
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLightweight foams of amine-rich organosilica and cellulose nanofibrils by foaming and controlled condensation of aminosilane†
Korneliya
Gordeyeva
a,
Hugo
Voisin
a,
Niklas
Hedin
 a,
Lennart
Bergström
a and
Nathalie
Lavoine
*ab
a,
Lennart
Bergström
a and
Nathalie
Lavoine
*ab
aDepartment of Materials and Environmental Chemistry, Stockholm University, Stockholm 10691, Sweden
bDepartment of Forest Biomaterials, North Carolina State University, Raleigh, North Carolina, USA. E-mail: nmlavoin@ncsu.edu
First published on 19th September 2018
Abstract
Organosilica foams are commonly formed by a multistep process involving hydrolysis and condensation of organosilanes followed by solvent exchange and e.g. supercritical CO2 drying. Here, we propose a straightforward route to synthesize lightweight hybrid foams from aqueous dispersions of a surface-active aminosilane (AS) and TEMPO-oxidized cellulose nanofibrils (TCNFs). Air bubbles were introduced in the TCNF/AS dispersion by mechanical blending, and the foam was solidified by oven-drying. Evaporative drying at mild temperature (60 °C) resulted in dry foams with low densities (25–50 kg m−3), high porosities (96–99%) and macropores of 150–300 μm in diameter. The foaming and foam stabilization were successful for a pH range of 10.4–10.8 for foams containing 55–65 wt% of organosilica in the dry state. The protonation of AS increased the ionic strength of the dispersion and enhanced the interparticle interactions with TCNFs and, in turn, the foam viscosity and foam stability upon drying. The evaporation of water catalyzed the condensation of the AS to form low-molecular linear polymers, which resulted in an increased stiffness and strength of the foam lamella. The crosslinking of the AS polymeric network with the TCNF matrix allowed lightweight and homogeneous macroporous foams to be obtained with controlled densities and high amine content (amine content >4.5 mmol g−1) using an environmentally friendly technique.
Introduction
Organosilica foams are lightweight porous materials (3–500 kg m−3)1 used e.g. as thermal and acoustic insulation materials,2 CO23 and oil sorbents, and water purification filters.4 Organosilica foams can be produced in several ways, including sol–gel processing of an organofunctional silane (OS),5–7 co-gelation of an OS with tetra-alkoxysilanes8,9 or post-modification of silica alcogels.10 The sol–gel processing of the OS is done in a multistep (i to iv) process involving the use of an alcohol–water mixed solvent.11 Typically, the pH of the silane solution is first (i) lowered to acidic values for providing a good hydrolysis yield. Then, the pH is (ii) raised to basic values to trigger the condensation of silanols and the formation of a gel.12 The aging of the gel is a necessary step (iii) to strengthen the structure of the wet gel and is followed by (iv) a solvent exchange to remove unreacted species and prepare for the drying step.2 In comparison, the post-modification approach consists of surface functionalization of a silica alcogel with an OS.13 Dry organosilica foams are commonly obtained by either freeze-drying4,14 or supercritical CO2 drying.9,10,15 These processes are rather time consuming (about a couple of weeks is usually required for completing the condensation, washing and drying of the gel2) or energy-intensive.2,16The chemical structure and composition of the OS (e.g. number of organic groups) define the properties and functionality of the organosilica foams such as hydrophobicity,9 mechanical elasticity,17 adsorption capacity of organic solvents18 and bacteria resistance.19 One of the main advantages for using OS is to reinforce the foams mechanical properties and reduce the drying cost and time. A simple solvent evaporation process at ambient conditions can, indeed, be performed to produce organosilica foams, instead of low-pressure drying techniques like supercritical drying.2 Alkyl-functionalized silanes7,20,21 or silanes with a reduced content of OH-groups (mono-, di- or tri-alkoxysilanes)22,23 are for example known to reduce the capillary pressure induced by solvent evaporation, thus minimizing the shrinkage, collapse and cracking of the foams upon drying.2,21,24
The use of OS with alkyl groups is yet not sufficient to fully preserve the foam structure and prevent its collapse upon drying.21 Other processing techniques have thus been investigated, which include the use of solvents with reduced surface tension (e.g. isopropanol/n-hexane,13N,N-dimethylformamide6 and ethanol/heptane25), in situ polymerization in organosilica gel8,26,27 or direct polymer addition28,29 to an OS solution.2In situ polymerization in wet silica gels can lead to the formation of a strongly cross-linked polymer/silica network, resulting in a monolithic aerogel with limited shrinkage (16%).8 By direct polymer addition, the chemical bonds formed between the OS and the polymer28 or tetraalkoxysilane and OS modified polymer30 can restrict the mobility of the wet silica network upon evaporation and limit the shrinkage and cracking of the material. However, the best control and preservation of the foam shrinkage and structure has so far been achieved by combining techniques that includes the use of polymers and organic solvents (in particular, isopropanol30 or acetone8) or the post-hydrophobization of the alcogel/polymer using an OS.28
One polymer in particular that has shown good potential in wet gel reinforcement is [nano]cellulose.4,28,31,32 Cellulose nanofibrils (CNFs) are nanoparticles of high aspect ratio (50–200)33 and high stiffness (20–50 GPa).34 CNFs have been successfully used as supporting matrix in the preparation of inorganic foams4 and as reinforcing additive in synthetic foams (polyurethane,35 polystyrene,36 resins37,38). The versatile surface chemistry and high aspect ratio of CNFs is an attractive feature for the preparation of functional cellulose-based foams using mechanical blending and evaporative drying of aqueous dispersions.39,40 For example, the crosslinking of negatively charged TEMPO-oxidized CNFs (TCNFs)41 by Ca2+ ions resulting from slow in situ dissolution of calcium carbonate, stabilized the air/water interfaces of surfactant foams upon drying, and resulted in lightweight (9–15 kg m−3) cellular materials with good compressive strength (0.9–1.4 MPa).39 In comparison to CNFs, short sulfonated cellulose nanocrystals did not provide wet foam stability sufficient enough to withstand the capillary compression arising upon foam drying.42 Other strategies involving air/water interface stabilization, such as Pickering foams, were also investigated using TCNFs, partially hydrophobized by electrostatically adsorbed octylamine.40 A careful control of the solvent evaporation and humidity during drying and covalent cross-linking of TCNFs resulted in lightweight cellulose-based foams with a homogeneous morphology.40,43 The surface modification of (nano)cellulose-based materials by selected OS was also successful for the production of stable emulsions44,45 and functional films and foams with e.g. high hydrophobicity,46 enhanced CO247 and oil adsorption capacity,4 improved thermal stability48 and antibacterial properties.49 Most of the surface modifications of CNFs by an OS are commonly performed as a pre-step before material elaboration (usually in organic solvent50), or as a post-modification of the CNF-based product.46 Many attempts to produce organosilica/cellulose foams were undertaken using OS gelation in [nano]cellulose fiber network,4,47,51 but few works only have presented the ambient dried foams.28 The direct blending of OS (or silane) and CNFs has also never been exploited for producing (organo)silica/polymer foams. This route has, nevertheless, been investigated to produce silica ceramic foams, but the use of surfactant was necessary to stabilize the air–water interface.52
Here, we present a new surfactant-free one-pot synthesis route of low density hybrid organosilica foams, which involves (i) the formation of a wet aqueous foam by mechanical blending of an organosilane/TCNFs dispersion followed by, (ii) the evaporation of water to produce a dry hybrid foam. Compared to previous studies, water is here the solely solvent used, and no surfactants were added for stabilizing the air/water interface.52 TCNFs and a commercially available 3-aminopropyl(methyl)diethoxysilane (AS) were used to prepare the wet and solid foams. The influence of TCNFs, AS concentration and pH of the dispersion on the foamability and stability of the wet foams upon drying were studied. The best conditions for foam processing and the key mechanisms involved in the foam formation and stabilization are proposed.
Experimental part
Materials
Never dried softwood sulfite pulp (Domsjö dissolving pulp) was provided by the Department of Fiber and Polymer Technology at KTH, the Royal Institute of Technology (Stockholm, Sweden) and used as the cellulose source. Sodium hypochlorite (NaClO, 11–14% available chlorine, Alfa Aesar), TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy free radical, ≥98%, Alfa Aesar), sodium hydroxide (NaOH, ≥99.2%, VWR Chemicals), sodium bromide (NaBr, BioUltra, ≥99.5%, Sigma Aldrich), concentrated hydrochloric acid (HCl, VWR Chemicals, 35%), 3-aminopropyl(methyl)diethoxysilane (AS, 97%, Sigma-Aldrich Sweden AB), and D-(+)-gluconic acid δ-lactone (GDL, ≥99%, Sigma Aldrich) were used as received.Preparation of TCNFs
Prior to the TEMPO-mediated oxidation, the cellulose pulp was washed using an HCl solution at pH = 2 under stirring at 500 rpm for 30 min. The excess of ions was thoroughly washed out by filtration using deionized water. 40 g of dry pulp, 0.64 g of TEMPO, 4 g of NaBr and 1.87 L of deionized water were mixed together until complete dissolution of the powders and adjusted to pH = 10 by the addition of 0.5 M of NaOH. The oxidation process was performed for 3 h under drop-wise addition of 60 mmol of NaClO solution while keeping the pH = 10 constant. The TEMPO-oxidized pulp was washed with deionized water and disintegrated using a high pressure Microfluidizer (M-110EH, Microfluidics) with a 200 μm and a 100 μm wide chamber connected in series. Three passes through each chamber were conducted to produce 1 wt% TCNF suspension.TCNFs with exchanged counter ion from Na+ to H+ were prepared as reference for IR spectroscopy as follows: the TCNF dispersion was first diluted at 0.2 wt%, then stirred for 30 min at pH = 1, adjusted by the addition of a 0.5 M HCl solution, and subsequently dialyzed against DI water until pH = 4.5.
Characterization of the TCNFs
The dimensions of the TCNFs were determined by Atomic Force Microscopy (AFM) using a Dimension 3100 SPM (Veeco, USA). A drop of TCNF suspension at 0.005 wt% was casted onto freshly cleaved mica substrate treated with (3-aminopropyl)triethoxysilane (Sigma Aldrich, 99%) and analyzed in tapping mode in air. Using the softwares NanoscopeAnalysis and Origin 2015, the average nanofibril diameter was 2.9 ± 0.9 nm for lengths up to 1 μm. The diameter was determined as the height of an individual nanofibril and was measured on 500 single nanoparticles. The height distribution histogram was plotted and the average nanofibril diameter was estimated using a Gaussian function.The total charge density of TCNFs was measured by conductometric titration using 0.5 M NaOH, as previously reported.53 The titration was performed twice per specimen and the average total charge was 1.60 ± 0.01 mmol of carboxyl groups per gram of cellulose.
Preparation of the TCNF/AS-based foams
The wet foams were prepared in glass beakers with a starting volume of the suspension or solution of 10 cm3. The TCNF suspension was dispersed in deionized water using a homogenizer at 7000 rpm for 3 min (Ultra-Turrax, IKA, Sweden). The desired volume of AS was added to the TCNF suspension, and the mixture was foamed for 3 min at 7000 rpm. The concentration of AS varied between 0.05 and 5 wt%, while the TCNF concentration was kept at 0.5 wt%. Abbreviations for the dispersions and foams are listed in Table 1. The pH of the TCNF/AS dispersions was adjusted during the last 1.5 min of the foaming process (i.e. right after the AS was homogeneously mixed and foamed with TCNFs) using a 0.5 M solution of either GDL or HCl. The dry foams were obtained after drying of the wet foams in an oven at 60 °C, for 72 h, covered with a perforated aluminum foil.39| Abbreviation | Description |
|---|---|
| ASxd(pH) | A wet dispersion (d), a wet foam (wf) or a dry foam (f) prepared from x wt% aqueous aminosilane (AS) solution at adjusted pH. |
| ASxwf(pH) | |
| ASxf(pH) | |
| TCNFd | A wet dispersion (d) or a freeze-dried (fd) TCNF dispersion at 0.5 wt% and neutral pH. |
| TCNFfd | |
| TCNF/ASxd | A dispersion (d), a wet foam (wf) or a dry foam (f) prepared from a mixture of 0.5 wt% TCNF dispersion and x wt% aqueous AS solution (pH > AS pKa = 10). |
| TCNF/ASxwf | |
| TCNF/ASxf | |
| TCNF/ASxd(pH) | A dispersion (d), a wet foam (wf), or a dry foam (f) prepared from a mixture of 0.5 wt% TCNF dispersion, x wt% aqueous AS solution, at a controlled pH. |
| TCNF/ASxwf(pH) | |
| TCNF/ASxf(pH) | |
Characterization of the wet TCNF/AS foams
The foaming behavior of the TCNF/ASwf at different AS concentrations and pH, and respective reference (3 wt% aqueous AS solution) was assessed by determining the volume of the wet foam, Vwet![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) foam (cm3), 10 s after foaming.
foam (cm3), 10 s after foaming.
The diameter of the air bubbles of the wet foams, which were prepared in transparent plastic containers (20 × 39 × 64 cm3) with flat walls to avoid optical distortions, were measured by a USB digital microscope (2 M pixels, magnification up to 500×). The bubble size distribution and average bubble diameter were estimated on at least 500 bubbles. Using the software Origin, the bubble size distribution data was fitted using a Gaussian distribution function.
The adsorption of AS on TCNFs was observed by electrophoretic mobility of 5 time-diluted dispersions of TCNFd and TCNF/AS1.5d at varying pH. The measurements were performed using a Zetasizer Nano series spectrometer (Malvern Instruments Ltd, England) in disposable cuvettes at 22 °C using deionized water as dispersant. Each measurement was an average of 62 scans.
The air/water interfaces of the TCNF/ASwf were observed by a Zeiss LSM 780 inverted confocal microscope system (Germany). Fluorescently labeled TCNFs were prepared by adding few droplets of a mixture of 1 g L−1 Calcofluor White Stain M2R and 0.5 g L−1 Evans blue solution (Sigma-Aldrich) to a 1 wt% TCNF suspension under homogenization at 6000 rpm. The suspension was left to react for 15 min and dialyzed against deionized water for two weeks to remove unreacted dye. A wet foam from the dyed TCNFs was prepared and mounted between a glass slide and a coverslip, surrounded by silicon oil to prevent evaporation and drying. The objective used for observation was a Plan-Apochromat 10×/0.45. Fluorescent excitation and emission were 405 and 415–505 nm, respectively. The pinhole size was set at 30 μm.
The average interfacial tension (IFT) of AS3d and TCNF/AS3d was measured by pendant drop testing using a Dataphysics Instruments OCA-40 micro dilatometer (Germany) and the software SCA20. The oscillation frequency was kept constant at 0.3 Hz. The diameter of the needle was set to 1.83 mm and the experiments were carried out at 21 °C and 50% RH.
The rheological properties of the TCNF/ASxd(pH) and TCNF dispersions were studied by oscillatory strain sweep test at 25 °C using a rheometer Anton Paar Physica MCR 301 (USA) equipped with a parallel-plate geometry (PP25-SN45236) with a 1 mm gap. The evolution of the storage (G′) and loss (G′′) moduli at a frequency of 1 Hz and a strain of 0.1% was monitored as a function of the time and temperature as follows: 15 min at 25 °C, heating at 2 °C min−1 to 60 °C, 3.6 h at 60 °C, cooling down at 2 °C min−1 to 25 °C and 15 min at 25 °C. A strain sweep between 0.01% and 100% at a constant frequency of 1 Hz was then applied. The storage modulus for each composition was estimated from the linear part of the strain sweep test at 2.15% (elastic region) of the strain and averaged over three samples. The rheology of the TCNF/ASxwf(pH) wet foams was assessed using a vane-in-cylinder geometry (C-CC27/T200/SS cylinder and ST14-4V-35/125 stirrer). G′ and G′′ of 30 mL wet foam were measured one minute after foaming and constantly recorded over time as a function of the temperature: 5 min at 25 °C, heating from 25 °C to 60 °C at 2 °C min−1 and 12 h at 60 °C. The strain and oscillation frequency were kept constant throughout the experiment at 0.1% and 5 Hz. The water evaporation was limited for all experiments by using a solvent trap and an oil cover.
The condensation of the AS at various conditions was studied by liquid state {1H}29Si NMR on a wet 3 wt% AS solution at room temperature, wet TCNF/AS3 dispersions at room temperature and 60 °C with and without pre-heating (12 h at 60 °C) and on TCNF/AS3(10.8) at 60 °C. The liquid state {1H}29Si NMR spectra were acquired with a refocused INEPT pulse program using composite pulse decoupling (waltz64) of the 1H contributions during the acquisition. A Bruker Advance spectrometer with a BBO probe head attached to a 9.4 T narrow bore magnet was used with a 29Si frequency of 79.5 MHz.
The chemical shift scale was calibrated with the chemical shifts for the dimeric compounds of the organosilane monomer 3-aminopropyl(methyl)diethoxysilane.54 The temperature scale was externally calibrated with the temperature dependent chemical shifts of methanol.55
Characterization of the dry TCNF/AS foams
The foam density, ρ (kg cm−3), and porosity, φ (%), were estimated at ambient conditions.The volume of the dry foam was measured using a caliper with a precision of ±0.01 mm. The foam porosity was calculated as follows using eqn (1):
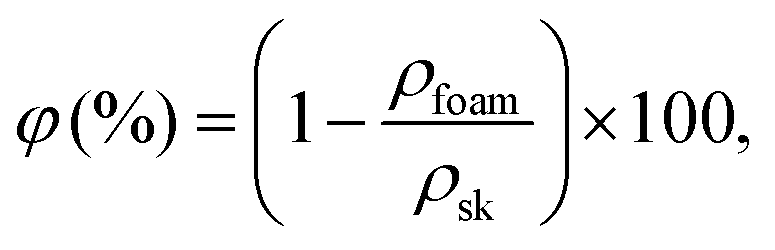 | (1) |
The porous structure of the dry foams was observed by tabletop scanning electron microscope (SEM) TM 3000 (Hitachi, Japan) at an accelerating voltage 15 kV and magnification range of ×30–50. Back-scattered electrons were detected using a 4-segment solid state semiconductor detector (TOPO mode). Prior to the SEM analysis, the sliced foams were coated with a thin layer of gold (35 s, 10 mA, 10 mbar of Ar gas). The pore diameters were measured and averaged on ca. 500 pores by SEM image analysis using the software ImageJ. The average pore size was determined by a non-linear regression analysis of the pore size distribution histogram using a Gaussian function. The polydispersity index (PDI) of the pores for each composition was calculated as follows (eqn (2)):
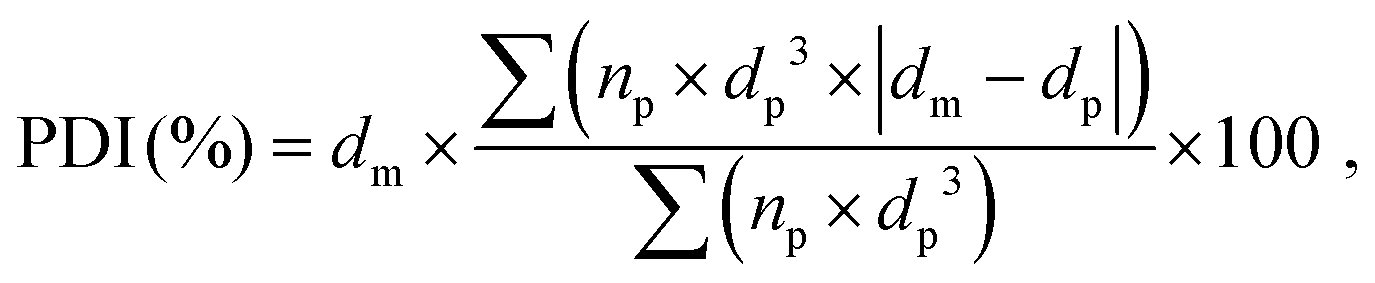 | (2) |
The compressive behaviour of the solid foams was tested using an Instron 5944 (USA), at 10% min−1, 50% of relative humidity and 23 °C. The Young's modulus was estimated from the linear part of the stress–strain curve at low strain, and was averaged over ten samples considering an average density of 33 ± 2 kg m−3.
The chemical composition of the foams was identified by ATR FTIR spectroscopy (Varian 600-IR, Varian Medical Systems, Inc., USA). Each measurement was performed on oven-dried TCNF/AS1.5f(pH) foams. 64 scans were recorded per foam with a resolution of 4 cm−1. Similar IR characterization was conducted on the TCNFf and 0.05 wt% ASf as reference.
The content of condensed silane was investigated by thermogravimetric analysis (TGA) using a TG-TA Instruments Discovery device (USA). The dry foams TCNF/AS2(11) and TCNF/AS2(10.8) were heated to 900 °C at a heating rate of 2 °C min−1 in an air flow of 20 mL min−1 and kept at constant temperature for 30 min.
The condensation degree of AS was studied by solid state {1H}29Si NMR on dry foam TCNF/AS3(10.8). Spectra were recorded under magic angle spinning (MAS) of 5 kHz on a Bruker Advance III spectrometer with a 7 mm probe head and an attached 9.4 T wide bore magnet. The 29Si frequency was 79.5 MHz. Spectra were recorded by acquiring 2560 transients using cross polarization (CP) contact times of 1, 2, 4 and 6 ms under 1H SPINAL decoupling. The CP matching and 29Si chemical shift scale were set externally by using spectra of tetrakis(trimethylsilyl)silane (Sigma Aldrich). The {1H}29Si spectra were deconvoluted with Matlab and matNMR routines, during which the fraction of Lorentzian contribution was held to 0.3. A Lorentzian line broadening of 100 Hz, significantly smaller than the linewidths, was applied. A recycling time of 5 s was used.
The content of N in dry foams was measured by MEDAC Ltd Analytical and Chemical Consultancy Services in UK using inductively coupled plasma-optical emission spectroscopy (ICP-OES). CHN analysis was performed on a FlashEA 1112 Analyzer (Thermo Scientific, USA).
Results and discussion
TCNF/AS solid foams composition and structure
Macroporous cellular materials (Fig. 1) of low density (25–70 kg m−3) and high porosity (above 96%) with an organosilica content of 55–65 wt% (Table 2) were obtained after oven-drying of wet aqueous foams made by blending the AS (3-aminopropyl(methyl)diethoxysilane, Fig. SI.1a, ESI†) and TCNFs (carboxyl contents of 1.6 mmol g−1, Fig. SI.1b, ESI†). The porous structure and the density of the foams were tuned by varying the AS concentration and pH of the wet dispersions (Fig. 1 and Table 2). Increase of the AS concentration from 2 to 4 wt% (Fig. 1a and b) or reduction of the pH from 11 to the measured pKa (=10) of the AS (Fig. 1a and c) generated foams with near spherical pores of relatively narrow size distribution, reduced pore diameters (from 1 mm to 150–360 μm), and no big cavities, as can be seen for TCNF/AS2f(11.1) vs. TCNF/AS2f(10.8) in Fig. 1a and c. When the pH reached or went below the pKa of the AS, the foam morphology degraded (Fig. 1d) with formation of bigger and polydisperse pores (295 μm with PDI 87%). | ||
| Fig. 1 SEM images of the dry foam cross-sections: (a) TCNF/AS2f(11.1), (b) TCNF/AS4f(11.1), (c) TCNF/AS2f(10.8), (d) TCNF/AS2f(10.0). The insets represent the pictures of the corresponding dry foams. | ||
| Solid foams | Density, ρ, kg m−3 | Porosity, φ, % | Average pore diameter,adporeav, μm | Pore size polydispersity, PDI, % | Composition of dry foams | |
|---|---|---|---|---|---|---|
| Concentration of condensed AS (Si),b wt% | Concentration of N, mmol g−1 (wt%) | |||||
| a The average pore diameters are presented only for the most frequent pore sizes (the most intense peak of the Gaussian fit). The less intense peaks describing pore size distribution histograms are neglected and only taken into consideration during calculation of the PDI. b Weight fraction of condensed AS was determined from the residues obtained after TGA in air and the structure of organosilica polymer determined from solid state {1H}29Si NMR. | ||||||
| TCNF/AS2f(11.1) | 14 ± 2 | 99.4 ± 0.07 | ≥1000 | n/a | 59 (13) | 4.7 ± 0.15 (6.6) |
| TCNF/AS4f(11.1) | 42 ± 17 | 98.3 ± 0.39 | 257 ± 81 | 7 | n/a | n/a |
| TCNF/AS2f(10.8) | 34 ± 3 | 98.4 ± 0.13 | 356 ± 162 | 23 | 62 (14) | 4.5 ± 0.14 (6.3) |
| TCNF/AS2f(10.5) | 26 ± 2 | 98.7 ± 0.11 | 151 ± 48 | 10 | n/a | n/a |
| TCNF/AS2f(10.0) | 67 ± 12 | 96.2 ± 0.66 | 295 ± 123 | 87 | n/a | n/a |
The foam density increased from 14 to 42 kg m−3 with the increasing AS concentration from 2 to 4 wt%, and decreased from 34 to 26 kg m−3 when the pH was reduced from 10.8 to 10.5 (Table 2). These densities (Si content >13 wt%) are much lower than the reported ones for silica aerogels prepared by supercritical drying using microcrystalline cellulose (∼100 kg m−3, Si content ∼20 wt%),31 and are, instead, in the range of the density of ultra-lightweight freeze-dried organosilica/CNF aerogels (10–20 kg m−3, Si content 10–38 wt%).4 The average compressive modulus of the TCNF/AS3f(10.7) foams was 54 ± 21 kPa for a foam density of 33 ± 2 kg m−3. This value is higher than that of freeze-dried CNF/methylsilane sponges (28 kPa for 20 kg m−3).4
Foaming behaviour of TCNF/AS dispersions at different AS concentration and pH
The AS had an interfacial tension (IFT) of 53 ± 0.1 mN m−1. Similar to short n-aminopentane molecules,56 the surface activity of the AS can be related to the amphiphilic structure of the molecule, consisting of hydrophobic methyl- and propyl-groups connected through silicon atom and hydrophilic amino-group (Fig. SI.1a, ESI†). The introduction of air into a 3 wt% AS aqueous solution by blending resulted in the formation of an unstable foam that rapidly drained and collapsed within one minute after preparation. The addition of TCNFs to the AS solution resulted, however, in a 3.5-times increase of the foam volume in comparison with the AS-foamed solution (Fig. 2a).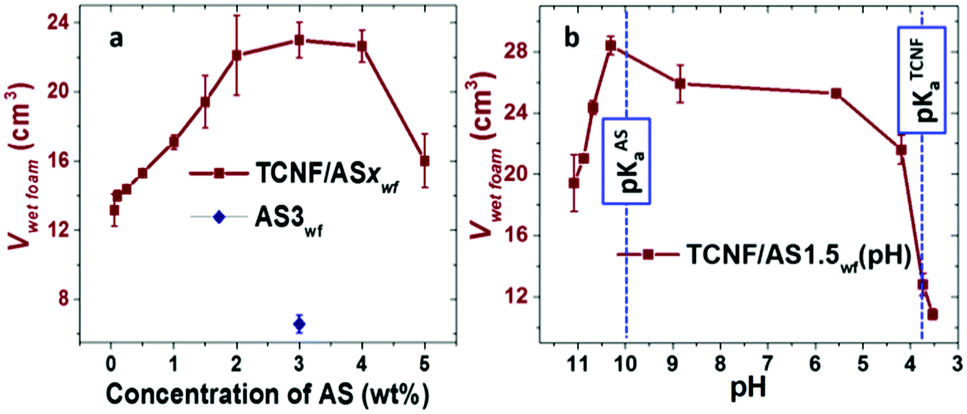 | ||
| Fig. 2 Foamability of TCNF/ASwf foams (Vwetfoam) as a function of (a) AS concentration and (b) pH of the dispersion for TCNF/AS1.5wf. The TCNF concentration was kept constant at 0.5 wt%. The blue diamond represents the volume of the wet foam prepared from 3 wt% aqueous solution of AS (AS3wf). The blue dashed lines on Fig. 2b show the pKa of AS (pKa = 10) and the pKa of TCNFs (pKa = 3.6). The volume of original dispersions was 10 cm3. | ||
The addition of 0.05 to 2 wt% of AS to 0.5 wt% TCNF dispersion resulted as well in a gradual increase of the wet foam volume from 13 ± 1 to 22 ± 2 cm3 (Fig. 2a).
The increase in the AS concentration also affected the air bubble diameters, which decreased from 97 ± 21 to 87 ± 25 μm for 0.05 and 0.5 wt% of AS (Fig. SI.2, ESI†). Between 2 to 4 wt% AS, the foam volume plateaued around 23 cm3, after which it significantly dropped to 16 ± 2 cm3 for AS concentration up to 5 wt% (Fig. 1a). The fibrillary structure and negative charges of the TCNFs generated a colloidal stable and entangled network, which has been reported to significantly enhance the foamability and stability of wet foams.39,40
The average IFT of the mixed TCNF/AS3d dispersion was close to the IFT of neat AS3d (58 ± 1 mN m−1), suggesting that the AS was mainly responsible for the surface activity and was probably adsorbed at the air/water interface. The surface activity of the AS was also demonstrated through the decrease in bubble size with increasing amount of added AS, consistent with an earlier study.57
The pH of the TCNF/AS dispersion did also have a significant influence on the foamability (Fig. 2b). By slightly reducing the pH from its intrinsic value of around pH = 11, a significant increase of the wet foam volume was observed with a maximum volume around pH 10.3, which was close to the pKa of the AS. The foamability was largely unaffected by pH in the range pH = 10–6, but dropped drastically for pH < 5.
Rheological behaviour of TCNF/AS dispersions and foams
The storage modulus of the TCNF/ASd dispersions, determined after pre-conditioning the dispersion at 60 °C for 3.6 h, increased strongly with an increasing AS concentration (Fig. 3a) or with a decreasing pH when the concentration of AS was kept constant at 1.5 wt% (Fig. 3b). For AS concentrations up to 2 wt% and pH = 11.1–10.3, the increased G′ correlated well with the observed changes in foamability (Fig. 2). This suggests that the viscoelastic reinforcement of the continuous phase, i.e. the foam lamellas, is important to promote the formation of a stable wet foam structure.58,59 A too high viscosity is, however, not desirable as well. When the AS concentration was above 4 wt% or for a pH ≤ 5, the formation of a strong gel limited the foam volume growth and resulted in solid foams of high density (Table 2).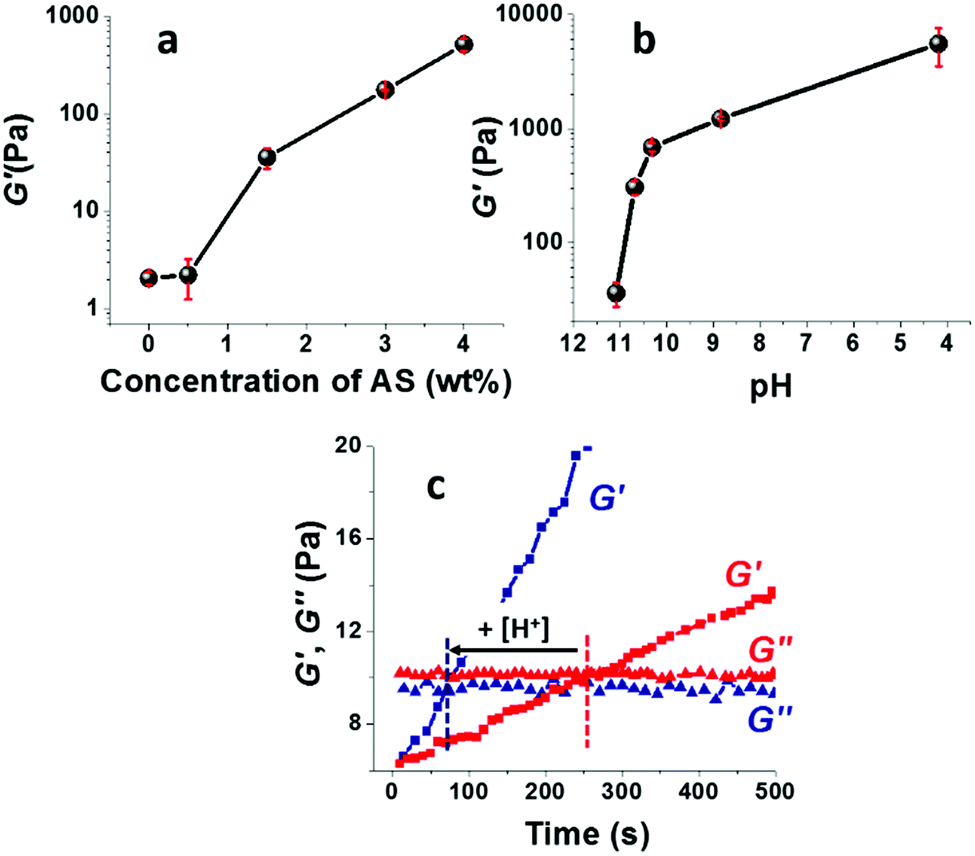 | ||
| Fig. 3 Viscoelastic properties of the TCNF/AS dispersions and foams at room temperature. Storage modulus, G′, of TCNF/ASd as a function of (a) varying concentration of AS (pH 10.6–11.2) and (b) of TCNF/AS1.5d at different pH. (c) Evolution of the storage (G′) and loss (G′′) moduli over time for TCNF/AS3wf at pH 11.1 (red) and pH 10.8 (blue) at room temperature, 1 min after foaming. | ||
The pH influenced the viscoelastic properties of the foams more than the AS concentration, with a 19-fold increase of G′ for TCNF/AS1.5d when the pH was decreased from 11.1 to 10.3 against a five-fold increase for a doubled AS concentration from 1.5 to 3 wt%. A slight pH decrease enabled to minimize the AS content in the wet foams, preserving the foam stability and decreasing the solid foam density. By optimizing the AS content and pH, the most homogeneous and lightweight solid foams could be produced at 2–3 wt% of AS at pH in the range 10.3–10.8, using 0.5 wt% of TCNFs.
Fig. 3c shows that G′ increased much faster with time at pH 10.8 than at pH 11.1. The formation of a percolating gel, defined by the time-course intersection of G′ and G′′, occurred 3 min earlier at pH 10.8 than at pH 11.1. The rapid (but not immediate) gelation was probably beneficial to inhibit drainage and foam collapse (see Fig. SI.3 (ESI†), showing the time and temperature dependent increase of G′ until a pseudo saturation state was reached approximately 45 min after foaming).
Protonation, hydrolysis and condensation of AS
Fig. 4 shows increasing intensities of the IR bands at 1606, 1500, and 1206 cm−1 with decreasing pH (<10). These IR bands are characteristic of the asymmetric and symmetric bending and rocking vibrations of NH3+, respectively.60–63 The amine groups of the AS became protonated as the pH was reduced below the pKa (= 10) of the AS.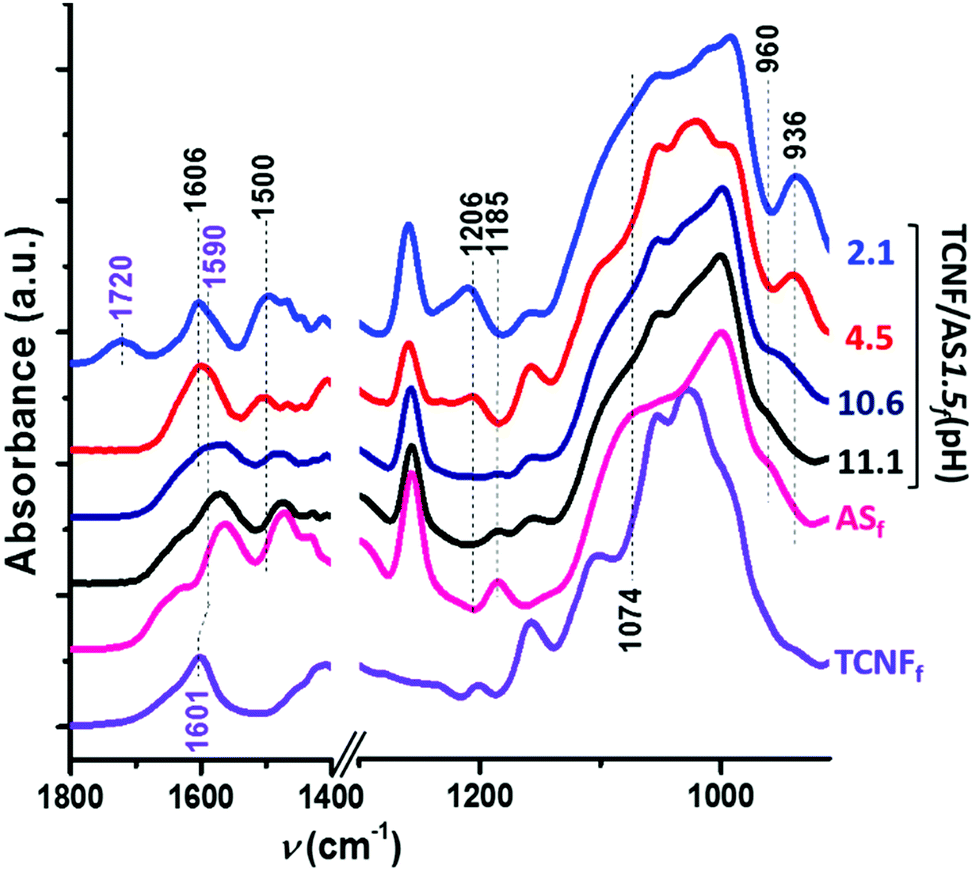 | ||
| Fig. 4 ATR FTIR spectra of TCNFfd(pH 7), ASf(pH 10.5) and TCNF/AS1.5f at pH: 11.1, 10.6, 4.5 and 2.1 (labeled on the right of the graph). | ||
The protonation of the AS amine groups increased the ionic strength of the system by contracting the electrical double layer64 and promoted interparticle interactions. The zeta-potential measurements showed, indeed, that the zeta potential of TCNF/AS1.5d became less negative with a decreased pH (Fig. SI.4, ESI†). Features of the interactions between AS and TCNFs were observed by ATR FTIR of dry foams through the red shift of the band from 1601 to 1592 cm−1 (Fig. 4), that is characteristic of the asymmetric bending of deprotonated TCNF carboxylate groups (COO−Na+ and COO−NH3+, respectively).65 TCNFs with protonated carboxyl groups were studied as well. The band at 1722 cm−1 (asymmetric deformation of CO group in protonated –COOH of TCNFs) observed in the TCNFfd spectrum disappeared when mixing TCNFs with AS in equimolar content of carboxyl and amino-groups (Fig. SI.5, ESI† TCNF/ASfd). Similarly, the band at 1592 cm−1 appeared in Fig. 4, confirming a change in the protonation and interactions between the TCNFs and the AS.66,67
The time- and temperature-dependent hydrolysis and condensation of AS is expected to have a significant influence on the colloidal stability and rheological properties of the dispersions and wet foams. Liquid state {1H}29Si NMR demonstrated that the condensation products (siloxane dimers) were produced at low content after mixing AS with water at pH 11.1 (Fig. 5a and Table SI.1, ESI†). None or very low amount of organosilica polymers were detected, which was in good agreement with previous studies.54
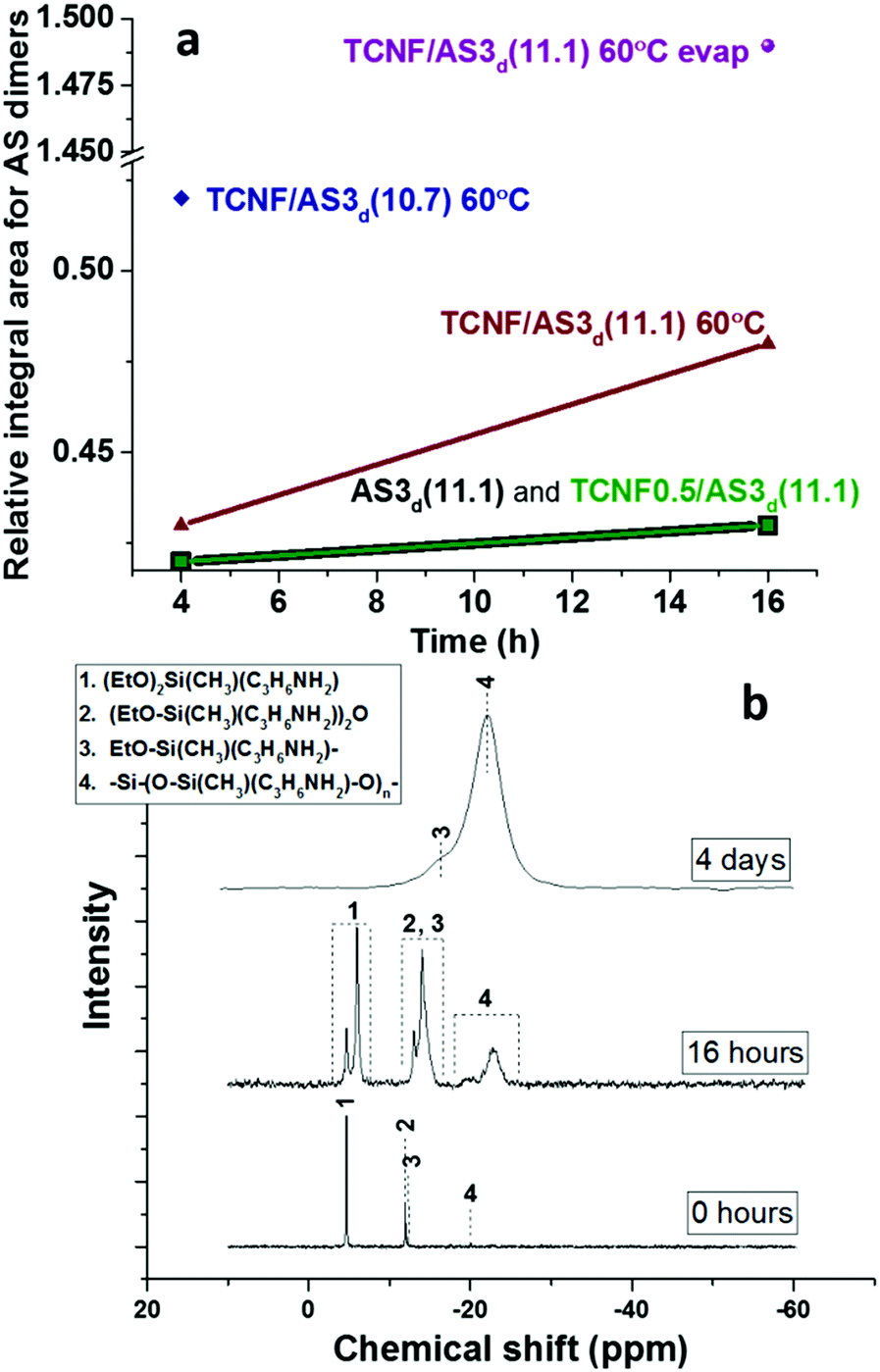 | ||
| Fig. 5 (a) Relative integral area for the bands of AS dimers as a function of time, determined from liquid state cross-polarization {1H}29Si NMR of wet dispersions: AS3d(11.1) (black squares), TCNF/AS3d(11.1) (green squares), TCNF/AS3d(11.1) (red triangles) and TCNF/AS3d(10.7) (blue diamonds) heated at 60 °C and TCNF/AS3d(11.1) (purple sphere) with solvent pre-evaporated in an oven at 60 °C for 16 h. (b) Liquid state cross-polarization {1H}29Si NMR spectra of TCNF/AS3d kept for 4 h at 60 °C without solvent evaporation (labelled 0 hours) and after 16 h of solvent evaporation at 60 °C in an oven (labelled 16 hours). Solid state cross-polarization {1H}29Si NMR of dry TCNF/AS3d(10.8) after 4 d of solvent evaporation at 60 °C in an oven (labelled 4 days). 1 refers to AS monomer, 2 – to AS dimer, 3 – end groups in organosilica chains and 4 – organosilica polymer. | ||
The addition of TCNFs to AS at basic pH did not affect the condensation of the AS since the relative concentration of dimers remained constant (see Fig. 5a and Table SI.1, ESI†). The NMR method was not fully quantitative and the analysis was limited to the study of the relative changes across the different specimen.
When the pH was decreased from 11.1 to 10.7, a 20% higher relative concentration of AS dimers was observed (TCNF/AS3d(11.1 vs. 10.7) at 60 °C on Fig. 5a and Table SI.1, ESI†). In the literature, the decrease of the pH below 10 is reported to promote the hydrolysis of silane but to decelerate the condensation with restricted formation of siloxanes.11,68 In this study, the IR spectra revealed, indeed, an increased intensity of the silanol band at 936 cm−1 and the disappearance of the ethoxy-group bands at 1185, 1074 and 960 cm−1 with decreasing pH (Fig. 4).60,62,69
Liquid state {1H}29Si NMR also demonstrated that the heating of TCNF/AS3d at 60 °C increased the relative condensation of AS dimers by 10%, after 16 h of heating (Fig. 5a).69 However, the evaporation of water had a stronger influence on the condensation of the AS than heating or slight pH adjustments (Fig. 5b). After 4 h of solvent evaporation from the TCNF/AS3d, the relative concentration of AS dimers increased by 20% (Table SI.1, ESI†). After 16 h, a 160% increase in the relative concentration content of the organosilica polymer was observed.
To clearly study the effect of drying, the same dispersion was pre-dried overnight in an oven at 60 °C before {1H}29Si NMR analysis. In the oven, the dispersion benefited of a much higher evaporation rate than in the narrow NMR tube. On such effective drying, the relative concentration of the AS dimers and polymers increased by 240% and 2060%. The corresponding {1H}29Si NMR spectrum in Fig. 5b (middle spectrum) exhibits a double peak for the monomers (peak 1) and the dimers (peaks 2 and 3), but not for the polymer (peak 4), which could indicate a two-phase situation with one polymer-rich phase in the studied oven-dried dispersion. After complete drying of the foam, no monomers or dimers were detected by NMR, which supported that complete condensation of the AS into organosilica polymers occurred during drying (Fig. 5b).
The oven-drying of the wet foams catalyzed the condensation of the poorly reactive AS into short linear polymers that might have created an additional supporting polymeric matrix for the foam stability. The majority of the organosilica is expected to be formed during the last hours of drying of the TCNF/ASwf.
Foamability and foam stability of TCNF/AS foams
Fig. 6 attempts to illustrate the main interactions and reactions responsible for the formation and stabilization of TCNF/AS foams. The air bubbles introduced by blending in the TCNF/AS dispersion were stabilized by the adsorption of AS and/or AS-TCNF complexes at the air/water interface. The accumulation of TCNFs close to the air/water interface could indeed be detected by fluorescent optical microscopy (Fig. 6a and b). The formation of a fibrillary TCNF network in the wet foam lamella also promoted the foam stability by decelerating the drainage of the wet foams (Fig. 6c). Evaporative drying at 60 °C resulted in the condensation of the AS into linear polymers, which contributed to a further enhancement of the strength and stiffness of the foam through the formation of an organosilica polymeric matrix built of linear low molecular siloxane chains [–Si(CH3)(C3H7NH2)O–] (deconvolution of the solid state CP {1H}29Si NMR spectrum in Fig. 5b indicated that the polymer consisted of about 15 units).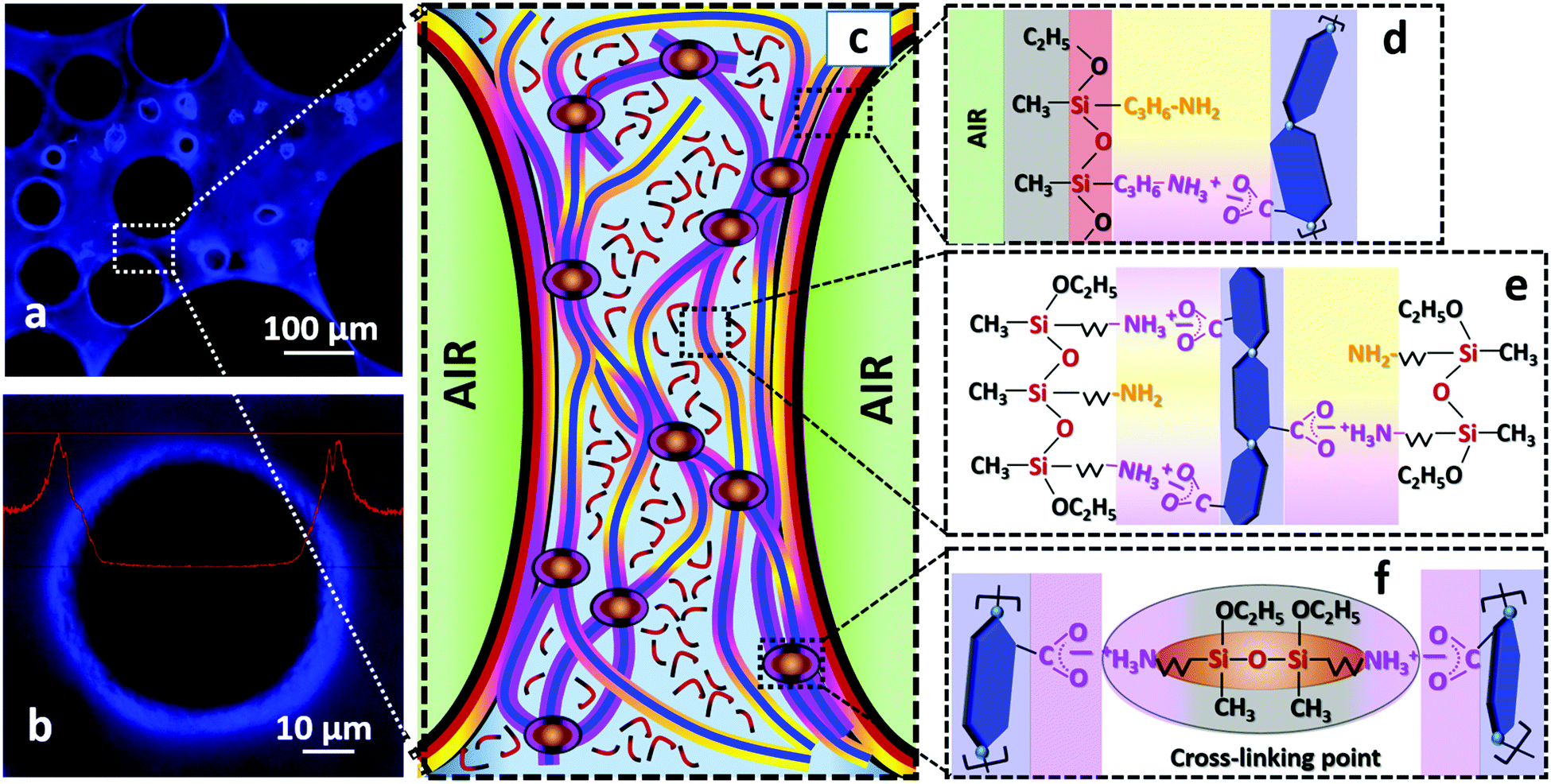 | ||
| Fig. 6 Stability mechanisms of the TCNF/AS foams. (a) Fluorescent optical microscopy images of TCNF/AS3wf(10.8) with (b) magnification of a single air bubble. The TCNFs are dyed with a blue fluorescent colorant. The red line on (b) represents the emission spectral profile. (c) Schematic illustration of the structure of the foam lamella after 12 h of drying: the green half spheres represent part of the air bubbles, the light blue background is the water medium, the blue continuous lines sketch the TCNFs and the red lines with black ends sketch short organosilica polymers. Molecular representation of (d) assembly of TCNF/AS at the air/water interface, (e) AS coated-TCNFs and (f) TCNFs and AS crosslinking points. The yellow color represents non-protonated amine groups of AS, pink – attractive electrostatic and van der Waals interactions, black – unreacted ethyl groups and methyl groups, red – siloxane bonds between neighbor AS molecules and in organosilica chains. The blue hexagons connected with light blue balls represent sugar rings with glycosidic bonds in a single cellulose chain as a simplified representation of TCNFs. Only a few deprotonated carboxylic groups are shown for simplicity. | ||
The resistance to excessive shrinkage during drying was highly dependent on the AS content and pH of the wet foams. With or without pH adjustment, addition of 1.5 or 3 wt% of AS together with 0.5 wt% TCNFs resulted in dry foams with a homogenous cellular structure. Adjustment of the pH in the range 10–10.8 triggered the adsorption of AS to TCNFs through electrostatic and van der Waals interactions between TCNFs and AS (Fig. 6d–f). These interactions resulted in an increase of the viscoelastic properties of both the dispersion and the wet foam, and enhanced further the foamability and resistance to drainage. Therefore, a slight pH adjustment is essential to generate homogeneous solid foams of low densities with a relatively low AS content.
Controlling the pH of the wet foams along with the water-evaporation resulted in the formation of a polymer/nanoparticle network, which was effective to produce stable organosilica foams (Fig. 6e and f).
In conclusion, stable wet and dry TCNF/AS foams can be achieved from an aqueous dispersion with: (i) 10.4 ≤ pH ≤ 10.8, (ii) a TCNF:AS weight ratio ≥1:6, and (iii) evaporation of water at 60 °C.
Conclusion
Lightweight hybrid organosilica foams (25–50 kg m−3) with high content of condensed silane (55–65 wt%) and amine groups (>4.5 mmol g−1) were produced from an aqueous dispersion of TEMPO-oxidized cellulose nanofibrils and aminosilane by mechanical blending and oven-drying (60 °C). The new preparation route used water as solvent and a surface-active silane as silica precursor and air/water interface stabilizer. The aminosilane condensation was significantly accelerated by the water evaporation. A slight reduction of the pH promoted the adsorption of aminosilane to TCNFs, which enhanced the storage modulus of the mixed TCNFs and organosilica matrixes. Tuning the pH of the AS/TCNF dispersion resulted in a significant improvement of the foamability and foam stability. The combination of the TCNF network and the organosilica polymer matrix and their connection through electrostatic and van der Waals interactions proved to be the key for producing stable foams upon drying. The demonstrated route to produce organosilica foams can be extended by mixing or post-treating the formed wet and dry foams, respectively, by silanes with different organic substitutes to yield functional foams for use e.g. as water purification scaffolds or packaging materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge Dr Maryam Ghanadpour from the Division of Fibre Technology, Royal Institute of Technology (KTH, Sweden) for providing the Domsjö dissolving pulp and conducting the high pressure Microfluidizer treatment to produce TEMPO-cellulose nanofibrils. We also acknowledge Dr Jan-Willem Benjamins from RISE Research Institute in Stockholm (Sweden) for his training and advice on the drop tension profilometer. We thank the Swedish Foundation for Strategic Research (RMA11-0065) and the Wallenberg Wood Science center (WWSC) for financial support.Notes and references
- C. E. Carraher, Polym. News, 2005, 30, 386–388 CrossRef.
- H. Maleki, L. Durães and A. Portugal, J. Non-Cryst. Solids, 2014, 385, 55–74 CrossRef.
- S. Cui, W. Cheng, X. Shen, M. Fan, A. (Ted) Russell, Z. Wu and X. Yi, Energy Environ. Sci., 2011, 4, 2070–2074 RSC.
- Z. Zhang, G. Sèbe, D. Rentsch, T. Zimmermann and P. Tingaut, Chem. Mater., 2014, 26, 2659–2668 CrossRef.
- Z. Wang, D. Wang, Z. Qian, J. Guo, H. Dong, N. Zhao and J. Xu, ACS Appl. Mater. Interfaces, 2015, 7, 2016–2024 CrossRef PubMed.
- L. Cai and G. Shan, J. Porous Mater., 2015, 22, 1455–1463 CrossRef.
- S. D. Bhagat, C. S. Oh, Y. H. Kim, Y. S. Ahn and J. G. Yeo, Microporous Mesoporous Mater., 2007, 100, 350–355 CrossRef.
- H. Yang, X. Kong, Y. Zhang, C. Wu and E. Cao, J. Non-Cryst. Solids, 2011, 357, 3447–3453 CrossRef.
- F. Scgwetfeger, W. Glaubitt and U. Schubert, J. Non-Cryst. Solids, 1992, 145, 85–89 CrossRef.
- A. V. Rao, M. M. Kulkarni, D. P. Amalnerkar and T. Seth, Science, 2003, 206, 262–270 Search PubMed.
- C. J. Brinker, J. Non-Cryst. Solids, 1988, 100, 31–50 CrossRef.
- A. C. Pierre and G. M. Pajonk, Chem. Rev., 2002, 102, 4243–4265 CrossRef PubMed.
- S. W. Hwang, H. H. Jung, S. H. Hyun and Y. S. Ahn, J. Sol–Gel Sci. Technol., 2007, 41, 139–146 CrossRef.
- J. L. Gurav, I. Jung, H. Park, E. S. Kang and D. Y. Nadargi, J. Nanomater., 2010, 1–11 Search PubMed.
- J. B. Peri, J. Phys. Chem., 1966, 70, 2937–2945 CrossRef.
- F. Schwertfeger, D. Frank and M. Schmidt, J. Non-Cryst. Solids, 1998, 225, 24–29 CrossRef.
- L. A. Capadona, M. A. B. Meador, A. Alunni, E. F. Fabrizio, P. Vassilaras and N. Leventis, Polymer, 2006, 47, 5754–5761 CrossRef.
- S. Štandeker, Z. Novak and Ž. Knez, J. Colloid Interface Sci., 2007, 310, 362–368 CrossRef PubMed.
- J. K. Oh, K. Perez, N. Kohli, V. Kara, J. Li, Y. Min, A. Castillo, M. Taylor, A. Jayaraman, L. Cisneros-Zevallos and M. Akbulut, Food Control, 2015, 52, 132–141 CrossRef.
- D. A. Loy and K. J. Shea, Chem. Rev., 1995, 95, 1431–1442 CrossRef.
- P. R. Aravind and G. D. Soraru, J. Porous Mater., 2011, 18, 159–165 CrossRef.
- L. Duffours, T. Woignier and J. Phalippou, J. Non-Cryst. Solids, 1995, 186, 321–327 CrossRef.
- D. M. Smith, R. Deshpande and J. Brinker, Mater. Res. Soc. Symp. Proc., 1992, 271, 567–572 CrossRef.
- G. W. Scherer, D. M. Smith, X. Qiu and J. M. Anderson, J. Non-Cryst. Solids, 1995, 186, 316–320 CrossRef.
- F. Shi, L. Wang and J. Liu, Mater. Lett., 2006, 60, 3718–3722 CrossRef.
- S. Yun, H. Luo and Y. Gao, J. Mater. Chem. A, 2014, 2, 14542 RSC.
- H. Maleki, L. Durães and A. Portugal, J. Phys. Chem. C, 2015, 119, 7689–7703 CrossRef.
- G. Markevicius, R. Ladj, P. Niemeyer, T. Budtova and A. Rigacci, J. Mater. Sci., 2017, 52, 2210–2221 CrossRef.
- J. Fu, S. Wang, C. He, Z. Lu, J. Huang and Z. Chen, Carbohydr. Polym., 2016, 147, 89–96 CrossRef PubMed.
- A. Fidalgo, J. P. S. Farinha, J. M. G. Martinho, M. E. Rosa and L. M. Ilharco, Chem. Mater., 2007, 19, 2603–2609 CrossRef.
- A. Demilecamps, G. Reichenauer, A. Rigacci and T. Budtova, Cellulose, 2014, 21, 2625–2636 CrossRef.
- J. Cai, S. Liu, J. Feng, S. Kimura, M. Wada, S. Kuga and L. Zhang, Angew. Chem., Int. Ed., 2012, 51, 2076–2079 CrossRef PubMed.
- R. J. Moon, A. Martini, J. Nairn, J. Simonsen and J. Youngblood, Chem. Soc. Rev., 2011, 40, 3941–3994 RSC.
- I. Usov, G. Nyström, J. Adamcik, S. Handschin, C. Schütz, A. Fall, L. Bergström and R. Mezzenga, Nat. Commun., 2015, 6, 1–11 Search PubMed.
- M. Özgür Seydibeyoǧlu and K. Oksman, Compos. Sci. Technol., 2008, 68, 908–914 CrossRef.
- P. Gatenholm, H. Bertilsson and A. Mathiasson, J. Appl. Polym. Sci., 1993, 49, 197–208 CrossRef.
- A. N. Nakagaito and H. Yano, Appl. Phys. A: Mater. Sci. Process., 2003, 80, 155–159 CrossRef.
- J. Leitner, B. Hinterstoisser, M. Wastyn, J. Keckes and W. Gindl, Cellulose, 2007, 14, 419–425 CrossRef.
- K. S. Gordeyeva, A. B. Fall, S. Hall and B. Wicklein, J. Colloid Interface Sci., 2016, 472, 44–51 CrossRef PubMed.
- N. T. Cervin, E. Johansson, J.-W. Benjamins and L. Wågberg, Biomacromolecules, 2015, 16, 822–831 CrossRef PubMed.
- A. Isogai, T. Saito and H. Fukuzumi, Nanoscale, 2011, 3, 71–85 RSC.
- Z. Hu, R. Xu, E. D. Cranston and R. H. Pelton, Biomacromolecules, 2016, 17, 4095–4099 CrossRef PubMed.
- N. T. Cervin, E. Johansson, P. A. Larsson and L. Wågberg, ACS Appl. Mater. Interfaces, 2016, 8, 11682–11689 CrossRef PubMed.
- M. Andresen and P. Stenius, J. Dispersion Sci. Technol., 2007, 28, 837–844 CrossRef.
- K. Xhanari, K. Syverud and P. Stenius, J. Dispersion Sci. Technol., 2011, 32, 447–452 CrossRef.
- A. Tejado, W. C. Chen, M. N. Alam and T. G. M. van de Ven, Cellulose, 2014, 21, 1735–1743 Search PubMed.
- C. Gebald, J. A. Wurzbacher, P. Tingaut, T. Zimmermann and A. Steinfeld, Environ. Sci. Technol., 2011, 45, 9101–9108 CrossRef PubMed.
- N. H. Mohd, N. F. H. Ismail, J. I. Zahari, W. F. B. Wan Fathilah, H. Kargarzadeh, S. Ramli, I. Ahmad, M. A. Yarmo and R. Othaman, J. Nanomater., 2016, 1–8 Search PubMed.
- S. Saini, M. N. Belgacem and J. Bras, Mater. Sci. Eng., C, 2017, 75, 760–768 CrossRef PubMed.
- C. Goussé, H. Chanzy, G. Excoffier, L. Soubeyrand and E. Fleury, Polymer, 2002, 43, 2645–2651 CrossRef.
- G. Hayase, K. Kanamori, K. Abe, H. Yano, A. Maeno and H. Kaji, ACS Appl. Mater. Interfaces, 2014, 6, 9466–9471 CrossRef PubMed.
- T. Tomita, S. Kawasaki and K. Okada, J. Porous Mater., 2004, 11, 107–115 CrossRef.
- Scand. Pulp, Pap. and Board Test. Comm., 2002, pp. 1–4.
- V. Bennevault-Celton, O. Maciejak, B. Desmazières and H. Cheradamea, Polym. Int., 2010, 59, 1273–1281 CrossRef.
- C. Ammann, P. Meier and A. Merbach, J. Magn. Reson., 1982, 46, 319–321 Search PubMed.
- L. R. Arriaga, W. Drenckhan, A. Salonen, J. A. Rodrigues, R. Íñiguez-Palomares, E. Rio and D. Langevin, Soft Matter, 2012, 8, 11085–11097 RSC.
- B. P. Binks, Curr. Opin. Colloid Interface Sci., 2002, 7, 21–41 CrossRef.
- M. Krzan, Tech. Trans., 2013, 10–27 Search PubMed.
- R. J. Pugh, Adv. Colloid Interface Sci., 1996, 64, 67–142 CrossRef.
- R. Peña-Alonso, F. Rubio, J. Rubio and J. L. Oteo, J. Mater. Sci., 2007, 42, 595–603 CrossRef.
- S. Naviroj, J. L. Koenig and H. Ishida, J. Adhes., 1981, 1–22 Search PubMed.
- J. Kim, P. Seidler, L. S. Wan and C. Fill, J. Colloid Interface Sci., 2009, 329, 114–119 CrossRef PubMed.
- G. Socrates, Infrared and Raman characteristic group frequencies, John Wiley and Sons, Ltd, 3rd edn, 2004 Search PubMed.
- R. Prathapan, R. Thapa, G. Garnier and R. F. Tabor, Colloids Surf., A, 2016, 509, 11–18 CrossRef.
- D. M. Byler and H. M. Farrell Jr., J. Dairy Sci., 1989, 72, 1719–1723 CrossRef.
- S. Fujisawa, Y. Okita, H. Fukuzumi, T. Saito and A. Isogai, Carbohydr. Polym., 2011, 84, 579–583 CrossRef.
- A. Sone, T. Saito and A. Isogai, ACS Macro Lett., 2016, 5, 1402–1405 CrossRef.
- B. Arkles, J. R. Steinmetz, J. Zazyczny and P. Mehta, J. Adhes. Sci. Technol., 1992, 6, 193–206 CrossRef.
- R. M. Pasternack, S. R. Amy and Y. J. Chaba, Langmuir, 2008, 24, 12963–12971 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Chemical formula of AS molecule; AFM topography of TCNFs; size distribution of air bubble in the wet TCNF/AS foams; time and temperature resolved rheology of TCNF/AS wet foam; zeta-potential of TCNF and TCNF/AS dispersions; ATR FTIR spectroscopy of TCNF/AS foam; NMR integral areas for AS and TCNF/AS dispersions. See DOI: 10.1039/c8qm00360b |
| This journal is © the Partner Organisations 2018 |