
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible light-induced water splitting in an aqueous suspension of a plasmonic Au/TiO2 photocatalyst with metal co-catalysts†
A.
Tanaka
 *a,
K.
Teramura
ab,
S.
Hosokawa
ab,
H.
Kominami
c and
T.
Tanaka
ab
*a,
K.
Teramura
ab,
S.
Hosokawa
ab,
H.
Kominami
c and
T.
Tanaka
ab
aDepartment of Molecular Engineering, Graduate School of Engineering, Kyoto University, Kyotodaigaku Katsura, Nishikyo-ku, Kyoto 615-8510, Japan. E-mail: atsu.tana@apch.kindai.ac.jp
bElements Strategy Initiative for Catalysts & Batteries (ESICB), Kyoto University, 1-30 Goryo-Ohara, Nishikyo-ku, Kyoto 615-8245, Japan
cDepartment of Applied Chemistry, Faculty of Science and Engineering, Kindai University, Kowakae, Higashiosaka, Osaka 577-8502, Japan
First published on 3rd January 2017
Abstract
We found that plasmonic Au particles on titanium(IV) oxide (TiO2) act as a visible-light-driven photocatalyst for overall water splitting free from any additives. This is the first report showing that surface plasmon resonance (SPR) in a suspension system effectively induces overall water splitting. Modification with various types of metal nanoparticles as co-catalysts enhanced the evolution of H2 and O2. Among these, Ni-modified Au/TiO2 exhibited 5-times higher rates of H2 and O2 evolution than those of Ni-free Au/TiO2. We succeeded in designing a novel solar energy conversion system including three elemental technologies, charge separation with light harvest and an active site for O2 evolution (plasmonic Au particles), charge transfer from Au to the active site for H2 production (TiO2), and an active site for H2 production (Ni cocatalyst), by taking advantage of a technique for fabricating size-controlled Au and Ni nanoparticles. Water splitting occurred in aqueous suspensions of Ni-modified Au/TiO2 even under irradiation of light through an R-62 filter.
Although hydrogen (H2) is primarily used in the chemical industry, it will become an important fuel in the near future. Production of H2 from water by a semiconductor photocatalyst under photoirradiation of solar light has attracted significant attention because it offers a promising way to produce a clean, low-cost and environmentally friendly energy source.1 Metal oxides as photocatalysts have been reported to be active for overall water splitting, but most of them require ultraviolet (UV) light (λ < 400 nm) because of the large bandgaps of semiconductor materials. However, UV light accounts for only about 5% of the total solar energy, whereas visible light accounts for about 50% of the total solar energy. The development of photocatalysts using visible light is an important topic from a practical point of view. Various visible light-responding photocatalysts, such as a solid solution of GaN and ZnO (GaN:ZnO)2 and Z-type systems based on Pt/WO3–Pt/ZrO/TaON3 and BiVO4–Ru/SrTiO3:Rh,4 have been reported for overall water splitting. To efficiently utilize solar energy, photocatalysts responding to longer wavelengths are required, and the design of photocatalysts working under green light (ca. 550 nm) and red light (ca. 620 nm) is a challenge. In addition, simple and additive-free water splitting systems, i.e., a system free from any chemicals except the photocatalyst and water, should be developed.
Nanoparticles of metals, such as copper (Cu), silver (Ag), and gold (Au), show strong photoabsorption (+light scattering) of visible light due to surface plasmon resonance (SPR). The resonance frequency of SPR is strongly dependent on the size, shape, interparticle interactions, dielectric properties, and local environment of the nanoparticles. Recently, plasmonic metal nanoparticles have been applied not only to stained glass but also to chemical sensors and biosensors,5 surface-enhanced Raman scattering (SERS),6 fluorescence enhancement,7 and photocurrent enhancement in photovoltaic cells.8 It has been reported that electron transfer from Au nanoparticles to a semiconductor occurred under irradiation of visible light (λ = ca. 550 nm) due to SPR.9 Supported Au materials have been used as visible-light-responding photocatalysts for various chemical reactions,10 including decomposition of organic substrates,11 selective oxidation of an aromatic alcohol to a carbonyl compound,12 H2 formation from alcohols,13 and reduction of organic compounds.14 The research groups that reported these photocatalytic reactions concluded that the reactions are induced by SPR of the Au nanoparticles. In our previous study, we found that TiO2 with both small metal nanoparticles and large Au particles shows activity for H2 formation15 and O2 formation16 with a sacrificial reagent such as 2-propanol and hexavalent chromium under irradiation of visible light.
We have focused on an SPR-based photocatalyst powder for overall water splitting to H2 and O2 in an aqueous suspension without any sacrificial reagents or pH adjustment under irradiation of visible light. Recently, Zhong et al. developed photoelectric conversion and photoelectrochemical water splitting systems that use Au nanoparticles loaded onto a SrTiO3 single-crystal photoelectrode irradiated by visible light.17 In their study, the stoichiometric evolution of H2 and O2 was simultaneously obtained from two separate solution chambers with a chemical bias by regulating the pH values of the chambers. In the present study, we used an Au/TiO2 sample for overall water splitting without using any reagents under irradiation of visible light. Here, we report that samples of Au/TiO2 with co-catalysts were successfully prepared by a combination of photodeposition (PD) or impregnation (IM) and colloid photodeposition in the presence of hole scavenger (CPH) methods, and the samples exhibited larger H2 and O2 evolution rates than those of a co-catalyst-free Au/TiO2 sample under the irradiation of visible light. Water splitting occurred in an aqueous suspension of Au/TiO2 with NiOx even under irradiation of light through an R-62 filter.
Fig. 1 shows the time courses of evolution of H2 and O2 from water over Au(1.0)/TiO2 under a dark condition (0–3 h) or irradiation of visible light (3–24 h) in the absence of any additives. No gas evolved in the dark between 0 and 3 h, indicating that no thermocatalytic H2 and O2 formation occurred in the case of Au(1.0)/TiO2. Just after irradiation with visible light, H2 and O2 evolved from the suspension of Au(1.0)/TiO2. Since H2 and O2 increased linearly with the photoirradiation time, the rates of H2 and O2 evolution were determined to be 0.94 and 0.46 μmol h−1, respectively. The overall photocatalytic reaction is expressed as eqn (1).
| H2O → H2 + ½O2 | (1) |
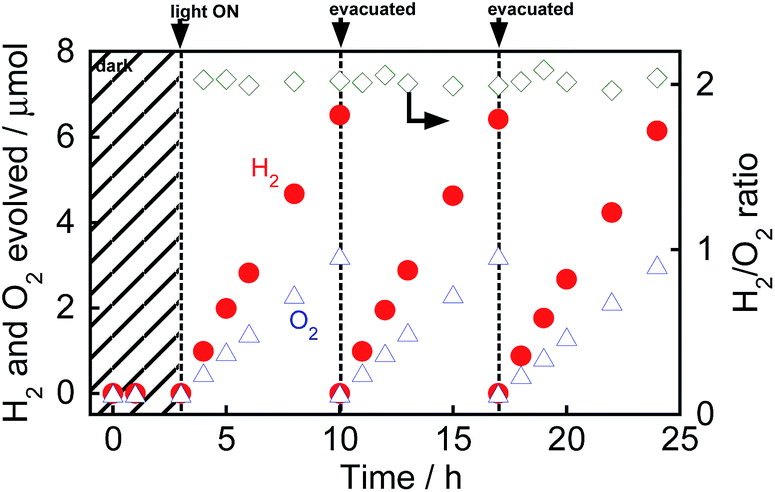 | ||
| Fig. 1 Time courses of evolution of H2 and O2 from water over Au(1.0)/TiO2 under irradiation with visible light from a Xe lamp with an L-42 filter. After 10 h and 17 h of irradiation and evacuation, the suspension was irradiated again. | ||
The ratio of moles of H2 and O2 (H2/O2 ratio) was calculated from eqn (2):
| H2/O2 ratio = n(H2)/n(O2), | (2) |
As shown in Fig. 1, the H2/O2 ratio was almost 2.0 regardless of the irradiation time, indicating that H2 and O2 evolution from water occurred with a high stoichiometry, as shown in eqn (1). To evaluate the stability of Au/TiO2 in H2 and O2 production from water, Au/TiO2 was used again. Irradiation with visible light of the reaction mixture again induced evolution of H2 and O2, and the formation continued from 10 to 17 h and from 17 to 24 h without deactivation.
It is known that the photocatalytic activity for overall water splitting over metal oxides is dependent on the presence of a co-catalyst.18 Various co-catalysts were loaded onto TiO2 using the PD method (Pt, Au, Pd, Rh, Ag) and IM method (Ni, Ru), and then, the Au particles were fixed on TiO2–M using the CPH method. Au(1.0)/TiO2–M(0.5) samples were used for evolution of H2 and O2 from water over various photocatalysts under visible light irradiation, and the effects of M on the reaction rates were compared. The amounts of H2 and O2 evolution for 4 h are shown in Table 1. The amounts of H2 and O2 evolution of all samples with co-catalysts, except for Pt, prepared in this study were larger than that of the M-free Au(1.0)/TiO2 sample. Among the co-catalysts used in this study, Ni had the greatest effect on H2 and O2 evolution.
| Cocat. | Modification method | Amount of products (4 h)/μmol | |
|---|---|---|---|
| H2 | O2 | ||
| a Reaction conditions: catalyst, 300 mg (co-catalyst-loaded); pure water, 300 cm3, Xe lamp (300 W) with an L-42 filter; reaction vessel, Pyrex side-irradiation type. | |||
| None | — | 3.9 | 1.9 |
| Pt | PD | 2.7 | 1.1 |
| Au | 10 | 5.2 | |
| Pd | 10 | 4.9 | |
| Rh | 9.3 | 4.7 | |
| Ag | 8.0 | 3.9 | |
| NiOx | IM | 22 | 11 |
| RuOx | 6.1 | 3.0 | |
Fig. 2 shows the absorption spectra of TiO2, TiO2–NiOx(0.5), Au(1.0)/TiO2, and Au(1.0)/TiO2–NiOx(0.5) samples. The bare TiO2 sample exhibited absorption only at λ < 400 nm because of the band gap excitation. Loading Ni species onto TiO2 resulted in an increase in the baseline of the photoabsorption, which has generally been observed. In the spectra of the Au(1.0)/TiO2 and Au(1.0)/TiO2–NiOx(0.5) samples, a strong photoabsorption was observed at around 550 nm, and it was attributed to SPR of the supported Au nanoparticles.9–17 Since photoabsorption due to Ni species was also included, the Au(1.0)/TiO2–NiOx(0.5) sample exhibited stronger photoabsorptions at 700–800 nm.
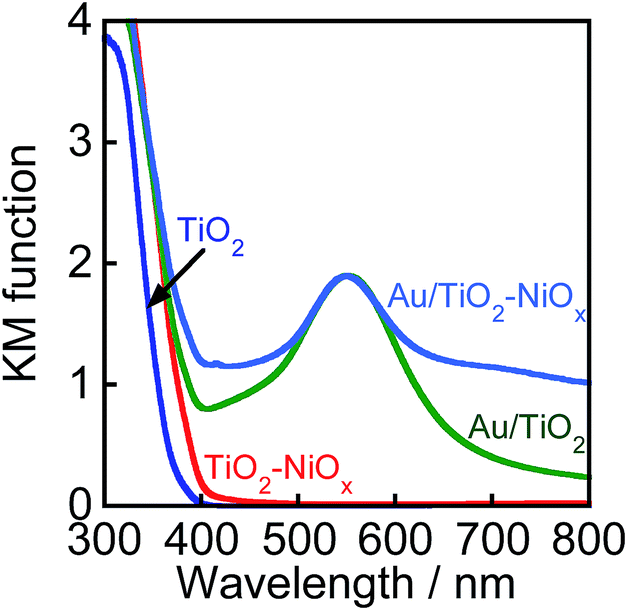 | ||
| Fig. 2 Absorption spectra of TiO2, TiO2–NiOx(0.5), Au(1.0)/TiO2, and Au(1.0)/TiO2–NiOx(0.5). | ||
Fig. 3(a) shows a TEM photograph of the TiO2–NiOx(0.5) sample simply prepared by the traditional IM method. NiOx particles were observed, and the average diameter was determined to be 9.3 nm (Fig. S1(a)†), indicating that the NiOx particles were successfully deposited on the surface of TiO2 using the IM method. Fig. 3(b) shows a TEM image of Au(1.0)/TiO2 prepared by the CPH method using an Au colloidal solution. Gold particles were observed in the image, indicating that Au nanoparticles were deposited on the TiO2 surface by the CPH method.
 | ||
| Fig. 3 TEM images of (a) TiO2–NiOx(0.5), (b) Au(1.0)/TiO2, and (c) Au(1.0)/TiO2–NiOx(0.5). | ||
The average diameter of the Au particles in the sample was determined to be 13 nm (Fig. S1(b)†), which is in good agreement with the average diameter of the original colloidal Au nanoparticles before Au loading (Fig. S2†). In the TEM photograph of the Au(1.0)/TiO2–NiOx(0.5) sample (Fig. 3(c)), both smaller and larger particles were observed, and the average diameters were determined to be 9.1 and 13 nm (Fig. S1(c and d)†), respectively. These results indicate that the CPH method induced no change in the NiOx particles during the loading of the Au particles and that the Au particles were successfully loaded onto the TiO2–NiOx without a change in the original particle size, as in the case of loading Au onto bare TiO2.15
Ni K-edge XANES spectra of Au(1.0)/TiO2–NiOx(0.5) and TiO2–NiOx(0.5) are shown in Fig. 4(a), for which Ni foil and NiO were used as references. The similar XANES spectra of Au/TiO2–NiOx, TiO2–NiOx and NiO clearly proves that the Ni species is NiO. X-ray photoelectron spectroscopy (XPS) was used to obtain information on the surfaces of the NiOx co-catalysts in the Au/TiO2–NiOx samples. Normalized Ni 2p XPS spectra of Au/TiO2–NiOx(0.5), TiO2–NiOx(0.5), Au(1.0)/TiO2 and TiO2 are shown in Fig. S3,† of which the Ni 2p spectra have an overlap with the Auger parameters of Ti.19 The subtracted XPS spectra obtained from the spectra of various samples (Au/TiO2–NiOx, TiO2–NiOx, Au/TiO2) and TiO2 are shown in Fig. 4(b). No peak is observed in the spectrum of Au/TiO2. In the spectra of TiO2–NiOx and Au/TiO2–NiOx, peaks due to Ni (2p3/2) that are assignable to Ni2+, such as NiO, are observed at 856.3 eV.20 On the other hand, peaks due to Ni (2p3/2) that are assignable to Ni0, such as Ni metal, are also observed at 852.7 eV (ref. 21) in the spectrum of Au/TiO2–NiOx. Although the corresponding change was not observed by XAFS measurement, partial formation of Ni metal is confirmed by the XPS spectrum, in which both signals assigned to Ni2+ and Ni0 are observed. It is expected that the surface of NiO in the TiO2–NiOx is reduced to Ni metal during the deposition of the Au particles by the CPH method under irradiation with UV light. Fig. S4† shows the time courses of evolution of H2 in methanol suspensions of TiO2–NiOx(0.5) and Au(1.0)/TiO2–NiOx(0.5) samples under irradiation with UV light. For the first hour, TiO2–NiOx showed a very low activity for H2 evolution, and the continuous formation of H2 at a constant rate was observed after the induction period. These results indicate that the state of the Ni species in TiO2–NiOx changes in the early stage of the photocatalytic reaction, i.e., the surface of NiO in TiO2–NiOx was reduced to the Ni0 species during the induction period. In contrast, there was no induction period for photocatalytic H2 production over Au/TiO2–NiOx, indicating that some of the NiOx particles had been reduced to metallic Ni.
 | ||
| Fig. 4 (a) Ni K-edge XANES spectra of Ni foil, NiO, TiO2–NiOx(0.5) and Au(1.0)/TiO2–NiOx(0.5); (b) XPS spectra of Au(1.0)/TiO2, TiO2–NiOx(0.5) and Au(1.0)/TiO2–NiOx(0.5) around Ni 2p components. | ||
In our previous study on the photocatalytic activity for H2 formation from 2-propanol over Au/TiO2–Pt samples, Pt exhibited the greatest effect on H2 evolution.15 The two types of metal particles had different functionalities, i.e., the large Au particles contributed to the strong light absorption, and the small Pt particles acted as reduction sites for the H2 evolution. However, the activity of the Au/TiO2–Pt samples was much smaller than that of the other samples in this study, and the reaction rate gradually decreased (Fig. S5†), suggesting that the H2–O2 consumption reaction occurred on the Pt nanoparticles. This was confirmed by testing the water formation reaction from a mixture of H2 and O2 in the dark using Au/TiO2–Pt, as shown in Fig. 5(a). The amounts of H2 and O2 in a closed-gas circulation system decreased with time, indicating that water formation occurs on Au/TiO2–Pt. On the other hand, Au/TiO2–NiOx and Au/TiO2 samples showed a smaller rate of backward reaction between H2 and O2 into H2O (consumption rates of H2 and O2) than that of the Au/TiO2–Pt samples, as shown in Fig. 5(b and c). The small rate of the reverse reaction over Au/TiO2 indicates that the formation of H2 and O2 was scarcely affected by the reverse reaction. Therefore, the reaction rate of H2 and O2 formation reflects the photocatalytic activity of Au/TiO2 for water splitting. A linear relationship between the yields and time in Fig. 1, i.e., zero-order kinetics, suggests that the reaction rate over Au/TiO2 was determined by photon flux and photon utilization efficiency because the concentration of water on the Au/TiO2 surface is constant. These results indicated that the actual rates of H2 and O2 evolution on the Au/TiO2–Pt sample should be higher than those recorded by monitoring the gas phase of the reaction system. TiO2, TiO2–Pt(0.5), TiO2–NiOx(0.5), Au(1.0)/TiO2, and Au(1.0)/TiO2–NiOx(0.5) samples were used for the evolution of H2 and O2 from water under visible light irradiation from a Xe lamp with an L-42 filter. The rates of H2 and O2 evolution are shown in Fig. S6.† No H2 or O2 was evolved in the case of TiO2, TiO2–Pt or TiO2–NiOx. These results indicate that the visible light coming from a filtered Xe lamp did not cause band gap excitation of TiO2 and that photocatalysis and/or thermocatalysis of TiO2–Pt and TiO2–NiOx were negligible under the present conditions. The Au(1.0)/TiO2 sample, which was active in the overall water splitting, showed H2 and O2 evolution rates of 0.94 and 0.46 μmol h−1, respectively. The Au(1.0)/TiO2–NiOx(0.5) sample exhibited much larger H2 and O2 formation rates of 5.5 and 2.7 μmol h−1, respectively, indicating that the NiOx co-catalyst loaded onto TiO2 effectively acted as reduction sites for H2 evolution. Loading both large Au particles and small co-catalyst particles on the TiO2 surface without alloying or nanoparticle coagulation has been difficult, as stated above. This requisite was successfully achieved, and a large reaction rate was obtained, as predicted, by the Au/TiO2–NiOx sample prepared by the combination of the traditional IM method for NiOx co-catalyst particles and the CPH method for large Au particles.
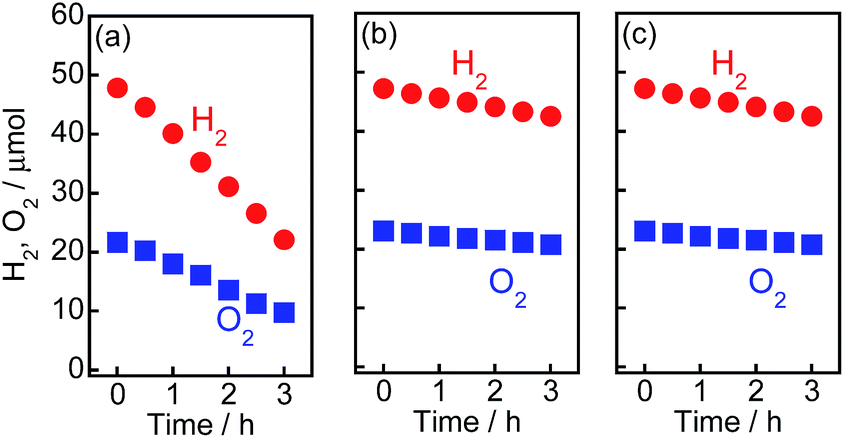 | ||
| Fig. 5 Water formation from H2 and O2 in the dark on (a) Au(1.0)/TiO2–Pt(0.5), (b) Au(1.0)/TiO2–NiOx(0.5) and (c) Au(1.0)/TiO2 samples. This experiment was carried out in pure water (300 cm3) using a closed-gas circulation system containing a stoichiometric mixture of H2 and O2 gases. | ||
To examine the effect of the amount of Au on the rates of H2 and O2 formation, various amounts of Au(X) were loaded onto TiO2–NiOx(0.5) and TiO2 samples using the CPH method. Au(X)/TiO2–NiOx(0.5) and Au(X)/TiO2 samples were used for water splitting under irradiation of visible light, and the rates of H2 and O2 formation are shown in Fig. 6(a and b), respectively. The amounts of H2 and O2 increased linearly with the photoirradiation time in all Au(X)/TiO2–NiOx(0.5) and Au(X)/TiO2 samples, and the formation rates of H2 and O2 were determined from the slopes of the time-H2 and O2 evolution plots. The formation rates of H2 and O2 increased almost linearly with an increase in X until X = 1.0 wt% and gradually increased after X = 1.0 wt%. In a previous study,15 the light absorption due to SPR at 550 nm of a sample prepared by the CPH method increased with an increase in Au loading until 1.0 wt% and gradually increased after X = 1.0 wt%. The Au loading dependency of the light absorption due to SPR was similar to that of the formation rates of H2 and O2, and a linear correlation was observed between the reaction rate and the light absorption due to SPR. We concluded that SPR photoabsorption by the Au particles was one of the important factors determining the photocatalytic activity in this study.
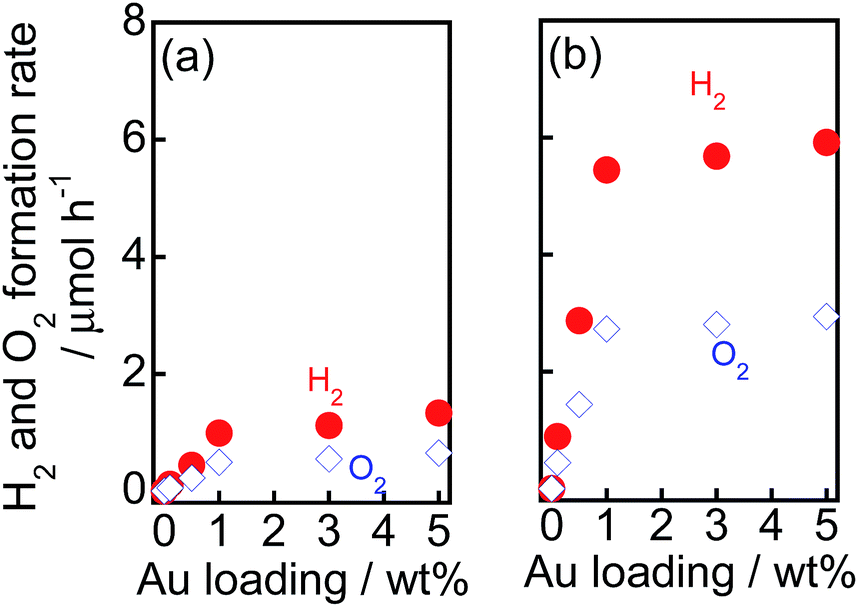 | ||
| Fig. 6 Effect of Au loading amounts (X) on the rate of evolution of H2 and O2 from water over (a) Au(X)/TiO2 and (b) Au(X)/TiO2–NiOx(0.5) samples under irradiation of visible light. | ||
To examine the effect of the amount of the NiOx co-catalyst on the rates of H2 and O2 formation, TiO2–NiOx(Y) samples with various Ni loadings (Y) were prepared, and Au particles (1.0 wt%) were introduced to the samples using the CPH method. The Au(1.0)/TiO2–NiOx(Y) samples were used for overall water splitting under irradiation of visible light; the rates of H2 and O2 formation are shown in Table 2. The Ni-free sample (Au(1.0)/TiO2) exhibited small rates of H2 and O2 formation (0.94 and 0.46 μmol h−1, respectively), as shown above. Only a small amount of NiOx loading (Y = 0.1 wt%) increased the formation rate, indicating that the NiOx co-catalyst effectively acted as the reduction center in the sample. The formation rate increased until Y = 0.5 wt%, and a further increase in Y decreased the formation rate. The maximum rates of H2 and O2 formation (5.5 and 2.7 μmol h−1, respectively) were obtained at Y = 0.5 wt%. The average size of the NiOx nanoparticles in Au(1.0)/TiO2–NiOx(Y) was determined by TEM (Fig. S7†). NiOx particles were loaded onto TiO2 when the amount of NiOx was small (<10 nm, Y = 0.1 and 0.5 wt%); however, the size of the NiOx particles drastically increased to 15 nm at Y = 1.0 wt%, indicating that the IM method was suitable for loading a small amount of small particles on TiO2. Since the particle size of NiOx with Y = 0.5 was smaller (<10 nm) than that with Y > 1.0 and the concentration of the Ni species with Y = 0.5 was higher than that with Y = 0.1, the photocatalytic activity of the Au(1.0)/TiO2–NiOx(0.5) samples was higher than those of the other samples. These results suggested that the NiOx particles acted as reduction sites in the Au(1.0)/TiO2–NiOx(Y) samples and that the reaction is sensitive to the structure of the NiOx particles.
| Entry | NiOx loading (Y)/wt% | Size of NiOx/nm | Formation rate/μmol h−1 | |
|---|---|---|---|---|
| H2 | O2 | |||
| 1 | — | — | 0.99 | 0.49 |
| 2 | 0.1 | 6.6 | 2.7 | 1.3 |
| 3 | 0.5 | 9.1 | 5.5 | 2.7 |
| 4 | 1.0 | 15 | 3.8 | 1.9 |
| 5 | 2.0 | 18 | 1.8 | 0.95 |
Fig. 7 shows time courses of the evolution of H2 and O2 from water over Au(1.0)/TiO2–NiOx(0.5) under a dark condition (0–5 h) or irradiation with visible light from a Xe lamp with L-42 (5–13 h, 29–37 h) or O-54 (13–21 h) or R-62 (21–29 h). Fig. 7 is different from an ordinary action spectrum, and the results indicate the effects of both the wavelength of light and amount of photons. No gas was evolved in the dark between 0 and 5 h, indicating that no thermocatalytic H2 and O2 formation occurred in the case of Au(1.0)/TiO2–NiOx(0.5). The value of H2/O2 was almost 2.0 regardless of the irradiation time and the cut-off filter used. The formation rates of H2 and O2 decreased with an increase in the filter number (L-42 > O-54 > R-62), corresponding to the decrease in photoabsorption (Fig. S8(a)†). It should be noted that water splitting continuously occurred, even under irradiation of light through an R-62 filter. To the best of our knowledge, this is the first report showing H2 and O2 formation from water splitting free from any additives over an Au plasmonic photocatalyst under irradiation of red light (>600 nm). Irradiation of visible light from a Xe lamp with L-42 to the reaction mixture again induced evolution of H2 and O2 from water, and the formation continued from 29 to 37 h without deactivation after irradiation of visible light from different cut-off filters.
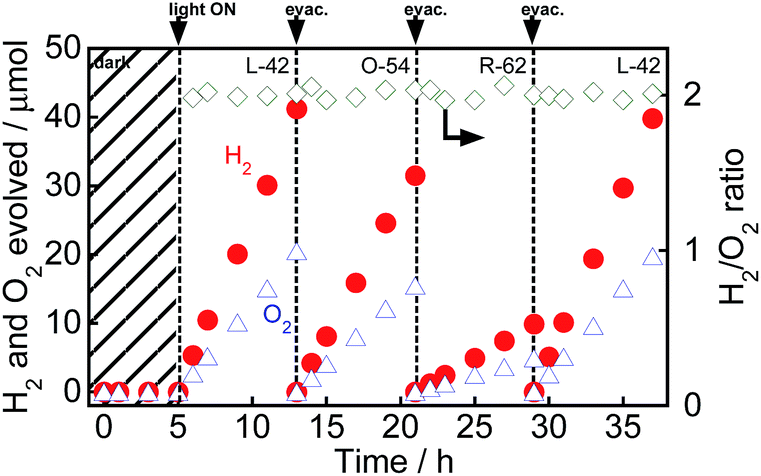 | ||
| Fig. 7 Time courses of evolution of H2 and O2 from water over Au(1.0)/TiO2–NiOx(0.5) under irradiation of visible light from a Xe lamp with various filters. After 13, 21 and 29 h of irradiation and evacuation, the suspension was irradiated again. | ||
Other research groups also reported H2 formation13a,22a and O2 formation22 over Au/TiO2 in the presence of a sacrificial reagent under irradiation of visible light, indicating that Au/TiO2-related materials have a sufficient potential for H2 formation (H+/H2: 0 V NHE pH 0) and oxidization of H2O (H2O/O2: 1.23 V NHE pH 0). Therefore, the simultaneous evolution of H2 and O2 by water splitting in this study is reasonable and is attributed to the acceleration of the positive reaction and suppression of the reverse reaction by co-catalysts introduced to Au/TiO2. Energy (ca. 2.5 or 2.1 eV) corresponding to light (λ = ca. 500 nm or 600 nm) that the Au/TiO2 materials absorb is reasonably large for both H2 and O2 formations even though some over-potentials are required for these formations. In our previous study, we examined the action spectra in H2 formation15 and O2 formation16 over Au/TiO2–Pt in the presence of sacrificial reagents under irradiation with visible light and reported that both spectra showed a similar tendency of absorption spectra. Since rates of water splitting were very small under the irradiation of weak monochromated light, we analyzed the results shown in Fig. 7, and the intensity of the light coming through the cut-off filters is shown in Fig. S8(a).† From the results, the difference in the intensity of light irradiated to Au(1.0)/TiO2–NiOx was calculated in three ranges (Fig. S8(b)†). Based on the incident photons from 520 to 650 nm and the difference in the formation rates in the case of O54 and R62, the value of the apparent quantum efficiency (AQE) was calculated to be 0.013%. In the same way, the values of AQE were calculated based on the incident photons in other ranges and plotted against the wavelength of light irradiated to Au(1.0)/TiO2–NiOx as well as that of AQE from 520 to 650 nm. As shown in Fig. S9,† the wavelength-dependency of AQE was roughly consistent with the spectrum of Au(1.0)/TiO2. We think that this action spectrum is direct evidence that the plasmon-induced water splitting is really induced over Au(1.0)/TiO2–NiOx. As shown in the action spectrum (Fig. S9†), the upper limit of light wavelength available for water splitting over Au/TiO2–NiOx seems to be ca. 700 nm (corresponding to ca. 1.8 eV).
Based on the proposed working mechanism for H2 and O2 formation in the presence of a sacrificial reagent, the expected energy diagram and working mechanism for H2 and O2 formation from water over Au/TiO2–NiOx under irradiation with visible light are shown in Scheme 1. Four processes would occur: (1) the incident photons are absorbed by Au particles through their SPR excitation,10 (2) electrons are injected from the Au particles into the conduction band of TiO2, (3) the resultant electron-deficient Au particles oxidize H2O to O2 and return to their original metallic state, and (4) electrons in the conduction band of TiO2 transfer to the NiOx nanoparticles as a co-catalyst at which reduction of H+ to H2 occurs. Rapid electron transfer from Au to the TiO2 film under visible light irradiation was observed using femtosecond transient absorption spectroscopy.9b In addition to O2 evolution in the presence of a sacrificial reagent,22 Shi et al. demonstrated enhancement in the photocurrent generation and photocatalytic water oxidation sensitized by Au showing SPR under irradiation with visible light.23 Process (3) is supported by these results. Reduction of H+ by electrons in the conduction band of TiO2 is difficult because the level is very close to the reduction potential. Therefore, continuous evolution of H2 is generally achieved by introducing a co-catalyst on TiO2. The co-catalyst effect observed in this study supports process (4). As shown in Fig. 1 and Table 1, Au(1.0)/TiO2 evolved H2 and O2, indicating that Au particles loaded by the CPH method also work as a co-catalyst. We also prepared Au-loaded tin(IV) oxide (SnO2) and Au/SnO2–NiOx using the CPH method, and used it for water splitting under the same conditions. Evolution of H2 and O2 over these samples was negligible because the level of the conduction band of SnO2 is positive to the potential for H+ reduction, i.e., insufficient for H+ reduction. The difference in the results between TiO2 and SnO2 indirectly supports process (4).
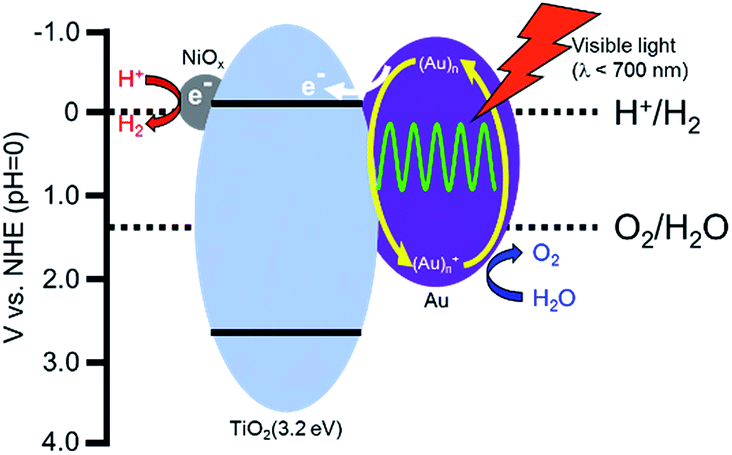 | ||
| Scheme 1 Expected reaction mechanism for the production of H2 and O2 from water over a Au/TiO2–NiOx sample. | ||
Conclusions
The Au/TiO2 sample continuously yielded H2 and O2 from water in the absence of additives under visible light irradiation. Au/TiO2 with NiOx particles was successfully prepared by the combination of photodeposition and impregnation methods. We observed that modification of the Au/TiO2 sample with Ni drastically improved the performance of the photocatalyst, and the samples continuously split water even under irradiation with red light. The backward reaction between the evolved H2 and O2 into H2O could be suppressed using NiOx, although the Pt co-catalyst enhanced the consumption rates and H2 and O2. These results suggest that separate loading of the NiOx co-catalyst without alloying with Au particles is effective for enhancing the activity of an Au plasmonic photocatalyst for overall water splitting.Experimental section
Commercial TiO2 powder (P25, 50 m2 g−1) with an anatase/rutile phase was supplied by Degussa.Loading of the co-catalysts (M: Pt, Au, Pd, Rh, Ag) on TiO2 (preparation of TiO2–M) was performed by the PD method. TiO2 powder was suspended in 10 cm3 of an aqueous solution of methanol (50 vol%) in a test tube, and the test tube was sealed with a rubber septum under argon (Ar). An aqueous solution of the co-catalyst source was injected into the sealed test tube and then photoirradiated for 2 h at λ > 300 nm by a 400 W high-pressure mercury arc (Eiko-sha, Osaka) with magnetic stirring in a water bath continuously kept at 298 K. The co-catalyst source was reduced by photogenerated electrons, and metal was deposited on the surface of the TiO2 particles. Analysis of the liquid phase after photodeposition revealed that the co-catalyst source had been almost completely (>99.9%) deposited on the TiO2 particles. The resultant powder was washed repeatedly with distilled water and then dried at 310 K overnight under air.
Loading of co-catalysts (M: Ni, Ru) on TiO2 (preparation of TiO2–M) was performed by the IM method. TiO2 powder was suspended in 10 cm3 of an aqueous solution of a co-catalyst source in a glass dish and was evaporated to dryness at 333 K. The product was dried overnight at room temperature and then calcined at 573 K for 1 h in air.
Colloidal Au nanoparticles were prepared using the method reported by Frens.24 To 750 cm3 of an aqueous tetrachloroauric acid (HAuCl4) solution (0.49 mmol dm−3), 100 cm3 of an aqueous solution containing sodium citrate (39 mmol dm−3) was added. The solution was heated and boiled for 1 h. After the color of the solution changed from deep blue to deep red, the solution was boiled for an additional 30 min. After the solution was cooled to room temperature, Amberlite MB-1 (ORGANO, 60 cm3) was added to remove excess sodium citrate. After 1 h of treatment, MB-1 was removed from the solution using a glass filter. Loading of Au particles on the TiO2–M samples was performed by the CPH method.25 Preparation of TiO2–M having 1.0 wt% Au as a typical sample is described. A TiO2–M sample (168 mg) was suspended in 20 cm3 of an aqueous solution of colloidal Au nanoparticles (0.085 mg cm−3) in a test tube, and the test tube was sealed with a rubber septum under Ar. An aqueous solution of oxalic acid (50 μmol) was injected into the sealed test tube. The mixture was photoirradiated at λ > 300 nm by a 400 W high-pressure mercury arc under Ar with magnetic stirring in a water bath continuously kept at 298 K. The resultant powder was washed repeatedly with distilled water and then dried at 310 K overnight under air. Co-catalyst-free samples (Au/TiO2) were also prepared by the same method using bare TiO2 samples. When samples with different Au contents were prepared, the amount of TiO2–M (or TiO2) was changed (volume and concentration of the Au colloidal solution being fixed). Hereafter, an Au-loaded TiO2–M sample having Y wt% of M and X wt% of Au is designated as Au(X)/TiO2–M(Y); for example, a sample having 0.5 wt% Ni and 1.0 wt% Au is shown as Au(1.0)/TiO2–NiOx(0.5). Oxalic acid is necessary for complete Au loading on TiO2 (ref. 25) and may protect citrate from covering Au particles. Since the numbers of co-catalyst particles was much larger than that of Au particles,15 the co-catalysts will act effectively.
Diffuse reflectance spectra of the samples were obtained with a JASCO Corporation V-670 spectrometer equipped with an integrating sphere. Spectralon, which was supplied by Labsphere Inc., was used as a standard reflection sample such as BaSO4. The morphology of the samples was observed under a JEOL JEM-2100F transmission electron microscope (TEM) operated at 200 kV at the Joint Research Center of Kindai University. X-ray photoelectron spectroscopy (XPS) measurements were conducted on an ESCA-3400 spectrometer (Shimadzu, Japan). A sample was mounted on a silver sample holder using conductive carbon tape and was analyzed using Mg Kα radiation in a vacuum chamber in 0.1 eV steps. The position of the carbon peak (284.6 eV) for C 1s was used to calibrate the binding energy for all the samples. Ni K-edge (8.3 keV) X-ray absorption fine structure (XAFS) measurements were made in the fluorescence mode at the BL01B1 beamline of the SPring-8 synchrotron radiation facility (Hyogo, Japan). A typical data reduction procedure (e.g., background removal or normalization) was carried out with Athena (version 0.9.20).
Reactions were conducted in a Pyrex side-irradiation vessel connected to a glass closed gas circulation system. A 300 mg sample of Au/TiO2–M powder was dispersed in pure water (300 cm3) using a magnetic stirrer, and this reactant solution was evacuated under vacuum several times to completely remove any residual air. Following this, a small amount of Ar gas was introduced into the reaction system prior to irradiation under a 300 W xenon lamp (Cermax, PE300BF) filtered with various filters with a 15 A output current. The amounts of H2 and O2 in the gas phase were measured using a Shimadzu GC-8A gas chromatograph equipped with an MS-5A column.
Acknowledgements
This study was partially supported by the Program for Element Strategy Initiative for Catalysts & Batteries (ESICB), commissioned by the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan; the Precursory Research for Embryonic Science and Technology (PRESTO), supported by the Japan Science and Technology Agency (JST); Grant-in-Aid for Scientific Research (No. 26289307, 15K18269) from the Japan Society for the Promotion of Science (JSPS); Program for the Strategic Research Foundation at Private Universities 2014–2018 from MEXT and Kindai University. The XAFS experiments were performed with the approval of SPring-8 (Proposal no. 2014B1439 and 2015A1487). Atsuhiro Tanaka is grateful to the JSPS for a Research Fellowship for young scientists.References
- (a) A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC; (b) R. Abe, J. Photochem. Photobiol., C, 2010, 11, 179 CrossRef CAS; (c) X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503 CrossRef CAS PubMed.
- (a) K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2006, 440, 295 CrossRef CAS PubMed; (b) K. Maeda, K. Teramura, D. Lu, N. Saito, Y. Inoue and K. Domen, Angew. Chem., Int. Ed., 2006, 45, 7806 CrossRef CAS PubMed.
- K. Maeda, M. Higashi, D. Lu, R. Abe and K. Domen, J. Am. Chem. Soc., 2010, 132, 5858 CrossRef CAS PubMed.
- (a) Y. Sasaki, H. Nemoto, K. Saito and A. Kudo, J. Phys. Chem. C, 2009, 113, 17536 CrossRef CAS; (b) Y. Sasaki, H. Kato and A. Kudo, J. Am. Chem. Soc., 2013, 135, 5441 CrossRef CAS PubMed.
- M. E. Stewart, C. R. Anderton, L. B. Thompson, J. Maria, S. K. Gray, J. A. Rogers and R. G. Nuzzo, Chem. Rev., 2008, 108, 494 CrossRef CAS PubMed.
- P. L. Stiles, J. A. Dieringer, N. C. Shah and R. P. Van Duyne, Annu. Rev. Anal. Chem., 2008, 1, 601 CrossRef CAS PubMed.
- J. R. Lakowicz, K. Ray, M. Chowdhury, H. Szmacinski, Y. Fu, J. Zhang and K. Nowaczyk, Analyst, 2008, 133, 1308 RSC.
- H. A. Atwater and A. Polman, Nat. Mater., 2010, 9, 205 CrossRef CAS PubMed.
- (a) Y. Tian and T. Tatsuma, J. Am. Chem. Soc., 2005, 127, 7632 CrossRef CAS PubMed; (b) A. Furube, L. Du, K. Hara, R. Katoh and M. Tachiya, J. Am. Chem. Soc., 2007, 129, 14852 CrossRef CAS PubMed.
- (a) M. Xiao, R. Jiang, F. Wang, C. Fang, J. Wang and J. C. Yu, J. Mater. Chem. A, 2013, 1, 5790 RSC; (b) S. C. Warren and E. Thimsen, Energy Environ. Sci., 2012, 5, 5133 RSC; (c) X. Zhou, G. Liu, J. Yu and W. Fan, J. Mater. Chem., 2012, 22, 21337 RSC; (d) S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911 CrossRef CAS PubMed; (e) T. Tatsuma, Bull. Chem. Soc. Jpn., 2013, 86, 1 CrossRef CAS.
- E. Kowalska, R. Abe and B. Ohtani, Chem. Commun., 2009, 241 RSC.
- (a) S. Naya, M. Teranishi, T. Isobe and H. Tada, Chem. Commun., 2010, 46, 815 RSC; (b) A. Tanaka, K. Hashimoto and H. Kominami, J. Am. Chem. Soc., 2012, 134, 14526–14533 CrossRef CAS PubMed.
- (a) H. Yuzawa, T. Yoshida and H. Yoshida, Appl. Catal., B, 2012, 115, 294 CrossRef; (b) A. Tanaka, S. Sakaguchi, K. Hashimoto and H. Kominami, Catal. Sci. Technol., 2012, 2, 907 RSC.
- (a) X. Ke, S. Sarina, J. Zhao, X. Zhang, J. Chang and H. Zhu, Chem. Commun., 2012, 48, 3509 RSC; (b) A. Tanaka, Y. Nishino, S. Sakaguchi, T. Yoshikawa, K. Imamura, K. Hashimoto and H. Kominami, Chem. Commun., 2013, 49, 2551 RSC.
- A. Tanaka, S. Sakaguchi, K. Hashimoto and H. Kominami, ACS Catal., 2013, 3, 79 CrossRef CAS.
- A. Tanaka, K. Nakanishi, R. Hamada, K. Hashimoto and H. Kominami, ACS Catal., 2013, 3, 1886 CrossRef CAS.
- Y. Zhong, K. Ueno, Y. Mori, X. Shi, T. Oshikiri, K. Murakoshi, H. Inoue and H. Misawa, Angew. Chem., Int. Ed., 2014, 53, 10350 CrossRef CAS PubMed.
- K. Maeda, J. Photochem. Photobiol., C, 2011, 12, 237 CrossRef CAS.
- G. M. Ingo, S. Dire and F. Babonneau, Appl. Surf. Sci., 1993, 70, 230 CrossRef.
- A. N. Mansour, Surf. Sci. Spectra, 1994, 3, 231 CrossRef CAS.
- A. N. Mansour, Surf. Sci. Spectra, 1994, 3, 211 CrossRef CAS.
- (a) C. G. Silva, R. Juarez, T. Marino, R. Molinari and H. Garcia, J. Am. Chem. Soc., 2011, 133, 595 CrossRef PubMed; (b) A. Primo, T. Marino, A. Corma, R. Molinari and H. Garcia, J. Am. Chem. Soc., 2011, 133, 6930 CrossRef CAS PubMed.
- X. Shi, K. Ueno, N. Takabayashi and H. Misawa, J. Phys. Chem. C, 2013, 117, 2494 CAS.
- G. Frens, Nature Phys. Sci., 1973, 241, 20 CrossRef CAS.
- A. Tanaka, A. Ogino, M. Iwaki, K. Hashimoto, A. Ohnuma, F. Amano, B. Ohtani and H. Kominami, Langmuir, 2012, 28, 13105 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc05135a |
| This journal is © The Royal Society of Chemistry 2017 |