
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImpact of mechanical bonding on the redox-switching of tetrathiafulvalene in crown ether–ammonium [2]rotaxanes†
Hendrik V.
Schröder
a,
Sebastian
Sobottka
b,
Maite
Nößler
a,
Henrik
Hupatz
a,
Marius
Gaedke
a,
Biprajit
Sarkar
b and
Christoph A.
Schalley
 *a
*a
aInstitut für Chemie und Biochemie, Freie Universität Berlin, Takustr. 3, 14195 Berlin, Germany. E-mail: christoph@schalley-lab.de
bInstitut für Chemie und Biochemie, Freie Universität Berlin, Fabeckstr. 34/36, 14195 Berlin, Germany
First published on 10th July 2017
Abstract
Switchable crown ether–ammonium [2]rotaxanes with a redox-active tetrathiafulvalene (TTF) unit implemented in their wheels were synthesised and fully characterised. Reversible operation in two modes is possible, in which the [2]rotaxane's axle is either charged or neutral. Cyclic voltammetry experiments reveal the effects of mechanical bonding on the electrochemical properties of TTF and show the [2]rotaxanes to perform a distinct function in both modes. In the charged mode, redox-switching is dominated by strong electrostatic repulsion in the [2]rotaxane which subsequently leads to a macrocycle translation along the axle. In the non-charged mode, a selective energetic stabilisation of TTF radical cations is observed, which can be attributed to an interplay of weak electrostatic interactions between wheel and axle.
Introduction
Mechanically interlocked molecules (MIMs) such as rotaxanes and catenanes have attracted increasing attention during the last decades not only because of their fascinating structure, but also because of unique properties caused by the mechanical bond.1 Even very weak interactions, which would be negligible for non-interlocked complexes, can become significant, when the mechanical bond prevents dissociation.2 As an early example, Sauvage and co-workers reported the “catenand effect” which describes the kinetic stabilisation of interlocked copper(I) complexes in comparison to their non-interlocked analogues.3 Similar effects were observed for the neutralisation of crown ether–ammonium rotaxanes:4 while a pseudorotaxane of this type is easily deprotonated, this is very challenging for the interlocked rotaxane without the aid of a second cationic station.5 Until today, many researchers have tried to fathom these not yet fully understood phenomena and use them to create different emergent properties in MIMs.A major aim in the field of MIMs is the design of switchable artificial molecular machines which can undergo dynamic conformational changes by applying external stimuli.6 Archetypical examples for molecular machines are switchable rotaxane shuttles in which the macrocycle can undergo a translational movement relative to its axle counterpart.7 Recently, molecular machinery based on shuttles was extended through more sophisticated architectures such as molecular elevators,8 muscles,9 pumps,10 or even molecular motors.11 However, the reported systems are often bistable and therefore limited for multi-purpose applications. The design and preparation of multistable molecular machines is therefore an appealing synthetic target for supramolecular chemists.12 Furthermore, possible applications of MIMs such as information processing require fast and clean switching as well as easily readable output signals. This renders electrochemical and optical inputs and output most promising.13
Here, we report a novel tri-stable rotaxane based on the widely used 24-crown-8/secondary ammonium motif.14 It incorporates a redox-switchable tetrathiafulvalene (TTF) unit in its wheel. TTF is a versatile organosulfur compound which can undergo two clean and reversible successive one-electron oxidations to generate the stable radical cation TTF+˙ and the dication TTF2+.15 TTF-crown ether macrocycles have been described as redox-responsive and switchable supramolecular receptors for different metal cations.16 Furthermore, Stoddart and co-workers reported pseudo[2]rotaxanes assembled from TTF-substituted crown ethers and dibenzylammonium hexafluorophosphate and concluded that the ammonium thread is not affecting the electronic properties of the TTF moiety.17 In contrast, we recently showed the redox properties of a TTF-substituted crown ether in a divalent charge-transfer rotaxane to be strongly affected by the axle, when using media of low dielectric constant and weakly coordinating anions (WCAs) such as tetrakis(3,5-bis(trifluoromethyl)phenyl)borate (BArF24−).18 Consequently, this observation opens the pathway for the construction of a novel class of redox-active MIMs which exhibit a strong effect on the electronic properties of TTF by the mechanical bond.
The rotaxanes reported here can be operated in two different modes in which the threaded axle is either charged or non-charged. In charged [2]rotaxane 1 (Fig. 1), redox-switching of the rotaxane can create strong charge repulsion effects between axle and oxidised macrocycle that subsequently lead to a translational motion of the macrocycle. The expulsive force and, thus, the conformational change can be adjusted by the stepwise oxidation of the TTF unit. In non-charged [2]rotaxane 2, a selective stabilisation of the tetrathiafulvalene radical cation state (TTF+˙) is observed, whereas the dicationic state (TTF2+) is energetically disfavoured in comparison to the free wheel.
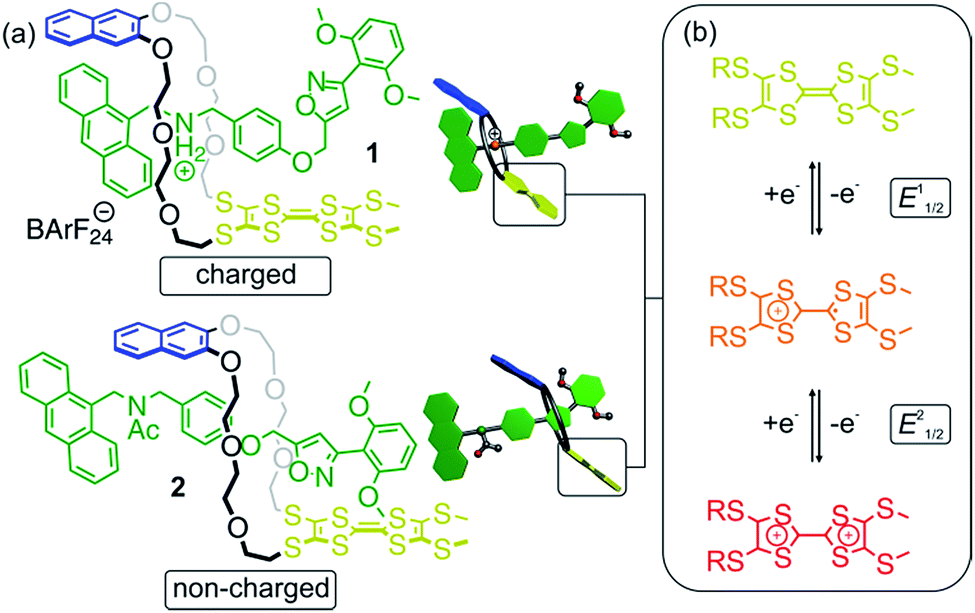 | ||
| Fig. 1 (a) Structures of [2]rotaxanes 1 and 2 and cartoons representing them. (b) Schematic representation of the stepwise and reversible oxidations of the thioalkyl-substituted tetrathiafulvalene moiety. | ||
Results and discussion
Pseudo[2]rotaxane formation and redox-induced axle expulsion
An equimolar mixture of ammonium axle 3 and TTF crown ether TTFC8 (Fig. 2, see Section 1.2 and 1.3 in the ESI† for synthetic procedures and characterisation data) in CH2Cl2 led to the quantitative formation of the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 pseudo[2]rotaxane 3@TTFC8 as demonstrated by 1H NMR. Typical downfield shifts for methylene protons Hf and Hg and a diastereotopic splitting of the macrocycle methylene protons confirm the interpenetrated structure as previously reported for similar intertwined structures (Fig. S4, ESI†).18,19 Proton Ha experiences a significant upfield shift (Δδ = −1.43 ppm) caused by π stacking of the anthracene stopper and the wheel's naphthalene unit (Fig. 2d).
:1 pseudo[2]rotaxane 3@TTFC8 as demonstrated by 1H NMR. Typical downfield shifts for methylene protons Hf and Hg and a diastereotopic splitting of the macrocycle methylene protons confirm the interpenetrated structure as previously reported for similar intertwined structures (Fig. S4, ESI†).18,19 Proton Ha experiences a significant upfield shift (Δδ = −1.43 ppm) caused by π stacking of the anthracene stopper and the wheel's naphthalene unit (Fig. 2d).
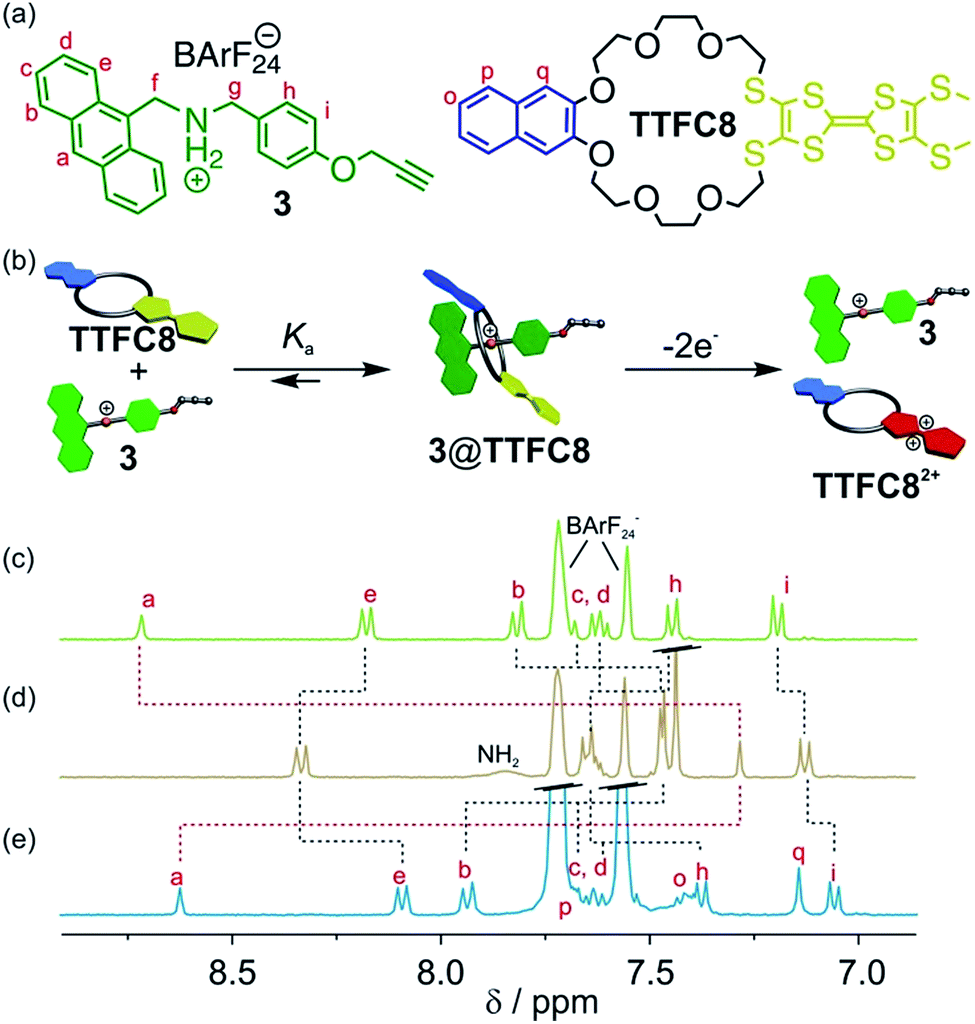 | ||
| Fig. 2 (a) Structures of macrocycle TTFC8 and ammonium axle 3. (b) Schematic representation of the association between ammonium axle 3 and macrocycle TTFC8 and the axle expulsion induced by double oxidation of the macrocycle. Stacked partial 1H NMR spectra (400 MHz, 2 mM, CD2Cl2, 298 K) of (c) ammonium axle 3, (d) pseudo[2]rotaxane 3@TTFC8 and (e) after oxidation by two equiv. of Fe(ClO4)3 and addition of 5 equiv. tetrabutylammonium BArF24 as stabilising electrolyte. | ||
The threading of TTFC8 onto axle 3 can also be monitored by fluorescence spectroscopy using the anthracene as a probe:20 if the macrocycle is in close proximity to the anthracene, its fluorescence (excitation wavelength λex = 370 nm) is strongly quenched by a distance-dependent electron transfer consistent with the strong emission decrease upon pseudo[2]rotaxane formation (Fig. S11, ESI†).
Isothermal titration calorimetry (ITC) experiments in 1,2-dichloroethane confirmed the 1:1 complex stoichiometry and provide quantitative data on the axle–wheel interaction in pseudo[2]rotaxane 3@TTFC8 (Fig. S12, ESI†). The thermodynamic parameters (ΔG = (−32.9 ± 0.3), ΔH = (−56.3 ± 1.0), and TΔS = (−23.4 ± 1.3) kJ mol−1) reveal a strong, enthalpy-driven complexation of axle and wheel. The binding constant Ka = (5.9 ± 0.6) 105 M−1 at 298 K is remarkably high in comparison to similar systems.14a We attribute this to a combination of π-stacking and strong hydrogen bonding between the ammonium axle and the wheel which is enhanced by the poor coordination of the weakly coordinating BArF24− counterion.21
As previously reported, the oxidation of TTF crown ether macrocycles in host–guest complexes can be used to trigger the expulsion of metal cations driven by charge repulsion.16a,16d,22 Chemical oxidation of 3@TTFC8 in CH2Cl2 with two equivalents of the one-electron oxidant Fe(ClO4)3 leads to a virtually complete disassembly of the system as nicely shown by a back-shifting of the signal for proton Ha (Fig. 2e). To the best of our knowledge, this is the first example of TTF-crown pseudorotaxane with a controlled assembly/disassembly by redox-switching. Unfortunately, the non-interlocked character of the pseudo[2]rotaxane thus prevents a detailed investigation of the interactions between axle and macrocycle in all three switching states (TTF, TTF+˙ and TTF2+). To gain more insight, we therefore investigated the redox-properties in a mechanically interlocked [2]rotaxane.
Synthesis and characterisation of switchable [2]rotaxanes
The synthesis of [2]rotaxanes 1 and 2 is shown in Scheme 1. Following the catalyst-free stoppering strategy of Takata et al.,23 a mixture of secondary ammonium axle 3 and TTFC8 in CH2Cl2 was treated with nitrile oxide 4 to obtain the [2]rotaxane 1 (79% yield). Its 1H NMR spectrum (Fig. 3b; for details of signal assignments, see Fig. S5–S7, ESI†) reveals similar characteristic chemical shifts: for methylene protons Hf and Hg and aromatic protons Ha and Hq as observed for the pseudorotaxane. In addition, a strong downfield shift of proton Hk (Δδ = +3.90 ppm) is observed due to isoxazole formation. Diastereotopic splitting of the macrocycle methylene proton signals confirms the threading of the unsymmetric axle. Comparison of [2]rotaxane 1 with a mixture of TTFC8 and free stoppered axle 5 (see ESI, Section 1.2 for details†) clearly shows the 1H NMR spectra not to be superimposable (Fig. S8, ESI†). The interlocked structure of rotaxane 1 was also confirmed by a HR-ESI mass spectrum and tandem MS experiments (Fig. S9 and S10, ESI†). The most prominent peak at m/z 1279, which corresponds to [2]rotaxane 1 after stripping off its counterion, was not observed for a 1:1 mixture of free stoppered axle 5 and macrocycle TTFC8. Therefore, we can safely conclude the interlocked structure of the rotaxanes to be intact.
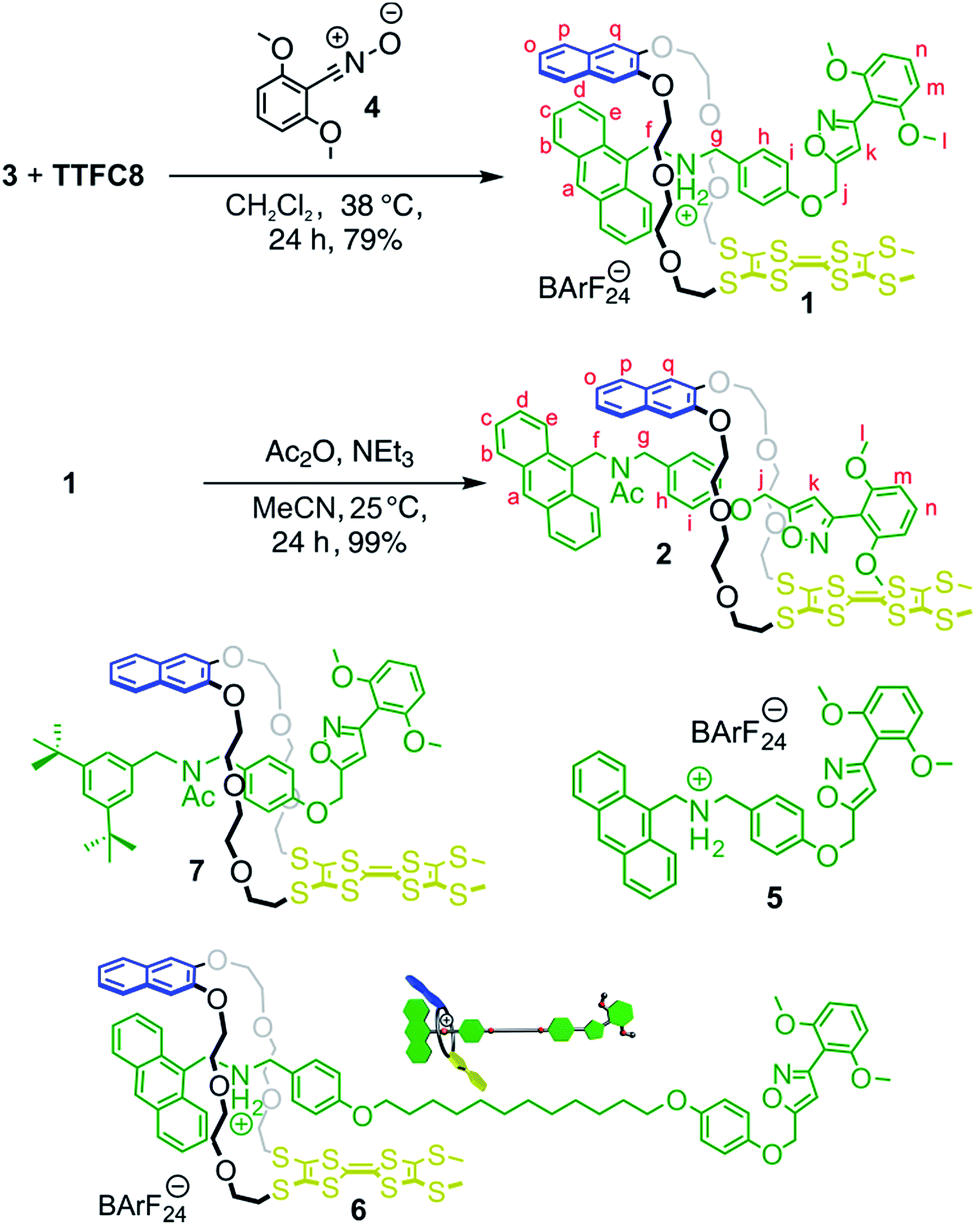 | ||
| Scheme 1 Preparation of charged and non-charged [2]rotaxanes 1 and 2; structures of free stoppered axle 5, [2]rotaxanes 6 and 7. | ||
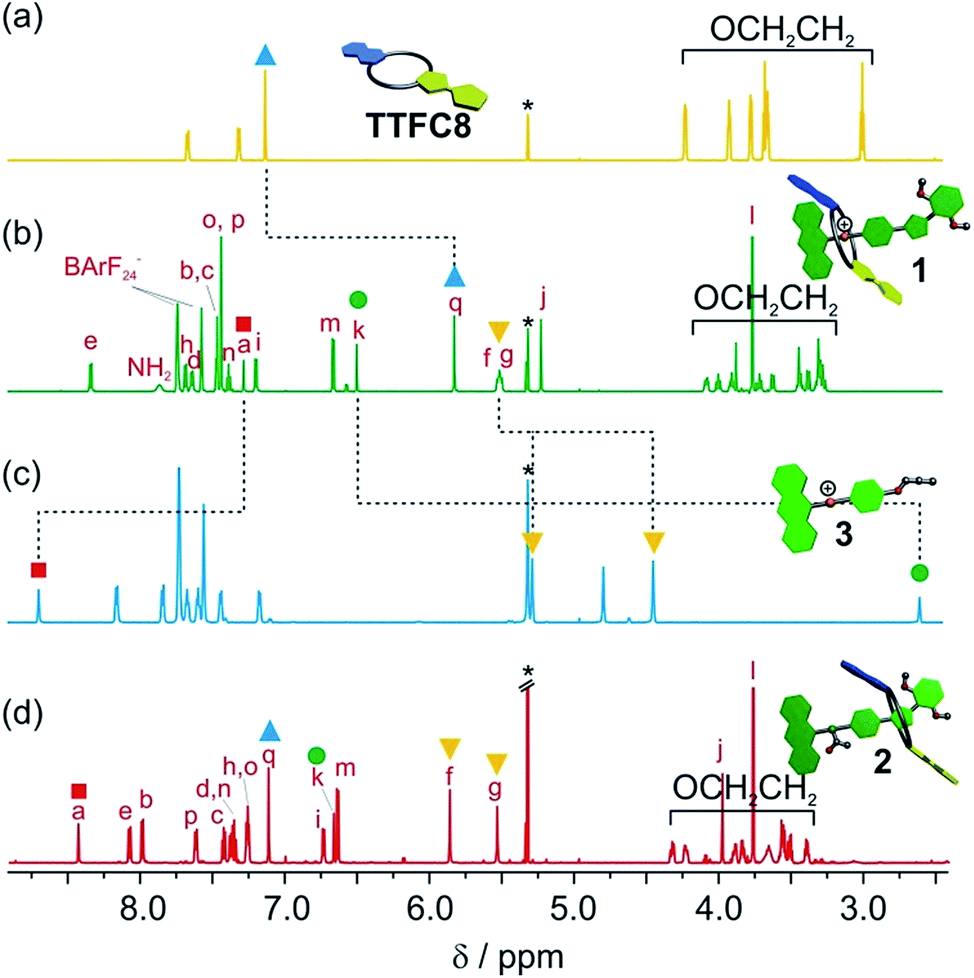 | ||
| Fig. 3 Stacked partial 1H NMR spectra (700 MHz, 2 mM, CD2Cl2, 298 K) of (a) macrocycle TTFC8, (b) charged [2]rotaxane 1, (c) axle 3, (d) neutral, acetylated [2]rotaxane 2. Selected signals are marked with coloured symbols. The solvent residue signal is marked with an asterisk. | ||
N-Acetylation24 of the ammonium station was achieved through the addition of acetic anhydride and triethylamine to obtain the non-charged rotaxane 2 in almost quantitative yield. As indicated by back-shifting of aromatic protons Ha and Hq, the 1H NMR of [2]rotaxane 2 suggests that the wheel has moved away from the former ammonium station (Fig. 3d). The highfield shifts of phenyl ether protons Hh and Hi (Δδ = −0.42 and −0.46 ppm, respectively) and axle protons Hj (Δδ = −0.83 ppm) point to a shielding effect by the macrocycle. The downfield shift of the isoxazole signal Hk (Δδ = +0.16 ppm) is probably caused by the anisotropic effect of the oxygen atoms in the crown ether.4a,24 Therefore, the chemical shifts are in good agreement with the wheel being located close to the isoxazole moiety. No cis–trans isomers were observed for the amide incorporated in the axle of 2 at 298 K.
Electrochemical properties of rotaxanes 1 and 2
Photometric titrations of 1 and 2 (Fig. 4a) with Fe(ClO4)3 reveal optoelectronic properties consistent with a switching between three redox states (TTF, TTF+˙, TTF2+).25 For each state, characteristic bands and clear-cut isosbestic points indicate clean and well-separated successive transitions. Furthermore, the characteristic absorptions are a straightforward optical output signal and make the switching states of the [2]rotaxanes easily identifiable by UV/Vis or even by the naked eye (Fig. 4b).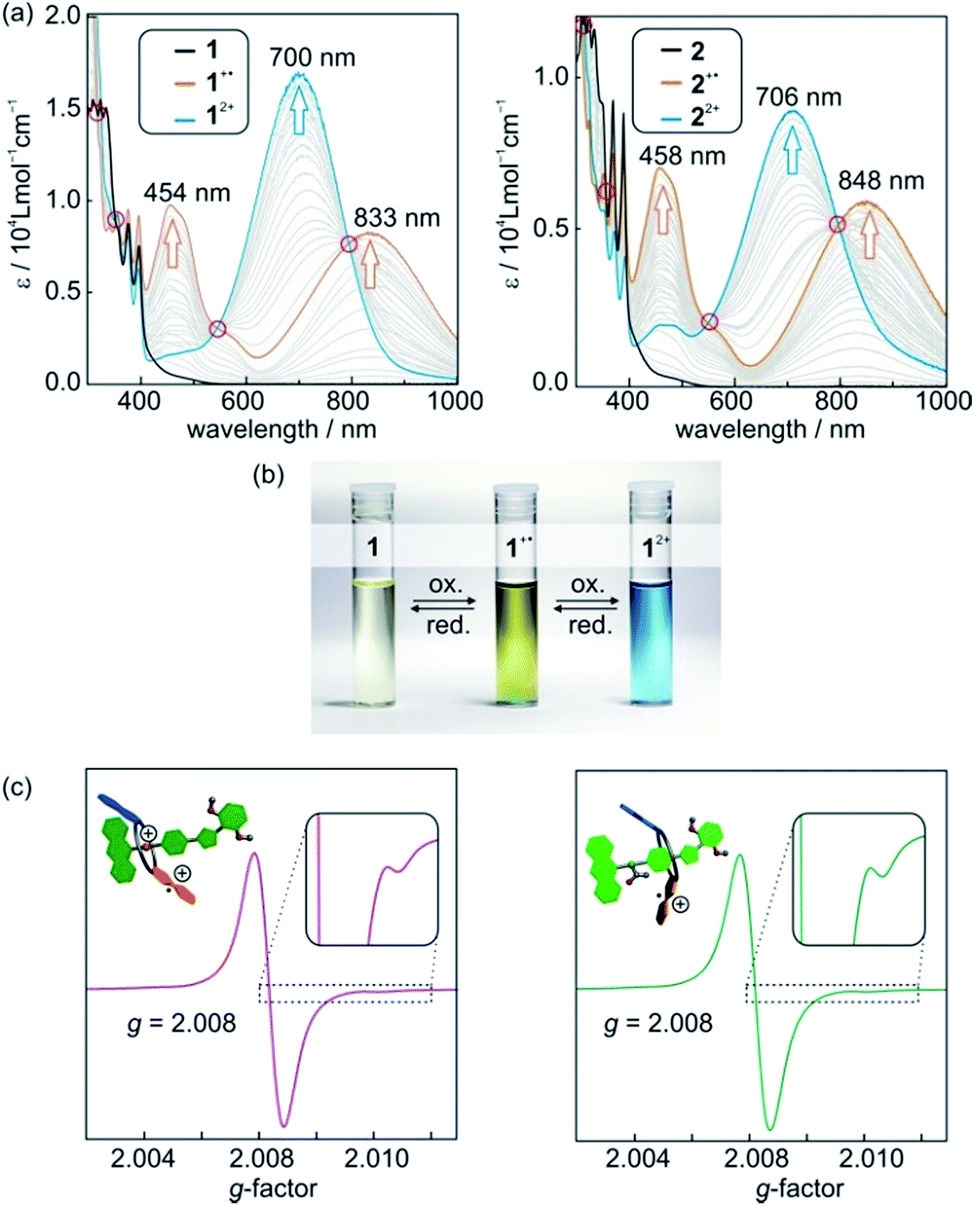 | ||
| Fig. 4 (a) Photometric titrations of [2]rotaxanes 1 (left) and 2 (right) with Fe(ClO4)3 as the oxidant (50 μM rotaxane solutions in CH2Cl2). The isosbestic points indicate two clean one-electron oxidations and are marked with red circles. (b) Photographs of 1 mM CH2Cl2 solutions of [2]rotaxane 1 before oxidation and after the addition of one or two equivalents of oxidant Fe(ClO4)3. (c) X-band EPR spectra of CH2Cl2 solutions (0.1 mM) of [2]rotaxanes 1+˙ (left) and 2+˙ (right) after addition of one equivalent of Fe(ClO4)3. | ||
The singly oxidised radical cations 1+˙ and 2+˙ show a symmetric signal in their EPR spectra (Fig. 4c) with small 33S couplings (0.35 mT) as expected for tetrathioalkyl-substituted TTF+˙ radical cations.25c This speaks in favour of a clearly TTF-centred oxidation for the first redox step. The g-factors for [2]rotaxane 1+˙ and 2+˙ of 2.008 are close to values reported for similar TTF compounds.25c,26
Cyclovoltammetric (CV) experiments with [2]rotaxanes 1 and 2 and, for comparison, with free macrocycle TTFC8 in CH2Cl2 reveal the influence of mechanical bonding on their electrochemical properties (Fig. 5 and S13 ESI†). In accordance with the UV/vis and EPR results, both [2]rotaxanes show two fully reversible redox processes which are clearly centred on the TTF moiety. Thus, the mechanically interlocked structures remain intact during the redox switching. The potentials for [2]rotaxane 1 (0.74 V and 1.42 V) and [2]rotaxane 2 (0.56 V and 1.14 V) are distinctly shifted as compared to those of free TTFC8 (0.61 V and 1.06 V), while such shifts are not observed for an equimolar mixture of free stoppered axle 5 and free TTFC8 (Table S1, ESI†). Therefore, these effects are clearly a consequence of the mechanically interlocked structure. The strong potential difference for the first (ΔE11/2 = +0.13 V) and second oxidation (ΔE21/2 = +0.36 V) of [2]rotaxane 1 compared to free TTFC8 demonstrates that the two TTF oxidations are both energetically disfavoured, likely because of charge repulsion between the TTF mono- and dications and the ammonium station. ΔE11/2 is remarkably high for TTF-crown ether macrocycles.22c,22d For [2]rotaxane 2, a more complex redox behaviour is observed in that E11/2 is decreased and E21/2 increased as compared to free TTFC8 (see below for the rationalisation of this observation).
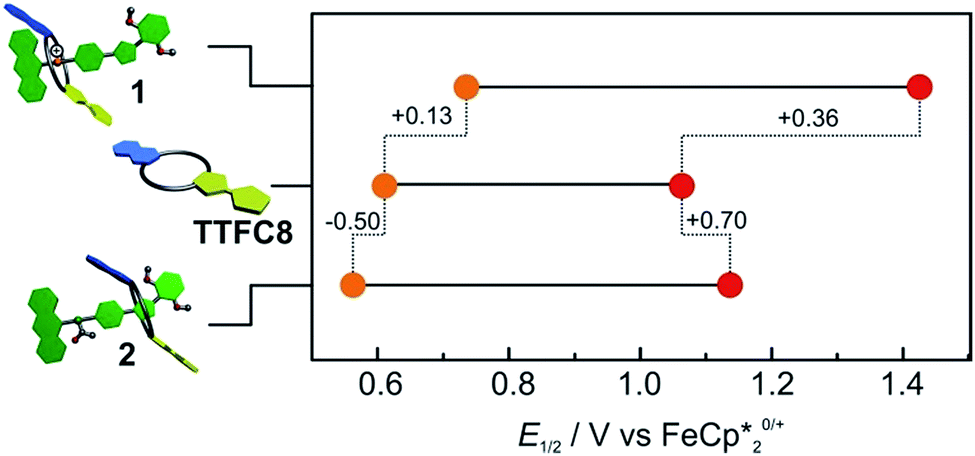 | ||
| Fig. 5 Correlation diagram of half-wave potentials for charged [2]rotaxane 1, macrocycle TTFC8 and neutral, acetylated [2]rotaxane 2. Half-wave potentials were determined by cyclic voltammetry measurements in CH2Cl2 against Fe(Cp*)20/+ with a 1 mM analyte concentration at a scan rate of 100 mV s−1 with tetrabutylammonium BArF24 (0.1 M) as the electrolyte. | ||
Oxidation-induced shuttling in charged [2]rotaxanes 1 and 6
Potentially, the repulsion in charged [2]rotaxane 1 between the ammonium station and the oxidised macrocycle TTFC8+˙ or TTFC82+ can be used to trigger a shuttling of the macrocycle towards the isoxazole moiety. In most of the reported non-interlocked host–guest systems, a guest expulsion can be detected by a largely unaffected second redox potential.16a,16d,22c For [2]rotaxane 1, however, the second oxidation is strongly affected which indicates that the repulsion between the ammonium station and TTFC8+˙ is not strong enough to trigger a translational movement. This is reasonable, as the potential difference (≙ repulsive force) for the first oxidation ΔE11/2 = +0.13 V (13 kJ mol−1) is obviously smaller than the crown/ammonium binding energy of −32.9 kJ mol−1 determined by ITC. Therefore, we focused on the doubly oxidised species which displayed a much higher potential difference of ΔE21/2 = +0.36 V (35 kJ mol−1) sufficient to overcome the binding energy.To obtain evidence for the shuttling in [2]rotaxane 1, fluorescence spectra of 1 and 12+ were recorded expecting that the anthracene fluorescence would become visible, when the wheel moves away from the ammonium station. However, fully oxidised [2]rotaxane 12+ showed no increase in fluorescence emission (Fig. 6a, left). This leads to the assumption that either no conformational change occurred or that the macrocycle is still close enough for an efficient quenching.
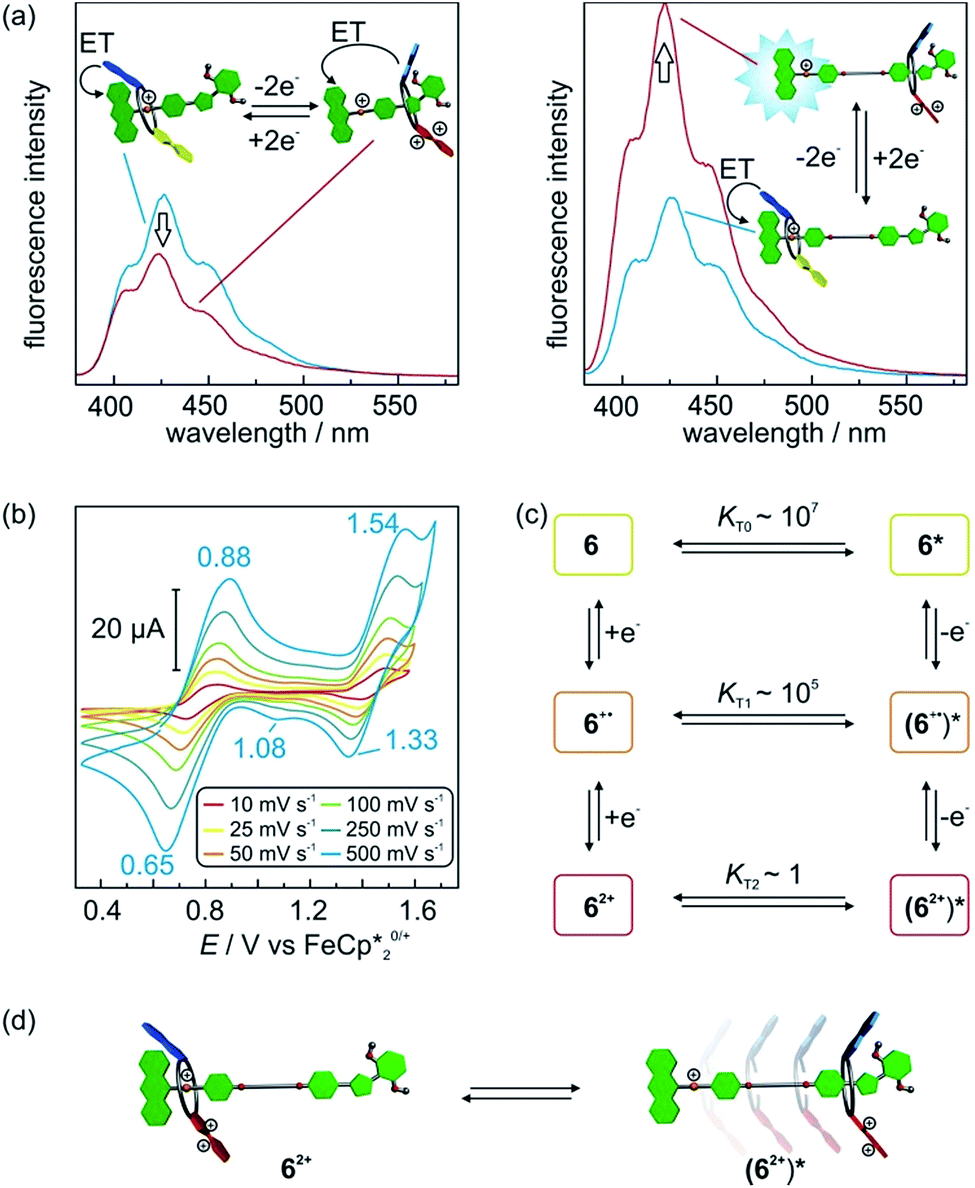 | ||
| Fig. 6 (a) Fluorescence spectra (CH2Cl2, 50 μM, λex = 370 nm) of [2]rotaxane 1 (left) and 6 (right) before and after double oxidation by addition of the oxidant Fe(ClO4)3. (b) Scan rate dependent cyclic voltammograms of [2]rotaxane 6 (5 mM) in CH2Cl2 against Fe(Cp*)20/+ with tetrabutylammonium BArF24− (0.1 M) as electrolyte. (c) Six-member square scheme with thermodynamic parameters obtained by digital simulations for translation equilibria of 6. (d) Schematic representation for the electrochemically triggered expulsion process in [2]rotaxane 62+. | ||
To clarify this question and to obtain a larger-amplitude shuttling, we synthesised [2]rotaxane 6 (Scheme 1, see Section 1.4 in the ESI for details†) that bears a longer alkyl spacer. Besides the increased spacer length allowing the wheel to undergo a larger translational motion, the binding motifs remain unchanged as compared to [2]rotaxane 1. After double oxidation, the fluorescence spectrum of rotaxane 62+ clearly shows a significant increase in fluorescence indicative of the wheel's motion to a position sufficiently distant from the anthracene stopper to at least partly restore its emission (Fig. 6a). It is expected that the shuttling also occurs in [2]rotaxane 1. Due to the smaller shuttling distance, however, this process cannot be detected by fluorescence spectroscopy.
The wheel shuttling in rotaxane 62+ was also evaluated by scan rate-dependent CV measurements (Fig. 6b). E11/2 of [2]rotaxane 6 is similar to [2]rotaxane 1 whereas E21/2 is slightly decreased by 0.03 V (Table S1, ESI†). A plot of the peak currents against the square root of the scan rates shows the first oxidation to be reversible whereas the second oxidation has a quasi-reversible character (Fig. S14, ESI†). Furthermore, a shoulder at 1.08 V is observed during the cathodic scan at high scan rates, which exhibits a peak potential similar to the reduction of acetylated [2]rotaxane 22+. These data point to a reaction mechanism in which the [2]rotaxane 6 undergoes two successive oxidations followed by a subsequent chemical reaction step. We identify this chemical reaction step with the translation of the wheel. The observed shoulder can then be attributed to the wheel residing in a position remote from the ammonium station and, thus, the TTF2+ moiety in 62+ shows a lower reduction potential.
We applied digital simulations to test the proposed mechanism and estimate the thermodynamic parameters of the translational motion. If equilibria between two distinguishable translational isomers 6n+ and (6n+)* (non-shuttled or shuttled) in all three oxidation states are considered, the system can be described by a six-member square scheme (Fig. 6c). The simulations based on this mechanism are in good agreement with the observed cyclic voltammograms (Fig. S15, ESI†). Fitting to the experimental data gives the intramolecular equilibrium constants KTn (KTn = [6n+]/[(6n+)*]) between the isomers in all oxidation states.276 and 6+˙ display high values of KT0 ∼ 107 and KT1 ∼ 105, respectively. In these oxidation states, the wheel is indeed almost completely located on the ammonium station. For 62+, the simulations point to a much lower equilibrium constant of KT3 ∼ 1. Hence, both translational isomers 62+ and (62+)* exist in significant amounts in equilibrium with each other at 298 K (Fig. 6d). Although a more exact determination of the equilibrium distribution is difficult to determine by digital simulations since the derived quantitative values of KTn are based on several assumptions and should be considered as estimates (see Section 6 in the ESI for details†), this is in good agreement with the energy values discussed above (32.9 kJ mol−1 for the axle binding in the pseudorotaxane and 35 kJ mol−1 for the potential shift in the doubly oxidised state). It may be worth mentioning that the present system allows an estimation of KT values across a range of 1–107 which is extremely wide for an intramolecular quantity.27,28
In the 1H NMR spectrum of [2]rotaxane 62+, only small shifts for the aromatic protons of axle and macrocycle are observed indicating π–stacking interactions to still play a significant role. This is reasonable, as 1H NMR spectroscopy is often not sensitive enough to determine intramolecular KT values.27 Shifts of the isoxazole and methoxy stopper signals are observed which can be explained by major conformational changes in the [2]rotaxane structure (Fig. S16, ESI†). Inferences about the macrocycle positioning in the second translational isomer (62+)* could potentially be drawn from a 1H NMR spectrum of non-charged acetylated [2]rotaxane 22+. However, the low solubility of this doubly oxidised species prevented an exact signal assignment.
The combination of all data obtained by fluorescence spectroscopy, CV and NMR spectroscopy confirms a translational motion in oxidised [2]rotaxane 62+. Therefore, the system can be operated as a novel type of switchable molecular shuttle reversibly driven by redox stimuli. However, only a partial expulsion is observed for 62+ due to a fine balance between charge repulsion and crown/ammonium binding. We assume that entropy effects and the lack of an alternative binding station energetically disfavours the translational isomer (62+)*. The sharp contrast to the efficient expulsion in the non-interlocked pseudo[2]rotaxane 3@TTFC8 underlines the impact of mechanical bonding on the interplay between axle and macrocycle.
Wheel/axle interactions in non-charged, acetylated [2]rotaxane 2
The cyclic voltammogram of non-charged [2]rotaxane 2 draws a contrasting picture of the TTF redox potentials (Fig. 5). A behaviour similar to the free macrocycle is expected, since strong [N–H⋯O] hydrogen-bonds are eliminated and, in addition, effects of Coulomb repulsion are avoided. The potential decrease of the first oxidation (ΔE11/2 = −0.05 V) indicates a TTF+˙ radical cation stabilisation by −5 kJ mol−1, whereas the second potential exhibits an increase of ΔE21/2 = +0.07 V and thus a destabilisation of 7 kJ mol−1 as compared to the free macrocycle.These findings imply a more complex interplay of different intermolecular forces between axle and wheel. Therefore, additional CV measurements were performed with both rotaxanes 1 and 2 using a series of tetrabutylammonium (TBA) salts with different WCAs (ClO4−, PF6−, BArF24−) as the electrolytes and two different solvents (dichloromethane and acetonitrile) to identify possible ion-pairing or solvent effects (Fig. 7).
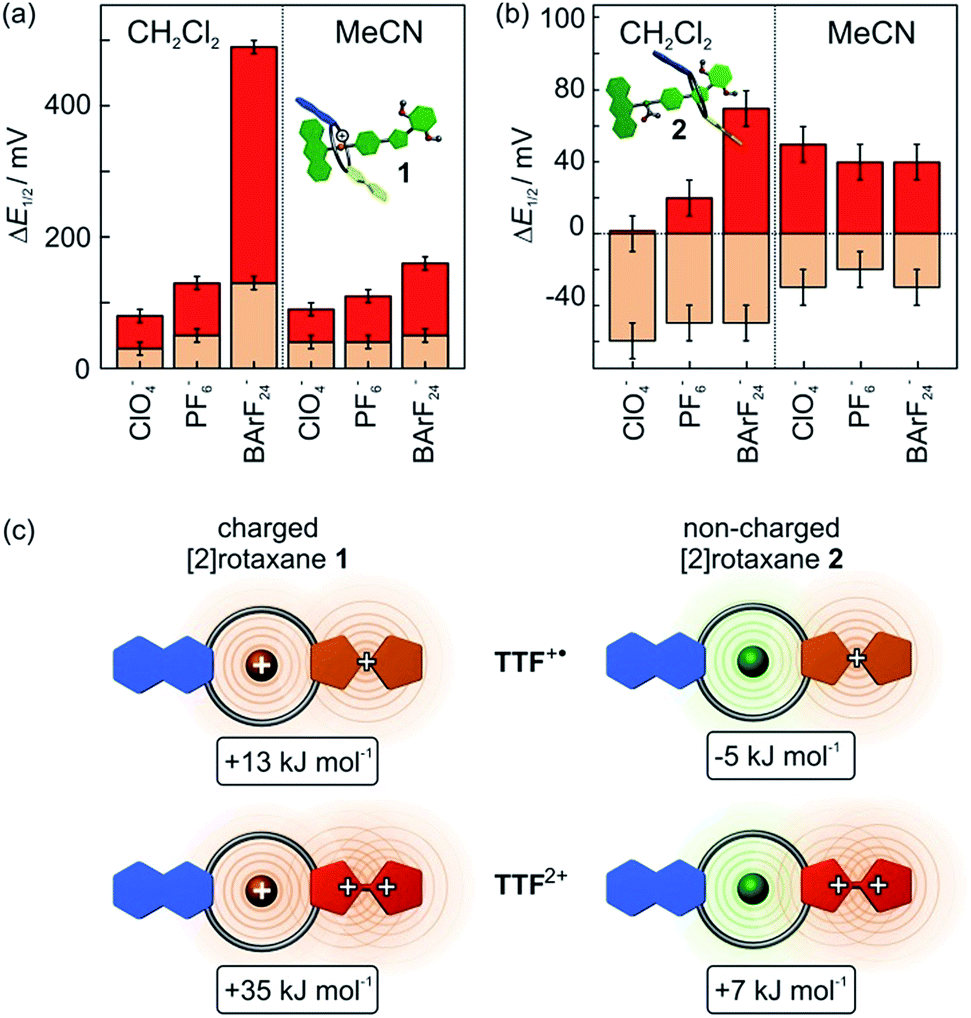 | ||
| Fig. 7 Difference in the first and second oxidation potential ΔE11/2 (orange) and ΔE21/2 (red) for (a) charged [2]rotaxane 1 and (b) non-charged [2]rotaxane 2 in comparison to the free macrocycle TTFC8. Half-wave potentials were obtained by CV measurements with a 1 mM analyte concentration, 0.1 M TBAX− electrolyte concentration and 100 mV s−1 scan rate referenced against Fe(Cp*)20/+. (c) Top view scheme to illustrate electrostatic forces in different redox states (CH2Cl2, TBABArF24) and resulting stabilisation and destabilisation energies between axle and macrocycle in [2]rotaxanes 1 (left) and 2 (right). | ||
In CH2Cl2, the choice of electrolyte has a strong impact on both redox potentials of charged [2]rotaxane 1 which identifies, as expected, charge repulsion as the most dominant interaction between axle and macrocycle. The order of potential increase (ClO4− < PF6− ≪ BArF24−) nicely reflects the coordination abilities of these WCAs29 and, thus, the ΔE11/2 values provide an excellent means to easily quantify them. Non-charged [2]rotaxane 2 shows no clear response of ΔE11/2 to the electrolyte which excludes strong contribution of ion-pairing effects. However, the ΔE21/2 value for [2]rotaxane 2 increases slightly when going from ClO4− to BArF24−. This means that the TTF2+ species enjoys poorer stabilisation as compared to free TTFC8. We assume that the steric demand of the axle diminishes the accessibility for counterions. As a consequence, the doubly charged species lacks charge stabilisation by the counterion. This effect is most pronounced when using TBABArF24 as the electrolyte.
The use of acetonitrile as a more polar solvent leads to a dampening effect on the charge repulsion effects for both oxidations in [2]rotaxane 1. In non-charged [2]rotaxane 2, ΔE11/2 is slightly decreased for all electrolytes in comparison to CH2Cl2 indicating that a more polar solvent weakens the TTF+˙ stabilising interaction. The ΔE21/2 values are in the similar range as in CH2Cl2, but the electrolyte effects are no longer present.
Besides solvent and counterion effects, the contribution of structural modifications on the TTF+˙ stabilising interaction was evaluated. We synthesised an acetylated [2]rotaxane 7 in which we replaced the anthracene stopper by a 3,5-di-tert-butyl benzyl stopper (Scheme 1, see Section 1.5 in the ESI for full synthesis†). We determined almost identical E11/2 and E21/2 values as for acetylated [2]rotaxane 2 (Table S1, ESI†) which indicates that the anthracene stopper has a minor effect for the observed stabilisation.
Overall, the electrochemical data reveals an interplay of different interactions during redox-switching for both [2]rotaxanes 1 and 2 as illustrated in Fig. 7c for the CH2Cl2/TBABArF24 measurement. Whereas charge repulsion hampers the oxidations in charged [2]rotaxane 1, the first oxidation in [2]rotaxane 2 is energetically favoured. An intramolecular interaction between the phenyl-ether/isoxazole/dimethoxybenzene axle part and radical cationic TTF+˙ moiety of the macrocycle is likely. Regarding solvent dependence, electrostatic forces such as cation–dipole interactions are a reasonable assumption.
Conclusions
In summary, we have synthesised and fully characterised the structural and optoelectronic properties of novel redox-switchable [2]rotaxanes which incorporate a redox-active tetrathiafulvalene unit in the wheel. The [2]rotaxanes can be electrochemically operated in two modes in which the threaded axle is either charged or neutral. We found by cyclic voltammetry, that this difference has a dramatic effect on the redox potentials of these [2]rotaxanes. In the charged mode, electrostatic repulsion is identified as the dominant interaction. It can be used to trigger a charge expulsion and subsequently a stimuli-responsive translation motion of the macrocycle which has been analysed by fluorescence, scan rate depended CV, and 1H NMR. In the non-charged mode, detailed CV investigations show a complex interplay of different stabilising and destabilising intramolecular effects in the interlocked system. This study nicely demonstrates that mechanical bonding is an outstanding strategy to create unpredictable emergent properties in MIMs. This novel type of redox-active rotaxane provides an excellent platform for the construction of switchable artificial molecular machines.Acknowledgements
The authors thank the Deutsche Forschungsgemeinschaft (CRC 765) and the Fonds der Chemischen Industrie (FCI) for funding. H. V. S. thanks the FCI for a Chemiefonds Ph.D. fellowship. We thank Constantin Stuckhardt and Sebastian Müller for help with measurements and synthesis.Notes and references
- (a) J. F. Stoddart, Chem. Soc. Rev., 2009, 38, 1802 RSC; (b) E. A. Neal and S. M. Goldup, Chem. Commun., 2014, 50, 5128 RSC; (c) J. E. Lewis, M. Galli and S. M. Goldup, Chem. Commun., 2016, 53, 298 RSC.
- (a) N. Kihara, M. Hashimoto and T. Takata, Org. Lett., 2004, 6, 1693 CrossRef CAS PubMed; (b) F. Coutrot, ChemistryOpen, 2015, 4, 556 CrossRef CAS PubMed.
- A. M. Albrecht-Gary, Z. Saad, C. O. Dietrich-Buchecker and J. P. Sauvage, J. Am. Chem. Soc., 1985, 107, 3205 CrossRef CAS.
- (a) K. Nakazono and T. Takata, Chem.–Eur. J., 2010, 16, 13783 CrossRef CAS PubMed; (b) N. Kihara, Y. Tachibana, H. Kawasaki and T. Takata, Chem. Lett., 2000, 29, 506 CrossRef.
- P. R. Ashton, R. Ballardini, V. Balzani, I. Baxter, A. Credi, M. C. T. Fyfe, M. T. Gandolfi, M. Gómez-López, M. V. Martínez-Díaz, A. Piersanti, N. Spencer, J. F. Stoddart, M. Venturi, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 1998, 120, 11932 CrossRef CAS.
- (a) S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081 CrossRef CAS PubMed; (b) V. Balzani, A. Credi, F. M. Raymo and J. F. Stoddart, Angew. Chem., Int. Ed., 2000, 39, 3348 CrossRef CAS.
- (a) P. L. Anelli, N. Spencer and J. F. Stoddart, J. Am. Chem. Soc., 1991, 113, 5131 CrossRef CAS PubMed; (b) J.-P. Collin, C. Dietrich-Buchecker, P. Gaviña, M. C. Jimenez-Molero and J.-P. Sauvage, Acc. Chem. Res., 2001, 34, 477 CrossRef CAS PubMed.
- (a) J. D. Badjic, V. Balzani, A. Credi, S. Silvi and J. F. Stoddart, Science, 2004, 303, 1845 CrossRef CAS PubMed; (b) Z. J. Zhang, M. Han, H. Y. Zhang and Y. Liu, Org. Lett., 2013, 15, 1698 CrossRef CAS PubMed.
- (a) C. J. Bruns and J. F. Stoddart, Acc. Chem. Res., 2014, 47, 2186 CrossRef CAS PubMed; (b) B. K. Juluri, A. S. Kumar, Y. Liu, T. Ye, Y. W. Yang, A. H. Flood, L. Fang, J. F. Stoddart, P. S. Weiss and T. J. Huang, ACS Nano, 2009, 3, 291 CrossRef CAS PubMed.
- (a) C. Cheng, P. R. McGonigal, S. T. Schneebeli, H. Li, N. A. Vermeulen, C. Ke and J. F. Stoddart, Nat. Nanotechnol., 2015, 10, 547 CrossRef CAS PubMed; (b) G. Ragazzon, M. Baroncini, S. Silvi, M. Venturi and A. Credi, Nat. Nanotechnol., 2015, 10, 70 CrossRef CAS PubMed.
- (a) M. R. Wilson, J. Sola, A. Carlone, S. M. Goldup, N. Lebrasseur and D. A. Leigh, Nature, 2016, 534, 235 CrossRef CAS PubMed; (b) J. V. Hernandez, E. R. Kay and D. A. Leigh, Science, 2004, 306, 1532 CrossRef CAS PubMed.
- W. Yang, Y. Li, H. Liu, L. Chi and Y. Li, Small, 2012, 8, 504 CrossRef CAS PubMed.
- K. Szacilowski, Chem. Rev., 2008, 108, 3481 CrossRef CAS PubMed.
- (a) P. R. Ashton, P. J. Campbell, P. T. Glink, D. Philp, N. Spencer, J. F. Stoddart, E. J. T. Chrystal, S. Menzer, D. J. Williams and P. A. Tasker, Angew. Chem., Int. Ed. Engl., 1995, 34, 1865 CrossRef CAS; (b) N. Yamaguchi and H. W. Gibson, Angew. Chem., Int. Ed., 1999, 38, 143 CrossRef CAS; (c) A. G. Kolchinski, D. H. Busch and N. W. Alcock, J. Chem. Soc., Chem. Commun., 1995, 1289 RSC.
- (a) D. Canevet, M. Salle, G. Zhang, D. Zhang and D. Zhu, Chem. Commun., 2009, 2245 RSC; (b) M. R. Bryce, J. Mater. Chem., 2000, 10, 589 RSC; (c) J. L. Segura and N. Martín, Angew. Chem., Int. Ed., 2001, 40, 1372 CrossRef CAS; (d) F. Wudl, G. M. Smith and E. J. Hufnagel, J. Chem. Soc., Chem. Commun., 1970, 1453 RSC.
- (a) F. Le Derf, M. Mazari, N. Mercier, E. Levillain, G. Trippé, A. Riou, P. Richomme, J. Becher, J. Garín, J. Orduna, N. Gallego-Planas, A. Gorgues and M. Sallé, Chem.–Eur. J., 2001, 7, 447 CrossRef CAS PubMed; (b) M. B. Nielsen and J. Becher, Liebigs Ann., 1997, 1997, 2177 CrossRef; (c) T. Jørgensen, T. K. Hansen and J. Becher, Chem. Soc. Rev., 1994, 23, 41 RSC; (d) T. K. Hansen, T. Joergensen, P. C. Stein and J. Becher, J. Org. Chem., 1992, 57, 6403 CrossRef CAS.
- P. R. Ashton, J. Becher, M. C. T. Fyfe, M. B. Nielsen, J. F. Stoddart, A. J. P. White and D. J. Williams, Tetrahedron, 2001, 57, 947 CrossRef CAS.
- H. V. Schröder, H. Hupatz, A. J. Achazi, S. Sobottka, B. Sarkar, B. Paulus and C. A. Schalley, Chem.–Eur. J., 2017, 23, 2960 CrossRef PubMed.
- W. Jiang, H. D. Winkler and C. A. Schalley, J. Am. Chem. Soc., 2008, 130, 13852 CrossRef CAS PubMed.
- (a) P. R. Ashton, R. Ballardini, V. Balzani, M. Gómez-López, S. E. Lawrence, M. V. Martínez-Díaz, M. Montalti, A. Piersanti, L. Prodi, J. F. Stoddart and D. J. Williams, J. Am. Chem. Soc., 1997, 119, 10641 CrossRef CAS; (b) S. Garaudee, S. Silvi, M. Venturi, A. Credi, A. H. Flood and J. F. Stoddart, ChemPhysChem, 2005, 6, 2145 CrossRef CAS PubMed.
- J. W. Jones and H. W. Gibson, J. Am. Chem. Soc., 2003, 125, 7001 CrossRef CAS PubMed.
- (a) C. Wartelle, P. M. Viruela, R. Viruela, E. Orti, F. X. Sauvage, E. Levillain, F. Le Derf and M. Salle, J. Phys. Chem. A, 2005, 109, 1188 CrossRef CAS PubMed; (b) M. R. Bryce, A. S. Batsanov, T. Finn, T. K. Hansen, J. A. K. Howard, M. Kamenjicki, I. K. Lednev and S. A. Asher, Chem. Commun., 2000, 295 RSC; (c) J. Lyskawa, F. Le Derf, E. Levillain, M. Mazari, M. Salle, L. Dubois, P. Viel, C. Bureau and S. Palacin, J. Am. Chem. Soc., 2004, 126, 12194 CrossRef CAS PubMed; (d) G. Trippé, E. Levillain, F. Le Derf, A. Gorgues, M. Sallé, J. O. Jeppesen, K. Nielsen and J. Becher, Org. Lett., 2002, 4, 2461 CrossRef.
- T. Matsumura, F. Ishiwari, Y. Koyama and T. Takata, Org. Lett., 2010, 12, 3828 CrossRef CAS PubMed.
- Y. Tachibana, H. Kawasaki, N. Kihara and T. Takata, J. Org. Chem., 2006, 71, 5093 CrossRef CAS PubMed.
- (a) S. V. Rosokha and J. K. Kochi, J. Am. Chem. Soc., 2007, 129, 828 CrossRef CAS PubMed; (b) M. B. Kirketerp, L. A. Leal, D. Varsano, A. Rubio, T. J. Jorgensen, K. Kilsa, M. B. Nielsen and S. B. Nielsen, Chem. Commun., 2011, 47, 6900 RSC; (c) V. Khodorkovsky, L. Shapiro, P. Krief, A. Shames, G. Mabon, A. Gorgues and M. Giffard, Chem. Commun., 2001, 2736 RSC.
- L. Avara, F. Gerson, D. O. Cowan and K. Lerstrup, Helv. Chim. Acta, 1986, 69, 141 CrossRef.
- G. Ragazzon, A. Credi and B. Colasson, Chem.–Eur. J., 2017, 23, 2149 CrossRef CAS PubMed.
- (a) A. C. Fahrenbach, J. C. Barnes, H. Li, D. Benitez, A. N. Basuray, L. Fang, C. H. Sue, G. Barin, S. K. Dey, W. A. Goddard III and J. F. Stoddart, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 20416 CrossRef CAS PubMed; (b) A. C. Fahrenbach, C. J. Bruns, D. Cao and J. F. Stoddart, Acc. Chem. Res., 2012, 45, 1581 CrossRef CAS PubMed.
- (a) M. G. Hill, W. M. Lamanna and K. R. Mann, Inorg. Chem., 1991, 30, 4687 CrossRef CAS; (b) R. J. LeSuer, C. Buttolph and W. E. Geiger, Anal. Chem., 2004, 76, 6395 CrossRef CAS PubMed; (c) W. E. Geiger and F. Barriere, Acc. Chem. Res., 2010, 43, 1030 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures and full characterisation of new compounds, tandem MS experiments, ITC data, full electrochemical experiments and additional NMR data. See DOI: 10.1039/c7sc02694c |
| This journal is © The Royal Society of Chemistry 2017 |