
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Co/NHPI-mediated aerobic oxygenation of benzylic C–H bonds in pharmaceutically relevant molecules†
Damian P.
Hruszkewycz
a,
Kelsey C.
Miles
a,
Oliver R.
Thiel
b and
Shannon S.
Stahl
*a
aDepartment of Chemistry, University of Wisconsin-Madison, 1101 University Avenue, Madison, Wisconsin 53706, USA. E-mail: stahl@chem.wisc.edu
bProcess Development, Drug Substance Technologies, Amgen Inc., One Amgen Center Drive, Thousand Oaks, California 91320, USA
First published on 7th October 2016
Abstract
A simple cobalt(II)/N-hydroxyphthalimide catalyst system has been identified for selective conversion of benzylic methylene groups in pharmaceutically relevant (hetero)arenes to the corresponding (hetero)aryl ketones. The radical reaction pathway tolerates electronically diverse benzylic C–H bonds, contrasting recent oxygenation reactions that are initiated by deprotonation of a benzylic C–H bond. The reactions proceed under practical reaction conditions (1 M substrate in BuOAc or EtOAc solvent, 12 h, 90–100 °C), and they tolerate common heterocycles, such as pyridines and imidazoles. A cobalt-free, electrochemical, NHPI-catalyzed oxygenation method overcomes challenges encountered with chelating substrates that inhibit the chemical reaction. The utility of the aerobic oxidation method is showcased in the multigram synthesis of a key intermediate towards a drug candidate (AMG 579) under process-relevant reaction conditions.
Introduction
Liquid-phase radical-chain autoxidation reactions are amongst the largest-scale oxidation reactions performed in industry (Scheme 1).1 Prominent examples include the Co/Mn/Br-catalyzed oxidation of p-xylene to terephthalic acid in variations of the Mid-Century process (Scheme 1B),2 autoxidation of cumene en route to phenol and acetone in the Hock process (Scheme 1C)3 and radical-chain autoxidation of cyclohexane to a mixture of cyclohexanone and cyclohexanol (“KA oil”, Scheme 1D).4 In contrast to these prominent large-scale applications, aerobic oxidations and radical autoxidation reactions, in particular, are rarely used for the production of pharmaceuticals or related complex molecules.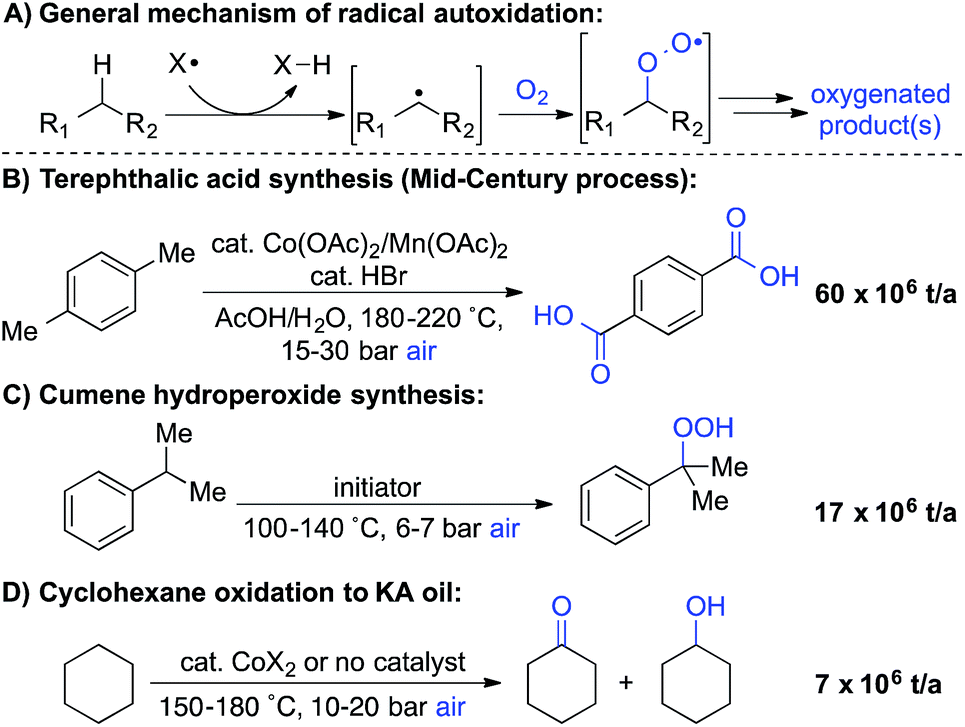 | ||
| Scheme 1 Summary of major industrial radical autoxidation processes. | ||
A number of groups have recently reported methods for aerobic oxygenation of benzylic C–H bonds5,6 (Scheme 2A). The reactions are often compatible with heterocycles and other heteroatom-containing functional groups, suggesting they could be well suited for use in pharmaceutical applications. Further studies have shown that heterocycles can play an important role in promoting the reaction by facilitating formation of an enamine or related tautomer that is more susceptible to aerobic oxygenation (Scheme 2B).7 In a representative example, Maes and coworkers developed a copper-catalysed oxygenation method, in which they contrasted successful oxygenation of 2- and 4-benzylpyridine with the poor reactivity of 3-benzylpyridine.6e A subsequent mechanistic study provided evidence for a heterolytic C–H cleavage pathway and the involvement of the benzylpyridine enamine tautomer (Scheme 2B).6l
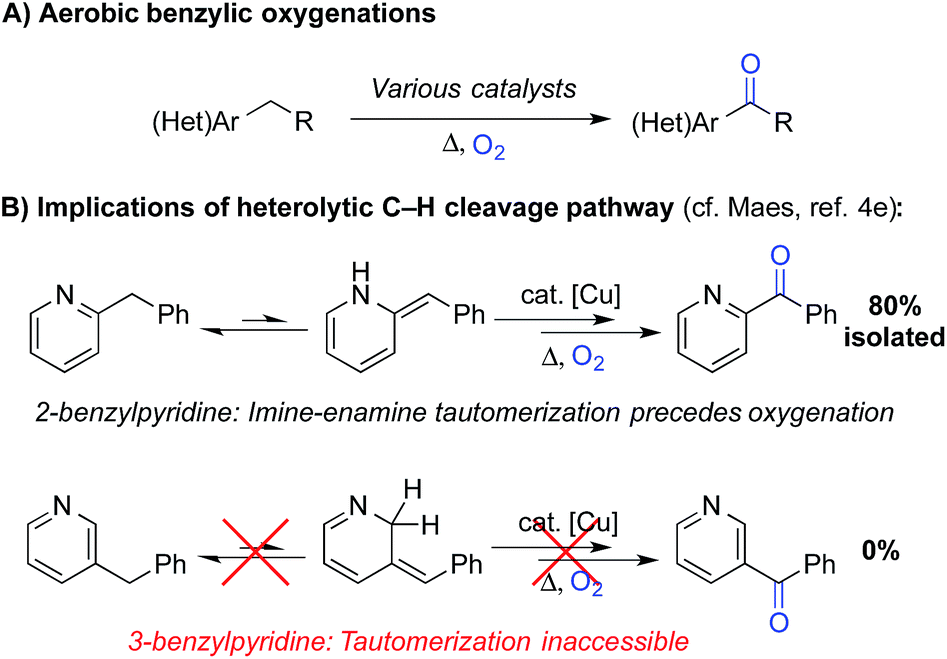 | ||
| Scheme 2 Recent work on aerobic benzylic oxygenation. | ||
The “heterolytic” C–H oxygenation reactions show broad synthetic appeal, but the mechanism limits their utility to substrates with relatively acidic benzylic C–H bonds. In this context, we sought an alternative method that could bypass this limitation, and our attention was drawn to the N-hydroxyphthalimide (NHPI)-based C–H oxygenation reactions.8 In the 1990s, Ishii and coworkers showed that use of redox-active metal salts, such as Co(OAc)2, in combination with O2 enable efficient conversion of NHPI into the phthalimido-N-oxyl (PINO) radical.9 PINO then mediates selective abstraction of weak C–H bonds (Scheme 3) to generate an organic radical that can react with O2 to afford the oxygenated products.10 This methodology has been demonstrated in numerous oxidation reactions, including industrial applications,8 but the vast majority of studies have focused on simple hydrocarbon substrates.11
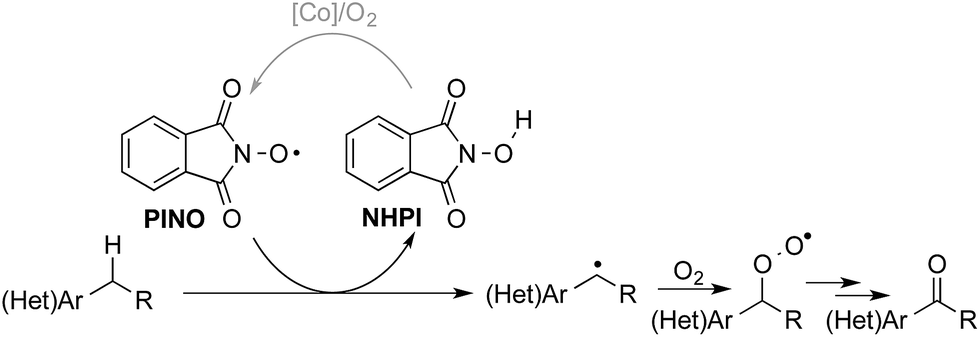 | ||
| Scheme 3 Simplified mechanism depicting C–H abstraction by phthalimido-N-oxyl (PINO) and radical oxygenation by O2. | ||
Here, we explore Co/NHPI-catalyzed benzylic12 oxygenation reactions of substrates bearing common heterocycles, such as pyridines, benzimidazoles and thiophenes, and benzylic ketones are often obtained in good-to-excellent yield. Notably, the radical autoxidation pathway enables oxygenation of non-acidic substrates that are ineffective with catalyst systems that mediate oxygenation via a heterolytic pathway. For reactions that appear to be inhibited by heterocycle chelation to the Co co-catalyst, we show that a Co-free, electrochemical NHPI-mediated method offers a promising alternative approach. Finally, we demonstrate the practicality and scalability of the Co/NHPI catalytic conditions in the multigram synthesis of a pharmaceutical intermediate.
Results and discussion
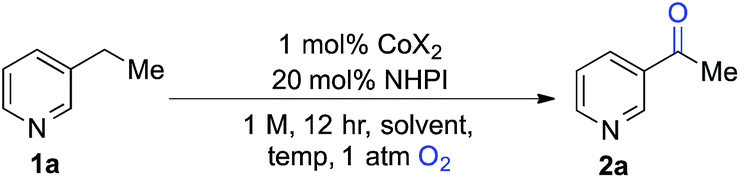
We began our studies by evaluating the oxygenation of 3-ethylpyridine (1a) to 3-acetylpyridine (2a) (Table 1). This reaction is expected to be problematic for many of the recently reported oxygenation methods. First, we used Co(OAc)2 as the cobalt source and compared solvents that have been considered previously in Ishii-type oxidation reactions (entries 1–4).13 A superior yield was obtained using EtOAc (entry 1), and a similar yield was obtained with the higher boiling ester solvent, n-BuOAc (entry 5). The identity of the counterion in the CoX2 co-catalyst had a marked effect on catalytic efficiency (entries 5–10). The best yields were obtained with Co(OAc)2·4H2O and other CoII-carboxylate salts (see Tables S1 and S2 in the ESI† for full screening data). Use of BuOAc solvent enabled access to higher reaction temperatures (entries 9 and 10), and an excellent yield of 2a was achieved at 90 °C (94%, entry 10).|
|
|||||
|---|---|---|---|---|---|
| Entry | CoX2 | Solvent | Temp (°C) | Conv.b (%) | Yieldb (%) |
| a 1 mmol scale, orbital mixing. b GC yields with benzonitrile as an internal standard. | |||||
| 1 | Co(OAc)2·4H2O | EtOAc | 70 | 60 | 59 |
| 2 | — | EtOAc | 70 | <1 | <1 |
| 3 | Co(OAc)2·4H2O | MeCN | 70 | 50 | 45 |
| 4 | Co(OAc)2·4H2O | AcOH | 70 | 35 | 30 |
| 5 | Co(OAc)2·4H2O | BuOAc | 70 | 61 | 59 |
| 6 | Co(NO3)2·6H2O | BuOAc | 70 | 35 | 35 |
| 7 | Co(acac)2 | BuOAc | 70 | <1 | <1 |
| 8 | CoCl2·6H2O | BuOAc | 70 | 14 | 14 |
| 9 | Co(OAc)2·4H2O | BuOAc | 80 | 95 | 89 |
| 10 | Co(OAc) 2 ·4H 2 O | BuOAc | 90 | 99 | 94 |
A comparison of the optimized Co/NHPI reaction conditions with previously reported benzylic oxygenation methods in Table 2 highlights the potential utility of the new conditions. Poor yields and mass balances were obtained using two Mid-Century-type radical autoxidation methods (entries 1 and 2),14 in which a bromine radical is proposed to participate in the H-atom abstraction step. We also tested representative catalyst systems6e for “heterolytic” aerobic C–H oxygenation, corresponding to the methods noted in Scheme 2. As expected from the precedents, 3-ethylpyridine 1a is not a viable substrate with these catalyst systems (entries 3 and 4).15
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst system | Ref. | Conv.b (%) | Yieldb (%) |
| a 1 mmol scale, orbital mixing. b GC yields with benzonitrile as an internal standard. c 1 mmol 1a, 1 mL AcOH, 10 mol% Co(OAc)2·4H2O, 10 mol% HBr, 12 h, 100 °C. d 1 mmol 1a, 1 mL AcOH, 5 mol% Co(OAc)2·4H2O, 5 mol% Mn(OAc)2·4H2O, 10 mol% HBr, 12 h, 100 °C. e 0.5 mmol 1a, 1 mL DMSO, 10 mol% CuI, 1 eq. AcOH, 1 atm O2, 24 h, 100 °C. f 0.5 mmol 1a, 1 mL DMSO, 10 mol% FeCl2·4H2O, 1 eq. AcOH, 24 h, 100 °C. g 1 mmol 1a, 1 mL BuOAc, 1 mol% Co(OAc)2·4H2O, 20 mol% NHPI, 12 h, 90 °C. | ||||
| 1 | Co/Brc | 14 | 51 | 19 |
| 2 | Co/Mn/Brd | 14 | 20 | <1 |
| 3 | CuI, 1 eq. AcOHe | 6e | 5 | <1 |
| 4 | FeCl2·4H2O, 1 eq. AcOHf | 6e | 7 | <1 |
| 5 | Co/NHPI | This work | 99 | 94 |
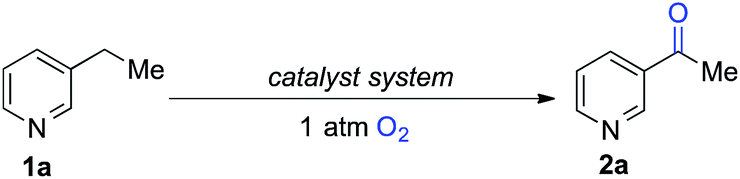
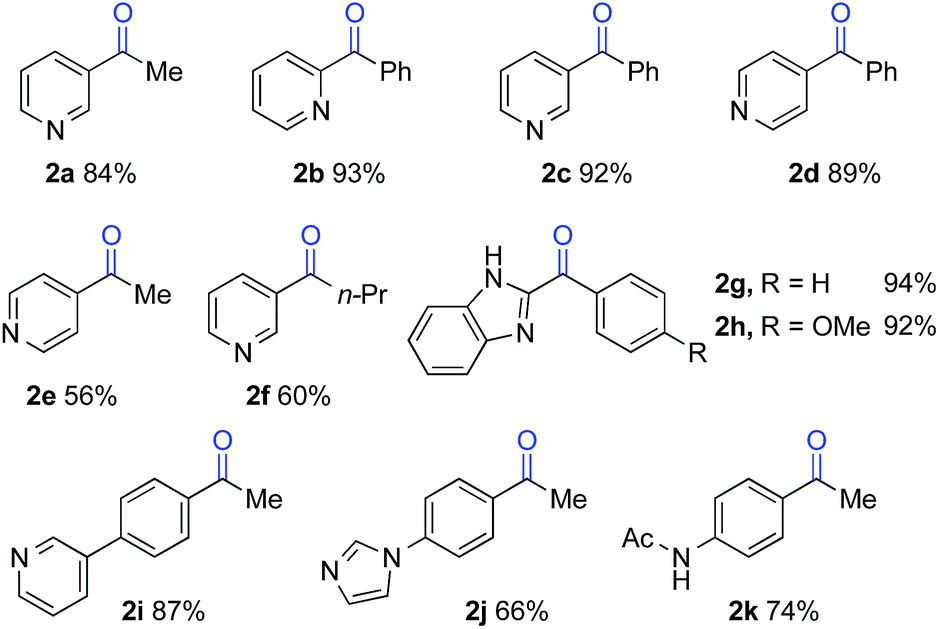
The optimized reaction conditions were then tested for aerobic oxygenation of a number of pharmaceutically relevant (hetero)arene substrates (Table 3).16 In general, pyridines are well tolerated, with good-to-excellent yields obtained for products 2a–f. This collection of substrates includes 3-substituted pyridine derivatives (1a, 1c, 1f), which are ineffective with heterolytic C–H oxygenation methods, as well as 2- and 4-substituted pyridines, which are compatible with the heterolytic methods. Excellent yields were obtained for the benzimidazole derivatives 1g and 1h. Ethylbenzene derivatives bearing remote heteroatom-containing functional groups, including a pyridyl, an imidazole and an acetamide group (1i, 1j and 1k), also underwent successful oxygenation.
|
|
|---|
| a 1 mmol scale, isolated yields. |
|
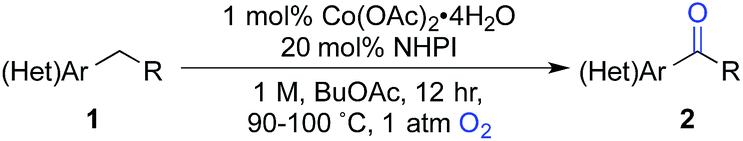
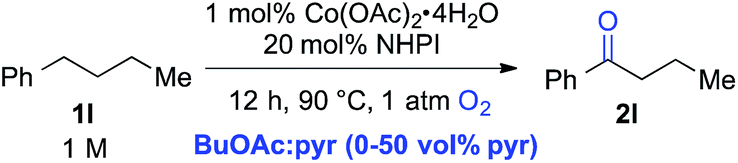
The standard reaction conditions in Table 3 proved to be ineffective for certain substrates,17 and two different approaches were identified to address some of these limitations. In the first case, the simple hydrocarbon, n-butylbenzene (1l) (Table 4), as well as the sulphur-containing heterocyclic substrates 1m, 1n and 1o (Table 5) led to poor results (42%, 3%, 7% and 31% yields, respectively). In the reaction of 1l, benzoic acid was observed as the major side product, arising from cleavage of the alkyl chain. Subsequent empirical studies showed that use of pyridine as a co-solvent attenuated this alkyl chain cleavage and enabled higher yields and mass balance in the oxygenation of 1l. The optimal 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 BuOAc:pyr solvent mixture (77% yield of 2l, entry 4) proved to be similarly beneficial for the oxygenation of the sulphur-heterocycle substrates 1m, 1n and 1o, as well as the N-methylated benzimidazole 1p (Table 5).18 The latter substrates underwent oxygenation in 65%, 50%, 72% and 94% yields, respectively. The reactions with pyridine co-solvent have a dark red color, consistent with pyridine coordination to cobalt.19 We speculate that this coordination might attenuate unproductive cobalt-mediated side reactions in these substrates.
:3 BuOAc:pyr solvent mixture (77% yield of 2l, entry 4) proved to be similarly beneficial for the oxygenation of the sulphur-heterocycle substrates 1m, 1n and 1o, as well as the N-methylated benzimidazole 1p (Table 5).18 The latter substrates underwent oxygenation in 65%, 50%, 72% and 94% yields, respectively. The reactions with pyridine co-solvent have a dark red color, consistent with pyridine coordination to cobalt.19 We speculate that this coordination might attenuate unproductive cobalt-mediated side reactions in these substrates.
|
|
|---|
| a 1 mmol scale. Yields were determined by 1H NMR spectroscopy. Isolated yields in parentheses. |
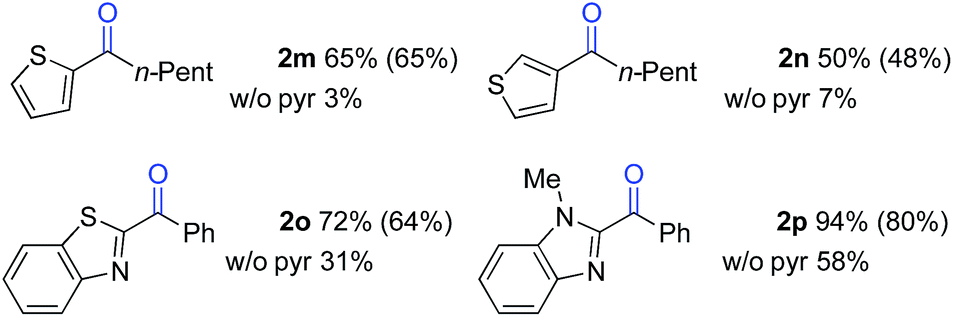
|

The second case involved 2-ethylpyridine (1q) and 2-ethylbenzimidazole (1r), in which the benzylic C–H bonds of the ethyl substituent is directly adjacent to a coordinating group. Under the standard conditions, the benzylic ketones 2q and 2r were obtained in only 48% and 15% yields, respectively (Scheme 4A). This poor reactivity contrasts the good reactivity observed with analogous doubly benzylic substrates 1b, 1g and 1h in Table 3. We hypothesized that, in these reactions, the product could chelate the cobalt co-catalyst and inhibit further product formation. To test this hypothesis, we investigated the reaction of an effective substrate, 4-ethylpyridine 1e, in the presence and absence of 4-acetylpyridine 2e or 2-acetylpyridine 2q (Scheme 4B). The results show that 2e has minimal impact on the reaction (Scheme 4B, entries 1–3), whereas the presence of 2q significantly lowers the yield in the oxygenation of 1e (Scheme 4B, entries 1, 4 and 5).
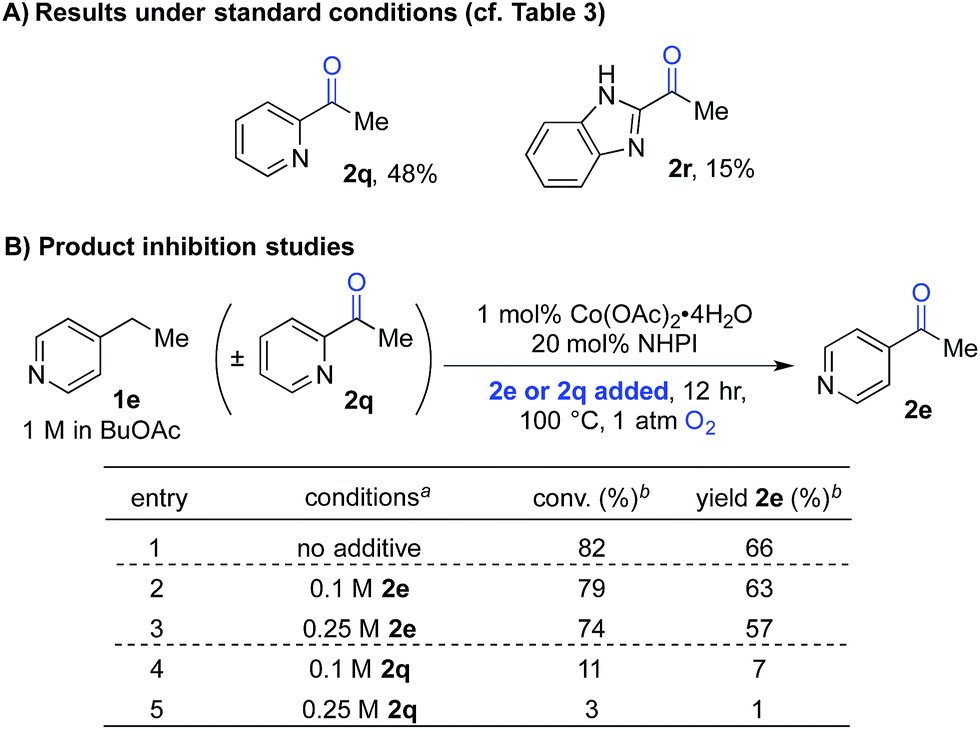 | ||
| Scheme 4 Product-inhibition studies in Co/NHPI-catalyzed oxygenation. a1 mmol scale, orbital mixing. bGC yields with chlorobenzene as an internal standard. | ||
The observations in Scheme 4 prompted us to consider cobalt-free electrochemical aerobic oxygenation reactions, originally reported by Masui and coworkers in the 1980s.20,21 These methods generate PINO via electrochemical oxidation of NHPI (i.e., in the absence of cobalt ions) and, therefore, should not be susceptible to chelate-inhibition by the product. Preliminary studies validated this hypothesis. Optimization of Masui's reported electrochemical conditions with 1q and 1r enabled formation of the ketone products 2q and 2r in 82% and 70% yields, respectively. These results represent substantial improvements over those obtained with the chemical conditions (Scheme 5). Further studies will be needed to explore the full scope and limitations of the electrochemical method. Preliminary studies with other substrates (e.g., with 1b and 1g) suggest that the chemical conditions will be superior to the electrochemical conditions in other cases. These observations together with the ease of reaction set up and performance of the Co/NHPI/O2 oxygenations suggest that the aerobic reactions will be advantageous in most cases. Nonetheless, the results with the oxygenation of 1q and 1r demonstrate that electrochemical NHPI-mediated reactions could be a valuable complement to Co/NHPI reactions in strategic situations.
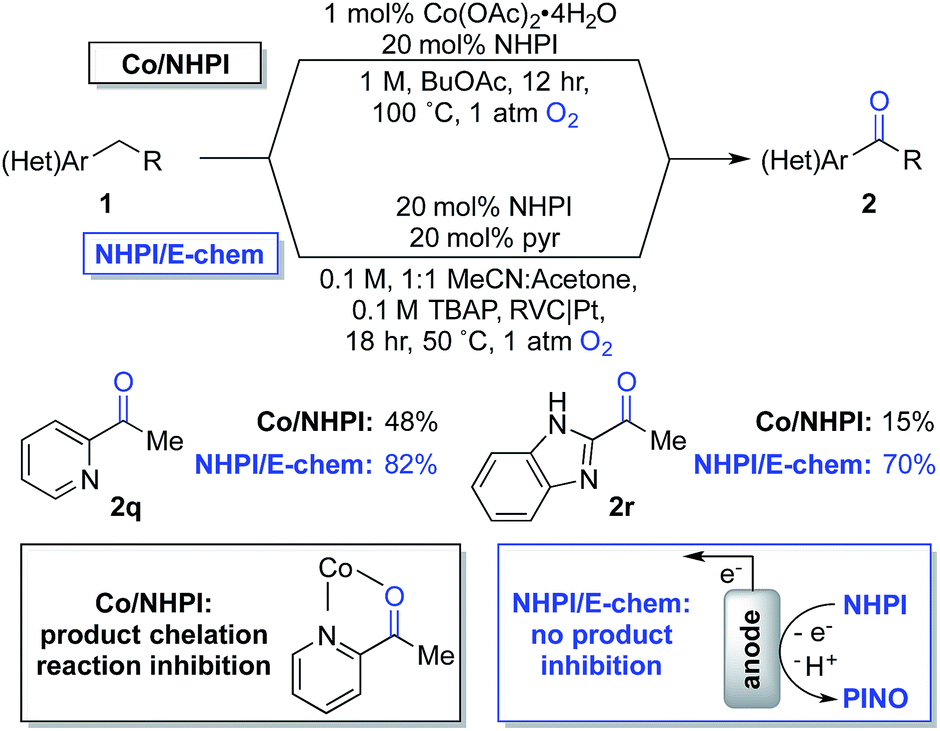 | ||
| Scheme 5 Overcoming a limitation in Co/NHPI chemistry through the electrochemical generation of PINO. | ||
The standard Co/NHPI/O2 conditions developed here exhibit a number of features that are appealing from a process chemistry perspective, including a low cobalt catalyst loading and a high reaction concentration, which minimizes solvent volume. The relatively high NHPI loading (20 mol%) is offset by its extraordinarily low cost (<$5 per kg on commercial scale). In an effort to demonstrate the potential utility of this method for larger-scale applications, we targeted a streamlined route to the heterocyclic ketone 2s (Scheme 6A), an intermediate en route to AMG 579.22 The latter compound is a clinical candidate for the treatment of neurological conditions such as schizophrenia and Huntington's disease. The reported multi-step route to 2s is effective, but it is operationally complex and requires careful control of reaction temperature during the deprotonation and addition (Scheme 6B).23 We envisioned an alternative, two-step route to 2s, starting from the inexpensive, commercially available precursors o-phenylene diamine and 4-fluorophenylacetic acid (Scheme 6C). The Co/NHPI-catalyzed oxygenation of 1s was performed in EtOAc (1 M) with 500 psi of 9% O2 in N2 to maintain the O2 concentration below the limiting oxygen concentration (LOC) of EtOAc.24 Operation of this reaction on 2 g scale resulted in near quantitative isolated yield of 2r (95%).
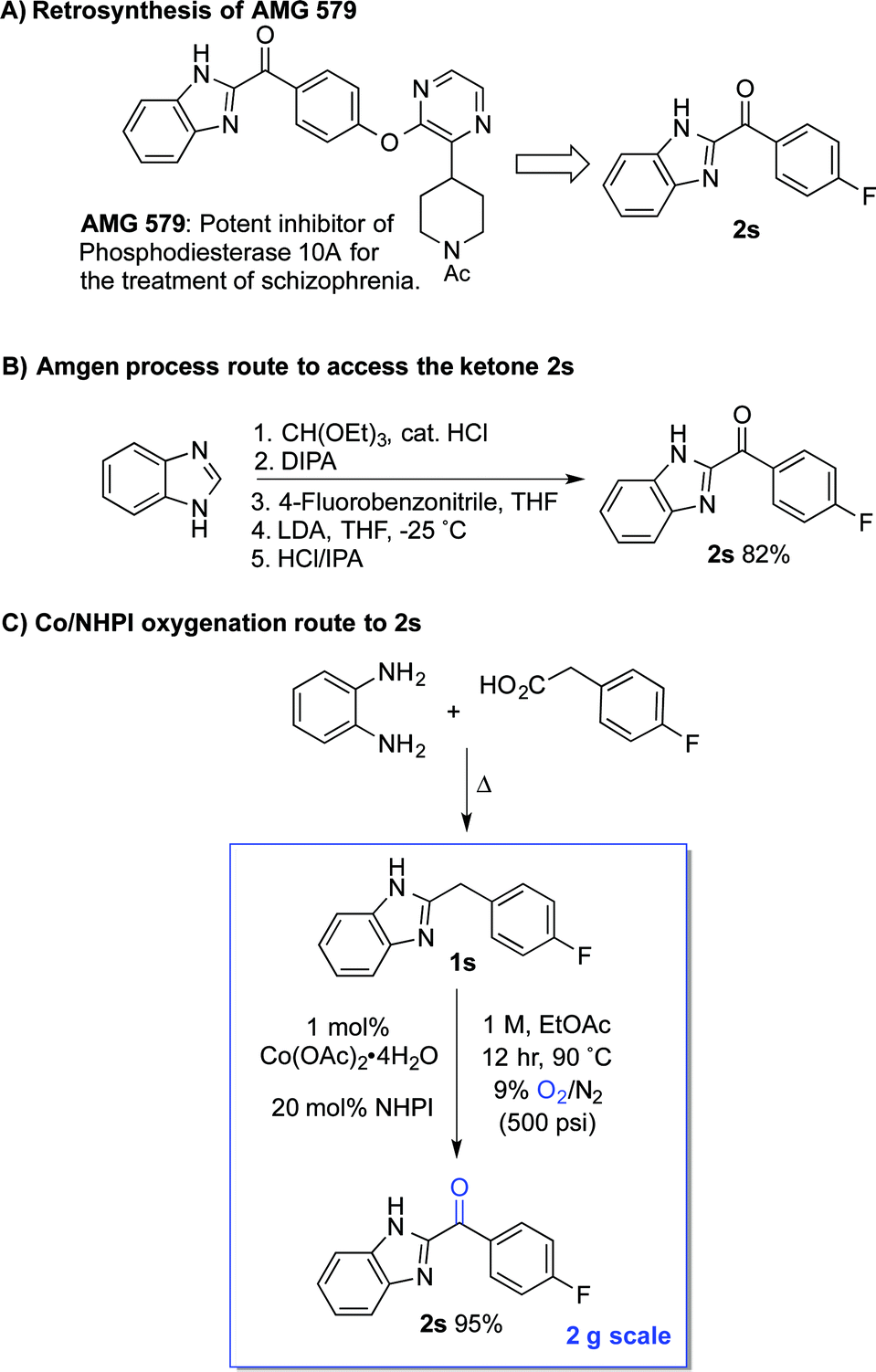 | ||
| Scheme 6 Streamlined synthetic route toward AMG 579via Co/NHPI-catalyzed benzylic oxygenation. | ||
This route to 2s incorporates a number established green chemistry principles:25 (i) waste prevention due to high volumetric efficiency, (ii) atom economy due to use of O2 as both the terminal oxidant and oxygen atom source, (iii) use of an environmentally benign ester solvent,26 (iv) use of an earth-abundant metal catalyst, (v) application of practical reaction conditions with respect to temperature and pressure, as well as (vi) application of inherently safe reaction conditions (i.e., below the LOC of EtOAc). Although more thorough analysis would be required to compare the overall process viability of the two routes, the two-step condensation/oxidation sequence in Scheme 6C should have a much smaller environmental footprint, owing to the formation of water as a only stoichiometric by-product in each of the two reactions.27 Reflecting this feature, the route in Scheme 6B has an estimated E-factor of >15, while the two-step condensation/oxidation sequence can be <5.28
Conclusions
The results described herein show that Co/NHPI-catalyzed, radical-mediated, benzylic aerobic oxygenation reactions are quite effective with pharmaceutically relevant heterocyclic substrates. The comparatively non-polar nature of the H-atom transfer step mediated by PINO plays an important role in expanding the scope of aerobic benzylic oxygenation to substrates that are ineffective with complementary heterolytic oxygenation methods.5,6 The chemistry and reaction conditions identified herein are sufficiently practical that these methods could be compelling for large-scale application. In addition to the low E-factors associated with these reactions, the homogeneous reaction conditions (i.e., lacking solid reagents or additives) suggest that they are excellent candidates for continuous-flow applications.29 This opportunity warrants attention in future studies.Acknowledgements
Financial support was provided by the NIH (F32GM113399, D. P. H.), the DOE (DE-FG02-05ER15690, S. S. S.), and Merck Research Laboratories–Rahway (K. C. M., electrochemical studies). Spectroscopic instrumentation was partially supported by the NIH (S10 OD020022) and the NSF (CHE-1048642).Notes and references
- H. J. Teles, I. Hermans, H. G. Franz and R. A. Sheldon, Oxidation in Ullmann's Encyclopedia of Industrial Chemistry, Electronic Release, Wiley-VCH, Weinheim, 2015 Search PubMed.
- (a) W. Partenheimer, Catal. Today, 1995, 23, 69 CrossRef CAS; (b) R. A. F. Tomas, J. C. M. Bordado and J. F. P. Gomes, Chem. Rev., 2013, 113, 7421 CrossRef CAS PubMed; (c) V. A. Adamian and W. H. Gong in Liquid Phase Aerobic Oxidation Catalysis: Industrial Applications and Academic Perspectives, ed. S. S. Stahl and P. L. Alsters, Wiley-VCH, Weinheim, 2016, pp. 41–66 Search PubMed.
- M. Weber, M. Weber and M. Kleine-Boymann, Phenol in Ullmann's Encyclopedia of Industrial Chemistry, Electronic Release, Wiley-VCH, 2004 Search PubMed.
- M. T. Musser, Cyclohexanol and Cyclohexanone in Ullmann's Encyclopedia of Industrial Chemistry, Electronic Release, Wiley-VCH, 2011 Search PubMed.
- For metal-free conditions: (a) K. K. Park, L. K. Tsou and A. D. Hamilton, Synthesis, 2006, 3617 CrossRef CAS; (b) C. Qi, H. Jiang, L. Huang, Z. Chen and H. Chen, Synthesis, 2011, 387 CAS; (c) C. Zhang, Z. Xu, L. Zhang and N. Jiao, Tetrahedron, 2012, 68, 5258 CrossRef CAS; (d) A. Dos Santos, L. El Kaim and L. Grimaud, Org. Biomol. Chem., 2013, 11, 3282 RSC; (e) L. Ren, L. Wang, Y. Lv, G. Li and S. Gao, Org. Lett., 2015, 17, 2078 CrossRef CAS PubMed; (f) K. Bao, F. Li, H. Liu, Z. Wang, Q. Shen, J. Wang and W. Zhang, Sci. Rep., 2015, 5, 10360 CrossRef PubMed.
- For metal-catalyzed examples: (a) F. M. Moghaddam, Z. Mirjafary, H. Saeidian and M. J. Javan, Synlett, 2008, 892 CrossRef CAS; (b) X. Fan, Y. He, X. Zhang, S. Guo and Y. Wang, Tetrahedron, 2011, 67, 6369 CrossRef CAS; (c) X. Fan, Y. He, Y. Wang, Z. Xue, X. Zhang and J. Wang, Tetrahedron Lett., 2011, 52, 899 CrossRef CAS; (d) Y.-F. Wang, F.-L. Zhang and S. Chiba, Synthesis, 2012, 44, 1526 CrossRef CAS; (e) J. De Houwer, K. Abbaspour Tehrani and B. U. W. Maes, Angew. Chem., Int. Ed., 2012, 51, 2745 CrossRef CAS PubMed; (f) C. Menendez, S. Gau, S. Ladeira, C. Lherbet and M. Baltas, Eur. J. Org. Chem., 2012, 409 CrossRef CAS; (g) B. Pieber and C. O. Kappe, Green Chem., 2013, 15, 320 RSC; (h) S. Cacchi, G. Fabrizi, A. Goggiamani, A. Iazzetti and R. Verdiglione, Synthesis, 2013, 45, 1701 CrossRef CAS; (i) Y. Jeong, Y. Moon and S. Hong, Org. Lett., 2015, 17, 3252 CrossRef CAS PubMed; (j) J.-W. Yu, S. Mao and Y.-Q. Wang, Tetrahedron Lett., 2015, 56, 1575 CrossRef CAS; (k) G. Zheng, H. Liu and M. Wang, Chin. J. Chem., 2016, 34, 519 CrossRef CAS; (l) H. Sterckx, J. De Houwer, C. Mensch, I. Caretti, K. A. Tehrani, W. A. Herrebout, S. Van Doorslaer and B. U. W. Maes, Chem. Sci., 2016, 7, 346 RSC; (m) H. Sterckx, J. De Houwer, C. Mensch, W. Herrebout, K. A. Tehrani and B. U. W. Maes, Beilstein J. Org. Chem., 2016, 12, 144 CrossRef CAS PubMed; (n) Q. Li, Y. Huang, T. Chen, Y. Zhou, Q. Xu, S.-F. Yin and L.-B. Han, Org. Lett., 2014, 16, 3672 CrossRef CAS PubMed; (o) Y. Huang, T. Chen, Q. Li, Y. Zhou and S.-F. Yin, Org. Biomol. Chem., 2015, 13, 7289 RSC; (p) M. Liu, T. Chen and S.-F. Yin, Catal. Sci. Technol., 2016, 6, 690 RSC; (q) H. Xie, Y. Liao, S. Chen, Y. Chen and G.-J. Deng, Org. Biomol. Chem., 2015, 13, 6944 RSC; (r) C. Zhang, Z. Xu, L. Zhang and N. Jiao, Angew. Chem., Int. Ed., 2011, 50, 11088 CrossRef CAS PubMed; (s) Z. Xu, C. Zhang and N. Jiao, Angew. Chem., Int. Ed., 2012, 51, 11367 CrossRef CAS PubMed; (t) C. Zhang, L. Zhang and N. Jiao, Adv. Synth. Catal., 2012, 354, 1293 CrossRef CAS.
- This pathway is analogous to aerobic oxygenation reactions adjacent to carbonyl groups. For examples, see: (a) F.-T. Du and J.-X. Ji, Chem. Sci., 2012, 3, 460 RSC; (b) X. Xu, W. Ding, Y. Lin and Q. Song, Org. Lett., 2015, 17, 516 CrossRef CAS PubMed; (c) X. Huang, X. Li, M. Zou, J. Pan and N. Jiao, Org. Chem. Front., 2015, 2, 354 RSC; (d) H. Cheng and C. Bolm, Synlett, 2016, 27, 769 CAS.
- (a) Y. Ishii, S. Sakaguchi and T. Iwahama, Adv. Synth. Catal., 2001, 343, 393 CrossRef CAS; (b) F. Recupero and C. Punta, Chem. Rev., 2007, 107, 3800 CrossRef CAS PubMed; (c) L. Melone and C. Punta in Liquid Phase Aerobic Oxidation Catalysis: Industrial Applications and Academic Perspectives, ed. S. S. Stahl and P. L. Alsters, Wiley-VCH, Weinheim, 2016, pp. 253–266 Search PubMed.
- (a) Y. Ishii, T. Iwahama, S. Sakaguchi, K. Nakayama and Y. Nishiyama, J. Org. Chem., 1996, 61, 4520 CrossRef CAS PubMed; (b) Y. Yoshino, Y. Hayashi, T. Iwahama, S. Sakaguchi and Y. Ishii, J. Org. Chem., 1997, 62, 6810 CrossRef CAS.
- For detailed discussion of the mechanism of aerobic Co/NHPI oxygenations, see ref. 8a.
- Studies of substrates potentially relevant to pharmaceutical applications are rare and have met with limited success: (a) A. Shibamoto, S. Sakaguchi and Y. Ishii, Org. Process Res. Dev., 2000, 4, 505 CrossRef CAS; (b) B. B. Wentzel, M. P. J. Donners, P. L. Alsters, M. C. Feiters and R. J. M. Nolte, Tetrahedron, 2000, 56, 7797 CrossRef CAS; (c) S. Sakaguchi, A. Shibamoto and Y. Ishii, Chem. Commun., 2002, 180 RSC; (d) L. Schmieder-van de Vondervoort, S. Bouttemy, F. Heu, K. Weissenbock and P. L. Alsters, Eur. J. Org. Chem., 2003, 578 CrossRef CAS.
- For simplicity, the term “benzylic” is used for sites adjacent to both aryl and heteroaryl rings.
- T. Iwahama, Y. Yoshino, T. Keitoku, S. Sakaguchi and Y. Ishii, J. Org. Chem., 2000, 65, 6502 CrossRef CAS PubMed.
- For similar reaction conditions for Mid Century-type autoxidation, see: (a) A. S. Hay and H. S. Blanchard, Can. J. Chem., 1965, 43, 1306 CrossRef CAS; (b) B. Gutmann, P. Elsner, D. Roberge and C. O. Kappe, ACS Catal., 2013, 3, 2669 CrossRef CAS.
- See also: J. Liu, X. Zhang, H. Yi, C. Liu, R. Liu, H. Zhang, K. Zhuo and A. Lei, Angew. Chem., Int. Ed., 2015, 54, 1261 CrossRef CAS PubMed.
- Pyridines and imidazoles are amongst the most commonly occurring N-heterocycles in F.D.A.-approved pharmaceuticals. See: E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257 CrossRef CAS PubMed.
- A “robustness screen” in which the reaction of 1a was carried out in the presence of additives bearing potentially interesting functional groups, together with independent reactions of 2-propylfuran, isobutylbenzene, and 2-ethylacetanilide, are presented in the ESI.† See Tables S4 and S5† and associated text for details.
- For a review on thiophene-containing pharmaceuticals, see: D. Gramec, L. Peterlin Masic and M. Sollner Dolenc, Chem. Res. Toxicol., 2014, 27, 1344 CrossRef CAS PubMed.
- (a) H. Henschel, J.-P. Kloeckner, I. A. Nicholls and M. H. Prosenc, J. Mol. Struct., 2012, 1007, 45 CrossRef CAS; (b) A. B. P. Lever and D. Ogden, J. Chem. Soc. A, 1967, 2041 RSC.
- M. Masui, S. Hara, T. Ueshima, T. Kawaguchi and S. Ozaki, Chem. Pharm. Bull., 1983, 31, 4209 CrossRef CAS.
- For electrochemical, PINO-mediated oxygenation of allylic C–H bonds, see: (a) M. Masui, K. Hosomi, K. Tsuchida and S. Ozaki, Chem. Pharm. Bull., 1985, 33, 4798 CrossRef CAS; (b) E. J. Horn, B. R. Rosen, Y. Chen, J. Tang, K. Chen, M. D. Eastgate and P. S. Baran, Nature, 2016, 533, 77 CrossRef CAS PubMed.
- E. Hu, N. Chen, M. P. Bourbeau, P. E. Harrington, K. Biswas, R. K. Kunz, K. L. Andrews, S. Chmait, X. Zhao, C. Davis, J. Ma, J. Shi, D. Lester-Zeiner, J. Danao, J. Able, M. Cueva, S. Talreja, T. Kornecook, H. Chen, A. Porter, R. Hungate, J. Treanor and J. R. Allen, J. Med. Chem., 2014, 57, 6632 CrossRef CAS PubMed.
- O. R. Thiel, J. R. Huckins, D. B. Brown, E. A. Bercot, J. T. Colyer, B. Riahi, R. R. Milburn, S. M. Shaw and J. Tomaskevitch, ACS Symp. Ser., 2014, 1181, 269 CrossRef CAS.
- The LOC is defined as the minimum partial pressure of oxygen that supports a combustible mixture. Below the LOC, combustion is not possible. P. M. Osterberg, J. K. Niemeier, C. J. Welch, J. M. Hawkins, J. R. Martinelli, T. E. Johnson, T. W. Root and S. S. Stahl, Org. Process Res. Dev., 2015, 19, 1537 CrossRef CAS PubMed.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, Oxford, 1998 Search PubMed.
- (a) R. K. Henderson, C. Jimenez-Gonzalez, D. J. C. Constable, S. R. Alston, G. G. A. Inglis, G. Fisher, J. Sherwood, S. P. Binks and A. D. Curzons, Green Chem., 2011, 13, 854 RSC; (b) D. Prat, O. Pardigon, H.-W. Flemming, S. Letestu, V. Ducandas, P. Isnard, E. Guntrum, T. Senac, S. Ruisseau, P. Cruciani and P. Hosek, Org. Process Res. Dev., 2013, 17, 1517 CrossRef CAS; (c) D. Prat, J. Hayler and A. Wells, Green Chem., 2014, 16, 4546 RSC.
- Another attractive feature of the reaction is that the NHPI and its decomposition products are readily separated from the reaction mixture through hydrolysis with aqueous NaOH.
- The condensation step was not optimized, but literature precedents suggest these reactions can proceed in near-quantitative yield under neat conditions (see ref. 5d). For E-factor analysis, see ESI.†.
- For leading references to continuous-flow aerobic oxidation chemistry, see ref. 6g and 14b, and the following: (a) X. Ye, M. D. Johnson, T. Diao, M. H. Yates and S. S. Stahl, Green Chem., 2010, 12, 1180 RSC; (b) H. P. L. Gemoets, V. Hessel and T. Noel, in Liquid Phase Aerobic Oxidation Catalysis: Industrial Applications and Academic Perspectives, ed. S. S. Stahl and P. L. Alsters, Wiley-VCH, Weinheim, 2016, pp. 399–417 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Screening data, experimental protocols, characterization data. See DOI: 10.1039/c6sc03831j |
| This journal is © The Royal Society of Chemistry 2017 |