
A thermally responsive host–guest conductive hydrogel with self-healing properties†
Yuting
Zhu
ab,
Sidi
Liu
b,
Xiaoli
Shi
*bc,
Dong
Han
bc and
Feng
Liang
*ab
aThe State Key Laboratory for Refractories and Metallurgy, Institute of Advanced Materials and Nanotechnology, School of Chemistry and Chemical Engineering, Wuhan University of Science and Technology, Wuhan 430081, China. E-mail: feng_liang@whu.edu.cn
bCAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology, Beijing 100190, P. R. China. E-mail: shixl@nanoctr.cn
cSchool of Future Technology, University of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 14th August 2018
Abstract
Conductive polymers with stimuli-responsive properties have significant potential for technological applications such as in electrical and biomedical devices, as well as stretchable electronics. In this study, we use the amphiphilic configuration property of α cyclodextrin (αCD) to demonstrate a facile strategy for the preparation of flexible polyaniline (PANI)-containing conductive hydrogel networks based on preorganized αCD-containing N-isopropylacrylamide (αCD-PNIPAM) hydrogels. These materials exhibit high conductivity (0.64 S m−1), excellent thermoresponsive and self-healing properties. The resulting hybrid hydrogels also contain homogeneous, interconnected macropores and exhibit perfect integration between the host phase (αCD-PNIPAM) and the PANI phase, which results in improved mechanical properties and remarkable stability.
1. Introduction
Owing to their environmental sensitivity, stimuli-responsive hydrogels have attracted significant attention across a range of applications such as electrochemical sensors, actuators, bioelectronics, and drug release.1–7 However, their poor mechanical properties and lack of electrical conductivity make practical applications difficult. Recently, several studies have focused on the preparation of conductive, responsive hydrogels with enhanced mechanical properties. These hydrogels were synthesized via the incorporation of additional ingredients such as conducting nanofillers,8 conductive polymers,9–11 or particles into the gel matrix.12,13 Use of simple blending methods for hydrogel conductivity enhancement requires that a considerable amount of conducting nanofiller be used to produce a continuous conductive network. This weakens the stimuli-responsive properties of the hydrogel.14–17 The most widely used conducting nanofillers such as carbon nanotubes and graphenes possess low solubilities and thus aggregate easily in the hydrogel matrix. This forces researchers to limit the dosages of these materials even when surface modification is used.18Moreover, the presence of strong interactions between the filler and polymer matrix restricts the motion of the polymer chains. This usually reduces the stimuli-responsiveness of the polymer.19 Thus, it is important to develop conducting, stimuli-responsive hydrogels with both enhanced mechanical strength and electrical properties.
Service life is another important parameter for these materials. The self-healing property of hydrogels aids in extending their duration of use and provides enhanced reliability, maintenance, and durability in important applications. However, it is really hard to realize all functions in one hydrogel and most reports only focus on two properties, which makes them comparatively easier to prepare.20–23
Poly(N-isopropylacrylamide) (PNIPAM) is one of the most common temperature-responsive polymers and thus has been studied extensively. The unique PNIPAM chain coil-to-globule transition allows this hydrogel to undergo a temperature-responsive volume phase transition at its lower critical solution temperature.24–27 PNIPAM hydrogels can exhibit several characteristic changes such as swelling/deswelling and absorption/desorption in response to temperature fluctuations. This makes it a promising smart hydrogel candidate.13,23,28,29 However, its inherent mechanical weakness and lack of conductivity limit its application to electrical devices. To achieve multifunctionality, a preorganized PNIPAM host phase with an interconnected pore system and high affinity can be designed to incorporate an electrically conductive phase. Conductive polymer polyaniline (PANI) is one of the most promising candidates in this area and has been incorporated in a broad range of gel systems.30,31 However, the hydrophobic properties of aniline monomers make achieving compatibility with the PNIPAM host phase difficult.32,33
In this paper, we present a facile and rapid strategy for the preparation of PANI-containing conductive hydrogel networks based on preorganized α-cyclodextrin-containing N-isopropylacrylamide (αCD-PNIPAM) hydrogels with homogeneous, interconnected macropores. Unlike in traditional hybrid hydrogels made from randomly dispersed conductive polymers and stimuli-responsive hydrogels, the introduction of αCD plays an important role in our materials. Due to the smart amphiphilic configuration of αCD, which contains a hydrophobic cavity and hydrophilic exterior, aniline monomers can easily use hydrophobic–hydrophobic interactions to accumulate in the hydrophobic cavity and self-assemble to form polyaniline (PANI) in an acidic environment.34,35 This promotes ideal integration between the host phase (αCD-PNIPAM) and the PANI phase via host–guest combination. The performance characteristics of PNIPAM and PANI are preserved, ensuring excellent, stable conductivity and temperature sensitivity. The introduction of αCD also contributes to the mechanical properties of hybrid hydrogels, via interactions between hydrogel networks. More interestingly, the resulting hybrid hydrogels exhibit self-healing properties when cut into two pieces. The repaired hydrogels exhibit excellent mechanical properties, electrical conductivities, and temperature sensitivities. These properties may make the hybrid hydrogels potential “smart” materials. In particular, their stimuli-responsive properties offer significant potential for drug release applications.
2. Materials and methods
2.1 Materials
N-Isopropylacrylamide (99%) was purchased from J&K Chemicals (Beijing, China). α-Cyclodextrin, aniline (ANI), and ammonium persulfate (APS) were purchased from Aladdin Industrial Corporation (Shanghai, China). N,N′-Methylenebisacrylamide (BIS) was purchased from Sigma-Aldrich (St. Louis, MO, USA). All other reagents were of the best grade available and used as received.2.2 Hydrogel preparation
In a typical synthesis, 282.5 mg of αCD and 1.13 g of NIPAM were mixed and completely dissolved in 10 mL deionized (DI) water under nitrogen at room temperature (20 °C). Then, the cross-linking agent (BIS) and a thermo-initiator (0.075% and 5% by mass, respectively, relative to NIPAM) were added to the solution. After vigorously stirring, the solution was transferred into a glass mould with the desired size and placed in an oven at 50 °C until solidification. Approximately 2 h later, the αCD-PNIPAM host hydrogel was produced.The as-prepared αCD-PNIPAM host hydrogel was immersed in DI water for 1 d to remove unreacted NIPAM molecules and then quickly rinsed twice. It was then transferred into an ANI/HCl solution at room temperature. This solution was prepared by mixing an ANI solution (1 mL of ANI in 30 mL of DI water) and 10 mL of a 1 M HCl solution. After swelling and loading with ANI for 24 h, followed by in situ polymerization into PANI, the dark-green αCD-PNIPAM/PANI hybrid hydrogel was fabricated. The determined loading mass of PANI was around 255 mg which was calculated as the mass difference of the freeze-dried hydrogels of αCD-PNIPAM and αCD-PNIPAM/PANI.
2.3 Physical and chemical characterization
The morphologies of the PNIPAM, αCD-PNIPAM, and αCD-PNIPAM/PANI hydrogels were characterized using a scanning electron microscope (SEM, Nova 200 Nanolab, FEI Instruments) operating at 6 kV. Before observation, the hydrogels were freeze-dried for 24 h and sputter-coated with a gold layer. The Fourier transform infrared (FTIR) spectra of the hydrogels were recorded using a FTIR spectrometer (Spectrum One, Perkin Elmer Instruments). The mechanical performances of the hydrogels were measured using a rheometer (Z010TE, Zwick/Roell) with parallel plate apparatus on a Peltier plate in frequency-sweep mode at 25 °C. The rheometer is also used to test the stability of the electrical properties of the hybrid hydrogels under mechanical strain. The tensile strength is measured using a universal testing machine (ISO 1798, Instron). All the samples are made in the form of rods with the same diameter of 4.5 mm with an initial tensile length of 12 mm. The velocity was constant at 100 mm min−1. A cylindrical αCD-PNIPAM/PANI hydrogel with a thickness of 5 mm was tested and compressed by 0.5 mm each time until the largest strain reaches 50% and the resistance was obtained by using a multimeter. Hydrogel swelling and deswelling behaviour was observed via immersing in water at 25–50 °C. The self-healing properties were tested by cutting the PNIPAM/PANI and αCD-PNIPAM/PANI samples into two pieces and immersing it in pure water or ANI/HCl solution for self-repair. The electrical conductivities of the hydrogels were measured with a linear probe head using a four-point probe technique and a sourcemeter (4200-SCS, Keithley Instruments).3. Results and discussion
The hybrid hydrogel synthesis process and gelation sequence are shown in Fig. 1. The host αCD-PNIPAM hydrogel is prepared via a one-pot sol–gel synthesis. Owing to the coil-to-globule transition found only among PNIPAM chains and participation by αCD, the preorganized networks are temperature responsive, highly porous and contain amphiphilic domains that can facilitate ANI monomer loading. The highly porous, temperature-responsive nature of the hydrogel ensures its swelling behaviour, which opens paths for ANI molecule diffusion. The αCD moieties can provide hydrophobic cavities for the easy collection of the ANI molecules via hydrophobic–hydrophobic interactions. Before ANI loading, the host αCD-PNIPAM hydrogel is placed in a 50 °C oven for a specific period. After the loss of a large amount of water, the deswelled hydrogel is immersed in an ANI solution, where it absorbs ANI monomers until it reswells to its original volume. Then the reswelled hydrogel that contains conductive polymer monomers is immersed into 1 M hydrochloric acid. In situ polymerization can be triggered directly without introducing the APS initiator in a separate step as the APS is dispersed on a molecular level in the preorganized αCD-PNIPAM matrix. This makes our strategy more efficient than those described in previous works that required an additional step for the introduction of the initiator. When the ANI monomers contact the host αCD-PNIPAM and diffuse throughout the network, the colour of the hydrogel changes from transparent to light yellow and then to dark green, indicating in situ PANI formation.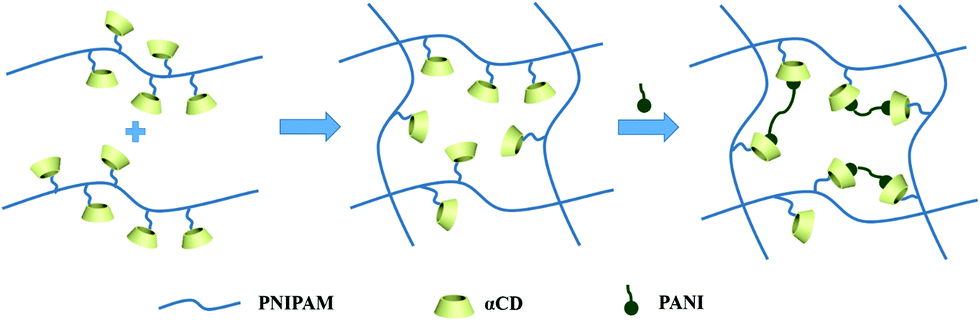 | ||
| Fig. 1 Schematic illustration of the synthesis process for the αCD-PNIPAM/PANI hybrid hydrogels composed of PNIPAM and conductive polymers. This synthesis is based on the preorganized αCD-containing N-isopropylacrylamide hydrogels that are amphiphilic and pH responsive. | ||
The chemical nature of the as-prepared αCD-PNIPAM/PANI network was analysed via its FTIR spectrum. Fig. 2a shows the FTIR spectra of PANI, PNIPAM, αCD-PNIPAM, and αCD-PNIPAM/PANI. The two characteristic absorption peaks at ∼1587 and ∼1489 cm−1 of the PANI spectrum (black) are attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibrations of the quinoid and benzenoid rings, respectively.36 Two strong peaks in the PNIPAM spectrum (red) at ∼1546 and ∼1653 cm−1 can be attributed to N–H bending and CO stretching vibrations. This is consistent with previous reports.13,23 The peaks at ∼2974 and ∼1020 cm−1 in the αCD-PNIPAM spectrum (blue) correspond to C–H stretching and O–H bending in αCD.36 The spectrum (pink) of αCD-PNIPAM/PANI shows the combined characteristic bands of the host structure and the PANI phase, again confirming the in situ formation of PANI in an αCD-PNIPAM network.
C stretching vibrations of the quinoid and benzenoid rings, respectively.36 Two strong peaks in the PNIPAM spectrum (red) at ∼1546 and ∼1653 cm−1 can be attributed to N–H bending and CO stretching vibrations. This is consistent with previous reports.13,23 The peaks at ∼2974 and ∼1020 cm−1 in the αCD-PNIPAM spectrum (blue) correspond to C–H stretching and O–H bending in αCD.36 The spectrum (pink) of αCD-PNIPAM/PANI shows the combined characteristic bands of the host structure and the PANI phase, again confirming the in situ formation of PANI in an αCD-PNIPAM network.
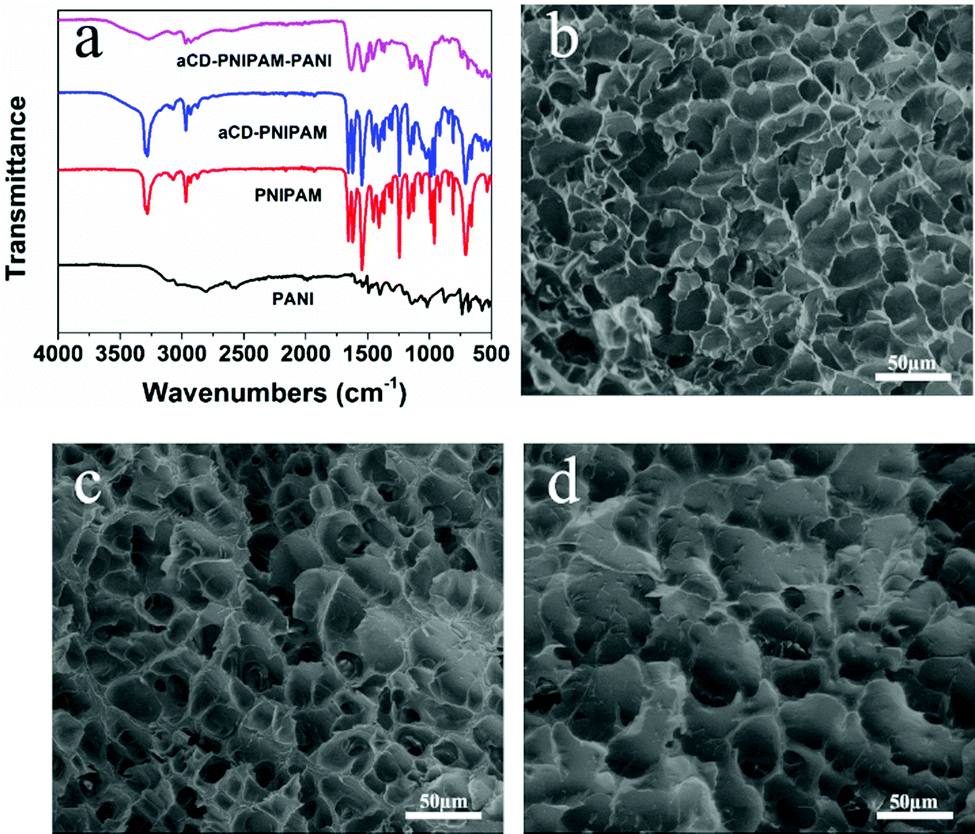 | ||
| Fig. 2 (a) FTIR spectra of the PNIPAM, αCD-PNIPAM, and αCD-PNIPAM/PANI hydrogels. SEM images of the (b) PNIPAM, (c) αCD-PNIPAM, and (d) αCD-PNIPAM/PANI hydrogels. | ||
The morphologies and internal structures of the freeze-dried hybrid hydrogels were investigated via (SEM). In Fig. 2b and c, the SEM images of the PNIPAM and αCD-PNIPAM hydrogels both reveal interconnected pores throughout the network. The pore sizes range from several to dozens of micrometres. However, the polymer chains in the αCD-PNIPAM network are thicker than those in the PNIPAM network because of αCD incorporation. Such a homogeneous, interconnected pore structure provides large open paths that facilitate water molecule transport and aniline loading. The pores in the αCD-PNIPAM/PANI network (Fig. 2d) appear irregular and the surface wrinkles formed by the polymer chains look thick, which indicates that PANI successfully fills the pores of the host hydrogel network. The fact that the pores are micrometres in size guarantees the high sensitivity of the PNIPAM network. Importantly, no phase transition between the host αCD-PNIPAM and the electrically conductive PANI phases is observed on the polymer network backbones. This indicates perfect integration of the two different polymer chains, which ensures a continuous electron transport path. Unlike in traditional configurations, our αCD-PNIPAM acts not only as a host structure (for PANI loading) but also as a spatial buffer that can protect the PANI from rapid degradation during long-term cycling, thus ensuring the electrical conductivity of the hybrid hydrogel.
For use in practical applications, the hybrid hydrogels should exhibit mechanical properties that allow them to resist environmental stimulation. Hydrogels are viscoelastic materials and their capacity to store and dissipate energy is an important parameter for the evaluation of their mechanical properties. The amounts of energy stored and dissipated via elastic and viscous deformation are indicated by the storage (G′) and loss (G′′) moduli, respectively.37 As shown in Fig. 3a and b, the dynamic frequency sweep experiments performed on the PNIPAM, αCD-PNIPAM, PNIPAM–PANI, and αCD-PNIPAM/PANI hydrogels show a wide linear viscoelastic region which reveals their gel states. They also confirm that the G′ value is significantly higher than the G′′ value in each case. It is interesting that the G′ values of the αCD-PNIPAM/PANI hybrid hydrogels are considerably higher than those of the pure PNIPAM and αCD-PNIPAM hydrogels and almost 1.5 times as much as PNIPAM/PANI. This indicates increased mechanical strength. This improvement is driven by the formation of a continuous, mechanically efficient network with rigid branched chains via the in situ polymerization of ANI, which significantly enhances the rheological properties of the hybrid hydrogels. The covalent bonds between the branched PANI chains and interactions between the networks can also ease hybrid hydrogel energy dissipation. We also find that the G′ value of αCD-PNIPAM/PANI is higher than that of PNIPAM–PANI. This may be due to the αCD/PNIPAM covalent bond and hydrophobic–hydrophobic interactions between αCD and PANI. The linking of the PNIPAM and PANI networks via host–guest combination results in an αCD-PNIPAM/PANI hybrid hydrogel with significantly improved mechanical strength. This has been confirmed via compression and tensile tests. Fig. 3c compares the compression stress–strain curves of the PNIPAM, αCD-PNIPAM, PNIPAM–PANI, and αCD-PNIPAM/PANI hydrogels. The compression strength of the αCD-PNIPAM/PANI hydrogel is more than 5 times higher than that of the pure PNIPAM hydrogel and its maximum tensile stress reaches 2.77 MPa. Moreover, the αCD-PNIPAM/PANI hydrogel is significantly more stretchable and tougher than its counterparts. It can be stretched as much as 4 times beyond its initial length without breakage and returns to its original state after 10 cycles of stretching, as shown in Fig. 3d and e. More detailed mechanical characterization of the as-prepared hydrogels was also tested, including the strain–stress behavior of breaking elongation and stretching–releasing cycles. In Fig. S1 (ESI†), the αCD-PNIPAM/PANI hydrogel exhibits the highest mechanical properties and excellent elasticity with a large ultimate strain of 490% at a tensile strength of 30 kPa and good cycling performance. This further proves that the host–guest interaction promotes the mechanical properties of the gels.
 | ||
| Fig. 3 (a and b) The storage (G′) and loss (G′′) moduli of the PNIPAM, αCD-PNIPAM, PNIPAM/PANI, and αCD-PNIPAM/PANI hydrogels tested in the frequency sweep mode. (c) Typical compression stress–strain curves of the PNIPAM, αCD-PNIPAM, PNIPAM–PANI, and αCD-PNIPAM/PANI hydrogels. (d and e) Stretching properties of the PNIPAM, αCD-PNIPAM, PNIPAM/PANI, and αCD-PNIPAM/PANI hydrogels over 10 stretching cycles. | ||
We further designed a test to demonstrate the thermal response properties of the αCD-PNIPAM/PANI hydrogels. We hypothesized that they should not be affected by the incorporation of conductive polymers. Fig. 4a shows that the coil-to-globule transitions of the PNIPAM chains cause the hydrogel volume to change quickly over the course of a few minutes when the temperature changes. This demonstrates good thermoresponsive properties. Fig. 4b describes the swelling and deswelling behaviours of the αCD-PNIPAM/PANI hybrid hydrogels in a more intuitive manner. A sharp decrease in mass occurs when the αCD-PNIPAM/PANI hydrogel is immersed in water at 50 °C and deswelling is maximized within 5 min. When the temperature is changed to 25 °C for the recovery half cycle, the hydrogel exhibits a sharp mass increase during the first several minutes. The recovery speed then slows dramatically such that the original swelled state is recovered after about 10 h. The swelling ratio of αCD-PNIPAM/PANI can reach 6.4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 which is similar to the results noted with the PNIPAM hydrogel. It confirms that the thermoresponsive property of the hybrid hydrogel is unaffected by the incorporation of the conductive polymers. The thermal-responsive behavior shown in Fig. 4c also demonstrated it. The good thermoresponsive behaviours of these hybrid hydrogels can be ascribed to the integrity of the PNIPAM backbone structure. The conductive polymers are incorporated like branched chains on the backbone, and thus affect little. Moreover, the interpenetrating binary network structure and interconnected macropores of the hybrid hydrogel minimize interference with the thermal motion of the PNIPAM chains and facilitate the flow of water molecules within the hydrogel network. This contrasts with blending methods in which the PNIPAM conductive hydrogels are prepared via the random dispersal of nanofillers and responsive behaviour is reduced. The excellent thermoresponsive behaviours of the αCD-PNIPAM/PANI hydrogels may support their use in responsive devices.
:1 which is similar to the results noted with the PNIPAM hydrogel. It confirms that the thermoresponsive property of the hybrid hydrogel is unaffected by the incorporation of the conductive polymers. The thermal-responsive behavior shown in Fig. 4c also demonstrated it. The good thermoresponsive behaviours of these hybrid hydrogels can be ascribed to the integrity of the PNIPAM backbone structure. The conductive polymers are incorporated like branched chains on the backbone, and thus affect little. Moreover, the interpenetrating binary network structure and interconnected macropores of the hybrid hydrogel minimize interference with the thermal motion of the PNIPAM chains and facilitate the flow of water molecules within the hydrogel network. This contrasts with blending methods in which the PNIPAM conductive hydrogels are prepared via the random dispersal of nanofillers and responsive behaviour is reduced. The excellent thermoresponsive behaviours of the αCD-PNIPAM/PANI hydrogels may support their use in responsive devices.
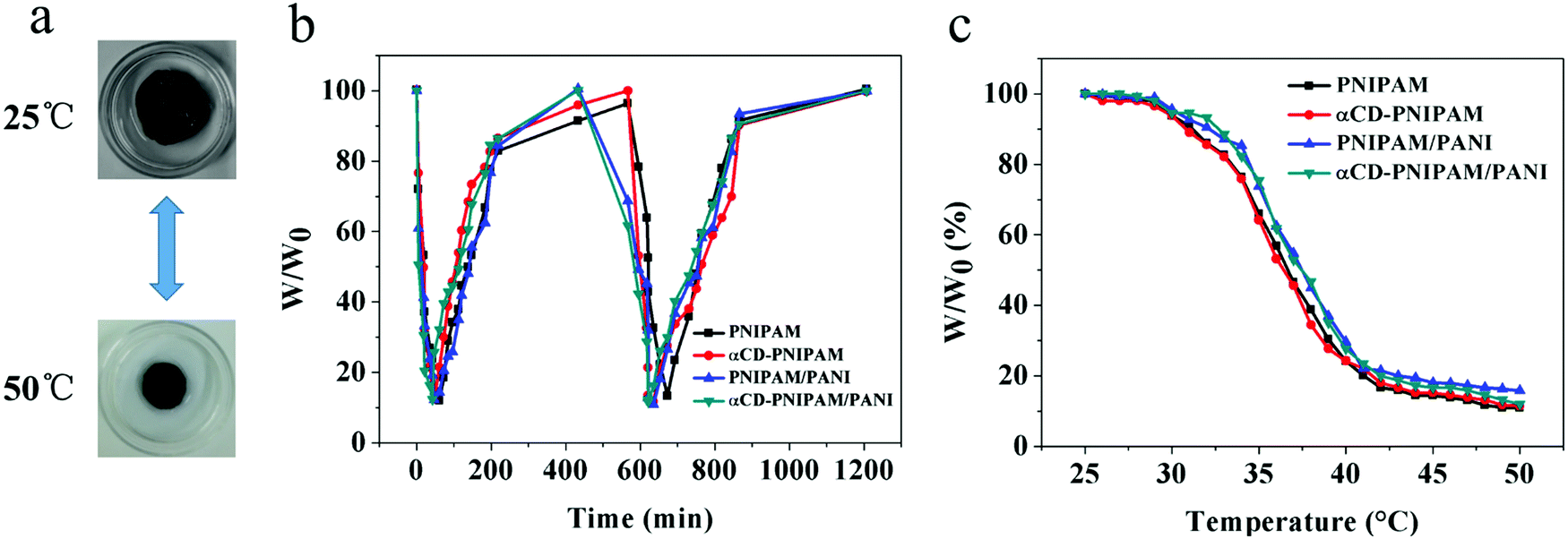 | ||
| Fig. 4 The swelling and deswelling behavior of (a) the αCD-PNIPAM/PANI hydrogel under 25 °C and 50 °C, and (b) the PNIPAM, αCD-PNIPAM, PNIPAM/PANI, and αCD-PNIPAM/PANI hydrogels under 25 °C and 50 °C. (c) The thermal-responsive behavior of the PNIPAM, αCD-PNIPAM, PNIPAM/PANI, and αCD-PNIPAM/PANI hydrogels at different temperatures. | ||
Another benefit from the host–guest interaction between the two hydrogel networks is that the hybrid hydrogels exhibit self-healing under some conditions. An αCD-PNIPAM/PANI hydrogel sample is cut into two pieces in Fig. 5a. The two separated pieces are then placed together and the freshly created fracture surfaces are put in contact with each other. Next, the samples are immersed in an ANI/HCl solution at room temperature for 3 h. The sample self-heals to form a single piece. The damage is repaired although the cut mark on the surface remains visible. The self-healed hydrogel also exhibits good mechanical strength and thermoresponsive properties. When we repeatedly stretch the self-healed hydrogel, it returns to its original state without fracturing. We further tested the breaking elongation and strain–stress behavior of stretching–releasing cycles of the repaired hydrogel as shown in Fig. S2 (ESI†). The repaired αCD-PNIPAM/PANI hydrogel still exhibits excellent elasticity with a large ultimate strain of 400% at a tensile strength of 21.9 kPa and good cycling performance. Fig. S3 (ESI†) shows the thermal-responsive behavior of the repaired hydrogel under different temperatures and no breakage has been found during the deformation.
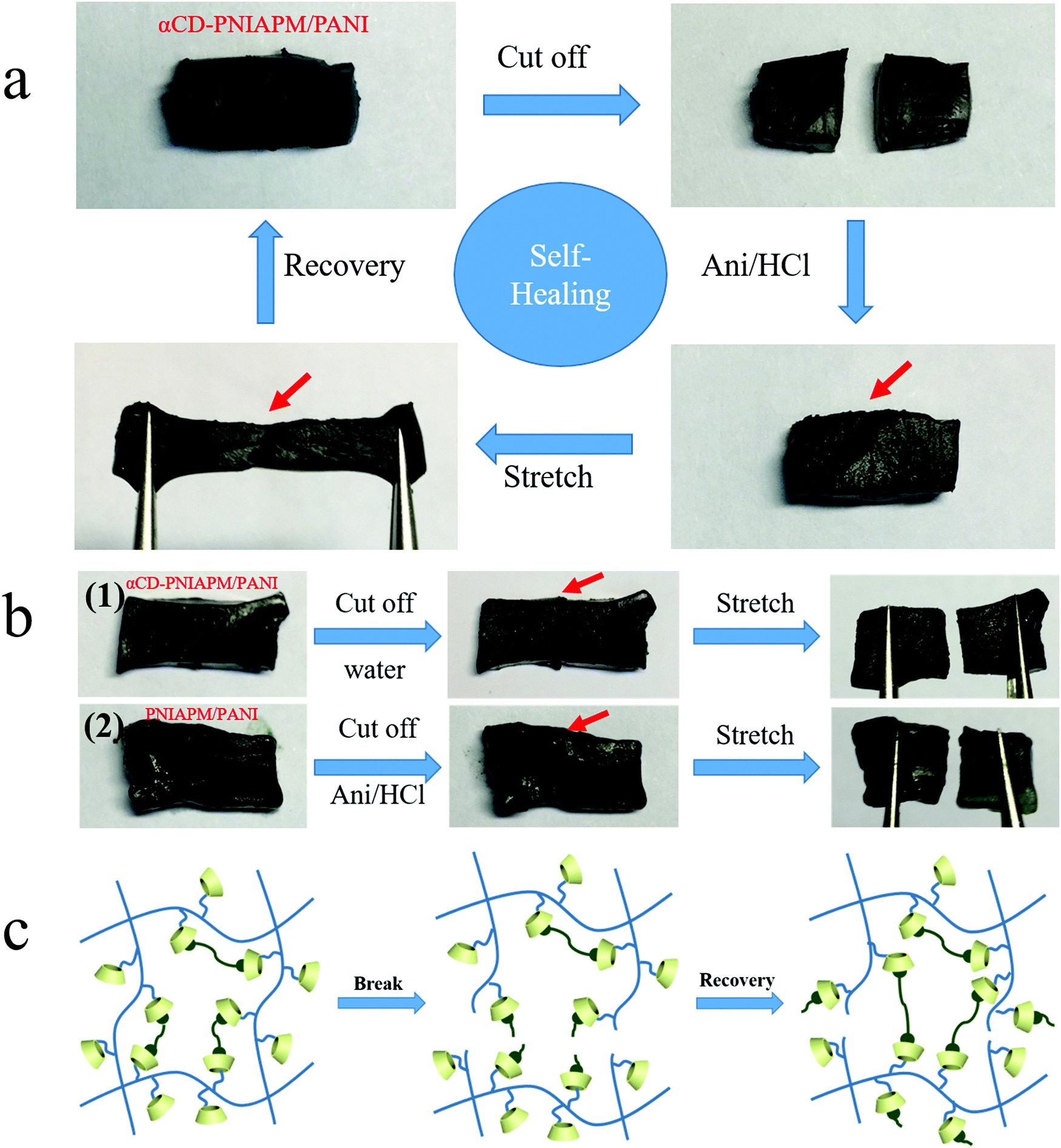 | ||
| Fig. 5 (a) A test sample of the αCD-PNIPAM/PANI hydrogel was cut into two pieces, which were then placed together and immersed in an ANI/HCl solution for self-healing. (b) Self-healing test of (1) the αCD-PNIPAM/PANI hydrogel in pure water, and (2) the PNIPAM/PANI hydrogel in the ANI/HCl solution. (c) The mechanism of αCD-PNIPAM/PANI hydrogel self-healing. | ||
The mechanism of self-healing was explored by some other experiments. First, we tested the αCD-PNIPAM/PANI hydrogel in a pure water environment instead of the ANI/HCl solution, and no self-healing occurs as shown in Fig. 5b(1). We also tested the prepared PNIPAM/PANI hydrogel without αCD in the ANI/HCl solution environment as a contrast, and it did not show any self-healing in Fig. 5b(2). The results demonstrated that the cutting process broke the covalent bonds of PANI instead of pulling the PANI out of the αCD and αCD plays the key role in the hydrogel self-healing by host–guest interaction with PANI.
As above, the mechanism of hybrid hydrogel self-healing is described in Fig. 5c. The PNIPAM backbone chains and host–guest interactions with PANI are broken after the αCD-PNIPAM/PANI hydrogel is cut into two pieces. When we immerse the two pieces into the ANI/HCl solution, the PANI chains are generated and the host–guest interaction with αCD-PNIPAM is rebuilt via interactions between the PANI chains and the vacant αCD moieties on the PNIPAM chains. This serves to repair the hybrid hydrogel. Our unique binary network structure supports the self-healing behaviour of the hybrid hydrogel. The homogeneous macroporous structure of the PNIPAM matrix facilitates molecular motion, and transportation at the cracks also plays an important role in hydrogel repair. This self-healing capacity can significantly enhance the life cycles of these materials and support sustainable development. For further practical application, we tested the cutting/self-healing cycle of the obtained hydrogel and found that the fracture surface of the hydrogel could be fully repaired at the same cut area even after 5 cycles, but partially repaired after 6 or more cutting/self-healing cycles. This may be due to the fact that the cavity of αCD at the fracture surface is gradually filled with the PANI chains.
In addition to its enhanced mechanical properties and well-preserved thermal sensitivity, the αCD-PNIPAM/PANI hydrogel prepared via our host–guest combination method exhibits excellent electrical properties. As shown in Fig. 6a, the electrical conductivities of ten samples were tested. The average conductivity is 0.64 S m−1 in the gel state. This high hydrogel conductivity can be ascribed to the large degree of aniline molecule loading and the in situ formation of conductive PANI in the hydrophobic cavity of αCD which also guarantee a high conductivity of 0.60 S m−1 of the hydrogel even after 5 cutting/self-healing cycles. PANI can act as a continuous electron transport path. In contrast, the hybrid hydrogels that consist of point-to-point connected conducting networks based on blending and random distribution of conductive nanofillers in the matrix result in poor electrical conductivity. The temperature dependence of the hybrid hydrogel conductivity was also measured. Fig. 6b shows that the electrical conductivity of the hydrogel decreases as the temperature increases. This may be caused by changes in the swelling states and microstructures.
 | ||
| Fig. 6 (a) The conductivities of ten αCD-PNIPAM/PANI hydrogel samples. (b) The temperature-dependent conductivity of the αCD-PNIPAM/PANI hydrogel in the gel state. (c) Cycling performance of the αCD-PNIPAM/PANI hydrogel. (d) Resistance of αCD-PNIPAM/PANI cylinder-shaped hybrid hydrogels at different strains. The consistency of the values across compression and relaxation cycles demonstrates excellent electrical stability under deformation. | ||
The stability of the hybrid-hydrogel electrical conductivity was also measured. Fig. 6c shows the results after 50 cycles of heating and cooling. The resistance of the hybrid hydrogel remains approximately 140 kΩ in the heated (deswelled) state and 20 kΩ in the cooled (swelled) state. We further designed a mechanical strain experiment in which a cylindrical αCD-PNIPAM/PANI hydrogel with a thickness of 5 mm was tested and compressed by 0.5 mm each time until the largest strain reaches 50%. As shown in Fig. 6d, little change in the resistance is noted for the αCD-PNIPAM/PANI hydrogel during the compressing test. The excellent cycling performance and electrical stability of the hybrid hydrogel can be attributed to its good thermal sensitivity and mechanical properties. We also tested the electrical properties and stability of PNIPAM/PANI for contrast. As shown in Fig. S4 (ESI†), the lower conductivity and stability compared with the αCD-PNIPAM/PANI hydrogel reveals that the presence of αCD in the hydrogel promotes the loading of the ANI molecules and protects the PANI from rapid degradation with long-term cycling.
Based on the properties demonstrated above, we designed an application using our hydrogels: the temperature-triggered electric switch shown in Fig. 7a. The on/off states of a light emitting diode (LED) can be controlled by the switch, whose state is determined by the temperature around it. When the temperature decreases, the circuit is closed by the swelled hydrogel. This activates the bright blue LED in Fig. 7b. When the temperature increases, the hydrogel loses contact with the electrode owing to deswelling. This induces an open-circuit state and deactivates the LED in Fig. 7c. Interestingly, separating the cylindrical shell such that the hydrogel contacts the electrode again rebuilds the closed circuit and a faint blue light is seen from the LED in Fig. 7d. This indicates that the hydrogel exhibits good electrical conductivity regardless of the temperature. This is attributed to the effective incorporation of PANI by the αCD moieties. The good thermoresponsive sensitivity and electrical performance of the host–guest hydrogels demonstrated here may provide a new approach to conductive hybrid hydrogel preparation.
 | ||
| Fig. 7 (a) Potential application of the conductive and temperature-responsive αCD-PNIPAM/PANI hydrogel as an electrical switch triggered by cooling/heating. (b and c) The on/off states of a LED that is controlled by a switch whose state is determined by the temperature. (d) The closed circuit can be rebuilt by separating the cylindrical shell and making the hydrogels contact the electrode again. | ||
4. Conclusions
In summary, this paper demonstrates a rapid, facile synthesis of highly conductive, temperature-sensitive hydrogel networks via the synergistic combination of hydrophobic–hydrophobic interactions and a pH-triggered opening effect. Due to the presence of the interpenetrating PANI chains governed by host–guest interactions, our hybrid hydrogels exhibit a unique combination of high electrical conductivity, high thermoresponsive sensitivity, and enhanced mechanical properties. In addition, their self-healing capabilities make them truly smart and lengthen their life cycles. We believe that this αCD-PNIPAM/PANI network synthesis strategy, as well as the high performance of these materials in terms of temperature sensitivity, self-healing properties, and conductivity may have a significant impact on advances in multifunctional hydrogel materials for next-generation smart devices. Our work demonstrates that the use of αCD to facilitate host–guest interactions plays an important role in determining the performance of the resulting hybrid materials. These host–guest hybrid hydrogels may be applied to stimuli-responsive electronic devices, self-adaptive electronics, and flexible bioelectronics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2017YFF0210003), the National Basic Research Program of China (No. 2014CB932200), the National Natural Science Foundation of China (21372183), and the Program for Innovative Teams of Outstanding Young and Middle–aged Researchers in the Higher Education Institutions of Hubei Province (T201702).Notes and references
- M. A. Stuart, W. T. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- Z. Lin, D. Mei, M. Chen, Y. Wang, X. Chen, Z. Wang, B. He, H. Zhang, X. Wang, W. Dai, Y. Yin and Q. Zhang, Nanoscale, 2015, 7, 14838–14847 RSC.
- P. Calvert, Adv. Mater., 2009, 21, 743–756 CrossRef.
- H. Jiang, G. Zhang, F. Li, Y. Zhang, Y. Lei, Y. Xia, X. Jin, X. Feng and H. Li, Nanoscale, 2017, 9, 15470–15476 RSC.
- X. Yang, L. Qiu, C. Cheng, Y. Wu, Z. F. Ma and D. Li, Angew. Chem., Int. Ed., 2011, 50, 7325–7328 CrossRef PubMed.
- C. Cheng and D. Li, Adv. Mater., 2013, 25, 13–30 CrossRef PubMed.
- C. Ma, Y. Shi, D. A. Pena, L. Peng and G. Yu, Angew. Chem., Int. Ed., 2015, 54, 7376–7380 CrossRef PubMed.
- C.-H. Zhu, Y. Lu, J. Peng, J.-F. Chen and S.-H. Yu, Adv. Funct. Mater., 2012, 22, 4017–4022 CrossRef.
- R. E. Rivero, M. A. Molina, C. R. Rivarola and C. A. Barbero, Sens. Actuators, B, 2014, 190, 270–278 CrossRef.
- F. Zhao, Y. Shi, L. Pan and G. Yu, Acc. Chem. Res., 2017, 50, 1734–1743 CrossRef PubMed.
- Y. Shi, Ming Wang, Ch. Ma, Y. Wang, X. Li and G. Yu, Nano Lett., 2015, 15, 6276–6281 CrossRef PubMed.
- S. Ahadian, J. Ramon-Azcon, M. Estili, X. Liang, S. Ostrovidov, H. Shiku, M. Ramalingam, K. Nakajima, Y. Sakka, H. Bae, T. Matsue and A. Khademhosseini, Sci. Rep., 2014, 4, 4271 CrossRef PubMed.
- L. Qiu, D. Liu, Y. Wang, C. Cheng, K. Zhou, J. Ding, V. T. Truong and D. Li, Adv. Mater., 2014, 26, 3333–3337 CrossRef PubMed.
- T. Dai, X. Qing, Y. Lu and Y. Xia, Polymer, 2009, 50, 5236–5241 CrossRef.
- T. Dai, X. Qing, H. Zhou, C. Shen, J. Wang and Y. Lu, Synth. Met., 2010, 160, 791–796 CrossRef.
- M. Karbarz, M. Gniadek, M. Donten and Z. Stojek, Electrochem. Commun., 2011, 13, 714–718 CrossRef.
- X. Zhang, V. Chechik, D. K. Smith, P. H. Walton, A.-K. Duhme-Klair and Y. Luo, Synth. Met., 2009, 159, 2135–2140 CrossRef.
- H. Kim, A. A. Abdala and C. W. Macosko, Macromolecules, 2010, 43, 6515–6530 CrossRef.
- K. Haraguchi and H. J. Li, Angew. Chem., Int. Ed., 2005, 44, 6500–6504 CrossRef PubMed.
- H. Chen, F. Yang, Q. Chen and J. Zheng, Adv. Mater., 2017, 29, 1606900 CrossRef PubMed.
- J. Liu, C. S. Y. Tan, Z. Yu, N. Li, C. Abell and O. A. Scherman, Adv. Mater., 2017, 29, 1605325 CrossRef PubMed.
- X. He, C. Zhang, M. Wang, Y. Zhang, L. Liu and W. Yang, ACS Appl. Mater. Interfaces, 2017, 9, 11134–11143 CrossRef PubMed.
- Y. Shi, C. Ma, L. Peng and G. Yu, Adv. Funct. Mater., 2015, 25, 1219–1225 CrossRef.
- G. Graziano, Int. J. Biol. Macromol., 1999, 27, 89–97 CrossRef.
- P. Kujawa, Macromolecules, 2001, 34, 4130–4135 CrossRef.
- K. Depa, A. Strachota, M. Šlouf, J. Brus and V. Cimrová, Sens. Actuators, B, 2017, 244, 616–634 CrossRef.
- Y. Shi, H. Ha, A. Al-Sudani, C. J. Ellison and G. Yu, Adv. Mater., 2016, 28, 7921–7928 CrossRef PubMed.
- C.-H. Lu, W. Guo, X.-J. Qi, A. Neubauer, Y. Paltiel and I. Willner, Chem. Sci., 2015, 6, 6659–6664 RSC.
- S. Naficy, J. M. Razal, G. M. Spinks, G. G. Wallace and P. G. Whitten, Chem. Mater., 2012, 24, 3425–3433 CrossRef.
- P. Pallavicini, S. Basile, G. Chirico, G. Dacarro, L. D'Alfonso, A. Dona, M. Patrini, A. Falqui, L. Sironi and A. Taglietti, Chem. Commun., 2015, 51, 12928–12930 RSC.
- J. Benson, I. Kovalenko, S. Boukhalfa, D. Lashmore, M. Sanghadasa and G. Yushin, Adv. Mater., 2013, 25, 6625–6632 CrossRef PubMed.
- Z. Tang, J. Wu, Q. Liu, M. Zheng, Q. Tang, Z. Lan and J. Lin, J. Power Sources, 2012, 203, 282–287 CrossRef.
- J. H. Wu, Z. Lan, J. M. Lin, M. L. Huang, S. C. Hao, T. Sato and S. Yin, Adv. Mater., 2007, 19, 4006–4011 CrossRef.
- E. M. M. Del Valle, Process Biochem., 2004, 39, 1033–1046 CrossRef.
- O. Peters and H. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8961–8963 CrossRef PubMed.
- G.-P. H. a. S. Kaskel, ACS Nano, 2014, 8, 7138–7146 CrossRef PubMed.
- P. Chakraborty, P. Bairi, B. Roy and A. K. Nandi, ACS Appl. Mater. Interfaces, 2014, 6, 3615–3622 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qm00324f |
| This journal is © the Partner Organisations 2018 |