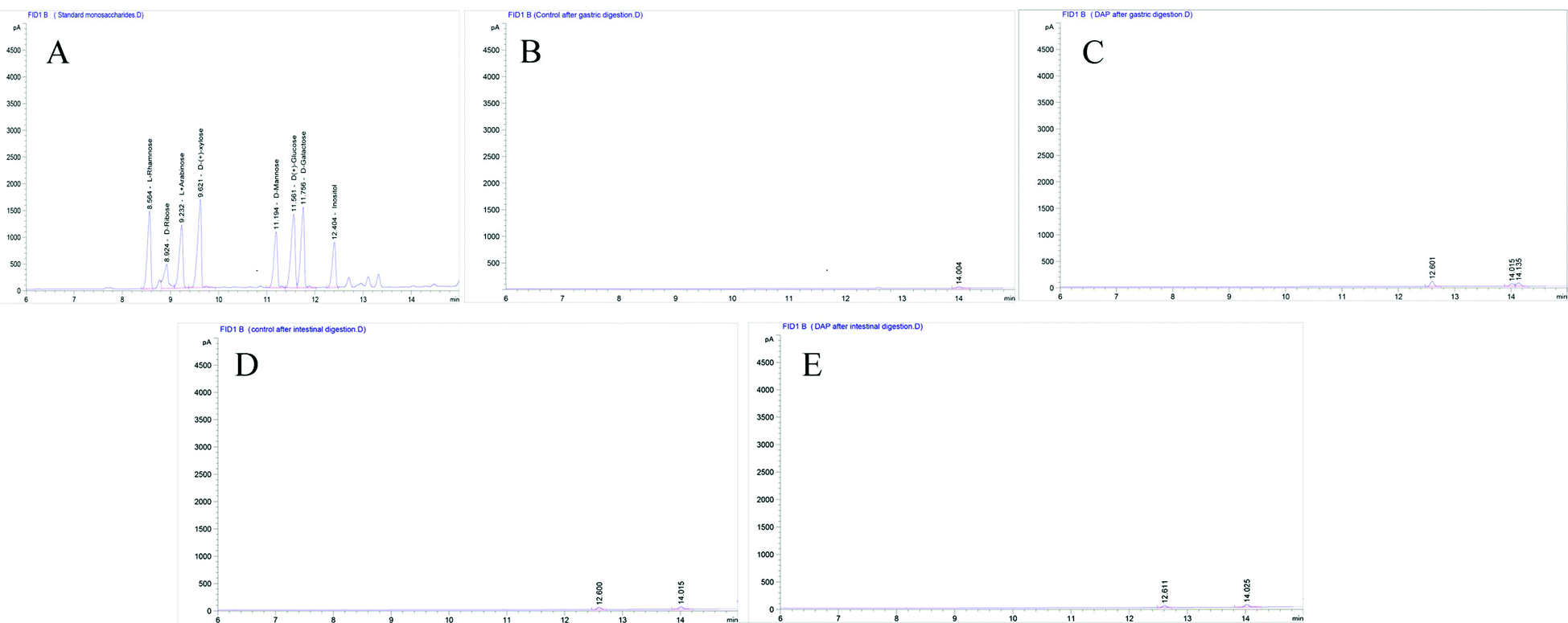 | ||
| Fig. 1 Monosaccharide compositions analysis by GC-MS. (A) Standard solution of monosaccharides including L-rhamnose, D-ribose, L-+-arabinose, D-(+)-xylose, D-mannose, D-(+)-glucose and D-galactose. Released free monosaccharides determination of D. aphyllum polysaccharides after stimulated gastric digestion (C) and stimulated intestinal digestion (D), and of control group after stimulated gastric digestion (B) and stimulated intestinal digestion (E) under a flame ionization detector with parameters of 230 °C at 300 mL min−1 for air rate, 30 mL min−1 for hydrogen rate, and 5 mL min−1 for nitrogen rate. | ||
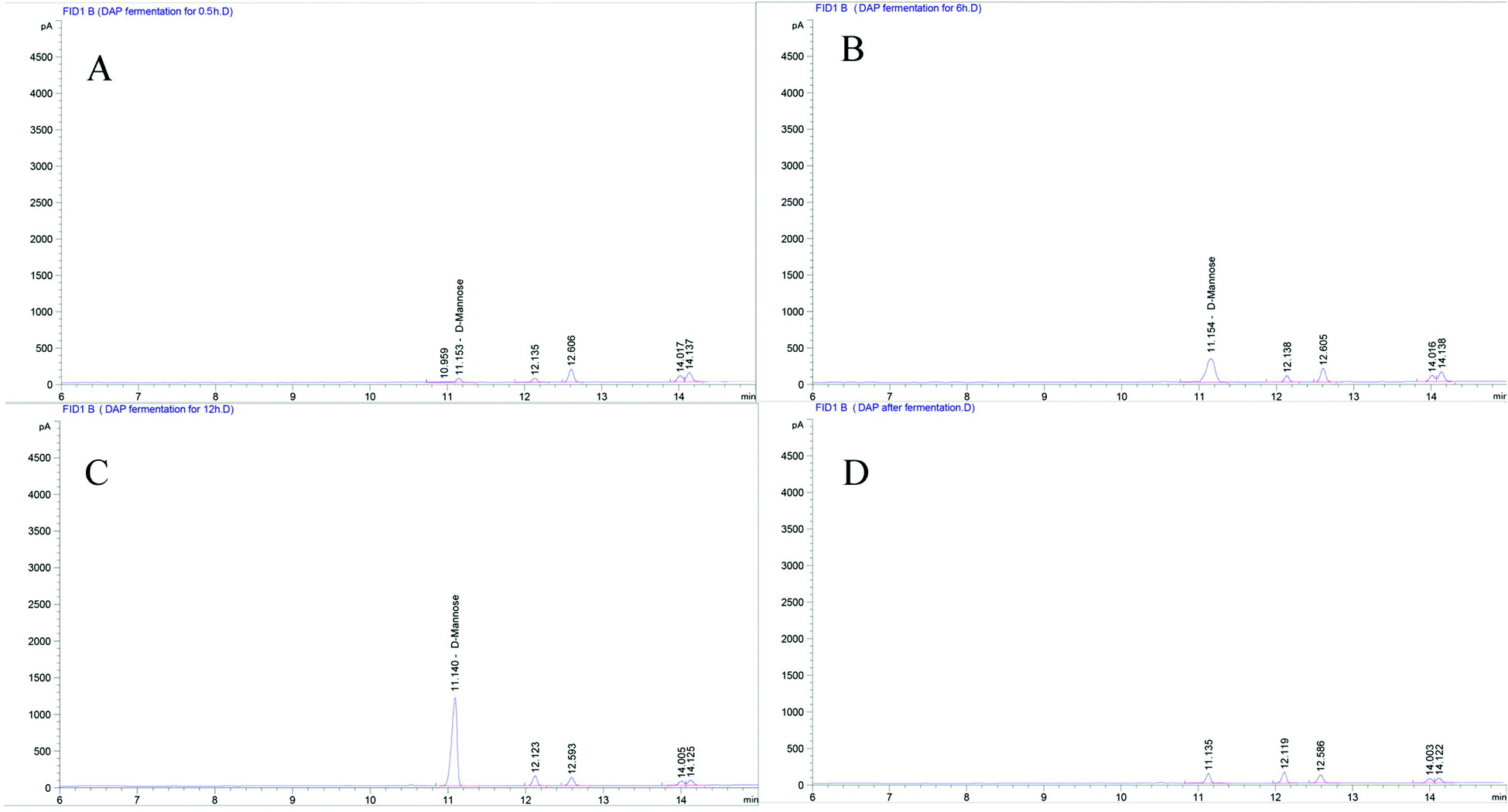 | ||
| Fig. 2 Monosaccharide compositions analysis by GC-MS. Released free monosaccharides determination of in vitro fermentation used D. aphyllum polysaccharides as substrate at different times within 0 h (A), 6 h (B), 12 h (C), 24 h (D) under a flame ionization detector with parameters of 230 °C at 300 mL min−1 for air rate, 30 mL min−1 for hydrogen rate, and 5 mL min−1 for nitrogen rate. | ||
3.4 Change in pH during fermentation
Some reports have shown decreased pH during polysaccharide fermentation,11,38 which were consistent with our study. According to Fig. 3, the pH of different groups after 48 h of fermentation by human fecal microbiota were all below 7. The initial acidic pH (5.25 ± 0.6) in each group was related to the initial contents of SCFAs in human fecal cultures as shown in Table 2. The pH of the controls were all higher than those of the polysaccharides because carbohydrate utilization by microbiota in the fermentation medium was much lower than that in the fermentation medium with polysaccharide sources; this produced more SCFAs to lower the pH. This was similarly observed with the comparison of the DAP-F, mannose, and glucose groups. Between 24 to 48 h of fermentation, there was no change in pH, indicating that there was no further production of SCFAs. In terms of the DAP-F fermentation pattern, the rapid drop in pH during the first 6 h indicates a great deal of SCFA generation during that period. Subsequently, slight increases in pH during the second 6 h implied a decrease in the speed of SCFA production, but for the third 6 h of fermentation, the speed obviously increased as the pH decreased again. Meanwhile, apart from the SCFA production, other factors influenced the change in pH, such as the culture medium variations, the types and amounts of microbiota in human fecal medium, and the types of SCFA generated. As we discussed above, pH is one of the most significant factors for altering the properties of polysaccharides; therefore, the molecular weight, reducing sugar content, and free monosaccharides were modulated by pH, mainly through the variation in SCFA production during the fermentation period.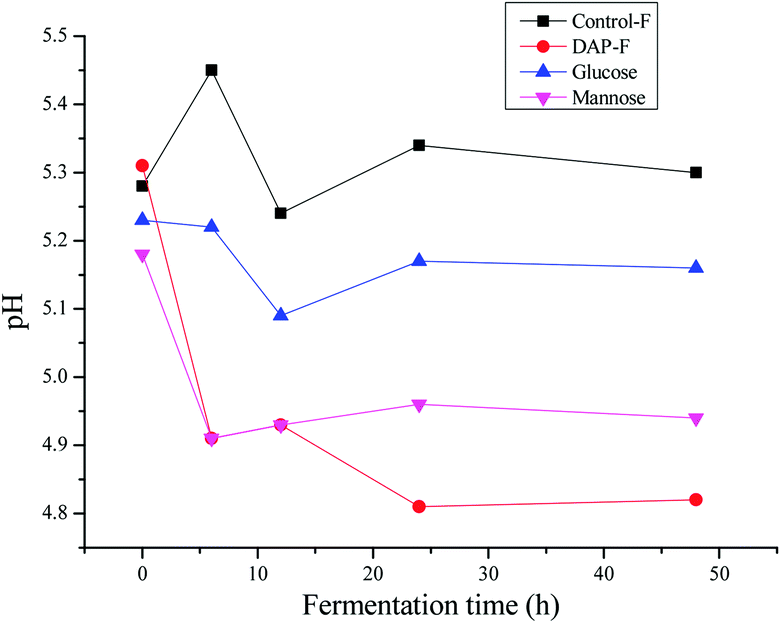 | ||
| Fig. 3 Changes of the pH in fermented cultures in vitro fermentation used D. aphyllum polysaccharides, mannose, glucose or water (Control) as substrate at different times within 0 h, 6 h, 12 h and 24 h. The data was shown as means ± SD (n = 3). | ||
| Samples | Time (h) | SCFAs (mM) | Total | |||||
|---|---|---|---|---|---|---|---|---|
| Acetic acid | Propionic acid | Butyric acid | i-Butyric acid | Valeric acid | i-Valeric acid | |||
| a Data are shown as mean ± standard (n = 3).b Mean values in the same column with different letters were significantly different (Tukey test, p < 0.05). | ||||||||
| DAP | 0.5 | 35.4682 ± 0.34ab | 21.4445 ± 0.79a | 6.5893 ± 0.12a | 5.0530 ± 0.53a | 8.2918 ± 0.37a | 5.9622 ± 1.06a | 82.8090 ± 0.38a |
| 6 | 41.2591 ± 0.56b | 26.7522 ± 0.63b | 8.1147 ± 0.23b | 5.5537 ± 0.55a | 8.8727 ± 0.45a | 4.2574 ± 0.65a | 94.8098 ± 0.43c | |
| 12 | 41.9415 ± 0.23b | 27.3576 ± 0.26b | 9.2107 ± 0.34c | 6.4534 ± 0.89a | 9.8632 ± 1.0a | 4.8503 ± 0.82a | 99.6767 ± 0.75d | |
| 24 | 50.5391 ± 0.56c | 28.1192 ± 0.85c | 10.9700 ± 0.54d | 7.3119 ± 0.98a | 11.9208 ± 0.65b | 6.3755 ± 0.66a | 115.2365 ± 0.81e | |
| Control | 0.5 | 36.9488 ± 0.78a | 25.7946 ± 0.78a | 6.3807 ± 0.22a | 5.2955 ± 0.45a | 7.0789 ± 0.72a | 4.4409 ± 0.37a | 85.9394 ± 0.69b |
| 6 | 34.3910 ± 0.39b | 26.9052 ± 0.82b | 5.9692 ± 0.45a | 4.7433 ± 0.51a | 6.5432 ± 0.51a | 5.3475 ± 0.66a | 83.8994 ± 0.79a | |
| 12 | 36.1859 ± 0.84a | 26.7541 ± 0.81b | 5.6494 ± 0.236a | 4.8263 ± 0.39a | 5.9959 ± 0.71a | 6.9415 ± 0.69a | 86.3531 ± 1.74b | |
| 24 | 35.9172 ± 0.45c | 26.9978 ± 0.56b | 6.2616 ± 0.67a | 6.0914 ± 0.12a | 5.8975 ± 0.34a | 5.3580 ± 0.62a | 86.5235 ± 1.07b | |
3.5 SCFA production
As shown in Fig. 4A, six standard SCFAs, including acetic acid, propionic acid, i-butyric acid, butyric acid, i-valeric acid, and valeric acid, were satisfactorily separated within 22 min of GC-MS analysis. These SCFAs were detected before DAP-F fermentation (Fig. 4B), but their content was much lower than that observed after 24 h of fermentation (Fig. 4C). SCFA peaks and related retention times by GC-MS analysis indicated that SCFA formation occurred during the fermentation period.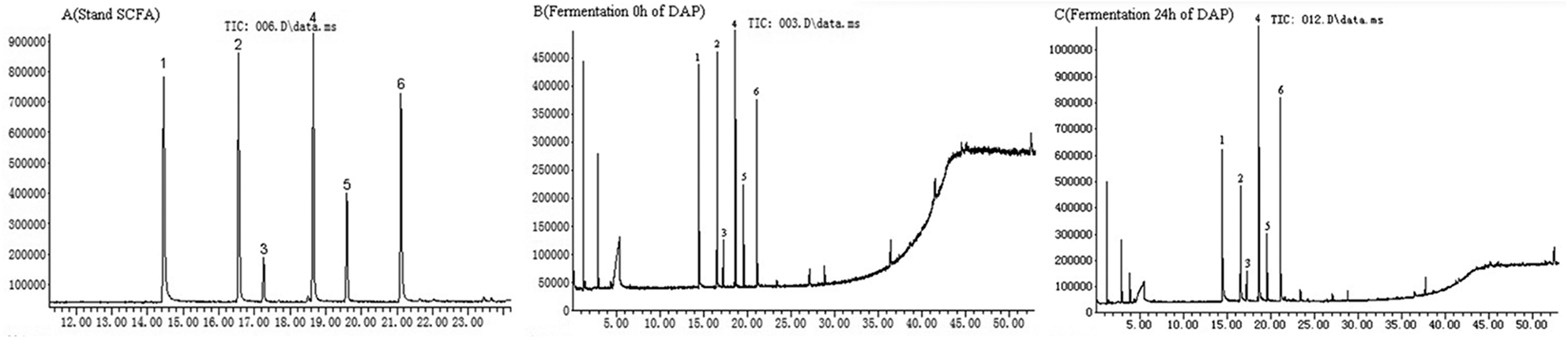 | ||
| Fig. 4 SCFAs analysis by GC-MS in standard solution (A) and in vitro fermentation used D. aphyllum polysaccharides as substrate before (B) and after (C) fermentation. Peaks followed by the order: 1, acetic acid; 2, propionic acid; 3, i-butyric acid; 4, butyric acid; 5, i-valeric acid; 6, valeric acid. | ||
As shown in Table 2, the production of these six types of SCFA in the control group was lower than that of the DAP-F group, which agreed with the pH results. In terms of the concentration of SCFAs in the DAP-F group, the production of all SCFAs increased with different tendencies; production of acetic acid, propionic acid, butyric acid, and valeric acid increased significantly from 35.4682 ± 0.34 to 50.5391 ± 0.56 mM, 21.4445 ± 0.79 to 28.1192 ± 0.85 mM, 6.5893 ± 0.12 to 10.9700 ± 0.54 mM, and 8.2918 ± 0.37 to 11.9208 ± 0.65 mM, respectively. These dominant SCFAs have been shown to have various advantages to human health and decrease the risk of chronic diseases.9,10
In general, various factors influence the content and distribution of SCFAs, including the type and amounts of microbiota, type and amounts of indigestible carbohydrates available, and the fermentation parameters such as time, temperature, and anaerobic environment.34 It has been reported that colonic microbiota obtains its energy mainly by oxidizing SCFAs in the order of butyric > valeric > propionic > acetic acid, which means that microbial metabolism activity also influences the proprieties of SCFAs.12 Moreover, the human diet contains a mixture of carbohydrates that influence fermentation patterns; thus, it is difficult to accurately determinate SCFA production values during the DAP fermentation process, but the tendency for their generation was confirmed by our results.
3.6 Carbohydrate contribution for SCFA production
Many studies have shown that improving the monosaccharide composition of polysaccharides significantly impacts the proprieties of SCFAs.6,39 Different types of monosaccharides varied the types and amounts of SCFAs; for example, the formation of acetic acid was largely attributed to non-starch, resistant starches, and certain oligosaccharides.10 When focusing on DAP fermentation, the generation of different types of SCFAs was affected by mannose and glucose contents (Fig. 5). Because increases in i-butyric acid and i-valeric acid after fermentation were not significant, only four types of SCFA, butyric acid, valeric acid, propionic acid, and acetic acid, were analyzed for their monosaccharide contribution. As the data shows, the fermentation time was not significantly related to the production of these SCFAs (Fig. 5D). By calculating the content of SCFAs produced when fermented with mannose or glucose only, we found that mannose was the prominent monosaccharide that promoted the generation of SCFA. However, it has been reported that glucose was the common major carbohydrate source contributing to the production of SCFAs.40,41 Considering that the amounts of mannose were higher than those of glucose in DAP (molar ratio 2.48:1), our results agreed with those reported above.
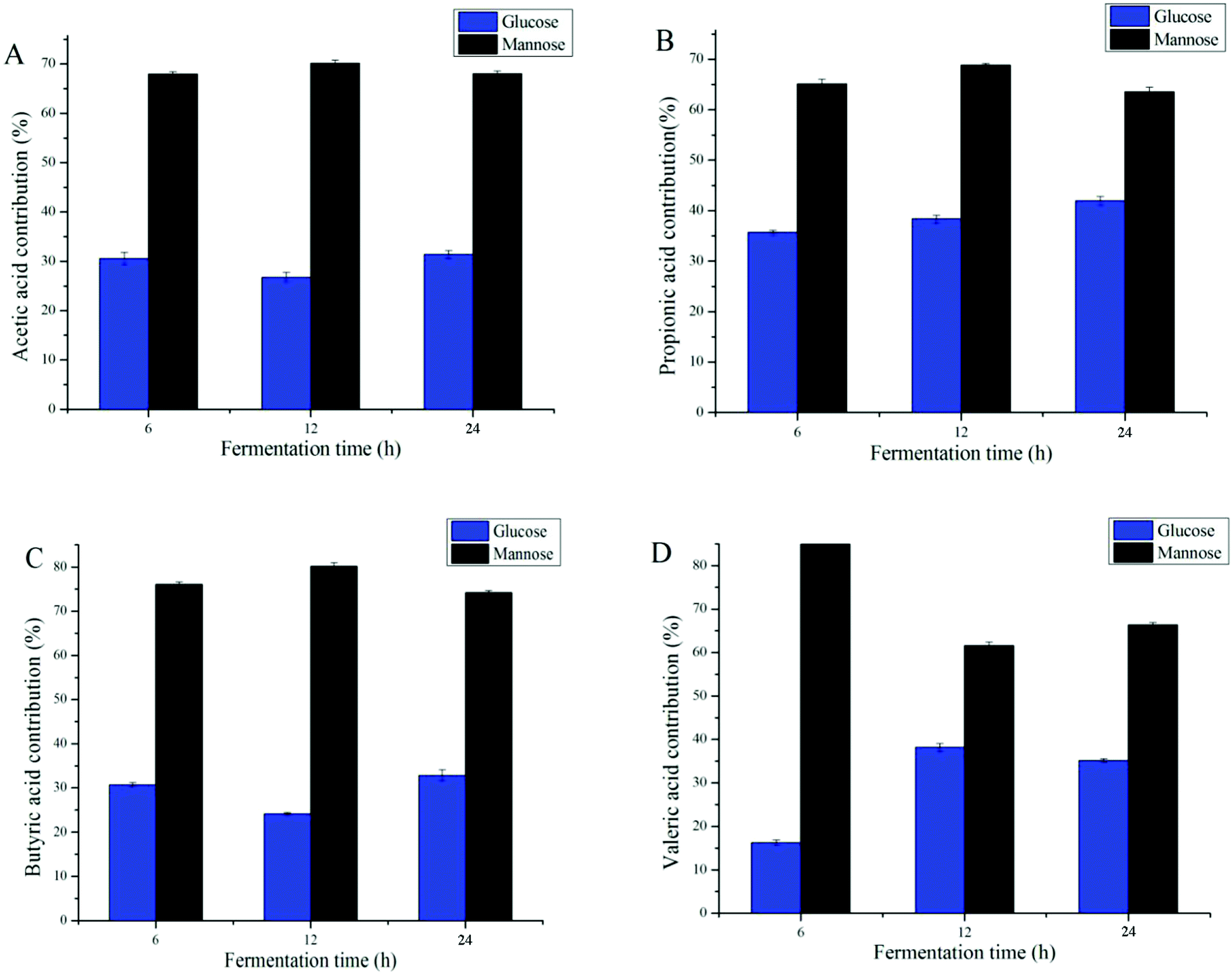 | ||
| Fig. 5 Contribution of glucose and mannose in the D. aphyllum polysaccharides for the production of acetic acid (A), propionic (B), butyric acid (C) and valeric acid (D) at different time during fermentation through GC-MS. | ||
4. Conclusion
The polysaccharide extracted from D. aphyllum was treated continuously with artificially simulated salivary, gastric, and intestinal digestion, and subsequently fermented in human fecal medium. A detailed flow chart (Fig. 6) shows the specific variations of each substance. To sum up briefly, salivary amylase showed no influence on DAP. However, the molecular weight decreased dramatically during the first 0.5 h of gastric digestion due to the acidic pH conditions, and then reduced steadily until it was undetectable during the subsequent GI tract consumption. The content of the reducing sugars increased steadily during GI digestion, indicating that the mode of molecular weight loss was due to breakage of glycosidic bonds. Meanwhile, the reducing sugar content increase and decrease during the fermentation was caused by the combined effects of degradation and utilization by fecal microbiota. Only released free mannose was detectable during the first 12 h of fermentation, which was contributed by the fecal microbiota metabolism. In terms of the fermentation pattern, the pH dropped significantly due to the formation of six types of SCFA, which were produced from mannose and glucose in different proportions. The above results show that polysaccharides extracted from D. aphyllum can be digested by the GI tract and are physiologically active in the human large bowel by lowering the pH of the large intestinal environment and promoting the production of SCFAs. This is the first report of artificial simulation of GI tract consumption in vitro of DAP, and may provide information on the in vitro digestion of other carbohydrates. However, because this study only stimulated the in vitro GI digestion and microflora fermentation of DAP, the relevant variations of bioactivity and the characterization of intestinal absorption and in vivo digestion are still unclear, and therefore further studies are needed.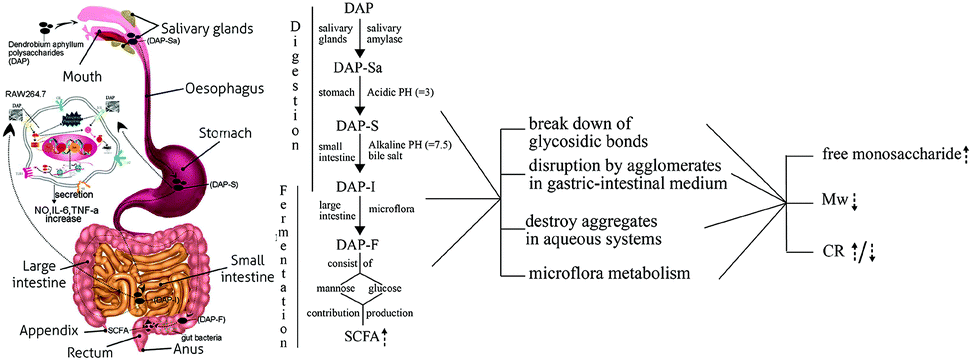 | ||
| Fig. 6 The flow chart of gastric-intestinal digestion and fermentation process of DAP consumption on human GI-tract and related variation. | ||
Conflicts of interest
There are no conflicts to declare.References
- L. Wang, W. U. Zi-Jian, A. G. Liu and L. I. Jian-Ying, Food Res. Dev., 2013, 34(6), 21–24 Search PubMed.
- S. S. Ferreira, C. P. Passos, P. Madureira, M. Vilanova and M. A. Coimbra, Carbohydr. Polym., 2015, 132, 378 CrossRef CAS PubMed.
- N. Binns, Probiotics Prebio Gut Microbio, 2013 Search PubMed.
- A. Barcenilla, S. E. Pryde, J. C. Martin, S. H. Duncan, C. S. Stewart, C. Henderson and H. J. Flint, Appl. Environ. Microbiol., 2000, 66, 1654 CrossRef CAS PubMed.
- U. S. Ramasamy, K. Venema, H. Gruppen and H. A. Schols, Bioact. Carbohydr. Diet. Fibre, 2014, 4, 48–57 CrossRef CAS.
- M. C. Jonathan, J. J. G. C. v. d. Borne, P. V. Wiechen, C. S. D. Silva and H. A. Scholsa, Food Chem., 2012, 133, 889–897 CrossRef CAS.
- C. M. Sawicki, K. A. Livingston, M. Obin, S. B. Roberts, M. Chung and N. M. Mckeown, Nutrients, 2017, 9, 125 CrossRef PubMed.
- M. H. M. C. V. Nuenen, K. Venema, J. C. J. V. D. Woude and E. J. Kuipers, Dig. Dis. Sci., 2004, 49, 485–491 CrossRef PubMed.
- G. Bosch, W. F. Pellikaan, P. G. Rutten, V. D. P. Af, M. W. Verstegen and W. H. Hendriks, J. Anim. Sci., 2008, 86, 2979–2989 CrossRef CAS PubMed.
- S. Macfarlane and G. T. Macfarlane, Proc. Nutr. Soc., 2003, 62, 67 CrossRef CAS PubMed.
- J.-L. Hu, S.-P. Nie, C. Li and M.-Y. Xie, Food Hydrocolloids, 2013, 33, 384–392 CrossRef CAS.
- Å. Henningsson, I. Björck and M. Nyman, Food Nutr. Res., 2001, 45 Search PubMed.
- T. B. Ng, J. Liu, J. H. Wong, X. Ye, S. C. W. Sze, T. Yao and K. Y. Zhang, Appl. Microbiol. Biotechnol., 2012, 93, 1795–1803 CrossRef CAS PubMed.
- X. M. Zi, C. L. Sheng, U. M. Goodale, S. C. Shao and J. Y. Gao, Mycorrhiza, 2014, 24, 487–499 CrossRef CAS PubMed.
- J. M. Wong, S. R. De, C. W. Kendall, A. Emam and D. J. Jenkins, J. Clin. Gastroenterol., 2006, 40, 235 CrossRef CAS PubMed.
- R. Crittenden, S. Karppinen, S. Ojanen, M. Tenkanen, R. Fagerström, J. Mättö, M. Saarela, T. Mattila-Sandholm and K. Poutanen, J. Sci. Food Agric., 2002, 82, 781–789 CrossRef CAS.
- H. Liu, J. Ma, F. Gong, F. Wei, X. Zhang and H. Wu, Int. J. Food Sci. Technol., 2017 DOI:10.1111/ijfs.13695.
- J. L. Hu, S. P. Nie, F. F. Min and M. Y. Xie, Carbohydr. Polym., 2013, 92, 1143 CrossRef CAS PubMed.
- C. Chen, B. Zhang, X. Fu, L. J. You, A. M. Abbasi and R. H. Liu, Food Hydrocolloids, 2016, 58, 171–178 CrossRef CAS.
- I. Asano, K. Hamaguchi, S. Fujii and H. Iino, Food Sci. Technol. Res., 2003, 9, 62–66 CrossRef CAS.
- N. L. Mkandawire, S. A. Weier, C. L. Weller, D. S. Jackson and D. J. Rose, LWT–Food Sci. Technol., 2015, 62, 662–667 CrossRef CAS.
- F. Sánchez-Patán, E. Barroso, d. W. T. Van, A. Jiménez-Girón, P. J. Martín-Alvarez, M. V. Moreno-Arribas, M. C. Martínez-Cuesta, C. Peláez, T. Requena and B. Bartolomé, Food Chem., 2015, 183, 273 CrossRef PubMed.
- C. J. V. Nevel, N. A. Dierick, J. A. Decuypere and S. M. D. Smet, Arch. Anim. Nutr., 2006, 60, 477 CrossRef PubMed.
- F. Bianchi, M. Dall'Asta, R. D. Del, A. Mangia, M. Musci and F. Scazzina, Food Chem., 2011, 129, 200–205 CrossRef CAS.
- S. M. V. Ruth and J. P. Roozen, Food Chem., 2000, 71, 339–345 CrossRef.
- B. P. Thornton, V. Vĕtvicka, M. Pitman, R. C. Goldman and G. D. Ross, J. Immunol., 1996, 156, 1235–1246 CAS.
- G. D. Ross, Immunol. Res., 2002, 25, 219–227 CrossRef CAS PubMed.
- W. Wei, L. Feng, W. R. Bao, D. L. Ma, C. H. Leung, S. P. Nie and Q. B. Han, Planta Med., 2016, 64, 881 CAS.
- C. C. Maningat and P. A. Seib, Cereal Chem., 2010 Search PubMed.
- W. Li, Q. Wang, S. W. Cui, X. Huang and Y. Kakuda, Food Hydrocolloids, 2006, 20, 361–368 CrossRef CAS.
- B. J. Savary, A. T. Hotchkiss, M. L. Fishman, R. G. Cameron and R. G. ShattersDevelopment of a Valencia Orange Pectin Methylesterase for Generating Novel Pectin Products, in Advances in Pectin and Pectinase Research, ed. F. Voragen, H. Schols and R. Visser, Springer, Dordrecht 2003 Search PubMed.
- A. Thorburn, J. Muir and J. Proietto, Metab., Clin. Exp., 1993, 42, 780–785 CrossRef CAS PubMed.
- A. G. Oomen, A. Hack, M. Minekus, E. Zeijdner, C. Cornelis, G. Schoeters, W. Verstraete, T. Van de Wiele, J. Wragg and C. J. M. Rompelberg, Environ. Sci. Technol., 2002, 36, 3326–3334 CrossRef CAS PubMed.
- S. A. Hughes, P. R. Shewry, L. Li, G. R. Gibson, a. M. L. Sanz and R. A. Rastall, J. Agric. Food Chem., 2007, 55, 4589–4595 CrossRef CAS PubMed.
- Y. Peng and L. Zhang, J. Biochem. Biophys. Methods, 2003, 56, 243–252 CrossRef CAS PubMed.
- Q. Wang, X. Huang, A. Nakamura, W. Burchard and F. R. Hallett, Carbohydr. Res., 2005, 340, 2637–2644 CrossRef CAS PubMed.
- U. Farooq, M. Mohsin, X. Liu and H. Zhang, Trop. J. Pharm. Res., 2013, 12, 189–194 Search PubMed.
- J. A. Arcila, S. A. Weier and D. J. Rose, Food Res. Int., 2015, 74, 217–223 CrossRef CAS PubMed.
- J. L. Hu, S. P. Nie, F. F. Min and M. Y. Xie, J. Agric. Food Chem., 2012, 60, 11525–11532 CrossRef CAS PubMed.
- S. Karppinen, K. Liukkonen, A. M. Aura, P. Forssell and K. Poutanen, J. Sci. Food Agric., 2000, 80, 1469–1476 CrossRef CAS.
- A. Pihlantoleppala, Trends Food Sci. Technol., 2000, 11, 347–356 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2018 |