
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organic oxidations promoted in vortex driven thin films under continuous flow†
Scott J.
Pye
 a,
Scott J.
Dalgarno
b,
Justin M.
Chalker
*a and
Colin L.
Raston
*a
a,
Scott J.
Dalgarno
b,
Justin M.
Chalker
*a and
Colin L.
Raston
*a
aCentre for NanoScale Science and Technology, College of Science and Engineering, Flinders University Sturt Road, Bedford Park, Adelaide, South Australia 5042, Australia. E-mail: colin.raston@flinders.edu.au; justin.chalker@flinders.edu.au
bInstitute of Chemical Sciences, Heriot-Watt University, Riccarton, Edinburgh, Scotland EH14 4AS, UK
First published on 27th November 2017
Abstract
With increasing concerns for the environmental impact of chemical manufacturing, reagents and processes that align with the principles of green chemistry are essential. The fundamental oxidation of organic substrates is no exception and in this report three distinct modes of green oxidation are demonstrated in a vortex fluidic device (VFD) under continuous flow: aerobic oxidation, oxidation using chlorine bleach, and oxidation using hydrogen peroxide. The VFD, which is a thin film microfluidic platform, revealed clear advantages in these oxidations in comparison to traditional batch reactor processing: Efficient mass transfer of gases in the dynamic thin film increased the rate of aerobic oxidations, and the intense micromixing allowed multi-phase oxidations to proceed efficiently, obviating the need for organic solvents and phase transfer catalysts. In addition, the rapid dissipation of heat in the VFD also improved the safety profile and stereoselectivity for exothermic oxidations.
Environmentally benign chemical transformations are essential in organic synthesis and sustainable processes for the future.1,2 Rudimentary oxidations are among the most widely used reactions in synthesis, so efforts to improve their green chemistry profile are especially important.3–5 However, many classic oxidations are still used routinely in both academic and industrial research despite their reliance on the stoichiometric use of toxic metals,6 high molecular weight reagents7 that suffer from poor atom economy,8,9 or highly energetic oxidants that present detonation risks.7,10 Alternative options such as air or molecular oxygen,11,12 hypochlorous acid and sodium hypochlorite (chlorine bleach),13 and hydrogen peroxide14,15 have been identified as green oxidants because of their attractive safety profile, high atom economy, and the generation of innocuous by-products such as water or sodium chloride.16 To this end, continuous flow reactors have been shown to address safety, scalability and efficiency of many transformations,17,18 including these oxidations.19,20 These advances notwithstanding, such continuous flow reactors often suffer from high overhead cost, complex operation, the use of organic solvents and channel blocking associated with the formation of emulsions, salts and other insoluble material.21–23 In this report, we examine how the previously mentioned green oxidants can be used ever more efficiently by pairing them with new methods of reaction processing that are efficient, safe, and operationally simple. Specifically, we studied these oxidations in a vortex fluidic device (VFD), with an aim to identify practical benefits that would encourage increased consideration and uptake of green oxidations in chemical synthesis.
The VFD (Fig. 1 and Fig. S1†) is a modular, “plug and play” continuous flow platform.24 Reaction mixtures are rotated at high speeds (0–9k rpm), producing a large surface area as the liquid forms a thin film. The unique conditions found provide many benefits in organic,25 biochemical26,27 and materials chemistry.28 Given the versatility of the VFD and its ease of use, we were motivated to explore the ways in which it could be integrated into oxidations that align with the tenets of green chemistry.1
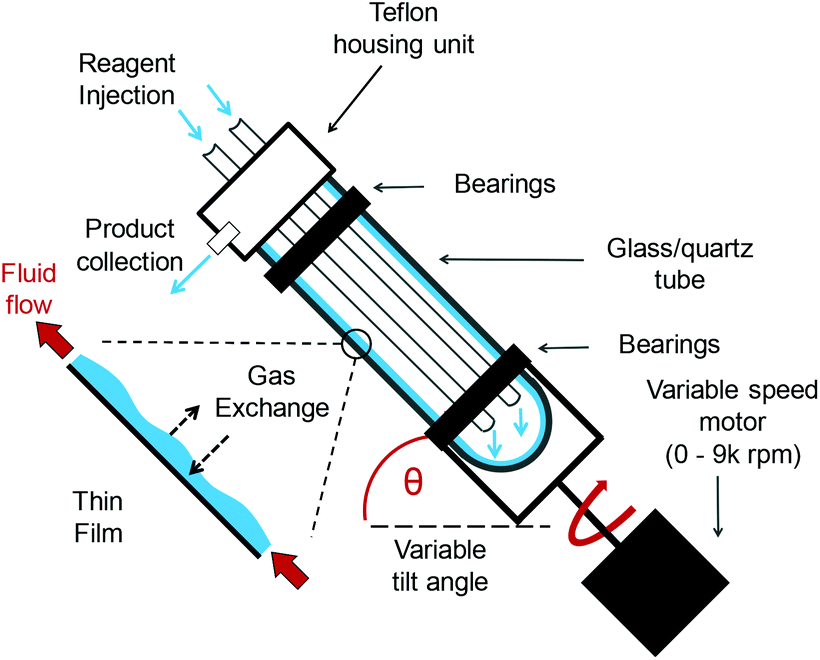 | ||
| Fig. 1 A schematic representation of the vortex fluidic device (VFD).24 | ||
Specifically, we designed our study around two key hypotheses. First, we posited that the thin film processing would improve gas exchange between the reaction medium and the atmosphere, perhaps improving the rate of aerobic oxidations and providing a simple and efficient method for deploying gaseous reagents in synthesis. Second, we suspected that the intense micro-mixing in the VFD that results from extremely rapid rotation of the inclined reaction tube, typically 45° for optimum processing,25–28 would enable efficient mixing of immiscible materials such as non-polar organic substrates and oxidants in aqueous solution. The problem of immiscibility limits the use of these aqueous oxidants, even in conventional flow reactors, as evident by the use of phase transfer catalysis to promote such reactions.19,29 Overcoming this requirement for a catalyst, through intense micro-mixing, could expand the scope of substrates for oxidation with aqueous hypochlorous acid or hydrogen peroxide. Pursuing these hypotheses, we examined three fundamental reactions in organic synthesis: aerobic oxidation of thiols, oxidation of sulfides with bleach, and epoxidation of alkenes with hydrogen peroxide, while eliminating the need for any added catalysts.
Aerobic oxidation in the VFD
Molecular oxygen (O2) is a ubiquitous and sustainable oxidant.11,12 Because the terminal product of aerobic oxidations is typically innocuous water, oxygen is commonly considered to be a desirable and benign reagent.30 Its green chemistry credentials notwithstanding, oxygen (as a gas) is often difficult to handle and can present risks for fire when used with organic substrates and flammable solvents.31,32 These risks are exacerbated at elevated temperatures and pressures—conditions that also introduce an energy penalty to the process. Therefore, it is useful to devise methods to carry out aerobic oxidations at atmospheric pressure, room temperature, and with non-flammable solvents. With this in mind, we examined the aerobic oxidation of N-acetyl-L-cysteine (1) in water. The conversion of thiols to disulfides is an important reaction in synthesis and biochemistry, with particular relevance to peptide and protein folding.33,34 Given that the VFD has been explored as a technology for folding proteins in-flow,26 the study of aerobic oxidations of cysteine to cystine also has broader relevance to bioorganic chemistry.As a starting point, a 1.0 mL solution of 1 in D2O was buffered to a pH of 9.8 and processed under a stream of oxygen both in a vial with simple stirring and also in the VFD in the confined mode of operation where a defined volume of liquid is placed in the tube at 45° tilt and rapidly rotated. The oxygen flow rate was set at a slow rate (no more than 0.5 L min−1) to maintain an oxygen atmosphere, and the pH was selected based on exploratory work that revealed an increase in the rate of oxidation with increasing pH. At pH 9.8, aerobic oxidation of 1 to 2 proceeded at a rate that was convenient for direct analysis of reaction conversion by 1H NMR. For this small-scale reaction, modest rate enhancement was observed using VFD processing with 40% conversion observed after 10 minutes in the VFD (7k rpm) and 25% conversion observed in the vial “batch reactor” (see page S6†). Apparently at this scale, there is sufficient O2 available to at least partially oxidise 1 using batch processing and mass transfer of the oxidant is not rate limiting. In contrast, when the batch processing was carried out at a 100 mL scale in a round bottom flask with stirring, very low levels of oxidation were observed (<5% conversion) even after several hours of reaction time. The resistance to oxidation was observed even when the headspace of the reaction flask was saturated with oxygen, and even when oxygen was bubbled directly into the reaction mixture (Schemes 1A and S11†). This experiment illustrates the common challenge of translating a batch reaction that occurs at small scales to one at larger scales where mass transfer (of gas in this case) is simply not as efficient. The VFD, as a microfluidic platform that can operate under continuous flow, overcomes this problem of scalability by design. As with other microfluidic reactors, a reaction is run locally on a small-scale in the VFD, with up-scaling a matter of simply running the process in continuous mode for a longer time. Gratifyingly, the oxidation of 1 to 2 could be carried out in the VFD operating in continuous mode, with a flow rate of 0.5 mL min−1 and a stream of oxygen supplied to the reaction at no more than 0.5 L min−1. The conversion was 55% (the average for product samples collected regularly from the reactor over a two-hour period). This conversion is slightly improved compared to that observed in confined mode, for the same rotational speed and tilt angle, indicating a reliable translation from small to large scale processing in the VFD (Scheme 1A).
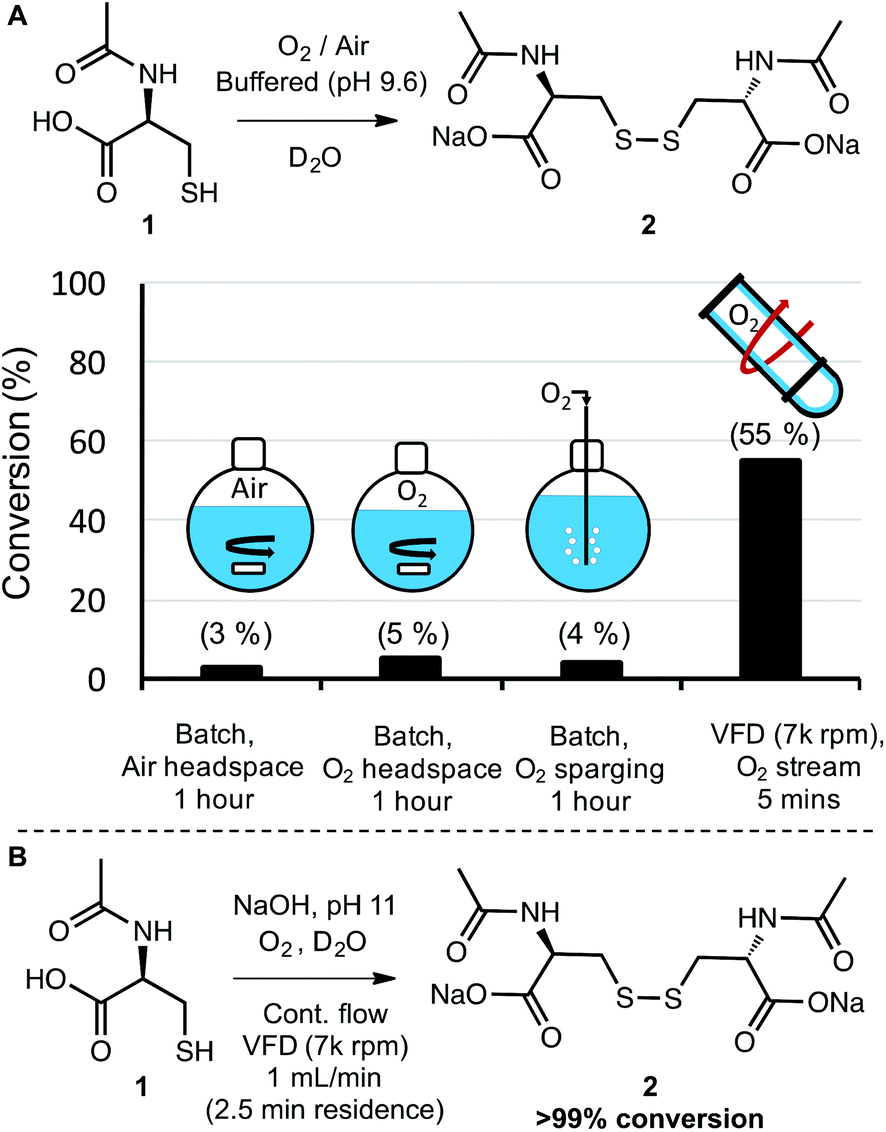 | ||
| Scheme 1 A. Large-volume (≥100 mL) aerobic oxidation of 1 to 2 is limited by inefficient mass transfer of oxygen to the reaction mixture, but this is more efficient using VFD processing (rotational speed 7k rpm, θ 45°). B. An optimised continuous flow oxidation of 1 to 2 provides full conversion of the disulfide over a short residence time. | ||
Finally, the continuous process was modified to illustrate that synthetically useful conversions could be obtained with this method. Simply raising the pH to 11 allowed the full conversion of 1 to 2, with 20 mL of reaction mixture processed in a mere 20 minutes (1 mL min−1).
Under these conditions, the residence time in the VFD (the time for a finite volume of liquid to enter the bottom of the tube and exit at the top) is 2.5 minutes, indicating a highly efficient aerobic oxidation (Scheme 1B). Because the oxidant (oxygen) produces water as the terminal by-product and the disulfide was formed in full conversion, there was no need for purification. In biochemical applications of this oxidation such as peptide modification and protein folding, the entire solution could be used directly. In this way, if the small mass of unreacted oxygen is not considered, no waste is generated and the E-factor is essentially 0. More generally, this series of experiments illustrated that gases such as oxygen can be delivered more efficiently to the thin film reaction mixtures in the VFD than they can to the bulk solutions in batch processing—with the high conversions in the VFD eliminating the need for purification and greatly reducing the E-factor in comparison to inefficient batch processing. Continuous processing in the VFD also revealed the ease at which reactions can be scaled relative to batch processing. More generally, the aerobic oxidation of thiols to disulfides is an important transformation in many areas such as organic synthesis35 and peptide and protein chemistry34,36 The results in Scheme 1 bode well for these applications and other aerobic oxidations in the VFD.
Oxidation of a hydrophobic substrate with bleach in the VFD
Aqueous hypochlorous acid and sodium hypochlorite (chlorine bleach) have long been used as non-flammable, non-explosive green oxidants.13 However, their direct use in chlorinating or otherwise oxidising non-polar organic substrates is often confounded by the immiscibility of the starting material in water. Accordingly, various surfactants, emulsifiers and phase transfer catalysts have been used to mediate these reactions.37 We were particularly intrigued by reports in which the non-polar substrate allyl phenyl sulfide was resistant to oxidation with bleach.38,39 This outcome was quite reasonably attributed to the low miscibility of the polar hypochlorite salts and the hydrophobic allyl phenyl sulfide, which prompted the development of various phase transfer catalysts to provoke this two phase reaction.38,39 As allyl sulfides and their corresponding sulfoxides and sulfones are highly useful in synthesis,40–45 we were curious as to whether or not the intense micro-mixing in the VFD would enable oxidation of organic sulfides in bleach without recourse to organic solvents, surfactants or phase transfer catalysts.First, allyl phenyl sulfide (3) was reacted directly with 10 and 20 equivalents of bleach oxidant in batch mode, where the mixture was simply stirred with a stirring bar for 40 minutes at room temperature. As seen on page S16,† allyl phenyl sulfide is clearly immiscible in the aqueous bleach.
Perhaps not surprisingly, limited oxidation was observed, with less than 10% total conversion to sulfoxide 4 and sulfone 5 (see pages S15–S18†). The major product in this control experiment was unreacted allyl phenyl sulfide 3 (Scheme 2A). In contrast, when these reaction mixtures were prepared in the same way and processed in the confined mode in the VFD operating at 7k rpm and using 20 equivalents of bleach, nearly all of the starting material was consumed within 40 minutes. The major products of the reaction were allyl phenyl sulfone 5 and its dichlorinated derivative 6. The formation of 6 was somewhat surprising, as this material has not been previously observed in the bleach oxidation of allyl phenyl sulfide.38,39,46 It is likely that the chlorination results from deprotonation of the acidic α protons of allyl phenyl sulfone (the pH of the bleach was 12), followed by direct reaction with an electrophilic chlorine species such as hypochlorous acid. This pathway is consistent with that observed in the α-chlorination of other aryl sulfones by reaction with hypochlorous acid,47 but we note that, to the best of our knowledge, this is the first reported route to 6 using chlorine bleach. Furthermore, other reports of α-chlorination of allyl aryl sulfones relied on phase-transfer catalysis and the toxic hexachloroethane as the electrophilic chlorine source.48 Clearly the VFD process in Scheme 2 is advantageous, as it requires only aqueous bleach. Furthermore, because the VFD processing dramatically improves conversion in the bleach oxidation of 3 in comparison to batch processing (Scheme 2Avs.2B), the E-factor also decreases accordingly. In Scheme 2A only 8% oxidation was observed in batch processing, corresponding to an E-factor of 96 whereas in VFD processing (Scheme 2B) 92% oxidation was observed, which corresponds to an E-factor of 6, with innocuous NaCl as the major waste product.
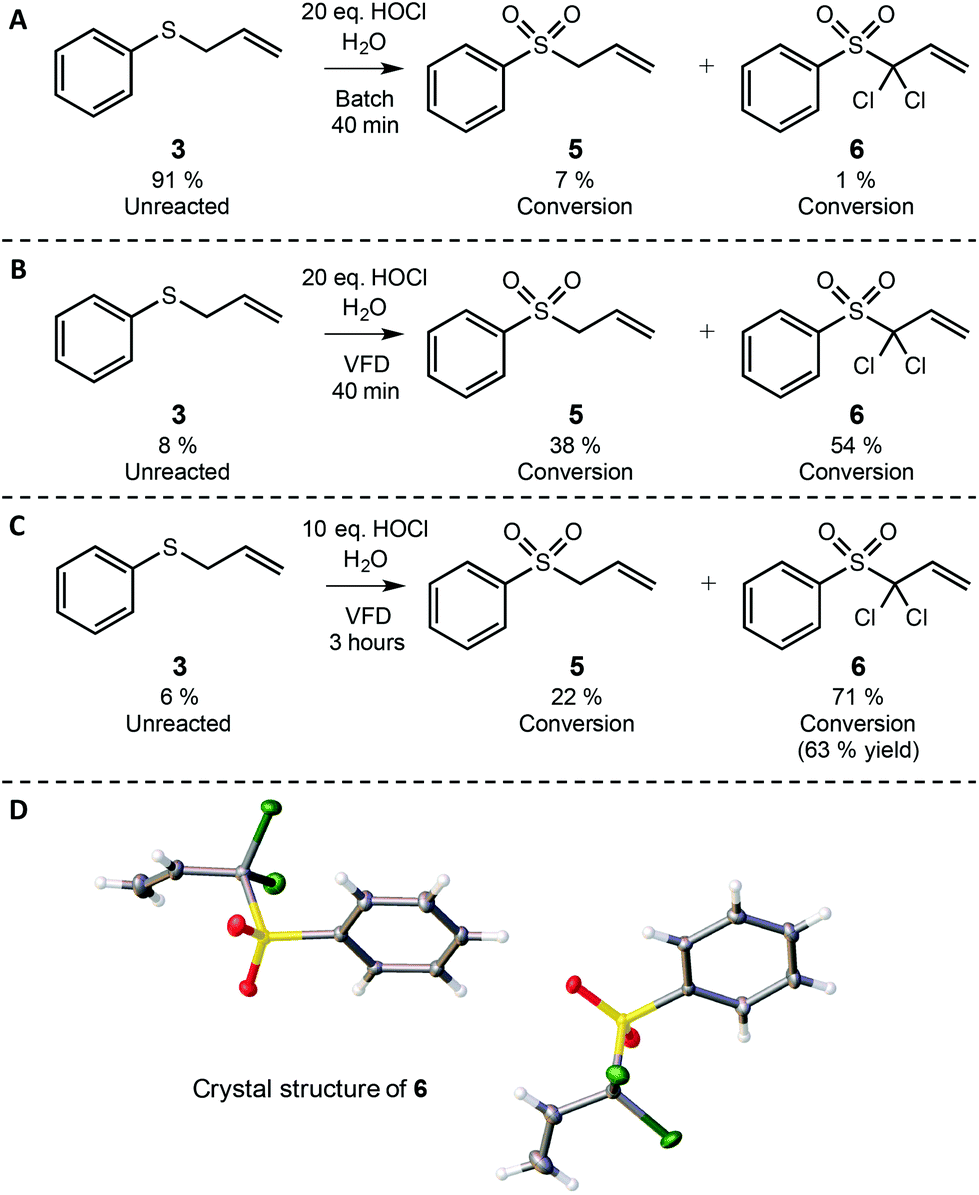 | ||
| Scheme 2 A. Allyl phenyl sulfide (3) is immiscible in bleach. Inefficient mixing in stirred batch reactions results in minimal oxidation. B. VFD processing promotes the bleach oxidation of 3, providing dichlorinated sulfone 6 as the major product. C. VFD processing provides sulfone 6 in useful isolated yields (VFD rotational speed 7k rpm, tilt angle (θ) 45°). D. Asymmetric unit found in the crystal structure of purified 6. | ||
To prepare 6 in synthetically useful yields, the reaction was simply run in the VFD for 3 hours in the confined mode. NMR analysis of the crude reaction mixture indicated approximately 71% conversion to 6, with purification by column chromatography providing this dichlorinated sulfone in a respectable 63% isolated yield. The column chromatography was carried out primarily for analytical purposes as compound 6 has not been previously reported. To confirm the structure unambiguously, single crystals of 6 that were suitable for diffraction studies were obtained from hexane and dichloromethane. The crystals were found to be in a monoclinic cell and structural analysis was carried out in the space group P21. Scheme 2D shows the asymmetric unit that contains two molecules of 6, unambiguously confirming the structure of this product. This reaction illustrates that the intense micro-mixing of the VFD enables efficient reaction of substrates that are largely immiscible, and indeed the promotion of reactions that have otherwise not been observed. This capability means that the use of organic solvents, phase transfer catalysts and emulsifiers commonly used in such transformations can be circumvented when using a VFD. Eliminating these materials simplifies the procedure and improves the green chemistry metrics—especially in the case of removing the requirement for organic co-solvents to facilitate mixing. Furthermore, immiscible liquids do not form emulsions in the VFD, thereby simplifying the processing.49 From a selectivity perspective, the unexpected chlorination is interesting in that it occurs only at the α position of the sulfone and not at the phenyl ring, so this can be considered a chemo- and regioselective oxidation. Importantly, this transformation is an exception to the rule that “the insolubility of hypochlorite salts in hydrocarbons and organic solvents has prevented the use of this material as a reagent for the selective oxidation of organic substrates.”38
Epoxidation of a hydrophobic alkene using aqueous hydrogen peroxide in the VFD
Aqueous hydrogen peroxide is a green oxidant that is relatively safe in comparison to organic peroxides and produces an innocuous water by-product after its reaction with reducing substrates.14 To explore this oxidant in the VFD, we examined the Weitz–Scheffer epoxidation of α,β-unsaturated ketones using hydrogen peroxide under basic conditions without the addition of a phase transfer catalyst (Scheme 3).50 Substrate 8 was selected because it is a hydrophobic liquid and insoluble in water at room temperature. Inspired by the efficient mixing of immiscible substrates observed in the VFD studies on bleach oxidations, we anticipated that the conversion of alkene 8 to epoxide 9 could be carried out without the need for organic solvents, surfactants, or phase-transfer catalysts. | ||
| Scheme 3 A. Without external temperature regulation, epoxidation of α,β-unsaturated ketone 8 proceeds in higher yield and diastereoselectivity in the VFD (7k rpm, 45° tilt angle). Both confined and continuous modes of operation provide high conversions. B. To determine the relative stereochemistry in 9, the epoxide was opened by the stereo- and regiospecific reaction with thiophenol. The product of this reaction was suitably crystalline for single crystal diffraction studies. C. Symmetry expanded crystal structure of 10, confirming the relative stereochemistry and showing a hydrogen-bonded dimer (H-bonds shown as dotted red lines). D. The VFD is efficient at dissipating the heat associated with the exothermic epoxidation. When the reaction was simply stirred (bottom left image), IR thermal imaging revealed temperature spikes to over 90 °C. The same reaction in the thin film of the VFD was maintained at 23 °C (bottom right image). | ||
Indeed, when 8 was reacted in the VFD in the confined mode with 5 equivalents of hydrogen peroxide in basic water, rapid consumption of starting material was observed. After a total of 1 hour of reaction time, epoxide 9 was isolated in 85% yield after chromatographic purification (Scheme 3A, entry 1). The epoxidation was also highly diastereoselective (dr = 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.0, as determined by GC and 1H NMR). To verify the relative stereochemistry in 9, it was converted to thioether 10 by the stereo- and regiospecific ring opening of the epoxide by thiophenol (Scheme 3B). Single crystals of 10 that were suitable for diffraction studies were obtained directly from this reaction mixture. The crystals were found to be in a monoclinic cell and structural analysis was carried out in the space group P21/n. The structure in Scheme 3C confirms the relative stereochemistry for the major diastereomer isolated upon opening of the epoxide with thiophenol and, by extension, the stereochemistry in epoxide 9.
:1.0, as determined by GC and 1H NMR). To verify the relative stereochemistry in 9, it was converted to thioether 10 by the stereo- and regiospecific ring opening of the epoxide by thiophenol (Scheme 3B). Single crystals of 10 that were suitable for diffraction studies were obtained directly from this reaction mixture. The crystals were found to be in a monoclinic cell and structural analysis was carried out in the space group P21/n. The structure in Scheme 3C confirms the relative stereochemistry for the major diastereomer isolated upon opening of the epoxide with thiophenol and, by extension, the stereochemistry in epoxide 9.
To obtain a direct comparison between batch and VFD processing, the epoxidation was carried out simultaneously over 30 minutes in the VFD at 7k rpm and in a batch process with simple magnetic stirring (Scheme 3A, entries 2 & 3). From this, slightly lower conversion was observed in batch (75% compared to 90% in VFD), with inferior dr (11:1.0 compared to 22:1 in VFD, as determined by GC). We attribute the eroded diastereoselectivty in batch processing to the uncontrolled exotherm that occurs when the reaction is run without external cooling. The conditions in the VFD result in high heat transfer and more uniform heat dissipation.24 This important feature of the microfluidic platform improves both safety and selectivity—important considerations in evaluating the green chemistry metrics and utility of a synthetic transformation. Examining the exotherm of the reaction further, it was revealed by infrared thermal imaging that the batch reaction reached temperatures over 90 °C (Scheme 3D), consistent with the original report of this reaction in which the liberation of heat was noted.50 In contrast, the thin film processing in the VFD dissipated thermal energy of the same reaction very efficiently, resulting in an internal temperature that did not exceed 23 °C over the course of the reaction, as indicated by IR thermal imaging (Scheme 3D). Therefore, while our initial interest in studying hydrogen peroxide oxidations in the VFD was centred on its ability to efficiently mix immiscible reagents, an additional benefit in exotherm control was revealed. Because peroxides present a detonation risk for runaway reactions,10 the VFD dramatically improves the safety profile for these reactions. To expand on this capability, the conversion of 8 to 9 was adapted to a continuous process in which the epoxidation was easily scaled with high reaction efficiency (84% conversion), while preventing runaway exotherms that would be associated with large scale batch processs (Scheme 3A, entry 4). Additionally, it should be noted that the E-factors for these epoxidations are very low (<0.4 in all cases, Scheme 3A). This is due to the fact that no extraction is required in the isolation of the epoxide with the water and product 9 spontaneously phase-separating (page S33†). Such phase separation of immiscible liquids after processing in the VFD is another advantage of the technology.24,49 The E-factor is also lower in the VFD as a consequence of improved diastereoselectivity which can potentially eliminate the requirement for tedious separation of stereoisomers. Additionally, the sodium hydroxide is catalytic and the aqueous solution can be recycled (page S33†). In cases where solvent was used for the isolation and purification of 9 (page S21†), this was purely for analytical purposes, as the stereochemical assignment of 9 has not been previously reported. The preparation of 10 as single crystals allowed the stereochemistry to be determined unambiguously using X-ray diffraction data.
Conclusions
Three distinct types of oxidation were studied in the vortex fluidic device, with thin film processing shown to be beneficial in aerobic oxidations, in which mass transfer of oxygen gas to the reaction medium is rate-limiting, as illustrated by the efficient oxidation of N-acetyl cysteine to its disulfide on a large volume. The intense micro-mixing and shorter diffusion paths in the VFD also benefits oxidations in which non-polar organic substrates are reacted with polar oxidants in water without phase transfer catalysis, as demonstrated in the oxidation and unexpected chlorination of allyl phenyl sulfide. Finally, the VFD is well-positioned to dissipate heat generated in the microfluidic thin film during exothermic reactions such as the epoxidation of α,β-unsaturated ketones using hydrogen peroxide, thereby making such reactions safer and more synthetically useful. Scaling up requirements using the VFD will depend on the product volume requirements, but for large volumes it is envisaged that a parallel array of VFDs will be effective, noting parallel processing features in conventional channel based microfluidics, albeit for much smaller volumes. Moreover, the just-in-time production possible using the VFD reduces storage requirements and improves safety of handling the products.We note that while the bleach oxidation of the sulfide to the sulfone is effective and provided a novel compound (6). The results, however, may heighten concern that using bleach as an oxidant may produce chlorinated organic impurities. Due to their potential toxicity, translating any findings using bleach into the market place should by accompanied by an analysis of the toxicity impact of such trace compounds, using assay systems such as TiPED.51 Accordingly, care needs to be taken in classifying bleach as a green oxidant, and should be done so only with consideration of potential chlorinated by-products.
Importantly, all of the oxidants used in the featured reactions benefit from high atom economy, low toxicity, and safer processing—especially when compared to other oxidants commonly employed in chemical synthesis. The methods proposed herein also minimise or eliminate the need for any organic solvents. Demonstrating ways in which these green transformations can be exploited in the VFD flow chemistry platform is an advance in improving the green chemistry aspects of the chemical processing.
Experimental procedures
Full experimental details and characterisations are provided in the ESI† for this document.Continuous flow production of disulfide 2
A buffered solution of D2O was prepared by the addition sodium carbonate (150 mg, 1.42 mmol) and sodium bicarbonate (420 mg, 5.00 mmol) to 50 mL of D2O. N-Acetyl-L-cysteine (82 mg, 0.50 mmol) was then added to the buffered solution and the initial pH was 9.9, as measured using a pH meter. This solution was then injected via syringe pump at the desired 0.5 mL min−1 into the base of the VFD tube, operating at 7k rpm with a 45° tilt angle. In another jet feed of the VFD, a stream of O2 gas was introduced so that flow rate was no more than 0.5 L min−1. The device was operated for a total of 2 h, over which time the collection vessel was changed every 30 min, giving 4 fractions of reaction product. Fractions were analyzed by 1H-NMR upon collection.VFD synthesis and isolation of compound 6
Allyl phenyl sulfide (3) (60 μL, 0.40 mmol), and a solution of 8–12% active chlorine bleach (2.45 mL, 2.6–3.9 mmol, 6.5–10 eq.) were transferred into a 20 mm VFD tube. This tube was then placed into a VFD and operated in confined mode at 7k rpm for 3 h. NMR analysis of crude product showed 71% conversion to compound 6. Flash column chromatography was performed with a mobile phase of 30% EtOAc in hexane. This resulted in isolation of compound 6 in yield of 63%. 1H NMR, 13C NMR, IR and MS spectra can be found in the ESI (S19–S20†).Continuous flow epoxidation of compound 8
Two syringe pumps were used to pump the liquid reagents through the continuous flow set up. A solution of NaOH (5 M, 3.6 mL, 18.3 mmol) and aqueous H2O2 (30% v/v, 7.14 mL, 91.4 mmol) was prepared and cooled to 0 °C over ice for 15 minutes. This solution was then removed from ice and transferred into 1 of the 2 syringes. This syringe was set to deliver contents at 0.051 mL min−1. The other syringe contained compound 8 (2 mL, 18.3 mmol) and was set to deliver contents at 0.009 mL min−1. This resulted in a total flow rate of 0.06 mL min−1, which relates to ≈30 minutes residence time (time for the liquid delivered to the base of the rotating tube to leave the top). Once exiting from the vortex, the reaction mixture was collected over ice. The total reaction time was 3.5 hours, over which time the collection vessel was changed every 30 min giving 7 fractions of reaction product. The organic material was extracted from these fractions with CDCl3 and analyzed by 1H-NMR upon collection. The average reaction conversion for all of these fractions was 84% and the average dr was 20:1. Representative 1H NMR spectra can be found in the ESI (S25†).
Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
This research was supported by Flinders University, the Government of South Australia and the Australian Research Council. Ashley Blythe, Joshua Cameron, and James Serotzki are acknowledged for completing preliminary experiments related to this project. The authors would also like to acknowledge Associate Professor Michael V. Perkins and Professor Gregory A. Weiss for helpful discussion.Notes and references
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- R. A. Sheldon, Green Chem., 2007, 9, 1273–1283 RSC.
- G.-J. ten Brink, I. W. C. E. Arends and R. A. Sheldon, Science, 2000, 287, 1636–1639 CrossRef CAS PubMed.
- Modern Oxidation Methods, ed. J.-E. Backvall, Wiley-VCH, Weinheim, Germany, 2nd edn, 2010 Search PubMed.
- S. K. Ritter, Chem. Eng. News, 2016, 94, 22–25 Search PubMed.
- G. Cainelli and G. Cardillo, Chromium Oxidants in Organic Chemistry, Springer-Verlag, Berlin Heidelberg, 1984 Search PubMed.
- H. Tohma and Y. Kita, Adv. Synth. Catal., 2004, 346, 111–124 CrossRef CAS.
- B. M. Trost, Science, 1991, 254, 1471–1477 CAS.
- B. M. Trost, Angew. Chem., Int. Ed. Engl., 1995, 34, 259–281 CrossRef CAS.
- Y.-S. Duh, X. H. Wu and C.-S. Kao, Process Saf. Prog., 2008, 27, 89–99 CrossRef CAS.
- N. Gunasekaran, Adv. Synth. Catal., 2015, 357, 1990–2010 CrossRef CAS.
- S. S. Stahl, Science, 2000, 309, 1824–1826 CrossRef PubMed.
- P. L. Anelli, C. Biffi, F. Montanari and S. Quici, J. Org. Chem., 1987, 52, 2559–2562 CrossRef.
- C. W. Jones, Applications of Hydrogen Peroxide and Derivatives, Royal Society of Chemistry, Cambridge, 1999 Search PubMed.
- B. Martin, J. Sedelmeier, A. Bouisseau, P. Fernandez-Rodriguez, J. Haber, F. Kleinbeck, S. Kamptmann, F. Susanne, P. Hoehn, M. Lanz, L. Pellegatti, F. Venturoni, J. Robertson, M. C. Willis and B. Schenkel, Green Chem., 2017, 19, 1439–1448 RSC.
- D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer Jr., R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411–420 RSC.
- J. Britton and C. L. Raston, Chem. Soc. Rev., 2017, 46, 1250–1271 RSC.
- M. Movsisyan, E. I. P. Delbeke, J. K. E. T. Berton, C. Battilocchio, S. V. Ley and C. V. Stevens, Chem. Soc. Rev., 2016, 45, 4892–4928 RSC.
- H. P. L. Gemoets, Y. Su, M. Shang, V. Hessel, R. Luque and T. Noel, Chem. Soc. Rev., 2016, 45, 83–117 RSC.
- A. Gavriilidis, A. Constantinou, K. Hellgardt, K. K. Hii, G. J. Hutchings, G. L. Brett, S. Kuhn and S. P. Marsden, React. Chem. Eng., 2016, 1, 595–612 CAS.
- R. L. Hartman, J. R. Naber, N. Zaborenko, S. L. Buchwald and K. F. Jensen, Org. Process Res. Dev., 2010, 14, 1347–1357 CrossRef CAS.
- M. Schoenitz, L. Grundemann, W. Augustin and S. Scholl, Chem. Commun., 2015, 51, 8213–8228 RSC.
- R. K. Shah, H. C. Shum, A. C. Rowat, D. Lee, J. J. Agresti, A. S. Utada, L.-Y. Chu, J.-W. Kim, A. Fernandez-Nieves, C. J. Martinez and D. A. Weitz, Mater. Today, 2008, 11, 18–27 CrossRef CAS.
- J. Britton, K. A. Stubbs, G. A. Weiss and C. L. Raston, Chem. – Eur. J., 2017, 23, 13270–13278 CrossRef CAS PubMed.
- J. Britton, J. M. Chalker and C. L. Raston, Chem. – Eur. J., 2015, 21, 10660–10665 CrossRef CAS PubMed.
- T. Z. Yuan, C. F. G. Ormonde, S. T. Kudlacek, S. Kunche, J. N. Smith, W. A. Brown, K. M. Pugliese, T. J. Olsen, M. Iftikhar, C. L. Raston and G. A. Weiss, ChemBioChem, 2015, 16, 393–396 CrossRef CAS PubMed.
- J. Britton, L. M. Meneghini, C. L. Raston and G. A. Weiss, Angew. Chem., Int. Ed., 2016, 55, 11387–11391 CrossRef CAS PubMed.
- K. Vimalanathan, J. R. Gascooke, I. Suarez-Martinez, N. A. Marks, H. Kumari, C. J. Garvey, J. L. Atwood, W. D. Lawrance and C. L. Raston, Sci. Rep., 2016, 6, 22865 CrossRef CAS PubMed.
- J. Jovanović, E. V. Rebrov, T. A. Nijhuis, V. Hessel and J. C. Schouten, Ind. Eng. Chem. Res., 2010, 49, 2681–2687 CrossRef.
- P. M. Osterberg, J. K. Niemeier, C. J. Welch, J. M. Hawkins, J. R. Martinelli, T. E. Johnson, T. W. Root and S. S. Stahl, Org. Process Res. Dev., 2015, 19, 1537–1543 CrossRef CAS PubMed.
- L. Vanoye, J. Wang, M. Pablos, R. Philippe, C. d. Bellefon and A. Favre-Réguillon, Org. Process Res. Dev., 2016, 20, 90–94 CrossRef CAS.
- C. A. Hone, D. M. Roberge and C. O. Kappe, ChemSusChem, 2017, 10, 32–41 CrossRef CAS PubMed.
- D. Givol, F. De Lorenzo, R. F. Goldberger and C. B. Anfinsen, Proc. Natl. Acad. Sci. U. S. A., 1965, 53, 676–684 CrossRef CAS.
- W. J. Wedemeyer, E. Welker, M. Narayan and H. A. Scheraga, Biochemistry, 2000, 39, 4207–4216 CrossRef CAS PubMed.
- I. V. Koval, Russ. Chem. Rev., 1994, 63, 735–750 CrossRef.
- M. Akcan and D. J. Craik, in Peptide Synthesis and Applications, ed. K. J. Jensen, P. Tofteng Shelton and S. L. Pedersen, 2013, pp. 89–101 Search PubMed.
- D. C. M. Albanese, F. Foschi and M. Penso, Org. Process Res. Dev., 2016, 20, 129–139 CrossRef CAS.
- J. H. Ramsden, R. S. Drago and R. Riley, J. Am. Chem. Soc., 1989, 111, 3958–3961 CrossRef CAS.
- N. Fukuda and T. Ikemoto, J. Org. Chem., 2010, 75, 4629–4631 CrossRef CAS PubMed.
- D. A. Evans and G. C. Andrews, Acc. Chem. Res., 1974, 7, 147–155 CrossRef CAS.
- B. M. Trost, N. R. Schmuff and M. J. Miller, J. Am. Chem. Soc., 1980, 102, 5979–5981 CrossRef CAS.
- N. S. Simpkins, Sulphones in Organic Synthesis, Pergamon, New York, 1993 Search PubMed.
- J. Clayden and M. Julia, J. Chem. Soc., Chem. Commun., 1994, 16, 1905–1906 RSC.
- K. Deng, J. M. Chalker, A. Yang and T. Cohen, Org. Lett., 2005, 7, 3637–3640 CrossRef CAS PubMed.
- J. M. Chalker, Aust. J. Chem., 2015, 68, 1801–1809 CrossRef CAS.
- T. Okada, H. Matsumuro, S. Kitagawa, T. Iwai, K. Yamazaki, Y. Kinoshita, Y. Kimura and M. Kirihara, Synlett, 2015, 26, 2547–2552 CrossRef CAS.
- C.-N. Hsiao and H. Shechter, J. Org. Chem., 1988, 53, 2688–2699 CrossRef CAS.
- A. Jonczyk and T. Radwan-Pytlewski, Pol. J. Chem., 1995, 69, 1422–1427 CAS.
- X. Luo, P. Smith, C. L. Raston and W. Zhang, ACS Sustainable Chem. Eng., 2016, 4, 3905–3911 CrossRef CAS.
- E. Weitz and A. Scheffer, Chem. Ber., 1921, 54, 2327–2344 CrossRef.
- T. T. Schug, R. Abagyan, B. Blumberg, T. J. Collins, D. Crews, P. L. DeFur, S. M. Dickerson, T. M. Edwards, A. C. Gore, L. J. Guillette, T. Hayes, J. J. Heindel, A. Moores, H. B. Patisaul, T. L. Tal, K. A. Thayer, L. N. Vandenberg, J. C. Warner, C. S. Watson, F. S. vom Saal, R. T. Zoeller, K. P. O'Brien and J. P. Myers, Green Chem., 2013, 15, 181–198 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details and analytical characterisation. CCDC 1552986 and 1552987. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7gc03352d |
| This journal is © The Royal Society of Chemistry 2018 |