
Rhodium-catalyzed intermolecular C(sp3)–H amination in a purely aqueous system†
Xunbo
Lu
,
Yufeng
Shi
and
Fangrui
Zhong
 *
*
Hubei Key Laboratory of Bioinorganic Chemistry & Materia Medica, School of Chemistry and Chemical Engineering, Huazhong University of Science and Technology (HUST), 1037 Luoyu Road, Wuhan 430074, China. E-mail: chemzfr@hust.edu.cn; Fax: +(0)86-(0)27-87543632
First published on 28th November 2017
Abstract
An efficient Rh-catalyzed intermolecular C(sp3)–H amination in a purely aqueous system is developed for the first time. This methodology features environmental benignity, broad substrate scope and versatility in late-stage functionalization of several biologically important molecules. Such an oxidation protocol provides easy access to various aliphatic amine derivatives in an efficient and sustainable manner.
In recent years, the synthetic community has been experiencing increasing pressure and motivation to improve the environmental benignity of chemical processes.1 However, despite considerable advances, there remain significant challenges for synthetic systems to perform transformations that nature can routinely achieve under physiological conditions. For instance, cytochrome P450 and other oxygenase enzymes are known for their capacity to selectively oxidize inert aliphatic C–H bonds into C–OH or carbonyl functional groups.2 Their remarkable catalytic performance under mild conditions represents a gold standard for developing novel synthetic methodologies. In particular, in light of the “green” nature of water, the aqueous medium adopted in enzymatic processes is of immense interest to the synthetic community.3 However, reactions mediated by synthetic catalysts, even for most prominent transition-metal complexes that have revolutionized industrial organic synthesis, are still primarily restricted to organic solvents and have met with limited success when used in water.4 Apparently, due to the poor solubility of catalysts and substrates, as well as the intrinsic sensitivity of metal catalysts, ligands and in situ generated reactive organometallic intermediates towards water, performing such reactions is often associated with various problems.4
Aliphatic amines represent a category of compounds particularly favored by nature, as validated by the wide occurrence of diverse biologically active alkaloids.5 In line with nature, synthetic aliphatic amine compounds are prevalent in pharmaceutical agents, as seen in selected examples of marketed drugs (Fig. 1).6 In this context, synthetic chemists hold long-standing interest in developing effective and convenient amination methods. Although conventional approaches mainly rely on the inherent reactivity of functional groups such as alcohol and carbonyl, the direct C–H amination provides a novel and straightforward synthetic strategy.7 In particular, C–H amination reactions via transition-metal catalyzed nitrene transfer, pioneered by Breslow8 and Mansuy,9 have been established as a reliable and powerful tool to create C–N bonds.7a–f Various metal catalysts, including Fe, Co, Ru, Mn and Rh complexes, are known to mediate such processes in combination with sulfonamides, sulfamates, azides, or carbamates.10 The resulting nitrenoids have proven to be reactive towards intra- and intermolecular C–H bonds to yield aliphatic amine or diamine derivatives as well as other aza-heterocycles.
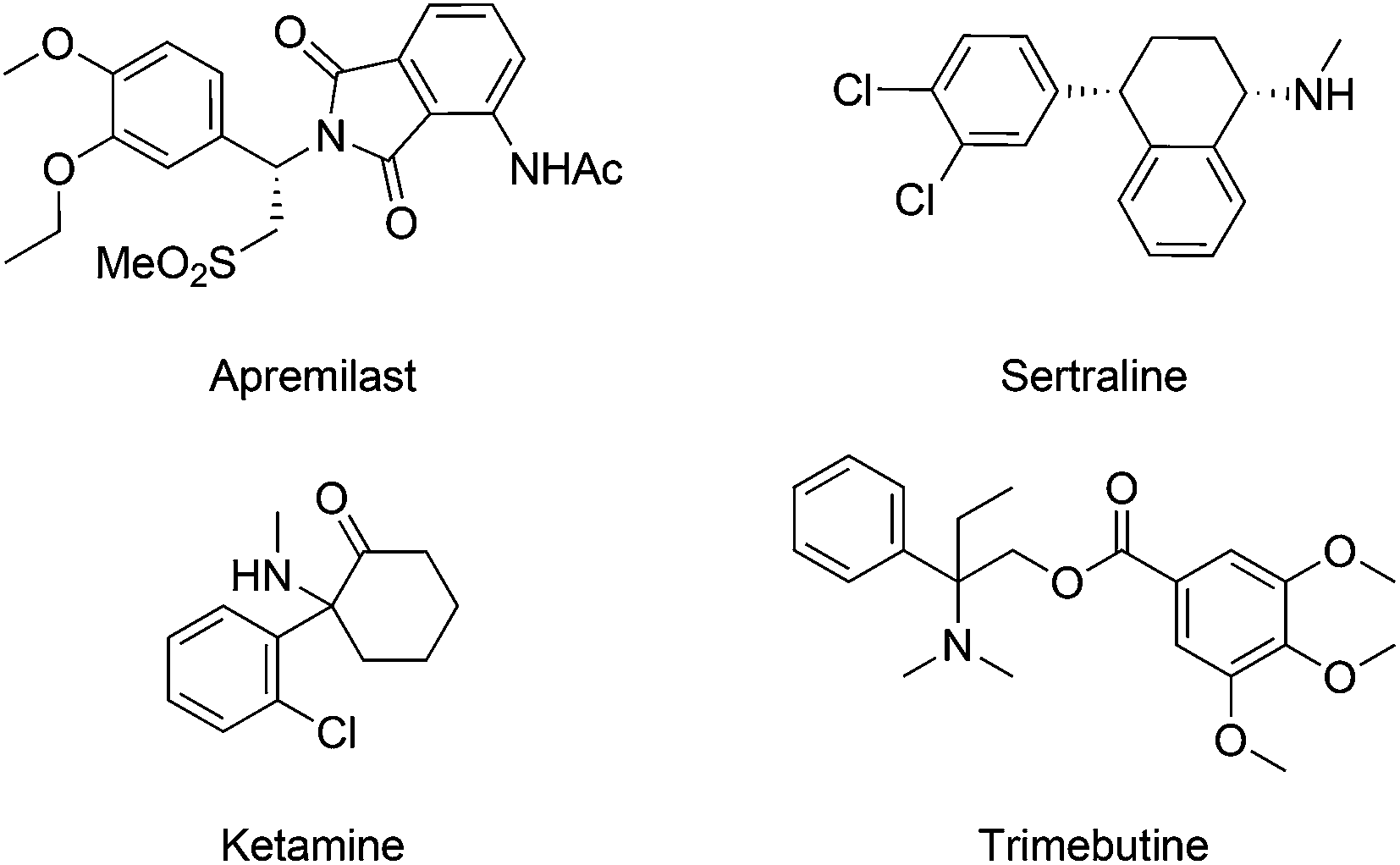 | ||
| Fig. 1 Representative marketed drugs derived from aliphatic amines. | ||
Given the high electrophilicity of metal nitrenoid intermediates, such amination processes are normally performed in inert but toxic solvents (e.g. benzene and dichloromethane).7a Moreover, it seems that water has been presumably considered as an inappropriate solvent for these transformations. Although water has been utilized successfully as the solvent for a number of C(sp2)–H amination reactions,11 to the best of our knowledge, the amination of C(sp3)–H in aqueous medium remains unknown. Notably, the former are often achieved in the presence of a directing group, the absence of which might make the nitrene transfer step highly sensitive in water. We hypothesized that a heterogeneous “on water” reaction12 might be able to overcome the decomposition problem of nitrenoid intermediates and result in a practical amination protocol that can be performed in aqueous medium.
Our initial experiments were aimed at identifying an appropriate metal complex and a nitrogen source. Binuclear rhodium tetracarboxylates are most frequently applied as catalysts, thus Rh2(OAc)4 was firstly used to catalyze the model reactions of 4-ethylbiphenyl 1a in combination with PhI(OAc)2 as the oxidant.13 The reactivities of nitrene precursors were found to be highly important and sulfamates displayed superior reactivity to sulfonamides (Table 1, entries 1–3). Among them, trichloroethyl sulfamate (TcesNH2) gave the best conversion, although only in a rather low yield (12%, entry 3). Then, we proceeded to test our hypothesis by performing the reaction in water. To our delight, such a purely aqueous system resulted in the desired amination product 3a in an even higher yield (entry 4). In contrast, other previously used metal complexes failed to afford any detectable desired products (entries 5–8). We further sought a more competent catalyst and used a designer robust rhodium dimer, bis[rhodium(a,a,a′,a′-tetramethyl-1,3-benzenedipropionate)] [Rh2(esp)2], developed by Du Bois,10a as the catalyst. Pleasingly, a significantly higher yield was recorded in this instance, which again overmatched those obtained in organic solvents (entries 9–11). Other parameters of the reaction conditions were further examined. While performing the reaction in diluted and chilled water (4 °C) led to a higher isolated yield of 3a without prolonging the reaction time (24 h), the presence of an inorganic base MgO or NaHCO3 indeed had no effect (entries 12–14). Notably, catalyst loading could be reduced to 1.5 mol% without a negative influence. A portion-wise addition of PhI(OAc)2 was found to be highly beneficial to elevate the yield of 3a to 89% (entries 15 and 16). This result represents by far one of the best yields attainable for intermolecular C(sp3)–H amination reactions.14 Moreover, altering stoichiometry by using excess 1a (1.5 equiv.) was proven to be helpful and eventually furnished 3a in 93% yield (entry 17). Given the easy availabilities of alkane substrates, the resulting modified protocol still holds significant synthetic utility. Under identical conditions, sulfamates with diverse steric and electronic properties were also identified to be suitable nitrogen precursors and furnished the corresponding aminated alkanes 3b–e in high yields.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. | T (°C) | RSO2NH2 | Solvent | Yieldb |
a Reaction conditions: 1a (0.15 mmol), 2 (0.15 mmol), catalyst (2.0 mol%) and PhI(OAc)2 (0.3 mmol) in 0.5 mL H2O were stirred at 25 °C for 24 h.
b Isolated yield.
c In 3.0 mL H2O.
d MgO or NaHCO3 (0.45 mmol) was added.
e In 3.0 mL H2O with 1.5 mol% of Rh2(esp)2.
f PhI(OAc)2 was added in three portions over 3 hours.
g ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1a (0.225 mmol) was used. N.D. = not detected. 1a (0.225 mmol) was used. N.D. = not detected. 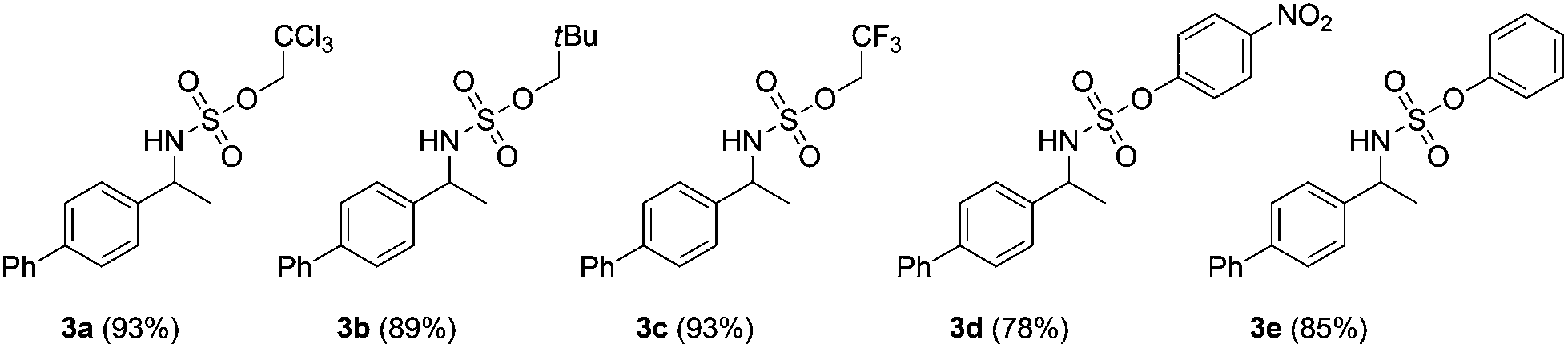
|
|||||
| 1 | Rh2(OAc)4 | 25 | TsNH2 | C6H6 | Trace |
| 2 | Rh2(OAc)4 | 25 | TfNH2 | C6H6 | <5% |
| 3 | Rh2(OAc)4 | 25 | TcesNH2 | C6H6 | 12 |
| 4 | Rh2(OAc)4 | 25 | TcesNH2 | H2O | 20 |
| 5 | Mn(TPP)Cl | 25 | TcesNH2 | H2O | N.D. |
| 6 | Cu(OTf)2 | 25 | TcesNH2 | H2O | N.D. |
| 7 | Fe(ClO4)2 | 25 | TcesNH2 | H2O | N.D. |
| 8 | Ru(TPP)CO | 25 | TcesNH2 | H2O | N.D. |
| 9 | Rh2(esp)2 | 25 | TcesNH2 | H2O | 57 |
| 10 | Rh2(esp)2 | 25 | TcesNH2 | C6H6 | 44 |
| 11 | Rh2(esp)2 | 25 | TcesNH2 | CH2Cl2 | 48 |
| 12 | Rh2(esp)2 | 4 | TcesNH2 | H2O | 71 |
| 13c | Rh2(esp)2 | 4 | TcesNH2 | H2O | 74 |
| 14d | Rh2(esp)2 | 4 | TcesNH2 | H2O | 70 |
| 15e | Rh2(esp)2 | 4 | TcesNH2 | H2O | 73 |
| 16e,f | Rh2(esp)2 | 4 | TcesNH2 | H2O | 89 |
| 17e,f,g | Rh2(esp)2 | 4 | TcesNH2 | H2O | 93 |
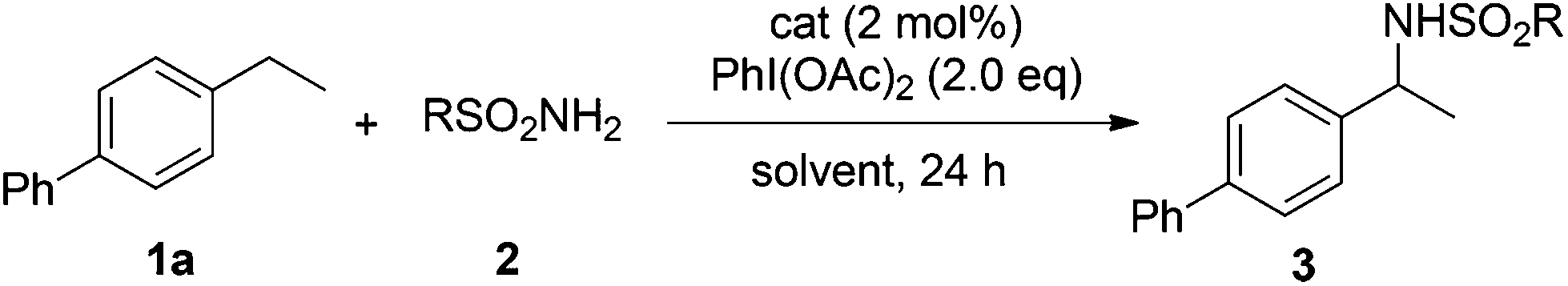
The optimized conditions were subsequently applied to different hydrocarbons (Table 2). Pleasingly, it turned out that different substrates bearing benzylic C–H bonds were well tolerated. Yields ranging from 56% to 86% were recorded for ethylbenzene and its para- and meta-substituted analogues (entries 1–6). Moreover, to our delight, reactions also proceeded smoothly with para-NO2 and ortho-substituted ethylbenzenes (entries 7–9), as such electronically or sterically disfavored alkanes are highly challenging substrates yet not addressed in C–H amination reactions.10b,14 Cyclic hydrocarbons such as indan and tetrahydronaphthalene were readily converted to the desired products 3o and 3p, respectively. Subsequent investigations revealed that functionalized starting materials possessing ester, amide, and ketone groups also nicely participated in this process and led to the formation of various amino acid, amino alcohol, diamine, and ketoamine derivatives in high yields (entries 12–18). Apart from benzylic C–H bonds, secondary and tertiary hydrocarbons could also be aminated in moderate to good yields, providing easy access to cyclohexyl, cycloheptyl as well as adamantyl amine products in aqueous medium (entries 19–21). Furthermore, when cyclohexene was applied as a substrate, its allylic site was selectively oxidized in 60% yield, albeit with the formation of a small amount of a separable aziridine side product (entry 22).
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Product | 3 | Yieldb (%) |
| a Reaction conditions: 1 (0.225 mmol), TcesNH2 (0.15 mmol), Rh2(esp)2 (1.5 mol%) and PhI(OAc)2 (0.3 mmol) in 3.0 mL H2O were stirred at 4 °C for 24 h. b Isolated yield. c 5.0 eq. of substrate 1 were used. d 3.0 eq. of substrate 1 were used. | ||||
| 1c |
|
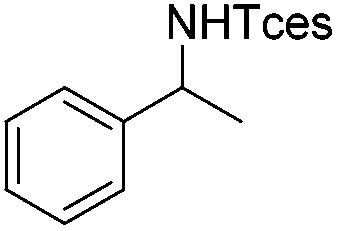
|
3f | 86 |
| 2 |
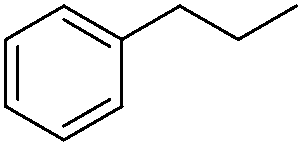
|
|
3g | 74 |
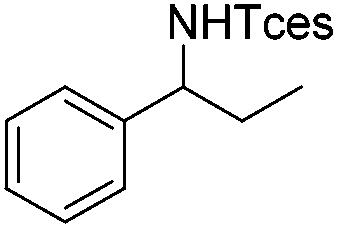
| 3 |
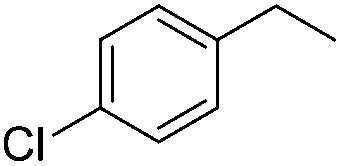
|
|
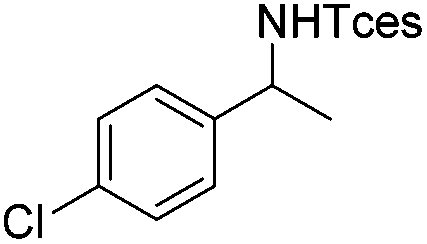
3h | 73 |
| 4 |
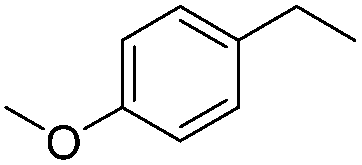
|
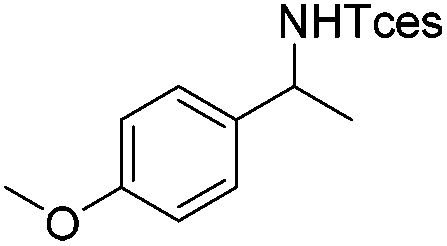
|
3i | 82 |
| 5 |
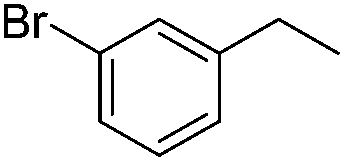
|
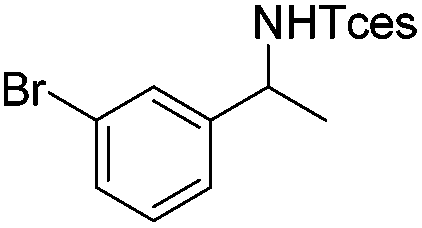
|
3j | 59 |
| 6 |
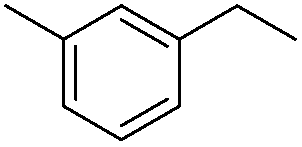
|
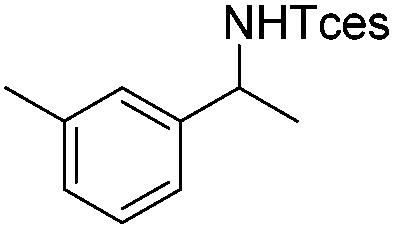
|
3k | 56 |
| 7 |
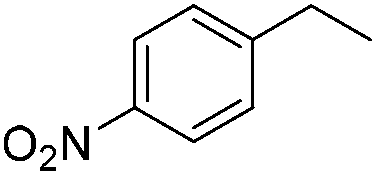
|
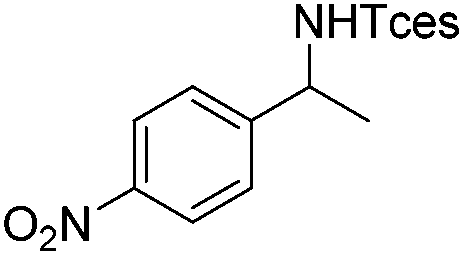
|
3l | 35 |
| 8 |
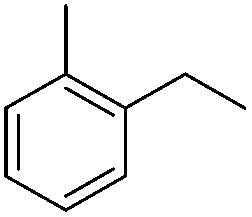
|
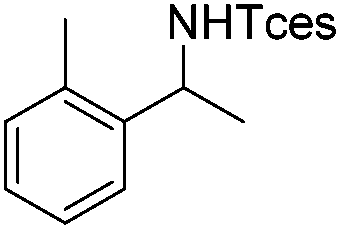
|
3m | 56 |
| 9 |
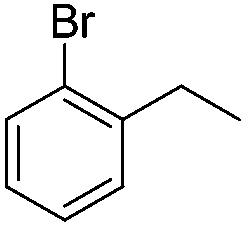
|
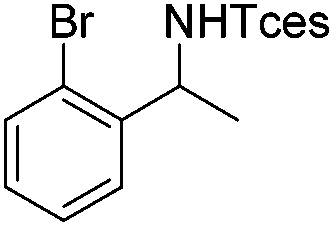
|
3n | 43 |
| 10d |
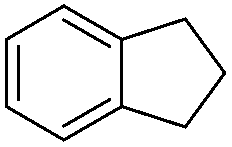
|
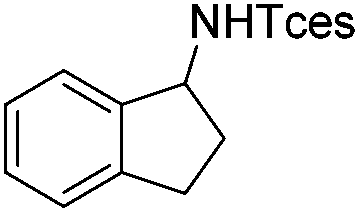
|
3o | 99 |
| 11 |
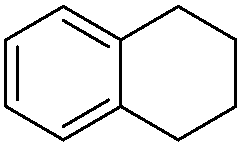
|
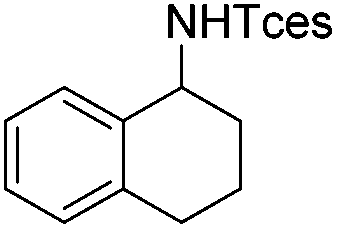
|
3p | 72 |
| 12 |
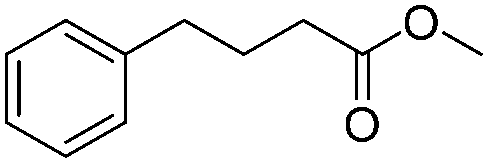
|
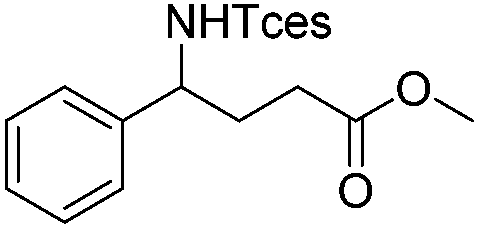
|
3q | 98 |
| 13 |
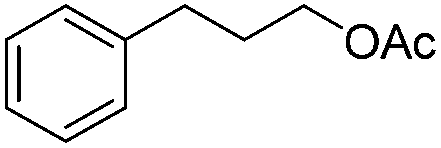
|
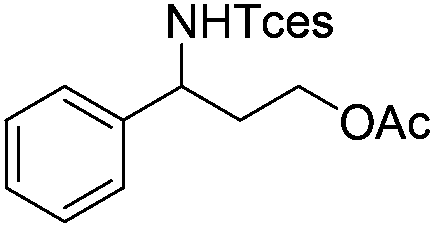
|
3r | 83 |
| 14 |
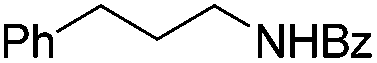
|
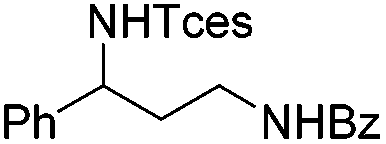
|
3s | 78 |
| 15 |
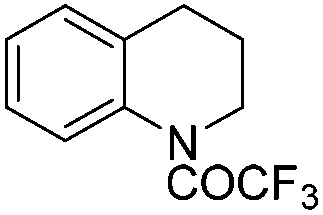
|
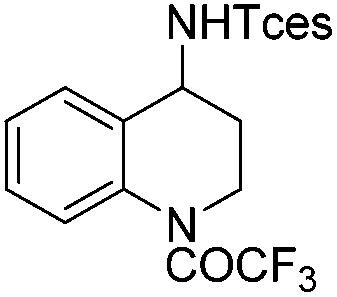
|
3t | 98 |
| 16 |
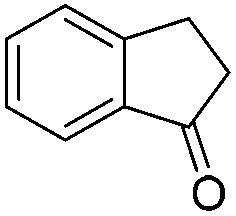
|
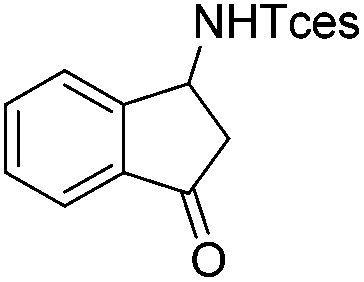
|
3u | 95 |
| 17 |
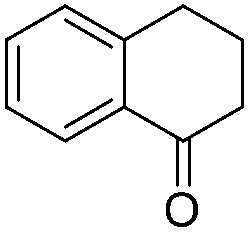
|
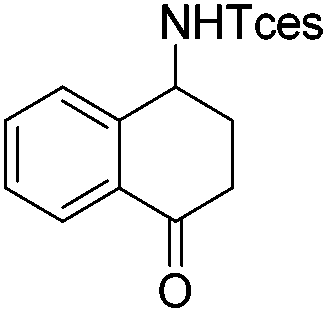
|
3v | 94 |
| 18 |
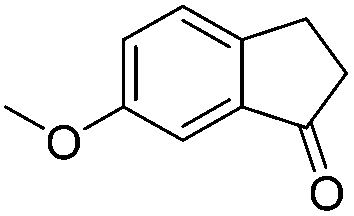
|
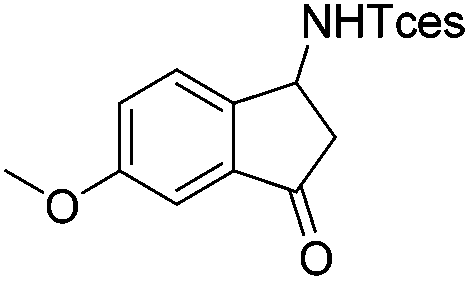
|
3w | 87 |
| 19 |
|
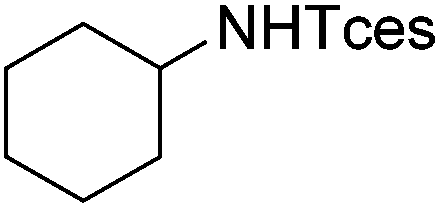
|
3x | 64 |
| 20 |
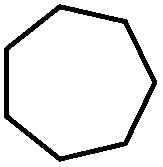
|
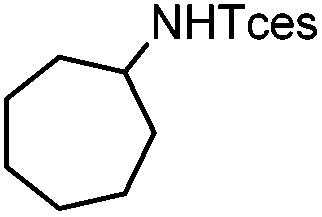
|
3y | 70 |
| 21 |
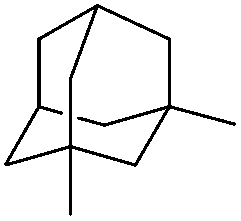
|
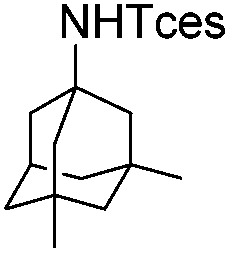
|
3z | 76 |
| 22 |
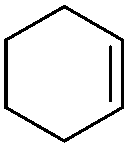
|
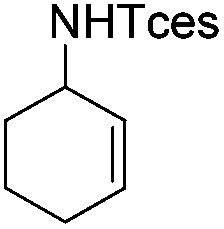
|
3a′ | 60 |
The broad generality of Rh-catalyzed C–H amination reactions in water suggests that such an amination protocol might hold potential for late-stage functionalization.15 We therefore investigated the functionalization of several biologically active compounds. Pleasingly, the derivatives of valine and phenylalanine underwent amination at the tertiary and benzylic C–H bonds, respectively, with high efficiency (Scheme 1a and b). Similar reactivity was also observed for the remote benzylic site-selective amination of a non-natural amino acid (homophenylalanine), albeit with negligible stereoselectivity (Scheme 1c). Nevertheless, the two diastereoisomers could be readily separated by column chromatography. Ibuprofen is a commonly prescribed nonsteroidal drug for treating pain, fever, and inflammation. The amination of ibuprofen methyl ester furnishes the product with a benzylic C–N bond in 74% yield (Scheme 1d).16 The synthetic practicality of this methodology was further demonstrated by a gram-scale preparation of compound 3v. It is noteworthy that in this case the catalyst loading could be reduced to 0.5 mol%, while a 90% yield was still attainable (Scheme 1e).
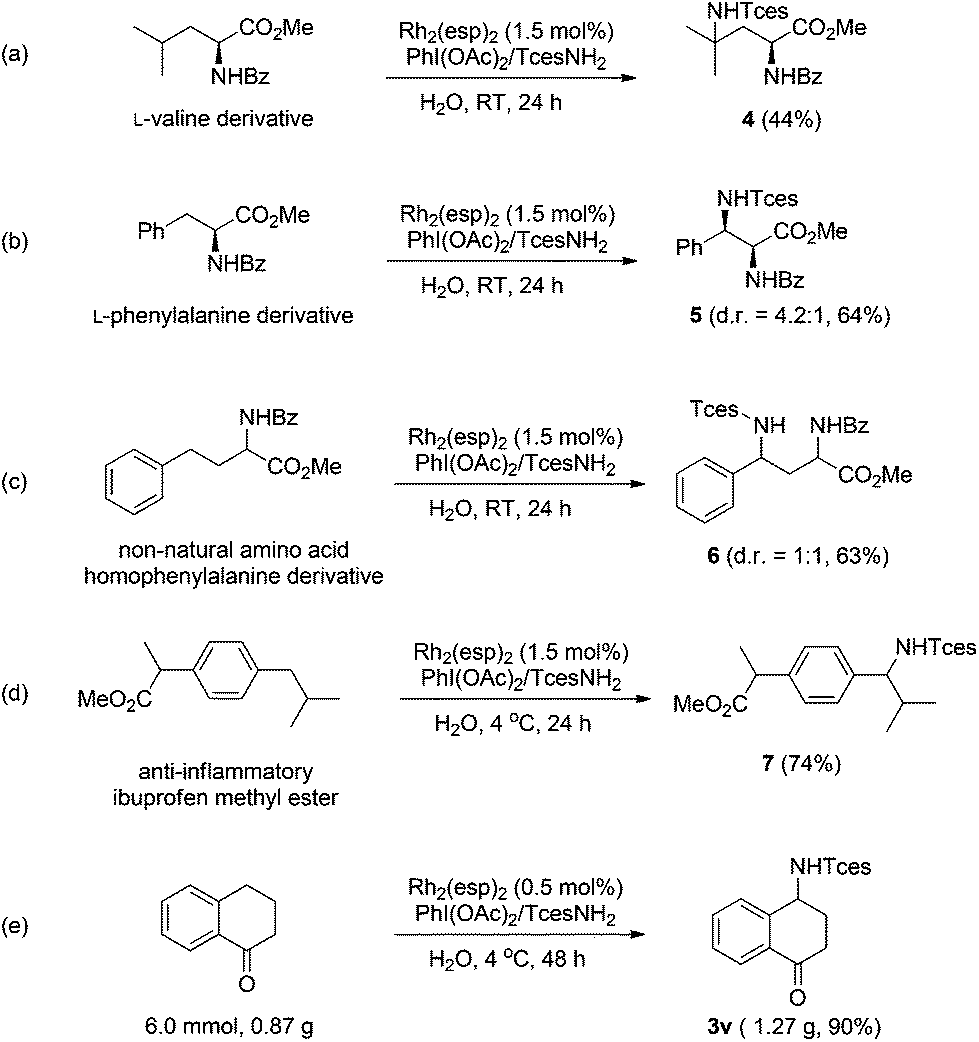 | ||
| Scheme 1 Synthetic utilities of Rh-catalyzed C–H amination reactions in water. | ||
The above reaction outcomes suggest that water plays important roles in the amination process. In previous cases, an inorganic base was frequently used to scavenge acid side products generated from oxidants.13b However, it was identified here that MgO or NaHCO3 did not affect the reaction outcome in aqueous medium (Table 1, entry 14) and the resulting aqueous phase after the completion of the reaction was found to be acidic (pH ≈ 4.0). Previous mechanistic studies confirmed that PhI(O2CtBu)2 was a superior oxidant with regard to solubility in benzene but led to the formation of the aziridine side product from tBuCO2H.17 Moreover, reaction rates have a first-order dependence on the oxidant, and the accumulation of AcOH is adverse due to the enhanced protonolysis of in situ generated iminoiodinane 2c′, which gave rise to highly electrophilic rhodium nitrenoid species and underwent a concerted asynchronous C–H insertion process (Scheme 2).18 Although it remains unclear in the present system whether water gets involved in stabilizing the transition state, it seems reasonable to assume that water is crucial for removing AcOH from the oil phase and meanwhile it promotes the formation of iminoiodinane possibly via hydrogen bonding with the sulfamate oxygen of 2c and PhI(OAc)2 at the oil–aqueous interface. Moreover, neither intramolecular amination in aqueous medium nor the model reaction (Table 1) under neat conditions was comparably efficient.19 It was further found that the addition of 20 eq. of water to the reaction carried out in benzene led to a significant deterioration in the yield.20 The above observations seem to suggest that a “on water” catalysis is likely in operation and we also speculate that hydrophobic interactions21 induced by water might additionally accelerate the intermolecular reaction.
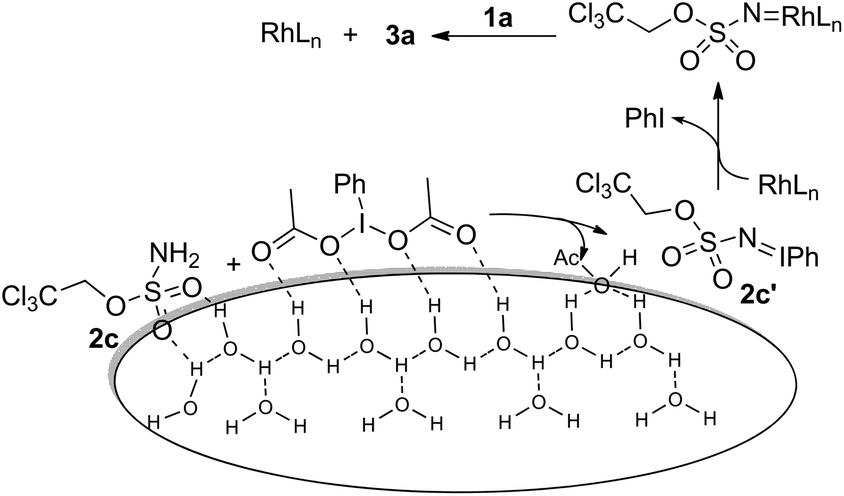 | ||
| Scheme 2 Plausible working model. | ||
In summary, we have successfully developed the first metal-catalyzed intermolecular C(sp3)–H amination reactions performed in a purely aqueous medium under mild conditions. This method features great environmental benignity and high efficiency towards a wide range of hydrocarbons bearing different functional groups. In light of its versatile synthetic utility demonstrated by the late-stage functionalization of several bioactive molecules, we anticipate that the methodology described here will find wide applications in the sustainable chemistry of catalytic C−H functionalization.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank financial support from the National Natural Science Foundation of China (21602067), a starting grant of the Huazhong University of Science and Technology. The authors are also grateful to the Analytical and Testing Centre of HUST, the Analytical and Testing Centre of School of Chemistry and Chemical Engineering (HUST) as well as the 100 Talents Program of the Hubei Provincial Government.Notes and references
- (a) S. C. Ameta and R. Ameta, Green Chemistry: Fundamentals and Applications, Apple Academic Press, New Jersey, 2013 Search PubMed; (b) M. B. Gawande, V. D. B. Bonifácio, R. Luque, P. S. Branco and R. S. Varma, Chem. Soc. Rev., 2013, 42, 5522 RSC.
- P. R. Ortiz, Cytochrome P450: Structure, Mechanism and Biochemistry, Springer, Berlin, 3rd edn, 2005 Search PubMed.
- (a) M. B. Gawande, V. D. B. Bonifacio, R. Luque, P. S. Branco and R. S. Varma, Chem. Soc. Rev., 2013, 42, 5522 RSC; (b) M. O. Simon and C. J. Li, Chem. Soc. Rev., 2012, 41, 1415 RSC; (c) R. N. Butler and A. G. Coyne, Chem. Rev., 2010, 110, 6302 CrossRef CAS PubMed.
- (a) B. Li and P. H. Dixneuf, Chem. Soc. Rev., 2013, 42, 5744 RSC; (b) C. Fischmeister and H. Doucet, Green Chem., 2011, 13, 741 RSC; (c) R. N. Butler and A. G. Coyne, Chem. Rev., 2010, 110, 6302 CrossRef CAS PubMed.
- T. Aniszewski, Alkaloids: Chemistry, Biology, Ecology, and Applications, Elsevier, Amsterdam, 2rd edn, 2015 Search PubMed.
- (a) R. S. Obach, L. M. Cox and L. M. Tremaine, Drug Metab. Dispos., 2005, 33, 262 CrossRef CAS PubMed; (b) H. T. Lee and B. J. Kim, Arch. Pharmacal Res., 2011, 34, 861 CrossRef CAS PubMed; (c) G. M. Keating, Drugs, 2017, 77, 459 CrossRef CAS PubMed; (d) M. W. Tyler, H. B. Yourish, D. F. Ionescu and S. J. Haggarty, ACS Chem. Neurosci., 2017, 8, 1122 CrossRef CAS PubMed.
- For selected reviews, see: (a) D. Hazelard, P.-A. Nocquet and P. Compain, Org. Chem. Front., 2017, 4, 2500 RSC; (b) Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247 CrossRef CAS PubMed; (c) B. Darses, R. Rodrigues, L. Neuville, M. Mazurais and P. Dauban, Chem. Commun., 2017, 53, 493 RSC; (d) T. G. Driver, Nat. Chem., 2013, 5, 736 CrossRef CAS PubMed; (e) J. L. Roizen, M. E. Harvey and J. Du Bois, Acc. Chem. Res., 2012, 45, 911 CrossRef CAS PubMed; (f) G. Dequirez, V. Pons and P. Dauban, Angew. Chem., Int. Ed., 2012, 51, 7384 CrossRef CAS PubMed; (g) K. Shin, H. Kim and S. Chang, Acc. Chem. Res., 2015, 48, 1040 CrossRef CAS PubMed; (h) M. L. Louillat and F. W. Patureau, Chem. Soc. Rev., 2014, 43, 901 RSC; (i) G. Y. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651 RSC.
- (a) R. Breslow and S. H. Gellman, J. Chem. Soc., Chem. Commun., 1982, 1400 RSC; (b) R. Breslow and S. H. Gellman, J. Am. Chem. Soc., 1983, 105, 6728 CrossRef CAS.
- D. Mansuy, J.-P. Mahy, A. Dureault, G. Bedi and P. Battioni, J. Chem. Soc., Chem. Commun., 1984, 1161 RSC.
- For recent examples, see: (a) C. G. Espino, K. W. Fiori, M. Kim and J. Du Bois, J. Am. Chem. Soc., 2004, 126, 15378 CrossRef CAS PubMed; (b) K. W. Fiori and J. Du Bois, J. Am. Chem. Soc., 2007, 129, 562 CrossRef CAS PubMed; (c) H. Lu, H. Jiang, L. Wojtas and X. P. Zhang, Angew. Chem., Int. Ed., 2010, 49, 10192 CAS; (d) M. E. Harvey, D. G. Musaev and J. Du Bois, J. Am. Chem. Soc., 2011, 133, 17207 CrossRef CAS PubMed; (e) Y. Liu, X. Guan, E. L.-M. Wong, P. Liu, J.-S. Huang and C.-M. Che, J. Am. Chem. Soc., 2013, 135, 7194 CrossRef CAS PubMed; (f) Q. Nguyen, T. Nguyen and T. G. Driver, J. Am. Chem. Soc., 2013, 135, 620 CrossRef CAS PubMed; (g) H. Lu, C. Li, H. Jiang, C. L. Lizardi and X. P. Zhang, Angew. Chem., Int. Ed., 2014, 53, 7028 CrossRef CAS PubMed; (h) S. M. Paradine, J. R. Griffin, J. Zhao, A. L. Petronico, S. M. Miller and M. C. White, Nat. Chem., 2015, 7, 987 CrossRef CAS PubMed; (i) M. J. T. Wilding, D. A. Iovan and T. A. Betley, J. Am. Chem. Soc., 2017, 139, 12043 CrossRef CAS PubMed.
- (a) M. A. Ali, X. Yao, H. Sun and H. Lu, Org. Lett., 2015, 17, 1513 CrossRef CAS PubMed; (b) M. A. Ali, X. Yao, G. Li and H. Lu, Org. Lett., 2016, 18, 1386 CrossRef CAS PubMed; (c) L. Yang, H. Li, H. Zhang and H. Lu, Eur. J. Org. Chem., 2016, 5611 CrossRef CAS; (d) L. Shi and B. Wang, Org. Lett., 2016, 18, 2820 CrossRef CAS PubMed; (e) T. Xiong, Y. Li, L. Mao, Q. Zhang and Q. Zhang, Chem. Commun., 2012, 48, 2246 RSC.
- S. Narayan, J. Muldoon, M. G. Finn, V. V. Fokin, H. C. Kolb and K. B. Sharpless, Angew. Chem., Int. Ed., 2005, 44, 3275 CrossRef CAS PubMed.
- (a) C. G. Espino, P. M. Wehn, J. Chow and J. Du Bois, J. Am. Chem. Soc., 2001, 123, 6935 CrossRef CAS; (b) C. G. Espino and J. Du Bois, Angew. Chem., Int. Ed., 2001, 40, 598 CrossRef CAS.
- (a) C. Liang, F. Robert-Peillard, C. Fruit, P. Müller, R. H. Dodd and P. Dauban, Angew. Chem., Int. Ed., 2006, 45, 4641 CrossRef CAS PubMed; (b) C. Liang, F. Collet, F. Robert-Peillard, P. Müller, R. H. Dodd and P. Dauban, J. Am. Chem. Soc., 2008, 130, 343 CrossRef CAS PubMed; (c) H. Lebel, C. Trudel and C. Spitz, Chem. Commun., 2012, 48, 7799 RSC; (d) Y. Nishioka, T. Uchida and T. Katsuki, Angew. Chem., Int. Ed., 2013, 52, 1739 CrossRef CAS PubMed; (e) C. K. Prier, R. K. Zhang, A. R. Buller, S. Brinkmann-Chen and F. H. Arnold, Nat. Chem., 2017, 9, 629 CrossRef CAS PubMed.
- For reviews on late stage C–H functionalization, see: (a) T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546 RSC; (b) J. He, L. G. Hamann, H. M. L. Davies and R. E. J. Beckwith, Nat. Commun., 2015, 6, 5943 CrossRef PubMed.
- As kindly suggested by one referee, the spatial separation between the two stereogenic centers across the phenyl ring of 7 might make its diastereoisomers not distinguishable in the NMR spectra. However, further analysis via HPLC confirmed that the diastereomeric ratio was approximately 1:1. See the ESI† for details.
- D. N. Zalatan and J. Du Bois, J. Am. Chem. Soc., 2009, 131, 7558 CrossRef CAS PubMed.
- (a) K. W. Fiori, C. G. Espino, B. H. Brodsky and J. Du Bois, Tetrahedron, 2009, 65, 3042 CrossRef CAS; (b) R. H. Perry, T. J. Cahill III, J. L. Roizen, J. Du Bois and R. N. Zare, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 18295 CrossRef CAS PubMed.
- An intramolecular reaction with 3-phenylpropyl sulfamate under these aqueous oxidative conditions furnished the amination product in 25% yield. A reaction between 1a and TcesNH2 under neat conditions afforded 3a in 52% yield, which was lower than 93% yield obtained in water (Table 1, entry 17) under otherwise identical conditions.
- For more details, see Table S1 in the ESI.†.
- (a) S. Otto, W. Blokzijl and J. B. F. N. Engberts, J. Org. Chem., 1994, 59, 5372 CrossRef CAS; (b) Y. Jung and R. A. Marcus, J. Am. Chem. Soc., 2007, 129, 5492 CrossRef CAS PubMed; (c) Q. Du, E. Freysz and Y. R. Shen, Science, 1994, 264, 826 CAS; (d) Y. R. Shen and V. Ostroverkhov, Chem. Rev., 2006, 106, 1140 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7gc03149a |
| This journal is © The Royal Society of Chemistry 2018 |