
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Indirect to direct bandgap transition in methylammonium lead halide perovskite†
Tianyi
Wang‡
a,
Benjamin
Daiber‡
 a,
Jarvist M.
Frost
b,
Sander A.
Mann
a,
Erik C.
Garnett
a,
Aron
Walsh
b and
Bruno
Ehrler
*a
a,
Jarvist M.
Frost
b,
Sander A.
Mann
a,
Erik C.
Garnett
a,
Aron
Walsh
b and
Bruno
Ehrler
*a
aCenter for Nanophotonics, FOM Institute AMOLF, Science Park 104, 1098 XG Amsterdam, The Netherlands. E-mail: b.ehrler@amolf.nl
bDepartment of Materials, Imperial College London, London SW7 2AZ, UK
First published on 9th December 2016
Abstract
Methylammonium lead iodide perovskites are considered direct bandgap semiconductors. Here we show that in fact they present a weakly indirect bandgap 60 meV below the direct bandgap transition. This is a consequence of spin–orbit coupling resulting in Rashba-splitting of the conduction band. The indirect nature of the bandgap explains the apparent contradiction of strong absorption and long charge carrier lifetime. Under hydrostatic pressure from ambient to 325 MPa, Rashba splitting is reduced due to a pressure induced reduction in local electric field around the Pb atom. The nature of the bandgap becomes increasingly more direct, resulting in five times faster charge carrier recombination, and a doubling of the radiative efficiency. At hydrostatic pressures above 325 MPa, MAPI undergoes a reversible phase transition resulting in a purely direct bandgap semiconductor. The pressure-induced changes suggest epitaxial and synthetic routes to higher efficiency optoelectronic devices.
Broader contextSolar cells made from hybrid perovskite semiconductors have seen an unprecedented rise in efficiency over the past few years, now reaching efficiency values close to the best silicon solar cells. However, the understanding as to why these materials perform so efficiently is lacking behind the device progress. One of the great mysteries is the unusually long charge carrier lifetime. Conventional semiconductors show either strong absorption and short charge carrier lifetime (direct bandgap semiconductors), or weak absorption and long lifetimes (indirect bandgap). Hybrid perovskites seem to offer both strong absorption and long lifetime, which is very convenient for solar cell operation. Here we show that the origin of the unusually long lifetime lies in the peculiar bandstructure of methylammonium lead iodide (MAPI). An indirect transition, arising from a relativistic spin–orbit splitting of the lower conduction band, is present just below the direct bandgap of the perovskites. This allows strong light absorption via the direct transition, then the generated charges relax into the indirect band where they are protected from recombination. We also show that the indirect bandgap disappears at high pressure. Then the lifetime is reduced and the radiative efficiency is doubled which could open the route towards more efficient light emitting devices such as LEDs and lasers. |
Solar cells based on methylammonium lead iodide perovskites (MAPI) have seen an unprecedented increase in efficiency over a short period of time,1–4 while other applications including lasers and photodetectors have shown promise.5,6 The high efficiency of these applications arises due to low defect density7 with long charge carrier lifetime8 and diffusion length,9 in spite of the material being solution processable. To date, MAPI has widely been considered a direct bandgap semiconductor according to both theoretical calculations and experimental observations.10,11 However, the unusually long minority carrier lifetime with values more similar to those of indirect bandgap semiconductors,12 and the associated long charge carrier diffusion length has been a long-standing mystery in the field. It evoked explanations based on long-lived trapping of charges,13 large polarons,14 and triplet exciton formation.15 Recently, theoretical calculations predicted a slightly indirect bandgap in this material.16 Brivio et al. calculated the band structure of MAPI using quasiparticle self-consistent GW theory and found that a Rashba-splitting of the conduction band should generate this slightly indirect bandgap.17 The same relativistic effect has been reported in other calculations.18–20
Recent experimental indications point towards a Rashba-split band in MAPbBr3.21 However, there is no direct experimental evidence that supports the theoretical predictions of the bandstructure of the prototypical solar cell material MAPI, or the dramatic consequences for the charge carrier dynamics. The bandstructure of a semiconductor can be altered by structural changes under external application of pressure.22,23 Pressure has been applied to MAPI to understand the structural changes24–26 and the recombination dynamics.27 It was found that MAPI undergoes a phase transition at around 325 MPa. The phase at ambient pressures is tetragonal23,25 and the high pressure phase has been subject to debate, assigned to orthorhombic24,26,27 and cubic25 crystal phases. A further phase transition is known to occur in the GPa regime.24–26
Here we find that MAPI has an indirect bandgap 60 meV below the direct bandgap both in absorption and emission spectra. This indirect gap is responsible for the unusually long carrier lifetime because the thermalized carriers are protected against recombination via the fast direct transition. The indirect transition arises from Rashba splitting of the conduction band. The band is split due to the local electric field generated by the absence of inversion symmetry around the Pb site, which acts on the 6p orbitals of the lead-atom where most of the conduction band minimum is located.17,28
We study the optoelectronic changes of thin polycrystalline MAPI films under mild hydrostatic pressure up to 400 MPa, below and just above the phase transition at 325 MPa. We show that the bandgap changes with pressure and that the direct transition is enhanced. Above the phase transition MAPI behaves like a purely direct bandgap semiconductor. As the bandgap becomes more direct, the charge carrier lifetime decreases drastically with increasing pressure and the photoluminescence quantum yield (PLQY) exhibits a two-fold increase. These changes can be understood in terms of an increase in inversion symmetry around the Pb site due to a reduction in the in-phase ordering of the MA ions under pressure. These changes lead to a reduction in Rashba splitting, resulting in a more direct bandgap of MAPI. Our results show that small structural changes can significantly improve relevant optoelectronic properties of MAPI.
Results and discussion
We apply hydrostatic pressure to 400 nm thin polycrystalline films of MAPI using a pressure cell filled with an inert, mechanically pumped pressure liquid (see Experimental methods for details). The material is continuously compressed with a maximum strain of 3% at 400 MPa, as calculated from a Young's modulus of 12.8 GPa.29 This value is in good agreement with the change of lattice volume up to 400 MPa, derived from powder X-ray diffraction (PXRD) data.25 The absorption spectrum remains constant in shape, but the onset of absorption clearly changes with pressure (Fig. 1a, and Fig. S1 for complete set of absorption spectra, ESI†). We used the linear part of Tauc plots30 of the absorption edge to extract the (direct) bandgap (see Fig. 1b and Section S1 for details, ESI†). For the pressure upstroke, a continuous red-shift of the absorption edge by 30 meV is observed until 325 MPa. This unusual trend arises due to the electronic band structure associated with the corner sharing PbI3− network.31 While many semiconductors have negative bandgap deformation potentials (the bandgap increases with increasing pressure), hybrid lead halide perovskites have positive values as the band extrema are found at the boundary of the Brillouin zone (R point) that corresponds to out-of-phase interactions between neighboring unit cells.32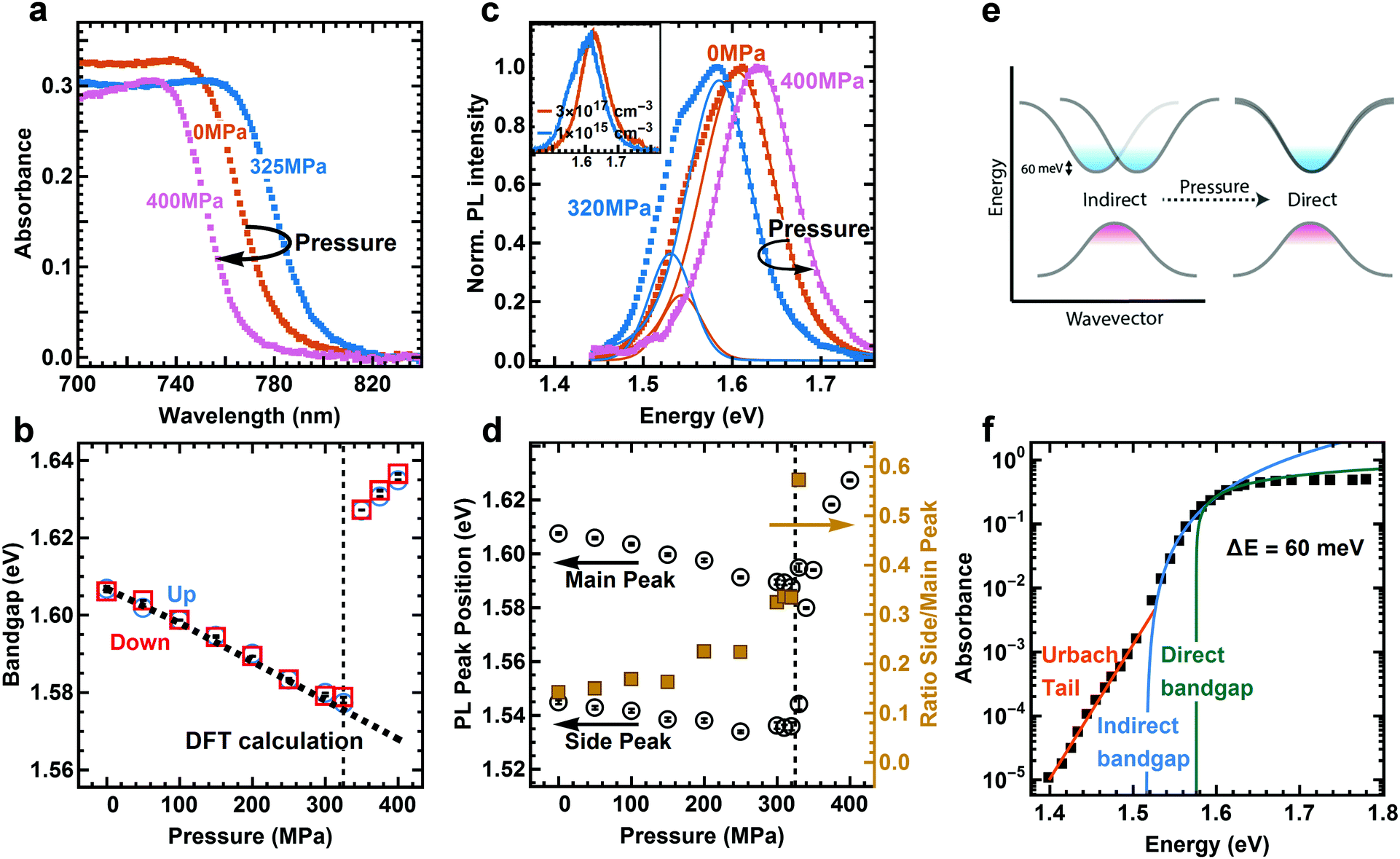 | ||
| Fig. 1 Absorbance and steady-state photoluminescence (PL) of MAPI at different pressures. (a) Absorbance spectra at three characteristic pressures. (b) Change of the direct bandgap under pressure for pressure up (blue circles) and down (red squares) stroke. DFT calculations (dashed line) predict the bandgap change before phase transition. Black bars inside symbols represent the error from the fit. (c) Photoluminescence (PL) spectra and fits at three different pressures. At pressures below phase transition (325 MPa) the PL fit requires two peaks while above the phase transition only one peak suffices. The inset shows PL spectra at different excitation densities under ambient pressure. (d) Position of main and side peak as extracted from fit to PL data. The ratio of side to main peak intensity increases with rising pressure. (e) Schematics of band structure, showing direct and indirect transitions and their change with pressure. (f) PDS data from literature43 fit with an exponential Urbach tail (orange) and indirect (blue) and direct (green) bandgap. | ||
An abrupt blue-shift occurs above 325 MPa which can be understood as a result of a phase transition from a tetragonal to an orthorhombic or cubic crystal structure.24–27 The phase transition is followed by a continuous blue-shift. Upon release of pressure from 400 MPa, i.e. the pressure downstroke, the change in bandgap is completely reversible, thus we can infer that the deformation of the crystal lattice is elastic and therefore the pressure-induced structural changes, including the phase transition, are reversible. The change in bandgap is in good agreement with the result of density functional theory (DFT) calculations before the phase transition (dashed line in Fig. 1b, see Experimental methods for details). Atomistic modeling was not attempted on the high-pressure structure that emerges above 325 MPa, so the secondary blue-shift is not recovered in the calculations.
The steady-state photoluminescence (PL) spectra of MAPI reveal peculiar behavior under pressure (see Fig. 1c, Fig. S2 for complete set of PL spectra, and Section S2 for details, ESI†). We find that the PL spectrum is composed of two distinct peaks before the phase transition, and that one peak is sufficient to fit the data after the phase transition. At 0 MPa, the dominating emission peak is located at around 1.61 eV, while a side emission peak is located at around 1.55 eV. The side peak disappears at high excitation densities (as shown in the inset of Fig. 1c). Many reports at high excitation densities hence do not resolve the side peak,15,33,34 and in reports at low excitation density the asymmetric PL spectrum is often ignored.27,35–38 The main peak has been assigned to band-to-band recombination and speculations about the origin of the side peak include phonon-assisted recombination34 and exciton–phonon interaction.39 A PL side peak has also been observed in the low-temperature orthorhombic phase of MAPI. However, that side peak has been assigned to trapped40,41 or bound5,15 charge-carrier pairs, which, as we show, is different compared to the side peak at room temperature. With increasing pressure up to 325 MPa, the main peak exhibits a red-shift by 10 meV and side peak exhibit a red-shift by 20 meV (Fig. 1d). The main peak shows an abrupt blue-shift at the phase transition followed up by a continuous blue-shift up to 400 MPa, which is consistent with the shift of absorption edge. Following previous work27 and from the PL peak position40 we assign the high-pressure phase to the orthorhombic phase. In the following we will show that the side peak in the low pressure phase arises from an indirect transition 60 meV below the direct bandgap.
Recent theoretical studies have predicted that a Rashba splitting of the conduction band is present for MAPI, leading to a slightly indirect transition 50 to 75 meV below the direct bandgap28 (Fig. 1e). This is due to strong spin–orbit coupling (SOC) in the MAPI crystal. The conduction band minimum in MAPI is mainly formed by the lead (Z = 82) 6p orbitals, while the valence band maximum is mainly formed by iodine (Z = 53) 5p orbitals. As the spin-splitting scales approximately with Z2, the valence band is less affected than the conduction band. Therefore, although a small splitting of the valence band is observed in the calculations, it is negligible compared to the large splitting in conduction band. The flattened valence band provides a large density of states that enables strong direct absorption. However, when MAPI is excited at low intensities, mainly the bottom of the conduction band will be filled, which means that (slow) indirect transitions will play a significant role.
In our PL spectra, the energy of the side peak is 60 meV below the main peak, which fits the theoretical prediction (the energy of the phonon required is small in comparison, see Section S3 for details, ESI†). The ratio between the area of side and main peak is increasing with increasing pressure up to 325 MPa (Fig. 1d). At the same time the difference in energy between the peak positions is reduced, from 60 meV to around 50 meV at 300 MPa (Fig. S3, ESI†). After the phase transition at 325 MPa only one PL peak is present. These phenomena can be understood as a result of changes in the ordering of MA ions. In-phase ordering of MA ions increases Rashba-splitting, while out-of-phase ordering decreases the splitting. The ordering of MA ions decreases at low pressure before the phase transition to decrease the occupied volume. The ambient tetragonal phase features in-phase ordering of the MA ions,42 hence the decrease in order at low pressure reduces the splitting. After the phase transition MAPI is in the orthorhombic phase, which features out-of-phase order,42 and the splitting vanishes and MAPI becomes a purely direct bandgap semiconductor.
The indirect bandgap is also apparent in the absorption spectrum below the direct bandgap. We find that data gathered from photothermal deflection spectroscopy (PDS, data taken from literature43–45) can only be fitted with both a direct bandgap and an indirect bandgap between 46 and 60 meV below the direct gap, in addition to the Urbach tail (one example in Fig. 1f, see Section S4 for details and Fig. S4–S6 for additional spectra, ESI†). Combining only a direct bandgap and the Urbach tail (dashed line in Fig. S4–S6, ESI†) yields a very poor fit. We note that the Tauc analysis is only valid in the absence of significant excitonic contribution to the absorption spectrum in the relevant region.46 An excitonic and a continuum contribution following the Elliott formula can also yield a reasonable fit to the absorption spectrum, though with much larger residuals in the region where we expect the indirect bandgap (see Section S5, ESI†). This indicates that accurately describing the below-bandgap absorption spectrum does require an indirect bandgap. Additionally, the dramatic changes we observe under pressure cannot be explained by an excitonic contribution alone, without invoking the indirect bandgap, since the change in excitonic contribution to the absorption spectrum under pressure is small (see below), in stark contrast to the strong changes in the optoelectronic properties.
We expect the transition towards a more direct bandgap under pressure to result in a shorter excited state lifetime. We use time-correlated single photon counting (TCSPC) to probe the excited state lifetime at different pressures. The sample is excited with a red laser at 640 nm (12 nJ cm−2), at a low initial charge carrier density of around 6 × 1014 cm−3 (Fig. 2a, see Section S6 for details, ESI†). This mild excitation density reduces the filling of the conduction band and thus enhances the contribution seen from the indirect transition. At a charge carrier density above 1017 cm−3, where bimolecular and monomolecular decay rates increase at higher pressure (Fig. 2b). We note that this is contrary to the trend recently measured for MAPI powders.27 To determine how well bimolecular recombination competes with monomolecular recombination we calculate the fraction of charge carriers that decayed via the bimolecular (radiative) and monomolecular (non-radiative) path (Fig. 2c) (see Section S6 for details of calculation, ESI†). The internal radiative efficiency increases from 20% at ambient pressure to 40% at 325 MPa and remains at around 40% from 325 to 400 MPa (Fig. 2c). The increasing fraction of radiatively decaying charge carriers and hence increase in PLQY is a result of an increasingly direct nature of the bandgap. The cross-section for vertical electron–hole recombination is increased as Rashba splitting is decreased. This model is consistent with the strong increase in PLQY with increasing excitation density47,48 as higher excitation density saturates the indirect transition and enhances the direct transition (numerically shown in literature28). Similar effects can also be seen in the low-temperature orthorhombic phase of MAPI where an ordered anti-ferroelectric structure is formed that suppresses internal electric fields.35 We therefore propose that the enhancement of PLQY at high pressure has the same microscopic origin as the recently reported low temperature optoelectronic behavior.
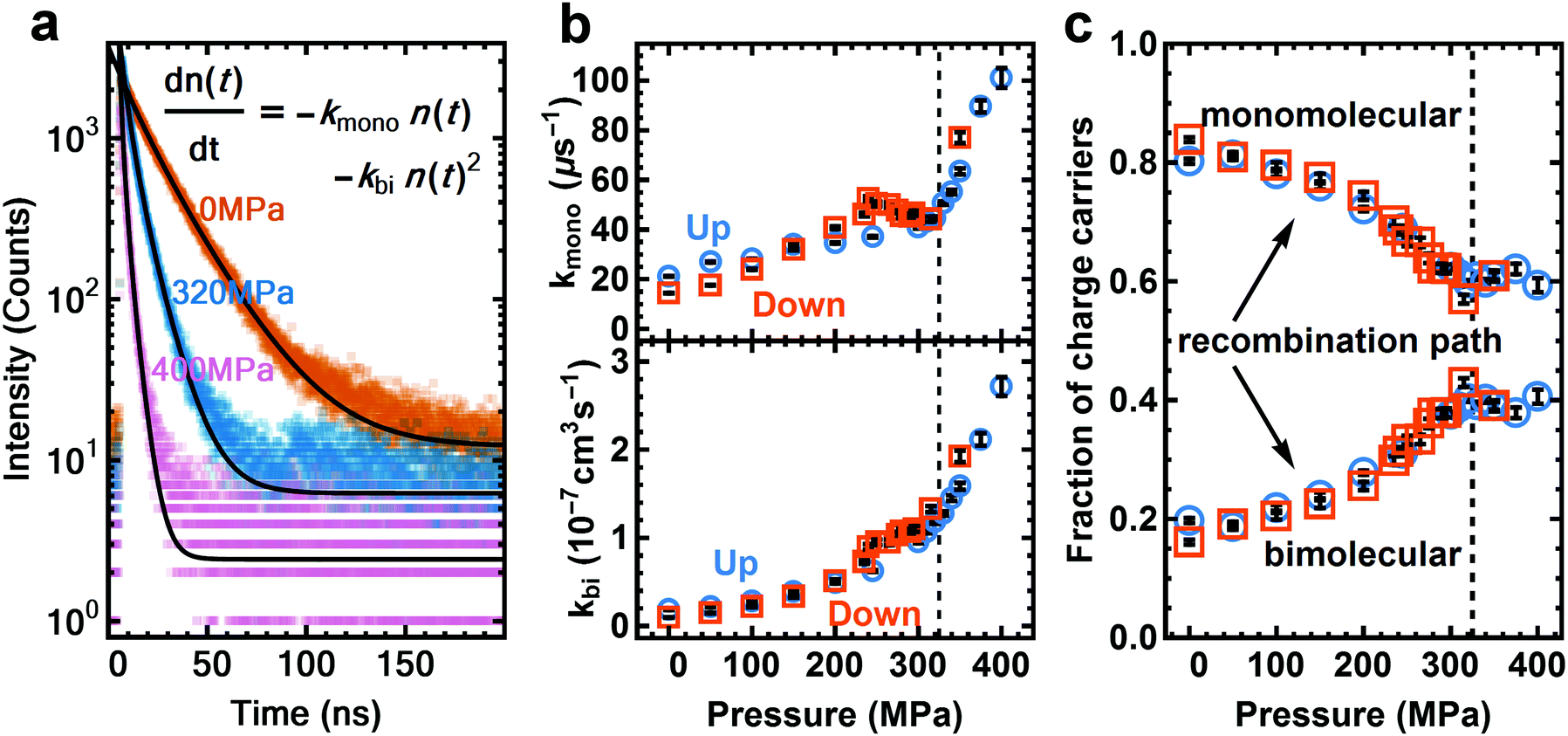 | ||
| Fig. 2 Time-Correlated Single Photon Counting (TCSPC) data of the PL of a MAPI film under pressure, excited with a 640 nm laser. (a) TCSPC data of 0 MPa, 320 MPa and 400 MPa pressure. The black line shows the fit from the model shown in the inset. (b) Monomolecular (top) and bimolecular (bottom) decay rates of the charge carriers, extracted from the fit. The bimolecular recombination rate (bottom) changes faster with pressure than the monomolecular recombination rate, with both changing slope after the phase transition at 325 MPa (dashed line). Black bars inside symbols represent the error from the fit. (c) Fraction of charge carriers that decayed through the monomolecular channel and bimolecular channel. | ||
The PL side-peak and the slow recombination have been associated with trap states40,41 and excitonic effects,5,15 and cation reorientation could also affect the PL spectrum.16 In the following we discuss our reasoning for assigning the effects observed under pressure to the indirect bandgap instead.
We can exclude trap states as a major factor that causes the effects under pressure as trap states cannot account for the simultaneous increase in the PLQY and in the side peak contribution of the PL spectra. If the trap states were to become more relevant under pressure one would expect the PLQY to decrease instead. To confirm that the charge carrier dynamics are not dominated by trap states, we measure the external PLQY of our films (see Section S7, ESI†), which is comparable to values measured for films showing very high solar cell performance.36 We also compare the PL spectrum of samples made by different processing methods, and compare several samples made by the same method. We observe the same PL side peak in all samples, with differences in the magnitude of the effect, and a slight difference in the energy difference between main and side peak (see Section S8, ESI†). These differences might arise from different doping levels induced by different preparation methods. We also observe the same behavior under pressure for a sample prepared by a different method (see Section S8, ESI†). It is unlikely that the same photoluminescence behaviour appears for such different samples if it came from deposition-induced traps or phase separation.
MA cation reorientation following excitation could also lead to a lower-energy state with a long lifetime. However, we think that it does not explain our dataset. For example, the absorption spectrum should show no significant features of such a low-energy transition, because the cation re-orientation only happens after charge generation. Also, the PL spectrum shows that the majority of the carriers decays via the high-energy transition, which can be easily explained by the Rashba-split conduction band, but would be very unlikely if the charge carrier protection would arise through the re-orientation of the cations. In that case the emission would come from the low-energy transition, with the exception of the very early-time PL, before the reorientation.
It is conceivable that the exciton in perovskites also stabilizes the excited state. We calculate the change of exciton binding energy over different pressures (see Section S9 and Fig. S8 for details, ESI†). Our data is consistent with no change in exciton binding energy over the entire pressure range, even across the phase transition, with an upper bound of a relative change of 26%. Such mild changes are unlikely to result in a 5-fold decrease in charge carrier lifetime and a 2-fold increase in PLQY.
Conclusions
In conclusion, our results show that the prototype hybrid perovskite MAPI has an indirect bandgap. This arises due to distortion of the lead iodide framework, which leads to an electric field across the Pb atom, splitting the conduction band (Rashba splitting). The position of the indirect transition 60 meV below the direct transition is consistent with an absorption spectrum that almost resembles a direct bandgap semiconductor. At the same time, charges thermalized in the conduction band are protected from recombination because recombination requires a change in crystal momentum. Hence, the slightly indirect bandgap explains the apparent contradiction between the long charge carrier lifetime and diffusion length, and the efficient absorption of perovskite semiconductors. Under pressure, the electric field across the Pb atom is reduced, increasing the relative strength of the direct transition, which reduces the charge carrier lifetime and increases the radiative efficiency. This tunability opens the path towards new, rational material and structural developments of perovskite semiconductors with engineered band structure. These materials could be even better suited for solar cells with a higher photovoltage, highly efficient LEDs, and lasers with low lasing threshold.Experimental methods
Methylammonium lead iodide (MAPI) thin films were prepared via solution-based methods, including spincoating, spincoating in combination with antisolvent precipitation and a dipping method. All chemicals were purchased from Sigma-Aldrich and used as received. Fused silica glass substrates were sonicated with detergent Micro 90, deionized water, acetone, and isopropanol sequentially for 10 min and oxygen plasma treated at 100 W for 15 min.For the data shown in the main text we use spincoating in combination with antisolvent precipitation following ref. 49. 1.2 g of PbI2 was first mixed with 2 mL of anhydrous N,N-dimethylformamide (DMF) and heated to 100 °C while stirring. After PbI2 was fully dissolved, 0.4 g of methylammonium iodide (MAI) was added to the solution. The solution was kept stirring at 100 °C until MAI was completely dissolved. For the formation of thin films, 100 μL of solution was deposited onto the glass substrates by spin-coating at 5000 rpm for 20 seconds. After 5 seconds of spinning, 100 μL of chlorobenzene was placed on the sample for the anti-solvent treatment. The sample was then transferred to a hotplate and annealed at 100 °C for 10 min. A dark brown film was obtained. A SEM image of the film can be found in Fig. S9 (ESI†).
A comparison of the experiments on films prepared by alternative deposition methods is presented in the Section S8 (ESI†).
Hydrostatic pressure was generated through a pressurizing liquid Fluorinert FC-72 (3M) inside a high pressure cell (ISS Inc.) using a manual pump. The pressure was first applied from ambient pressure to 400 MPa (pressure upstroke) and then down from 400 MPa to ambient pressure (pressure downstroke), both in steps of 25 MPa. A 5–7 min waiting step after application of pressure and before the measurement was chosen to allow the material to equilibrate under pressure. The pressure liquid FC-72 started to scatter a fraction of light (∼5%) from 300 MPa onwards (flat spectral response, see inset Fig. S10, ESI†), which we corrected for in the absorbance spectra (see below). The PL spectra and lifetimes were unaffected. From the measurement-to-measurement variability we estimate the upper limit on the error of the pressure reading (following the 5–7 min waiting step) to be 20 MPa.
Absorbance spectra of MAPI thin films were measured with a LAMBDA 750 UV/Vis/NIR Spectrophotometer (Perkin Elmer) from 550 nm to 850 nm. A correction of the absorption spectra was done for all the spectra obtained above 300 MPa by subtracting the background signal from scattering in the liquid. The background was obtained from a fit in the region of 820 nm to 850 nm where MAPI does not absorb.
Steady-state photoluminescence (PL) was measured with a home-built setup equipped with a 640 nm continuous-wave laser as source of excitation (PicoQuant LDH-D-C-640) at a power output of 0.7 mW. The PL spectra were collected at an angle of 45°. Two Thorlabs FEL-700 highpass filters were used to remove the excitation laser from the signal. The PL was coupled in a fiber connected to a OceanOptics USB4000 spectrometer, set to an integration time of 2000 ms for each measurement. The PL spectra were fitted with either one or two Voigt profiles (see Section S2, ESI†).
Time-correlated single photon counting (TCSPC) measurements were performed with a home-built setup equipped with PicoQuant PDL 828 “Sepia II” and a PicoQuant HydraHarp 400 multichannel picosecond event timer and TCSPC module. A 640 nm pulsed laser (PicoQuant LDH-D-C-640) with a repetition rate of 5 MHz was used as source of excitation and a single-photon avalanche diode (SPAD) detector (Micro Photon Devices, MPD-5CTD) was used for the detection of photoemission. A Thorlabs FEL-700 long-pass filter was used to remove the excitation laser. The TCSPC data was collected over the course of 60 seconds per measurement.
The photoluminescence quantum yield (PLQY) measurement was performed with a home-built setup. A supercontinuum laser (Fianium WL-SC-390-3) was fiber coupled into an acousto-optical tunable filter (AOTF), which has a visible output for wavelengths 400–750 nm. A Mitutoyo objective focused the light on the sample inside the integrating sphere. Light inside the integrating sphere is monitored by the integrating sphere photodetector. The incident intensity was controlled by the AOTF RF power. Thorlabs PDA100 calibrated photodiodes were used as photodetectors. The acousto-optical tunable filter (AOTF) output was modulated with a 50 percent duty cycle at 3 kHz. The absorption and photoluminescence measurements were done with Thorlabs FELH0650 and FESH0650 long pass and short pass filters, and the excitation wavelength was 600 nm.
First-principles materials modelling was employed to assess the bulk response of the electronic structure to external pressure. Calculations were performed within the Kohn–Sham Density Functional Theory (DFT) formalism as implemented in VASP.50 Projector-augmented-wave core potentials were combined with a kinetic energy cutoff of 600 eV for the plane wave basis set describing the valence electrons including spin–orbit coupling. A scalar-relativistic description of Pb[Xe] was used with the 5d semi-core electrons included as valence. The phonon-stable crystal structures reported previously51 were taken as the starting point for a series of isobaric calculations with a stress tensor ranging from 0 to 500 MPa. A positive bandgap deformation was found (bandgap decrease with increasing pressure) in agreement with initial calculations.31 No attempt was made to model the phase change above 325 MPa, which would require sampling changes in spatial and temporal structural disorder.
Corroborating work on the Rashba splitting of MAPI38 was recently reported by Hutter et al. during review of this paper.
Acknowledgements
This work is part of the research program of the Foundation for Fundamental Research on Matter (FOM), which is part of The Netherlands Organization for Scientific Research (NWO). The work at ICL was supported by the EPSRC (grant no. EP/K016288/1 and EP/M009580/1) and the ERC (grant no. 277757). The authors thank Sarah Brittman for comments on the manuscript. T. W. thanks Dr. Michiel Petrus, Henk-Jan Boluijt, Ricardo Struik, Marc Duursma and mechanical workshop at AMOLF for help with the experiments. B. D. thanks Gede Adhyaksa for help with sample preparation.Notes and references
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- L. Etgar, P. Gao, Z. S. Xue, Q. Peng, A. K. Chandiran, B. Liu, M. K. Nazeeruddin and M. Gratzel, J. Am. Chem. Soc., 2012, 134, 17396–17399 CrossRef CAS PubMed.
- H. S. Kim, J. W. Lee, N. Yantara, P. P. Boix, S. A. Kulkarni, S. Mhaisalkar, M. Gratzel and N. G. Park, Nano Lett., 2013, 13, 2412–2417 CrossRef CAS PubMed.
- J. H. Im, I. H. Jang, N. Pellet, M. Gratzel and N. G. Park, Nat. Nanotechnol., 2014, 9, 927–932 CrossRef CAS PubMed.
- G. C. Xing, N. Mathews, S. S. Lim, N. Yantara, X. F. Liu, D. Sabba, M. Gratzel, S. Mhaisalkar and T. C. Sum, Nat. Mater., 2014, 13, 476–480 CrossRef CAS PubMed.
- S. Yakunin, M. Sytnyk, D. Kriegner, S. Shrestha, M. Richter, G. J. Matt, H. Azimi, C. J. Brabec, J. Stangl, M. V. Kovalenko and W. Heiss, Nat. Photonics, 2015, 9, 444–U444 CrossRef CAS.
- D. Shi, V. Adinolfi, R. Comin, M. J. Yuan, E. Alarousu, A. Buin, Y. Chen, S. Hoogland, A. Rothenberger, K. Katsiev, Y. Losovyj, X. Zhang, P. A. Dowben, O. F. Mohammed, E. H. Sargent and O. M. Bakr, Science, 2015, 347, 519–522 CrossRef CAS PubMed.
- Y. Bi, E. M. Hutter, Y. J. Fang, Q. F. Dong, J. S. Huang and T. J. Savenije, J. Phys. Chem. Lett., 2016, 7, 923–928 CrossRef CAS PubMed.
- Q. F. Dong, Y. J. Fang, Y. C. Shao, P. Mulligan, J. Qiu, L. Cao and J. S. Huang, Science, 2015, 347, 967–970 CrossRef CAS PubMed.
- H. S. Kim, C. R. Lee, J. H. Im, K. B. Lee, T. Moehl, A. Marchioro, S. J. Moon, R. Humphry-Baker, J. H. Yum, J. E. Moser, M. Gratzel and N. G. Park, Sci. Rep., 2012, 2, 591 Search PubMed.
- T. Baikie, Y. N. Fang, J. M. Kadro, M. Schreyer, F. X. Wei, S. G. Mhaisalkar, M. Graetzel and T. J. White, J. Mater. Chem. A, 2013, 1, 5628–5641 CAS.
- A. Cuevas and D. Macdonald, Sol. Energy, 2004, 76, 255–262 CrossRef CAS.
- D. W. deQuilettes, S. M. Vorpahl, S. D. Stranks, H. Nagaoka, G. E. Eperon, M. E. Ziffer, H. J. Snaith and D. S. Ginger, Science, 2015, 348, 683–686 CrossRef CAS PubMed.
- X. Y. Zhu and V. Podzorov, J. Phys. Chem. Lett., 2015, 6, 4758–4761 CrossRef CAS PubMed.
- H. H. Fang, R. Raissa, M. Abdu-Aguye, S. Adjokatse, G. R. Blake, J. Even and M. A. Loi, Adv. Funct. Mater., 2015, 25, 2378–2385 CrossRef CAS.
- C. Motta, F. El-Mellouhi, S. Kais, N. Tabet, F. Alharbi and S. Sanvito, Nat. Commun., 2015, 6, 7026 CrossRef CAS PubMed.
- F. Brivio, K. T. Butler, A. Walsh and M. van Schilfgaarde, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 155204 CrossRef.
- T. Etienne, E. Mosconi and F. De Angelis, J. Phys. Chem. Lett., 2016, 7, 1638–1645 CrossRef CAS PubMed.
- M. Kepenekian, R. Robles, C. Katan, D. Sapori, L. Pedesseau and J. Even, ACS Nano, 2015, 9, 11557–11567 CrossRef CAS PubMed.
- F. Zheng, L. Z. Tan, S. Liu and A. M. Rappe, Nano Lett., 2015, 15, 7794–7800 CrossRef CAS PubMed.
- D. Niesner, M. Wilhelm, I. Levchuk, A. Osvet, S. Shrestha, M. Batentschuk, C. Brabec and T. Fauster, Phys. Rev. Lett., 2016, 117, 126401 CrossRef PubMed.
- F. J. Manjon, A. Segura, V. Munoz-Sanjose, G. Tobias, P. Ordejon and E. Canadell, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 125201 CrossRef.
- D. Errandonea, E. Bandiello, A. Segura, J. J. Hamlin, M. B. Maple, P. Rodriguez-Hernandez and A. Munoz, J. Alloys Compd., 2014, 587, 14–20 CrossRef CAS.
- T. J. Ou, J. J. Yan, C. H. Xiao, W. S. Shen, C. L. Liu, X. Z. Liu, Y. H. Han, Y. Z. Ma and C. X. Gao, Nanoscale, 2016, 8, 11426–11431 RSC.
- A. Jaffe, Y. Lin, C. M. Beavers, J. Voss, W. L. Mao and H. I. Karunadasa, ACS Cent. Sci., 2016, 2, 201–209 CrossRef CAS PubMed.
- S. J. Jiang, Y. A. Fang, R. P. Li, H. Xiao, J. Crowley, C. Y. Wang, T. J. White, W. A. Goddard, Z. W. Wang, T. Baikie and J. Y. Fang, Angew. Chem., Int. Ed., 2016, 55, 6540–6544 CrossRef CAS PubMed.
- L. Kong, G. Liu, J. Gong, Q. Hu, R. D. Schaller, P. Dera, D. Zhang, Z. Liu, W. Yang, K. Zhu, Y. Tang, C. Wang, S.-H. Wei, T. Xu and H.-k. Mao, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 8910–8915 CrossRef CAS PubMed.
- P. Azarhoosh, S. McKechnie, J. M. Frost, A. Walsh and M. van Schilfgaarde, APL Mater., 2016, 4, 091501 CrossRef.
- J. Feng, APL Mater., 2014, 2, 081801 CrossRef.
- J. Tauc, Mater. Res. Bull., 1968, 3, 37–46 CrossRef CAS.
- S. H. Wei and A. Zunger, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 13605–13610 CrossRef CAS.
- J. M. Frost, K. T. Butler, F. Brivio, C. H. Hendon, M. van Schilfgaarde and A. Walsh, Nano Lett., 2014, 14, 2584–2590 CrossRef CAS PubMed.
- R. L. Milot, G. E. Eperon, H. J. Snaith, M. B. Johnston and L. M. Herz, Adv. Funct. Mater., 2015, 25, 6218–6227 CrossRef CAS.
- K. W. Wu, A. Bera, C. Ma, Y. M. Du, Y. Yang, L. Li and T. Wu, Phys. Chem. Chem. Phys., 2014, 16, 22476–22481 RSC.
- G. Grancini, A. R. S. Kandada, J. M. Frost, A. J. Barker, M. De Bastiani, M. Gandini, S. Marras, G. Lanzani, A. Walsh and A. Petrozza, Nat. Photonics, 2015, 9, 695–701 CrossRef CAS PubMed.
- J. T. W. Wang, Z. P. Wang, S. Pathak, W. Zhang, D. W. deQuilettes, F. Wisnivesky-Rocca-Rivarola, J. Huang, P. K. Nayak, J. B. Patel, H. A. M. Yusof, Y. Vaynzof, R. Zhu, I. Ramirez, J. Zhang, C. Ducati, C. Grovenor, M. B. Johnston, D. S. Ginger, R. J. Nicholas and H. J. Snaith, Energy Environ. Sci., 2016, 9, 2892–2901 CAS.
- D. H. Li, G. M. Wang, H. C. Cheng, C. Y. Chen, H. Wu, Y. Liu, Y. Huang and X. F. Duan, Nat. Commun., 2016, 7, 11330 CrossRef CAS PubMed.
- E. M. Hutter, M. C. Gelvez-Rueda, A. Osherov, V. Bulovic, F. C. Grozema, S. D. Stranks and T. J. Savenije, Nat. Mater., 2016 DOI:10.1038/nmat4765.
- V. D'Innocenzo, G. Grancini, M. J. P. Alcocer, A. R. S. Kandada, S. D. Stranks, M. M. Lee, G. Lanzani, H. J. Snaith and A. Petrozza, Nat. Commun., 2014, 5, 3586 Search PubMed.
- C. Wehrenfennig, M. Z. Liu, H. J. Snaith, M. B. Johnston and L. M. Herz, APL Mater., 2014, 2, 081513 CrossRef.
- X. X. Wu, M. T. Trinh, D. Niesner, H. M. Zhu, Z. Norman, J. S. Owen, O. Yaffe, B. J. Kudisch and X. Y. Zhu, J. Am. Chem. Soc., 2015, 137, 2089–2096 CrossRef CAS PubMed.
- M. T. Weller, O. J. Weber, P. F. Henry, A. M. Di Pumpo and T. C. Hansen, Chem. Commun., 2015, 51, 4180–4183 RSC.
- A. Sadhanala, F. Deschler, T. H. Thomas, S. E. Dutton, K. C. Goedel, F. C. Hanusch, M. L. Lai, U. Steiner, T. Bein, P. Docampo, D. Cahen and R. H. Friend, J. Phys. Chem. Lett., 2014, 5, 2501–2505 CrossRef CAS PubMed.
- W. Zhang, M. Saliba, D. T. Moore, S. K. Pathak, M. T. Hörantner, T. Stergiopoulos, S. D. Stranks, G. E. Eperon, J. A. Alexander-Webber, A. Abate, A. Sadhanala, S. Yao, Y. Chen, R. H. Friend, L. A. Estroff, U. Wiesner and H. J. Snaith, Nat. Commun., 2015, 6, 6142 CrossRef CAS PubMed.
- S. De Wolf, J. Holovsky, S.-J. Moon, P. Löper, B. Niesen, M. Ledinsky, F.-J. Haug, J.-H. Yum and C. Ballif, J. Phys. Lett., 2014, 5, 1035–1039 CAS.
- M. A. Green, Y. J. Jiang, A. M. Soufiani and A. Ho-Baillie, J. Phys. Chem. Lett., 2015, 6, 4774–4785 CrossRef CAS PubMed.
- M. Saba, M. Cadelano, D. Marongiu, F. P. Chen, V. Sarritzu, N. Sestu, C. Figus, M. Aresti, R. Piras, A. G. Lehmann, C. Cannas, A. Musinu, F. Quochi, A. Mura and G. Bongiovanni, Nat. Commun., 2014, 5, 5049 CrossRef CAS PubMed.
- F. Deschler, M. Price, S. Pathak, L. E. Klintberg, D. D. Jarausch, R. Higler, S. Huttner, T. Leijtens, S. D. Stranks, H. J. Snaith, M. Atature, R. T. Phillips and R. H. Friend, J. Phys. Chem. Lett., 2014, 5, 1421–1426 CrossRef CAS PubMed.
- M. L. Petrus, T. Bein, T. J. Dingemans and P. Docampo, J. Mater. Chem. A, 2015, 3, 16874 CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- F. Brivio, J. M. Frost, J. M. Skelton, A. J. Jackson, O. J. Weber, M. T. Weller, A. R. Goni, A. M. A. Leguy, P. R. F. Barnes and A. Walsh, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 144308 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ee03474h |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2017 |